
Multi-scale in silico analysis of the phase separation behavior of FUS mutants†
Kalindu S.
Fernando
 and
Ying
Chau
*
and
Ying
Chau
*
Chemical and Biological Engineering Department, Hong Kong University of Science & Technology, Hong Kong SAR, China. E-mail: keychau@ust.hk
First published on 4th November 2024
Abstract
Fused in sarcoma (FUS) is an intrinsically disordered RNA-binding protein that helps to regulate transcription and RNA transport while reversibly assembling into membraneless organelles (MLOs). Some mutations of FUS can promote irreversible aggregation, contributing to neurodegenerative diseases. We previously reported a multi-scale computational framework combining a series of molecular dynamics simulations (MD) followed by lattice Monte Carlo (MC) simulations to describe the tendency and dynamics of the assembly and disassembly of intrinsically disordered proteins (IDPs) using wild-type (WT)-FUS as an illustrative example. In this study, we utilized our computational model to simulate three FUS mutants widely experimented with glycine point mutation G156E, arginine point mutation R244C, and deletion of the C-terminal nuclear localization signal (ΔNLS). MD simulation results conveyed that G156E has improved sticker contact probability compared to WT-FUS, while R244C has slightly lower contact probability, which is also complemented by change of net interactions according to the molecular mechanics Poisson Boltzmann surface area (MMPBSA) method. The MC simulation results revealed that G156E has a higher aggregation propensity than the WT-FUS, while ΔNLS has more liquid-like assemblies. R244C demonstrated higher dynamics at the beginning, while over the evolution of MC simulations, it tends to aggregate compared to WT-FUS. In addition, the G156E mutant has more stable protein aggregates, lacking the rapid dynamics shown in all other scenarios. From the peak height of radial distribution functions (RDFs) of the assemblies, the phase separation propensity in ascending order is ΔNLS < FUS-WT < R244C < G156E. Moreover, interpreting the dynamic assembly propensity (DAP) parameter over time, the fluidity of the assemblies in ascending order is G156E < FUS-WT < R244C < ΔNLS. The results obtained from this study support that the computational model is able to predict the effect of mutation down to single amino acid substitution on the phase separation behavior of FUS. This efficient in silico method can be generalized to investigate the phase separation propensity of other IDPs and their mutants.
Introduction
Intrinsically disordered proteins (IDPs) can dynamically and reversibly assemble into membraneless organelles (MLOs),1,2 which can regulate cell functions efficiently, and their mutations can induce fibril-like aggregations leading to neurodegenerative diseases.3,4 Fused in sarcoma (FUS) is one of the most studied IDPs for liquid–liquid phase separation (LLPS) and their mutants for pathological aggregation in vitro and in vivo.5–7 We selected three mutations widely reported in experimental studies for our in silico analysis.The FUS G156E mutation has been found to have an increased tendency to assemble into aggregates compared to FUS.4,5,8,9 This mutation introduces a phosphomimetic, a negatively charged residue, into the FUS low complexity domain, which can accelerate the aggregation propensity of FUS.4In vitro and in vivo studies have shown that the G156E mutant increases the tendency of FUS for aggregation. The aggregated protein fibrils serve as a structural template that facilitates the fibrillation of disordered proteins, supporting the model of a “seeding reaction”.10 The decrease in the positive charges not only induces the generation of condensates but causes accelerated loss of fluidity.11,12 The results contribute to the understanding of how the molecular-level change is related to neurodegenerative disease-associated FUS aggregation.
Secondly, R244C mutation is also reported as an aggregate-induced mutation13,14 contributing to neurodegenerative diseases.9 The R244C mutation can disrupt one of the arginine–glycine–glycine (RGG) motifs, reported to be essential for FUS LLPS3 and pathological fibril formation,15 which may reduce the aggregation intensity. However, arginine replaced with cysteine can improve pi–pi and cation–pi interactions.16 The loss of the positively charged arginine leads to defective binding between FUS and RNA. The aberrant configuration has more potential to grow into large condensates with less fluidity.9,11
Other than point mutations, the deletion of domains of FUS can also alter its LLPS behavior. The FUS protein consists of several domains, including the QSGY rich region, three RGG domains, RNA recognition motif (RRM), and nuclear localization signal (NLS). Among them, C-terminal NLS helps to deliver FUS into the nucleus. The deletion of NLS in FUS has been shown to decrease nuclear import and cause cytoplasmic accumulation of the mutant protein.17–19 The nuclear import of FUS is regulated by nuclear import receptors (NIRs) in NLS, which play a critical role in preventing and reversing aberrant phase transitions of FUS.20 NIRs prevent irregular phase separation by the chaperone and disggregase activity,21 interactions with prion-like domains,22 oligomerization disruption,23 and nucleoporin interactions with phenylalanine–glycine repeats (FG-Nups).23,24 In addition, Wang et al. revealed that ΔNLS of wild-type (WT) FUS lowers the condensation, remaining dynamic inside the stress granules as supported by fluorescence recovery after photobleaching (FRAP) experiments on Hela cells.25
Even though there are computational models for the WT IDPs, in silico studies on mutated proteins are limited. Bonet et al. have studied the FUS mutations using MD simulations combined with prediction algorithms to analyze the FUS mutant's frustration26 selecting only 20 amino acid sequences in the NLS domain. Chatterjee et al. have studied FUS and its P525R mutation using MD simulations using a single chain of 20 amino acids in the NLS domain in FUS.27 Dignon et al. have developed a coarse-grained (CG) computational model to construct the phase diagram of FUS and its phosphomimetic mutations in an implicit solvent environment.28 A minimal multiscale model has been developed by Ren et al.29 using classical associative polymer theory30 to construct phase diagrams. It can be extended for mutants by changing the number of multivalent interaction sites.
Nevertheless, previously reported computational studies on mutant IDPs do not involve simulations across hundreds of chains and are not built on multi-scale modeling. We have recently introduced a versatile multi-scale computational framework capable of studying the dynamic assembly/disassembly of MLOs formed via LLPS. It effectively employs MD simulations, which can be costly to optimize, while preventing complete reliance on MC simulations, which restricts the depiction of actual dynamic properties.31 Our multi-scale model, although simplified, is sufficiently comprehensive to simulate the dynamic behavior of IDPs as chains of CG beads composed of stickers and spacers. Both homotypic and heterotypic interactions of interactive motifs (stickers) known as low-complexity amyloid-like reversible kinked segments (LARKS) extracted from the LARKS32,33 database are incorporated in determining chain movements. LARKS are multivalent interaction sites contribute for reversal binding IDPs that undergo LLPS.33 The discrete binding energy distributions for all possible sticker pairs are considered to represent the random nature of the assembly/disassembly process of the IDP, as observed in lattice-based MC stimulations. To account for spacer involvement more precisely, the number of CG beads representing stickers and spacers was determined based on the sequence-dependent Kuhn length estimation. In this study, we report the simulation results from applying this model to the three FUS mutants, G156E, R244C, and ΔNLS, to study the different levels of aggregation behaviors of FUS mutants.
Methods
Sticker identification
We followed the procedure described in Fernando et al.'s study to determine the stickers and spacers using LARKSdb.31 Briefly, into the relevant sequences of mutant FUS G156E, R244C, and NLSΔFUS were input LARKSdb32,34 to identify the potential stickers, and the fibrillogenic propensity profiles were obtained. If overlapping LARKS were found, the most energetically favorable sequence (with the lowest Rosetta energy) was chosen.MD simulations and pairwise interaction potential estimation
We performed AA-MD simulations and molecular mechanics Poisson Boltzmann surface area (MMPBSA) calculations to estimate the pairwise binding energy of additional sticker pairs in mutant FUS G156E and R244C. AA-MD simulations were performed for additional spacers found for the FUS G156E mutant. WT-FUS protein sticker pairwise binding energies were obtained from Fernando et al.'s study.31Pymol (v1.7.4 (for academics)) software was used to construct the initial structures of stickers and spacers found for mutant FUS. The termini of starting and ending residues were capped with acetyl and amide groups to mitigate the artificial effects caused by the charges. Docking simulations were conducted using Haddock 2.4,35 to identify the best complex among all possible pairs of stickers. The default parameters available in Haddock 2.4 were employed for all docking simulations. The resulting peptide configuration obtained from docking simulations was used as the input for the subsequent AA-MD simulations. The extended configuration was directly employed as the input for AA-MD simulations for spacers.
AA-MD simulations with explicitly presented water were performed using GROMACS36 (v2019.5.1). OPLS/AA force field37 and tip5p model38 were employed for modeling peptides and water, respectively. Two hexapeptides (LARKS) stickers or single spacer peptides were inserted in a dodecahedron box. The system was solvated with NaCl to mimic the physiological conditions as much as possible. All MD simulations were performed at 300 K temperature and 1 atm pressure. The molecular mechanics Poisson Boltzmann surface area (MMPBSA) method was used to estimate the pairwise binding energy distribution of mutant sticker pairs. Docking analysis, MD simulations, and MMPBSA computations were performed for all additional pairs of LARKS structures from selected FUS mutants. The convergence of MD simulations was assessed using the stability of the radius of gyration of peptide complexes (Fig. S1(A) and (B), ESI†). Furthermore, contact probability and buried solvent accessible surface area (SASA) were estimated using MD trajectories of sticker pairs.
Lattice MC simulations for phase separation estimation
Then, we performed lattice MC simulations separately for three FUS mutants by configuring protein sequences as the strings of CG sticker and spacer beads, as shown in Table 1. They were randomly placed in a three-dimensional simple cubic lattice with a constant lattice spacing (a = 1) as a canonical ensemble. We utilized four types of MC trial moves: local move, end rotational move, translational move, and slithering snake move with equal statistical ratios.39 Periodic boundary conditions were applied in all x, y, z cartesian directions to minimize the edge effects. We employed the fixed bond model features, allowing CG beads to move along the lattice sides but not diagonally. Local and end rotation moves were implemented to move single lattice site while translation and slithering snake moves were allowed to move a whole chain of CG beads per single move. We performed over two million steps of hypothetical trial MC moves in total as self-avoiding random walks on the lattice for each simulation. Additionally, WT-FUS simulations from our previous study were extended up to two million MC steps for a fair comparison with mutant simulations as a reference. After discarding the initial one hundred thousand MC steps of mutant simulations, a quasi-equilibrated system was obtained using the Metropolis–Hastings algorithm as move acceptance criteria, and discretized pairwise energies were estimated using the MMPBSA method. All lattice MC simulations were performed at 300 K temperature. Radial distribution function (RDF) and dynamic assembly propensity (DAP) parameters were used to estimate the phase separation propensity and dynamic characteristics of phase separation of each system using eqn (1) and (2), respectively. The DAP parameter can quantitatively evaluate the degree of change in RDF to estimate the assembly and disassembly states of each system, as it is a function of spatiotemporal changes of the CG bead clusters on the lattice, which provide insights into the dynamic assembly of IDP droplets.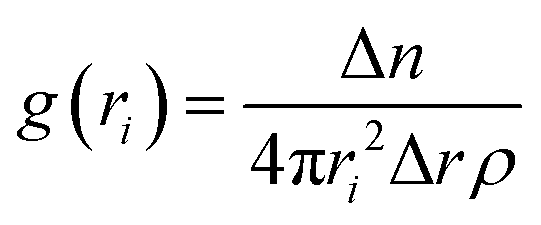 | (1) |
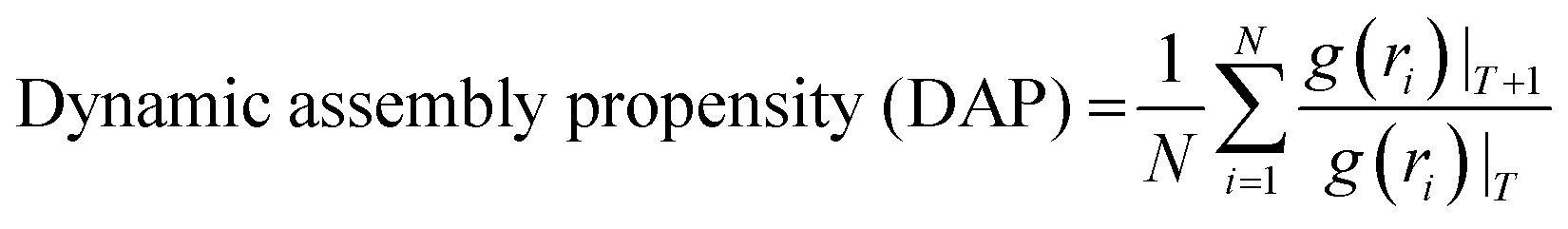 | (2) |
| FUS G156E CG bead representation | |||
| No of stickers | 17 | L2SL5S2L2SL2SL10SLSL7S2LSL4S2L7SL13SL14SLSL3SL4 | 93 CG beads |
| No of spacers | 15 | ||
| FUS R244C CG bead representation | |||
| No of stickers | 16 | L2SL5S2L2SL2SL12SL7S2LSL4S2L7SL13SL14SLSL3SL4 | 93 CG beads |
| No of spacers | 14 | ||
| NLSΔFUS CG bead representation | |||
| No of stickers | 15 | L2SL5S2L2SL2SL12SL7S2LSL4S2L7SL13SL14SLSL2 | 87 CG beads |
| No of spacers | 13 | ||
Each lattice MC simulation used six hundred CG chains for all three systems considered. All three systems simulated in simple cubic lattices having the total number of lattice sites varies from 1.30 × 107–1.38 × 107 while keeping the fraction of lattice sites filled as 0.004 for all systems to closely mimic the physiological concentration that can observe phase separation as reported in Wang et al.'s in vitro study.25 The simulation method described in our previous study31 was the same procedure if not explicitly stated.
Results and discussion
In the first step of the model, we conceptually abstracted an IDP as a sequence of interactive stickers and non-interacting spacers per the associative polymer theory. We describe the FUS protein as a chain of randomly distributed stickers with pairwise interactions and energetically neutral spacers. In our previous report, we identified sixteen unique stickers for WT-FUS from LARKSdb.31 Here, we identified an additional sticker (QGYEQQ) for the G156E mutant using LARKSdb. For the R244C, the RGGGRG sticker in the FUS wild-type was replaced by the CGGGRG sticker. Mutant ΔNLS is one DRGGFG sticker short from the WT-FUS. Due to the introduction of the new sticker into the G156E mutant, the number of interactions was increased by 17 overall.We summarized the pairwise binding energies related to each mutation, as shown in Table 2 and Fig. S2, S3 (ESI†). G156E mutation has the highest number of pairwise attractive interactions compared to WT-FUS, supporting that there is an increased driving force for G156E to undergo phase separation. In contrast, ΔNLS has the lowest number of attractive interactions. Due to the replacement of the RGGGRG sticker in WT-FUS by CGGGRG in R244C mutation, the number of repulsive pair interactions was reduced to the lowest among all the systems considered.
| G156E | R244C | ΔNLS | FUS-WT31 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| No of stickers | 17 | 16 | 15 | 16 | |||||
| Sticker–sticker interactions | Attractive | 92 | 60.13% | 83 | 61.03% | 73 | 61.43% | 78 | 66.1% |
| Repulsive | 23 | 15.03% | 15 | 11.03% | 22 | 18.49% | 22 | 12.8% | |
| Mixed | 38 | 24.84% | 38 | 27.94% | 24 | 20.17% | 36 | 21.1% | |
| Total | 153 | 100% | 136 | 100% | 119 | 100% | 136 | 100% | |
Next, we analyzed the MD simulation results of sticker pair complexes. We summarized each pair of stickers’ buried solvent-accessible surface area (SASA) with heat maps (Fig. 1). We observed relatively high buried SASA in the sticker complexes formed with the additional QGYEQQ sticker in the G156E mutant, implying that hydrophilicity is introduced into FUS by the mutation (Fig. 1(B) and (A)). On the other hand, the complexes formed with the mutant sticker CGGGRG from R224C have lower buried SASA than those with the original RGGGRG sticker from WT-FUS, implying that R224C is more hydrophobic than the wild-type protein (Fig. 1(C)). The complexes formed with DRGGFG sticker in WT-FUS are missing from ΔNLS (Fig. 1(D)) and the buried SASA of these complexes are slightly hydrophobic improving the overall hydrophilicity due to deletion of NLS compared to wild-type.
 | ||
| Fig. 1 Average buried solvent accessible surface area of sticker–sticker complexes estimated using 200 ns production of AA-MD simulation trajectories performed at 300 K and 0.15 M NaCl concentration of all possible sticker pairs using the eqn (3). Heat maps are shown for WT-FUS(A), G156E (B) (complexes with the additional sticker bracketed by red lines), R244C (C) (complexes with the substituted sticker bracketed by red lines), and ΔNLS (D) (with the DRGGFG sticker short from WT-FUS). | ||
The buried SASA was estimated using the eqn (3)
 | (3) |
We summarized the contact probability of sticker–sticker complexes, indicating each sticker pair's binding strength. The heat maps indicated in Fig. 2 show the contact probability of WT-FUS and the three mutants. The additional sticker in G156E (QGYEQQ) has a higher contact probability with multiple stickers than most stickers do in WT-FUS (Fig. 2(A) and (B)). A slight decrease in contact probability was observed for the complexes with the mutant sticker CGGGRG of R244C in comparison to the complexes with the original RGGGRG of WT-FUS (Fig. 2(C)). Mutant ΔNLS contains a subset sticker–sticker complex of WT-FUS, missing the interactions with the DRGGFG sticker, and thus has an overall reduction in contact probability (Fig. 2(D)).
 | ||
| Fig. 2 Contact probability maps of stickers from WT-FUS and the three mutants estimated using AA-MD simulations at 300 K, and 0.15 M NaCl concentration as the ratio of contacts less than 0.5 nm between the center of mass of each sticker pair to the total number of contacts. The maps are shown for FUS wild type (A), G156E (B) (complexes with the additional sticker bracketed by red lines), R244C (C) (complexes with the substituted sticker bracketed by red lines), and ΔNLS (D) (with one sticker short from WT-FUS). | ||
The results from AA-MD were fed into lattice MC simulations. The complete CG bead representations of three mutant protein sequences are summarized in Fig. 3.
 | ||
| Fig. 3 Complete CG sticker–spacer representation of the FUS mutants. Yellow-highlighted hexapeptides are sticker CG beads, and spacer CG beads are colored alternatively in red and pale blue. Green color highlighted stickers are additional or replaced stickers compared to WT-FUS. Purple-highlighted amino acids within the green highlighted sticker are the relevant point mutations. The gray color highlighted amino acids are deleted for the ΔNLS mutant. The sequences are shown for G156E (A), R244C (B), and ΔNLS (C). | ||
We observed that the G156E mutant reached the lowest potential energy. The ΔNLS system obtained the highest energy potential, having the lowest number of stickers per chain of CG beads. The R244C system has gained a slight decrease in potential energy plateau compared to WT-FUS (Fig. 4). These results can be explained by the replaced sticker interactions in the mutant R244C system. As Table 2 results suggest, the overall repulsive interactions were reduced by ∼1.8% while having the same net number of total interactions as WT-FUS. Furthermore, the change in potential energy of each mutant is affected by both sticker interactions and spacer arrangement on the lattice. Besides the ΔNLS system, the spacer contribution is almost identical to the other systems.
 | ||
| Fig. 4 Evolution and gradual stabilization of potential energy of three repeated MC simulations for each mutant and WT-FUS. These simulations were performed up to 2 × 106 MC trial moves at 300 K temperature. The shown results are 600 CG chains of G156 E mutant (red), WT-FUS (black) R244C mutant (green), and NLSΔFUS mutant (blue) in simulation box sizes of (240 × 240 × 240), (240 × 240 × 240) and (235 × 235 × 235), respectively. | ||
Compared to our previous study, we extended the lattice MC simulation steps nearly twice in this study as mutation simulations may introduce new local minima and energy barriers that the simulations need to encounter, altered interaction network which can reduce the stability of the simulation system, and additional uncertainty of systems arisen due to mutation that need to be overcome. The lattice size was varied in a narrow range from (235 × 235 × 235) to (240 × 240 × 240) and consistent potential energy plateaus were observed.
We also generated an average radial distribution function (RDF) for each simulation of 2.5 × 104 MC move steps (as a period) to investigate the three mutant systems’ relative phase separation propensity. The average RDF is estimated by the ratio of the CG bead density in a hollow sphere with a thickness of one lattice unit at a variable radial distance from the origin to the average density of CG beads in the whole lattice within the period of MC moves. A spike in the average RDF represents the local density hike of CG beads in a 3-dimensional lattice space, indicating the phase separation of CG bead chains in the lattice space.31
The RDF plots suggest that all mutants can undergo LLPS. Mutant G156E has the highest RDF sharp peak, indicating the highest phase separation propensity out of the systems considered. The high-resolution graph shows a continuous increment in the peak height with lowering its rate of change of RDF peak height in latter simulation steps, indicating the increasing assembly states leading to aggregation (Fig. 5(A)). In contrast, mutant ΔNLS has the lowest RDF peak, illustrating a lower phase separation propensity. Both R244C and ΔNLS systems showed ups and downs in RDF peak heights at equilibrium, meaning the alternative assembly and disassembly states, which are characteristics of dynamic phase separation. Similar fluctuation was noted in WT-FUS in our previous report.31
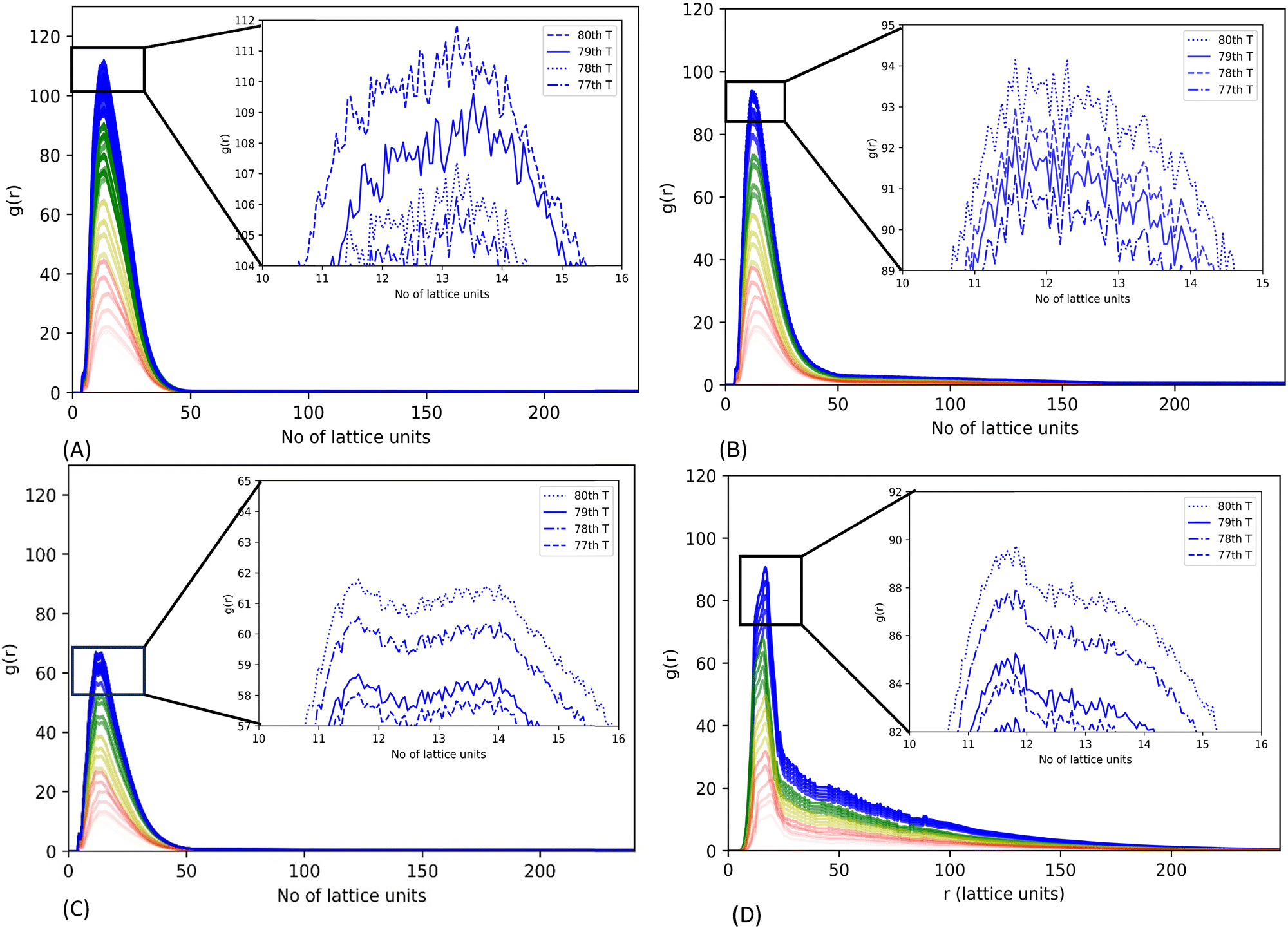 | ||
| Fig. 5 Radial distribution function (RDF) for WT-FUS and the three mutants (G156E, R244C, and ΔNLS). RDFs were generated using trajectories of CG lattice simulations of 600 chains, run up to 2 × 106 MC trial moves. The evolution of the RDFs was calculated using 2.5 × 104 MC move steps and colored by each set with red, yellow, green, and blue, indicating the progression from the beginning to the end of the simulation. The zoomed-in graph at the upper right corner shows the last four RDFs estimated for each simulation system, (A) for G156E, (B) for R244C, (C) for ΔNLS and (D) FUS WT. | ||
Dynamic assembly propensity (DAP) is a parameter we define as the ratio of the average RDFs of two successive periods to track the assembly/disassembly states over the progress of the simulation. It indicates how RDF changes from one period to the following period of MC steps. A DAP value of greater than 1.0 suggests an increase in assembly, while a value of less than 1.0 indicates a disassembly of CG bead clusters. The magnitude of DAP fluctuation signifies the dynamics or fluidity of the condensates.31
The DAP of G156E converges to a value above one in the last periods of the simulation with decreasing fluctuations (Fig. 6(A)), indicating that the phase separation is continuously progressing. Additionally, and more prominently towards the end of the simulation, the G156E mutant exhibits a relatively lower fluctuation of DAP compared to WT-FUS and other mutants, implying that the assembled mutants are less fluidic and form more stable aggregations. Thus, the G156E mutant has slower dynamics of CG bead clusters over time, indicating more of a larger solid-like assembly at a higher density, as seen in the graphical visualization of the simulation results (Video S1, ESI†). In various experimental investigations, the G156E mutation in the prion-like domain of FUS has been shown to increase the tendency of FUS to form aggregates.8,40–42 This mutation introduces a negatively charged residue into the FUS low-complexity (LC) domain, which can accelerate the aggregation kinetics of FUS.4 Additionally, expression of FUS with the G156E mutation in neurons has resulted in the formation of intranuclear foci, further supporting the increased aggregation propensity of this mutation.43
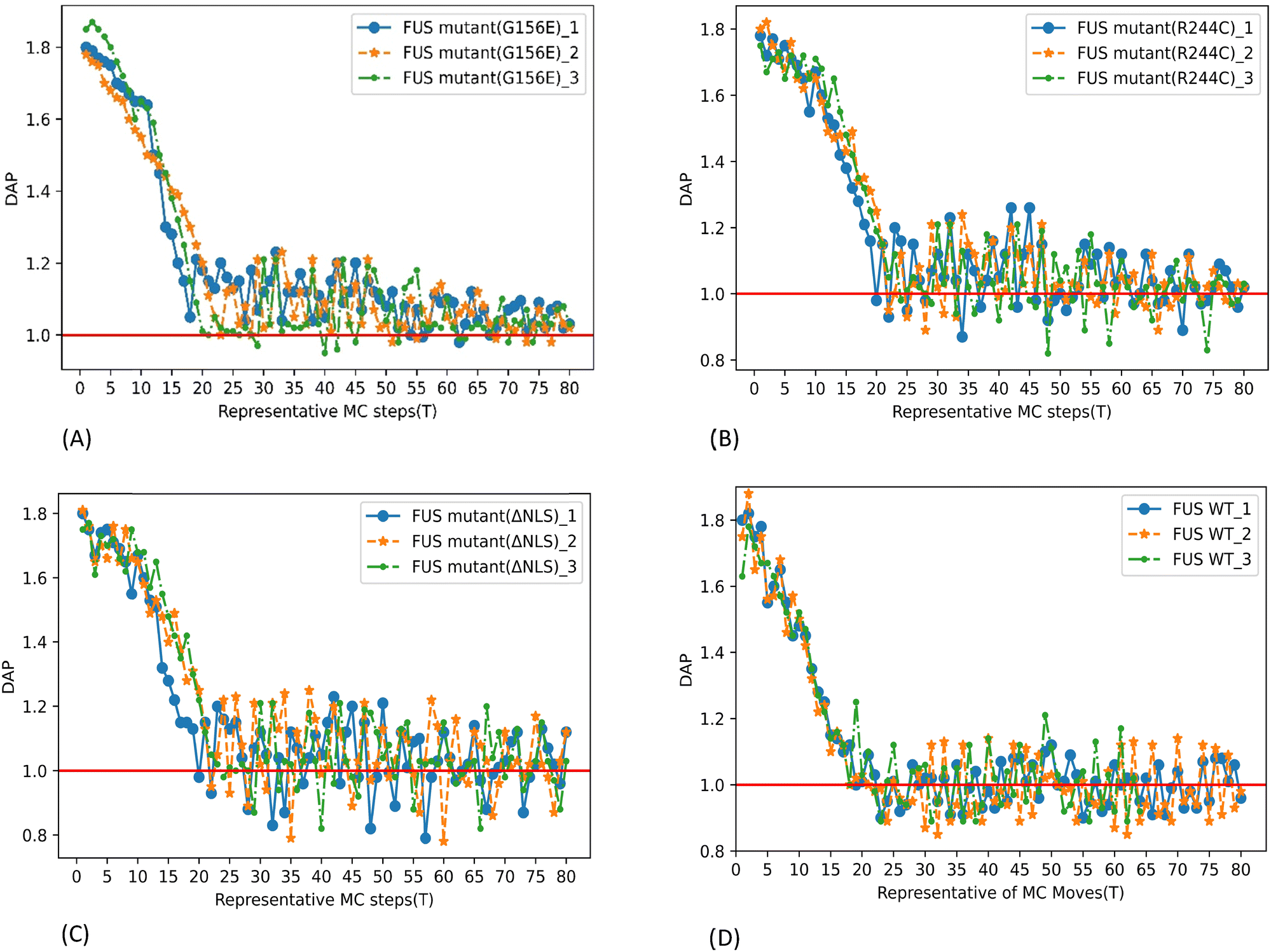 | ||
| Fig. 6 DAP parameter as a function of representative MC steps. The results showed three repeated simulations performed up to 2 × 106 MC trial moves with 600 CG chains at 300 K temperature. (A) FUS mutant G156E, (B) FUS mutant R244C, (C) FUS mutant ΔNLS, and (D) WT FUS. | ||
The higher RDF peak of R244C compared to WT-FUS suggests that the mutant has a higher phase separation propensity than WT-FUS (Fig. 5B and D). Initially, the DAP of R244C has higher fluctuations compared to WT-FUS. It hovers above one instead of around one. The results illustrate the faster kinetics of R244C to form fluidic assemblies in the beginning. Towards the end of the simulation, the DAP of R244C converges around one. At the same time, the fluctuation becomes less than that of WT-FUS, signifying the stabilization of phase-separated droplets into aggregates less fluidic than the wild-type (Fig. 6B and D). Replacing positively charged arginine with uncharged polar cysteine creates a new sticker with a lower strength (Fig. S3 (ESI†) and Table 2) but having higher attractive interactions, forming an altered configuration with the potential to aggregate into condensates with less fluidity. Additionally, the lower hydrophilicity of the new sticker contributed to the aggregation propensity of the R244C mutant compared to WT FUS. The phenomenon can be visualized from the simulation of the CG bead clusters representing R244C in Video S2 (ESI†).
As shown in the DAP plot in Fig. 6C, mutant ΔNLS has a more dynamic phase separation, spanning between 0.8 and 1.24, which represents the highest magnitude among all the systems studied here. The fast dynamics of the CG bead clusters of ΔNLS were observed in MC lattice simulation (Video S3, ESI†), compared to WT-FUS,31 consistent with the interpretation of the DAP fluctuation. The condensates can be seen as more diminutive in size and lower in density, illustrating the results of the lowest RDF peak among all the systems tested.
All MD simulations of stickers and spacers were performed using the OPLS/AA force field only; other relevant force fields for proteins can also be employed to estimate pairwise binding energy distributions. However, using alternative force fields may affect the results of lattice MC simulations. This study highlights the ability of the multiscale model to demonstrate the alteration of phase separation behavior by replacing a single amino acid or deletion of a domain of an IDP sequence, which leads to changes in molecular interactions. The model is adaptable for other LLPS-related applications as it effectively combines MD simulations, which can be costly to utilize completely, and it avoids relying solely on MC simulations, which have limitations in representing actual dynamic properties.
The study underscores the significance of the strength of the sticker in determining the behaviors of three selected mutants of FUS. Replacement, changing, and deleting stickers can significantly alter the phase separation behavior as their binding strength crucially influences the protein's phase separation and aggregation behaviors. It highlights the sequence-dependent behavior of LLPS of IDPs. This understanding could provide valuable insights into how the residue-level properties are propagated towards the macroscopic phase separation behavior of IDPs, which are important considerations for developing drug targets for therapeutic intervention.
Conclusions
In conclusion, this study connects the detailed information on molecular interactions at the residue level to the macroscopic understanding of LLPS phenomena that can lead to aggregation, using three different FUS mutants (G156E, R244C, and ΔNLS) as examples. The study underscores the potential of the multi-scale computational model to differentiate the phase separation behaviors of IDPs due to mutations, with resolution down to single amino acid substitution. It offers an efficient in silico pipeline for studying the phase separation propensity of IDPs and their mutants. The study shows the importance of sticker strength to drive phase separation, and differences in molecular interactions can be exemplified by contact probability and SASA. The peak height of RDFs and the magnitude of DAP fluctuation provide insights into the dynamic phase separation and macromolecular aggregation. Our computational framework can successfully capture the reversible assembly leading to stable aggregation of G156E and R244C, as well as a lesser propensity for assembly of ΔNLS and its fluidic assembly. To this end, the computational model is validated by experimental results reported by others. Overall, this study contributes valuable knowledge and tools for analyzing residue level information and macroscopic observation of aggregation propensities in IDPs and their mutants, paving the way for speeding up experimental investigations. Our protocols are adaptable to other natural or engineered protein systems to study their assembly behavior with limited computational cost.Author contributions
Y. Chau and K. S. Fernando initiated and conceptualized the project. K. S. Fernando performed all simulations and wrote the original draft. Y. Chau and K. S. Fernando edited, reviewed, and wrote the manuscript. Y. Chau provided the supervision and guidance.Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the funding support from the Hong Kong Research Grants Council (GRF 16102520). K. S. Fernando received financial support from the Hong Kong PhD Fellowship Scheme. The authors thank the ITSC at HKUST for providing high-performance cluster (HPC3) resources to facilitate the running of simulations for this research study.References
- V. N. Uversky, Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder, Curr. Opin. Struct. Biol., 2017, 44, 18–30 CrossRef.
- R. J. Wheeler and A. A. Hyman, Controlling compartmentalization by non-membrane-bound organelles, Philos. Trans. R. Soc., B, 2018, 373, 20170193 CrossRef.
- S. Alberti, A. Gladfelter and T. Mittag, Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates, Cell, 2019, 176, 419–434 CrossRef.
- R. F. Berkeley, M. Kashefi and G. T. Debelouchina, Real-time observation of structure and dynamics during the liquid-to-solid transition of FUS LC, Biophys. J., 2021, 120, 1276–1287 CrossRef PubMed.
- T. Nomura, et al., Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis, J. Biol. Chem., 2014, 289, 1192–1202 CrossRef PubMed.
- Y. Zhou, S. Liu, G. Liu, A. Öztürk and G. G. Hicks, ALS-Associated FUS Mutations Result in Compromised FUS Alternative Splicing and Autoregulation, PLoS Genet., 2013, 9, 1–17 Search PubMed.
- M. Kamelgarn, et al., Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS, Biochim. Biophys. Acta, Mol. Basis Dis., 1862, 2004–2014, 2016 Search PubMed.
- A. Patel, et al., A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation, Cell, 2015, 162, 1066–1077 CrossRef CAS.
- K. Rhine, et al., ALS/FTLD-Linked Mutations in FUS Glycine Residues Cause Accelerated Gelation and Reduced Interactions with Wild-Type FUS, Mol. Cell, 2020, 80, 666–681 CrossRef CAS e8.
- E. N. Guerrero, et al., TDP-43/FUS in motor neuron disease: Complexity and challenges, Prog. Neurobiol., 2016, 145–146, 78–97 CrossRef CAS PubMed.
- Y. Ji, F. Li and Y. Qiao, Modulating liquid-liquid phase separation of FUS: mechanisms and strategies, J. Mater. Chem. B, 2022, 10, 8616–8628 RSC.
- A. G. Niaki, et al., Loss of Dynamic RNA Interaction and Aberrant Phase Separation Induced by Two Distinct Types of ALS/FTD-Linked FUS Mutations, Mol. Cell, 2020, 77, 82–94 CrossRef PubMed e4.
- A. Patel, et al., A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation Article A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation, Cell, 2015, 162, 1066–1077 CrossRef.
- A. Aguzzi and M. Altmeyer, Phase Separation: Linking Cellular Compartmentalization to Disease, Trends Cell Biol., 2016, 26, 547–558 CrossRef PubMed.
- E. Bertrand, et al., FUS fibrillation occurs through a nucleation-based process below the critical concentration required for liquid–liquid phase separation, Sci. Rep., 2023, 13, 1–13 CrossRef PubMed.
- R. M. C. Vernon, et al., Pi-Pi contacts are an overlooked protein feature relevant to phase separation, eLife, 2018, 7, 1–90 CrossRef.
- P. Convertini, et al., Biochimica et Biophysica Acta Genome wide array analysis indicates that an amyotrophic lateral sclerosis mutation of FUS causes an early increase of CAMK2N2 in vitro, Biochim. Biophys. Acta, Mol. Basis Dis., 2013, 1832, 1129–1135 CrossRef PubMed.
- J. Gal, et al., Nuclear localization sequence of FUS and induction of stress granules by ALS mutants, Neurobiol. Aging, 2011, 32, 2323.e27–2323.e40 CrossRef PubMed.
- R. Xia, et al., Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS, Mol. Neurodegener., 2012, 7, 1–17 Search PubMed.
- B. Khalil, M. Linsenmeier, C. L. Smith, J. Shorter and W. Rossoll, Nuclear-import receptors as gatekeepers of pathological phase transitions in ALS/FTD, Mol. Neurodegener., 2024, 19, 1–29 CrossRef.
- J. H. Weishaupt, T. Hyman and I. Dikic, Common Molecular Pathways in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia, Trends Mol. Med., 2016, 22, 769–783 CrossRef PubMed.
- K. Kapeli, F. J. Martinez and G. W. Yeo, Genetic mutations in RNA-binding proteins and their roles in ALS, Hum. Genet., 2017, 136, 1193–1214 CrossRef PubMed.
- A. F. Harrison and J. Shorter, RNA-binding proteins with prion-like domains in health and disease, Biochem. J., 2017, 474, 1417–1438 CrossRef PubMed.
- O. Spead, B. L. Zaepfel and J. D. Rothstein, Nuclear Pore Dysfunction in Neurodegeneration, Neurotherapeutics, 2022, 19, 1050–1060 CrossRef PubMed.
- J. Wang, et al., A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins, Cell, 2018, 174, 688–699 CrossRef PubMed e16.
- L. F. S. Bonet, J. P. Loureiro, G. R. C. Pereira, A. N. R. Da Silva and J. F. De Mesquita, Molecular dynamics and protein frustration analysis of human fused in Sarcoma protein variants in Amyotrophic Lateral Sclerosis type 6: An in silico approach, PLoS One, 2021, 16, 1–16 CrossRef.
- S. Chatterjee, S. Abbas and J. Y. Lee, Insights into amyotrophic lateral sclerosis linked Pro525Arg mutation in the fused in sarcoma protein through in silico analysis and molecular dynamics simulation, J. Biomol. Struct. Dyn., 2021, 39, 5963–5976 CrossRef.
- G. L. Dignon, W. Zheng, Y. C. Kim, R. B. Best and J. Mittal, Sequence determinants of protein phase behavior from a coarse-grained model, PLoS Comput. Biol., 2018, 14, 1–23 CrossRef.
- C.-L. Ren, P. Zhang and Y. Ma, Minimal Multi-Scale Model for Liquid-Liquid Phase Separation Regulated by Small, Molecules, 2021, 1–6 Search PubMed.
- M. Rubinstein and A. N. Semenov, Thermoreversible gelation in solutions of associating polymers. 2. Linear dynamics, Macromolecules, 1998, 31, 1386–1397 CrossRef CAS.
- K. S. Fernando, G. Jahanmir, I. C. Unarta and Y. Chau, Multiscale Computational Framework for the Liquid–Liquid Phase Separation of Intrinsically Disordered Proteins, Langmuir, 2024, 40, 7607–7619 CrossRef CAS.
- LARKSdb. https://srv.mbi.ucla.edu/LARKSdb/.
- M. P. Hughes, L. Goldschmidt and D. S. Eisenberg, Prevalence and species distribution of the low-complexity, amyloid-like, reversible, kinked segment structural motif in amyloid-like fibrils, J. Biol. Chem., 2021, 297, 101194 CrossRef CAS.
- M. P. Hughes, L. Goldschmidt and D. S. Eisenberg, The prevalence and distribution in genomes of low-complexity, amyloid-like, reversible, kinked segment (LARKS), a common structural motif in amyloid-like fibrils, bioRxiv, Biochem., 2020, 1–21 Search PubMed.
- G. C. P. Van Zundert, et al., The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes, J. Mol. Biol., 2016, 428, 720–725 CrossRef CAS.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, GROMACS: A message-passing parallel molecular dynamics implementation, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- Y. Khalak, B. Baumeier and M. Karttunen, Improved general-purpose five-point model for water: TIP5P/2018, J. Chem. Phys., 2018, 149, 224507 CrossRef PubMed.
- K. S. Fernando, G. Jahanmir, J. Liu, I. C. Unarta and Y. Chau, Associate polymer theory-inspired modelling of intrinsically disordered protein-mimicking polymer-oligopeptide hybrids for the formation of artificial membraneless organelles, in MRS spring meeting and exhibit 2021, 2021.
- E. N. Guerrero, et al., TDP-43/FUS in motor neuron disease: Complexity and challenges, Prog. Neurobiol., 2016, 145–146, 78–97 CrossRef CAS PubMed.
- K. A. Burke, A. M. Janke, C. L. Rhine and N. L. Fawzi, Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II, Mol. Cell, 2015, 60, 231–241 CrossRef CAS PubMed.
- K. Fernando and Y. Chau, A multi-scale model for reversible assembly of FUS and irreversible aggregation of its G156E mutant, in Controlled Release Society Annual meeting & Expo 2023 DOI:10.13140/RG.2.2.20120.98563.
- Y. Furukawa and E. Tokuda, Aggregation of FET proteins as a pathological change in amyotrophic lateral sclerosis, Adv. Exp. Med. Biol., 2017, 925, 1–12 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Supporting Video 1: Illustration of slow dynamics of G156E. A part of the CG MC lattice simulation production run of 600 G156E mutant FUS protein chains after reaching equilibrium. The protein clusters are colored in blue and visualized using VMD (v1.9.4) software. (2) Supporting Video 2: Illustration of dynamic assembly and disassembly of R244C. A part of the CG MC lattice simulation production run of 600 R244C mutant FUS protein chains after reaching equilibrium. The protein clusters are colored in blue and visualized using VMD (v1.9.4) software. (3) Supporting Video 3: Illustration of fast dynamic assembly and disassembly of ΔNLS. A part of the CG MC lattice simulation production run of 600 ΔNLS mutant FUS protein chains after reaching equilibrium. The protein clusters are colored in blue and visualized using VMD (v1.9.4) software. See DOI: https://doi.org/10.1039/d4tb01512f |
| This journal is © The Royal Society of Chemistry 2024 |