DOI:
10.1039/D4TC03740E
(Paper)
J. Mater. Chem. C, 2024,
12, 19506-19514
Enhanced oxygen electrode performance in solid oxide fuel cells via La-doping of Pr2NiO4+δ-based Ruddlesden–Popper perovskites†
Received
30th August 2024
, Accepted 21st October 2024
First published on 22nd October 2024
Abstract
Solid oxide fuel cell (SOFC) is a high-efficiency, low-carbon power generation device that directly converts the chemical energy of fuel directly into electricity at elevated temperatures. However, the performance of the oxygen electrode, which is critical for the oxygen reduction reaction (ORR), remains limited by high polarization losses and limited long-term stability. Ruddlesden–Popper (R–P) perovskites, such as Ln2NiO4+δ (Ln = La, Pr, Nd), offer excellent oxygen transport due to their unique A–O rock salt layers. In this study, La3+-doped Pr2−xLaxNi0.8Cu0.2O4+δ (x = 0, 0.5, 1.0, PLxNC) oxygen electrode materials are synthesized by the sol–gel method, and their electrochemical properties are systematically investigated. While the electrical conductivity decreases with increasing La3+ content, PL0.5NC exhibits optimal oxygen surface exchange and bulk diffusion properties. SOFC using PL0.5NC as the oxygen electrode achieved excellent performance at 800 °C, with a polarization resistance of 0.245 Ω cm2 and a peak power density of 0.864 W cm−2, a 64% improvement over PNC. This study highlights the potential of La3+ doping to enhance PNO-based oxygen electrodes and provides insight into the development of high performance SOFC materials.
1. Introduction
Solid oxide fuel cells (SOFC) efficiently convert chemical energy into electrical energy using hydrocarbon fuels such as H2, CH4, and CH3OH to generate electricity.1,2 The oxygen electrode, a key component in SOFC, drives the oxygen reduction reaction (ORR).3 Ideal oxygen electrode materials require high thermodynamic stability, mixed ionic–electronic conductivity, enhanced catalytic activity, and adequate porosity.4 Currently, conventional ABO3-type perovskite oxides, such as LaMnO3-based compounds (LSM),5,6 La1−xSrxCo1−yFeyO3 (LSCF),7,8 and Ba1−xSrxCo1−yFeyO3 (BSCF),9,10 are commonly used as oxygen electrodes. However, the low oxygen ion conductivity of LSM limits its ORR efficiency, while the long-term stability of LSCF and BSCF is compromised by the presence of alkaline earth metals such as Ba and Sr.11 Therefore, the development of new oxygen electrode materials with enhanced performance and stability remains a pressing challenge.
Ruddlesden–Popper (R–P) perovskites, represented by the formula Ln2NiO4+δ (Ln = La, Pr, Nd), are a remarkable class of mixed ionic electronic conductors (MIECs) with the general structural formula An+1BnO3n+1 or AO(ABO3)n. Unlike conventional perovskites, R–P perovskites have a distinctive layered structure in which A–O layers alternate with rock salt and ABO3 perovskite layers along the c-axis. This unique A–O layer can accommodate excess interstitial oxygen, thereby enhancing oxygen ion conductivity. R–P perovskites are characterized by high oxygen ion diffusion coefficients and surface exchange rates, making them promising candidates for oxygen electrode materials.12–14 These materials have demonstrated favorable electrocatalytic activity for the ORR. For example, the La1.5Pr0.5Ni0.8Co0.2O4+δ oxygen electrode achieved a peak power density of 0.4 W cm−2 at 700 °C in fuel electrode-supported SOFCs.15 Similarly, Pr2NiO4+δ electrodes exhibited a peak power density of 0.95 W cm−2 at 750 °C, with a degradation rate of less than 3%/1000 h at a constant voltage of 0.8 V.16 Among the Ln2NiO4+δ series, Pr2NiO4+δ (PNO) stands out for its exceptional oxygen surface exchange (k*) and bulk diffusion (D*) coefficients, recorded at 2.5 cm s−1 and 5.0 × 10−3 cm2 s−1, respectively,17,18 at 750 °C. Notably, the D* of PNO is an order of magnitude higher than that of Nd2NiO4+δ at 500 °C.19 However, PNO is less thermally stable, prone to decomposition into heterogeneous phases such as Pr4Ni3O10 and Pr6O11 at elevated temperatures, and has a lower electronic conductivity.20,21 In contrast, La2NiO4+δ offers better thermal stability within the Ln2NiO4+δ family, although its electrochemical activity remains suboptimal.22 Substitution at the B-site of PNO with transition metal elements such as Cu can lower the phase formation temperature while improving the electronic conductivity and oxygen permeability of the material.23 Among these, Pr2Ni0.8Cu0.2O4+δ (PNC) has shown the lowest polarization impedance over different Cu2+ doping levels.24,25
In this study, we focused on PNC-based Ruddlesden–Popper perovskite materials and synthesized Pr2Ni0.8Cu0.2O4+δ (PNC), Pr1.5La0.5Ni0.8Cu0.2O4+δ (PL0.5NC), and Pr1.0La1.0Ni0.8Cu0.2O4+δ (PLNC) by A-site doping with 25% and 50% La3+, respectively. The synthesis was carried out by the sol–gel method. The influence of La3+ doping on the material properties was systematically investigated by phase analysis and physical and chemical characterization. Subsequently, the electrochemical performances of half-cell and full cells based on these materials were evaluated, allowing us to assess the impact of La3+ doping on the oxygen electrode performance.
2. Experimental
2.1 Materials synthesis
The materials PNC (Pr2Ni0.8Cu0.2O4+δ), PL0.5NC (Pr1.5La0.5Ni0.8Cu0.2O4+δ), PLNC (Pr1.0La1.0Ni0.8Cu0.2O4+δ), and LDC (Ce0.6La0.4O1.8) materials were synthesized by the sol–gel method. Taking PL0.5NC as an example, the preparation involved dissolving stoichiometric amounts of Pr(NO3)3·6H2O, La(NO3)3·6H2O, Ni(NO3)2·6H2O, and Cu(NO3)2·3H2O in deionized water to form a homogeneous nitrate solution. Citric acid (CA) monohydrate and ethylenediaminetetraacetic acid (EDTA) were added as complexing agents in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.5 for metal ions, EDTA, and CA. The solution was stirred thoroughly and the pH was adjusted to 7–8 with ammonia to ensure complete dissolution. The mixture was stirred continuously at 80 °C for about 10 h until a homogeneous gel was formed. This gel was dried at 240 °C for 10 h to obtain a fluffy carbonaceous precursor, which was then ground to a fine powder and calcined at 950 °C for 3 h to obtain the RP-structured PL0.5NC.
:1:1.5 for metal ions, EDTA, and CA. The solution was stirred thoroughly and the pH was adjusted to 7–8 with ammonia to ensure complete dissolution. The mixture was stirred continuously at 80 °C for about 10 h until a homogeneous gel was formed. This gel was dried at 240 °C for 10 h to obtain a fluffy carbonaceous precursor, which was then ground to a fine powder and calcined at 950 °C for 3 h to obtain the RP-structured PL0.5NC.
2.2 Cell preparation
YSZ electrolyte sheet prepared by the casting method was used as a half-cell support. To prevent unwanted chemical reactions between the electrode and the YSZ, a GDC barrier layer was screen printed on the YSZ surface. PNC, PL0.5NC, PLNC with a solids content of 60 wt% were then prepared. These pastes were screen-printed symmetrically onto the GDC|YSZ|GDC structure, with an area of approximately 0.5 cm2 at the central position of the barrier layer on both sides. The half-cells were then obtained by sintering at 950 °C for 2 h. For the full cell, La0.8Sr0.2Ga0.8Mg0.2O3−δ (LSGM) was used as the electrolyte support. Rectangular (24 × 6 × 2.5 mm3) dense bars of PNC, PL0.5NC, and PLNC powders were prepared by uniaxial pressing (15 MPa for 1 min) of 1.0–1.5 g of the corresponding materials in a rectangular mold, followed by sintering at 1250 °C in air for 5 h. The LSGM powder was homogenized by high-energy ball milling at 250 rpm for 24 h, then pressed into a cylindrical mold at 10 MPa pressure for 2 min and sintered at 1450 °C for 10 h to produce dense LSGM electrolyte sheets (thickness ∼240 μm). The electrode paste was screen printed onto one side of the LSGM sheet as an oxygen electrode, while the opposite side was used as a fuel electrode with Ni-GDC, with the LDC acting as a barrier layer to prevent reactions between Ni and the LSGM electrolyte. Pt paste was used as the current collector for all electrodes.
2.3 Cell test and characterization
The samples were characterized by X-ray diffraction (XRD) using a PANalytical Empyrean diffractometer (Netherlands) with a 2θ range of 20° to 80° and a scan rate of 5° per min. XRD data were refined using GSAS software for peak position correction, data fitting, and phase analysis. Microscopic morphology and elemental distribution were examined using a GeminiSEM 300 field emission scanning electron microscope (FSEM). Thermal expansion in air was measured using a Netzsch DIL 402C dilatometer, heated from room temperature to 900 °C at 10 °C per min. Electrical conductivity was assessed by the standard four-probe method in air, and the response to changes in oxygen concentration was evaluated by electrical conductivity relaxation (ECR) tests. Thermogravimetric analysis (TGA) was performed using a Netzsch STA 449F3A in nitrogen, heating from 50 °C to 800 °C at 5 °C min−1, to study weight loss patterns and oxygen vacancy evolution at elevated temperatures. Oxygen adsorption capacity was characterized by temperature-programmed desorption (TPD) using a Hiden DECRA 5080B instrument. Oxygen vacancy peak intensities were measured at room temperature using a Bruker A300 electron paramagnetic resonance (EPR) spectrometer. Electrochemical impedance spectroscopy (EIS) tests were performed on half-cells and full-cells with an applied perturbation amplitude of 10 mV, over a frequency range of 106 Hz to 10−1 Hz. Voltage sweeps were conducted from 1.2 V to 0 V in SOFC mode at a scan rate of 10 mV s−1.
3. Results and discussion
3.1 Phase and composition analysis
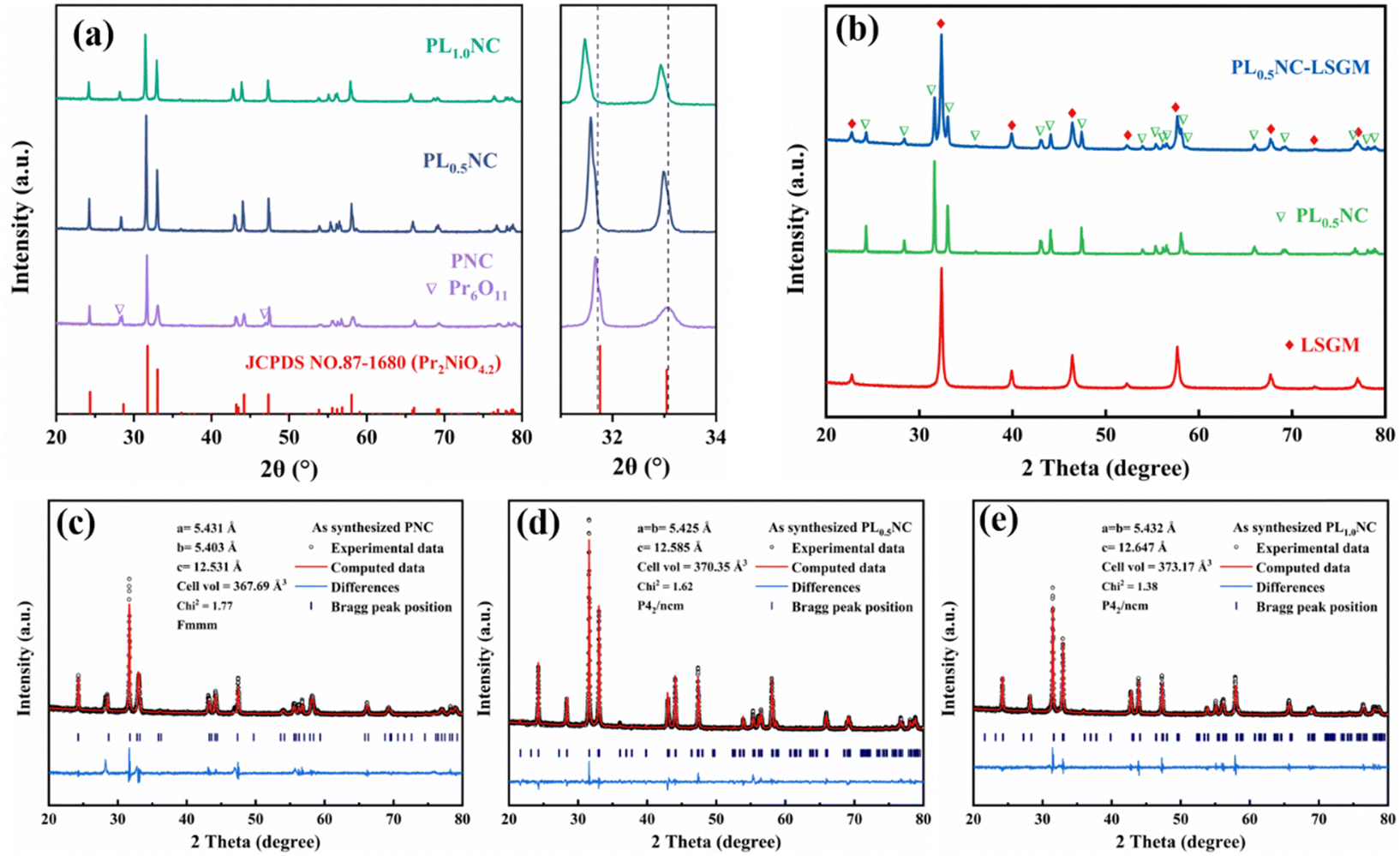
Fig. 1(a) shows the XRD patterns of Pr2−xLaxNi0.8Cu0.2O4+δ (x = 0, 0.5, 1.0) powders after sintering at 950 °C for 3 h. All samples show diffraction peaks that match the standard PDF card (#87-1680) for Pr2NiO4.2. Notably, the undoped PNC sample exhibits secondary peaks at 2θ = 28.2° and 47.0°, which are attributed to the formation of Pr6O11 due to the thermal instability of PNC at elevated temperatures. However, these secondary peaks disappear upon partial substitution of Pr3+ with La3+, suggesting that La3+ doping at the A-site improves the thermal stability of the material. To further investigate the effect of La3+ doping on the crystal structure, the main diffraction peaks in the 31° to 34° range were examined in detail. The results show a systematic shift of the diffraction peaks towards lower angles with increasing La3+ content. This shift is due to lattice expansion caused by the larger ionic radius of La3+ (1.061 Å) compared to Pr3+ (1.013 Å), leading to an overall increase in the lattice volume. Elemental diffusion and interfacial chemical reactions at the electrode/electrolyte interface are critical factors influencing ion transport. To assess the chemical compatibility of PL0.5NC with LSGM, a 1:1 mixture of PL0.5NC and LSGM powders was co-fired at 1000 °C for 10 h. As shown in Fig. 1(b), XRD analysis of the co-fired mixture shows no significant secondary phases, indicating that no undesirable interfacial reactions occur between the LSGM electrolyte and the PL0.5NC electrode within the operating temperature range of 650–800 °C. The effect of La3+ doping on the lattice parameters and crystal structure was further elucidated by Rietveld refinement of the XRD patterns for PNC, PL0.5NC, and PLNC (Fig. 1(c)–(e)). The PNC sample has an orthorhombic structure (space group Fmmm) with lattice constants a = 5.431 Å, b = 5.403 Å, c = 12.531 Å, and a unit cell volume of 367.69 Å3. In contrast, PL0.5NC has lattice constants a = b = 5.425 Å, c = 12.585 Å, and a unit cell volume of 370.35 Å3. For PLNC, the lattice constants are a = b = 5.432 Å, c = 12.647 Å (Table S1, ESI†), with a corresponding unit cell volume of 373.17 Å3. The increase in lattice constants and unit cell volume with higher La3+ doping levels is consistent with lattice expansion due to the larger ionic radius of La3+. In addition, the crystal structure transitions from an orthorhombic (space group Fmmm) to a tetragonal (space group P42/ncm) system upon La3+ doping. This transformation is mainly driven by the expansion along the c-axis, induced by the larger La3+ occupying the A–O rock salt layer in the structure.26,27 The Rietveld refinement results, with Chi2 values below 2 for all samples, confirm the high accuracy of these structural analyses.
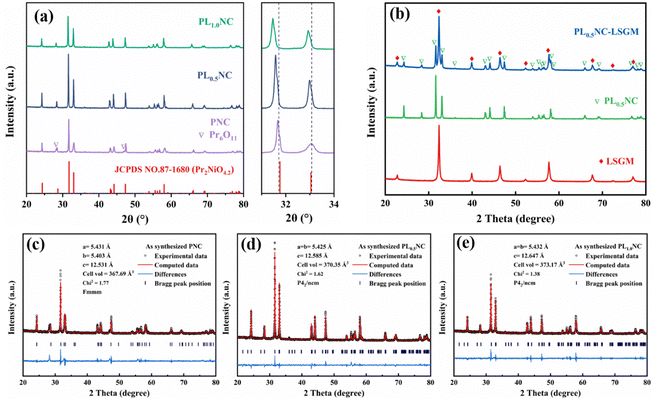 |
| | Fig. 1 XRD patterns and local magnification of Pr2−xLaxNi0.8Cu0.2O4+δ (x = 0, 0.5, 1.0) electrode powder sintered at 950 °C (a), PL0.5NC mixed with LSGM and calcined at 1000 °C for 10 h (b), Rietveld refinement of the XRD patterns of (c) PNC, (d) PL0.5NC, (e) PL1.0NC. | |
3.2 Physical and chemical properties
The thermal expansion coefficients (TECs) of PNC, PL0.5NC, and PLNC were measured, as shown in Fig. 2(a). Over the temperature range of 50 to 800 °C, the average TECs were 13.55 × 10−6 K−1, 14.44 × 10−6 K−1, and 13.89 × 10−6 K−1, respectively, with the materials exhibiting linear expansion with temperature. The increase in TEC with La3+ doping at the A-site is primarily attributed to the inverse relationship between the thermal expansion coefficient and lattice energy, which is determined by the ionic bond length; a longer bond length results in a lower bond energy.28 As the La–O bond length is greater than that of the Pr–O bond, La3+ doping reduces the lattice energy, thereby increasing the TEC. However, at higher levels of La3+ doping, the TEC decreases slightly due to an increase in oxygen vacancies, the introduction of additional oxygen vacancies creates lattice distortions in the R–P structure, resulting in shorter metal–oxygen bond lengths compared to undoped PrNiO4+δ. This increase in lattice energy may cause the reduction in TEC in PLNC. For SOFC applications, it is crucial that the TECs of the electrode and electrolyte materials are closely matched to prevent mechanical problems such as electrode detachment, interfacial cracking, and delamination during high temperature operation. In this study, YSZ was used as the electrolyte for half-cells and LSGM for full-cells. The TEC of LSGM was found to be approximately 12.13 × 10−6 K−1 in the 50–800 °C range, as shown in Fig. 2(b). Table S2 (ESI†) shows a comparison of the TECs for common electrolyte materials such as YSZ,29 GDC30 and SDC.31 The results indicate that Pr2Ni0.8Cu0.2O4+δ-based Ruddlesden–Popper (R–P) perovskites have TECs that are well matched with those of commonly used electrolyte materials in SOFC. Thermogravimetric analysis (TGA) of PNC, PL0.5NC, and PLNC in a nitrogen atmosphere, conducted between 30 °C and 800 °C, is shown in Fig. 2(c). All samples show varying degrees of weight loss with increasing temperature. The weight loss observed at lower temperatures (∼300 °C) is mainly due to the volatilization and desorption of impurities such as H2O, O2, and CO2 adsorbed on the sample surface. In contrast, the weight loss at higher temperatures (300–800 °C) is mainly due to the release of lattice oxygen and the formation of oxygen vacancies.32,33 The respective weight losses for PNC, PL0.5NC, and PLNC in the 300–800 °C range were 0.51%, 0.41%, and 0.65%. While low levels of La3+ doping had little effect on weight loss, a doping level of 50% resulted in a 0.14% increase in high temperature weight loss for PLNC compared to undoped PNC, suggesting that higher La3+ doping enhances lattice oxygen release and oxygen vacancy formation at elevated temperatures. However, the total weight loss of all samples from room temperature to 800 °C remained below 3%, indicating good thermal stability of Pr2Ni0.8Cu0.2O4+δ-based R–P perovskites, which is beneficial for maintaining electrode material stability during device operation and high temperature testing. Conductivity measurements of PNC, PL0.5NC, and PLNC in air, shown in Fig. 2(d), show an initial increase followed by a decrease in conductivity with increasing temperature in the 100–800 °C range. Initially, the materials exhibit semiconducting behavior, with thermally activated carriers (electrons and holes) contributing to the increased conductivity.34 As the temperature continues to rise, the materials transition to metallic behavior, but continued heating reduces lattice oxygen and increases oxygen vacancies, leading to a decrease in carrier concentration and conductivity.35 This decrease in conductivity at higher temperatures correlates with the increased weight loss observed in Fig. 2(c), indicating a significant effect of oxygen vacancy formation on conductivity. The peak conductivity for PNC, PL0.5NC, and PLNC occurs at approximately 450 °C, with peak values of 117.2, 105.7, and 93.7 S cm−1, respectively. The decrease in conductivity with increasing La3+ doping is mainly due to the expansion of the lattice parameters a and b, which lengthen the Ni(Cu)–O bond. This increased bond length reduces the carrier transport rate and the overall conductivity of the material.
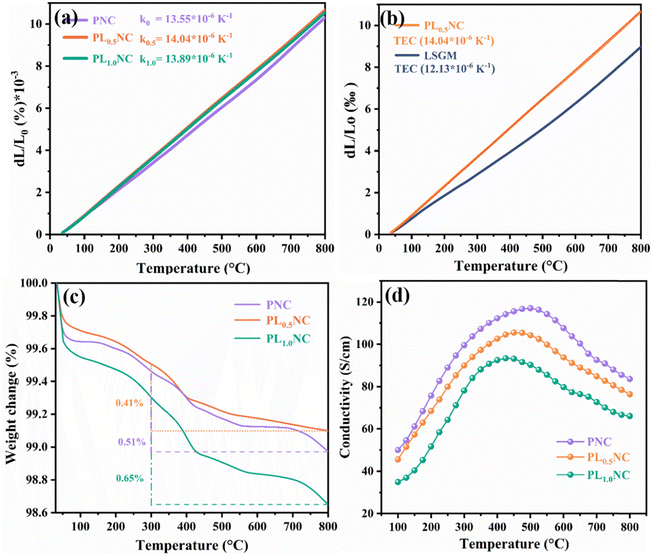 |
| | Fig. 2 Thermal expansion coefficient of (a) PNC, PL0.5NC and PLNC (b) PL0.5NC and LSGM, TG curves (c) and conductivity (d) of PNC, PL0.5NC and PLNC. | |
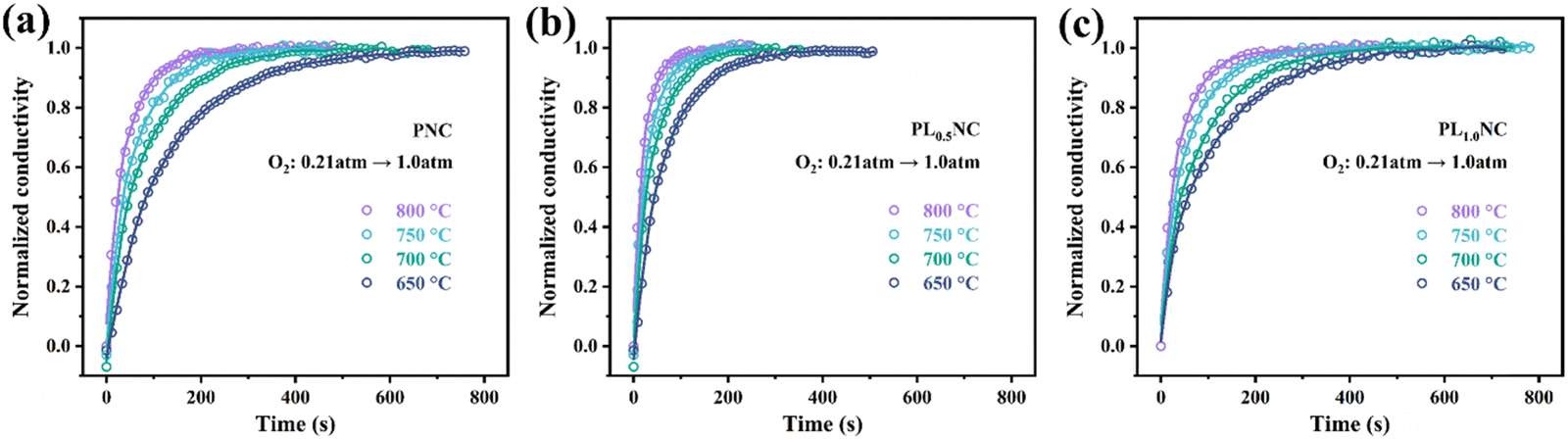
To further elucidate the effect of La3+ doping on the oxygen exchange capacity, conductivity relaxation (ECR) tests were conducted. Fig. 3(a)–(c) shows the normalized conductivity curves for PNC, PL0.5NC, and PLNC. As the oxygen electrode facilitates ORR in SOFC, the ECR test assesses the material's response to changes in oxygen partial pressure. A shorter equilibrium time indicates increased sensitivity to oxygen partial pressure, reflecting improved oxygen surface exchange and bulk diffusion capabilities. Table S3 (ESI†) summarizes the equilibrium times required at different temperatures for PNC, PL0.5NC, and PLNC. The equilibrium time decreases with increasing temperature, consistent with the improved catalytic activity of the electrode materials at elevated temperatures. After La3+ doping, the equilibrium times for PL0.5NC were significantly reduced. The oxygen surface exchange coefficients (Kchem) and bulk diffusion coefficients (Dchem) for the three sample groups were determined by fitting calculations of the ECR test data (Tables S4 and S5, ESI†). These results suggest that La3+ doping effectively enhances the oxygen surface exchange and bulk diffusion properties of Pr2Ni0.8Cu0.2O4+δ-based R–P perovskites, thereby improving their sensitivity to oxygen partial pressure. In particular, PL0.5NC exhibits the shortest equilibrium times, highest Kchem and Dchem values, highlighting its superior oxygen transport capabilities.
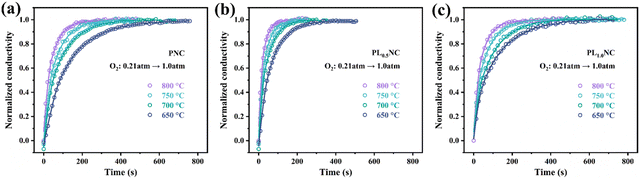 |
| | Fig. 3 ECR curves of (a) PNC (b) PL0.5NC (c) PLNC. | |
3.3 Half-cell performance
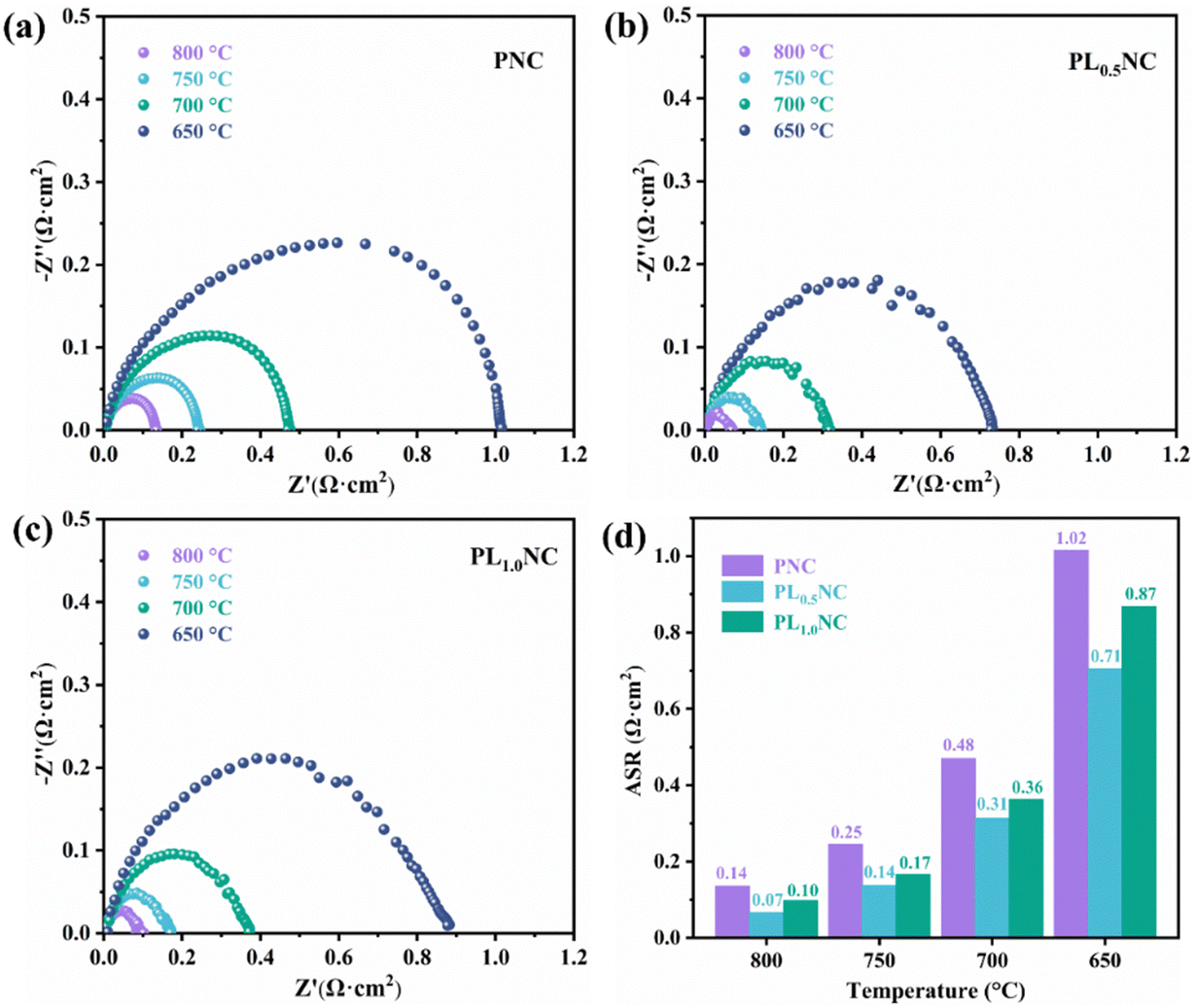
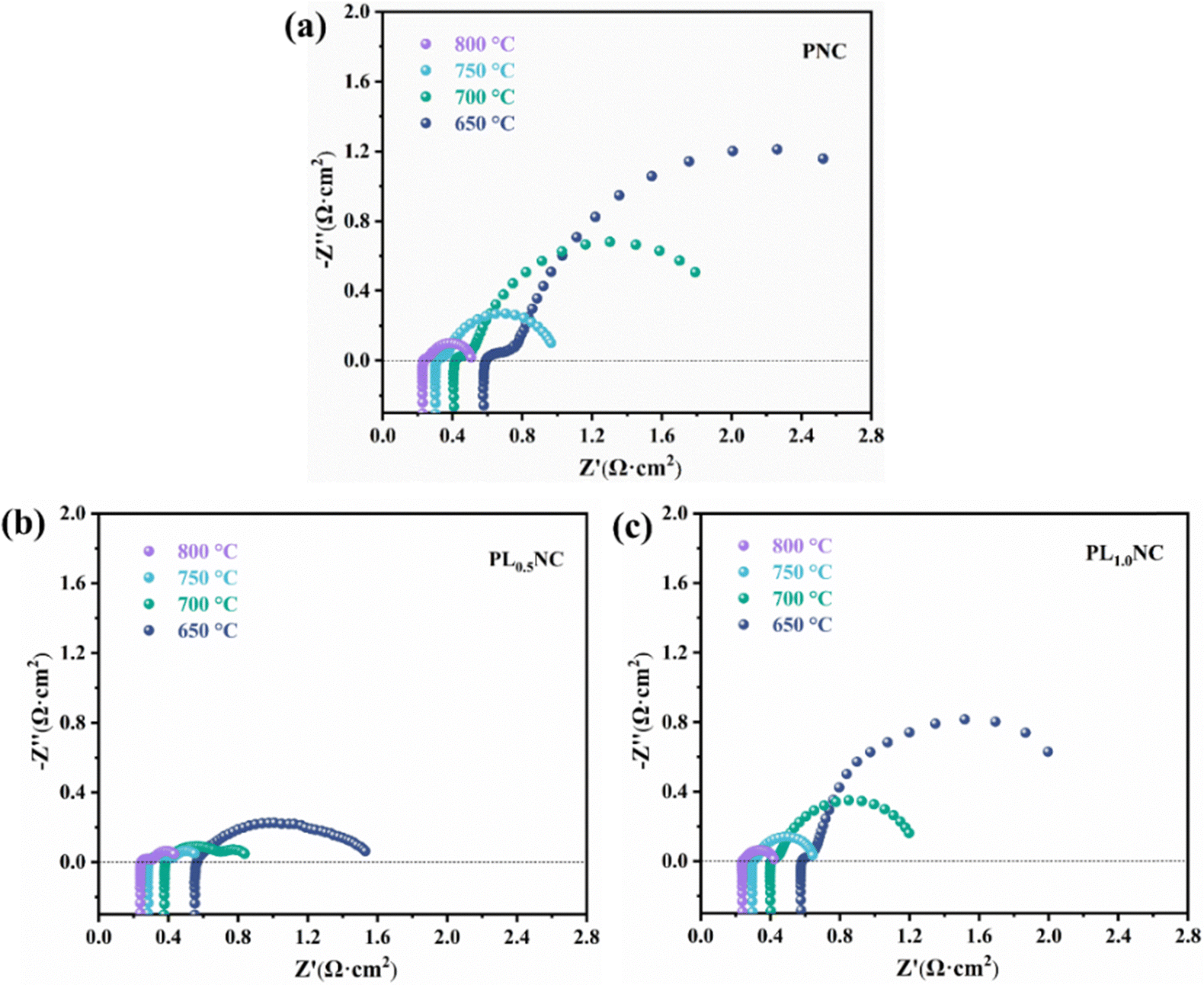
To further investigated the effect of La3+ doping on the electrochemical performance of the electrode materials, EIS was performed on half-cells with the configuration Pr2−xLaxNi0.8Cu0.2O4+δ|GDC|YSZ|GDC|Pr2−xLaxNi0.8Cu0.2O4+δ (x = 0, 0.5, 1.0). The results shown in Fig. 4 represent the polarization impedance (Rp) after taking into account the ohmic resistance for ease of comparison. As illustrated in Fig. 4(a), the Rp values for the PNC electrode were 0.14, 0.25, 0.48, and 1.02 Ω cm2 at 800, 750, 700, and 650 °C, respectively. The La3+ doped electrodes, specifically PL0.5NC and PLNC, exhibited significantly lower Rp compared to PNC. In particular, PL0.5NC showed the most significant reduction in Rp with values of 0.07, 0.14, 0.31, and 0.71 Ω cm2 at the corresponding temperatures (Fig. 4(b)), while PLNC showed Rp values of 0.10, 0.17, 0.36, and 0.87 Ω cm2 (Fig. 4(c)). Fig. 4(d) highlights the comparative polarization impedance of all three electrode materials at different temperatures, showing that La3+ doping significantly enhances the ORR catalytic activities, with the most pronounced improvement observed at a doping level of 0.5. The Arrhenius plots and activation energies for the three materials shown in Fig. S2 (ESI†) indicate that the activation energies for PNC, PL0.5NC, and PLNC are 136.29, 129.68, and 132.34 kJ mol−1, respectively. The activation energy decreases with La3+ doping up to a doping level of 0.5, beyond which it increases slightly. This suggests that La3+ doping effectively lowers the energy barriers for ORR, with the largest reduction in activation energy occurring at a doping level of 25%. Consequently, PL0.5NC shows enhanced electrocatalytic activity, highlighting its potential for improved performance in fuel cells.
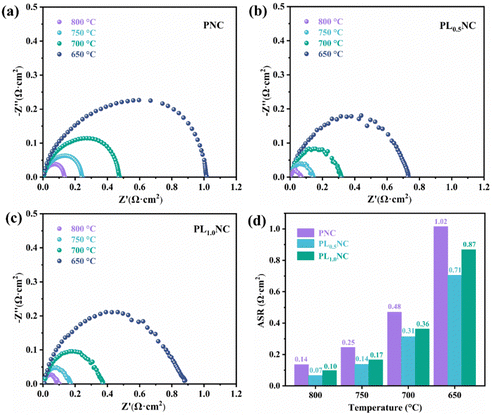 |
| | Fig. 4 Comparation of the EIS of PNC (a), PL0.5NC (b) and PLNC (c) electrode and histogram (d) in half-cell mode. | |
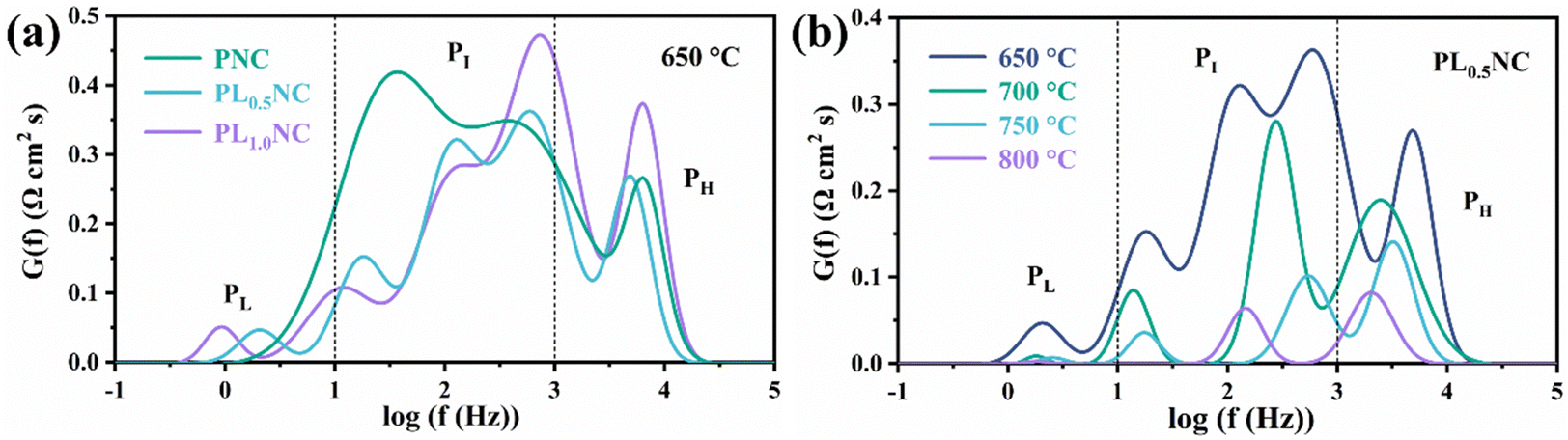
To further analyze the effect of La3+ doping on the electrode reaction processes, the impedance spectra of the half-cells were deconvoluted into three different frequency regions using the distribution of relaxation time (DRT) method: the low-frequency peak (PL) (0.1–10 Hz), the mid-frequency peak (PI) (10–103 Hz), and the high-frequency peak (PH) (103–105 Hz), as shown in Fig. 5. Typically, the high-frequency region reflects the ion exchange process at the three-phase boundary, the mid-frequency region corresponds to charge transfer, and the low-frequency region is associated with gas adsorption and dissociation processes.36,37Fig. 5(a) illustrates the DRT peak fitting results for the PNC, PL0.5NC, and PLNC half-cells at 650 °C. The total peak area in the DRT analysis decreases with increasing La3+ doping and then increases again, with PL0.5NC exhibiting the smallest peak area, consistent with the EIS results shown in Fig. 4. It is evident that La3+ doping has minimal effect on the peak position, shape, and width in the high-frequency region. However, it significantly affects the low and mid frequency regions by shifting the peak positions to lower frequencies and reducing the peak areas. This suggests that La3+ doping mainly enhances the oxygen diffusion process on the electrode surface and the charge transfer process in the electrode response. Fig. 5(b) shows the DRT analysis results of the impedance spectrum of PL0.5NC half-cell at different temperatures. The total peak area progressively decreases with increasing temperature, indicating the positive effect of elevated temperatures on the electrode reaction kinetics. In contrast to Fig. 5(a), temperature changes significantly affect the peak positions, numbers, and areas across all frequency bands. At 800 °C, no distinct low-frequency peaks are observed due to the forward shift and reduction in peak area, leaving only two prominent peaks in the mid and high-frequency regions. This highlights the accelerating effect of high temperature on the electrode reaction processes and kinetics.
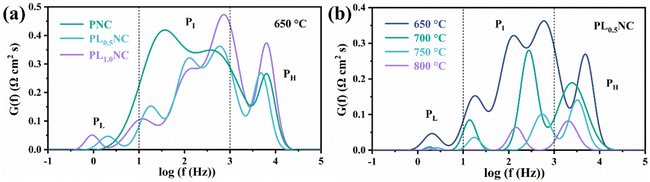 |
| | Fig. 5 DRT analysis of the impedance spectrum of half cells: (a) DRT analysis of the impedance spectrum of PNC, PL0.5NC, PLNC at 650 °C (b) DRT analysis of the impedance spectrum of PL0.5NC at 650–800 °C. | |
3.4 Full cell performance
A full cell with the structure NiO-GDC|LDC|LSGM| Pr2−xLaxNi0.8Cu0.2O4+δ (x = 0, 0.5, 1.0) was tested in SOFC mode, with the fuel electrode was supplied with humidified hydrogen (3% H2O) and the oxygen electrode was exposed to air. EIS and current–voltage–power (I–V–P) characteristic measurements were carried out. The EIS curves obtained at 650–800 °C for cells using PNC, PL0.5NC, and PLNC as oxygen electrodes are shown in Fig. 6. The total ohmic resistance (RΩ), which is mainly determined by the ionic conduction in the electrolyte and the contact resistance between the cell components, and the Rp, which reflects the electrode reaction kinetics, were quantified. Table S6 (ESI†) summarizes the RΩ and Rp values at different temperatures for the full cells. Both RΩ and Rp decrease with increasing temperature due to enhanced ion and electron transport within the electrolyte and electrodes. At 650, 700, 750, and 800 °C, the RΩ values for the PNC full cells were 0.591, 0.415, 0.310, and 0.234 Ω cm2, respectively. For PL0.5NC, these values were 0.564, 0.385, 0.288, and 0.245 Ω cm2, while PLNC had RΩ values of 0.587, 0.405, 0.297, and 0.240 Ω cm2. The RΩ values suggest a trend of initial decrease followed by a slight increase with higher La3+ doping. As the electrolyte primarily determines the RΩ in these cells, the RΩ shows limited variation. The Rp values for the PNC full cell were 1.933, 1.375, 0.655, and 0.272 Ω cm2 at 650, 700, 750, and 800 °C, respectively. For PL0.5NC, the Rp values were significantly lower at 0.965, 0.452, 0.263, and 0.189 Ω cm2, and for PLNC, they were 1.414, 0.791, 0.345, and 0.218 Ω cm2. The results indicate that La3+ doping significantly reduces the Rp, with the most significant reduction observed at a doping level of 25%. This suggests that PL0.5NC has superior electrocatalytic activity.
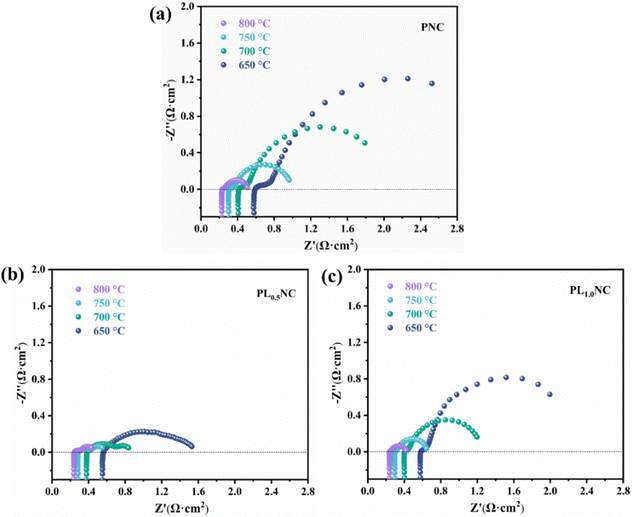 |
| | Fig. 6 EIS curves of different oxygen electrodes Pr2−xLaxNi0.8Cu0.2O4+δ (x = 0, 0.5, 1.0) single cells (a) PNC (b) PL0.5NC (c) PLNC. | |
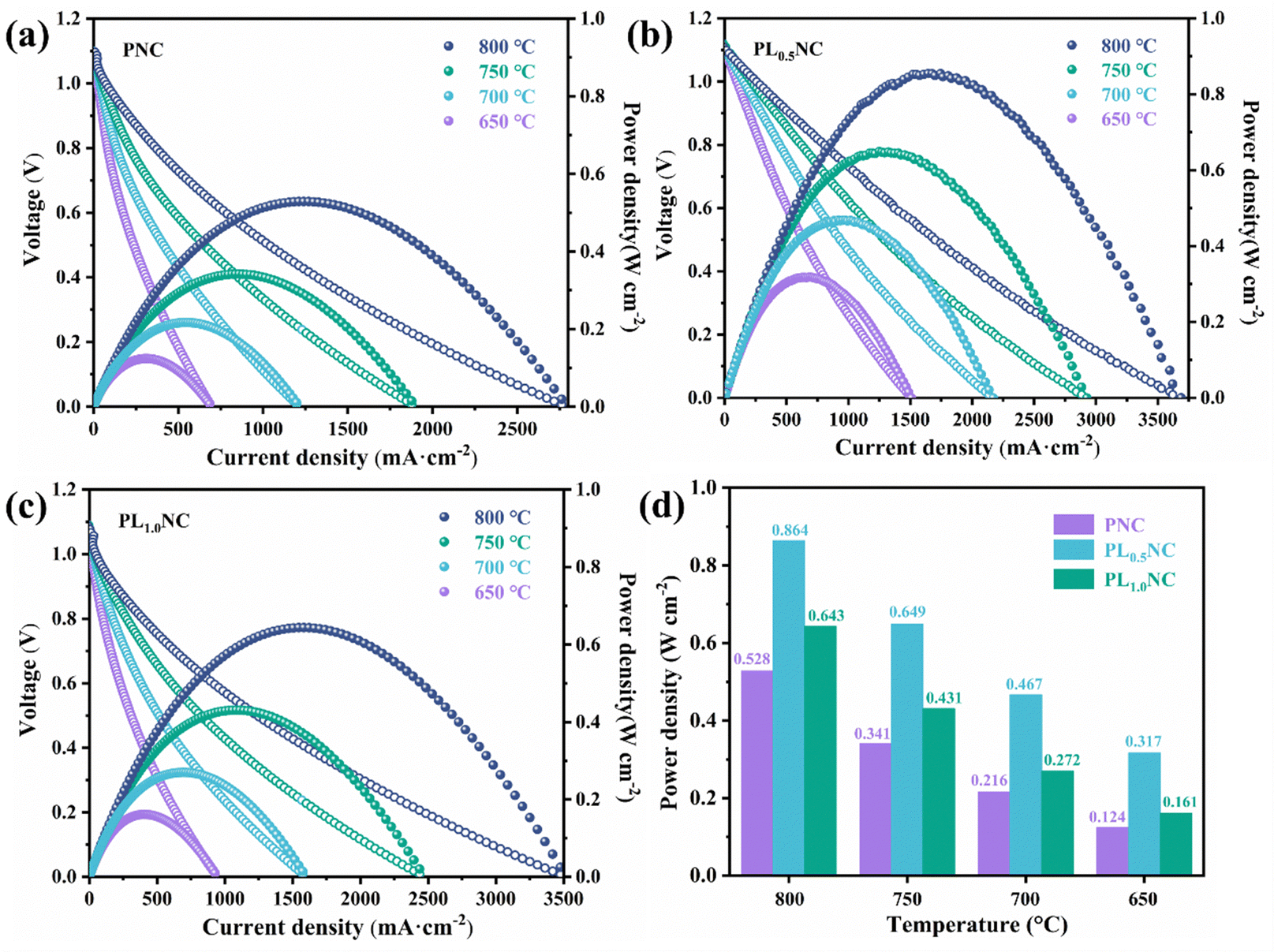
Fig. 7(a)–(c) shows the I–V–P curves for PNC, PL0.5NC, and PLNC full cells measured at 650–800 °C. The open circuit voltages (OCVs) for all cells are around 1.1 V, with a slight decrease observed with increasing temperature, indicating that the cells maintain good air tightness and have dense electrolytes. Fig. 7(d) compares the peak power densities (PPD) of the three full cells at different temperatures. The PPD values follow the trend PL0.5NC > PLNC > PNC, demonstrating that La3+ doping effectively enhances cell output performance. At 650, 700, 750, and 800 °C, the PPDs of the PNC full cell were 0.124, 0.216, 0.341, and 0.528 W cm−2, respectively. In contrast, the PL0.5NC full cell achieved PPDs of 0.317, 0.467, 0.649, and 0.864 W cm−2, while the PLNC cell exhibited PPDs of 0.161, 0.272, 0.431, and 0.643 W cm−2 at the corresponding temperatures. Compared to PNC, the PL0.5NC full cell shows a remarkable improvement in PPD of approximately 156%, 116%, 90% and 64% respectively at these temperatures. These results suggest that La3+ doping significantly improves cell performance, with the optimal doping level being 0.5 for the PL0.5NC oxygen electrode, which exhibits the highest electrocatalytic activity. Coupled with impedance spectral DRT analysis, the performance enhancement by La3+ doping is likely due to the promotion of oxygen surface diffusion and charge transfer processes in the electrode reaction.
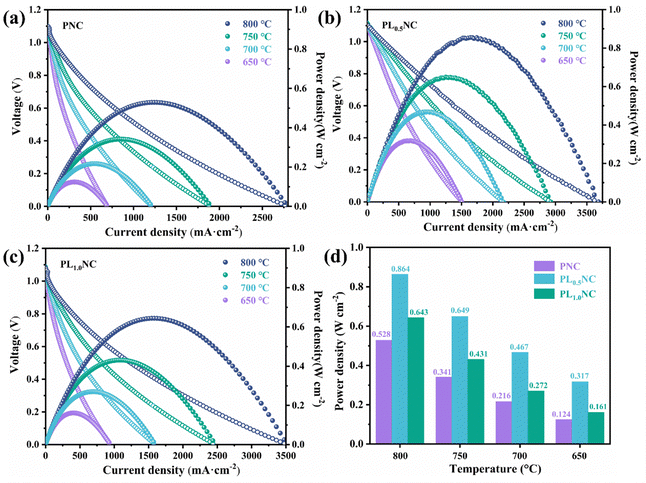 |
| | Fig. 7
I–V–P curves of different oxygen electrode Pr2−xLaxNi0.8Cu0.2O4+δ (x = 0, 0.5, 1.0) single cells in SOFC (a) PNC (b) PL0.5NC (c) PLNC, and (d) comparison of peak power density at 650–800 °C. | |
To further investigated the effect of La3+ doping on the electrode morphology, the microstructures of PNC, PL0.5NC, and PLNC oxygen electrodes are shown in Fig. 8(a)–(c). All three electrodes exhibit an optimal pore distribution, which facilitates oxygen adsorption and diffusion on the electrode surface, thus providing more active sites for electrochemical reactions. However, the electrode morphology changes significantly with different La3+ doping levels. In particular, the surface of PL0.5NC becomes rougher, while the particle size of PLNC decreases. Integrating these observations with the results of the half-cell and full-cell performance tests, PL0.5NC shows superior electrocatalytic activity compared to PNC and PLNC. This enhancement can be attributed to its relatively rough microstructure. The rough, continuous skeleton of PL0.5NC is likely to increase the specific surface area of the electrode, providing more active reaction sites and extending the three-phase boundary, thus accelerating the electrode reaction process.
 |
| | Fig. 8 SEM cross-section of (a) PNC (b) PL0.5NC (c) PLNC electrodes after performance test. | |
4. Conclusion
Three materials—PNC, PL0.5NC, and PLNC—with different levels of La3+ doping was successfully synthesized by the sol–gel method. The results reveal that increasing La3+ doping leads to a gradual decrease in electrical conductivity, with peak values of 117.2, 105.7, and 93.7 S cm−1 for PNC, PL0.5NC, and PLNC, respectively. Among these, PL0.5NC exhibits the fastest conductivity relaxation, indicating enhanced oxygen surface exchange and bulk diffusion capabilities. La3+ doping effectively lowers the energy barrier for electrode reactions, as evidenced by the comparison of Rp and activation energy, following the trend: PL0.5NC < PLNC < PNC. The enhancement is primarily attributed to improved oxygen surface exchange and charge transfer processes. The PL0.5NC-based cell demonstrated superior electrochemical performance, with a Rp as low as 0.245 Ω cm2 at 800 °C and PPD of 0.864 W cm−2, a 64% improvement over PNC. In conclusion, La3+ doping significantly enhances the catalytic activity of PNC-based electrodes, with PL0.5NC emerging as a promising candidate for high-performance SOFC oxygen electrodes due to its excellent physicochemical and electrochemical properties.
Author contributions
Zihao Liao: investigation, methodology, writing – original draft. Yiping Yang: data curation, formal analysis, investigation, methodology. Dingrong Ou: methodology, data curation. Yuan Tang: methodology, funding acquisition. Bo Wang: methodology, funding acquisition. Binbin He: methodology, project administration, funding acquisition. Yu Zeng: investigation, methodology. Yunfeng Tian: supervision, conceptualization writing – review & editing. Bo Chi: methodology, funding acquisition, project administration.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We acknowledge the financial support from the National Key R&D Program of China (2022YFB4002205), Southern Power Grid Corporation Science and Technology Project Funding (030100KC23020007), National Natural Science Foundation of China (52172199, 52302334), Jiangsu Province (BZ2022027, BE2023092). Changzhou City (CZ20230010). The project was also supported by the State Key Laboratory of Materials Processing and Die & Mould Technology, Huazhong University of Science and Technology (P2023-025).
References
- S. He, Y. Zou, K. Chen and S. P. Jiang, Interdiscip. Mater., 2023, 2, 111–136 Search PubMed.
- M.-Y. Zhang, Y.-F. Tian, L. Zou, J. Pu and B. Chi, Rare Met., 2024, 1–11 Search PubMed.
- H. Su, D. Wu, C. Li, C. Li and C. Zhang, Prog. Nat. Sci.: Mater. Int., 2023, 33(3), 309–319 CrossRef.
- W. Li, J. Sunarso, Y. Yang, Y. Chen, C. Ge, W. Wang, Y. Guo, R. Ran and W. Zhou, Energy Rev., 2024, 100085 CrossRef.
- W. Wang, Y. Huang, S. Jung, J. M. Vohs and R. J. Gorte, J. Electrochem. Soc., 2006, 153, A2066 CrossRef.
- Y. Liu, Y. Tian, W. Wang, Y. Li, S. Chattopadhyay, B. Chi and J. Pu, ACS Appl. Mater. Interfaces, 2020, 12, 57941–57949 CrossRef.
- S. P. Jiang, Int. J. Hydrogen Energy, 2019, 44, 7448–7493 CrossRef.
- R. Yang, Y. Tian, Y. Liu, J. Pu and B. Chi, J. Rare Earths, 2023, 41, 599–604 CrossRef CAS.
- X. Xu, C. Su and Z. Shao, Energy Fuels, 2021, 35, 13585–13609 CrossRef.
- V. Sivtsev, E. Lapushkina, I. Kovalev, R. Guskov, M. Popov and A. Nemudry, Green Carbon, 2023, 1, 154–159 CrossRef.
- Y. Tian, N. Abhishek, C. Yang, R. Yang, S. Choi, B. Chi, J. Pu, Y. Ling, J. T. Irvine and G. Kim, Matter, 2022, 5, 482–514 CrossRef.
- Z. Li, P. Shan, W. Tang, Q. Ni, B. Qian, S. Wang, Y. Zheng, L. Ge, H. Chen and C. Zhang, J. Alloys Compd., 2023, 932, 167646 CrossRef.
- D. Mishchenko, M. Arapova, Y. Bespalko, Z. Vinokurov and A. Shmakov, J. Alloys Compd., 2023, 967, 171693 CrossRef.
- A. Staykov, T. Nguyen, T. Akbay and T. Ishihara, J. Phys. Chem. C, 2022, 126, 7390–7399 CrossRef.
- L. Navarrete, M. Fabuel, C.-Y. Yoo and J. Serra, Int. J. Hydrogen Energy, 2020, 45, 15589–15599 CrossRef.
- X.-D. Zhou, J. W. Templeton, Z. Nie, H. Chen, J. W. Stevenson and L. R. Pederson, Electrochim. Acta, 2012, 71, 44–49 CrossRef CAS.
- N. Porotnikova, A. Khodimchuk, M. Ananyev, V. Eremin, E. Tropin, A. Farlenkov, E. Y. Pikalova and A. Fetisov, J. Solid State Electrochem., 2018, 22, 2115–2126 CrossRef CAS.
- S. Saher, J. Song, V. Vibhu, C. Nicollet, A. Flura, J.-M. Bassat and H. J. Bouwmeester, J. Mater. Chem. A, 2018, 6, 8331–8339 RSC.
- E. Boehm, J.-M. Bassat, P. Dordor, F. Mauvy, J.-C. Grenier and P. Stevens, Solid State Ionics, 2005, 176, 2717–2725 CrossRef CAS.
- X. Xu, Y. Pan, Y. Zhong, R. Ran and Z. Shao, Mater. Horiz., 2020, 7, 2519–2565 RSC.
- A. Kovalevsky, V. Kharton, A. Yaremchenko, Y. Pivak, E. Naumovich and J. Frade, J. Eur. Ceram. Soc., 2007, 27, 4269–4272 CrossRef CAS.
- A. Kovalevsky, V. Kharton, A. Yaremchenko, Y. Pivak, E. Tsipis, S. Yakovlev, A. Markov, E. Naumovich and J. Frade, J. Electroceram., 2007, 18, 205–218 CrossRef CAS.
- W. Wang, Y. Li, Y. Liu, Y. Tian, B. Ma, J. Li, J. Pu and B. Chi, ACS Sustainable Chem. Eng., 2021, 9, 10913–10919 CrossRef CAS.
- J. Hyodo, K. Tominaga, Y.-W. Ju, S. Ida and T. Ishihara, Solid State Ionics, 2014, 256, 5–10 CrossRef CAS.
- A. P. Tarutin, Y. G. Lyagaeva, A. I. Vylkov, M. Y. Gorshkov, G. K. Vdovin and D. A. Medvedev, J. Mater. Sci. Technol., 2021, 93, 157–168 CrossRef CAS.
- M. Khoshkalam, Đ. Tripković, X. Tong, M. A. Faghihi-Sani, M. Chen and P. V. Hendriksen, J. Power Sources, 2020, 457, 228035 CrossRef CAS.
- S. R. Maity, M. Ceretti, L. Keller, J. Schefer, T. Shang, E. Pomjakushina, M. Meven, D. Sheptyakov, A. Cervellino and W. Paulus, Phys. Rev. Mater., 2019, 3, 083604 CrossRef CAS.
- M. Hrovat, S. Bernik, J. Holc, D. Kuscer and D. Kolar, J. Mater. Sci. Lett., 1997, 16, 143–146 CrossRef CAS.
- T. Jiang, Z. Wang, B. Ren, J. Qiao, W. Sun and K. Sun, J. Power Sources, 2014, 247, 858–864 CrossRef CAS.
- Y. Wang, X. Zhao, S. Lü, B. Yu, X. Meng, Y. Zhang, J. Yang, C. Fu and Y. Ji, Ceram. Int., 2014, 40, 7321–7327 CrossRef CAS.
- Q. Zhou, Y. Zhang, Y. Shen and T. He, J. Electrochem. Soc., 2010, 157, B628 CrossRef CAS.
- W. Jia, Y. Wang, J. Huang, M. Li, B. Xiang, Y. Wang, L. Wu, L. Zheng and L. Ge, Appl. Energy, 2024, 353, 122096 CrossRef CAS.
- H. Gong, D. Zhou, X. Zhu, N. Wang, J. Bai, L. Hu, Y. Zhang, Y. Chen, C. Luo and W. Yan, Int. J. Hydrogen Energy, 2024, 50, 1492–1502 CrossRef CAS.
- N. Dasgupta, R. Krishnamoorthy and K. T. Jacob, Solid State Ionics, 2002, 149, 227–236 CrossRef CAS.
- C. Yang, Y. Tian, J. Pu and B. Chi, ACS Sustainable Chem. Eng., 2022, 10, 1047–1058 CrossRef CAS.
- M. Ghamarinia, A. Babaei, C. Zamani and H. Aslannejad, Chem. Eng. J. Adv., 2023, 15, 100503 CrossRef CAS.
- J. H. Park, C. H. Jung, K. J. Kim, D. Kim, H. R. Shin, J.-E. Hong and K. T. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 2496–2506 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *c and
Bo
Chi
*c and
Bo
Chi