
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and large crystal growth of a family of mixed-anionic methanesulfonate salts by anionic site-substitution: Na5(SO3CH3)4(X) (X = BF4−, ClO4−, PF6−, I−)†
Eric A. Gabilondo and
P. Shiv Halasyamani *
*
University of Houston, Department of Chemistry, Houston, TX 77204-5003, USA. E-mail: psh@uh.edu
First published on 6th March 2025
Abstract
Four new mixed-anionic sodium methanesulfonate compounds were synthesized – Na5(SO3CH3)4(X) (X = (I) BF4−, (II) ClO4−, (III) PF6−, and (IV) I−). Compounds I–III were grown as centimeter-size crystals under ambient conditions. Metal methanesulfonates (i.e., MSO3CH3 or M(SO3CH3)2) are proposed as potential high-performance nonlinear optical materials owing to their large calculated bandgaps, local anisotropy, and large polarization. However, there are few experimental reports in the literature. The present study involves using anionic substitution as a structure building strategy to discover new complex metal methanesulfonates. The new compounds, Na5(SO3CH3)4(X), where X = (I) [BF4]−, (II) [ClO4]−, (III) [PF6]−, and (IV) [I]− crystallize in the monoclinic space groups P2/n or I2/m, and are prepared as (I–III) high-quality and centimeter-sized crystals grown at ambient conditions, whereas millimeter-sized crystals of I, II, and IV could be prepared in high yields by hydrothermal synthesis. I–III have wide optical transparency from 400–1100 nm and λabs of ≤210 nm. I–III are each highly soluble in water, air stable for several months, and thermally stable below 350 °C. These data suggest that ionic-substitution is a viable method of discovering new and complex metal methanesulfonate salts with diverse properties.
1. Introduction
Compounds containing mixed-anionic groups are of essential importance in materials applications owing to the remarkable tuneability of both crystal structure and properties.1–4 Some of these recent applications include using functional groups with stereoactive lone pairs5,6 (e.g., [SeO3]2−, [BiO6]9−) or polar tetrahedra7,8 (e.g., [PO3F]2− and [BO3F]4−) to break symmetry and form acentric crystal structures for applications as nonlinear optical (NLO) materials.9,10 Similarly, anionic substitution facilitates the improvement of materials such as the improved crystal growth and reduced toxicity in Cs3Zn6B9O21,11 the AZn2BO3X2 family,12 or Rb3Ba3Li2Al4B6O20,13 versus the famous NLO crystal KBe2BO3F2 that requires the use of toxic BeO in the synthesis.14 As a result, the substitution of anionic groups within structure classes is an effective method for materials discovery, and may be used to create new compounds with improved properties.Metal methanesulfonates, i.e., MSO3CH3 or M(SO3CH3)2, have been recently predicted as a potential new class of NLO materials owing to their calculated larger optical bandgaps and polarizability of the [SO3CH3]− anion compared to commonly used functional groups such as [IO3]−, [SO4]2−, and [BO3]3−.15 This has been empirically demonstrated in the case of Ba(SO3CH3)2 as a NLO candidate crystal.16 Most reported metal methanesulfonates are centrosymmetric (55 out of 65 as of Jan. 2025),17 however, and cannot be used as NLO materials. As previously mentioned, adding additional anionic groups could lower the symmetry and allow NLO properties to flourish. There are currently only three mixed-metal methanesulfonates reported, M[Au(SO3CH3)4] (M = Li+, Na+, Rb+),18 and only five mixed-anionic methanesulfonates – (Bi(Se/Te)O3)(SO3CH3), Ba(SO3CH3)(BF4), Ba2(SO3CH3)3(BF4)(H2O)3,19 and Na(S(OH)3CH3)(BO3).20 Each of these compounds exhibit unique structures and properties in contrast to their simple binary salts and respective hydrates, e.g. Ba(SO3CH3)2, Ba(SO3CH3)2·1.5H2O, and NaSO3CH3. The dearth of reported complex metal methanesulfonates requires further exploration.
Many binary metal methanesulfonates, e.g., LiSO3CH3 and Ba(SO3CH3)2,15,21 crystallize as layered structures that are ripe targets for ion-substitution or -exchange as a rapid discovery method for lamellar compounds.22,23 For example, sodium methanesulfonate (NaSO3CH3), is structurally comprised of [Na5(SO3CH3)4]n+ 2D-layers with an intercalated [SO3CH3]n− anion.24 In the present work, we investigate NaSO3CH3 as an example structure-class to perform interlayer site-substitution to discover new mixed-anionic metal methanesulfonates. NaSO3CH3 is simple and low-cost to prepare as large crystals and is generally easier to handle compared to its more hygroscopic counterparts. We discuss below the discovery of four new and isostructural mixed-anionic metal methanesulfonates, Na5(SO3CH3)4(X), where X = (I) [BF4]−, (II) [ClO4]−, (III) [PF6]−, and (IV) [I]−. The known NaSO3CH3 compound, or (V) X = SO3CH3− for brevity, was similarly prepared for direct comparison. The new compounds crystallize in centrosymmetric monoclinic space groups P2/n (no. 13) (I, II, IV) or I2/m (no. 12) (III) derived from the structure of (V) that crystallizes in the orthorhombic space group Pbma (no. 57). Each compound could be prepared in high purity through hydrothermal methods, whereas compounds I–III could be grown as high-quality centimeter-sized crystals under ambient conditions. This manuscript discusses their preparation, crystal structure, optical properties, and thermal stabilities.
2. Experimental
2.1. Reagents
Na2CO3 (98%, Alfa Aesar), NaCl (99%, EM Chemicals), HSO3CH3 (70 w/w% solution, Sigma-Aldrich), NaBF4 (97%, Alfa Aesar), HClO4 (60–62% solution, Alfa Aesar), KPF6 (99% min, Alfa Aesar), and NH4I (99%, Sigma-Aldrich), and acetonitrile (VWR) were used as reagents with no additional purification.Caution: Perchloric acid (CAS#: 7601-90-3) is a strong oxidizer, potentially explosive, and should be handled with great care. All usage should be carried out in a fume hood specially rated for HClO4 in case of spills.
2.2. Synthesis
Compounds I, II, and IV were synthesized using hydrothermal methods. Nonstoichiometric mixtures of Na2CO3, HSO3CH3 solution, and X (i.e., (I) NaBF4, (II) HClO4, or (IV) NH4I) were required for each reaction. The loadings were (I) 1.2 g (11.3 mmol) Na2CO3, 0.84 g (7.7 mmol) NaBF4, and 1.5 mL (14.9 mmol) HSO3CH3; (II) 1.15 g (10.9 mmol) Na2CO3, 0.34 mL (3.1 mmol) HClO4, and 0.5 mL (5.0 mmol) HSO3CH3; (IV) 1.00 g (9.4 mmol) Na2CO3, 0.547 g (3.8 mmol) NH4I, and 1.5 mL (14.9 mmol) HSO3CH3. The reagent mixtures each had 1 mL of water added and were loaded into separate 23 mL Teflon-lined autoclaves. The autoclaves were heated at a rate of 1 °C min−1 to 220 °C for 96 h (4 days), and a cooled to room temperature at a rate of 0.1 °C min−1. The products were vacuum filtered and rinsed with 5 mL of acetonitrile to remove excess solvent adhered to the surface prior to air drying. Each reaction produced mm-scale crystals that could be mechanically separated and then cut for single-crystal X-ray diffraction. Compounds I, II, and IV each had yields of 78%, 79%, and 70%, respectively, with respect to the X-containing reagent. Compound III could not be prepared under similar reaction conditions.Large crystal growth of compounds I–III and V was performed through solvent evaporation at ambient conditions by mixing Na2CO3, and X (i.e., (I) NaBF4, (II) HClO4, or (III) KPF6) in 10 mL of HSO3CH3 and 30 mL of deionized water giving a total volume of approximately 45 mL in each reaction. Loadings were (I) 2.428 g (22.9 mmol) Na2CO3 and 1.6836 g (15.3 mmol) NaBF4; (II) 2.3 g (21.7 mmol) Na2CO3 and 0.679 mL (6.2 mmol) HClO4; (III) 2.04 g (34.9 mmol) NaCl, 1.28 g (7.0 mmol) KPF6; (V) 2.5 g (23.6 mmol) Na2CO3. Plastic containers were used for compounds I and III as the [BF4]− and [PF6]− anions attack glass. Each liquid mixture was placed uncovered on a benchtop in air except compound III which was in a fume hood rated for HClO4. Centimeter-scale (I, II, V) plate and (III) rectangular prism crystals appeared after 1–2 weeks. Compound IV could not be prepared under similar conditions. Crystals were mechanically separated from the mother liquor before vacuum filtration and rinsing with 10 mL of acetonitrile to remove the excess. All large crystals are stable in air for several months.
2.3. Structural determination
Single-crystal X-ray diffraction (SCXRD) for compounds I–IV was performed with crystals obtained from hydrothermal synthesis described above. Data was collected using a Bruker SMART X-ray diffractometer using with a CCD detector and graphite monochromator, controlled through the APEX2 software.25 Data collection was obtained with a Mo Kα source (λ = 0.71073 Å) at 298 K. The SAINT and SADABS programs were used to integrate the collected frames and to correct for absorption, respectively, and the space group confirmed by the XPREP software.26,27 The structure solution was obtained by using the ShelXT direct method and intrinsic phasing prior to structural refinement with ShelXL in the Olex2-1.5 software package.28–31 The PLATON and ADDSYM software packages were used to check for missing symmetry elements.32–34 Bond-valence summations (BVS) were calculated using method described by Brown35 with parameters obtained from the International Union of Crystallography and reported crystal structures containing X-group motifs.36–40 The structure refinement summary for compounds I–IV, BVS, bond lengths, and bond angles, are contained in Table 1 and Tables S1–S5 in the ESI.† The crystallographic information files (cifs) for compounds I–IV are provided as part of the electronic ESI.† The crystal structure for compound V has been previously reported elsewhere (CCDC no. 1105562).24 All structural models were constructed using the VESTA software.41| a R1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = {∑w[(Fo)2 − (Fc)2]2/∑w[(Fo)2]2}1/2. | ||||
|---|---|---|---|---|
| Formula | Na5(SO3CH3)4(BF4) | Na5(SO3CH3)4(ClO4) | Na5(SO3CH3)4(PF6) | Na5(SO3CH3)4(I) |
| Crystal size (mm) | 0.20 × 0.30 × 0.50 | 0.40 × 0.40 × 0.35 | 0.15 × 0.13 × 0.05 | 0.05 × 0.10 × 0.12 |
| Formula weight (g mol−1) | 582.14 | 594.78 | 640.30 | 622.23 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P2/n (13) | P2/n (13) | I2/m (12) | P2/n (13) |
| a (Å) | 12.524(4) | 12.6653(7) | 12.9030(3) | 12.4118(4) |
| b (Å) | 5.534(1) | 5.5400(3) | 5.5449(1) | 5.5158(2) |
| c (Å) | 15.001(4) | 15.0037(9) | 15.1892(6) | 14.9258(5) |
| β (°) | 102.878(4) | 103.263(3) | 103.946(2) | 102.489(2) |
| V (Å3) | 1013.5(5) | 1024.67(10) | 1054.69(5) | 997.66(6) |
| Z | 2 | 2 | 2 | 2 |
| Dcalc (g cm−1) | 1.907 | 1.928 | 2.016 | 2.071 |
| μ (mm−1) | 0.664 | 0.773 | 0.734 | 2.181 |
| GooF on F2 | 1.050 | 1.049 | 1.046 | 1.009 |
| R1/wR2 [I > 2σ(I)]a | 0.0288/0.0776 | 0.0269/0.0781 | 0.0311/0.0710 | 0.0293/0.0716 |
| R1/wR2 (all data)a | 0.0358/0.0819 | 0.0281/0.0813 | 0.0407/0.0760 | 0.0575/0.0716 |
| F(000) | 584 | 600 | 640 | 608 |
| Largest diff. peak/hole (e Å−3) | 0.56, −0.43 | 0.45, −0.67 | 0.38, −0.35 | 0.60, −0.50 |
| Reflections total, unique | 9448, 2352 | 35337, 2567 | 8438, 1246 | 31164, 2734 |
| Rint, Rσ | 0.0294, 0.0215 | 0.0286, 0.0104 | 0.0349, 0.0221 | 0.063, 0.0296 |
All powder X-ray diffraction (PXRD) data was collected on ground crystals with a PANalytical Empyrean diffractometer using Cu Kα radiation (λ1 = 1.54059 Å, λ2 = 1.54432 Å, I2/I1 = 0.5) in Bragg–Brentano geometry on a fixed glass plate. Indexing of the primary crystal facet was observed by mounting a crystal flat on the sample stage position and rotating the crystal 90° before subsequent scans. PXRD data sets from the indexing method are provided in Fig. S7 in the ESI.†
Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) were used to confirm crystal homogeneity and elemental composition. SEM/EDS was performed using an Axia-ChemiSEM (Thermo Fisher) equipped with EDS and cathodoluminescense detectors. SEM images with EDS elemental mapping as well as the raw EDS spectra are contained in Fig. S1–S6 in the ESI.†
2.4. Thermal stability and optical characterization
The thermal stability of compounds I–V were determined using thermogravimetric analysis (TGA) on an EXSTAR 6300 TG/DTA. Data was collected under N2 flow from 25–600 °C and a ramp rate of 10 °C min−1. Thermal decomposition was performed by annealing each compound in an evacuated and sealed quartz tube at 600 °C for 2 h. Partial pyrolysis was investigated by annealing single-crystals of I–IV at 300 °C for 2 h under static vacuum. Diffuse reflectance spectroscopy was collected from 200–1200 nm on a JASCO V-770 UV-Visible/NIR spectrophotometer with a Spectralon reference. Optical bandgaps were estimated using the Tauc method and extrapolating the square of the remission function to the baseline.42–44 Fourier-Transformed Infrared (FT-IR) spectroscopy was performed over the range of 400–4000 cm−1 using a Thermo Scientific Nicolet iS10 FT-IR spectrophotometer.3. Results and discussion
3.1. Synthesis and crystal structures
The anionic groups [BF4]−, [ClO4]−, and [PF6]−, and [I]− were chosen as interlayer substituents for NaSO3CH3 as they have similar ionic radii and charge to the [SO3CH3]− anion although different coordination environments. Each listed anion is considered a “non-coordinating ion”45 and possess similar charge densities (r = 2.2–2.24 Å).46–48 We anticipate the four anionic groups have similar bonding affinities as the mesylate group. Therefore, isostructural substitution is a reasonable approach to new metal methanesulfonates. Compounds I, II, and IV were prepared via hydrothermal synthesis and large crystals of I–III and V were grown at room temperature through slow evaporation of solvent. IV could not be grown by solution evaporation owing to the instability of the solvated [I]− anion in the presence of oxygen. PXRD of ground crystals and photographs of I–IV crystals are shown in Fig. 1 and the crystal structure refinements in Table 1. As shown in Fig. 1, each crystal is single-phase and the PXRD data are in good agreement with the calculated Bragg-peaks obtained from SCXRD. The reported (V) NaSO3CH3 typically grows as long, rectangular platelets (Fig. S7†), while I and II have blade and angular plate habits. III and IV grow as rectangular prisms and irregular prisms, respectively. The largest single-crystals obtained for each compound were (I) 1.4 × 0.5 × 0.1 (II) 1.0 × 0.5 × 0.1, (III) 1 × 0.4 × 0.4, all in cm3, and (IV) = 1.4 × 1.5 × 1 in mm3. The primary facets of the cm-scale crystals for I–III and V were indexed (Fig. S7†) to the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) (I, II), (001) (III), or (020) (V) interlayer planes, similar to their PXRD. Crystals of IV were too small to index, however PXRD suggests the crystal orientation is similarly layered in preference. EDS (Fig. S1–S6†) agree with the XRD-determined composition. Compounds I, II, and IV are highly oriented in a layered habit, increasing the intensity of the PXRD peaks corresponding to the {10} planes. This is especially true in IV, where the (30
) (I, II), (001) (III), or (020) (V) interlayer planes, similar to their PXRD. Crystals of IV were too small to index, however PXRD suggests the crystal orientation is similarly layered in preference. EDS (Fig. S1–S6†) agree with the XRD-determined composition. Compounds I, II, and IV are highly oriented in a layered habit, increasing the intensity of the PXRD peaks corresponding to the {10} planes. This is especially true in IV, where the (30![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ) is significantly more intense than calculated. Crystals of I–III are air stable, however IV and all the powders are significantly hygroscopic. IV in general appears to leak iodine slowly into its surroundings and produces red-brown stains.
) is significantly more intense than calculated. Crystals of I–III are air stable, however IV and all the powders are significantly hygroscopic. IV in general appears to leak iodine slowly into its surroundings and produces red-brown stains.
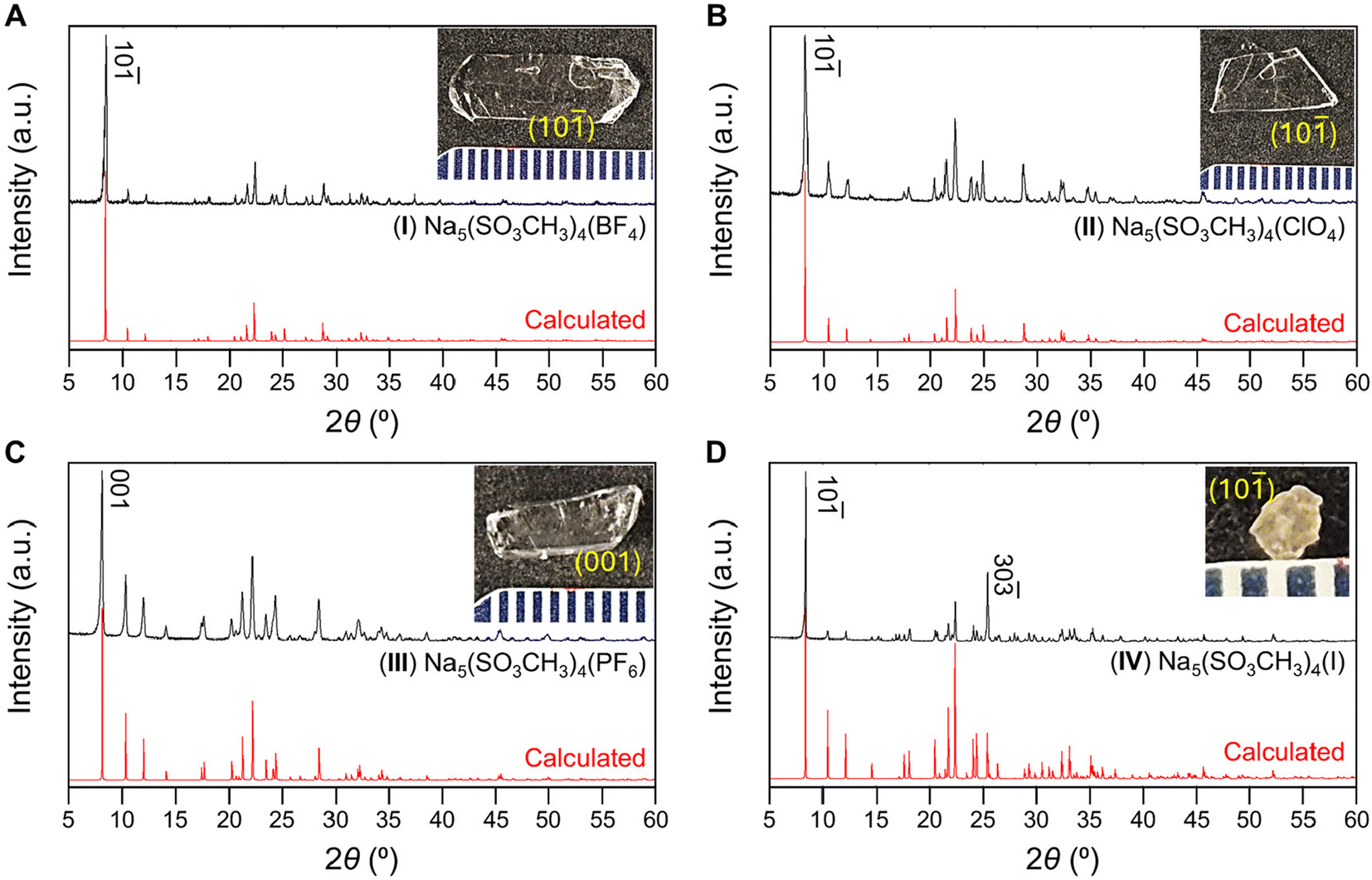 | ||
| Fig. 1 PXRD of compounds (A) I, (B), II, (C) III, and (D) IV with calculated PXRD patterns (red) below. (Inset) photographs of the as-grown crystals of I–IV with the primary face indexed. | ||
The unit cells of I–V as determined by SCXRD are shown in Fig. 2 with a schematic representation of the structural similarities and the local coordination environments in Fig. 3, while full crystallographic data including atomic coordinates bond lengths and angles are available in Table 1 and Tables S1–5.† The known V (Fig. 2A) crystallizes in the orthorhombic space group Pbma (no. 57) with lattice parameters a = 17.0713(6) Å, b = 22.0217(6) Å, and c = 5.6058(3) Å, and V = 2107.4(1) Å3. Compounds I, II, and IV crystallize in the monoclinic space group P2/n (no. 13) while III crystallizes in I2/m (no. 12) with similar lattice parameters. Boxes drawn in Fig. 2A illustrate the structural evolution. The higher symmetry substituent-anions [BF4]−, [ClO4]−, [PF6]−, and [I]− (Fig. 2B–E) do not require multiple layers to describe since they disorder over a single site as opposed to the ordered [SO3CH3]−. Their coordination will be discussed further below. The resulting unit cells have a reduction in volume of ∼½, i.e., (V) 2107 Å3 → (I) 1013 Å3, and a reduction of overall symmetry, orthorhombic → monoclinic. The resulting lattice parameters and β-angle of I–IV are comparable to each other as shown in Table 1. BVS calculations (Table S1†) are in agreement with the anticipated oxidation states for each structural unit S(VI), Na(I), O2−, C(IV), B(III), F−, P(V), I−, and Cl(VII).
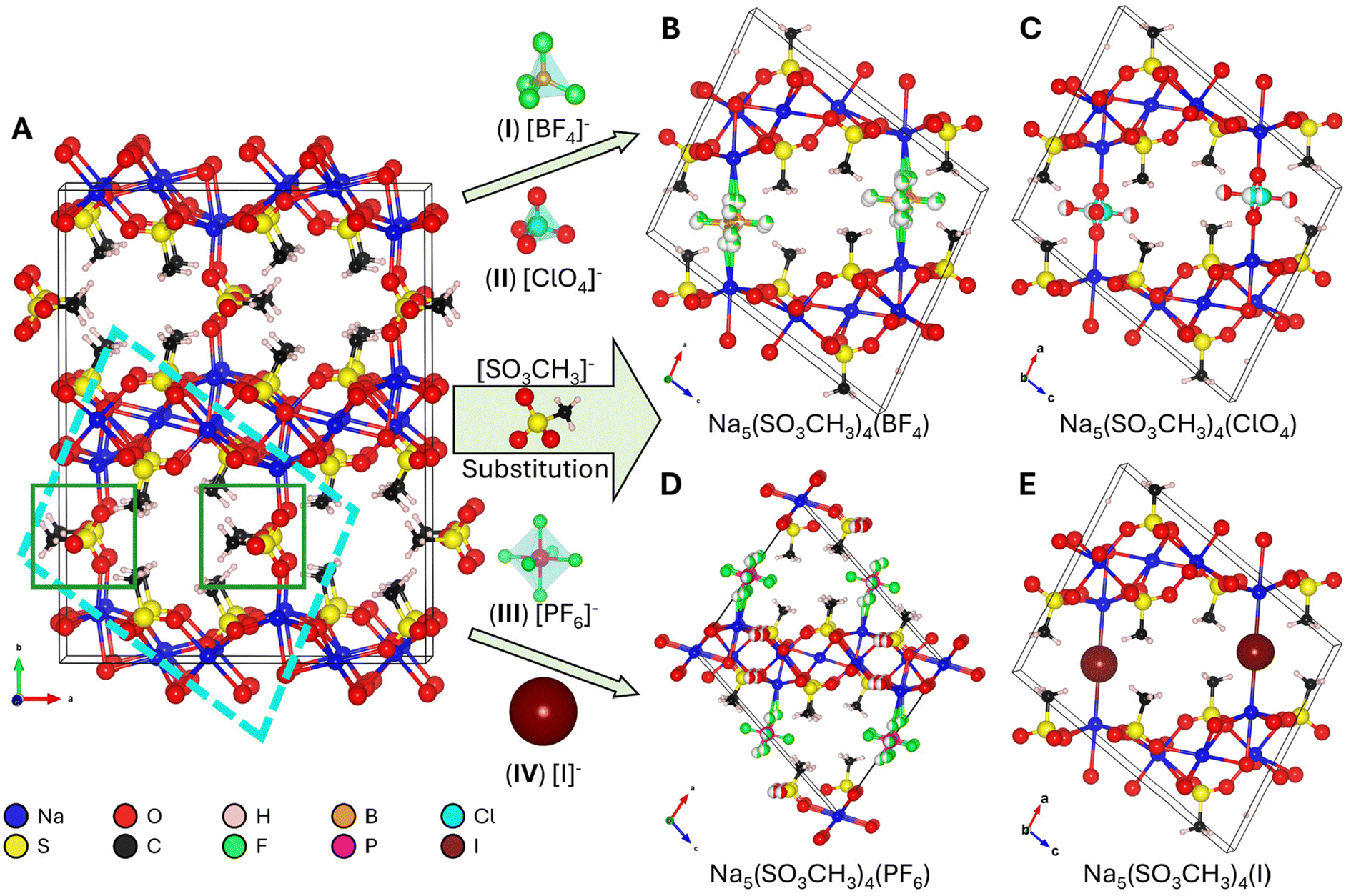 | ||
| Fig. 2 Unit cells and structural evolution of Na5(SO3CH3)4(X) where X = (A) (V) [SO3CH3]−, (B) (I) [BF4]−, (C) (II) [ClO4]−, (D) (III) [PF6]−, and (E) (IV) [I]− through anionic substitution of the interlayer anion site. The cyan box on A shows the prototype unit cell of I–IV and the green boxes are the substituted crystallographic site. | ||
 | ||
| Fig. 3 (A) Interlayer anion coordination for Na5(SO3CH3)4(X) where X = (I) [BF4]−, (II) [ClO4]−, (III) [PF6]−, and (IV) [I]−, (B) deconvolution of the disordered tetrahedral (top) and octahedral (bottom) fragments, and (C) a partial 1D-slice of the edge-sharing [Na5(SO3CH3)4]n+ layers that comprise I–V. | ||
The primary structural motif in I–V are the [Na5(SO3CH3)4]n+ layers that are comprised of edge-sharing and distorted NaO6 octahedra with bridging [SO3CH3]−. A 1D-slice of this motif is shown in Fig. 3A. In I–IV, the [Na5(SO3CH3)4]n+ layers remain largely unchanged. For example, the shortest, longest, and average Na–O bond lengths in I–V are all within 2.30(2) Å, 2.84(2) Å, and 2.49(1) Å, respectively. The intralayer [SO3CH3]− anions are similarly unaffected by the substitutions with S–O and S–C bond lengths of 1.453(4) Å and 1.753(3) Å, all consistent with the reported crystal structure for V.24 In V, the distorted and polar [SO3CH3]− tetrahedra align in an antipolar arrangements between each layer yielding an overall centrosymmetric crystal structure. The disordered anionic groups fall into two categories. The first are the tetrahedral (I) [BF4]−, (II) [ClO4]−, units, shown in Fig. 3A (top). In I and II, the nonpolar tetrahedra are only bonded on one axis and are free to rotate as there is no hydrogen bonding nor crystalline waters by SCXRD to prevent rotation. The two disordered fragments superimposed form a pseudo-octahedron (Fig. 3B, top), and fill the interlayer space. In I, the average Na–F and B–F bond distances are 2.22(8) Å and 1.38(2) Å, respectively, slightly shorter to that of the crystal structure of NaBF4 (ICSD no. 1607866, 2.359 Å and 1.389 Å, respectively). The average Na–O and Cl–O bond distances in II are 2.305 Å and 1.362 Å, respectively, similarly slightly shorter to that of the NaClO4 crystal structure (CCDC no. 8103733, 2.45(8) Å and 1.435(3) Å, respectively).
Similarly, III which contains the [PF6]− octahedral anion that is disordered over two positions forming a pseudo-spherical group (Fig. 3A and B, bottom). The higher symmetry pseudo-spherical disorder in the [PF6]− group gives the lattice an additional body-centering (I) symmetry element. The average Na–F and P–F bond lengths are 2.4(3) Å and 1.56(2) Å, which is significantly longer than the Na–F bonds but close to the P–F bond lengths in NaPF6 (ICSD no. 90615, 2.262(4) Å and 1.545(4) Å). Therefore, the [PF6]− group is not internally distorted and retains its stableconfirmation. In compound IV there is no crystallographic disorder since [I]− is already spherical, however it does not possess the I symmetry since it is slightly distorted off the center position in the interlayer site (Wyckoff position 2c in I2/m or 2e in P2/n). This could be owing to partial iodine sublimation, however, as has been observed in other iodide containing compounds and mentioned above.40,49,50 In IV, the Na–I distance is noticeably longer than the Na–X length in I–III and V at 3.1975(1) Å but this is consistent with the stable Na–I bond length in the NaI salt (ICSD no. 53823, 3.237(3) Å).
3.2. Thermal stability
There is generally limited available literature data on the thermal decomposition pathways of methanesulfonate salts owing to their complexity and postulated radical intermediates.51 Presently, there are some decomposition temperatures and residue PXRDs of simple binary metal methanesulfonates and generally decompose from 350–500 °C.52,53 Owing to this complexity, only the net decomposition and temperatures will be discussed herein for characterization purposes. In each of the compounds I–V, the primary mass component is the [SO3CH3]− group, accounting for 60–65 mass%, with each X-group accounting for 15–22 mass%. I–IV behave similarly to V, albeit at varying temperatures and decomposition pathways likely owing to the enhanced molecular complexity, discussed below (Fig. 4). | ||
| Fig. 4 TGA results for (A–D) I–IV from 50–600 °C under N2 flow. PXRD of each residue and the TGA data for V is shown in the ESI Fig. S8 and S9.† | ||
The internal standard, V (Fig. S8†) is somewhat hygroscopic and loses ∼0.2 equivalents of water at 55–60 °C. It decomposes at 435–450 °C into Na2CO3 and loses 52.68% of its mass. This is attributed to the nonstoichiometric decomposition of the methanesulfonate group into primarily sulfur in the form of SO2, consistent with previous literature.54 PXRD plots of each residue for I–IV are shown in Fig. S9.† Similarly, compound I is more hygroscopic and loses ∼1.6 molar equivalents of adsorbed water from 50–200 °C. Compound I decomposes from 450–490 °C into NaF. Therefore, the corresponding mass loss of 67.48% is likely the entire [SO3CH3]− group as well as the NaBF4 → NaF(s) + BF3(g) in a concerted step. Compound II is significantly more hygroscopic, losing ∼3.2 equivalents of water at 80–100 °C. It decomposes at 420–435 °C into a mixture of NaCl, Na2CO3, and Na6(CO3)(SO4)2 owing to the complete decomposition of the [ClO4]− subunit and partial decomposition (18.78 mass%) of the mesylate group. The rate of decomposition of the mesylate unit observed in I and V is significantly reduced in II over the measured range (24.24 mass% vs. > 50 mass%), perhaps inhibited by the NaCl presence or a semi-stable intermediate.
Compound III is not measurably hygroscopic by TGA but it has three decomposition steps in the range of 360–520 °C. The residue contains Na2CO3, Na7(PO4)2F, and NaF, suggesting a complete release of sulphur while retaining most of the phosphorous and some fluorine. The decomposition is likely initiated (−36.31%) by the [PF6]− group since NaPF6 has lower thermal stability than NaSO3CH3 (285 °C vs. 435 °C, respectively).55 The second (−6.59%) and third (−19.07%) steps begin at 460 °C and 510 °C, respectively. The evolved gases are therefore likely rich in CO2, SO2, HF, and H2O. The pyrolysis products of IV were poorly crystallized and more challenging to determine well. Compound IV is similarly hygroscopic to II and loses 2.9 molar equivalents of water from 50–120 °C. A rapid mass loss of 20.12 mass% occurs at 390 °C which is likely the I (20.4 mass%) as the residue appears to primarily be a mixture of Na2CO3–Na2SO4 solid solutions, with no crystalline I-containing phases. Additional decomposition of the mixed carbonate-sulfates continues for an additional 5.5 mass% over the measured temperature range. In summary, the relative thermal stability of I–IV is similar to V and previously reported metal methanesulfonates although the pathways change depending on the X-group. Lastly, the thermal stability of I–IV was examined after annealing crystals of each at 300 °C under vacuum. PXRD of the ground crystals are shown in Fig. S11.† II and III showed no visual change nor significant change by PXRD. Crystals of I became opaque and there are some minor changes in peak intensities. IV has nearly completely changed structure to another unknown phase.
3.3. Optical properties
The diffuse reflectance spectroscopy results from 200–1200 nm for compounds I–IV are shown in Fig. 5 and compound V in Fig. S10.† I–III and V have wide transparencies in the visible region from 400–1100 nm. All four compounds share a weak absorption band at 1170 nm which is common amongst metal methanesulfonates56 and is also present in V. The absorption cutoff edges (λabs) were estimated from the data as <200 nm for I and III, 210 nm for II, and 230 nm for IV. The bandgaps (Eg, Fig. 5 insets) were estimated to be (I) 5.4, (II) 5.5, (III) 5.2, and (IV) 4.5, all in eV. Compound IV has one lower energy absorption bands from the [I]− centered at 365 nm, and the cause of the yellow coloration of the crystals. It is likely that the 365 nm band is due to the partial oxidation of the [I]− in air to I2 or IOx−.61 For comparison, the λabs and Eg of V were measured to be 210 nm and 5.3 eV, respectively. Despite no change in the [Na5(SO3CH3)4]+ structural layers, the absorption cutoffs have decreased into the deep-UV (λ < 200 nm) from substitution of [SO3CH3]− for [BF4]− and [PF6]−, no change with [ClO4]−, and increased to 230 nm with [I]−.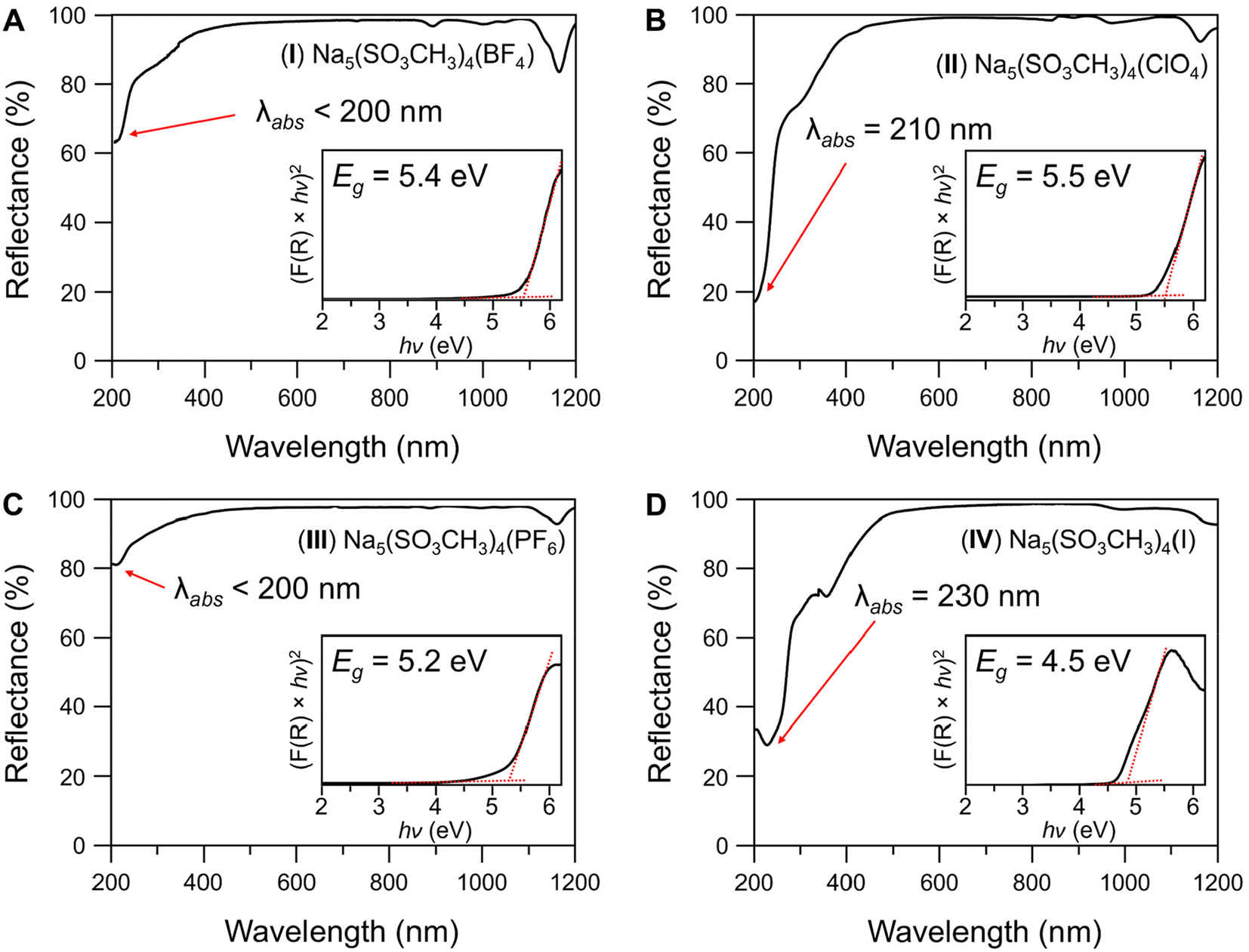 | ||
| Fig. 5 UV-Vis-NIR diffuse reflectance spectroscopy results for (A–D) I–IV from 200–1200 nm. (insets) Tauc plots of the remission function squared. Bandgaps (Eg) were determined by extrapolating to the baseline (red lines). | ||
The FT-IR spectra of compounds I–IV over the range of 400–4000 cm−1 are provided in Fig. 6 and Fig. S10† for V. A full list of spectral peak assignments and frequencies has been tabulated in Table S6 in the ESI.† The characteristic peaks from the sodium methanesulfonate structural unit (i.e., C–H, S–O, etc.) produce similar FT-IR spectra with minor shifts in frequencies between I–V and have been thoroughly investigated elsewhere.57 In brief, the SO3 asymmetric and symmetric stretches occur at 1180–1250 cm−1 and 1050–1100 cm−1, respectively. The CH3 asymmetric and symmetric stretches occur at 3000–3050 cm−1 and 2940 cm−1, respectively. The bending modes for SO3 and the SO3 + CH3 combination bands are in the 530–800 cm−1 region, while the CH3 bends are centered at 1330–1350 cm−1.
 | ||
| Fig. 6 FT-IR spectroscopy results for I–IV (top to bottom, respectively) from 4000–400 cm−1. FT-IR spectrum for V is in Fig. S10B in the ESI.† | ||
For the new IR peaks observed, I has new peaks corresponding to B–F stretches at 1039 and 534 cm−1 which overlap with the mesylate vibrations.58 II has a new sharp peak at 635 cm−1 from ClO4 asymmetric bending.59 III similarly has new peaks owing to the PF6 stretching 858 and 829 cm−1.60 New peaks emerge from at ∼850 and 890 cm−1 in II and IV that are proposed to be water libration, similarly observed in multiple metal sulfonates and hydrates.57 This is consistent with compounds II and IV having higher hygroscopicity. Broad bands at 3400–3450 cm−1 attributed to water are present in all but compound III, which was also observed to be the least hygroscopic and had no water loss in the TGA as described above. No new peaks were observed, nor expected, for Na–I stretching in compound IV. These data confirm the crystallographic structure and purity of the crystals obtained and the X-group substitution.
4. Conclusions
The present work investigated using anionic site-substitution as a structure building method to discover new metal methanesulfonates with mixed-anionic groups. Experiments resulted in large crystals of four new compounds, Na5(SO3CH3)4(X) (X = (I) BF4, (II) ClO4, (III) PF6, and (IV) I), each isostructural with the known NaSO3CH3 salt (X = SO3CH3). Compounds I, II, and IV each crystallize in the monoclinic space group P2/n (no. 13) whereas III crystallizes in the monoclinic space group I2/m (no. 12). The four anionic groups were chosen as they have similar charge density to the [SO3CH3]− anion, and were successfully fully substituted seamlessly into the host [Na5(SO3CH3)4]n+ layered substructure. I–III have wide optical transparency in the visible-NIR range (400–1200 nm) and I and III have deep-UV λabs < 200 nm and II has λabs = 210 nm. IV has significant absorption at 350–400 nm due to iodide oxidation in air. I–IV have large optical bandgaps of 5.4, 5.5, 5.2, and 4.5, all in eV, respectively. Compounds I–III are thermally stable up to 350 °C while IV undergoes a phase transition prior to decomposition. The simple synthesis method and resulting >1 cm optical-quality crystals of I–III highlights the tuneability of metal methanesulfonate structures and is a promising method for discovering more new compounds with diverse properties.Author contributions
E.A.G. – conceptualization, data curation, investigation, methodology, writing – original draft, review & editing. P.S.H. – conceptualization, funding acquisition, resources, writing – review & editing.Data availability
Crystallographic data for I–IV has been deposited at the CCDC under accession numbers 2147057–2417060.† The data supporting this article, such as: crystallographic information for I–IV including atomic coordinates, BVS calculations, anisotropic parameters, bond lengths, and bond angles; SEM and EDS for I–V; PXRD indexing of crystal facets; TGA for V and PXRD for residues of I–V; UV-Vis DRS for V and FT-IR for I–V; have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
E. A. G. would like to thank Prof. Xiqu Wang for assistance and discussions with crystal structure refinements. E. A. G. and P. S. H. thank the Welch Foundation (grant E-1457) for their support.References
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Expanding frontiers in materials chemistry and physics with multiple anions, Nat. Commun., 2018, 9, 772 CrossRef PubMed.
- K. Maeda, F. Takeiri, G. Kobayashi, S. Matsuishi, H. Ogino, S. Ida, T. Mori, Y. Uchimoto, S. Tanabe, T. Hasegawa, N. Imanaka and H. Kageyama, Recent progress on mixed-anion materials for energy applications, Bull. Chem. Soc. Jpn., 2022, 95(1), 26–37 CrossRef CAS.
- C. Chen, Y. Wu and R. Li, The anionic group theory of the non-linear optical effect and its applications in the development of new high-quality NLO crystals in the borate series, Int. Rev. Phys. Chem., 1989, 8(1), 65–91 Search PubMed.
- J. Cui, C. Li and F. Zhang, Development of mixed-anion photocatalysts with wide visible-light absorption bands for solar water splitting, ChemSusChem, 2018, 12(9), 1872–1888 CrossRef PubMed.
- G. Laurita and R. Seshadri, Chemistry, structure, and function of lone pairs in extended solids, Acc. Chem. Res., 2022, 55, 1004–1014 CrossRef CAS PubMed.
- S. D. Nguyen, J. Yeon, S. H. Kim and P. S. Halasyamani, BiO(IO3): A new polar iodate that exhibits an aurivillius-type (Bi2O2)2+ layer and a large shg response, J. Am. Chem. Soc., 2011, 133(32), 12422–12425 CrossRef CAS PubMed.
- L. Xiong, J. Chen, J. Lu, C.-Y. Pan and L.-M. Wu, Monofluorophosphates: a new source of deep-ultraviolet nonlinear optical materials, Chem. Mater., 2018, 30(21), 7823–7830 CrossRef CAS.
- H. Qiu, F. Li, C. Jin, Z. Yang, J. Li, S. Pan and M. Mutailipu, Fluorination strategy towards symmetry breaking of boron-centered tetrahedron for poly-fluorinated optical crystals, Angew. Chem., 2024, 136(4), e202316194–e202316202 CrossRef.
- T. T. Tran, H. Yu, J. M. Rondinelli, K. R. Poeppelmeier and P. H. Halasyamani, Deep Ultraviolet nonlinear optical materials, Chem. Mater., 2016, 28(15), 5238–5258 Search PubMed.
- K. M. Ok, Towards the rational design of novel noncentrosymmetric materials: factors influencing the framework structures, Acc. Chem. Res., 2016, 49, 2774–2785 CrossRef CAS PubMed.
- H. Yu, H. Wu, S. Pan, Z. Yang, X. Hou, X. Su, Q. Jing, K. R. Poeppelmeier and J. M. Rondinelli, Cs3Zn6B9O21: A chemically benign member of the KBBF family exhibiting the largest second harmonic generation response, J. Am. Chem. Soc., 2014, 136, 1264–1267 CrossRef CAS PubMed.
- G. Yang, P. Gong, Z. Lin and N. Ye, AZn2BO3X2 (A = K, Rb, NH4; X = Cl, Br): New members of KBBF family exhibiting large SHG response and the enhancement of layer interaction by modified structures, Chem. Mater., 2016, 28, 9122–9131 CrossRef CAS.
- Y. Shen, S. Zhao, Y. Yang, L. Cao, Z. Wang, B. Zhao, Z. Sun, Z. Lin and J. Luo, A new KBBF-family nonlinear optical material with strong interlayer bonding, Cryst. Growth Des., 2017, 17, 4422–4427 CrossRef CAS.
- C. T. Chen, T. Sasaki, R. K. Li, Y. C. Wu, Z. S. Lin, Y. Mori, Z. G. Hu, J. Wang, G. Aka and M. Yoshimura, Nonlinear optical borate crystals: principals and applications, John Wiley & Sons, Weinheim Germany, 2012 Search PubMed.
- B. Xu, P. Gong, F. Liu, X. Zhang, H. Huo and Z. Lin, (SO3CF3)−: A non-pi-conjugated motif for nonlinear optical crystals transparent into the deep-ultraviolet region, Adv. Opt. Mater., 2024, 12(5), 2301725 CrossRef CAS.
- H. Tian, C. Lin, X. Zhao, F. Xu, C. Wang, N. Ye and M. Luo, Ba(SO3CH3)2: A deep-ultraviolet transparent crystal with excellent − optical nonlinearity based on a new polar non-π-conjugated NLO building unit SO3CH3−, CCS Chem., 2023, 5(11), 2497–2505 CrossRef CAS.
- D. Zagorac, H. Muller, S. Ruehl, J. Zagorac and S. Rehme, Recent developments in the inorganic crystal structure database: theoretical crystal structure data and related features, J. Appl. Crystallogr., 2019, 52, 918–925 CrossRef CAS PubMed.
- C. Logemann and M. S. Wickleder, The [Au(CH3SO3)4]− Anion in the crystal structures of M[Au(CH3SO3)4] (M = Li, Na, Rb), Z. Anorg. Allg. Chem., 2012, 638(10), 1468–1472 CrossRef CAS.
- M. Liang, Y. Zhang, E. Izvarin, M. J. Waters, J. M. Rondinelli and P. S. Halasyamani, Metal methanesulfonates with mixed anionic groups with large band gaps and enhanced birefringence, Chem. Mater., 2024, 36(4), 2113–2123 CrossRef CAS.
- E. A. Gabilondo and P. S. Halasyamani, Synthesis and large crystal growth of a noncentrosymmetric sodium methylsulfanetriol borate lews-adduct: Na(S(OH)3CH3)(BO3), Inorg. Chem., 2024, 63(37), 17208–17214 CrossRef CAS PubMed.
- S. F. Parker, E. J. Revill-Hivet, D. W. Nye and M. J. Gutmann, Structure and vibrational spectroscopy of lithium and potassium methanesulfonates, R. Soc. Open Sci., 2020, 7, 200776–200787 CrossRef CAS PubMed.
- R. Uppuluri, A. S. Gupta, A. S. Rosas and T. E. Mallouk, Soft chemistry of ion-exchangeable layered metal oxides, Chem. Soc. Rev., 2018, 47, 2401–2430 RSC.
- E. A. Gabilondo, S. O'Donnell, R. Newell, R. Broughton, M. Mateus, J. L. Jones and P. A. Maggard, Renaissance of topotactic ion-exchange for functional solids with close packed structures, Chem. – Eur. J., 2022, 28(33), e202200479–e202200484 CrossRef CAS PubMed.
- C. H. Wei and B. E. Hingerty, Structure of sodium methanesulfonate, Acta Crystallogr., Sect. B, 1981, 37, 1991–1997 Search PubMed.
- Bruker APEX2, Bruker AXS Inc., Madison, WI, USA, 2003 Search PubMed.
- G. M. Sheldrick, SAINT, XPREP (Version 2008/2), Bruker AXS Inc., Madison, WI, USA, 2008 Search PubMed.
- R. H. Blessing, An empirical correction for absorption anisotropy, Acta Crystallogr., Sect. A:Found. Crystallogr., 1995, 51, 33 CrossRef PubMed.
- G. M. Sheldrick, SHELXT-Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, A Short History of SHELX, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: A complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Single-Crystal Structure Validation with the Program PLATON, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- Y. Le Page, Computer derivation of the symmetry elements implied in a structure description, J. Appl. Crystallogr., 1987, 20, 264–269 CrossRef CAS.
- Y. Le Page, MISSYM 1.1 – a flexible new release, J. Appl. Crystallogr., 1988, 21, 983–984 CrossRef.
- I. D. Brown, Recent developments in the methods and applications of the bond valence model, Chem. Rev., 2009, 109(12), 6858–6919 CrossRef CAS PubMed.
- IUCr Crystallographic Data on Bond Valence Parameters page. https://www.iucr.org/resources/data/datasets/bond-valence-parameters, (accessed 2024-06-06).
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–197 CrossRef CAS PubMed.
- G. Brunton, Refinement of the structure of NaBF4, Acta Crystallogr., Sect. B, 1968, 24, 1703–1704 CrossRef CAS.
- R. Wartchow and H. J. Berthold, Crystal structure of sodium perchlorate, Z. Kristallogr., 1978, 147, 307–317 CrossRef CAS.
- K. F. Tebbe and U. Georgy, Die kristallstrukturen von rubidiumtriiodid und thalliumtriiodid, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 1986, 42, 1675–1678 CrossRef.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric, and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- P. Kubelka and F. Munk, An article on optics of paint layers, Z. Tech. Phys., 1931, 12, 593–603 Search PubMed.
- E. L. Simmons, Reflectance spectroscopy: application of the Kubelka-Munk theory to the rates of photoprocesses of powders, Appl. Opt., 1976, 15, 951–954 CrossRef CAS PubMed.
- J. Tauc, Amorphous and Liquid Semiconductors, Springer International Publishing, London, UK, 1974 Search PubMed.
- I. Krossing and I. Raabe, Noncoordinating anions – fact or fiction? A survey of likely candidates, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- L. H. Ahrens, Geochim. Cosmochim. Acta, 1952, 2(3), 155–169 CrossRef CAS.
- H. D. B. Jenkins and K. P. Thankur, Reappraisal of thermochemical radii for complex ions, J. Chem. Educ., 1979, 56(9), 576–577 CrossRef CAS.
- A. L. Rohl and D. M. P. Mingos, The size and shape of molecular ions and their relevance to the packing of the hexafluorophosphate salts, Dalton Trans., 1992, 24, 3541–3552 RSC.
- R. C. L. Mooney, The configuration of the triiodide group in ammonium triiodide crystals, Z. Kristallogr. – Cryst. Mater., 1937, 90(1–6), 143–150 Search PubMed.
- G. H. Cheesman and A. J. T. Finney, Refinement of the structure of ammonium triiodide, NH4I3, Acta Crystallogr., Sect. B, 1970, 26, 904–906 CrossRef CAS.
- J. Cao, W.-L. Wang, L.-J. Gao and F. Fu, Mechanism and thermodynamic properties of CH3SO3 decomposition, Acta Phys.-Chim. Sin., 2013, 29(6), 1161–1167 CAS.
- F. Charbonnier, Thermal behavior of some compounds of methanesulfonic acid with transition metals, Thermochim. Acta, 1979, 33, 31–39 CrossRef CAS.
- M. Wang, Z. G. Song, H. Hiang and H. Gong, Thermal decomposition of metal methanesulfonates in air, J. Therm. Anal. Calorim., 2009, 98, 801–806 CrossRef CAS.
- F. Charbonnier, Study of some alkaline salts of alcanemonosulfonic and alcanedisulfonic acids, Ann. Chim., 1971, 6(6), 405–411 CAS.
- T. C. Ehlert and M.-M. Hsia, Thermal decomposition of alkali metal hexafluorophosphates, J. Chem. Eng. Data, 1972, 17(1), 18–21 CrossRef CAS.
- G. Andreev, N. Budantseva and A. Fedoseev, Crystal structure and spectral properties of Np(VI) and U(VI) methanesulfonates, J. Radioanal. Nucl. Chem., 2019, 320, 485–490 CrossRef CAS.
- S. F. Parker and L. Zhong, Vibrational spectroscopy of metal methanesulfonates: M = Na, Cs, Cu, Ag, Cd, R. Soc. Open Sci., 2018, 5, 171574–171573 CrossRef PubMed.
- O. Zavorotynska, M. Corno, A. Damin, G. Spoto, P. Ugliengo and M. Baricco, Vibrational properties of MBH4 and MBF4 crystals (M = Li, Na, K): a combined DFT, infrared, and raman study, J. Phys. Chem. C, 2011, 115, 18890–18900 CrossRef CAS.
- Y. Chen, Y. H. Zhang and L. J. Zhao, ATR-FTIR spectroscopic studies on aqueous LiClO4, NaClO4, and Mg(ClO4)2 solutions, Phys. Chem. Chem. Phys., 2004, 6(3), 537–542 RSC.
- A. M. Heyns, The I.R. and raman spectra of sodium hexafluorophosphate monohydrate, NaPF6˙H2O, Spectrochim. Acta, 1977, 33, 315–322 CrossRef.
- N. N. Kazantseva, A. Ernepesova, A. Khodjamamedov, O. A. Geldyev and B. S. Krumgalz, Spectrophotometric analysis of iodide oxidation by chlorine in highly mineralized solutions, Anal. Chim. Acta, 2002, 456, 105–119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2417057–2417060. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00223k |
| This journal is © The Royal Society of Chemistry 2025 |