
Structural and mechanistic insights into oxidative biaryl coupling to form the arylomycin core by an engineered CYP450†
Vandana
Kardam
 ,
Vaibhav
Bhatt
and
Kshatresh Dutta
Dubey
*
,
Vaibhav
Bhatt
and
Kshatresh Dutta
Dubey
*
Department of Chemistry, School of Natural Sciences, Shiv Nadar Institution of Eminence, Delhi-NCR, NH91, Tehsil Dadri, Greater Noida, Uttar Pradesh 201314, India. E-mail: kshatresh.dubey@snu.edu.in; kshatresh@gmail.com
First published on 6th November 2024
Abstract
Arylomycin, a potent antibiotic targeting bacterial signal peptidase, is difficult to synthesize experimentally due to its poor to moderate yields and the formation of a mixture of compounds. A recent experimental bioengineering work shows that the core of arylomycin can be efficiently synthesized by engineering the cytochrome P450 enzyme from Streptomyces sp.; however, the mechanism of the same was not elucidated. Herein, we have thoroughly investigated the mechanism behind the evolution of the enzyme for the synthesis of the arylomycin core via C–C bond formation in the CYP450 enzyme using hybrid QM/MM calculations, MD simulations, and DFT calculations. We show that strategic mutations such as (a) G-101 → A facilitate biaryl coupling by subtly pushing the substrate and (b) the Q-306 → H mutation creates a strong pi–pi interaction with the substrate that brings the two phenol rings of the substrate closer to undergo C–C coupling. Importantly, our QM/MM calculations show that for efficient C–C formation, the reaction should proceed via the biradical mechanism rather than hydroxylation.
1. Introduction
Enzymes are the ultimate catalysts since they catalyze reactions in a highly selective and environmentally benign way.1–4 However, enzymes are selective and have evolved for specific reactions, which limits their scope as catalysts for industrial applications. In recent years, it has been well established that enzymes can be tuned to catalyze desired reactions for modern applications using directed evolution and site-directed mutagenesis techniques. Such bioengineered enzymes, particularly cytochrome P450, have efficiently shown several new-to-nature reactions, such as carbene transfer, nitrene transfer, C–H activation, and many more reactions.5,6 The functions and mechanisms of CYP450 enzymes are well studied and documented in several reviews and monographs.7–10 These studies show that the shapes and sizes of substrate binding pockets are highly diverse and are crucial for reaction selectivity and substrate specificity.11–14 In fact, several rationale-based bio-engineering studies on CYP450 enzymes have shown that prior knowledge of the mechanism can pave the way for designing new and efficient enzymes.15–17A recent experimental study shows that the bacterial cytochrome P450 monooxygenase from Streptomyces sp. can be engineered to synthesize the arylomycin core via C–C coupling between two biaryl groups.18 This study is of significant interest as it synthesizes the arylomycin core, a potent antibacterial agent. Arylomycin inhibits the function of bacterial type I signal peptidases, which play a vital role in the bacteria's normal processes. Interestingly, arylomycin is the only known inhibitor of bacterial type I signal peptidases, making it a promising target for new antibiotics.18–21 So far, the synthesis of arylomycin has posed a challenge due to poor to moderate yields and the formation of a mixture of compounds that are difficult to separate. Therefore, the study by Molinaro et al. provided a greener, highly selective, and environmentally benign way to synthesize arylomycin. A very recent work has revealed a C–C coupling reaction initiated by another bacterial CYP450 from Mycobacterium tuberculosis leading to the biosynthesis of fluorine-substituted mycocyclosin derivatives.22 Another CYP450 mediated biosynthesis of cihanmycins has been reported via intramolecular C–O coupling.23 Despite the plethora of experimental and theoretical studies showing that CYP450 enzymes perform C–C coupling and C–N bond formation as their native function,24–28 the study by Molinaro et al.,18 is novel since it provides a way to engineer a monooxygenase CYP450 into a C–C bond forming CYP450 to synthesize the arylomycin core.29 Therefore, we believe that a mechanistic insight could pave the way for bioengineering several other CYP450 scaffolds that are designed for such C–C coupling reactions, leading to the synthesis of more natural products like arylomycin.
In the present study, we focus on mechanistic insight into how two mutations (i.e., G101A and Q306H) could evolve a natural CYP450 scaffold that performs hydroxylation as a native function for a non-native C–C coupling reaction as a secondary function. The site of the mutation, the catalytic site, and the 2D structure of the substrate, arylomycin, are shown in Fig. 1. To this end, we have performed comprehensive MD simulations to analyze structural and conformational changes due to the mutations and hybrid QM/MM calculations to study the mechanism of C–C bond formation.
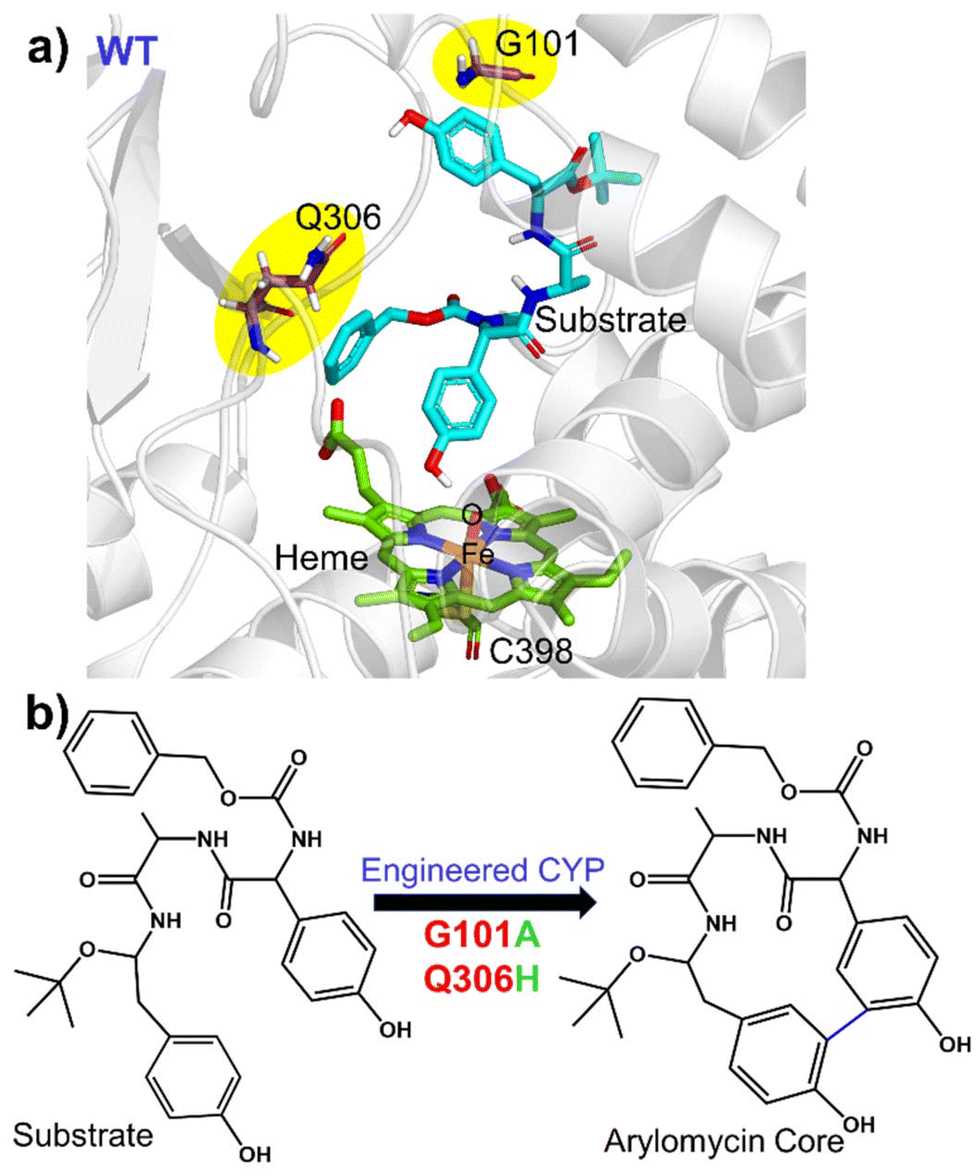 | ||
| Fig. 1 (a) Structure of the WT showing the active site region along with the residues undergoing mutation in the WT. (b) C–C coupling reaction catalysed by engineered CYP450. | ||
2. Methodology
2.1. System setup
The study focuses on the bacterial CYP450 from Streptomyces sp., which performs hydroxylation mainly in its native form. But after certain mutations, it transforms into engineered CYP to catalyse C–C bond formation between biaryl rings to form arylomycin as its major product (see Fig. 1). Homology modelling was employed using the Swiss Model to construct a three-dimensional structure for a target protein for the WT. The target protein sequence was obtained from the GenBank accession database (Id: OK585091). The modelling process involved aligning the target protein sequence with a suitable template structure to generate a reliable structural prediction. In this case, the template structure with the Protein Data Bank (PDB ID: 4UBS)30 was selected as the reference. After modelling the initial structure, a mutant was generated from a WT carrying the G101A and Q306H mutations with the help of pyMoL.31 These mutations were introduced to investigate their impact on the functionality and catalytic activities of the CYP450 enzyme. The substrate was docked inside the active site of the modelled structure using UCSF Chimera.32 The missing hydrogen atoms and some heavy atoms were added by the leap module of Amber 20.33 The force fields for the heme moiety were taken from the already published parameters for the Cpd I state.34 The Amber ff19SB force field was employed for all the enzymatic systems. The associated partial atomic charges and missing parameters for the substrate were generated by applying the restraint electrostatic potential (RESP) method of QM calculated charges at the HF/6-31G(d) level of theory.35,36 The corresponding parameters for the substrate were generated using Generalized Amber Force Fields (GAFF2) in the Antechamber module of Amber 20. A few Na+ ions were added to the protein surfaces to neutralize the total charge of the system. Finally, the resulting systems were solvated in an octahedral box with the TIP3P water model37 extended up to a minimum cut-off of 10 Å from the protein boundary. The catalytic cycle of CYP450 has many states. However, here we focus on the Cpd I state, which is the ultimate oxidant species that leads to the catalysis of the substrate.8,92.2. MD simulation
After proper parametrization of the system, to remove bad contacts, minimization was performed in two stages using a combination of steepest descent (5000 steps) and conjugate gradient (5000 steps) methods. In the first stage, the water position and conformations were relaxed keeping the protein fixed. Thereafter, the whole complex was minimized. Subsequently, the system was gently annealed up to 300 K under an NVT ensemble for 50 ps. After that, 1 ns of density equilibration was performed under an NPT ensemble at a target temperature of 300 K and a pressure of 1 atm by using a Langevin thermostat38 and a Berendsen barostat39 with a collision frequency of 2 ps and a pressure relaxation time of 1 ps. This 1 ns density equilibration is a weakly restrained MD simulation, in which the system is slowly released to achieve uniform density after heating under periodic boundary conditions. Then, after we removed all the restraints applied before, the system was equilibrated for 3 ns to get a well-settled pressure and temperature for chemical and conformational analyses. Thereafter, a productive MD simulation was performed using the Monte Carlo barostat for 100 ns for each complex system. We used three different replicas starting from different initial velocities each for 300 ns. Therefore, we performed a total of 300 ns simulations for each system including the wild type and the mutant. During all the MD simulations, covalent bonds containing hydrogens were constrained using the SHAKE algorithm,40 and the Particle Mesh Ewald (PME) method41 was used to treat long-range electrostatic interactions with the cut-off set as 10 Å. All MD simulations were performed with the GPU version of the AMBER 22 package.33 The MD trajectory analysis was done with the CPPTRAJ module of AMBER 22. The visualization of the MD trajectories was performed by VMD.422.3. SPM analysis
Shortest Mean Path (SPM) is a Python-based tool, introduced by Romero-Rivera et al. in 2017.54 It generates a path by calculating mean distances and correlation values. Then, using the Dijkstra algorithm from the igraph module, it identifies the shortest paths. The algorithm systematically examines all nodes in the graph, determining the shortest route from the first to the last protein residue. This approach highlights the graph's edges with shorter lengths, which signify stronger correlations and greater centrality in the communication/interaction pathway. Only those edges with a significant contribution are displayed, with their representation weighted according to their level of contribution.2.4. QM/MM methodology
The reaction mechanism was investigated using QM/MM calculations. The QM-region involves the substrate, truncated heme porphyrin ligated with C398. The active region in QM/MM calculations in all the systems involves the protein residues and water molecules present within a cutoff of 8 Å from the active oxidant heme. The atoms in the selected “active region” (mainly from the MM part) interact with the QM zone through electrostatic and van der Waals interactions and the corresponding polarization effects were considered in the subsequent QM/MM calculations. All QM/MM calculations were performed with ChemShell,43,44 by combining Turbomole45 for the QM part and DL_POLY46,47 for the MM part. The MM part was described using the ff19SB forcefield. To account for the polarizing effect of the protein environment on the QM region, an electronic embedding scheme was used. Hydrogen link atoms with the charge shift model48 were employed for treating the QM/MM boundary. During QM/MM geometry optimization, the QM region was treated using the hybrid UB3LYP functional with two basis sets, B1 and B2, where B1 stands for def2-SVP49 and B2 stands for def2-TZVP. The B1 basis set was used for geometry optimization, potential energy surface scanning, and frequency calculations. The energies were further corrected with the Grimme dispersion correction. All of the QM/MM transition states were located by relaxed potential energy surface (PES) scans, followed by full TS optimization using the P-RFO50 optimizer implemented in the HDLC code. The energies were further corrected with a large all-electron basis set, designated as def2-TZVP. The zero-point energies (ZPEs) were calculated for all the species and the corresponding final energies were reported as UB3LYP/B2+ZPE. Since s = 1/2 (doublet) and s = 3/2 (quartet), the spin states of Cpd I generally exhibit similar reactivities in CYP450 enzymes. We have performed the calculation in the same spin state, i.e., s = 1/2 (doublet) used in earlier studies of CYP450.3. Results and discussion
3.1. MD simulation studies of WT and mutant enzymes
To explore the conformational changes due to site mutation in the wild type (WT) enzyme, we performed extensive molecular dynamics (MD) simulations for the WT and the mutant enzymes. Substrate binding and recognition are crucial in enzymatic reactions, particularly in C–C coupling reactions. As such, the proper alignment of the two phenol rings of the substrate is necessary to generate aromatic ring radicals.51,52 In addition, the C–C coupling reaction involves the formation of two radicals on each phenol ring, and therefore, precise positioning of the two phenol rings becomes of utmost importance.26 Therefore, we primarily focused on the positioning of the phenyl ring core within the active site in the WT and mutant enzymes. The simulation of the WT enzyme shows that both phenolic rings are far apart from each other for the majority of the time, which leads us to believe that the WT enzyme does not prefer the C–C coupling reaction. However, for a brief period, the two phenolic rings settle in a conformation primed for efficient C–C coupling. On the other hand, the two phenolic groups in the mutant enzyme are found in close proximity of each other during much of the simulation time and thus are in the optimum position for C–C coupling (see Fig. 3b). The corresponding population of the reactive conformations for both WT and mutant enzymes can be seen in Fig. 2. Fig. 3c quantitatively portrays the positioning of the two phenolic rings by depicting the variation between the distances of C7 (ring 1) and C23 (ring 2) atoms in both the WT and mutant enzymes. Interestingly, the hydroxyl group of ring 1 forms a stable H-bond with the O–Fe of Cpd I during entire simulations for the WT and mutant enzymes. This is in contrast to a previous study on CYP121, another enzyme known to perform C–C coupling reactions, where such a hydrogen bond between the substrate and Cpd I was absent due to the presence of axial water molecules in the active site.53 | ||
| Fig. 2 The population of the reactive conformation in the WT (blue) and mutant (orange) enzymes. The reactive conformation is defined as the one in which the distance between C7 (ring 1) and C23 (ring 2) is equal to or less than 4 Å in both WT and mutant enzymes. | ||
 | ||
| Fig. 3 Positioning of phenol rings in (a) the WT and (b) mutant enzymes. (c) Distance plot between the phenol –OH of ring 1 of the substrate and O1 of heme, and between C7–C23 of the substrate, respectively. | ||
So, how do the mutations bring ring 1 and ring 2 close to each other to enable C–C coupling? To study this intriguing question, we thoroughly monitored the substrate–protein interactions in the WT and mutant enzymes. Let us first discuss what makes ring 1 and ring 2 stay apart in the WT enzyme. Fig. 4a shows the interactions of the substrate with the enzymatic residues for the WT enzyme. We see that R84 forms a hydrogen bond with L107; on the other hand, Q100 interacts with the backbone of P87, which pulls the side chain of Q100 away from the substrate. This makes the active site more open, and the substrate adopts an entropically more favourable conformation where ring 1 and ring 2 are far apart. At this juncture, though weakly, the Q306 stabilizes the ring 1 conformation via interacting its polar side chain with the pi-cloud of ring 1. Therefore, the substrate adopts a more staggered conformation, which is entropically driven.
 | ||
| Fig. 4 (a). MD snapshots of the WT and mutant enzymes. Wheat coloured: wild type. Green coloured: mutant. (b) Water occupancy is around 5 Å of G/A 101 for the WT and mutant enzymes. (c) Water population in the mutant and the wild type complex. | ||
Let us see what happens when we mutate G101 → A and Q306 → H for efficient C–C coupling. A representative snapshot of the mutant from the MD simulation is shown in Fig. 4b. Since alanine has an additional methyl group relative to glycine, the mutation of G101 → A applies a steric kick to the active site (Fig. S4†). Moreover, Q100 curls inward and forms a water-mediated hydrogen bond with R84. Since R84 is now partnering with Q100, it releases ring 1, which, in turn, swings inward towards ring 2. At the same instant, H306, which replaces Q306 in the mutated complex, stabilizes the substrate, and hence, the substrate adopts a more compact conformation where the two rings are close and in optimal orientation for C–C coupling. In addition, we found increased water inflow in the mutated enzyme, as can be seen in Fig. 4c.
As can be seen, these mutations are far apart from each other (Fig. 5a); therefore, we performed the Shortest Mean Path (SPM) analysis, a correlation dependent methodology that facilitates the analysis of allosteric interactions between two remote substructures to see if these mutations have any inter-correlations. The SPM analysis provided an energy-based algorithm to calculate the most feasible connection network between several protein residues. In this method, the residues participating in distant interactions are represented by a thread (black), and the magnitude of the corresponding interaction is represented by the size of the spherical beads. As can be seen in Fig. 5b, both Q100 and H306 are directly connected to each other via a strong network, and Q100 is connected to A101. This shows that mutating at one end can create a local disturbance at the other end via this network.
 | ||
| Fig. 5 (a) A representative snapshot that shows the allosteric interaction, (b) SPM analysis for G101A and Q306H mutations in the mutant enzyme. | ||
In a nutshell, the MD simulation of the WT and the mutant complex shows that on mutations, the entropically dominant staggered conformation of the substrate turns into an enthalpic-driven compact conformation, which brings two rings closer for the C–C coupling.
3.2. Mechanism for hydroxylation versus C–C bond formation
We propose a reaction mechanism as shown in Scheme 1. As can be seen, both reactions, i.e., hydroxylation and C–C coupling, are initiated by Cpd I via a hydrogen atom transfer (HAT) reaction from ring 1 of the substrate, which forms a Cpd II-like intermediate. The bifurcation in C–C coupling and the hydroxylation reaction started with the positioning of ring 1 and ring 2. When both rings are far apart, a typical Cpd II-mediated rebound mechanism occurs, which forms the hydroxylated product at ring 1 of the substrate (see green lines in Scheme 1). In contrast, when two rings are close, the Cpd II intermediate gets another hydrogen (HAT-2) from the alcoholic group of ring 2 and quickly converts it into the resting state. This makes the substrate a biradical system, where each of the rings occupies one radical, which couples together to form a C–C bond, resulting in the formation of the arylomycin core.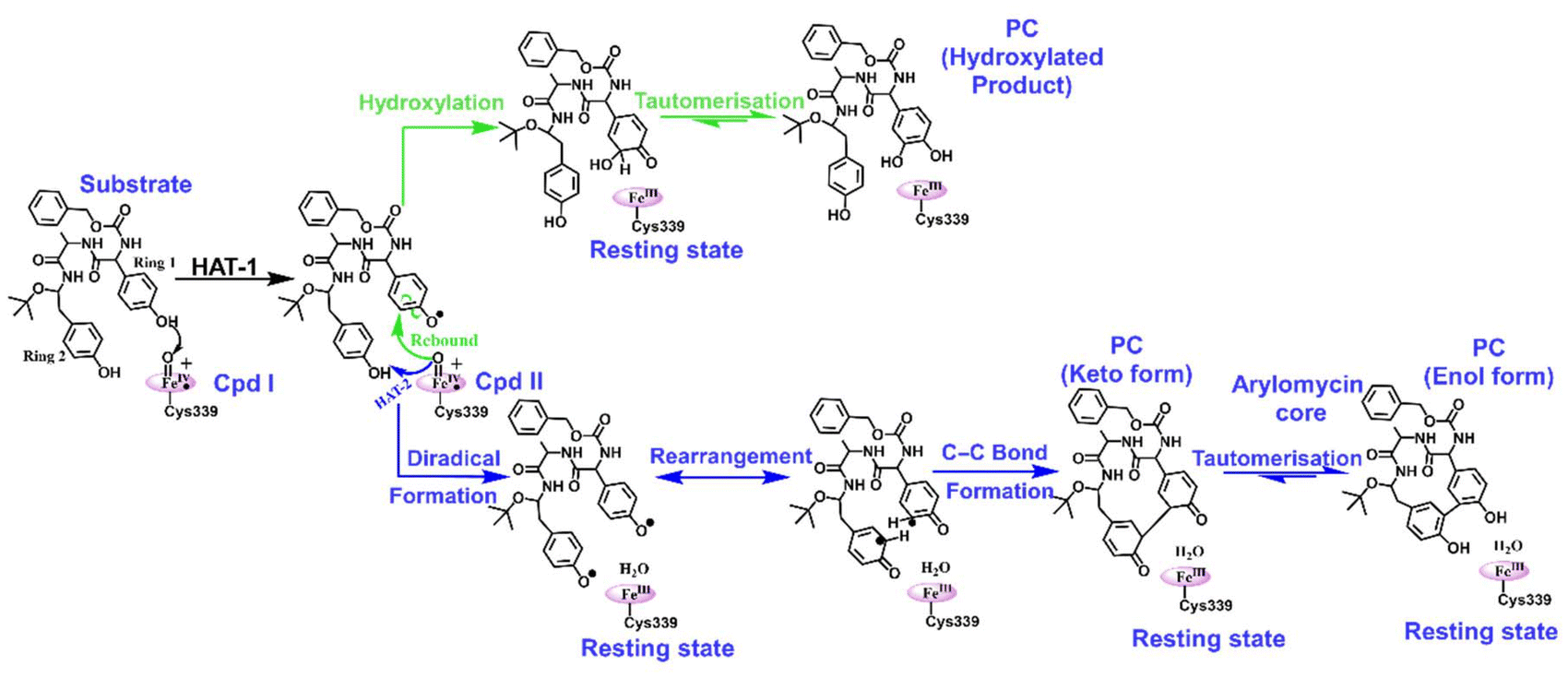 | ||
| Scheme 1 A reaction scheme for hydroxylation and C−C bond-forming reactions for the substrate. Note that the green coloured pathway underlines the hydroxylation mechanism and the blue coloured pathway underlines the C–C bond forming reaction. | ||
 | ||
| Fig. 6 (a) The reaction profile for the wild type enzyme calculated by hybrid QM/MM calculations at the B3LYP/def2-TZVP level of theory. Reported energies are ZPEs corrected from the subsequent frequency calculation at the B3LYP/def2-SVP level of theory. Energies are in kcal mol−1 and relative to the reactant complex (RC). (b) Spin density and charges of species obtained during the mechanism. The optimized geometries of all the species obtained during (c) the hydroxylation mechanism and (d) the C–C bond forming reaction. Respective bond distances are in Å units. Note that RC, TS and IM indicate the reactant complex, transition state and intermediate, respectively. | ||
Thus, from the reaction profile that is obtained, we can infer that the C–C coupling reaction is the major reaction that is taking place, whereas the hydroxylation pathway with its acute barrier accounts for the very minor hydroxylation products as found in the experimental data.18 Therefore, our theoretical calculations qualitatively agree with the experimental finding that C–C coupling is more feasible than hydroxylation in the WT enzyme.
To check the feasibility of both reactions in the mutant enzyme, we have also carried out another set of QM/MM calculations for the mutant enzyme, which has been discussed in the section below.
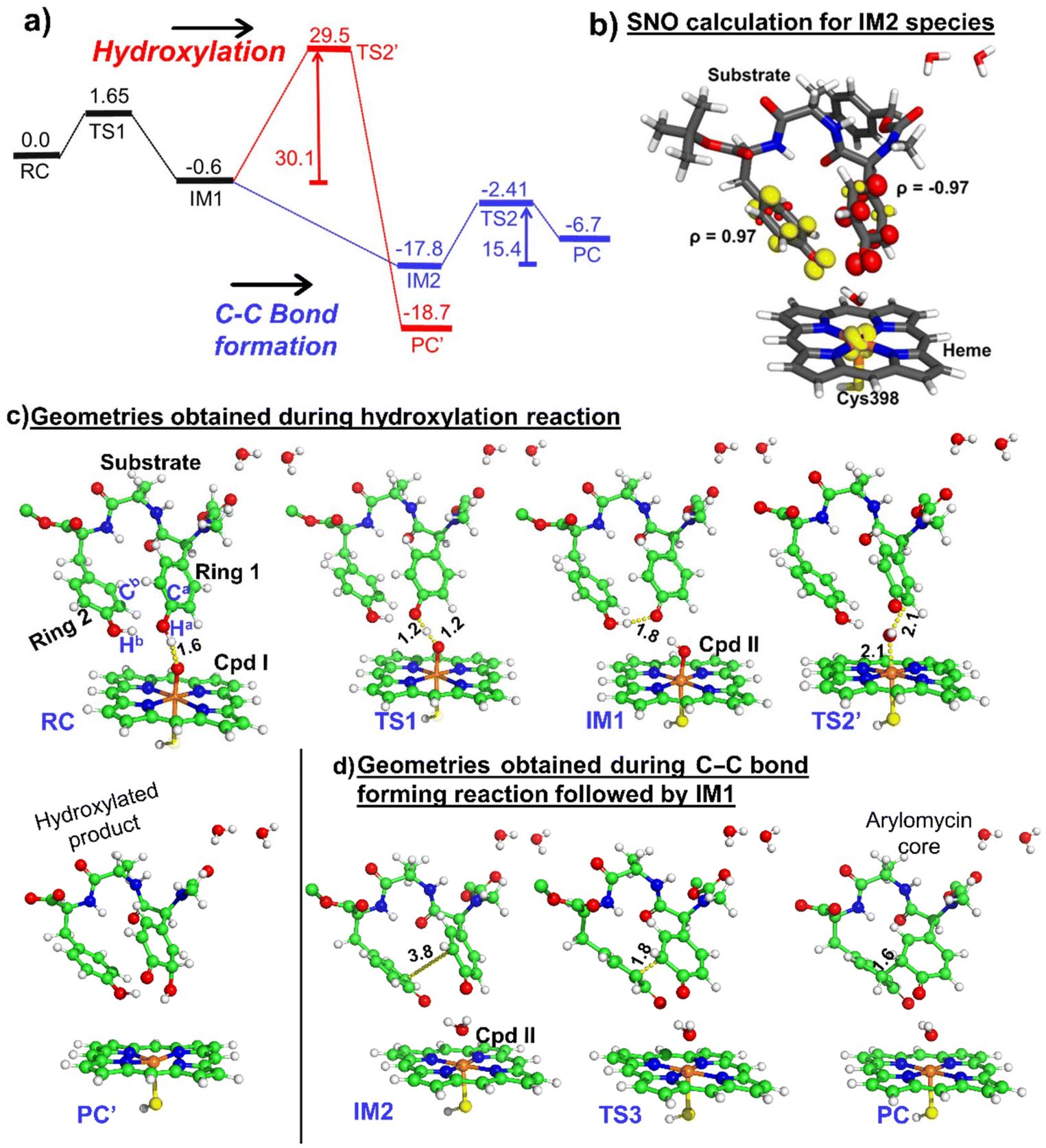 | ||
| Fig. 7 (a) The reaction profile for the mutant calculated by hybrid QM/MM calculations at the B3LYP/def2-TZVP level of theory. Reported energies are ZPEs corrected from the subsequent frequency calculation at the B3LYP/def2-SVP level of theory. Energies are in kcal mol−1 and relative to the reactant complex (RC). (b) Spin natural orbital calculation of IM2 of the mutant enzyme. Yellow and red indicate the positive and negative spin density iso-surfaces, respectively. The optimized geometries of all the species obtained during (c) the hydroxylation mechanism and (d) the C–C bond forming reaction. Respective bond distances are in Å units. Note that RC, TS, and IM indicate the reactant cluster, transition state, and intermediate, respectively. | ||
From the above QM/MM calculations, it is clear that in both the wild type and the mutant enzyme, the predominant reaction is the C–C coupling reaction, although double mutations contribute to an overall increase in selectivity towards C–C coupling as can be deduced from the experimental data.18 The study is crucial since it is focused on an engineered CYP, which catalyzes the biaryl coupling for the formation of the antibiotic drug arylomycin.
4. Conclusion
In conclusion, we have investigated an engineered cytochrome P450-catalyzed oxidative biaryl coupling reaction for the formation of an arylomycin core, which is an antibiotic-targeting signal peptidase. With MD simulation studies, we have studied the impact of two-point mutations on the conformational stability of the substrate that facilitates the C–C coupling reaction. We show that the C–C coupling in the mutant enzyme is entropically driven, which dominates the entropic penalties due to contraction of the two phenolic rings. We show that strategic mutations such as (a) G-101 → A facilitate biaryl coupling by subtly pushing the substrate and (b) the Q-306 → H mutation creates a strong pi–pi interaction with a substrate that brings two phenol rings of the substrate closer to undergo C–C coupling. Importantly, our QM/MM calculations show that for efficient C–C formation, the reaction should undergo the biradical mechanism rather than hydroxylation.These findings will pave the way for creating more bioengineered enzymes that can act as green catalysts for antibiotic synthesis.
Data availability
Data necessary to reproduce the work are provided in the ESI available for free in the online version of the article.† It includes all the coordinates of the optimized geometries of the reactant, transition state, and product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KDD acknowledges the Shiv Nadar Foundation for providing financial support and computational resources to conduct this project.References
- S. Shoda, H. Uyama, J. Kadokawa, S. Kimura and S. Kobayashi, Enzymes as Green Catalysts for Precision Macromolecular Synthesis, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS
.
- J. Chapman, A. E. Ismail and C. Z. Dinu, Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks, Catalysts, 2018, 8, 238 CrossRef
- R. Mu, Z. Wang, M. C. Wamsley, C. N. Duke, P. H. Lii, S. E. Epley, L. C. Todd and P. J. Roberts, Application of Enzymes in Regioselective and Stereoselective Organic Reactions, Catalysts, 2020, 10, 832 CrossRef CAS
- G. M. Whitesides and C. Wong, Enzymes as Catalysts in Synthetic Organic Chemistry [New Synthetic Methods (53)], Angew. Chem., Int. Ed. Engl., 1985, 24, 617–638 CrossRef
- S. Yadav, S. Shaik and K. D. Dubey, On the Engineering of Reductase-Based-Monooxygenase Activity in CYP450 Peroxygenases, Chem. Sci., 2024, 15, 5174–5186 RSC
- S. Shaik, H. Hirao and D. Kumar, Reactivity Patterns of Cytochrome P450 Enzymes: Multifunctionality of the Active Species, and the Two States–Two Oxidants Conundrum, Nat. Prod. Rep., 2007, 24, 533–552 RSC
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Heme-Containing Oxygenases, Chem. Rev., 1996, 96, 2841–2888 CrossRef CAS PubMed
- B. Meunier, S. P. de Visser and S. Shaik, Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes, Chem. Rev., 2004, 104, 3947–3980, DOI:10.1021/cr020443g
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, P450 Enzymes: Their Structure, Reactivity, and Selectivity—Modeled by QM/MM Calculations, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS PubMed
- V. Kardam, S. Kalita and K. D. Dubey, Computations Reveal a Crucial Role of an Aromatic Dyad in the Catalytic Function of Plant Cytochrome P450 Mint Superfamily, J. Inorg. Biochem., 2022, 237, 111990 CrossRef CAS PubMed
- T. L. Poulos, Cytochrome P450 Flexibility, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13121–13122 CrossRef CAS
- P. Urban, T. Lautier, D. Pompon and G. Truan, Ligand Access Channels in Cytochrome P450 Enzymes: A Review, Int. J. Mol. Sci., 2018, 19, 1617 CrossRef PubMed
-
T. L. Poulos and E. F. Johnson, Structures of Cytochrome P450 Enzymes, Cytochrome P450 Struct. Mech. Biochem, 2015, pp. 3–32 Search PubMed
- S. Yadav, V. Kardam, A. Tripathi, S. T. Guruvayurappan and K. D. Dubey, The Performance of Different Water Models on the Structure and Function of Cytochrome P450 Enzymes, J. Chem. Inf. Model., 2022, 62, 6679–6690 CrossRef CAS
- F. Zhao, P. Bai, T. Liu, D. Li, X. Zhang, W. Lu and Y. Yuan, Optimization of a Cytochrome P450 Oxidation System for Enhancing Protopanaxadiol Production in Saccharomyces Cerevisiae, Biotechnol. Bioeng., 2016, 113, 1787–1795 CrossRef CAS PubMed
-
V. B. Urlacher and S. Schulz, Multi–Enzyme Systems and Cascade Reactions Involving Cytochrome P450 Monooxygenases, in Cascade Biocatal. Integr. stereoselective Environ. friendly React, 2014, pp. 87–132 Search PubMed
- S. E. Carrera-Pacheco, A. Mueller, J. A. Puente-Pineda, J. Zúñiga-Miranda and L. P. Guamán, Designing Cytochrome P450 Enzymes for Use in Cancer Gene Therapy, Front. Bioeng. Biotechnol., 2024, 12, 1405466 CrossRef PubMed
- C. Molinaro, Y. Kawasaki, G. Wanyoike, T. Nishioka, T. Yamamoto, B. Snedecor, S. J. Robinson and F. Gosselin, Engineered Cytochrome P450-Catalyzed Oxidative Biaryl Coupling Reaction Provides a Scalable Entry into Arylomycin Antibiotics, J. Am. Chem. Soc., 2022, 144, 14838–14845 CrossRef CAS
- S. I. Walsh, D. S. Peters, P. A. Smith, A. Craney, M. M. Dix, B. F. Cravatt and F. E. Romesberg, Inhibition of Protein Secretion in Escherichia Coli and Sub-MIC Effects of Arylomycin Antibiotics, Antimicrob. Agents Chemother., 2019, 63, 10–1128 CrossRef
- A. Craney and F. E. Romesberg, The Inhibition of Type I Bacterial Signal Peptidase: Biological Consequences and Therapeutic Potential, Bioorg. Med. Chem. Lett., 2015, 25, 4761–4766 CrossRef CAS PubMed
- P. A. Smith and F. E. Romesberg, Mechanism of Action of the Arylomycin Antibiotics and Effects of Signal Peptidase I Inhibition, Antimicrob. Agents Chemother., 2012, 56, 5054–5060 CrossRef CAS
- S.-H. Li, X. Zhang, Z.-L. Mei, Y. Liu, J.-A. Ma and F.-G. Zhang, Chemoenzymatic Synthesis of Fluorinated Mycocyclosin Enabled by the Engineered Cytochrome P450-Catalyzed Biaryl Coupling Reaction, J. Am. Chem. Soc., 2024, 146(29), 19962–19973 CrossRef CAS
- C. Fang, L. Zhang, Y. Wang, W. Xiong, Z. Yan, W. Zhang, Q. Zhang, B. Wang, Y. Zhu and C. Zhang, Discovery and biosynthesis of Cihanmycins Reveal Cytochrome P450-Catalyzed Intramolecular C-O Phenol Coupling Reactions, J. Am. Chem. Soc., 2024, 146(24), 16478–16489 CrossRef CAS
- S.-S. Gao, T. Zhang, M. Garcia-Borràs, Y.-S. Hung, J. M. Billingsley, K. N. Houk, Y. Hu and Y. Tang, Biosynthesis of Heptacyclic Duclauxins Requires Extensive Redox Modifications of the Phenalenone Aromatic Polyketide, J. Am. Chem. Soc., 2018, 140, 6991–6997 CrossRef CAS
- L. E. Zetzsche, J. A. Yazarians, S. Chakrabarty, M. E. Hinze, L. A. M. Murray, A. L. Lukowski, L. A. Joyce and A. R. H. Narayan, Biocatalytic Oxidative Cross-Coupling Reactions for Biaryl Bond Formation, Nature, 2022, 603, 79–85 CrossRef CAS
- F. P. Guengerich and F. K. Yoshimoto, Formation and Cleavage of C–C Bonds by Enzymatic Oxidation–Reduction Reactions, Chem. Rev., 2018, 118, 6573–6655 CrossRef CAS
- M. Makino, H. Sugimoto, Y. Shiro, S. Asamizu, H. Onaka and S. Nagano, Crystal Structures and Catalytic Mechanism of Cytochrome P450 StaP That Produces the Indolocarbazole Skeleton, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11591–11596 CrossRef CAS
- V. V. Shende, Y. Khatri, S. A. Newmister, J. N. Sanders, P. Lindovska, F. Yu, T. J. Doyon, J. Kim, K. N. Houk and M. Movassaghi, Structure and Function of NzeB, a Versatile C–C and C–N Bond-Forming Diketopiperazine Dimerase, J. Am. Chem. Soc., 2020, 142, 17413–17424 CrossRef CAS PubMed
- H. Aldemir, S. Shu, F. Schaefers, H. Hong, R. Richarz, S. Harteis, M. Einsiedler, T. M. Milzarek, S. Schneider and T. A. M. Gulder, Carrier Protein–Free Enzymatic Biaryl Coupling in Arylomycin A2 Assembly and Structure of the Cytochrome P450 AryC, Chem. – Eur. J., 2022, 28, e202103389 CrossRef CAS
- L.-H. Xu, H. Ikeda, L. Liu, T. Arakawa, T. Wakagi, H. Shoun and S. Fushinobu, Structural Basis for the 4′-Hydroxylation of Diclofenac by a Microbial Cytochrome P450 Monooxygenase, Appl. Microbiol. Biotechnol., 2015, 99, 3081–3091 CrossRef CAS
- W. L. DeLano, Pymol: An Open-Source Molecular Graphics Tool, CCP4 Newsl. Protein Crystallogr., 2002, 40, 82–92 Search PubMed
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera–a Visualization System for Exploratory Research and Analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS
- K. Shahrokh, A. Orendt, G. S. Yost and T. E. Cheatham III, Quantum, Mechanically Derived AMBER–compatible Heme Parameters for Various States of the Cytochrome P450 Catalytic Cycle, J. Comput. Chem., 2012, 33, 119–133 CrossRef CAS
- W. D. Cornell, P. Cieplak, C. I. Bayly and P. A. Kollman, Application of RESP Charges to Calculate Conformational Energies, Hydrogen Bond Energies, and Free Energies of Solvation, J. Am. Chem. Soc., 1993, 115, 9620–9631 CrossRef CAS
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- J. A. Izaguirre, D. P. Catarello, J. M. Wozniak and R. D. Skeel, Langevin Stabilization of Molecular Dynamics, J. Chem. Phys., 2001, 114, 2090–2098 CrossRef CAS
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular Dynamics with Coupling to an External Bath, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS
- T. Darden, D. York and L. Pedersen, Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS
-
J. Kendrick, P. Sherwood, A. H. De Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjoevoll, A. Fahmi, A. Schäfer and C. Lennartz, QUASI: A General Purpose Implementation of the QM/MM Approach and Its Application to Problems in Catalysis, 2003 Search PubMed
- S. Metz, J. Kästner, A. A. Sokol, T. W. Keal and P. Sherwood, ChemShell—a Modular Software Package for QM/MM Simulations, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 101–110 CAS
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, TURBOMOLE: Modular Program Suite for Ab Initio Quantum-Chemical and Condensed-Matter Simulations, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS
- W. Smith and T. R. Forester, DL_POLY_2.0: A General-Purpose Parallel Molecular Dynamics Simulation Package, J. Mol. Graphics, 1996, 14, 136–141 CrossRef CAS PubMed
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Electronic Structure Calculations on Workstation Computers: The Program System Turbomole, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed
- A. D. Becke, Density–functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- J. Kästner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, DL-FIND: An Open-Source Geometry Optimizer for Atomistic Simulations, J. Phys. Chem. A, 2009, 113, 11856–11865 CrossRef PubMed
- X. Chen, K. M. Engle, D. Wang and J. Yu, Palladium(II)–catalyzed C–H Activation/C–C Cross–coupling Reactions: Versatility and Practicality, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS
- C. Liu, H. Zhang, W. Shi and A. Lei, Bond Formations between Two Nucleophiles: Transition Metal Catalyzed Oxidative Cross-Coupling Reactions, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS
- V. G. Dumas, L. A. Defelipe, A. A. Petruk, A. G. Turjanski and M. A. Marti, QM/MM Study of the C-C Coupling Reaction Mechanism of CYP121, an Essential Cytochrome P450 of Mycobacterium Tuberculosis, Proteins: Struct., Funct., Bioinf., 2014, 82, 1004–1021 CrossRef CAS
- A. Romero-Rivera, M. Garcia-Borras and S. Osuna, Role of Conformational Dynamics in the Evolution of Retro-Aldolase Activity, ACS Catal., 2017, 7, 8524–8532 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt02197e |
| This journal is © The Royal Society of Chemistry 2025 |