DOI:
10.1039/D3TA06790D
(Paper)
J. Mater. Chem. A, 2024,
12, 3501-3512
Pendent carboxylic acid-fuelled high-performance uranium extraction in a hydrogen-bonded framework and prolifically improved water oxidation via post-metalation-actuated composite fabrication†
Received
6th November 2023
, Accepted 2nd January 2024
First published on 3rd January 2024
Abstract
Pore functionality engineering in metal–organic frameworks (MOFs) benefits monitoring of uranium contamination via fluoro-sensing, and further promises its effective extraction. Alternatively, mixed-metal-based MOF-composites aid high-performance electrochemical oxygen evolution reaction (OER). Herein, we strategically built a fish-bone-shaped Co(II)-framework that contains pendent carboxylic acid moieties and displays high hydro-chemical stability. The activated MOF demonstrates selective and ultra-fast turn-off fluoro-detection of UO22+ in uranium-spiked water and simulated seawater with high recyclability. The detection limit (0.13 μM) outperforms that of the majority of sensory MOFs. The layer-stacked framework shows regenerable extraction of U(VI) ion with fast kinetics. Besides notable saturated uptake (129.8 mg g−1), uranium adsorption capacity reveals minor alteration over multiple cycles or in the presence of interfering cations. Apart from in-depth experimental support, the sensing and scavenging mechanisms are explicitly validated through a unique molecular scissoring approach by employing a pendent-functionality-truncated isostructural framework. The results mutually confirm the free –COOH group as a task-specific site for uranium detection and extraction. Redox-active Co(II) nodes further assist this MOF in alkaline medium electrochemical OER via a quasi-reversible Co2+/Co3+ couple with 331 mV overpotential and 62 mV dec−1 Tafel slope. Suitably oriented carboxylic acids benefit anchoring of Ni(II) to yield a hetero-bimetallic composite with increased active sites. Interestingly, the overpotential of this post-metalated material (308 mV) is far less than that of the pristine MOF. Significantly, the Tafel slope is 1.6 times reduced (38 mV dec−1) and ranks among top-tier OER catalysts, including benchmark (RuO2 and IrO2)/commercial (Co3O4 and NiO) ones. Both the pristine and post-modified MOFs divulged >97% faradaic efficiencies with 30 h chronopotentiometric stability and reusability up to 1500 cycles. Remarkably, the turnover frequency displays a 4-fold increment, subsequently boosting charge-transfer resistance by 3.5 times due to hetero-bimetallic synergy. The results set an example to drastically improve the OER performance in hydrogen-bonded MOFs via post-metalation of free carboxylic acids.
Introduction
As one of the foremost elementary substances in the nuclear industry, environmental contamination from uranium sources has become a critical concern owing to the rapid expansion of nuclear science and technology.1 The half-life of uranium and its isotopes ranges from one hundred thousand to 4.5 billion years, which has rendered uranium a persistent contaminant.2 Consequently, improper disposal of nuclear wastes and nuclear safety accidents frequently result in uranium leakage into the open environment.3 The most chemically stable species of uranium is the uranyl ion (UO22+), which is soluble in water4 and enters easily into the human body through the food chain. Awfully, its excessive ingestion causes acute damage to the liver, kidney, bones and even DNA, as well as induces severe red blood cell reduction; resulting in various diseases.5 At present, conventional methods for uranium detection include inductively coupled plasma-mass spectrometry (ICP-MS), ion chromatography, laser-induced kinetic phosphorescence analysis (KPA), and radioactivity measurement, which are costly and require time-consuming pre-treatment processes.6 In contrast, the fluorometric method attracts much attention as it involves specific interaction between uranium and the probe containing diverse functional groups. Despite being much more facile than direct methods, fluoro-detection often suffers from less sensitivity or selectivity. In addition to uranium detection, its efficient extraction is proven to ensure sustained power generation for years and is considered one of the seven most demanding chemical separations to bring revolutionary global benefits. This founded the development of porous adsorbents, including zeolites, clays, carbon materials, silica materials, and so forth.7,8 However, such materials generally possess the disadvantages of short active periods, reduced sorption capacity, slow kinetics, and poor regenerability. Therefore, fabrication of highly efficient state-of-the-art materials capable of detecting and adsorbing uranyl is of utmost necessity. Although both uranium sensing and sorption have been separately realized in some reports, their astute combination over a single platform is hitherto unexplored and likely to reduce the cost as well as save time and energy.
On the other hand, from the perspective of developing carbon-free fuels, implementation of hydrogen technology is considered the most advantageous over alternative energy sources.9 Among various H2 production methods, water electrolysis is considered superior due to its carbon-free feature and has become a research hotspot, which partly solves the exhaustion of fossil energy.10 Such electrolysis consists of the anodic oxygen evolution reaction (OER) and the cathodic hydrogen evolution reaction (HER), where the complex, multistep four-electron transfer OER is a bottleneck in water splitting because of its sluggish kinetics and adverse thermodynamics.11,12 Therefore, the most challenging part of the OER is to find an effective electrocatalyst with good activity, long-term stability, faster kinetics, and comparatively low overpotential. As contemporary materials, ruthenium and iridium oxides are yardstick electrocatalysts for the OER.13 However, their low natural abundance and high cost limit commercial usage. To alleviate these issues, the development of non-noble metal-based, active, and stable electrocatalysts is required. In this milieu, metal–organic frameworks (MOFs), constructed from metal ions and organic struts, have emerged as a fascinating class of novel porous materials owing to their high surface area, tunable pore functionalization, remarkable physico-chemical stability and prospects to establish structure–property synergies.14–18 These amazing structural features instigated researchers to check the applicability of MOFs in diverse areas such as fluorescence sensing, adsorption, heterogeneous catalysis, batteries, solar cells and so on.19–27 Specifically, pore-functionalization in MOFs via immobilizing carboxylic sites can profoundly facilitate their detection and scavenging performances towards targeted analytes. However, they hardly remain as task-specific sites due to the extreme tendency of coordination with metal ions. In addition, MOFs are extensively studied as electrocatalysts for water oxidation, owing to their accessible channels, which can assist in diffusion of the reactants, provide sufficient space for electrolytes and facilitate transportation of the evolved oxygen gas.28 Nevertheless, only a few examples are known where MOFs are directly employed as OER electrocatalysts.29,30 Clearly, further development of approaches to devise hetero-bimetallic MOF composites is much demanding to drive high current densities at low overpotentials via creation of more number of active sites and hetero-metallic synergy.
Herein, we synthesized a Co(II)-based, layer-stacked framework CSMCRI-21 (CSMCRI = Central Salt & Marine Chemicals Research Institute) from the 4,4′,4′′-tricarboxytriphenylamine (H3TCA) acid ligand and (E)-1,2-di(pyridin-4-yl)diazene (dpa) as the auxiliary linker. The MOF features a hydrogen-bonded two-dimensional structure and contains the in situ grafted dangling –COOH moiety as cation interaction sites.31–33 Alternatively, the redox-active Co2(COO)4 secondary building units (SBUs) facilitate electrocatalytic activity. The activated MOF (21a) demonstrates highly selective and fast responsive sensing of uranyl with a low detection limit (0.13 μM) and high reusability. Batch experiments show fast kinetics (∼98% adsorption within 4 h) in uranium scavenging while the sorption isotherm reveals excellent uptake capacity (129.8 mg g−1). Moreover, uranyl uptake and sensing were found to be highly recyclable, and the sorption mechanism was explicitly authenticated via diverse analytical tools and by employing a novel molecular scissoring approach (Scheme 1). Furthermore, 21a revealed decent OER activity in an alkaline medium with 331 mV overpotential and 62 mV dec−1 Tafel slope. The electrocatalytic OER parameters are drastically improved via fabrication of a hetero-bimetallic composite (Ni2+@21a) through grafting of the secondary redox-active Ni2+ ion to the pristine MOF (Scheme 1). Interestingly, the turnover frequency for Ni2+@21a was found to be four times higher (12.7 s−1) than that of 21a (3.45 s−1) while the Tafel slope of 38 mV dec−1 ranges among the top-ranked OER electrocatalysts. Noticeably, both the catalysts exhibit remarkable chronopotentiometric stability over 30 h with notable recyclability up to 1500 times for the OER.
 |
| | Scheme 1 Schematic overview of functionality-actuated uranium sensing/scavenging and improved electrochemical water oxidation through hetero-bimetallic composite fabrication. | |
Results and discussion
Synthesis and the crystal structure
The solvothermal reaction of H3TCA, dpa and Co(NO3)2·6H2O in water/N,N-dimethylformamide (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 72 h at 100 °C produces block shaped orange crystals. Single crystal X-ray diffraction (SCXRD) analysis revealed that CSMCRI-21 crystallizes in triclinic space group P
:1) for 72 h at 100 °C produces block shaped orange crystals. Single crystal X-ray diffraction (SCXRD) analysis revealed that CSMCRI-21 crystallizes in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , where the asymmetric unit consists of Co(II) ions, partially deprotonated H3TCA ligands (HTCA2−: L) and dpa linkers (Fig. S1a†), three each. Though disordered lattice solvent molecules were not crystallographically defined, their presence was ascertained from a collective outcome of thermogravimetric analysis (TGA) and PLATON34 calculation, which represents the molecular formula as [Co3(L)3(dpa)3]·3DMF·7H2O. In the crystal structure, each Co(II) ion is attached to four carboxylate oxygen atoms of three different L (average Co–O: 2.09 Å). One of them shows chelating bidentate mode while the other two L display bridging bidentate ligation, generating a dinuclear Co2(COO)4 secondary building unit (SBU). Interconnection of the SBUs via L forms ellipsoidal units, which are interlinked to generate a one-dimensional (1D) chain. The remaining two apical coordination sites of Co(II)-ion are satisfied with the pyridyl nitrogen atoms from two dpa linkers (average Co–N distance: 2.17 Å), which further connect the one-dimensional (1D) chains to a fish-bone type 2D layered network (Fig. S1b†). Strong π⋯π stacking interactions from the aromatic rings of HTCA2− moieties (centroid-to-centroid distance: 4.2 Å) interdigitate these 2D layers with each other (Fig. 1a). Interestingly, neighbouring 2D layers are stacked in such a fashion that two uncoordinated –COOH groups of L are oriented in opposite directions, and connected through hydrogen bonding interaction (O–H⋯O: 2.59 Å) (Fig. 1b). Structural simplification by TOPOS35 shows a 3,5-connected 2-nodal net (Fig. S1c†) with the Schlafli symbol {42.67.8}{42.6}.
, where the asymmetric unit consists of Co(II) ions, partially deprotonated H3TCA ligands (HTCA2−: L) and dpa linkers (Fig. S1a†), three each. Though disordered lattice solvent molecules were not crystallographically defined, their presence was ascertained from a collective outcome of thermogravimetric analysis (TGA) and PLATON34 calculation, which represents the molecular formula as [Co3(L)3(dpa)3]·3DMF·7H2O. In the crystal structure, each Co(II) ion is attached to four carboxylate oxygen atoms of three different L (average Co–O: 2.09 Å). One of them shows chelating bidentate mode while the other two L display bridging bidentate ligation, generating a dinuclear Co2(COO)4 secondary building unit (SBU). Interconnection of the SBUs via L forms ellipsoidal units, which are interlinked to generate a one-dimensional (1D) chain. The remaining two apical coordination sites of Co(II)-ion are satisfied with the pyridyl nitrogen atoms from two dpa linkers (average Co–N distance: 2.17 Å), which further connect the one-dimensional (1D) chains to a fish-bone type 2D layered network (Fig. S1b†). Strong π⋯π stacking interactions from the aromatic rings of HTCA2− moieties (centroid-to-centroid distance: 4.2 Å) interdigitate these 2D layers with each other (Fig. 1a). Interestingly, neighbouring 2D layers are stacked in such a fashion that two uncoordinated –COOH groups of L are oriented in opposite directions, and connected through hydrogen bonding interaction (O–H⋯O: 2.59 Å) (Fig. 1b). Structural simplification by TOPOS35 shows a 3,5-connected 2-nodal net (Fig. S1c†) with the Schlafli symbol {42.67.8}{42.6}.
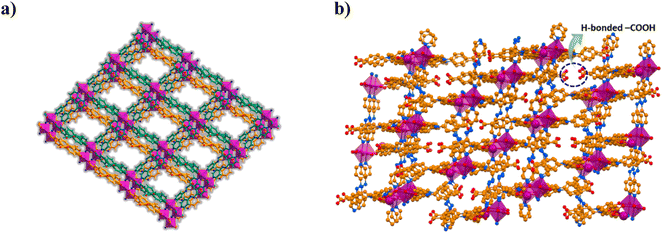 |
| | Fig. 1 (a) Layer-stacked architecture of CSMCRI-21. (b) View of the oppositely faced, hydrogen-bonded pair of pendent –COOH groups within the MOF. | |
Characterization and thermochemical stability
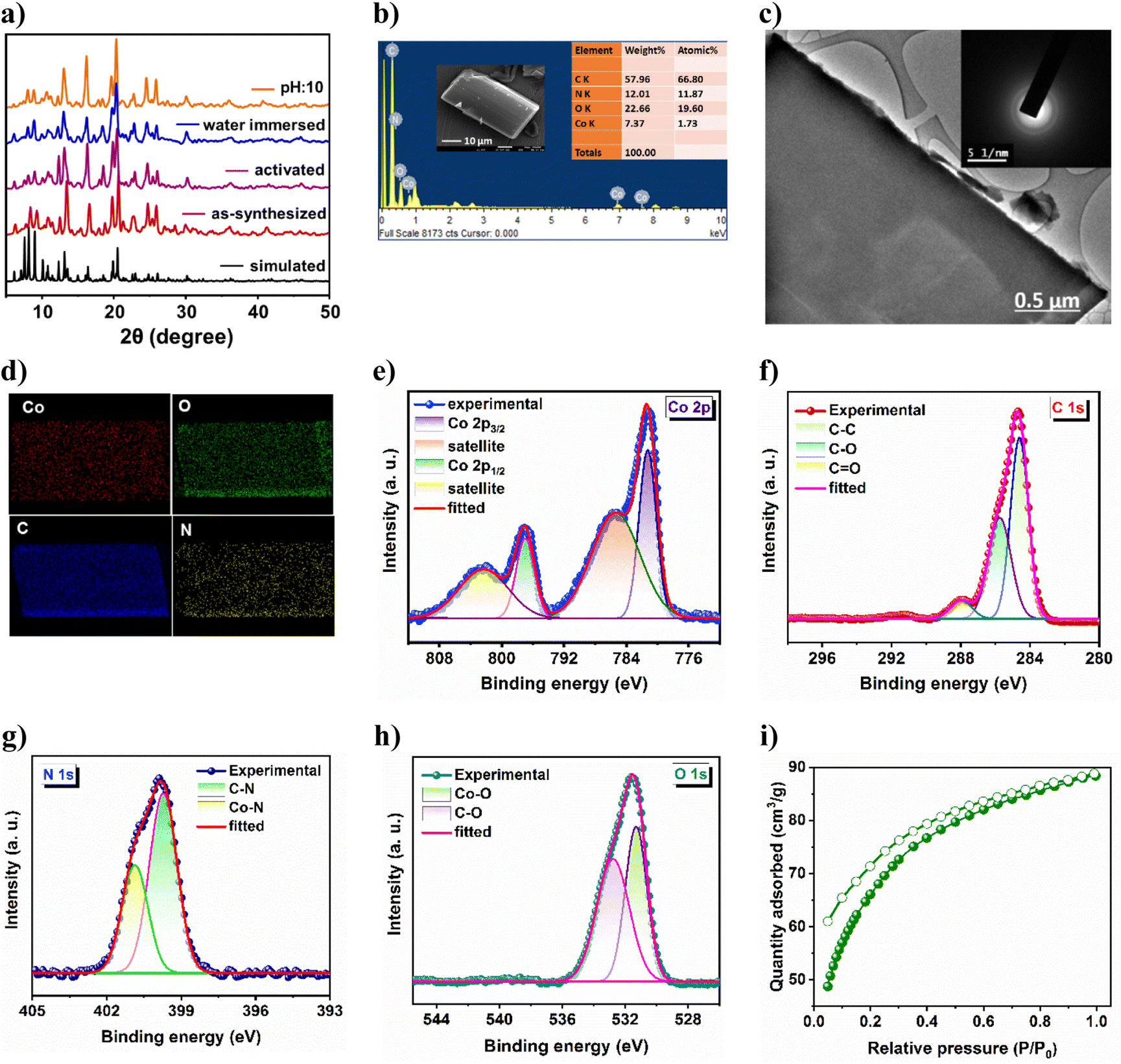
High crystallinity and phase purity of the as-synthesized crystals were confirmed from good agreement of powder X-ray diffraction (PXRD) peaks with those of the simulated one, derived from SCXRD (Fig. 2a). The lattice guest molecules in the framework were exchanged by immersing the as-synthesized crystals in lower boiling dichloromethane (DCM) for three days while the solvent was replenished with fresh DCM thrice daily. The guest-free framework (21a) was obtained by heating these solvent-exchanged crystals at 80 °C for 2 h followed by overnight degassing under a dynamic vacuum. From Fig. 2a, it is evident that the activated framework exhibits good agreement of PXRD pattern with CSMCRI-21, and supports retention of structural integrity after activation. The MOF was separately immersed in water and boiling water for 3 days and 24 h, respectively; and it was found to sustain its structural integrity under these conditions (Fig. 2a). In addition, MOF stability has also been observed in different pH media and in some common organic solvents (Fig. S2a and b†). Thermogravimetric analysis of CSMCRI-21 under a N2 atmosphere (30 to 800 °C) exhibited 15.19% (calc. 15.67%) weight loss up to 370 °C (Fig. S3†). Thereafter, an abrupt weight loss occurs, indicating framework decomposition. Field emission-scanning electron microscopy (FE-SEM) analysis of the as-synthesized crystals shows a rectangular block-shaped morphology with well-defined edges (Fig. 2b), which was also confirmed from transmission electron microscopy (TEM) of the crystal (Fig. 2c). Energy dispersive X-ray (EDX) analysis revealed uniformly distributed MOF constituting elements throughout the crystal surface (Fig. 2d). Furthermore, the selected area electron diffraction (SAED) pattern of the MOF endorses highly crystalline nature (Fig. 2c).
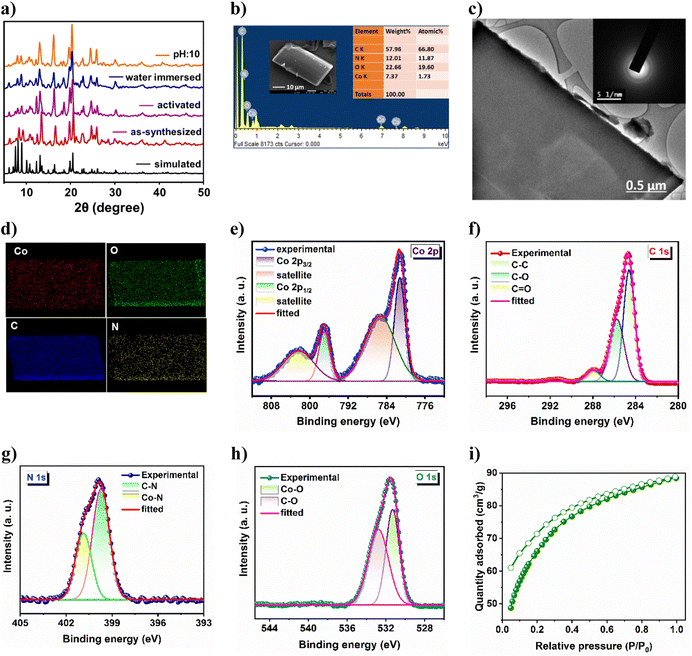 |
| | Fig. 2 (a) PXRD pattern of the MOF under several drastic conditions. (b) FE-SEM image and EDX analysis, (c) elemental mapping pattern and (d) TEM image and the corresponding SAED pattern of CSMCRI-21. High-resolution XPS spectrum of (e) Co 2p, (f) C 1s, (g) N 1s and (h) O 1s. (i) CO2 adsorption–desorption isotherm of the activated MOF at 195 K. | |
X-ray photoelectron spectroscopy (XPS) provided insights into the electronic structure as well as the surface composition of the as-synthesized material. The survey spectrum (Fig. S4†) endorses the presence of Co, C, N, and O in the sample. The high-resolution Co 2p spectrum (Fig. 2e) shows two binding energy peaks at 781.69 and 797.41 eV, corresponding to Co 2p3/2 and Co 2p1/2, respectively.36 The related shake-up satellites were also observed at 785.62 and 802.51 eV. The deconvoluted C 1s spectrum (Fig. 2f) exhibits three peaks at 284.63 eV, 285.75 eV and 287.97 eV, assigned to C–C, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species, respectively.37 The high-resolution N 1s spectrum (Fig. 2g) displayed two peaks with binding energies of 399.7 and 400.8 eV which are ascribed to C–N and Co–N moieties.38 Furthermore, the O 1s spectrum (Fig. 2h) reveals two deconvoluted peaks at 531.32 and 532.76 eV, corresponding to Co–O and C–O species, respectively.39 The activated framework showed negligible N2 uptake (5.45 cm3 g−1) at 77 K (Fig. S4b†). Although the interlayer framework channels are accessible towards N2 (kinetic diameter: 3.6 Å), the insignificant sorption is primarily because of the strong interaction of N2 molecules with the one-dimensional aperture (Fig. 1a) at very low temperature, which prevents further passage of gas, as also realized in earlier reports.40,41 Hence, the CO2 adsorption isotherm was recorded at 195 K,42 which revealed a maximum uptake of 88.39 cm3 g−1 (Fig. 2i), corresponding to a Brunauer–Emmett–Teller (BET) surface area of 309 m2 g−1.
O species, respectively.37 The high-resolution N 1s spectrum (Fig. 2g) displayed two peaks with binding energies of 399.7 and 400.8 eV which are ascribed to C–N and Co–N moieties.38 Furthermore, the O 1s spectrum (Fig. 2h) reveals two deconvoluted peaks at 531.32 and 532.76 eV, corresponding to Co–O and C–O species, respectively.39 The activated framework showed negligible N2 uptake (5.45 cm3 g−1) at 77 K (Fig. S4b†). Although the interlayer framework channels are accessible towards N2 (kinetic diameter: 3.6 Å), the insignificant sorption is primarily because of the strong interaction of N2 molecules with the one-dimensional aperture (Fig. 1a) at very low temperature, which prevents further passage of gas, as also realized in earlier reports.40,41 Hence, the CO2 adsorption isotherm was recorded at 195 K,42 which revealed a maximum uptake of 88.39 cm3 g−1 (Fig. 2i), corresponding to a Brunauer–Emmett–Teller (BET) surface area of 309 m2 g−1.
Selective detection of uranium (UO22+) by 21a
Although Co(II)-complexes are generally weakly fluorescent owing to d7 configuration, presence of highly conjugated H3TCA leads to marked photo-luminescent (PL) attributes in 21a. The UV-visible spectrum of the aqueous MOF dispersion revealed an absorption maximum at 337 nm (Fig. S5†). Upon excitation at this particular wavelength, 21a shows a strong luminescence signature at 454 nm, which is accredited to the mixed contribution of the π–π*/n–π* transition and intra-ligand charge transfer of two conjugated ligands, perturbed by the coordination effect to the metal center.43 Surmising on fluorescent nature coupled with good hydrolytic stability, we checked the detection capability of 21a towards the uranyl ion (UO22+) along with a series of environmentally abundant metal ions including Na+, K+, NH4+, Ca2+, Ni2+, Cu2+, Zn2+, Cd2+ and Sr2+. Luminescence spectra were collected for 21a with different concentrations of uranyl in deionized water solution (3 to 400 mg L−1, solid/liquid ratio = 1 g L−1). Delightedly, 96% quenching of the MOF emission intensity emerged (Fig. 3a) upon addition of 400 ppm of UO22+ solution, while moderate to negligible quenching was observed for the other cations (Fig. 3b). This phenomenon indicates that the MOF sensor is highly selective towards UO22+ over interfering ions. Interestingly, apart from PL quenching of 21a at 454 nm, additional weaker peaks appeared around 515 nm, which are assigned to the characteristic emission peaks of UO22+.44
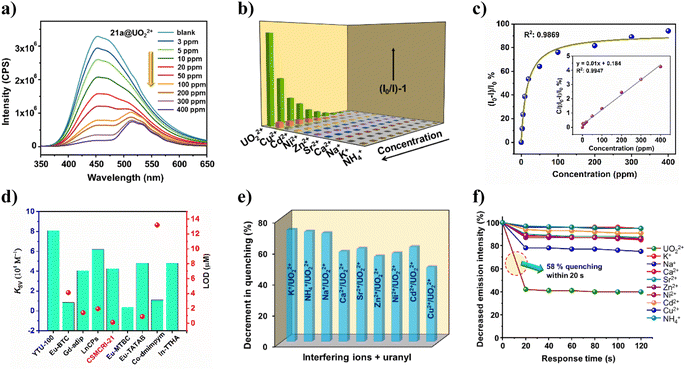 |
| | Fig. 3 (a) Luminescence quenching of 21a upon addition of uranyl nitrate solution (0 to 400 ppm). (b) Stern–Volmer plot for the studied cationic species. (c) Simulated correlation between (I0 − I)/I0% and the uranyl concentration using the Langmuir model. The inset shows the correlation between the uranyl concentration and C/[(I0 − I)/I0]%. (d) Comparison of KSV and LOD values for some uranium sensory probes. (e) Detection selectivity plot for UO22+ in the presence of interfering ions. (f) Decrease in fluorescence intensity (%) at different time intervals for uranyl, revealing its selective fast responsive sensing in the presence of other ions. | |
The correlation between the luminescence quenching ratio [denoted by (I0 − I)/I0%] and uranyl concentration could be successfully fitted with the Langmuir model (R2 = 0.9869) (Fig. 3c), which is generally used to describe the adsorption behavior. This signifies that the concentration-dependent luminescence quenching is highly correlated with the uranium uptake process.2 When the equation was mathematically transformed into the correlation between the uranyl concentration and C/[(I0 − I)/I0%] (C: uranyl concentration), a linear correlation with R2 = 0.9947 was established (inset of Fig. 3c). It is worth mentioning that 21a is also capable of detecting uranyl ions from simulated seawater with a nearly similar magnitude of quenching efficiency (82%). To quantitatively assess the sensitivity of UO22+ detection by 21a, the quenching constant (KSV) was measured from the Stern–Volmer (S–V) equation.45 The magnitude of KSV was found to be 4.26 × 104 M−1 (Fig. S7a†), which surpasses some recently reported MOF-based UO22+ sensors and is comparable to other probes as well (Fig. 3d).4,46 Additionally, a pronounced curvature of the S–V plot at higher concentration (Fig. 3b) points to the concurrent existence of static and dynamic quenching. Furthermore, the limit of detection (LOD)47 for UO22+ turned out to be as low as 0.13 μM/0.24 ppm, which is much less than that of most of the previously reported uranium sensory probes (Table S4†). While a handful of reports exist for uranium sensing using a MOF, such a low detection limit essentially endorses 21a as an ultrasensitive probe towards the minute presence of this radioactive element in water.
Anti-interfering and fast-responsive detection of uranium
To evaluate the anti-interference ability of 21a for uranyl detection, we performed competitive fluorescence titration experiments in the presence of related congeners. The UO22+ solution was gradually added to the MOF dispersion that contains interfering cations. Delightedly, no significant interference could be witnessed from other metal ions; but drastic quenching was perceived in each case upon the addition of uranyl ions (Fig. 3e). Such outcomes reveal that the present probe is highly selective toward UO22+ even in the presence of eight different cationic species. Given that the quick response of any sensory probe is highly desirable for in-field application, we next checked the optical response of 21a towards uranyl as well as other ions in a time-variable fashion. For this, after recording the PL intensity of bare MOF dispersion, 50 μL of uranyl solution was added and 58% quenching of MOF emission was realized within 20 s that remained unaffected for a further 100 s (Fig. 3f). The same protocol was followed for other metal ions as well, but trivial responses were observed. To check the recyclability of 21a for uranyl sensing, we recovered the probe through centrifugation and washed it with DCM before using it for the next sensing cycle. Gratifyingly, the used MOF restored pristine luminescence intensity, and consequent quenching efficacy upon uranyl addition up to five successive cycles (Fig. S8†). These outcomes mutually validate the selective, recyclable and fast-responsive detection of UO22+ by the present MOF.
Uranium uptake studies
Building on highly effective uranyl sensing together with the pendent carboxylic acid functionalization, we envisaged the ability of 21a in uranium ion adsorption in the aqueous phase. At the outset, extraction was checked in solutions of different pH values. As portrayed in Fig. S9†, the uranium removal rate gradually increased from pH 4 (46%) to pH 5 (97%), and remained nearly constant up to pH 7, followed by a decrease at pH 8. On the basis of framework stability and speciation of uranium in different pH media, the influence of pH on adsorption is explained. Below pH 4, the MOF structure may start collapsing that leads to minor adsorption. It is worth mentioning that the hydrolysed oligomeric/colloidal species, like (UO2)(OH)+, (UO2)2(OH)22+, and (UO2)3(OH)5+ are predominant in the pH range 6–8, while uranium exists mostly as UO22+ in the aqueous solution at pH less than 5.32 Considering these, pH 5 was used as optimum for further experiments. Adsorption kinetics as a function of time (using 10 ppm uranyl nitrate solution) revealed that (Fig. 4a) uptake by 21a increases with an increase in time, and 98% uranium gets adsorbed within 4 h. Between the two kinetic models, the experimental adsorption data was best fitted (R2: 0.999) to pseudo-second-order (inset of Fig. 4a). These results indicate that the rate-determining step for the pseudo-second-order model is chemisorption, which involves valence forces through sharing or exchange of electrons between the adsorbent and adsorbate.
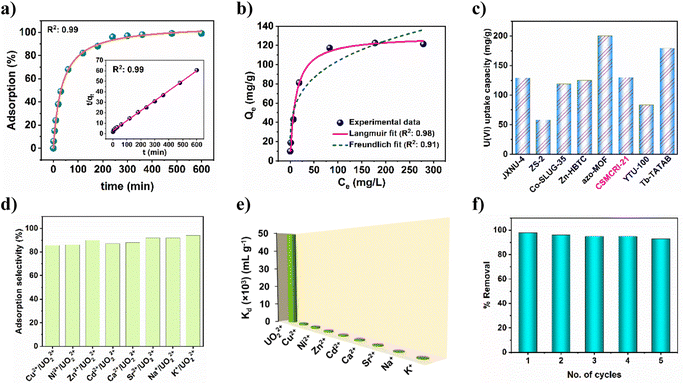 |
| | Fig. 4 (a) Uranium adsorption kinetics using 10 ppm initial concentration. The inset shows the corresponding pseudo-second-order fitting model. (b) Adsorption isotherm of uranyl by 21a fitted with the Langmuir and Freundlich models. (c) Comparison of maximum uptake capacities for some uranyl adsorbents. (d) Adsorption selectivity of 21a among interfering cations. (e) Distribution coefficients of diverse cations. (f) Recyclability of uranyl adsorption by 21a. | |
To determine maximum uptake capacity, we conducted batch adsorption experiments with different initial concentrations of uranium(VI) ions. As displayed in Fig. 4b, uranium adsorption capacity intensifies as the concentration increases. However, after a certain extent, the uptake capacity remains negligibly altered. In-depth analysis of the experimental data using Langmuir and Freundlich adsorption isotherms suggests that the former model is more appropriate to describe the adsorption behaviour of 21a, as confirmed by the relatively higher R2 value (0.98). The maximum adsorption capacity was calculated to be 129.8 mg g−1, which approaches the experimental value (121.3 mg g−1). It is imperative to stress that the magnitude of maximum uptake capacity surpasses many earlier reports (Fig. 4c and Table S5†).2,33,48 In a separate experiment, we observed 56% uranium removal efficiency by 21a from simulated seawater (Fig. S10a†).
Selective and regenerable UO22+ adsorption
To study the selectivity of 21a in UO22+, adsorption, we performed additional experiments using a series of competing metal ions (Na+, K+, Ca2+, Ni2+, Cu2+, Zn2+, Cd2+ and Sr2+). The results show that the sorption capacity of uranium is still high and varies between 86 and 94% (Fig. 4d) in these mixed solutions of diverse metal ions. Clearly, this represents excellent selective uptake capacity of uranyl by 21a even under such high ionic strength conditions. To further evaluate the affinity of this MOF towards uranium ions, the distribution coefficient (Kd) was calculated. In general, materials with a Kd value of >104 mL g−1 are considered good adsorbents for uranium recovery.49 The Kd value for 21a was calculated to be 4.9 × 104 mL g−1, thereby indicating its high affinity towards uranyl. Furthermore, selective adsorption of uranyl from a binary mixture of competitive metal ions was also carried out (Fig. 4e), and the selectivity (SU/M) was calculated from the ratio of Kd(U):Kd(M). Table S6† reveals that 21a offers superior selectivity towards the uranyl ion over other competitive cations in the water, and suggests the potential of this material for promising uranium extraction. For practical application purposes, regeneration and reusability of an adsorbent is crucial from an environmental and economic viewpoint. The adsorbed uranium could be completely removed by 0.1 M aqueous Na2CO3 solution. Subsequently, the recovered material could be recycled up to five times with maintenance of a high uranium removal percentage (Fig. 4f). In addition, PXRD analysis of this recycled material divulged comparable pattern to that of the pristine MOF (Fig. S11a†) while the FE-SEM image revealed unaltered morphology (Fig. 5b), corroborating the retention of structural integrity. Furthermore, ICP-MS of the supernatant revealed negligible presence of Co2+ ions, thereby nullifying any framework leaching. All these analyses indicate the excellent durability and regenerability of 21a in uranium extraction.
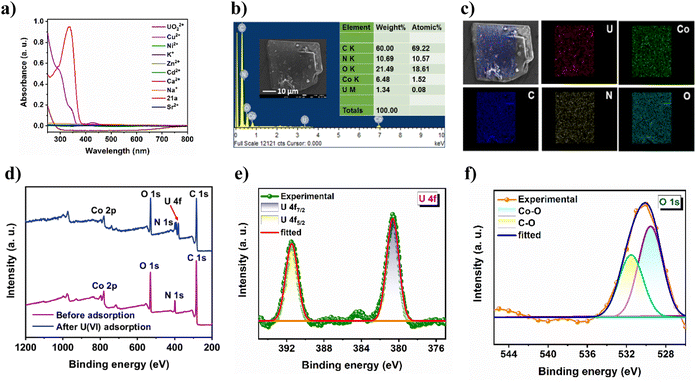 |
| | Fig. 5 (a) UV-vis spectral overlap of 21a with uranyl, substantiating absorption of excitation light by U(VI). (b) FE-SEM image and EDX pattern and (c) elemental mapping of UO22+@21a. (d) XPS survey spectra of the MOF before and after U(VI) adsorption. High-resolution (e) U 4f and (f) O 1s spectrum of UO22+@21a. | |
Mechanistic aspects of luminescence quenching and adsorption of uranium
PXRD results (Fig. S11a†) clearly show that fluorescence quenching of 21a by the uranyl ion is not caused by framework degradation. Therefore, the luminescent response of the MOF toward the uranyl ion can only be attributed to the strong interaction between the analyte and the framework. For comparison, the emission of the ligand (H3TCA) in the presence of U(VI) was also investigated (Fig. S11b†), which shows significantly less quenching to that of MOF dispersion. This can be attributed to the spatial separation of pendent carboxylic acid groups within the rigid framework architecture that leads to increased interaction with U(VI), which is otherwise not possible in the ligand because of self-aggregation via strong intermolecular hydrogen-bonding interaction. Furthermore, it is clear from Fig. 5a that the absorption spectra of the MOF show a significant overlap to that of uranyl nitrate in the region 285–450 nm, and points that the uranyl ion absorbs the excitation light, leading to such efficient turn-off PL.50 To acquire deeper insight into the uranium uptake phenomenon, we conducted a series of post-adsorption experiments. For example, the FT-IR spectrum of the uranium-loaded MOF (Fig. S13†) displayed a prominent new peak at 934 cm−1, which is characteristic of the antisymmetric stretching vibration of uranyl, and suggests a strong interaction between the framework skeleton and UO22+.51 Moreover, FE-SEM analysis of the reused material showed identical surface morphology to that of the as-synthesized CSMCRI-21 (Fig. 5b), discarding any structural breakdown.
Interestingly, SEM-EDX analysis revealed the presence of uranium along with MOF constituents (Fig. 5b) while elemental mapping shows the uniform distribution of uranium all over the crystal surface (Fig. 5c). In addition, the wide-scan XPS spectrum of UO22+@21a shows additional binding energy peaks of U along with the MOF elements (Fig. 5d). The high-resolution U 4f spectrum (Fig. 5e) shows two binding energy peaks at 392.26 and 381.57 eV, corresponding to U 4f5/2 and U 4f7/2, respectively. Notably, both the values are lower than those of uranyl nitrate at 392.94 and 382.10 eV,31 demonstrating the direct interaction between uranyl and 21a. To further verify such interaction, we analyzed the high-resolution XPS spectrum of C 1s, N 1s, and O 1s. While C 1s did not show any shift after the adsorption of U(VI) (Fig. S14a†), the C–O moiety in the O 1s spectrum (Fig. 5f) as well as C–N species in the N 1s spectrum (Fig. S14b†) of UO22+@21a are markedly shifted towards lower binding energies as compared to that of the pristine sample, indicating that both carboxyl groups and N-functionalities participate in chelation of U(VI) with the MOF structure.
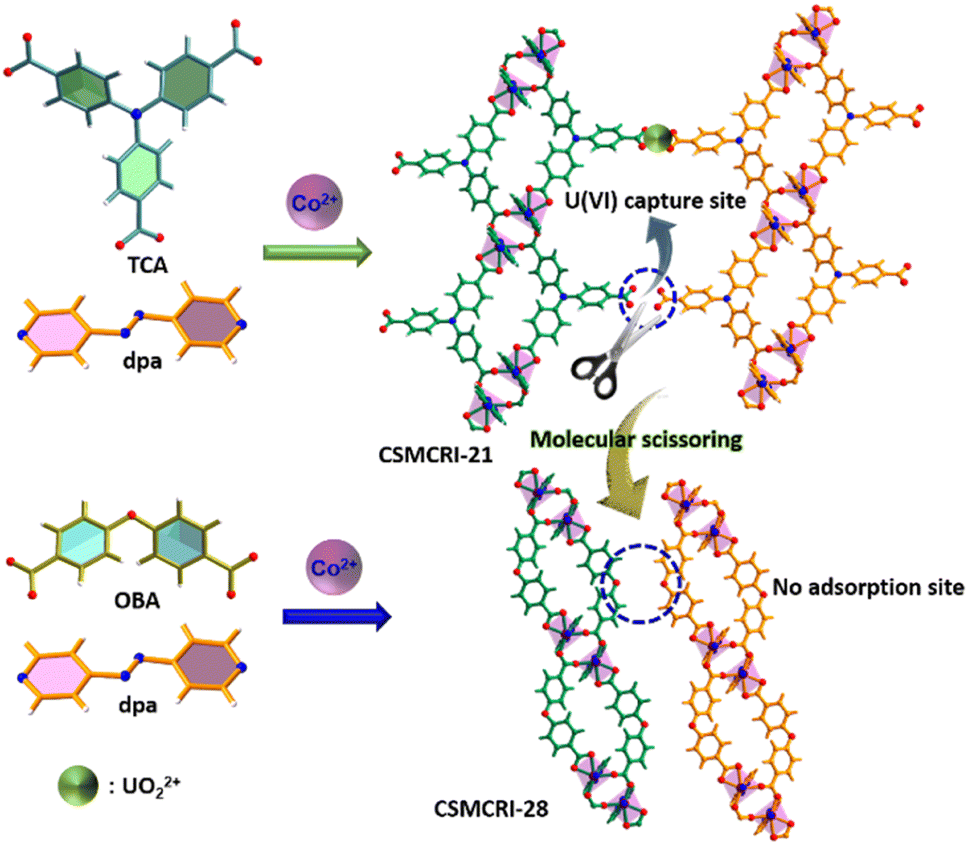
To obtain more explicit proof of the fact that free –COOH groups act as U(VI) binding sites in the present MOF, we employed a molecular-scissoring approach, which includes deliberate trimming of flanked acid groups. In principle, this linker-sheared unfunctionalized MOF should impede sensing and/or adsorption of UO22+ if pendent carboxylates acted as the sole interaction site for these applications. Fortunately, we were able to grow the single crystals of CSMCRI-28 from 4,4′-oxybis(benzoic acid) (H2oba) and the dpa linker that possesses exactly a similar structure to that of CSMCRI-21 but lacks the pendant –COOH group (Scheme 2). To our delight, this new framework showed neither uranium sensing nor uptake (Fig. S10†), owing to the absence of a third carboxylic acid in its arm of oba2−. In a separate control experiment, we blocked the available carboxylic acid by performing a post-synthetic Ni2+ grafting (vide infra), and found no uranium adsorption for the post-modified material (Ni2+@21a) (Fig. S10†). These results unequivocally corroborate the role of dangling carboxylic acid moieties as the prime task-specific functionality for uranium extraction.
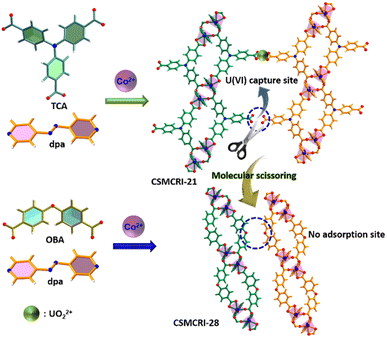 |
| | Scheme 2 Schematic representation of mechanistic elucidation for uranyl capture via the molecular-scissoring strategy. | |
Electrochemical water oxidation
Activated CSMCRI-21 has the potential to perform as an electrocatalyst in the oxygen evolution reaction (OER) because of its multiple structural features such as presence of (i) ample redox-active Co(II) ions, (ii) highly-electron rich and conjugated aromatic struts and (iii) ample stability in an alkaline medium. For the OER study, the activated MOF was used as the working electrode, Hg/HgO was used as the reference electrode, and carbon cloth as the counter electrode. The OER activity of 21a was studied via linear sweep voltammetry (LSV) in 1.0 M KOH (scan rate: 5 mV s−1) to reach 10 mA cm−2 current density. The overpotential turned out to be 331 mV (Fig. 6a), which is lower than that of commercial catalysts (Fig. 6a) or some Co(II) MOFs (Table S7†). Further investigation on the OER kinetics from Tafel plot analysis revealed a Tafel slope of 62 mV dec−1 (Fig. 6b), which outperforms that of benchmark RuO2 and commercial Co3O4, NiO (84 mV dec−1), and IrO2 (100.47 mV dec−1) and denotes high rate of electron transfer. A quick cycling study with a scan rate of 200 mV s−1 up to 1500 cycles displayed only an 18 mV change in the overpotential (Fig. 6a), thereby pointing to the great stability of this MOF. Furthermore, the E–t curve for chronopotentiometry showed negligible alteration up to 30 h (Fig. 6e), again certifying a high degree of MOF stability. The faradaic efficiency (FE) for water oxidation was evaluated by gas chromatographic analysis to measure the amount of evolved oxygen gas from the electrochemical cell while chronoamperometry was conducted for 1 h at a specific time interval of 12 min (Fig. S21 and S22†). The FE value is calculated to be 97%, which exceeds that of many MOF materials and confirms this framework as an efficient OER catalyst. Succeedingly, pH dependence OER activity was examined which revealed enhancement of electrocatalytic performance with the increased pH of the electrolyte solution (Fig. 6c).
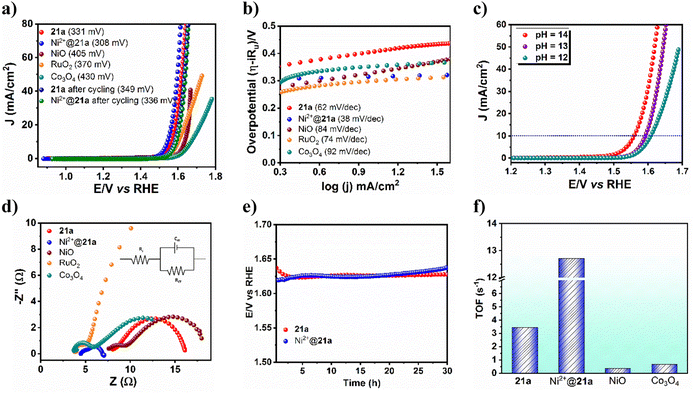 |
| | Fig. 6 (a) Linear sweep voltammetry (LSV) and (b) Tafel plot for 21a, Ni2+@21a as well as other catalysts. (c) pH-dependent LSV plot for 21a. (d) Nyquist plots of 21a, Ni2+@21a and other commercial as well as benchmark catalysts. (e) Chronopotentiometric study of 21a and Ni2+@21a for 30 h. (f) TOF values of 21a, Ni2+@21a and other commercial catalysts. | |
Moreover, Nyquist plot-derived charge-transfer resistance (Rct) was found to be 8.06 Ω (Fig. 6d), which showed marginal shift upon post-cycling, meaning that 21a can endure the OER activity with a high degree of stability and can sustain its long-term usability. Note that these values are lower than that of the remaining catalysts like commercial NiO (10.37 Ω), commercial Co3O4 (8.35 Ω) and RuO2 (34.17 Ω), and designates faster charge transfer in this fish-bone-shaped structure. Further insights into the intrinsic catalytic activity of the MOF were obtained from the electrochemically active surface area (EASA), roughness factor (Rf), and turnover frequency (TOF). The EASA has been evaluated from cyclic voltammetry (CV) in the non-faradaic potential region by varying the scan rate and found to be 0.17625 cm2. The TOF for the 21a-catalyzed OER is assessed to be 3.45 s−1 (Fig. 6f). As the long-term durability of any electrocatalyst is very important, we checked the stability of 21a after the OER. The PXRD pattern of the recovered catalyst revealed good agreement with that of the pristine one (Fig. S26†), confirming the retention of structural integrity. While FE-SEM images show similar morphology (Fig. S27†), XPS analysis revealed almost unaltered peak patterns and intensities (Fig. S28†) to that of CSMCRI-21. Moreover, negligible leaching of metal ions was evinced from ICP-MS analysis. These outcomes mutually confirm the excellent robustness of the present catalysts in the OER.
A plausible mechanism for 21a-catalysed alkaline oxygen evolution is proposed (eqn (1)–(4)) based on experimental outcomes and literature reports.52 At the onset, hydroxide ions (OH−) interact with the metal-centre (M: Co/Ni) to generate M⋯O–H species. CV of 21a revealed an obvious quasi-reversible couple for the Co2+/Co3+ oxidation process (Fig. S17†). Subsequently, reaction with another OH− forms the M⋯O intermediate through the elimination of H2O. In the next step, a metal-oxyhydroxide (M⋯OOH) intermediate is formed via concurrent attack of a third OH−. Finally, the last OH− abstracts a proton from oxyhydroxy species to release H2O while oxygen is liberated from the metal centre with regeneration of the pristine MOF. As per earlier reports, the Tafel slopes of 120, 60, and 40 mV dec−1 of a catalyst subjected to the OER resemble the first step (one-electron transfer), second step (two-electron transfer), and third step (three-electron transfer), if this were the rate-determining step (RDS).53 Therefore, the given Tafel slope of 21a indicates the second step (two-electron transfer) as the RDS.
| | | M + OH− → M···O–H + e− | (1) |
| | | M···O–H + OH− → M···O + H2O + e− | (2) |
| | | M···O + OH− → M···O–OH + e− | (3) |
| | | M···O–OH + OH− → M + O2 + H2O + e− | (4) |
Having attained such proficient OER activity for 21a, we endeavoured further boosting the water oxidation potential of the catalyst through heterobimetallic composite fabrication. In principle, the pendent –COOH groups, perfectly oriented in an end-on fashion, provide a platform to graft another redox-active and non-noble Ni2+ ion. The obtained hetero-bimetallic composite (Ni2+@21a) (Scheme 3) was thoroughly characterized. While FE-SEM analysis showed preservation of block-shaped morphology (Fig. 7a), the PXRD pattern (Fig. S15†) supported the maintenance of the basic structural integrity of CSMCRI-21. It may be stressed that we performed SEM-EDX (Fig. 7b) elemental mapping in a cross-section of Ni2+@21a crystals to confirm that grafted Ni is present in the interior of the MOF as well. Furthermore, XPS analysis of Ni2+@21a (Fig. 7c and Fig. S16†) revealed successful grafting of Ni within the material. Comparative LSV for Ni2+@21a under similar measurement conditions (scan rate: 5 mV s−1, 1 M KOH) revealed 308 mV overpotential (Fig. 6a) to reach a current density of 10 mA cm−2, which is lower than that of 21a and corresponds to hetero-bimetallic synergy that facilitates creating more number of active sites (Scheme 1).54 Importantly, the magnitude of the Tafel slope is 1.6 times less for Ni2+@21a (38 mV dec−1) than that of the pristine MOF, indicating its highest rate of electron transfer among all at the said interface. What is more, Co3O4 furnished a Tafel slope of 92 mV dec−1, which is 2.4 times higher than that of Ni2+@21a, describing the superiority of this hetero-bimetallic MOF composite over commercial catalysts in water oxidation. A pH-dependent LSV measurement for Ni2+@21a showed that higher current densities are produced with an increase in the pH of the solution (Fig. S18†), and suggests improvement in the efficiency of oxygen evolution with increasing OH− concentration, as also realized in previous reports.55,56 Further, the EIS study divulged that Ni2+@21a possesses nearly 3.5 times lower Rct value (2.47 Ω) than 21a (8.06 Ω), implying much smoother charge transfer in the former. A pH-dependent EIS study (Fig. S20†) denoted that the Rct value becomes lower at higher pH, corresponding to faster charge transfer.
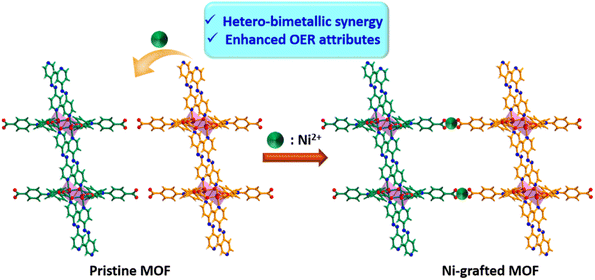 |
| | Scheme 3 Schematic representation of improved OER activity of Ni2+@21a through hetero-bimetallic synergy. | |
 |
| | Fig. 7 (a) FE-SEM image/EDX analysis and (b) elemental mapping of Ni2+@21a, showing uniform distribution of all the respective elements. (c) XPS survey spectra of the MOF before and after Ni2+ incorporation. | |
The long-standing static stability for Ni2+@21a was determined using chronopotentiometric studies for 30 h (Fig. 6e), which revealed negligible degradation in the activity after such a long time of continuous potentiostatic analysis. In addition, only a 28 mV change in overpotential was observed even after 1500 cycles (Fig. 6a), confirming the high-scale durability of the post-metalated 21a. Furthermore, FE measurement showed 98.3% for Ni2+@21a, suggesting that the applied current is almost fully utilized to produce O2. Magnitude of EASA is found to be improved to 0.22175 cm2 for Ni2+@21a compared to 21a, indicating highly exposed surface areas. Finally, the turnover frequency (TOF) for Ni2+@21a was calculated to be 12.7 s−1, which is nearly four times higher as compared to 21a (3.45 s−1) (Fig. 6f). These results essentially demonstrate much superior performance of the hetero-bimetallic analogue over the pristine MOF in electrochemical water oxidation and set an example to drastically improve the catalytic performance via hitherto unexplored post-metalation of free carboxylic acid groups.
Conclusion
In summary, we purposefully devised a fish-bone-shaped two-dimensional (2D) Co(II) framework, CSMCRI-21 using a mixed ligand approach. The layer-stacked structure contains dangling Brønsted acidic sites and exhibits sufficient hydrolytic and chemical stability. The luminescent feature of this MOF is effectively harnessed in ultra-fast, selective detection of uranyl ions (UO22+) in the aqueous phase and simulated seawater. The quenching constant (4.26 × 104 M−1) and limit of detection (0.13 μM) rank among the best-reported values. The MOF further acts as a recyclable adsorbent for U(VI) ions with 98% uptake in just 4 h. The experimental kinetic data correlate with the pseudo-second-order model (R2: 0.99), and the maximum adsorption capacity turned out to be 121.3 mg UO22+ per g of MOF. The results outperform the majority of the MOF reports for uranium ion scavenging. The prime role of the free –COOH group as a task-specific-site in uranium sensing and capture is unambiguously validated from the performance comparison of an isostructural MOF without a pendent moiety and deliberate blocking of free carboxylic-acid functionality. The presence of redox-active Co2(COO)4 nodes renders the activated MOF (21a) an excellent electrocatalyst for the oxygen evolution reaction (OER) in an alkaline medium. The MOF requires a low overpotential of 331 mV to reach an anodic current density of 10 mA cm−2, and exhibits an admirable 62 mV dec−1 Tafel slope. Interestingly, post-metalation of the dangling carboxylic-acid moiety by Ni2+ ions leads to devising a hetero-bimetallic composite (Ni2+@21a). Both the materials display excellent chronopotentiometric stability up to 30 h, along with more than 97% faradaic efficiencies. Remarkably, the overpotential (308 mV), Tafel slope (38 mV dec−1), and turn-over frequency (12.7 s−1) for water oxidation are prolifically boosted compared to those of the pristine MOF and signifies a very high rate of the OER in the composite. It is worth mentioning that the electrochemical parameters of Ni2+@21a surpass those of commercial catalysts and rank among the best-reported values owing to hetero-bimetallic synergy. Our design principle sheds light on diverse modes of functionality engineering in H-bonded 2D MOFs for efficient detection and extraction of uranium as well as drastic augmentation of the OER potential for promising renewable energy generation.
Author contributions
Nilanjan Seal: material synthesis & characterization, data curation, formal analysis, writing original draft. Arun Karmakar: experimentation of electrochemical water oxidation, writing original draft. Subrata Kundu: writing – review and editing, supervision, funding acquisition. Subhadip Neogi: writing – review and editing, supervision, funding acquisition.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
N. S. acknowledges the CSIR, New Delhi, India, for a research fellowship (CSIR Grant No. 31/028(0252)/2019-EMR-I). A. K. acknowledges CSIR, New Delhi for SRF (CSIR Grant No. 31/020(0190)/2019-EMR-I). S. K. acknowledges the Department of Science and Technology (DST) for CRG (Core Research Grant) funding number #CRG/2021/001089 dated November 20, 2021, with CSIR-CECRI project number GAP 22/2021. S. N. acknowledges DST-SERB (CRG/2021/002529) for financial support. The analytical and instrumental facility from AESD & CIF (CSMCRI) is gratefully acknowledged. CSMCRI communication no. 124/2023.
References
- S. Mollick, S. Saurabh, Y. D. More, S. Fajal, M. M. Shirolkar, W. Mandal and S. K. Ghosh, Energy Environ. Sci., 2022, 15, 3462–3469 RSC.
- M. Lei, Y. Jia, W. Zhang, J. Xie, Z. Xu, Y. Wang, W. Du and W. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 51086–51094 CrossRef CAS PubMed.
- X.-F. Wang, Y. Chen, L.-P. Song, Z. Fang, J. Zhang, F. Shi, Y.-W. Lin, Y. Sun, Y.-B. Zhang and J. Rocha, Angew. Chem., Int. Ed., 2019, 58, 18808–18812 CrossRef CAS PubMed.
- M. Wang, Z. Liu, X. Zhou, H. Xiao, Y. You and W. Huang, Inorg. Chem., 2020, 59, 18027–18034 CrossRef CAS PubMed.
- J. Q. Li, L. L. Gong, X. F. Feng, L. Zhang, H. Q. Wu, C. S. Yan, Y. Y. Xiong, H. Y. Gao and F. Luo, Chem. Eng. J., 2017, 316, 154–159 CrossRef CAS.
- W. Liu, X. Dai, Z. Bai, Y. Wang, Z. Yang, L. Zhang, L. Xu, L. Chen, Y. Li, D. Gui, J. Diwu, J. Wang, R. Zhou, Z. Chai and S. Wang, Environ. Sci. Technol., 2017, 51, 3911–3921 CrossRef CAS PubMed.
- H. Zhang, W. Liu, A. Li, D. Zhang, X. Li, F. Zhai, L. Chen, L. Chen, Y. Wang and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 16110–16114 CrossRef CAS PubMed.
- Y. Yuan, Q. Meng, M. Faheem, Y. Yang, Z. Li, Z. Wang, D. Deng, F. Sun, H. He, Y. Huang, H. Sha and G. Zhu, ACS Cent. Sci., 2019, 5, 1432–1439 CrossRef CAS PubMed.
- A. De, A. Karmakar and S. Kundu, ACS Appl. Energy Mater., 2023, 6, 5761–5773 CrossRef CAS.
- A. Karmakar, R. Jayan, A. Das, A. Kalloorkal, M. M. Islam and S. Kundu, ACS Appl. Mater. Interfaces, 2023, 15, 26928–26938 CrossRef CAS PubMed.
- N. Seal, K. Karthick, M. Singh, S. Kundu and S. Neogi, Chem. Eng. J., 2022, 429, 132301 CrossRef CAS.
- A. Karmakar and S. Kundu, Mater. Today Energy, 2023, 33, 101259 CrossRef CAS.
- A. Karmakar, D. Mahendiran, R. Madhu, P. Murugan and S. Kundu, J. Mater. Chem. A, 2023, 11, 16349–16362 RSC.
- H. Furukawa, E. Cordova Kyle, M. O'Keeffe and M. Yaghi Omar, Science, 2013, 341, 1230444 CrossRef PubMed.
- X. Zhang, Z. Chen, X. Liu, S. L. Hanna, X. Wang, R. Taheri-Ledari, A. Maleki, P. Li and O. K. Farha, Chem. Soc. Rev., 2020, 49, 7406–7427 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- N. Seal and S. Neogi, ACS Appl. Mater. Interfaces, 2021, 13, 55123–55135 CrossRef CAS PubMed.
- N. Seal and S. Neogi, Chem. Commun., 2023, 59, 4954–4957 RSC.
- S. C. Pal, R. Ahmed, A. K. Manna and M. C. Das, Inorg. Chem., 2022, 61, 18293–18302 CrossRef CAS PubMed.
- L.-H. Liu, L. Liu, H.-R. Chi, C.-N. Li and Z.-B. Han, Chem. Commun., 2022, 58, 6417–6420 RSC.
- D. Mei and B. Yan, ACS Appl. Mater. Interfaces, 2023, 15, 16882–16894 CrossRef CAS PubMed.
- T. K. Pal, Mater. Chem. Front., 2023, 7, 405–441 RSC.
- H. Lv, H. Chen, L. Fan and X. Zhang, Inorg. Chem., 2022, 61, 15558–15568 CrossRef CAS PubMed.
- N. Seal and S. Neogi, Mater. Today Chem., 2022, 26, 101064 CrossRef CAS.
- N. Seal, A. S. Palakkal, R. S. Pillai and S. Neogi, Inorg. Chem., 2023, 62, 11528–11540 CrossRef CAS PubMed.
- N. Seal, P. P. Mondal and S. Neogi, J. Environ. Chem. Eng., 2023, 11, 111219 CrossRef CAS.
- N. Seal, P. P. Mondal, A. S. Palakkal, R. S. Pillai and S. Neogi, ACS Appl. Mater. Interfaces, 2023, 15, 54397–54408 CrossRef CAS PubMed.
- N. Seal, A. Karmakar, S. Kundu and S. Neogi, ACS Sustainable Chem. Eng., 2023, 11, 979–993 CrossRef CAS.
- S. Gutiérrez-Tarriño, J. L. Olloqui-Sariego, J. J. Calvente, M. Palomino, G. Mínguez Espallargas, J. L. Jordá, F. Rey and P. Oña-Burgos, ACS Appl. Mater. Interfaces, 2019, 11, 46658–46665 CrossRef PubMed.
- L. Zhong, J. Ding, X. Wang, L. Chai, T.-T. Li, K. Su, Y. Hu, J. Qian and S. Huang, Inorg. Chem., 2020, 59, 2701–2710 CrossRef CAS PubMed.
- X. Qin, W. Yang, Y. Yang, D. Gu, D. Guo and Q. Pan, Inorg. Chem., 2020, 59, 9857–9865 CrossRef CAS PubMed.
- R. Liu, Z.-Q. Wang, Q.-Y. Liu, F. Luo and Y.-L. Wang, Eur. J. Inorg. Chem., 2019, 2019, 735–739 CrossRef CAS.
- L. L. Wang, F. Luo, L. L. Dang, J. Q. Li, X. L. Wu, S. J. Liu and M. B. Luo, J. Mater. Chem. A, 2015, 3, 13724–13730 RSC.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- V. A. Blatov, M. O'Keeffe and D. M. Proserpio, CrystEngComm, 2010, 12, 44–48 RSC.
- R.-L. Zhang, J.-J. Duan, J.-J. Feng, L.-P. Mei, Q.-L. Zhang and A.-J. Wang, J. Colloid Interface Sci., 2021, 587, 141–149 CrossRef CAS PubMed.
- Z. Han, J.-J. Feng, Y.-Q. Yao, Z.-G. Wang, L. Zhang and A.-J. Wang, J. Colloid Interface Sci., 2021, 590, 330–340 CrossRef CAS PubMed.
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS.
- L. Ai, X. Gao and J. Jiang, J. Power Sources, 2014, 257, 213–220 CrossRef CAS.
- M. Mohan, M. Essalhi, D. Durette, L. K. Rana, F. K. Ayevide, T. Maris and A. Duong, ACS Appl. Mater. Interfaces, 2020, 12, 50619–50627 CrossRef CAS PubMed.
- L. Xu, C.-Y. Xing, D. Ke, L. Chen, Z.-J. Qiu, S.-L. Zeng, B.-J. Li and S. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 3032–3041 CrossRef CAS PubMed.
- X.-L. Yang, Y.-T. Yan, W.-J. Wang, Z.-Z. Hao, W.-Y. Zhang, W. Huang and Y.-Y. Wang, Inorg. Chem., 2021, 60, 3156–3164 CrossRef CAS PubMed.
- N. Seal, A. S. Palakkal, M. Singh, R. Goswami, R. S. Pillai and S. Neogi, ACS Appl. Mater. Interfaces, 2021, 13, 28378–28389 CrossRef CAS PubMed.
- N. Du, J. Song, S. Li, Y.-X. Chi, F.-Y. Bai and Y.-H. Xing, ACS Appl. Mater. Interfaces, 2016, 8, 28718–28726 CrossRef CAS PubMed.
- N. Seal, M. Singh, S. Das, R. Goswami, B. Pathak and S. Neogi, Mater. Chem. Front., 2021, 5, 979–994 RSC.
- L. Li, S. Shen, J. Su, W. Ai, Y. Bai and H. Liu, Anal. Bioanal. Chem., 2019, 411, 4213–4220 CrossRef CAS PubMed.
- N. Seal, R. Goswami, M. Singh, R. S. Pillai and S. Neogi, Inorg. Chem. Front., 2021, 8, 296–310 RSC.
- T. Zheng, Z. Yang, D. Gui, Z. Liu, X. Wang, X. Dai, S. Liu, L. Zhang, Y. Gao, L. Chen, D. Sheng, Y. Wang, J. Diwu, J. Wang, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017, 8, 15369 CrossRef CAS PubMed.
- W.-R. Cui, C.-R. Zhang, W. Jiang, F.-F. Li, R.-P. Liang, J. Liu and J.-D. Qiu, Nat. Commun., 2020, 11, 436 CrossRef CAS PubMed.
- W. Liu, Y. Wang, L. Song, M. A. Silver, J. Xie, L. Zhang, L. Chen, J. Diwu, Z. Chai and S. Wang, Talanta, 2019, 196, 515–522 CrossRef CAS PubMed.
- W. Yang, Z.-Q. Bai, W.-Q. Shi, L.-Y. Yuan, T. Tian, Z.-F. Chai, H. Wang and Z.-M. Sun, Chem. Commun., 2013, 49, 10415–10417 RSC.
- M. Singh, A. Karmakar, N. Seal, P. P. Mondal, S. Kundu and S. Neogi, ACS Appl. Mater. Interfaces, 2023, 15, 24504–24516 CrossRef CAS PubMed.
- R. Madhu, A. Karmakar, K. Karthick, S. Sam Sankar, S. Kumaravel, K. Bera and S. Kundu, Inorg. Chem., 2021, 60, 15818–15829 CrossRef CAS PubMed.
- J.-J. Duan, Z. Han, R.-L. Zhang, J.-J. Feng, L. Zhang, Q.-L. Zhang and A.-J. Wang, J. Colloid Interface Sci., 2021, 588, 248–256 CrossRef CAS PubMed.
- J. Jiang, L. Huang, X. Liu and L. Ai, ACS Appl.
Mater. Interfaces, 2017, 9, 7193–7201 CrossRef CAS PubMed.
- X. Zhou, X. Shen, Z. Xia, Z. Zhang, J. Li, Y. Ma and Y. Qu, ACS Appl. Mater. Interfaces, 2015, 7, 20322–20331 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Physical measurements, experimental details, structural and topological figures, characterization data, crystal data and tables. CCDC 2222321 and 2240355. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ta06790d |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Arun
Karmakar
*ab,
Arun
Karmakar