
Enzyme-mimicking of copper-sites in metal–organic frameworks for oxidative degradation of phenolic compounds†
Ainara
Valverde‡
ab,
Eneko
Alkain‡
c,
Natalia Ahiova
Rio-López
a,
Luis
Lezama
d,
Arkaitz
Fidalgo-Marijuan
 ad,
José Manuel
Laza
b,
Stefan
Wuttke
*ae,
José María
Porro
ae,
Itziar
Oyarzabal
ae,
Mónica
Jiménez-Ruiz
f,
Victoria
García Sakai
g,
Pedro Luis
Arias
c,
Iker
Agirrezabal-Telleria
c and
Roberto
Fernández de Luis
*a
ad,
José Manuel
Laza
b,
Stefan
Wuttke
*ae,
José María
Porro
ae,
Itziar
Oyarzabal
ae,
Mónica
Jiménez-Ruiz
f,
Victoria
García Sakai
g,
Pedro Luis
Arias
c,
Iker
Agirrezabal-Telleria
c and
Roberto
Fernández de Luis
*a
aBCMaterials, Basque Center for Materials, Applications and Nanostructures, UPV/EHU Science Park, 48940 Leioa, Spain. E-mail: roberto.fernandez@bcmaterials.net; stefan.wuttke@bcmaterials.net
bDepartment of Physical Chemistry, Faculty of Science and Technology, Macromolecular Chemistry Group (LABQUIMAC), University of the Basque Country (UPV/EHU), Barrio Sarriena s/n 48940 Leioa, Spain
cDepartment of Chemical and Environmental Engineering, School of Engineering, University of the Basque Country (UPV/EHU), 48013 Bilbao, Spain
dDepartment of Inorganic and Organic Chemistry. Faculty of Science and Technology, University of the Basque Country (UPV/EHU), Barrio Sarriena s/n 48940 Leioa, Spain
eIKERBASQUE, Basque Foundation for Science, 48009 Bilbao, Spain
fInstitut Laue Langevin, 71 Avenue des Martyrs, CS 20156, 38042 Grenoble, France
gISIS Neutron and Muon Facility, Science & Technology Facilities Council, Rutherford Appleton Laboratory, Didcot, UK
First published on 9th January 2024
Abstract
Two on the main factors that control the activity and selectivity of the metal sites within metalloenzymes are: (i) their coordination environments, and the (ii) number and connectivity of the metal ions in the active site (i.e. nuclearity). This is the archetypal example of copper metalloproteins, as specifically of laccases and copper oxidases. Here we show that metal–organic frameworks can be used to install amino acid-copper sites with a partial control over their coordination environment and nuclearity of the final site. The activity of our bioinspired MOF-808@(amino)acid-copper catalysts have been assessed over the wet oxidation of phenolic pollutants. Our results demonstrate a clear modulation of the catalytic efficiency and selectivity of the copper-sites controlled by the coordination-sphere and their clustering degree. We anticipate that the approach presented in this work can be the starting point for more sophisticated reconstruction of enzyme active sites with regioselective activities.
1. Introduction
Nature, through millions of years of evolution, has found sophisticated ways to coordinate metal ions in precise and unconventional ways in order to reach outstandingly effective and selective biocatalysts.1–3 Through the sequential assembly of simple repetitive units, biology has built up complex three-dimensional scaffolds of metalloproteins, which deposit functional amino acid residues able to coordinate the metal-site in specific spatial configurations.4 The chemical arrangement of the amino acid residues that conforms the first and second coordination environment of the metal-sites plays a key role in defining the activity and selectivity of metalloenzymes.5,6 Thus, a mutation or alteration of any of the amino acid residues that coordinate the metal placed into the heart of the metalloenzyme, affects drastically their efficiency, selectivity and function. In fact, metalloenzymes are artificially modified by surgically altering the amino acid sequence that controls metal coordination environment.7,8In this respect, an important example is the copper metalloenzymes, where the coordination of copper ions is usually completed by histidine, cysteine and carboxyl residues, or a combination of these.9–12 Depending on the copper-biocatalyst, CuII/I ions are stabilized as isolated and/or clustered sites able to carry out oxidative catalytic reactions based on the activation of oxygen (e.g. laccases13–15) or hydrogen peroxide (e.g. peroxidase) molecules.16,17 The overall selectivity and oxidoreductive efficiency of copper-enzymes over a specific substrate is partially defined by the coordination modes of the copper ions.18–23 Furthermore, the oxidative capacity of copper bio-catalysts has important technological implications in water remediation, since it can be adapted to degrade highly concerning and persistent phenolic compounds via Catalytic Wet Peroxide Oxidation (CWPO).24–26 CWPO is a Fenton process able to drive the degradation of phenolic compounds via the activation of hydrogen peroxide to hydroxyl radicals27 with the participation of a catalyst.24,28,29
Extensive research is being carried out to replicate and expand the metalloenzymes' pockets and catalytic activity to robust porous materials.30–32 In this regard, the chemical encoding of a particular class of porous ordered materials called Metal–Organic Frameworks (MOFs) holds an enormous promise.32–39 MOFs are crystalline solids built from metal ions or clusters that are connected by organic linkers to form extended, ordered, and highly porous networks.40–43 Thanks to their (i) impressive and ordered porosity metrics (i.e. record surface area, tuneable pore volume/window…), and (ii) their versatility to be encoded with functionalities placed surgically at specific positions of their frameworks,44–50 MOFs have been successfully employed as matrixes to install amino acids51–53 and peptides.54–57 Once the pore space has been decorated with specific amino acid moieties, metal-sites can be easily installed by adsorbing them from aqueous or non-aqueous solutions.58–61 The functionality of these copper sites have been proved for varied reactions,62 as the low temperature methane to methanol oxidation,62,63 or the peroxidase oxidative degradation of phenols by copper doped UiO-67-Bpy.64,65 In addition, MOFs directly assembled with copper ions, as copper-trimesate66 or copper-benzene dicarboxylate,67 also exhibit outstanding oxidative functions to degrade phenols, but their hydrolytic instability may prevent their application in water remediation processes.
In this work, we have decorated the mesopore space of the MOF-808 with amino acid and carboxylic acid molecules, to later, stabilize coppers ions with varied clustering degrees and coordination modes in order to unravel the impact of the metal-sites characteristics on the catalytic wet oxidation (CWPO) of phenolic compounds. The features of the copper-sites installed within the framework have been unravelled by a combination of infrared (IR), Raman, X-ray photoelectron (XPS), electron paramagnetic (EPR) and inelastic neutron scattering (INS) spectroscopies. Local structural models for the copper sites have been proposed after an crystallochemical analysis of the copper coordination modes with (amino)acid molecules found in the Cambridge Structural Database68 The efficiency and selectivity of our system to oxidize phenolic pollutants (i.e. phenol, 2,4-dichlorophenol, catechol, hydroquinone or dopamine) through CWPO have been duly assessed, confirming that both the coordination modes and the clustering degree of the copper sites modulate its efficiency and selectivity to oxidize phenols through the hydrogen peroxide activation. The results presented in this work represent a milestone for the installation and artificial evolution of metal-amino catalytic sites into the robust architectural backbone of MOFs, which successfully mimic those oxidative functions of macromolecules such as metalloenzymes.
2. Experimental
2.1 Assembly of MOF-808 based bioinspired catalysts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 225 mL) solutions, and mixed in a 1 L screw-capped glass jar. Afterwards, the reaction was performed at 130 °C for 24 h. The white precipitate was collected by centrifugation and washed two times with water (12 h each) and two times with MeOH (12 h each) over a period of 24 h. The sample was then activated at 80 °C for 24 h.
:1 to 225 mL) solutions, and mixed in a 1 L screw-capped glass jar. Afterwards, the reaction was performed at 130 °C for 24 h. The white precipitate was collected by centrifugation and washed two times with water (12 h each) and two times with MeOH (12 h each) over a period of 24 h. The sample was then activated at 80 °C for 24 h.
2.2 Catalytic wet peroxide oxidation of phenolic compounds
The catalytic activity of MOF-808@His doped with a copper solution of 100 ppm (MOF-808@His-Cu100) was measured via a chromogenic reaction of phenol, 2,4-dichlorophenol, catechol, hydroquinone and dopamine phenolic compounds with 4-aminoantypirine (4-AP) (Scheme 1).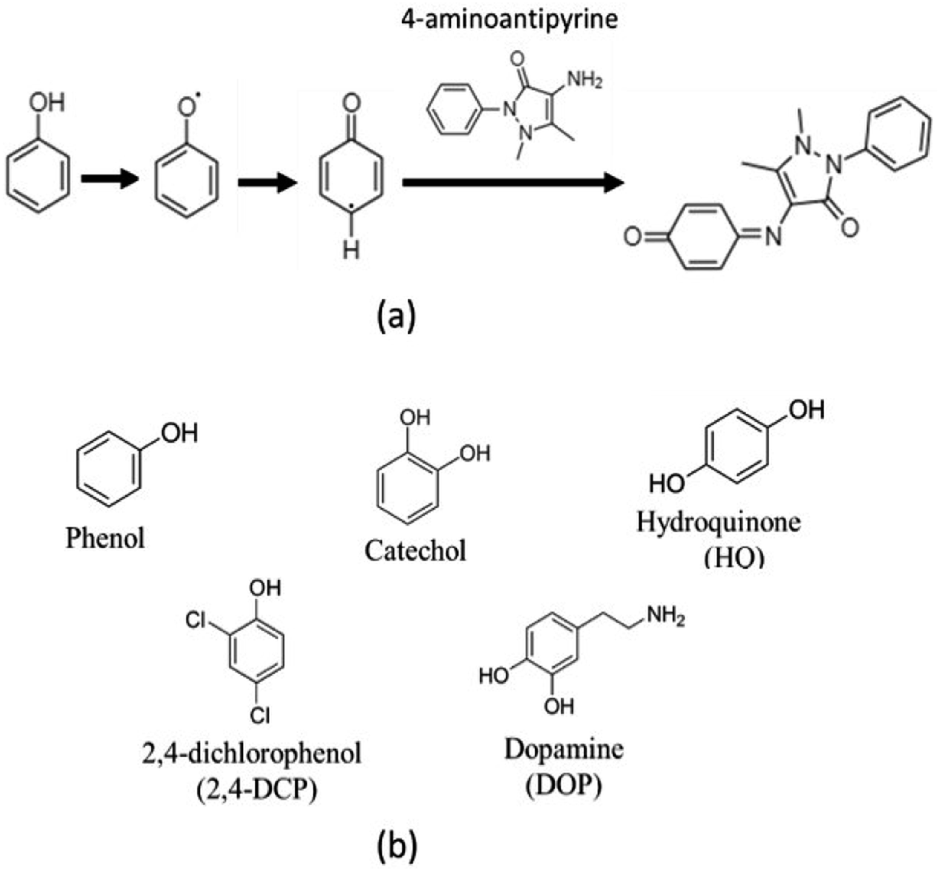 | ||
| Scheme 1 (a) Chromogenic reaction for phenols oxidations with 4-aminoantipyrine. (b) Chemical structure of the phenolic substrates employed in this work. | ||
In brief, for laccase like activity, 100 μL of an aqueous solution of the phenolic pollutant (1 mg mL−1), 400 μL of 4-AP (1 mg mL−1), 100 μL of the bioinspired catalyst (1 mg mL−1) were added. To test the CWPO activity, 20 μL of H2O2 were added as well. The volume of the reaction was completed with distilled water until a total of 1 mL. The reactor was shaken during specific periods of time, centrifuged at 10000 rpm during 1 min and the absorbance of the supernatant was measured by UV-Vis at 510 nm. Each of the reactions were fitted to a first order kinetic model to obtain the k-rate of the substrate oxidation.
For the study of the influence of the pollutant concentration, 100 μL of an aqueous solution of the phenolic pollutant of 0.1 to 2 mg mL−1 were used. The quantities of the rest of the materials/reagents were maintained the same. For these reactions, the KM and Vmax kinetic parameters were obtained with the Michaelis–Menten fitting described by the eqn (1):
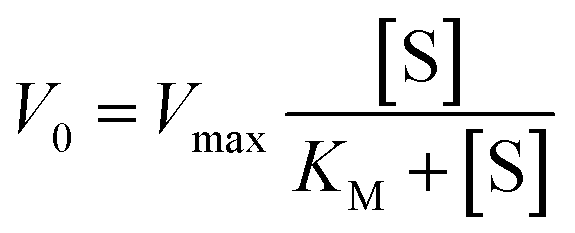 | (1) |
The stability of the MOF-808@His-Cu100 catalysts was studied by screening the (a) ionic strength of the media, (b) the reaction temperature from 30 to 120 °C, and (c) the pH of the media between 1 and 11. In addition, the reusability after washing it with distilled water, or after exposing the catalyst to 70 and 90 °C in water were studied as well.
Once the CWPO activity for MOF-808@His-Cu100 was studied, the impact of (i) the copper loading within the same host porous matrix and (ii) the coordination environment of the copper ions on the catalytic activity for the oxidation of hydroquinone and 2,4-DCP was studied. To this end, MOF-808@His with different CuII loadings and MOF-808 functionalized with various (amino) acids were used as catalysts. The following reaction conditions were employed to compare the oxidation reactions starting from batch solutions/dispersions of 1 mg mL−1 concentration of the different MOF-808(amino) acid-Cu catalysts. Hydroquinone: 100 μL of catalyst, 200 μL of hydroquinone and 400 μL of 4-AP solutions, 20 μL of H2O2 and 280 μL of H2O. 2,4-DCP: 100 μL of catalyst, 300 μL of 2,4-DCP and 400 μL of 4-AP solutions, 20 μL of H2O2 and 180 μL of H2O. The aliquots taken from the reactions at 0, 5, 10, 15, 30, 45, 60, 90 and 120 min were treated as above described for the previous reactions.
It is important to note that measuring the UV-Vis abs. at 510 nm arising from the complex formed between the oxidation product of the substrates and the 4-AP does not enable quantifying the reaction product. The molar extinction coefficient of the complexes may be different depending on the starting substrate. Thus, a protocol for the semiquantitative estimation of the product concentration has been developed. First, we have assumed that a high concentrated (100 mg L−1) copper chloride solution employed as a homogeneous catalyst would induce almost all the substrate conversion into the final product. Hence, the CWPO was performed increasing progressively the concentration of the substrates, obtaining a linear concentration vs. 510 nm-absorbance (Fig. S37a†). The absorbance values obtained when employing 100 mg per L copper solution as homogeneous catalyst exceed by far the ones of MOF-808(amino) acid-Cu system. This finding points towards a high conversion degree of the substrates when employing CuCl2-100 ppm homogeneous catalyst. When fitting the data a relatively good correlation is obtained. Still, significant background absorbance arising from the secondary oxidation of 4-aminoantipyrine is obtained in the calibration curve (Fig. S37b†). That is, the absorbance value at 510 nm obtained for a reaction without substrate but with 4-AP (i.e. 0.186) is similar to the one read for all the substrates except phenol. In any case, this background signal can be slightly enhanced or attenuated when performing the reaction in the presence of the substrates, so we have opted to overlook it during the calculation of the product concentration. All in all, the fittings obtained from the Abs. vs. substrate concentration curves, give semiquantitative estimations of the conversion, but they are still more accurate than comparing the performance and selectivity of the catalyst on the basis of the absorbance values recorded in the UV-Vis spectra. In addition, the conversion efficiencies and rates of the different substrates by MOF-808@His-Cu100 after translating absorbance values into product concentration are in line with the expected ones considering their chemical structure.
In order to follow the by-product generation of the phenol substrate after the first oxidation step, an extra experiment was performed using liquid chromatography. To this end, the reaction was scaled up to 20 mL but avoiding the addition of 4-AP co-reagent. Aliquots were taken and filtered with 0.2 μm hydrophilic PTFE syringe filter at 0, 15, 30, 60, 90, 120, 180, 240 and 1200 min. The complete mineralization of phenol through oxidative processes involves an initial step where catechol, hydroquinone or benzoquinone by-products, whose toxicity excesses the one of phenol, are formed. At this stage, it is key that the catalysts further oxidize these by-products until the rupture of C–C bond in benzoquinones led to the formation of environmentally friendly carboxylic acids. Further oxidation of these acids would lead to the complete mineralization of phenolic compounds.
The formation of the reaction sub products was monitored by liquid chromatography (HPLC) with an Agilent 1200 chromatographer and a ZORBAX SB-C18 (3.0 mm × 150 mm × 3.5 μm) column in reverse phase. Water was employed as the mobile phase. The measurements were carried out with a 0.4 mL min−1 flux rate, column temperature of 30 °C and 10 μL injection. A UV-Vis detector at 210 nm was used to monitor the by-products of the reaction. Calibration curves for phenol, catechol and hydroquinone molecules were obtained with solutions of 0, 20, 50, 100, 250 and 500 mg L−1 prepared from a parent solution at a concentration of 1 g L−1. The measurements were done by triplicate for each of the solutions.
3. Results and discussion
3.1 MOF-808@(amino) acid functionalization and characterization
As described by Furukawa et al.69,70 for the first time, the crystal structure of MOF-808 meets the pore-space metrics parameters to install amino-acid and metal-amino-acid sites after its post-synthetic functionalization and metalation.51 Just as a brief review, the crystal structure of MOF-808 is built up from the coordination of the archetypal hexanuclear Zr6O4(OH)4(–COO)6(HCOO)6 clusters.71 Each of these inorganic secondary building blocks (SBU) has twelve exchangeable coordination sites, six of them bonded to the trimesate (BTC) organic linkers. The peripheral sites of the equatorial plane of the clusters are occupied by easily exchangeable modulator molecules (i.e. formate). Indeed, in this work we have decorated these positions with amino acids as histidine (MOF-808@His), cysteine (MOF-808@Cys), histidine–cysteine (MOF-808@HisCys), mercaptosuccinic acid (MOF-808@Msc), or natural acids as malic acid (MOF-808@Mal) or citric acids (MOF-808@Cit) following the water based solvent assisted installation (SALI) protocol recently reported by A. Valverde et al.,51 to later, install copper ions with different coordination environments and clustering degrees via adsorption from water solutions.MOF-808@(amino) acid samples after SALI and before metalation were fully characterized by X-ray powder diffraction, Raman spectroscopy, thermogravimetric analysis and N2 adsorption at 77 K. As concluded from the XRD data, the crystallinity, the position, and the relative intensity of the diffraction maxima for MOF-808@(amino) acids remain similar to the ones of the parent material (Fig. 1a). As expected, the disorder of the installed (amino) acid function slightly modifies the main XRD signature of the compounds. This fact is specially observed in the intensity ratio between the diffraction maxima located at low 2θ (°) values. We have tried to obtain some information from the Rietveld refinement of the XRD data of the parent and (amino)acid decorated materials, but the intensity differences are too small to enable a reasonable structural refinement including the atomic positions coming from the MOF-808 framework, together with the ones accounting the (amino)acid molecules.
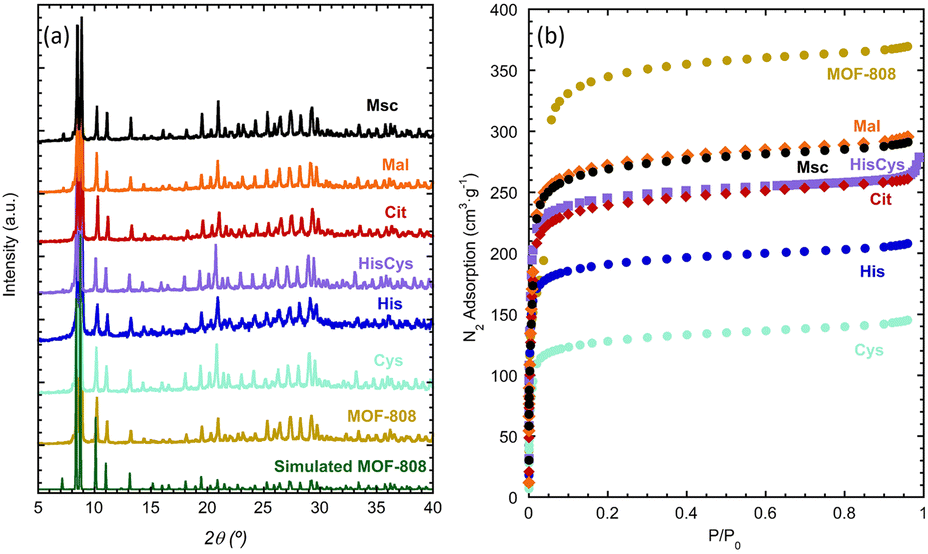 | ||
| Fig. 1 (a) X-ray powder diffraction patterns and (b) N2 adsorption isotherms (77 K) of parent MOF-808 and MOF-808 (amino) acid functionalized materials. | ||
In addition, the degree of hydration of each of the materials can vary, affecting as well to the XRD-data. For instance, the XRD patterns simulated from MOF-808 models with C1, C2 and C3 modulator molecules, or varying the hydration degree of the material, show that both parameters affect very weakly the intensity ratio of the low 2θ (°) diffraction maxima (Fig. S40†). All in all, the weak XRD signature arising from the amino acids, together with their high degree of disorder once installed in the pore matrix, hinder their location via powder X-ray diffraction data. Thus, we move to the spectroscopic characterization of our system, in order to gain complementary information.
Raman spectroscopy confirms the presence of the characteristic bands for the amino groups for amino-acid molecules. For di and tricarboxylic functions as Msc and Cit, the Raman band of non-coordinated carboxyl groups is also observed after their installation into the MOF-808 (Fig. S20–S26†). This evidence points out that in addition from the carboxyl bridging, there are non-coordinated –COOH groups pointing to the pore space of the material. Significant variations in the Raman bands associated to the parent materials after its modification are not observed. A slight reduction of the surface area after the SALI was found (Fig. 1b).51 Linker defects on the MOF-808 material were determined by analysing the weight loss associated to the trimesate linker (BTC) calcination step in the thermogravimetric analysis, as explained in detail in our previous work and in the ESI file (Fig. S12†). The same protocol could not be applied to the functionalized samples, due to the high overlapping of the weight losses associated to the amino acid/carboxyl acid functionalities, and trimesate organic molecules (Fig. S13†). Indeed, the thermal decomposition of MOF-808 is significantly affected by the incorporation of the (amino) acid groups, as detailed in the ESI file (section S1.3†). Although we do not dismiss that BTC could be partially replaced by the (amino) acids during SALI, the BTC:(amino) acid molar ratio for each sample was obtained from the integration of the proton signatures of BTC and (amino) acid in the 1H-NMR spectra of the digested samples (section S1.2†). Considering all the above-explained characterization, the general empirical formula: [Zr6O4.08(OH)4.08(BTC)1.92((amino) acid)x(OH)6−x(H2O)6−x], can be proposed for our system; where x is the amino acid molecules installation per zirconium hexanuclear cluster (Table 1).
| Before Cu-metalation | ||
|---|---|---|
| Formula | Cluster/AAa ratio | |
| a AA = amino acid or carboxyl acid molecules employed to decorate the MOF-808. b Concentration of the copper ions in the water solution employed for the MOF metalation. c These data are obtained from the normalization of the sum of weight percentages for Zr and Cu ions to 100% in the FRX measurements. | ||
| MOF-808 | Zr6O4(OH)4(C9O6H3)1.9(CH3COO)6.0 | — |
| His | Zr6O4(OH)5.6(H2O)5.6(C9O6H3)2(C6H8N3O2)4.4(3) | 1:4.4(3) |
| Cys | Zr6O4.0(OH)7.2(H2O)3.2(C9O6H3)2(C3H6NO2S)2.8(2) | 1:2.8(2) |
| His/Cys | Zr6O4(OH)4(C9O6H3)2(C6H8N3O2)2.7(4)(C3H6NO2S)3.3(4) | 1:2.7(4) to 3.3(4) |
| Msc | Zr6 O4.0(OH)6.1(H2O)2.1(C9O6H3)2(C4H6O4S2)3.9(4) | 1:3.9(4) |
| Mal | Zr6O4.0(OH)6.0(H2O)2.0(C9O6H3)2(C4H5O5)4.0(4) | 1:4.0(4) |
| Cit | Zr6O4.0(OH)7.2(H2O)3.2(C9O6H3)2(C6H7O7)2.8(4) | 1:2.8(4) |
Starting from the crystal and chemical structures of the MOF-808 and of the (amino) acid molecules, a qualitative description of their disposition within the pore space of the material can be done by building up tentative structural models where the (amino) acid molecules are coordinated to the defective positions of the zirconium clusters of MOF-808 (Fig. 2a.1–3). It is well known that the anchoring mechanism of the (amino) acid molecules to MOF-808 is mainly given through their coordinative binding to the equatorial plane of the Zr6O4(OH)4 secondary building units (Fig. 2a.3). After SALI,72 the side chains of (amino) acids are foreseen to point towards the centre of the 12 Å pore-window that give access to the adamantane 18 Å pores of MOF-808. Hence, (amino) acid functionalized MOF-808 will show a decoration of its pore space with imidazole, thiol, amine or carboxyl groups, or of their combination. Our qualitative structural models enable evaluating qualitatively the d1 (5–7 Å) and d2 (7–10 Å) distances between the metal-coordination groups decorating the pore space of the MOF-808@(amino) acids (Fig. 2b.1 and b.2). Although it is a rough estimation that needs further computational polishing, d2 is within the range of distances found for the coordination of both a single or dimeric copper units in the Cambridge Structural Database. In parallel, d1 distances are too long to involve the coordination of single copper ions.
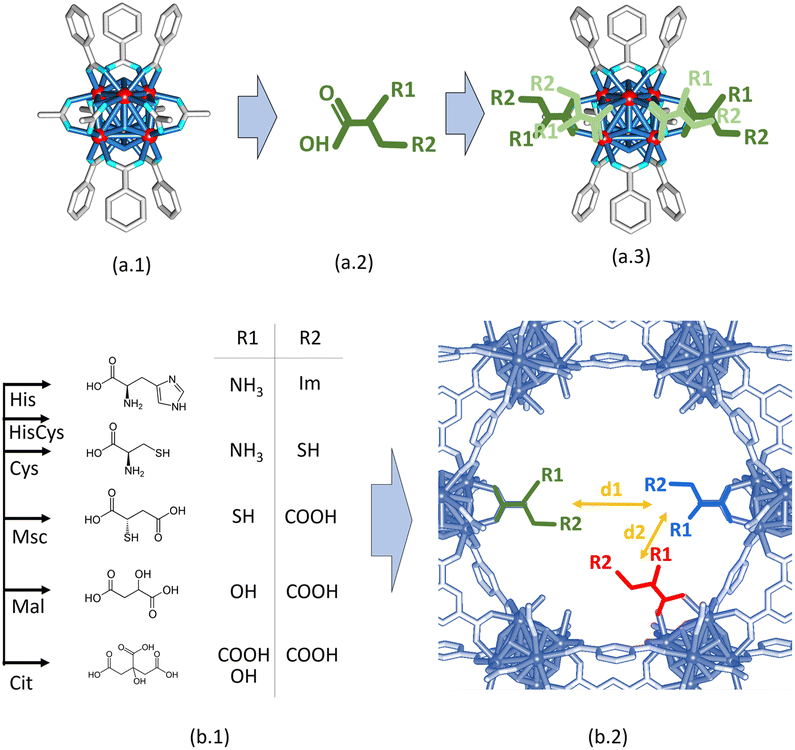 | ||
| Fig. 2 (a.1) Local structure of zirconium hexanuclear clusters in MOF-808. (a.2) Simplified chemical structure of (amino) acid molecules. (a.3) Tentative local structure of zirconium hexanuclear clusters after their functionalization. Colour code: red – zirconium. Light blue – oxygen. Grey – carbon. (b.1) Chemical structure and metal coordination groups of (amino) acid functions installed into MOF-808. (b.2) Simplification of the pore structure of MOF-808 once functionalized with (amino) acids. | ||
This general rule is partially broken for His and Cit, since the d2 can be shortened due to their chemical structure.
3.2 Copper immobilization
The metal immobilization in MOF-808@(amino)acid materials was carried out by the direct adsorption of copper ions from aqueous solutions of 10, 50, 100 or 1000 mg L−1 concentration. Fluorescence X-ray spectroscopy measurements of the solid samples were employed to estimate the copper loading of our biomimetic catalysts (Table 1). The concentration of the copper loading for the other MOF-808@(amino) acid samples was controlled to get a final CuII doping similar to the one found for MOF-808@His-Cu100 (Table 1). Immediately after the MOF-808@(amino) acid materials made contact with the copper solution, it turned an intense light blue to green colour. For instance, the Mal and Cit variants of MOF-808 exhibit a pale blue-greenish colour, MOF-8080@His gains an intense dark blue colour, whilst the Cys and Msc variants exhibit a light to dark brown coloration that suggests the existence of mixed valence CuI and CuII species after metalation. As will be alluded to later, the colour of the copper coordination complexes is closely related to their electronic structure, and hence, to their spectroscopic signature.3.3 Combined characterization of the copper catalytic sites
In general terms, the MOF-808@(amino) acid materials are robust enough to resist the copper metalation (Fig. 3a) except for His and Mal variants, which doping with 1000 ppm copper solutions induces a significant loss of crystallinity of their XRD patterns (Fig. 3b and S3†). Scanning electron microscopy and energy-dispersive X-ray spectroscopy were employed as complementary characterization to assess that the amino acid functionalization and copper metalation does not induce any surface effect on the particle size and morphology of parent MOF-808 material. As expected, the particle size distribution and the octahedral like morphology of MOF-808 crystals is maintained during their encoding (Fig. S34†). | ||
| Fig. 3 X-ray powder diffraction patterns of (a) MOF-808@Cys and (b) MOF-808@His materials before and after their doping with copper ions with water copper(II) nitrate solutions of 10, 100 and 1000 ppms concentrations. | ||
The metal complexation within the MOF-808@(amino) acid pore space induces a significant tuning on the electronic structure and spectroscopic properties of the copper sites in comparison to the hexa-aquo complex found in solution. The benefits of selecting copper as the ion-probe is that its UV-Vis, EPR and XPS spectroscopic fingerprints are intimately linked to (i) the groups conforming its coordination sphere, and (ii) the monomeric or clustered nature of the copper-complexes formed during its binding to the (amino) acid residues installed into MOF-808. In addition, there is a vast background on spectroscopy and magnetism of Cu-complexes that can be employed to understand such a complex and disordered system of our bioinspired materials. In addition, copper installation also induces a variation of the spectroscopic signature of the parent MOF-808@(amino) acid hosts.
Raman spectra of the pre and post-functionalized and metalated compounds show significant variations of the absorption bands involved or affected in/by the metal-coordination (Fig. 4 and S20–S26†). MOF-808 decorated with di (Mal, Msc) and tricarboxylic acids (Cit), shows the characteristic adsorption band associated to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond, which indirectly indicates the presence of protonated carboxyl groups at the pore space of the material (Fig. S21–S26†).73 After copper adsorption, this band is drastically attenuated, indicating the deprotonation of the free-carboxyl groups, and indirectly, their coordination to copper ions.
O double bond, which indirectly indicates the presence of protonated carboxyl groups at the pore space of the material (Fig. S21–S26†).73 After copper adsorption, this band is drastically attenuated, indicating the deprotonation of the free-carboxyl groups, and indirectly, their coordination to copper ions.
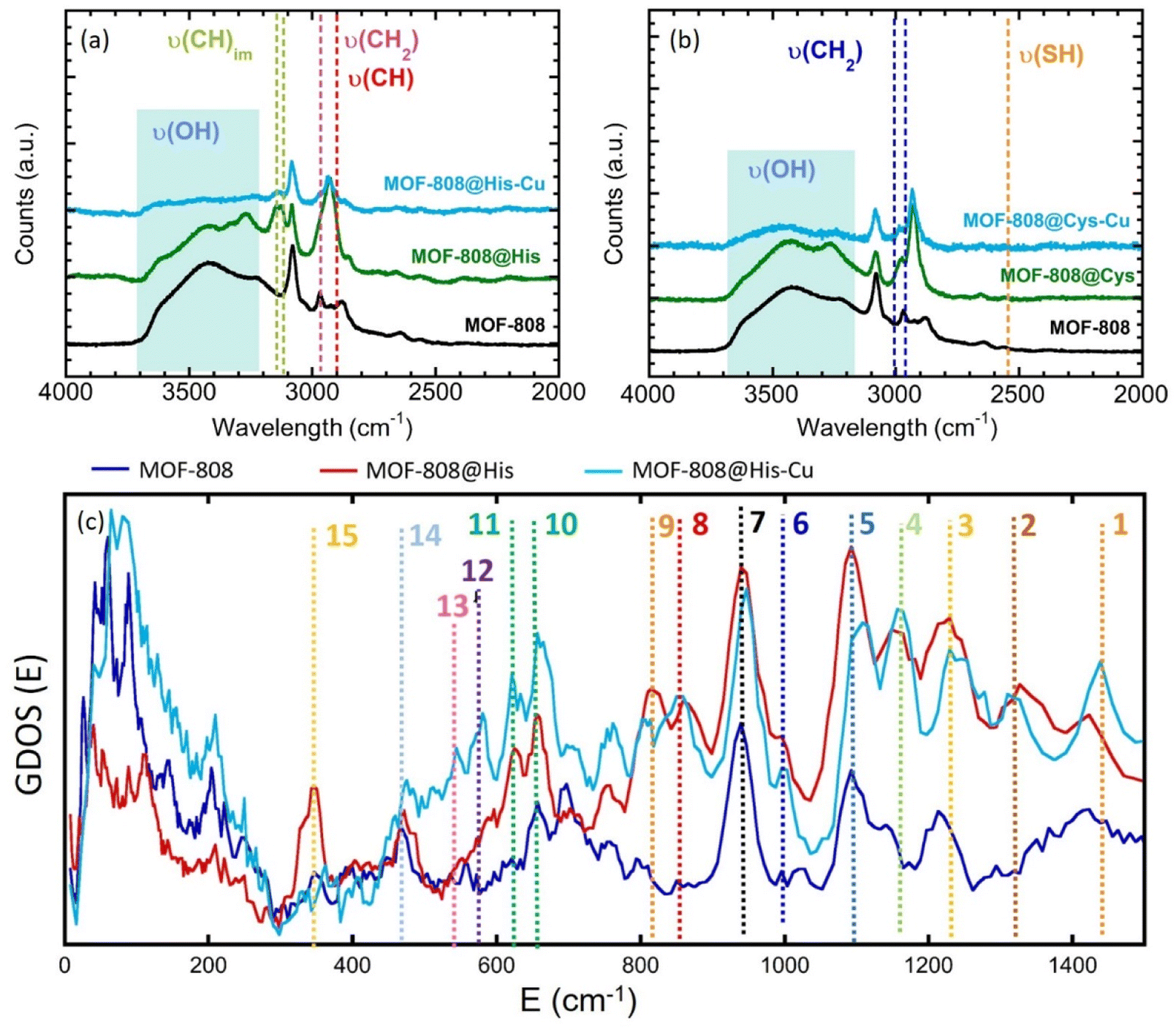 | ||
| Fig. 4 Raman spectra of MOF-808-His (a) and (b) MOF-808@Cys samples before and after copper metalation. (c) Inelastic neutron scattering spectra of MOF-808@His sample before and after its copper metalation. | ||
For Msc and Cys variants, the weak Raman bands associated to the –SH stretching vibration (∼2500 cm−1), have been identified (Fig. 4b, S25 and S26†). However, in Cys sample the signal is almost suppressed, probably due to Cys–Cys interactions through sulphide groups. Contrary, in Msc sample, the signal corresponding to the S–H bond is clearly visible. As expected, the νs(SH) is blocked after the adsorption of copper ions by MOF-808@Msc because of the deprotonation of SH groups resulting from the formation of Cu-thiolate bridges. Finally, the HisCys variant shows also a weak signal corresponding to νs(SH). This band does not loss intensity after the copper adsorption, which indicates that the CuII is preferably immobilized within the imidazole or amino groups. In fact, both HisCys and His variants show significant modification of the Raman bands associated to the stretching vibration of C–H bonds of the imidazole rings after copper immobilization (Fig. 4a and S20†). Therefore, it seems that the vibrational freedom of imidazole residues is as well attenuated, but not suppressed, after copper installation, either because they are directly in the copper binding, or cause the vibrational mode is attenuated after the copper-complexation by the His molecules.
Spectroscopic information obtained from Raman was completed with inelastic neutron scattering (INS) data for the histidine variant (Fig. 4c). The vibrational bands of the INS data of MOF-808 and MOF-808@His samples were identified on the basis of the bibliographic data based on the combination of experimental and computational studies (de Sousa et al.74 and Freire et al.75), as well as taking into account our preliminary studies in the MOF-808@Cys system. For instance, the assignation of the Raman and IR bands found in literature have been summarized in the Fig. S35 of the ESI.† The bands assignation for the MOF-808@His compounds have been summarized as well in the Fig. 4c and the Table 2.
| Band | IR | Raman | INS | Assignment |
|---|---|---|---|---|
| 1 | 1416 | 1449 | 1416 → 1435 | ν(CN)im + ν(CN)im |
| 1416 | ||||
| 2 | 1337 | 1334 | 1330 → 1315 | δ(NH)im + ν(CN)im |
| 3 | — | 1262 | 1225 → 1240 | δ(CH2) + δ(NH3) |
| 4 | 1144 | 1143 | 1160 → 1160 | τ(NH2) + δ(CH) + ν(NC)im |
| 5 | 1120 | — | 1095 → 1110 | ω(NH3) + δ(CH) |
| 6 | 959 | 977 | 996 → 998 | r(NH3) + δ(CH) |
| 7 | 875–1000 | 940 → 945 | ν(CC) MOF + His | |
| 8 | 868 | 874 | 860 → 860 | Mixed vibr. NH3 + δ(CH) |
| 9 | — | 803 | 817 → 810 | Def. (Im) + δ(NH3) + δ(CH) |
| 10 | 696 | 694 | 660 → 660 | δ-ofp (CH)im |
| 11 | 631 | 645 | 630 → 639 | δ-ofp (CH)im |
| 12 | — | 604 | — → 580 | def.(Im) + δ(CCN) |
| 13 | 528 | 540 | — → 552 | def.(Im) + δ(CH2) |
| 14 | — | — | 475 → 475 | δ(COH) |
| 15 | — | — | 350 → — | Skeletal vibrations of His + torsion (NH3) |
Firstly, the incorporation of histidine molecules into the MOF induces the disappearance of the INS bands associated to the COOH vibrations of the amino acid. Although in this study we could not measure the base histidine sample, it is well known that the CO2 rocking and COH bending modes of amino acids are usually located around 530 and 500 cm−1.51 However, these bands are not observed in MOF-808@His. These variations agree with the coordination of the His to the zirconium clusters via the carboxyl groups.
It is important to highlight the variation of the band number 15, which can be tentatively ascribed to two possible vibrational modes: the torsion vibration of NH3+ and the skeletal vibration of the His molecule. In contrast to the MOF-808@Cys homologue studied previously by the authors, the NH3+ torsion or skeletal vibration of His (∼350–400 cm−1) are not attenuated after its incorporation into the MOF. Thus, strong intermolecular interactions between His molecules through the –NH3+ groups are not expected within the MOF. This vibrational mode, located at 350 cm−1, is one of the most affected by the copper installation. This can be easily understood, since independently on the coordination mode of His to copper ions, their mobility may be severely restricted once copper ions are directly linked to them. In parallel, amino functions are prompt to coordinate copper ions forming Cu–N bonds. Therefore, any coordination of His to copper via amino groups is likely to suppress some of the vibrational modes associated to this group, but also to restrict the overall skeletal vibration of the hole molecule (ν(sk)).
In parallel, there are several signals of the INS spectra that do not disappear during metalation, but that experience a slight attenuation or a shift of their positions. These are the cases of the bands 1, 2, 3 and 5. Indeed, the bands 1 and 2 are closely related to the stretching vibrational modes of the C–N bonds within the imidazole groups. In parallel, the bands 3 and 5 are linked to the bending and wagging vibrations of NH3. Although all these slight variations do not give a direct evidence of the copper coordination to the imidazole or the amino groups of the histidine, it is clear that the metalation process induces some adaptation of their vibrational freedom.
Moreover, we do not observe any intensity reduction of the band 2, which is directly related to the deformation vibration of the NH group within the imidazole. This does not discard the copper coordination to the imidazole, since it can remain protonated whilst coordinated to this metal, as observed in several imidazole–copper coordination complexes found in the Cambridge Structural Database.68 An indirect evidence of the Cu–Im coordination could be the intensity gain of the out of plane vibration of C–H bonds within the ring (bands number 10 and 11), and the appearance of the 12 and 13 bands, also related with the deformation of the imidazole. Since the coordination of the copper ions by the NH3+ groups of His does not involve their deprotonation, a complete disappearance of the INS vibrational modes after the metalation of the MOF-808@His is not expected. For instance, the slight variations on the relative intensities of the rocking vibration of NH3+ and stretching vibration of CN bonds further indicate that amino groups have an active role during the immobilization of copper ions within the material. All in all, it can be concluded that both the imidazole rings and the NH3+ groups can participate in the coordination of copper ions.
Copper is most usually stabilized in biocatalysts as CuII paramagnetic ions with a (3d9) electronic configuration. Nevertheless, it is important to note that cysteine rich peptides or metalloenzymes can isolate it as 3d10 CuI diamagnetic centres. CuII prefers tetragonally distorted (compressed or elongated) octahedral, tetra- and penta-coordination environments showing Jahn–Teller effect. In addition to this, the nature of the amino acid residues forming part of the copper coordination sphere significantly alters the electronic structure of this 3d9 ion, and hence, its UV/Vis and EPR spectra.
Regarding their electronic structure, [Cu(H2O)6]2+ ideal complex exhibits three transitions that collapse into a single UV-Vis absorption band located approximately at 780 nm. The replacement of coordinated water molecules in [Cu(H2O)6]2+ by stronger electron donors, as peptide N > NH2 > imide > COO− > H2O, displaces the UV-Vis absorption maximum to higher energy wavelengths. This is the case of our MOF-808@(amino) acid biomimetic systems, for which the CuII – 2Eg → 2T2g transition is found between 800 and 600 nm (i.e. Cit = 770 nm, Mal = 795 nm, Cys = 670, His/Cys = 630, His = 620 nm) (Fig. 5a).
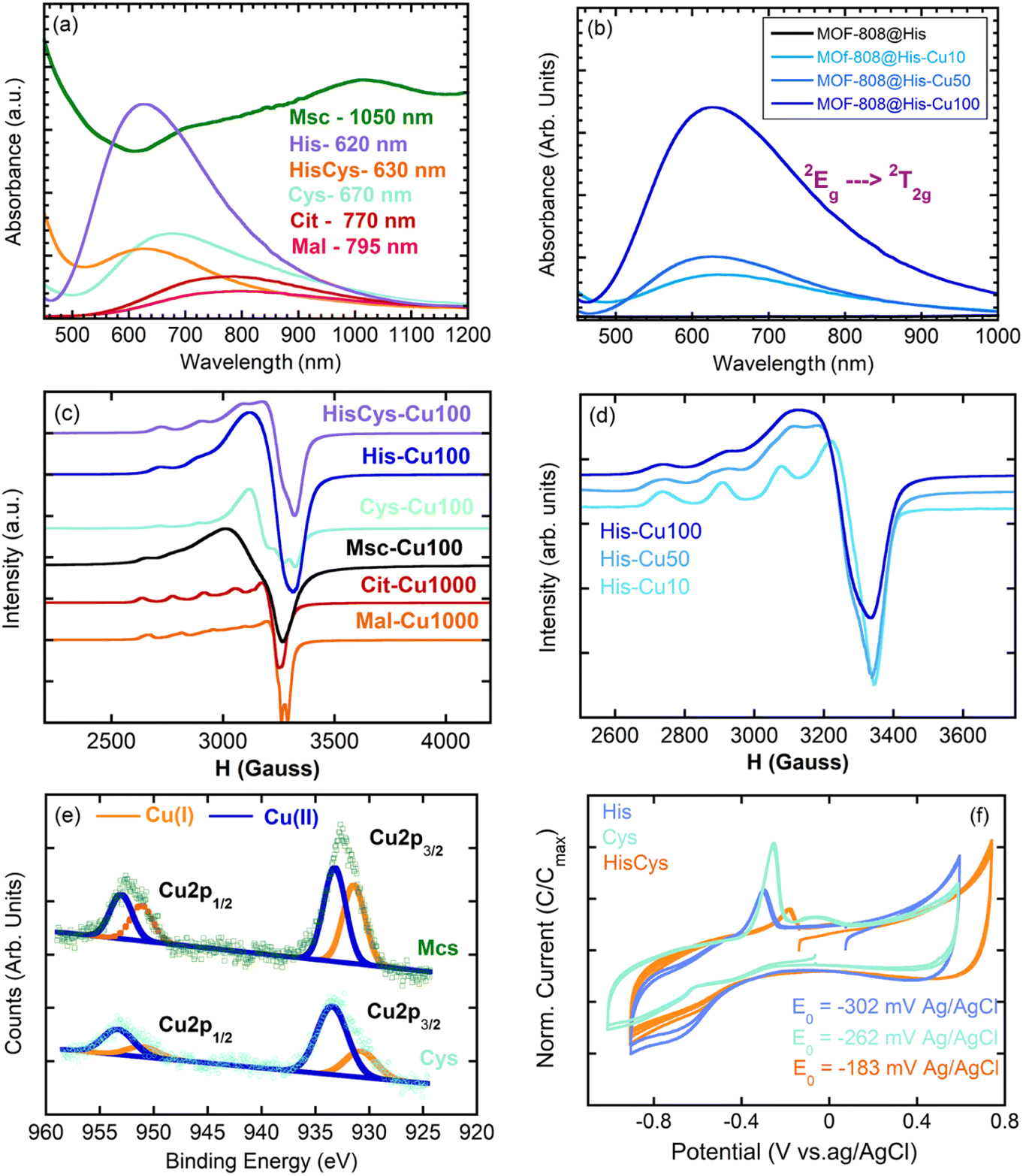 | ||
| Fig. 5 UV-Vis spectra of (a) MOF-808@His variant with increasingly copper content and (b) MOF-808@(amino) acid compounds with similar copper loadings. EPR spectra of (c) MOF-808@His variant with increasingly copper content and (d) MOF-808(amino) acids with similar copper loadings. (e) Fitting of the XPS spectra for MOF-808@Cys and MOF-808@Msc materials. (f) Cyclic voltamperograms of MOF-808@His-Cu, MOF-808@Cys-Cu and MOF-808@HisCys-Cu. | ||
In addition, the position of the copper UV-Vis adsorption maxima (λmax) can be qualitatively calculated based on the nature and the number of the groups forming part of the CuII coordination environment if the empirical equation obtained by Sigel and Martin76 is applied. The λmax for Mal (795 nm) and Cit (770 nm) compounds fits with copper's equatorial plane formed by two water molecules and two carboxyl groups (calc. λmax = 790 nm). When analysing the UV-Vis signature of the Cys variant (λmax = 690 nm), the equatorial plane of copper seems to consist of two amine and two water molecules (calc. λmax 663 nm). At first sight, this conclusion seems counterintuitive given the great chemical affinity of thiols to form copper thiolate bonds. The increase of the overall UV-Vis background signal for Cys and Msc variants points in this direction, since it suggests the co-existence of CuII and partially reduced CuI-thiolate species. This conclusion is as well in agreement with the CuII/CuI mixed valence speciation found in many copper-thiolates.77,78 In the specific case of His and multivariate His/Cys variant, the displacement of the UV-Vis λmax to higher energies suggests the participation of at least an imidazole and an amino group (calc. λmax 648 nm) or of two imidazole groups (calc. λmax 634 nm) in the copper coordination.
The configuration of the copper ions within the MOF-808@(amino) acid systems is not only shaped by the coordinating environment, but by the degree of copper loading, as corroborated for MOF-808@His. The copper loading affects: (i) the λmax of the UV-Vis spectra that is related with the coordination environment of copper (Fig. 5b), and (ii) the broadening of the EPR signal related with the clustering of the copper sites (Fig. 5d).
The MOF-808@His-Cu10 exhibits the characteristic EPR fingerprint of magnetically isolated CuII complexes with tetragonal ligand field environments consisting of a central EPR adsorption signal (g⊥ = 2.274 and g‖ = 2.063) completed by four less intense signals associated to the hyperfine structure lines (A‖ = 175 × 10−4 and A⊥ = 10 × 10−4 cm−1) (Fig. 5d). Given the g-values obtained for the fitting of the EPR spectra, the copper ions are stabilized in an axially highly elongated system that could involve a square-planar coordination, or an octahedral or pyramid-square coordination with two long Cu–O bonds (>2.3–2.4 Å). When the concentration of the copper solution employed to immobilize the ions is increased to 50 ppms (MOF-808@His-Cu50), the EPR spectrum shows the presence of two copper ions with slightly different g values, and hence, a slight variation on the distortion of the coordination environment. This tendency is further accentuated for the MOF-808@His-Cu100, where the EPR spectrum fits with the usual signature observed for copper magnetically clustered systems. The g values estimated by EPR for the MOF-808@His-Cu systems agree with a highly symmetric equatorial square planar coordination geometry for copper ions formed by two oxygen and two nitrogen atoms. Therefore, the copper clustering process can be achieved by two histidine–copper complexes with copper ions coordinated in a bidentate mode by the amino and imidazole groups linked by water or hydroxyl bridges. This model explains both the information coming from INS and UV-Vis spectroscopies together with the clustering process observed by EPR.
EPR was employed as well to study the MOF-808@(amino) acid materials decorated with similar copper loadings (Fig. 5c). It is clear that Cit and Mal variants stabilize copper ions as magnetically isolated species. In the case of MOF-808@Cit-Cu1000 only one type of copper is observed by EPR, whilst MOF-808@Mal-Cu1000 exhibits two types of magnetically isolated copper ions stabilized in slightly different coordination environments. For Msc and Cys functionalization, copper ions are stabilized as magnetically clustered species, or as a mixture of different magnetically isolated ions with different coordination environments. This occurs as well when a multivariate combination of His and Cys functions is included within the framework (Fig. 5c). This clustering of copper centres could be even more accentuated than as suggested by EPR, since in addition to the CuII species detected by this technique, XPS spectra of Msc, Cys and HisCys variants confirm the additional existence of CuI ions (Fig. 5e). In fact, the bimodal peaks of the XPS spectra, which are related to copper binding energies, can be fitted to two contributions centred at 952.6/953.0 eV (CuII) and 951.0/950.8 eV (CuI) for the Cu1p1/2 electronic level in Cys/Msc variants.79 In parallel, the same splitting of the signal is observed for Cu2p2/3 binding energies of CuII/CuI in Msc (933.0/931.0 eV) and Cys (933.4/931.1 eV) (Fig. 5e). These values agree as well with the binding energies reported for CuII oxides and coordination compounds and CuI thiolates. From the intensity of each contribution, it can be concluded that the ¼ and ½ of the copper ions stabilized within the Cys and Msc variants have been reduced to CuI. In this line, the voltamperometric measurements confirm the reversibility of the CuII ↔ Cu0 redox pairs, and the influence of the (amino) acid coordination on the redox potentials observed for each of the studied materials (Fig. 5f). This modulation of the redox characteristics of metal-sites is also observed in metalloenzymes when the amino acid residues participating in the coordination of the metal-sites are altered.80
Even though our combined spectroscopic characterization has enabled identifying the coordinating groups participating in the copper stabilization, it is not possible to detail their local structure. In order to gain further insights, a systematic search of the coordination complexes and polymers having Cu-(amino) acid fragments has been performed in the Cambridge Structural Database and summarized in the Fig. 6 and ESI Cu-Cit.cif, Cu-Mal.cif, Cu-His.cif, Cu-Cys1.cif, Cu-Cys2.cif, Cu-Msc.cif files.†68,81
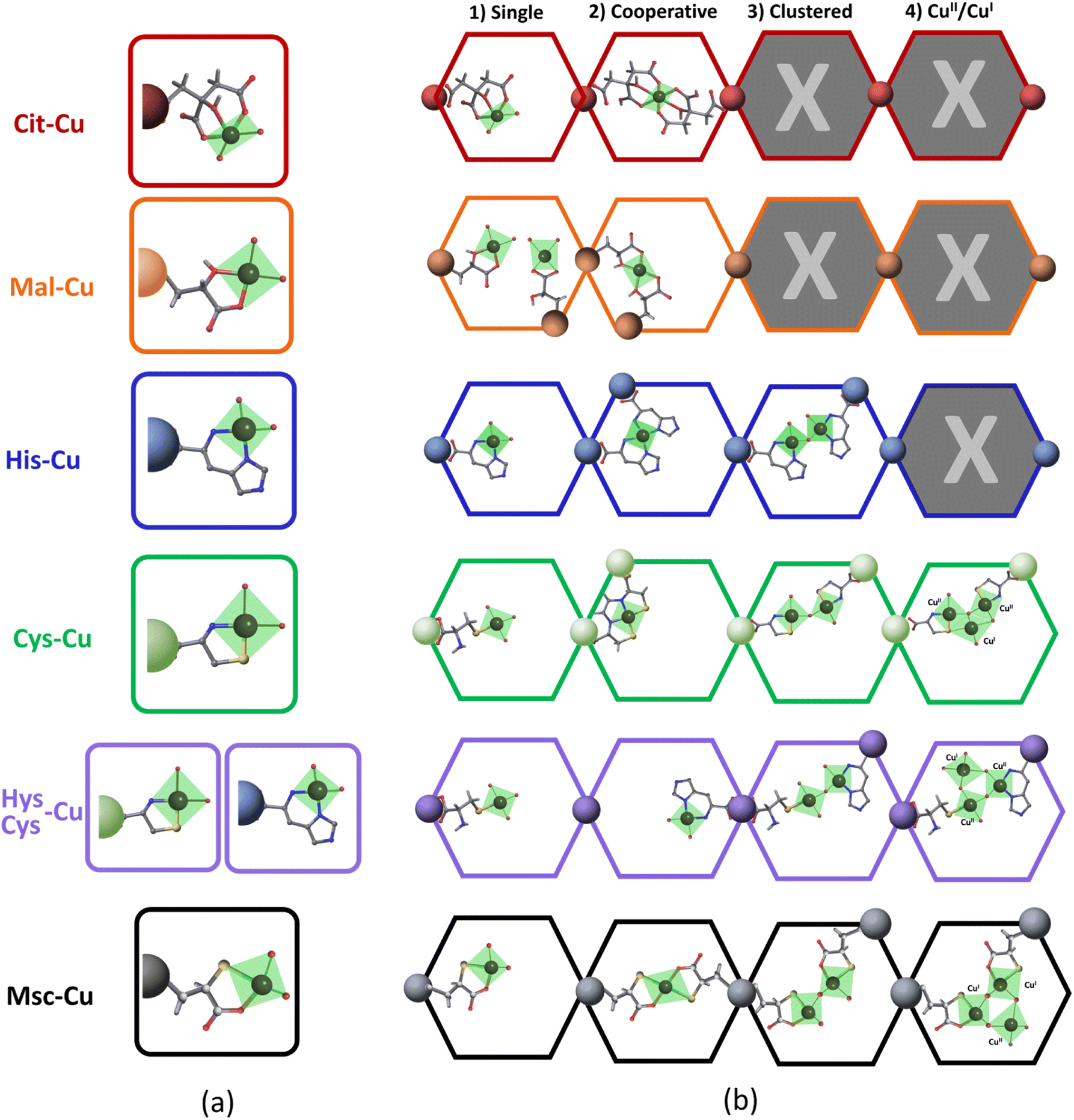 | ||
| Fig. 6 (a) Most representative CuII-(amino)acid coordination modes identified in the CSD database. (b) Possible stabilization of copper-amino acid coordination complexes within the pore space of MOF-808. | ||
It is important to have in mind that the carboxylate group of His and Cys, or one of the them in Cit, Mal or Msc is not-functional to bind CuII, since it is the anchoring point of the (amino)acid to the zirconium clusters. When considering the coordination complexes built up from CuII found in the CSD, the bidentate mode is the most common coordination of His, Cys, Mal and Msc functions (Fig. 6a). In most of them, two (amino)acid molecules complete the coordination of the equatorial plane of CuII. In addition to these general trends, in some cases Cys is able to immobilize copper via sulphide bonds. For CuII-citrate compounds, tridentate and bidentate modes have been observed as well (Fig. 6a). Even though the installation of the copper ions into the (amino)acid functionalized pore space of MOF-808 is constrained by the framework, our spectroscopic data fit the coordination modes stabilizing isolated copper ions, as these observed in the CSD (Fig. 6b). This does not preclude that the stabilization of the copper ions could occur via the participation in a cooperative fashion of two (amino)acid molecules to bind one metal, or in a clustered fashion by the participation of two Cu-(amino)acid complexes connected by an oxo, hydroxo or water bridges. In the specific case of His, Cys, His/cys and Msc, the clustering of the copper ions observed by EPR spectroscopy can be explained by the concurrent presence of two CuII-His complexes within the MOF-808 pore space, as depicted in the Fig. 6b. The cooperative mode to stabilize a single CuII between two His or Cys molecules is discarded in this case if the conclusions extracted from EPR, UV-Vis and INS spectroscopies are considered. In addition, for Cys and Msc, the partial reduction of CuII to CuI can further promote the generation of complex clusters within the pore space of the MOF-808, as suggested by the many CuII–CuI polynuclear discrete units found in the CSD when mixed valence copper species are stabilized. It is important to remind at this point that a mixture of the Cu-speciation depicted in the Fig. 6 is the most plausible of the scenarios, although some of the coordination modes could be favoured depending on the pore spaces' characteristics, and the copper doping process. Overall, as depicted in the Fig. 6, the amino acid functionalization of MOF-808 and the copper doping process does not only enable controlling the coordination environment of the copper ions immobilized within the framework, but their clustering degree and mixed oxidation state. All these features of the catalytic sites are foreseen to significantly affect the catalytic activity and affinity of the system.
3.4 Catalytic wet peroxide oxidation (CWPO) of phenolic model pollutants
This section assesses the performance of the biomimetic systems studied for the Catalytic Wet Peroxide Oxidation (CWPO) of phenolic pollutants as phenol, catechol, hydroquinone, 2,4-dichlorophenol and dopamine. MOF-808@His-Cu100 catalyst was selected as the starting point for the study to later on move to other MOF-808@(amino) acid-Cu catalysts with copper-sites exhibiting different coordination environments and clustering degrees.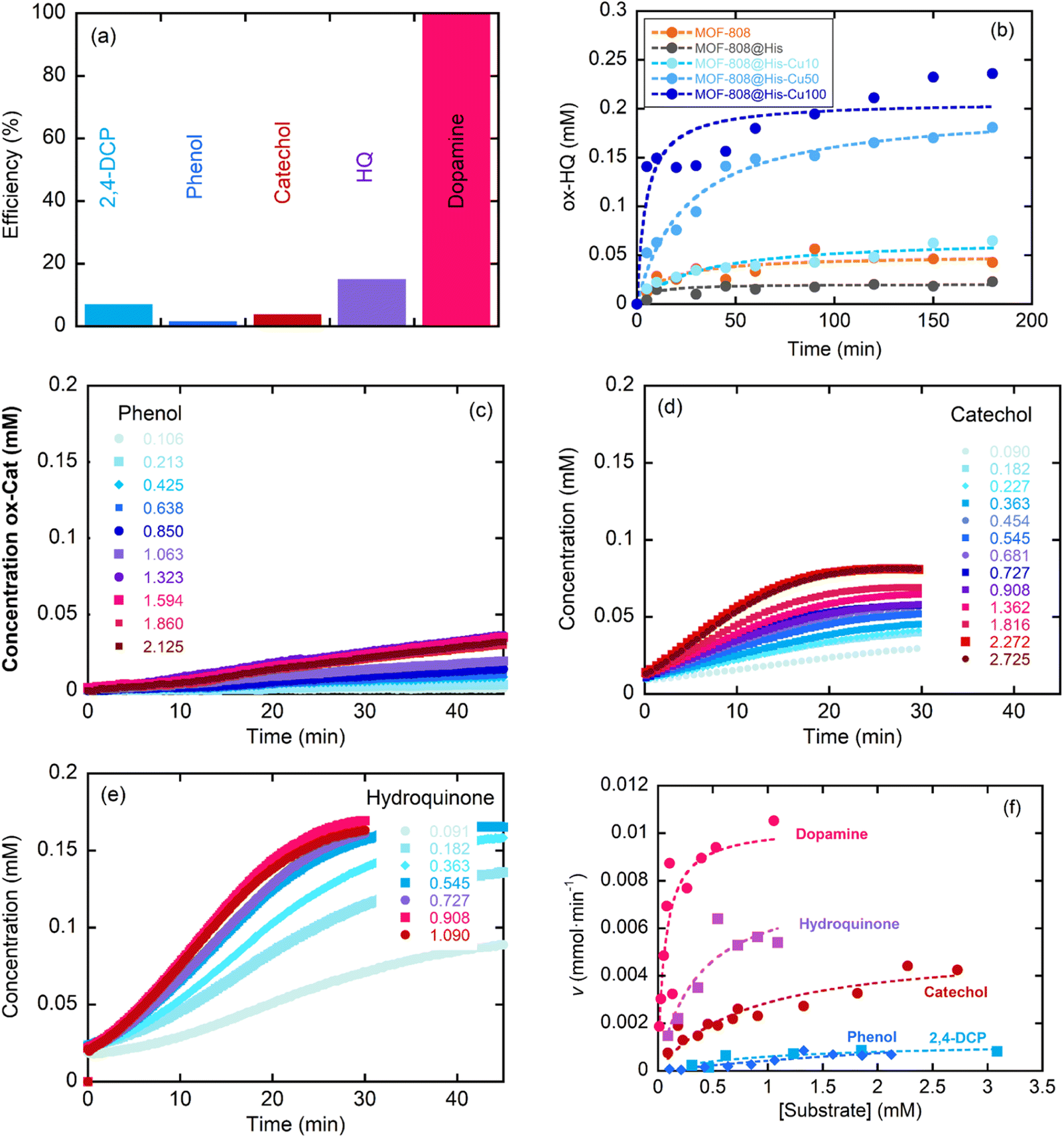 | ||
| Fig. 7 (a) Concentration of the oxidized products after the CWPO of different model pollutants by MOF-808@His-Cu100. (b) Evolution of the oxidation for hydroquinone by MOF-808, MOF-808@His, MOF-808@His-Cu10, MOF-808@His-Cu50 and MOF-808@His-Cu100. (c–e) Concentration dependence on the CWPO oxidation of phenol, catechol and hydroquinone. (f) Michael–Menten plot for the CWPO of catechol, phenol, 2,4-dichlorophenol, hydroquinone and dopamine with MOF-808@His-Cu100. | ||
The response of the MOF-808@(amino)acid-Cu biomimetic catalysts was also studied for 2,4-dichlorophenol, phenol, catechol, hydroquinone and dopamine oxidations at varying concentrations. The reaction kinetics to oxidize phenol (Fig. 7c) or 2,4-DCP (Fig. S36b†) are slower than the ones obtained for the transformation of catechol (Fig. 7d), hydroquinone (Fig. 7e) and dopamine (Fig. S36c†). In addition, the sigmoidal profile for the hydroquinone oxidation indicates a negative allosteric effect due to the substrate binding to the copper-sites. Therefore, the activity of MOF-808@His-Cu depends on the number and position of the hydroxyl groups within the aromatic ring of the studied substrates, but it is influenced as well by the existence of other substituent groups (as is the case of the dopamine). Overall, the MOF-808@His-Cu exhibits enzymatic like kinetics, as concluded from the Michaelis–Menten plot and the fitting of the experimental data shown in the Fig. 7f. For instance, the KM and Vmax values obtained from the Michaelis–Menten fittings (eqn (1)) confirm that our biomimetic systems are more efficient for the oxidation of catechol or hydroquinone substrates in comparison to phenolic based ones (Table 3). In parallel, when comparing the figure of merits of our biomimetic catalysts with other laccase and peroxidase like nanozymes, it usually overperforms the KM and Vmax parameters reported for TMB and H2O2 substrates in contrast with the substrates studied in this study (Table S3†).
| MOF-808@His-Cu100 | ||||||
|---|---|---|---|---|---|---|
| Substrate | ρ e (g L−1) | [E] (mM) | K M (mM) | V max (mM min−1) | k cat (min−1) | k cat/KM (mM−1 min−1) |
| 2,4-DCP | 0.1 | 3.586 × 10−7 | 1.0(7) | 1.2(4) × 10−3 | 3.34 × 103 | 3.34 × 103 |
| PHE | 0.1 | 3.586 × 10−7 | 5 ± 2 | 3.3(3) × 10−3 | 9.21 × 103 | 1.84 × 103 |
| CAT | 0.1 | 3.586 × 10−7 | 0.81 ± 0.24 | 5.2(7) × 10−3 | 1.45 × 104 | 1.79 × 104 |
| HQ | 0.1 | 3.586 × 10−7 | 0.36 ± 0.20 | 7.9(1.6) × 10−3 | 2.20 × 104 | 6.12 × 104 |
| DOP | 0.1 | 3.586 × 10−7 | 0.062 ± 0.029 | 10.3(1.2) × 10−3 | 2.87 × 104 | 4.63 × 105 |
Even if the CWPO is a green and highly appealing approach to face the mineralization of phenolic compounds in polluted waters, the oxidative routes for phenol can accelerate the generation of catechol, hydroquinone and benzoquinone products at the first stage of the reaction. These intermediates exhibit even higher environmental risks than the parent pollutant. So, as reported by A. Azizi et al.,83 an efficient oxidation of these intermediates is necessary to induce the aromatic ring opening, and the subsequent generation of linear and environmentally friendly carboxylic acids. Thus, it is important to follow up the oxidation to identify the intermediate products generated during the reaction. The chromogenic protocol employed in the initial stage of this work allows a rapid and easy experimental access to evaluate the CWPO, but it is limited to the first oxidation step within the oxidative degradation path of phenolic compounds. Liquid chromatography is necessary to monitor the sub-products generation, as is the case of the phenol CWPO shown in the Fig. 8.
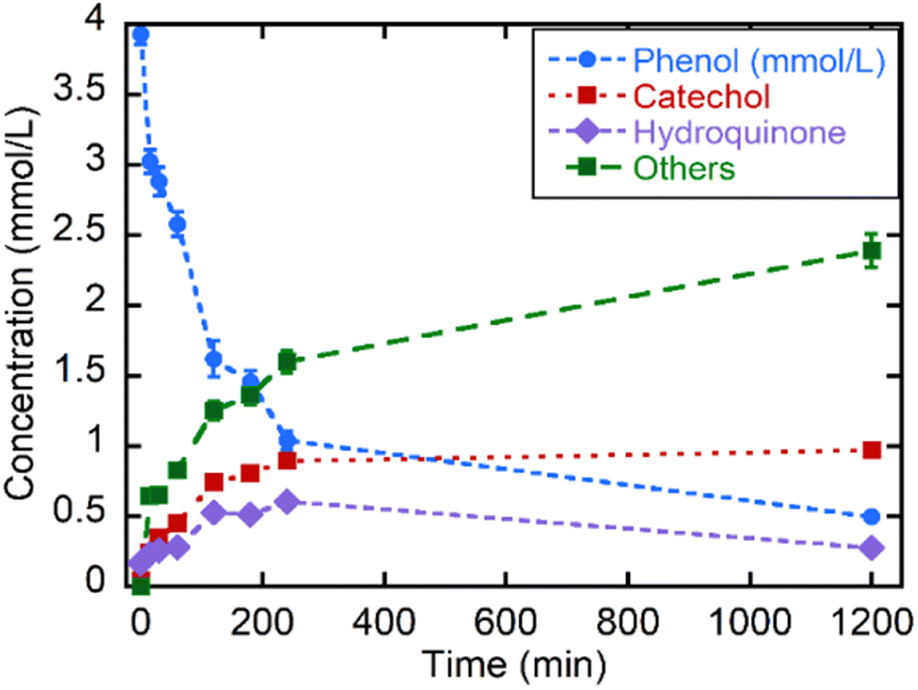 | ||
| Fig. 8 Kinetics of the catalytic wet oxidation of phenol and of its degradation products with MOF-808@His-Cu100 followed by liquid chromatography. | ||
First, it is important to note that the MOF-808@His-Cu100 is able to oxidize nearly 75% of the phenol in less than three hours, reaching an 87.5% conversion in 20 h at RT. Nearly half of the by-products generated during the first 4 h cannot be retained by the chromatographic column employed for catechol and hydroquinone. This fraction of the reaction was tentatively ascribed to the carboxyl-molecules generated after the ring-opening. Interestingly, the oxidation of phenol to catechol is slightly more favourable than the one to hydroquinone. Nevertheless, after 4 h, the catalyst is still able to degrade hydroquinone and phenols, whilst the concentration of catechol remains unaltered. At this point, it is important to note that higher catalyst loadings, temperature of the reaction or H2O2 addition could contribute to further displace the oxidation into non-environmentally hazardous by-products as carboxyl acids.
 | ||
| Fig. 9 Stability dependence of MOF-808@His-Cu50 catalyst over (a) ionic strength, (b) temperature and (c) pH. (d) Reusability of the catalyst. | ||
The chemical stability was further assessed by measuring the copper leaching of the different catalysts after one day of exposure to the reaction media by ICP analyses. All of the catalysts show a partial metal release ranging from 4.5 to the 20.7% of the copper installed in the initial material (Table S2†). This leaching gives rise to four different concentrations of dissolved-copper ions in the reaction media depending on the metal loading in the initial catalyst (0.5 ppm for Mal and Cit, near 0.5 ppms for CysHis, close to 1 ppm for His and Cys, and 4 ppm for Msc). For the shake of comparison, we have compared the performance of the His-Cu-100 catalysts with the one obtained by incorporating, 0.1, 0.5 and 1 and 4 ppms of copper dissolved in the reaction media for the dopamine CWPO. Our heterogeneous catalyst overperform by far the conversion rate of the copper solutions of 0.1 to 1 ppm concentrations, and is comparable to the one obtained with the Cu-solution of 4 ppms. As the leaching of His-Cu-100 system leads only to a final copper concentration of 0.8 ppms, the dopamine transformation can be mainly ascribed to the heterogeneous component. In parallel, it is interesting how the performance of the copper solution skyrockets when a concentration of 4 ppms is employed in the media, a fact that suggest that copper ions can initiate clustering in solution above a given threshold (Fig. S38†).At this point, it is clear that the recyclability and copper stabilization in our system need to be improved significantly to make the catalysts competitive in comparison to the ones reported in previous studies. Up to date, copper MOFs as HKUST-1 and copper loaded zeolites, clays or alumina, have been employed to face the phenols oxidation by CWPO.84–86 Although it is difficult to quantitatively compare catalysts with such different characteristics (specially copper loading) and operated under varied conditions, it is important to note that the performance of MOF-808@His-Cu100 is comparable in terms of % of phenol oxidation to catalysts with similar copper contents operated at room temperature conditions.
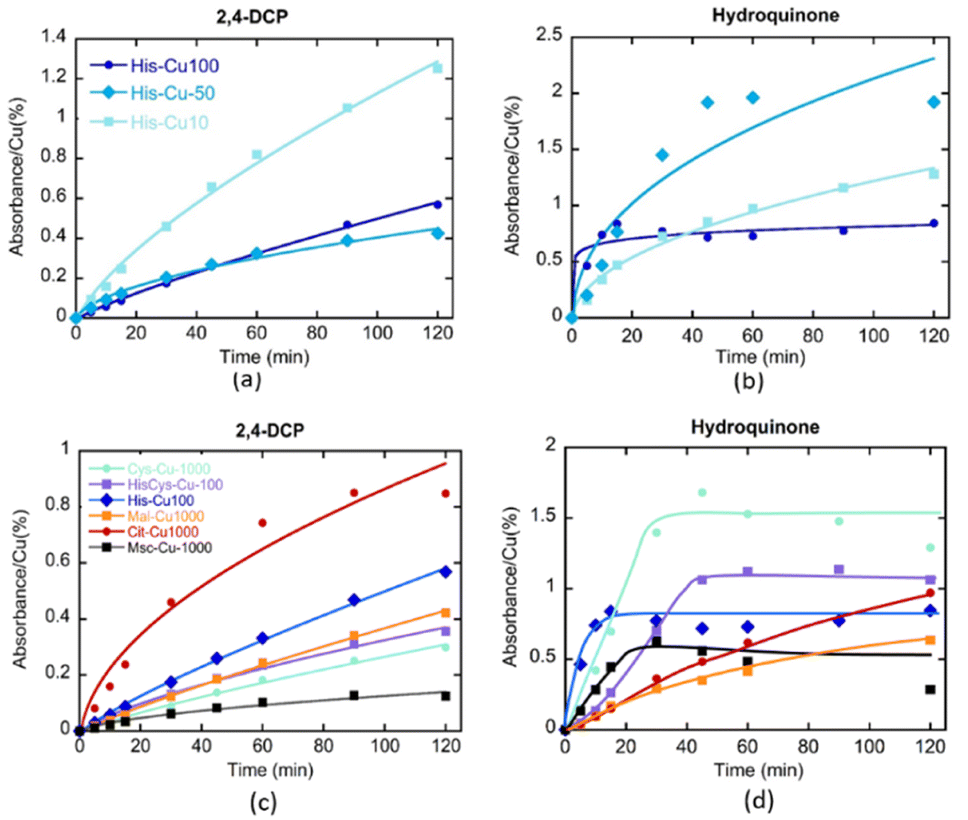 | ||
| Fig. 10 Kinetics of CWPO of (a and c) 2,4-dichlorophenol and (b and d) hydroquinone of (a and b) MOF-808@His-Cu system with increasing doping of copper, and (c and d) MOF-808@(amino) acid-Cu catalysts exhibiting Cu-catalytic sites stabilized by different amino acid and acid residues. For all the reactions, the absorbance value recorded at 510 nm was normalized to the copper content of each material to obtain the turnover frequency (TOF). | ||
As previously demonstrated, the clustering of the copper ions installed into MOF-808@His increases progressively when employing copper solutions of higher concentrations (i.e. Cu-10, Cu-50 and Cu-100) for its metalation. The kinetics of 2,4-DCP oxidation strongly depends on the presence of single-copper metal-sites in the catalysts, since the TOF is significantly greater for MOF-808@His-Cu10 than for Cu50 and Cu100 variants. That is, the activity per catalytic site seems inversely related to the copper clustering. In contrast, for the CWPO of hydroquinone some degree of copper-clustering is beneficial, since the conversion for MOF-808@His-Cu50 is better than for Cu-10 and Cu-100 catalysts.
In a second step, the effect on their catalytic activity of the coordination environment of the copper ions installed into MOF-808@(amino) acids was studied for the abovementioned two reactions (Fig. 10c and d). In agreement with the results obtained for MOF-808@His-Cu, the MOF-808@(amino) acid materials featuring less copper clustering (i.e. Cit > His – His/Cys > Mal > Cys > Msc) exhibit better efficiencies for the 2,4-DCP oxidation. Even if the MOF-808@(amino) acid catalysts show similar degree of copper clustering in the active sites, small variations of their coordination environment induces significant changes on their oxidative activity, as is the case of His, His/Cys and Cys variants. The tendency observed for the 2,4-DCP oxidation is reversed when studying the CPWO of hydroquinone. In this case, the best conversion is achieved by the Cys-Cu variant, followed by the Cys/His and His based compounds. It is interesting to note the significant differences observed in the transformation rate of hydroquinone by the different catalysts. His, Cys, HisCys and Msc variants show a fast transformation of the substrate in the first 20 minutes until the evolution is frozen, whilst Mal and Cit based compounds exhibit slower kinetics but a constant hydroquinone oxidation along the 120 min of the experiment. This fact points out that the coordination sphere of copper ions does not only modulate its overall efficiency to oxidize hydroquinone, but also the kinetics of the transformation at the initial stages of the reaction.
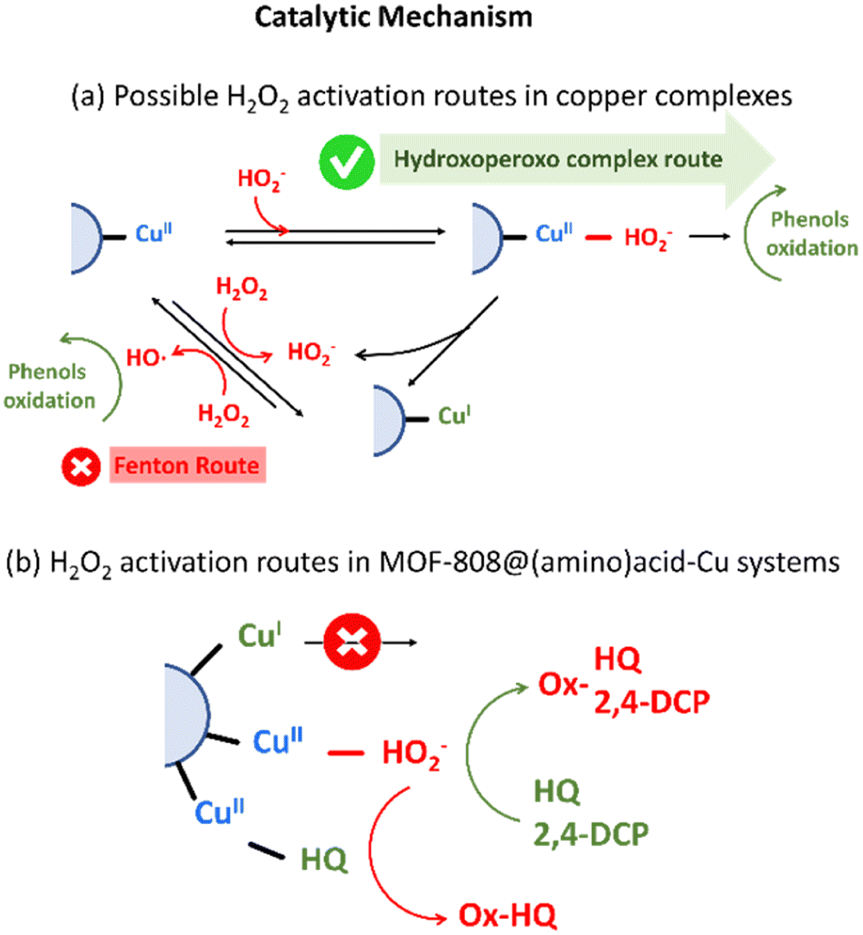 | ||
| Fig. 11 Catalytic mechanisms of H2O2 activation. | ||
On one hand, the generation of hydroxoperoso or peroxo species coordinated to copper is highly dependent on the reversible dissociation of water molecules from the distorted octahedral Cu(II) complex, and specially, of those located on the equatorial coordination plane. For homogeneous Cu-amino acid/H2O2 system, an increase of the ligand to metal ratio stabilizes the formation of CuL2 complexes. In this scenario, amino acid molecules bind four of the six coordination positions of copper ions, hindering the access and posterior activation of hydrogen peroxide in the equatorial plane of this acid site. Contrary, CuL complexes enable a faster and more efficient activation of hydrogen peroxide as highly reactive hydroxo/peroxo species directly coordinated to the copper sites.
This trend is consistent with the activity of copper-species stabilized in MOF-808@(amino) acid-Cu system. In general terms, the copper clustering gives rise to a decrease of the turn over frequency per active-site for 2,4-DCP oxidation. That is, a slower dissociation of a water molecules and a hindered accessibility of H2O2 molecules to the coordination sphere of copper-clustered sites is expected, and hence, a lower efficiency per copper ion. It is true that for hydroquinone oxidation certain degree of copper-clustering is beneficial, together with the presence of Cys and His amino acids in its coordination sphere. It is important as well to note that polydentate citrate acts as a shield protecting the access of HQ substrate to the active site, slowing down its oxidation in comparison to other copper-centres. Negative allosteric effect in the first stage of HQ-oxidation, together with the poor reusability of the catalysts, suggest that HQ is able to access the coordination environment of copper-sites. Although copper-clustering could attenuate the activation of H2O2via -hydroxoperoxo formation, may be beneficial to prevent the HQ access to the copper sites (Fig. 11b).27,87
On the other hand, when the back-electron transfer of Cu(I) occurs, hydrogen peroxide may dissociate to ˙OH. As a result, the active ˙OH reacts rapidly with the substrate. Hydroxyl and electron scavengers were employed to assess whether this route could be active for MOF-808@His-Cu100 catalyst (Fig. S41†). In both cases, the reactivity of the catalysts was not significantly altered, and thus, it seems that hydroxyl radicals' generation is not the main transformation path for our biomimetic system. Further work is ongoing to fully discard this reactive route by quantifying the ROS generation.
Nonetheless of the type of H2O2 activation mechanisms, the catalytic activity per site of MOF-808@(amino) acid variants with a significant weight of copper stabilized as CuI (i.e. Msc or Cys is significantly lower than for these catalysts where copper is immobilized as CuII) (Fig. 11b).90
4. Conclusions
The chemical encoding of MOFs with amino acid and acid functions enables the subsequent adsorption-driven immobilization of copper sites with controlled coordination environments and clustering degrees within these porous and ordered materials. Similarly, than metalloenzymes, the catalytic activity and selectivity of our biomimetic systems depend on: (i) the amino acid and acid residues coordinating the copper metal sites and (ii) the clustering degree of the copper catalytic centres installed within the pore space of MOF-808. Further research is needed to fully understand the activation of hydrogen peroxide by the copper-sites, and the evolution of the oxidation state and of the coordination environments of the catalytic centres, especially for reactions where a negative allosteric effect has been observed during the first stage of the phenolic wet oxidation. In return, this work opens the perspective of copper enzyme mimicking and fine tuning in MOFs, as well as for their future application in added value enantioselective catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank financial support from the Spanish Agencia Estatal de Investigación (AEI) through EVOLMOF PID2021-122940OB-C31 (AEI/FEDER, UE) (including FEDER financial support), and Tailing23Green-ERAMIN projects. Basque Government Industry and Education Departments under the IKUR, ELKARTEK and PIBA (PIBA-2022-1-0032) programs, are also acknowledged. Ainara Valverde thanks the Basque Government (Education Department) for her PhD grant (PREB_2018_1_004). The MSCA-RISE-2017 (No. 778412) INDESMOF, and H2020-RIA-4AirCraft (No. 101022633) projects, which received funding from the European Union's Horizon 2020 research and innovation programme are also acknowledged. The SUPREN group at UPV/EHU acknowledges the funding from Basque Government (IT1554-22). The authors thank the technical and human support provided by SGIker (UPV/EHU). The authors are really grateful to Prof. Francisco Javier del Campo for his support during the electrochemical testing.Notes and references
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5, 1120–1136 CrossRef CAS PubMed.
- Z. Thompson and J. A. Cowan, Small, 2020, 16, 2000392 CrossRef CAS PubMed.
- Y. Yu, X. Liu and J. Wang, Acc. Chem. Res., 2019, 52, 557–565 CrossRef CAS PubMed.
- V. C. C. Wang, S. Maji, P. P. Y. Chen, H. K. Lee, S. S. F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- Y.-M. Li, J. Yuan, H. Ren, C.-Y. Ji, Y. Tao, Y. Wu, L.-Y. Chou, Y.-B. Zhang and L. Cheng, J. Am. Chem. Soc., 2021, 143, 15378–15390 CrossRef CAS PubMed.
- H. Eom and W. J. Song, J. Biol. Inorg. Chem., 2019, 24, 517–531 CrossRef CAS PubMed.
- M. L. Zastrow and V. L. Pecoraro, Coord. Chem. Rev., 2013, 257, 2565–2588 CrossRef CAS PubMed.
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855–862 CrossRef CAS PubMed.
- M. Fontecave and J. L. Pierre, Coord. Chem. Rev., 1998, 170, 125–140 CrossRef CAS.
- D. A. Quist, D. E. Diaz, J. J. Liu and K. D. Karlin, JBIC, J. Biol. Inorg. Chem., 2017, 22, 253–288 CrossRef CAS PubMed.
- M. Fontecave and J. L. Pierre, Coord. Chem. Rev., 1998, 170, 125–140 CrossRef CAS.
- B. E. R. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS PubMed.
- M. Alcalde, Industrial Enzymes: Structure, Function and Applications, 2007, pp. 461–476 Search PubMed.
- H. Liang, F. Lin, Z. Zhang, B. Liu, S. Jiang, Q. Yuan and J. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 1352–1360 CrossRef CAS PubMed.
- W. S. Hernández-Monjaraz, C. Caudillo-Pérez, P. U. Salazar-Sánchez and K. L. Macías-Sánchez, Braz. J. Microbiol., 2018, 49, 269–275 CrossRef PubMed.
- E. N. Mirts, A. Bhagi-Damodaran and Y. Lu, Acc. Chem. Res., 2019, 52, 935–944 CrossRef CAS PubMed.
- J. Dawson, Science, 1988, 240, 433–439 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- O. K. Farha, A. M. Shultz, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 5652–5655 CrossRef CAS PubMed.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed.
- R. Balasubramanian, S. M. Smith, S. Rawat, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, 115–119 CrossRef CAS PubMed.
- E. I. Solomon, P. Chen, M. Metz, S. K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570–4590 CrossRef CAS PubMed.
- N. S. Inchaurrondo, P. Massa, R. Fenoglio, J. Font and P. Haure, Chem. Eng. J., 2012, 198–199, 426–434 CrossRef CAS.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. Dekrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS PubMed.
- R. M. Liou and S. H. Chen, J. Hazard. Mater., 2009, 172, 498–506 CrossRef CAS PubMed.
- K. Maduna, N. Kumar, A. Aho, J. Wärnå, S. Zrnčević and D. Y. Murzin, ACS Omega, 2018, 3, 7247–7260 CrossRef CAS PubMed.
- G. Charles Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935–1943 CrossRef PubMed.
- B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- M. Zhao, S. Ou and C. de Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed.
- X. Wang, P. C. Lan and S. Ma, ACS Cent. Sci., 2020, 6, 1497–1506 CrossRef CAS PubMed.
- P. J. Deuss, R. Denheeten, W. Laan and P. C. J. Kamer, Chem.–Eur. J., 2011, 17, 4680–4698 CrossRef CAS PubMed.
- J. R. Bour, A. M. Wright, X. He and M. Dincǎ, Chem. Sci., 2020, 11, 1728–1737 RSC.
- C. Doonan, R. Riccò, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res., 2017, 50, 1423–1432 CrossRef CAS PubMed.
- M. Hartmann, Chem. Mater., 2005, 17, 4577–4593 CrossRef CAS.
- Z. Ji, H. Wang, S. Canossa, S. Wuttke, O. M. Yaghi, Z. Ji, H. Wang, O. M. Yaghi, S. Canossa and S. Wuttke, Adv. Funct. Mater., 2020, 30, 2000238 CrossRef CAS.
- N. Landaluce, M. Perfecto-Irigaray, J. Albo, G. Beobide, O. Castillo, A. Irabien, A. Luque, A. S. J. Méndez, A. E. Platero-Prats and S. Pérez-Yáñez, Sci. Rep., 2022, 12(1), 1–7 CrossRef PubMed.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Wöll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS PubMed.
- W. Schrimpf, J. Jiang, Z. Ji, P. Hirschle, D. C. Lamb, O. M. Yaghi and S. Wuttke, Chemical diversity in a metal–organic framework revealed by fluorescence lifetime imaging, Nat. Commun., 2018, 9, 1647 CrossRef PubMed.
- H. Lyu, Z. Ji, S. Wuttke and O. M. Yaghi, Chem, 2020, 6, 2219–2241 CAS.
- S. Canossa, Z. Ji, C. Gropp, Z. Rong, E. Ploetz, S. Wuttke and O. M. Yaghi, Nat. Rev. Mater., 2022, 2022, 1–10 Search PubMed.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- J. Hao, X. Xu, H. Fei, L. Li and B. Yan, Adv. Mater., 2018, 30, 1–22 Search PubMed.
- P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS PubMed.
- B. Moulin, F. Salles, S. Bourrelly, P. L. Llewellyn, T. Devic, P. Horcajada, C. Serre, G. Clet, J. C. Lavalley, M. Daturi, G. Maurin and A. Vimont, Microporous Mesoporous Mater., 2014, 195, 197–204 CrossRef CAS.
- P. G. Saiz, A. Valverde, B. Gonzalez-Navarrete, M. Rosales, Y. M. Quintero, A. Fidalgo-Marijuan, J. Orive, A. Reizabal, E. S. Larrea, M. I. Arriortua, S. Lanceros-Méndez, A. García and R. Fernández de Luis, Catalysts, 2021, 11, 51–60 CrossRef CAS.
- R. G. P. Saiz, N. Iglesias, B. Gonzalez-Navarrete, M. Rosales, Y. M. Quintero, A. Reizabal, J. Orive, A. Fidalgo-Marijuan, S. Lanceros-Mendez, M. I. Arriortua and F. de Luis, Chem.–Eur. J., 2020, 26, 1–13 CrossRef PubMed.
- Y. Peng, H. Huang, Y. Zhang, C. Kang, S. Chen, L. Song, D. Liu and C. Zhong, A versatile MOF-based trap for heavy metal ion capture and dispersion, Nat. Commun., 2018, 9, 187 CrossRef PubMed.
- A. Valverde, G. I. Tovar, N. A. Rio-López, D. Torres, M. Rosales, S. Wuttke, A. Fidalgo-Marijuan, J. M. Porro, M. Jiménez-Ruiz, V. G. Sakai, A. García, J. M. Laza, J. L. Vilas-Vilela, L. Lezama, M. I. Arriortua, G. J. Copello and R. F. de Luis, Chem. Mater., 2022, 34, 9666–9684 CrossRef CAS.
- R. Newar, N. Akhtar, N. Antil, A. Kumar, S. Shukla, W. Begum and K. Manna, Angew. Chem., Int. Ed., 2021, 60, 10964–10970 CrossRef CAS PubMed.
- J. Canivet, S. Aguado, G. Bergeret and D. Farrusseng, Chem. Commun., 2011, 47, 11650–11652 RSC.
- G. Chen, S. Huang, X. Kou, F. Zhu and G. Ouyang, Angew. Chem., Int. Ed., 2020, 59, 13947–13954 CrossRef CAS PubMed.
- S. Wu, Z. Sun, Y. Peng, Y. Han, J. Li, S. Zhu, Y. Yin and G. Li, Biosens. Bioelectron., 2020, 169, 112613 CrossRef CAS PubMed.
- T. Schnitzer, E. Paenurk, N. Trapp, R. Gershoni-Poranne and H. Wennemers, J. Am. Chem. Soc., 2021, 143, 644–648 CrossRef CAS PubMed.
- A. Mantion, L. Massüger, P. Rabu, C. Palivan, L. B. McCusker and A. Taubert, J. Am. Chem. Soc., 2008, 130, 2517–2526 CrossRef CAS PubMed.
- A. Valverde, D. Payno, L. Lezama, J. M. Laza, S. Wuttke and R. Fernández de Luis, Adv. Sustainable Syst., 2022, 6, 2200024 CrossRef CAS.
- A. N. Hong, E. Kusumoputro, Y. Wang, H. Yang, Y. Chen, X. Bu, P. Feng, A. N. Hong, E. Kusumoputro, Y. Wang, Y. Chen, P. Feng, H. Yang and X. Bu, Angew. Chem., Int. Ed., 2022, 61, e202116064 CrossRef CAS PubMed.
- J. D. Evans, V. Bon, I. Senkovska, H. C. Lee and S. Kaskel, Nat. Commun., 2020, 11(1), 1–11 CrossRef PubMed.
- L. Feng, G. S. Day, K. Y. Wang, S. Yuan and H. C. Zhou, Chem, 2020, 6, 2902–2923 CAS.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y. S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- T. Ikuno, J. Zheng, A. Vjunov, M. Sanchez-Sanchez, M. A. Ortuño, D. R. Pahls, J. L. Fulton, D. M. Camaioni, Z. Li, D. Ray, B. L. Mehdi, N. D. Browning, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 10294–10301 CrossRef CAS PubMed.
- C. Yi Hu, Z. Wei Jiang, C. Zhi Huang and Y. Fang Li, Microchim. Acta, 2021, 188, 272 CrossRef PubMed.
- Y. Pan, S. Jiang, W. Xiong, D. Liu, M. Li, B. He, X. Fan and D. Luo, Microporous Mesoporous Mater., 2020, 291, 109703 CrossRef CAS.
- S. Shams, W. Ahmad, A. H. Memon, Y. Wei, Q. Yuan and H. Liang, RSC Adv., 2019, 9, 40845–40854 RSC.
- O. Gholipoor and S. A. Hosseini, New J. Chem., 2021, 45, 2536–2549 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- H. Furukawa, F. Gándara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- J. Jiang, F. Gándara, Y. B. Zhang, K. Na, O. M. Yaghi and W. G. Klemperer, J. Am. Chem. Soc., 2014, 136, 12844–12847 CrossRef CAS PubMed.
- J. Pascual-Colino, B. Artetxe, G. Beobide, O. Castillo, M. L. Fidalgo-Mayo, A. Isla-López, A. Luque, S. Mena-Gutiérrez and S. Pérez-Yáñez, Inorg. Chem., 2022, 61, 4842–4851 CrossRef CAS PubMed.
- P. Deria, W. Bury, I. Hod, C. W. Kung, O. Karagiaridi, J. T. Hupp and O. K. Farha, Inorg. Chem., 2015, 54, 2185–2192 CrossRef CAS PubMed.
- K. I. Hadjiivanov, D. A. Panayotov, M. Y. Mihaylov, E. Z. Ivanova, K. K. Chakarova, S. M. Andonova and N. L. Drenchev, Chem. Rev., 2021, 121, 1286–1424 CrossRef CAS PubMed.
- G. P. de Sousa, P. T. C. Freire, J. M. Filho, F. E. A. Melo and C. L. Lima, Braz. J. Phys., 2013, 43, 137–144 CrossRef.
- P. T. C. Freire, F. M. Barboza, J. A. Lima, F. E. A. Melo, J. M. Filho, P. T. C. Freire, F. M. Barboza, J. A. Lima, F. E. A. Melo and J. M. Filho, in Raman Spectroscopy and Applications, IntechOpen, 2017 Search PubMed.
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS.
- P. K. Bharadwaj, E. John, J. A. Potenza, H. J. Schugar, C. L. Xie, D. Zhang and D. N. Hendrickson, Inorg. Chem., 1986, 25, 4541–4546 CrossRef CAS.
- N. Yoshinari, K. Tatsumi, A. Igashira-Kamiyama and T. Konno, Chem.–Eur. J., 2010, 16, 14252–14255 CrossRef CAS PubMed.
- T. M. Ivanova, K. I. Maslakov, A. A. Sidorov, M. A. Kiskin, R. v. Linko, S. v. Savilov, V. v. Lunin and I. L. Eremenko, J. Electron Spectrosc. Relat. Phenom., 2020, 238, 146878 CrossRef CAS.
- A. Alessandrini, S. Corni and P. Facci, Phys. Chem. Chem. Phys., 2006, 8, 4383–4397 RSC.
- C. R. Groom and F. H. Allen, Angew. Chem., Int. Ed., 2014, 53, 662–671 CrossRef CAS PubMed.
- J. Wang, R. Huang, W. Qi, R. Su, B. P. Binks and Z. He, Appl. Catal., B, 2019, 254, 452–462 CrossRef CAS.
- A. Azizi, M. Abouseoud and A. Amrane, Nat., Environ. Pollut. Technol., 2017, 16, 321–330 CAS.
- N. S. Inchaurrondo, P. Massa, R. Fenoglio, J. Font and P. Haure, Chem. Eng. J., 2012, 198–199, 426–434 CrossRef CAS.
- L. A. Lozano, A. Devard, M. A. Ulla and J. M. Zamaro, J. Environ. Chem. Eng., 2020, 8, 104332 CrossRef CAS.
- X. Zhong, Y. Lu, F. Luo, Y. Liu, X. Li and S. Liu, Chem.–Eur. J., 2018, 24, 3045–3051 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117(17), 11302–11336 CrossRef CAS PubMed.
- M. H. Robbins and R. S. Drago, J. Catal., 1997, 170, 295–303 CrossRef CAS.
- K. S. Banu, T. Chattopadhyay, A. Banerjee, S. Bhattacharya, E. Zangrando and D. Das, J. Mol. Catal. A: Chem., 2009, 310, 34–41 CrossRef CAS.
- M. Langerman and D. G. H. Hetterscheid, ChemElectroChem, 2021, 8, 2783–2791 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06198a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |