
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-cost, resilient, and non-flammable rechargeable Fe-ion batteries with scalable fabrication and long cycle life†
Yufan
Zhang‡
a,
Chi Ho
Lee‡
bc,
Md Zahidul
Islam
d,
Joseph Sang-Il
Kwon
 *bc and
Choongho
Yu
*ad
*bc and
Choongho
Yu
*ad
aDepartment of Materials Science and Engineering, Texas A&M University, College Station, Texas 77843, USA. E-mail: chyu@tamu.edu
bDepartment of Chemical Engineering, Texas A&M University, College Station, Texas 77843, USA. E-mail: kwonx075@tamu.edu
cTexas A&M Energy Institute, College Station, Texas 77843, USA
dDepartment of Mechanical Engineering, Texas A&M University, College Station, Texas 77843, USA
First published on 23rd December 2024
Abstract
Aqueous Fe-ion batteries remain largely unexplored owing to their short cycle life despite their extremely low material cost. Furthermore, their working mechanisms are mostly undisclosed with only a few experimental studies available. In this study, we fabricated Fe-ion batteries, which delivered an impressive specific capacity of 225 mA h g−1 at a relatively low rate of 5C and exhibited an extremely long cycle life of up to 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles with a capacity retention of 82% at 15C. Furthermore, the overall setup included a carbon steel foil anode (moderate-purity Fe source), along with scalable cathodes and low-cost FeSO4 electrolyte, offering cost-effective solutions. Our theoretical study revealed that the Fe incorporation processes in the cathode, along with the corresponding voltage profiles during cycling, are primarily influenced by the formation energy of Fe on the emptied N sites of polyaniline and the structural deformations caused by Fe attachment. Notably, our batteries were shown to be free from fire hazard and failure due to short circuits. As manufacturing-friendly sandwich-type or 3D cylindrical cathodes eliminate multi-stack electrodes, our batteries are cost-effective, long-lasting, and safe for stationary energy storage systems.
000 cycles with a capacity retention of 82% at 15C. Furthermore, the overall setup included a carbon steel foil anode (moderate-purity Fe source), along with scalable cathodes and low-cost FeSO4 electrolyte, offering cost-effective solutions. Our theoretical study revealed that the Fe incorporation processes in the cathode, along with the corresponding voltage profiles during cycling, are primarily influenced by the formation energy of Fe on the emptied N sites of polyaniline and the structural deformations caused by Fe attachment. Notably, our batteries were shown to be free from fire hazard and failure due to short circuits. As manufacturing-friendly sandwich-type or 3D cylindrical cathodes eliminate multi-stack electrodes, our batteries are cost-effective, long-lasting, and safe for stationary energy storage systems.
Broader contextRecently, aqueous metal-ion batteries have garnered significant attention owing to their low cost and safety. Among them, zinc-ion batteries have been the primary focus of research. However, iron, being cheaper and more abundant in the Earth's crust than zinc, holds promise as an alternative metal anode, though it remains relatively unexplored. A reliable cathode for iron-ion batteries is essential for advancing their research and commercialization, which require a simple preparation process and a well-understood mechanism. Herein, we introduced sandwich-type and cylindrical cathodes using polyaniline as the active material for iron-ion batteries. This cathode was binder-free and fabricated through a simple low-pressure press process. It delivered a high capacity of 225 mA h g−1 (compared to the theoretical capacity of 300 mA h g−1 for polyaniline) at a 5C rate. Additionally, our high-loading battery demonstrated a long cycle life of 27000 cycles with an 82% capacity retention at a 15C rate. We also conducted a systematic theoretical study to elucidate the electrochemical behaviors upon the incorporation of iron into polyaniline during the charge and discharge processes. Thus, this work presents a reliable and promising solution for utilizing iron-ion batteries in stationary energy storage applications, potentially outperforming lead-acid and lithium-ion batteries.
|
Introduction
Renewable energy sources, such as solar and wind power, are essential for reducing greenhouse gases. However, intermittent power generation necessitates energy storage systems, which are often made of rechargeable Li-ion batteries (LIB). Although the LIBs are attractive owing to their high energy densities, their expensive raw materials (Li, Co, and Ni) and the high risk of fire due to the use of flammable electrolytes and metal oxides demand alternatives. Recent studies have shown a glimpse into more suitable energy storage systems, such as Zn-ion batteries (ZIB)1–4 and redox flow batteries that mitigate costly materials and fire hazards with water-based electrolytes.5 However, despite the recent surge in interest for rechargeable aqueous Fe-ion batteries (FIB), their utility remain largely unexplored. FIB can offer significant advantages over other batteries owing to the extremely low cost of iron (∼200- and ∼40-times cheaper than lithium and zinc, respectively6) and no fire hazard (non-flammable electrolyte and metal oxides). Furthermore, the theoretical capacities of Fe (960 mA h g−1 and 7557 mA h cm−3) are higher than those of Zn. Therefore, FIB can be a game-changer in the domain of stationary energy storage.7,8Over the past few years, several cathode materials have been tested for Fe-ion batteries. For instance, a Prussian blue analog was used as a cathode material, demonstrating 1000 cycles with an 80% capacity retention.7 However, the cycle life was much shorter (∼150 cycles) for Fe–sulfur batteries.9 When Fe-ion was hosted by VOPO4 with a capacity of 100 mA h g−1, a large capacity fading (68%) was observed only after 800 cycles.10 However, for stationary energy storage, a lifespan of more than 3000 cycles (equivalent to one cycle per day over 10 years) with 80% capacity retention is necessary. The aforementioned batteries were made of inorganic materials, which are popular in rechargeable batteries, but several organic materials also have been employed as cathode materials, including LIB,11,12 Na-ion batteries,13,14 K-ion batteries,15 and ZIB.16
In particular, polymers are advantageous for avoiding active material loss from the electrode, attaining molecular-level controllability and structural diversity, and reducing capacity fading17 owing to their long molecular chains intermingling with the supporting carbon materials. Also, it enables avoidance of the use of expensive materials or costly processing for cost-effective solutions. For example, ZIB with a polyaniline (PANI)-based cathode displayed 92% capacity retention after 3000 cycles, proposing a supercapacitor-like mechanism to enhance the operating voltage, capacity, and cycle life.16 Zn-ion insertion/extraction on PANI were conjectured through ex situ X-ray photoelectron spectroscopy. However, despite these advancements, there is still a gap in comprehensive theoretical studies. Specifically, it is necessary to unveil the role of metal ions in the charge/discharge processes in batteries to clarify their working mechanisms and corresponding performances and characteristics. Recently, although aqueous ZIB have attracted significant attention, FIB with an anode is made of Fe are desirable compared to Zn because the redox potential of Fe/Fe2+ (−0.44 V vs. standard hydrogen electrode (SHE)) is more positive than that of Zn/Zn2+ (−0.76 V vs. SHE) to circumvent water splitting in aqueous batteries. This can result in better electrolyte stability to extend the cycle life.
Recently, cross-linked PANI was used in Fe-ion batteries,18 demonstrating a specific capacity of 110 mA h g−1 and 39000 charge/discharge cycles at an undesirably high C rate (∼83C where 1C = 300 mA per g of PANI). However, the use of the costly Fe(OTf)2 electrolyte significantly diminishes the primary benefit of low cost. Thus, many aspects of this study still need to be explored or further investigated. For example, the impractically high 83C rate can mask side reactions generally observed in aqueous batteries. Aqueous batteries typically suffer from degradation and oxidation at low C rates, which are common operating conditions for stationary energy storage systems. They used 1.3 V as the cut-off voltage for the charge and discharge process, whereas other reported Fe-ion batteries used 0.9 V or 1 V.7,10 At high voltage, Fe2+ oxidation can occur in the electrolyte, but the Fe2+/Fe3+ redox peak may have been attributed to the PANI redox peak, which can lead to a misunderstanding of the mechanism. Also, the cross-linked PANI may enhance the cathode stability, but there is no evidence that it improves the capacity.
Herein, we fabricated FIB employing a sandwich-type PANI cathode, low-cost and readily available carbon steel anode, and inexpensive aqueous FeSO4 electrolyte. Consequently, our FIB delivered a remarkable discharge specific capacity of 225 mA h g−1 at 5C (1C = 300 mA per g) with 8 wt% active material in the entire cathode. Furthermore, they showed good charge/discharge cycling performances, maintaining 82% capacity retention after 27000 cycles at 15C. The sandwich-type cathode was fabricated simply by sandwiching PANI between porous carbon nanotube (CNT) layers, eliminating complicated and time-consuming processing steps. Despite the simplicity of the fabrication process, the performance of our FIB was significantly better than that with conventional cathodes made of electroplated PANI on conductive substrates. Furthermore, we performed density functional theory (DFT) calculations to understand the charge/discharge processes of PANI with Fe-ions by examining the diverse properties of PANI structures (including geometric, electronic, and thermodynamic changes with varying Fe concentrations). Finally, an FIB whose dimensions are similar to an AA battery was designed with a three-dimensional (3D) CNT as a host for the cathode, serving as a proof of concept for scalability. Finally, we demonstrated that our aqueous batteries are immune to even direct contact (short-circuit) of the cathode and anode.
Results and discussion
Our batteries were composed of a carbon steel anode and a composite anode, in which as-purchased PANI powder was simply sandwiched between interconnected CNT layers (Fig. 1(a)) with an aqueous FeSO4 electrolyte. The CNT electrode19 was obtained by slicing a sponge-like cylindrical CNT,20 where the fibrous CNTs were intertwined, maintaining the porous structure without any binders.21 The central PANI layer between the two CNT sponges, as shown in the scanning electron microscopy (SEM) image, is distinct from the PANI-based electrode in the literature22,23 although all the PANI materials appear to be similar according to the Fourier transform infrared spectroscopy results (ESI,† Fig. S1). The sandwiched structure is advantageous for increasing the loading of PANI, in addition to fast and simple fabrication given that the electrodeposition process tends to clog the pores in the electrode, limiting the material loading.24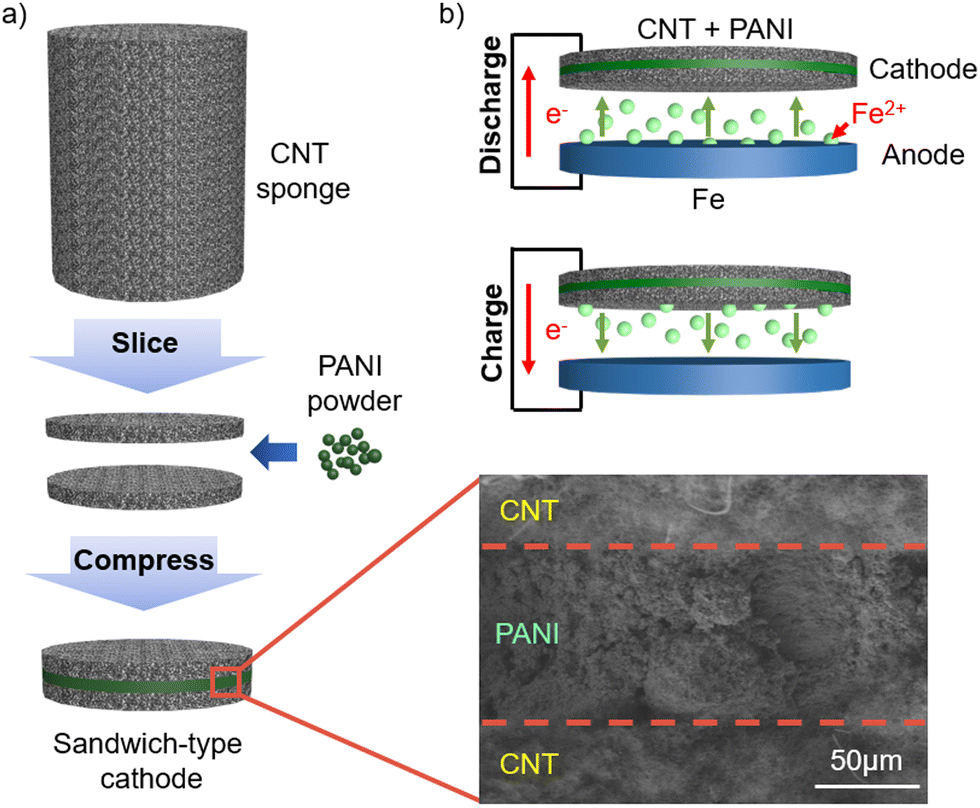 | ||
| Fig. 1 (a) Fabrication of the cathode by slicing cylindrical sponge-like CNT and then sandwiching PANI powder. The SEM image shows a PANI-impregnated layer between the CNT layers. (b) Illustration of discharging and charging processes and corresponding flow of Fe ions and electrons. | ||
During the discharge process, Fe ions migrate from the anode towards the cathode (Fig. 1(b)), and Fe ions move back to the anode for charge. Thus, the sponge-like porous CNT promises not only electrical conduction through the intertwined CNTs but also the movement of ions to the PANI located in the middle of the cathode. Here, carbon steel foil was employed as the anode because it is readily available at an extremely low price compared to pure iron. The battery performances with the carbon steel anode were superior to that with an anode made of Fe powder (99.9%+), conductive carbon (Super P), and a polymer binder using the conventional wet slurry coating method, implying that the performances are not limited by the high Fe purity and large surface area of the anode (ESI,† Fig. S2).
Initially, we carried out experimental and computational studies to identify how PANI interacts with Fe, together with the corresponding electrochemical potentials. Fig. 2(a) shows the cyclic voltammetry (CV) results using a PANI-deposited CNT electrode in an aqueous 1 M FeSO4 solution and Pt counter electrode (without an Fe anode unlike full cells). Two noticeable reduction and oxidation peaks were observed, labeled as Red1 and Red2 and Ox1 and Ox2, respectively. A prominent shift was only observed for peak Red1, where the reduction potential was around −0.293 V (vs. Ag/AgCl) in the 1st CV cycle. This peak shifted gradually until −0.050 V in the 5th cycle. A small shift in peak Ox1 was also observed, whereas that for Red2 and Ox2 was negligible. We identified that Red1 and Ox1 are from PANI because these redox peaks were found in the CV results with PANI in 1 M H2SO4 (substituted FeSO4, green curve in ESI,† Fig. S3). When PANI was displaced from the working electrode in the FeSO4 solution (blue curve in ESI,† Fig. S3), only Red2 and Ox2 peaks were observed, suggesting that Red2 and Ox2 are the Fe2+/Fe3+ redox peaks. These peaks can be easily mistaken for PANI redox peaks, as documented in a recent paper.18
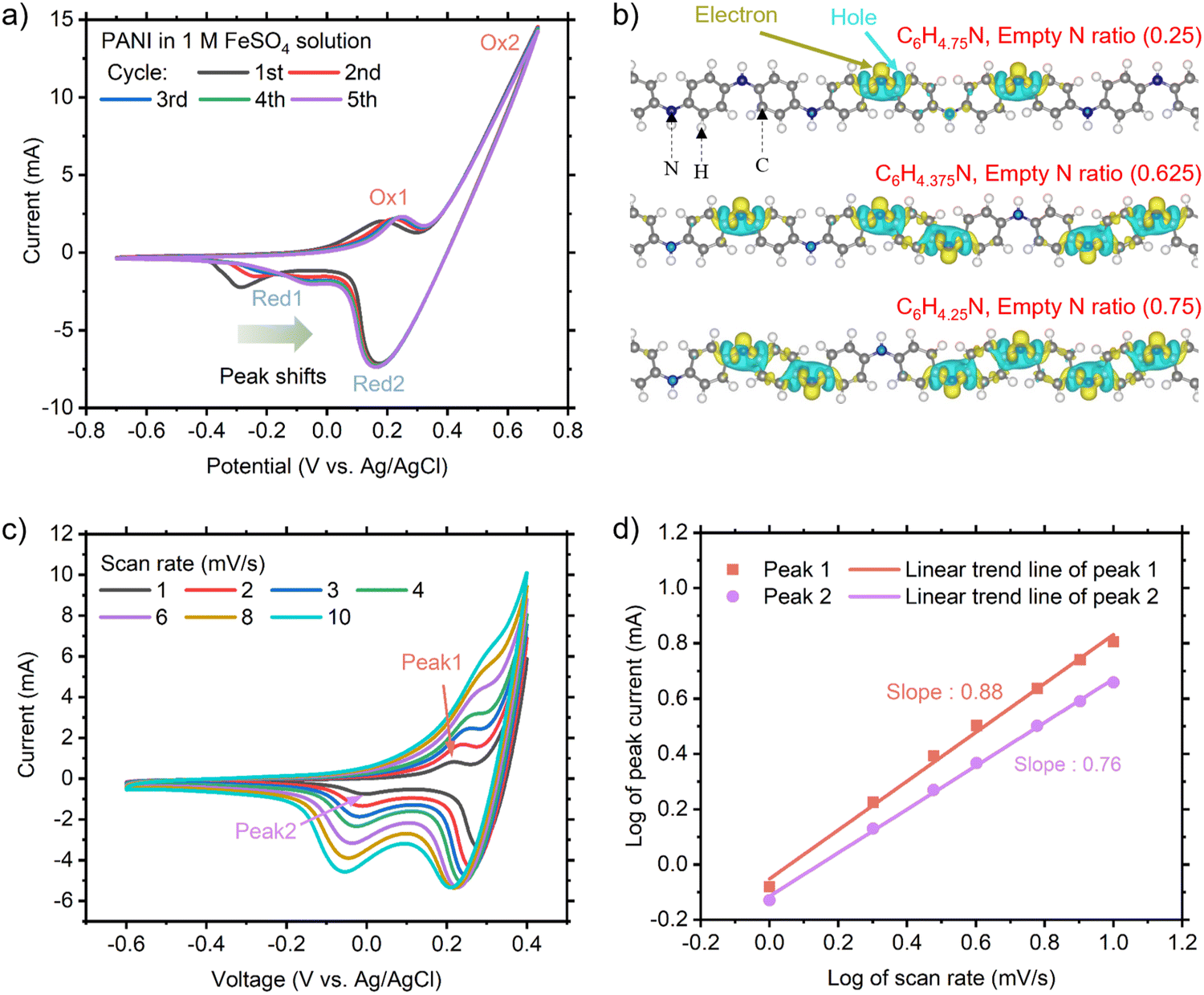 | ||
| Fig. 2 (a) CV curves of the PANI-embedded CNT electrode for the first five cycles at a scan rate of 10 mV s−1. (b) Charge density difference map of empty N sites (i.e., N without having H) within the stable intermediates that can be generated during CV. Isosurface levels for all structures are 0.004 e Å−3. Cyan and yellow colors represent holes and electrons, respectively. (c) CV curves of the PANI-embedded CNT electrode at different scan rates. (d) Corresponding log (peak current) and log (scan rate) for the redox peaks. The R square values of the slopes for peaks 1 and 2 are 0.995 and 0.998, respectively. | ||
To understand the noticeable shift in Re1 during the initial CV cycles, DFT calculations were performed to identify stable Fe-incorporated PANI structures ranging from fully H-incorporated to fully Fe-incorporated structures. We examined the relative formation energies (Ef) across varying Fe concentrations during the reductive process using the cluster approach to statistical mechanics (CASM), which simplifies the modeling of complex systems by efficiently screening potential intermediate structures of Fe-incorporated PANI. Also, this method allows the exploration of all the possible geometries regarding Fe doping by simulating only the most critical interactions within clusters of atoms. Specifically, we considered all possible configurations (total of 256) of intermediate structures in the Fe concentration range of 0 ≤ x ≤ 1, as shown in ESI,† Fig. S4a. Each point in ESI,† Fig. S4b represents Ef of a C1.2FexH(1−x)N0.2 configuration at a specific Fe concentration, following the formula Ef = E(one-phasesolidsolution) − E(two-phasecomposite), which is equivalent to Ef = E(C1.2FexH(1−x)N0.2) − [xE(C1.2FeN0.2)] + (1 − x)E(C1.2HN0.2). The calculations revealed that C1.2FexH(1−x)N0.2 structures favor the one-phase solid solution, given that the majority of Ef values are negative, indicating higher stabilities of the intermediate structures compared to fully H-incorporated and fully Fe-incorporated structures. According to these findings, we identified three stable intermediates with varying Fe concentrations (0.25, 0.625, and 0.75), as indicated by the convex hull diagram (ESI,† Fig. S4b). Moreover, we deduced that the steady increase in the Fe concentration within the PANI structure is likely due to the efficient interaction between the Fe2+ cations and the exposed nitrogen, facilitated by hydrogen detachment during the oxidative process. To substantiate this, we analyzed the charge density difference map for the three intermediates, revealing that the exposed nitrogen bears a negative charge (yellow region in Fig. 2(b)), which encourages Fe2+ attraction to the nitrogen through effective electrostatic attraction.
Leveraging the three stable structures with Fe attachment, we analyzed the activation energy barriers necessary for the transfer of Fe2+ cations from the SO42− anions in the solvent to the nitrogen of the PANI structure. This was done to elucidate the marked shift in Red1 that occurred with changes in the Fe concentration. To identify a probable pathway for the Fe transfer, we initially explored both the consecutive mechanism, where Fe attaches to one atom at a time, and the concerted mechanism, where multiple Fe atoms attach simultaneously (ESI,† Fig. S5). Upon investigating the four activation energy barriers for each mechanism, we discovered that the consecutive mechanism is more favorable, exhibiting lower energy levels than the concerted mechanism (ESI,† Fig. S6).
Next, we compared the calculated activation energy barriers based on the Fe concentration, with shifts in experimental potentials. Specifically, we observed that the shifts in the experimental potentials were progressively larger as the number of reductive cycles increased compared to the potential from Red1 (i.e., first cycle). We compared these experimental potential changes, occurring in the initial stage of the reductive process (i.e., 1st–5th cycles), with the theoretical potential changes (i.e., activation barrier changes) necessary for the increase in Fe concentration. As shown in ESI,† Fig. S7, we calculated the potential changes corresponding to the increase in the Fe concentration within the PANI structure. These increments included changes in the Fe concentration from 0.25 to 0.625, 0.75, and 1.0, with the potential changes calculated to be 0.05, 0.22, and 0.34 V, respectively. Interestingly, there is a similarity between these calculated potentials (0.22 V for Fe concentration of 0.75) and the potential shifts (∼0.2 V) observed during the 1st–5th reductive CV cycles. However, the potential change is not consistent with the calculated value of 0.34 V for the transition from a Fe concentration of 0.25 to 1.0, which implies that the incorporation of Fe in the PANI structure may be limited to a concentration of 0.75 during the CV cycles.
To gain further insight into the constraints imposed by the Fe concentration during the reductive process, we explored the correlation between potential values and structural changes, particularly in response to an increase in Fe concentration. Leveraging the consecutive mechanism, we evaluated the average dihedral angles between the two benzene rings centered around the nitrogen site at which Fe binds. We sought to understand the influence of specific Fe concentrations, potentially encountered during battery cycles, on the magnitude of structural changes. As shown in ESI,† Fig. S8, it is evident that the structural deformations became more pronounced with an increase in the Fe concentration. Specifically, the PANI structure with Fe0.75 exhibited a substantially larger dihedral angle change compared to Fe0.625, which is consistent with the observed trend of the experimental potential shift. Furthermore, we conducted theoretical predictions on the post-5-cycle PANI structure, aiming to understand how it evolves. Specifically, we investigated the structural changes that occurred during the oxidative process when the PANI structures were loaded with Fe up to an Fe0.75 ratio after the 5th cycle. In particular, we considered two possible scenarios that could occur during the oxidative process following the 5th reductive cycle, i.e., Fe or H detachment. We found that the calculated corresponding reaction energy value for H detachment (0.67 eV) is relatively more favorable than that involving Fe detachment (0.88 eV). This implies that as the cycles progressed, the H ratio decreased significantly, suggesting that only Fe attachment and detachment would play a role in the reductive and oxidative processes.
Based on these theoretical findings, we further investigated the kinetics of the PANI redox in FeSO4 solution through CV measurement, as presented in Fig. 2(c). The relationship between peak current and scan rate can be described as follows:25,26
| i = avb | (1) |
Although the aforementioned results show that H on the nitrogen sites of the pristine PANI is replaced by Fe during the initial reduction process of the CV cycles, the following assessment unveiled the characteristics of the full cells. Here, we first investigated the compositions where Fe can stably exist during repeated charge and discharge processes, and subsequently examined whether these structures are favorable to coexist with H. Our theoretical findings suggest that H detachment is more favorable than Fe detachment, and thus it may not be easy for H to coexist with Fe within the PANI structure. To systematically study the Fe composition, we evaluated all the plausible configurations (total of 256) of the intermediate structures that could form in the Fe concentration range of 0 ≤ x ≤ 1, as indicated by the convex hull diagram (Fig. 3(a)). In this diagram, each point represents the Ef of a C6FexN configuration at a specific Fe concentration. The calculated results reveal that solid solutions are favored instead of two separate composites where x = 0 and 1, as indicated by the negative Ef values. This analysis allowed us to identify four stable intermediates at different Fe concentrations (0.25, 0.375, 0.625, and 0.875), as illustrated in Fig. 3(b). Based on these four intermediates, we systematically compared the formation energy by binding H to the empty N sites within these structures to verify whether they are indeed stable compared to the structures coexisting with H. The results indicate that in the PANI structures coexisting with Fe and H, the formation energy is significantly higher than in the cases where only Fe is present, as shown in Fig. 3(c), together with the corresponding Fe and Fe + H structures (ESI,† Fig. S9). This suggests that after extended battery cycles, one can envision that only Fe would be involved without having H on the N sites.
 | ||
| Fig. 3 (a) Convex hull diagram of the formation energy when Fe is attached to PANI during discharge in the full cell. All considered phases are shown as black symbols, and stable phases are highlighted with red symbols. (b) PANI structures without hydrogen (C6H4N) and with Fe (C6H4FexN, x = 0.25, 0.375, 0.625, 0.875, and 1.0) during discharge in the full cell. (c) Formation energies of the two different PANI structures, where the nitrogen sites are occupied by only Fe or Fe + H (mixed Fe and H). (d) Theoretical voltage profiles when the Fe concentration in PANI is 0.25, 0.375, 0.625, 0.875, and 1.0 along with the experimental one at 5C. (e) Average dihedral angles of the PANI structure corresponding to the Fe concentration. | ||
Based on the confirmed Fe-incorporated structures, we decided to explore the value of the discharging voltage using the Nernst equation (V = ΔGf/(zF)), where z is the charge carried by Fe in the electrolyte, F is the Faraday constant, and ΔGf is the change in Gibbs free energy, defined by ΔGf = ΔEf + PΔVf − TΔSf, where ΔEf, P, ΔVf, T, and ΔSf are the formation energy of the PANI structure, pressure, volume change, temperature, and entropy change, respectively. In this equation, the volume and entropy changes can be neglected given that they are of a very small order compared to ΔEf due to the absence of variations in pressure and temperature, which can transform into the formation energy defined by ΔEf = E(C6Fex2N) − E(C6Fex1N) − (x2 − x1)E(Fe) with x2 > x1. According to this understanding, we constructed a theoretical voltage profile by assigning specific values at distinct Fe concentrations (0, 0.25, 0.375, 0.625, 0.875, and 1). We found that the calculated points are consistent with the trend of the experimental voltage profile (Fig. 3(d)), thus confirming the validity of using PANI without hydrogens as the initial configuration. Moreover, this result demonstrates that the discharging voltage drop initiated at Fe0.625 and exhibited a substantial decrease upon reaching Fe0.875. The comparison of this finding with the experimental voltage drop suggests that starting from Fe0.625, the further addition of Fe into PANI faces increasing difficulty. This challenge is particularly pronounced when the concentration is close to Fe1.0, at which point the integration of Fe into PANI encounters significant difficulty.
To understand the reason for the voltage drop, we investigated the structural changes associated with an increasing Fe concentration and found that there is a strong correlation between the voltage drop and variations in dihedral angles centered on the nitrogen sites. As shown in Fig. 3(e) and ESI,† Fig. S10, it is evident that the changes in dihedral angles became more pronounced with an increase in Fe concentration. Notably, starting from Fe0.625 and moving closer to Fe1.0, the change in the dihedral angle became much larger, which aligns with the explored tendency of the theoretical voltage profile. Furthermore, in the case of the notably symmetric structure of Fe1.0, the accumulated change in dihedral angles up to Fe0.875 suddenly returned to its original state. This reversal resulted in the most pronounced change in dihedral angles observed, leading to a considerable voltage drop.
To further validate our theoretical calculations indicating that the concentration of Fe in the PANI structure varies at the different states of charge/discharge, we carried out ex situ X-ray photoelectron spectroscopy (XPS) experiments to substantiate the PANI reaction mechanisms. Our Fe-ion cells were disassembled at three different states of discharge (ESI,† Fig. S11a), and the cathodes were washed and dried before the XPS experiments. ESI,† Fig. S11b show the XPS spectra of Fe 2p at the three different discharge states, revealing higher Fe 2p peak intensities at lower cell voltages. As shown in ESI,† Fig. S11c–e and Table S4, the atomic Fe:N ratios are 0.365 at 0.95 V, 0.436 at 0.6 V, and 0.581 at 0.1 V, respectively. This implies that more Fe ions are attached to PANI as the cell discharges (i.e., at lower cell voltage). The observed trend from the XPS results is consistent with our theoretical findings although the Fe concentrations in PANI are not identical, which is presumably caused by the ex situ experiments involving washing of the FeSO4 electrolyte and drying of the electrode in an ambient air environment during the sample preparation as well as long degassing processes under ultra-high vacuum environment during the XPS experiments. The resolved N 1s spectra at 0.95 V, 0.65 V, and 0.1 V (ESI,† Fig. S11i–k, respectively) highlight the changes in the nitrogen environment of PANI,27 as illustrated in ESI,† Fig. S11l. Notably, the hydrogen bonded to nitrogen in PANI may be replaced by Fe in this system. Specifically, N1 (neutral imine) and N4 (cation amine) correspond to the oxidized states of PANI, whereas N2 (neutral amine) and N3 (cation radical amine) represent the reduced states.28 In the range of 0.95 V to 0.1 V, the N1 + N4 intensities decrease, while the N2 + N3 intensities increase, reflecting a progressive reduction in the concentration of Fe. This observed trend in the XPS data is consistent with the theoretical findings.
The rate capability tests at 5–20C rates (1C = 300 mA per g) were carried out with an 8 wt% loading of the active material in the entire cathode, as displayed in Fig. 4(a), and the corresponding charge/discharge voltage profiles are shown in Fig. 4(b) with a cut-off voltage of 1.2 V. Our battery with an 8 wt% loading exhibited an outstanding specific capacity of ∼225 mA h g−1 at a rate of 5C. It should be noted that this high specific capacity was obtained with an affordable FeSO4 electrolyte. For example, a previous study29 showed that an FeSO4 electrolyte in PANI-based Fe-ion cells delivered only ∼10 mA h g−1. Therefore, the costly Fe(OTf)2 electrolyte was required to enhance the capacity, presenting a significant challenge to achieving cost-effective batteries. Accordingly, the outstanding performance of our economical batteries represents a notable breakthrough in this area. Although an active material loading of 10 wt% or less is typical in aqueous batteries, we tested higher loadings of 24 and 39 wt% with a cut-off voltage of 1.2 V (ESI,† Fig. S12). Our results indicate that the capacity decreased as the active material loading increased. Alternatively, with a lower (2 wt%) active material loading, the specific capacity increased, reaching 362 mA h g−1 at 5C due to the pseudo-capacitive effects resulting from the large surface area of CNT.22 Additionally, we evaluated the capacity of the CNT cathode without PANI in the full cell configuration (ESI,† Fig. S13). The capacity of only ∼1.2 mA h g−1 was obtained, confirming that CNT had a minimal contribution to the overall capacity.
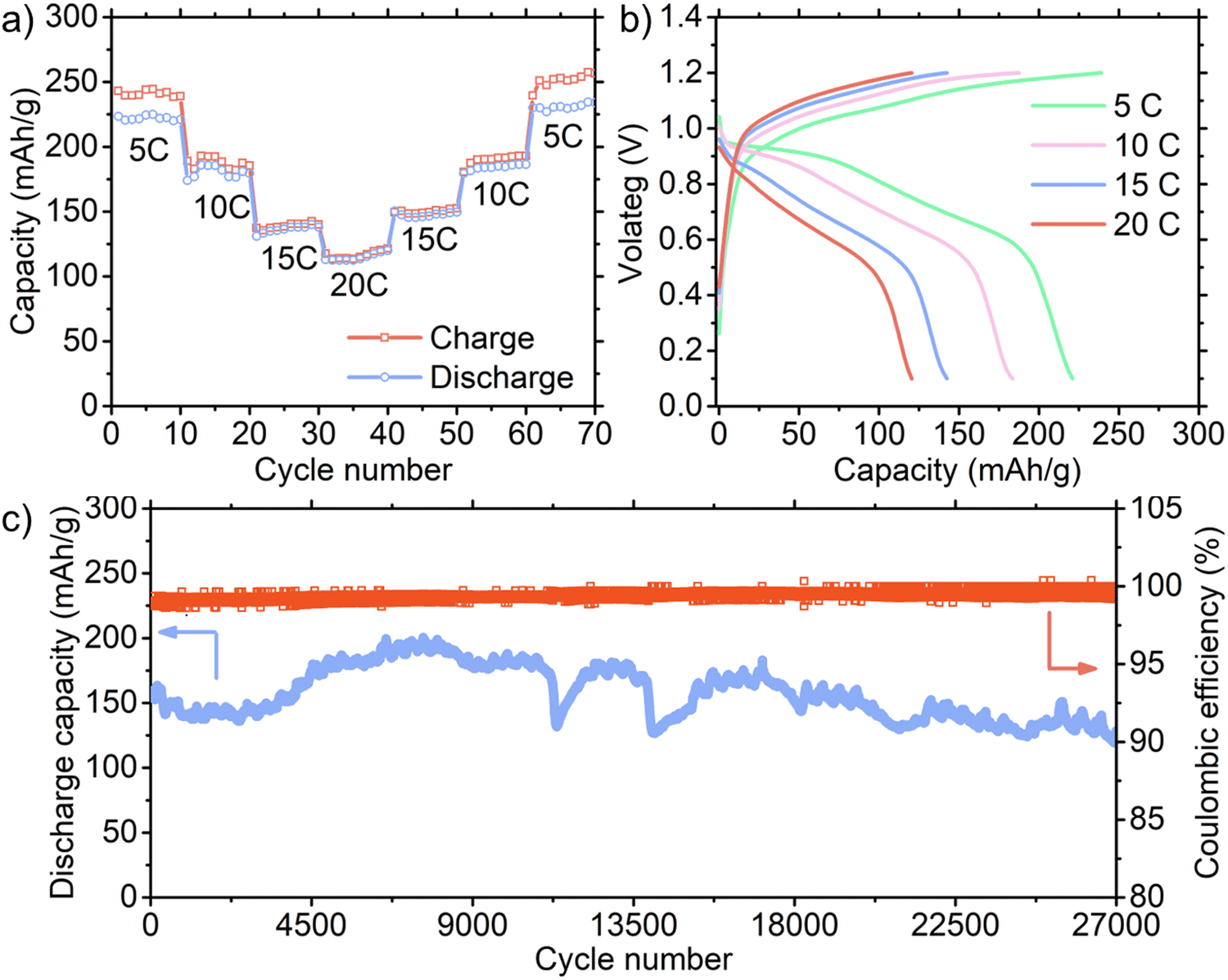 | ||
| Fig. 4 (a) Specific capacity and of the full cell with 8% active material loading of PANI-sandwiched CNT cathode at different C rates (1C = 300 mA per g). (b) Corresponding charge/discharge voltage profiles at different C rates. (c) Specific discharge capacity and Coulombic efficiency as a function of cycle number at 15C for 8 wt% active material loading cell. | ||
Although the aforementioned rate capability tests focused on maximizing the specific capacity, it is more important to maximize the Coulombic efficiency for long-term cycling by limiting the upper voltage limits. For example, the upper voltage was set to 1.15 V for 39 wt% loading when the Coulombic efficiency was maintained over 97%, as displayed in ESI,† Fig. S14a and b. It should be noted that the cell voltage dropped during the charge process due to the oxidation of Fe2+. The Fe2+/Fe3+ redox reaction occurred at 0.75 V vs. SHE, but the actual oxidation potential varied depending on the C rate, which was identified by charging the full cell up to 200 mA h g−1 to observed the saturation and/or drop in the voltage (ESI,† Fig. S14c). The actual potentials to avoid the Fe2+/Fe3+ redox reaction are displayed in the Pourbaix diagram (ESI,† Fig. S14d). A higher C rate necessitates a higher rate of reactant (Fe2+ ions) movement to the electrode, where a limited supply of Fe2+ for oxidation suppresses the unwanted oxidation of Fe2+, which can result in better performances.
The long-term cycling performance of our full cell was tested at 15C for the 8 wt% loading and the cut-off voltage of 1.2 V (Fig. 4(c) and ESI,† Fig. S15) and 5C for the 39 wt% loading and the cut-off voltage of 1.15 V (ESI,† Fig. S16). The specific capacity for the 8 wt% loading gradually increased from ∼157 mA h g−1 (which is close to the value of the rate capability test) to ∼200 mA h g−1 presumably due to the initial activation processes. At the 27000th cycle, the capacity was ∼129 mA h g−1, demonstrating an outstanding capacity retention of 82%. In the case of the 39 wt% loading cell, the reduced upper voltage of 1.15 V voltage maintained a high Coulombic efficiency even at a low 5C rate by regulating side reactions, as shown in ESI,† Fig. S17. As the initial specific capacity rapidly increased in the first ∼100 cycles, we used 53.6 mA h g−1 at the 100th cycle to calculate the capacity retention. Surprisingly, this cell demonstrated 94.7%, 88.0%, and 68.0% capacity retention at the 10000th, 20000th, and 30000th cycle, respectively. Furthermore, we tested the long-term cycling performance for the 2 wt% loading at 15C rate (ESI,† Fig. S18). The high capacity retention after extensive cycling indicates that PANI was securely anchored in the sandwich-type electrodes (ESI,† Fig. S19 shows SEM image of the sandwich-type electrode after 5000 cycles). It is worth noting that the capacity fluctuated during cycling, a phenomenon also observed in other aqueous batteries. This behavior is particularly prevalent under high current conditions, which is likely caused by the unstable charge transfer resistance under these demanding conditions.30,31
We closely investigated the carbon steel anode, focusing on its morphology and the stability of the Fe plating/stripping process. Fig. 5(a) displays the XRD peaks of the pristine carbon steel, carbon steel anode from the coin cell without cycling (rested for 14 days), and after 500 and 5000 cycles, respectively. In all these samples, only peaks at 44.7°, 65.1°, and 82.4° were observed, indicating an absence of crystalline iron corrosion products during the charge and discharge processes in the FeSO4 electrolyte. It should be noted that typical iron corrosion products are crystalline structures, including Fe2O3,32 FeO,33 Fe3O4,34 and FeOOH.35 We also conducted XPS analyses on the carbon steel anodes after 500 and 5000 cycles, as shown in ESI,† Fig. S20. The Fe 2p spectra for both cycling conditions are similar. To gain deeper insights into the surface components, we performed spectral fitting of the Fe 2p spectra (ESI,† Fig. S21). The XPS results revealed the presence of Fe2+ and Fe3+ species, which typically indicate the formation of iron oxide although the corrosion products were not detected by XRD (Fig. 5(a)). This discrepancy arises from the presence of amorphous products or the difference in depth sensitivity between XPS (nanometer scale) and XRD (micron scale).
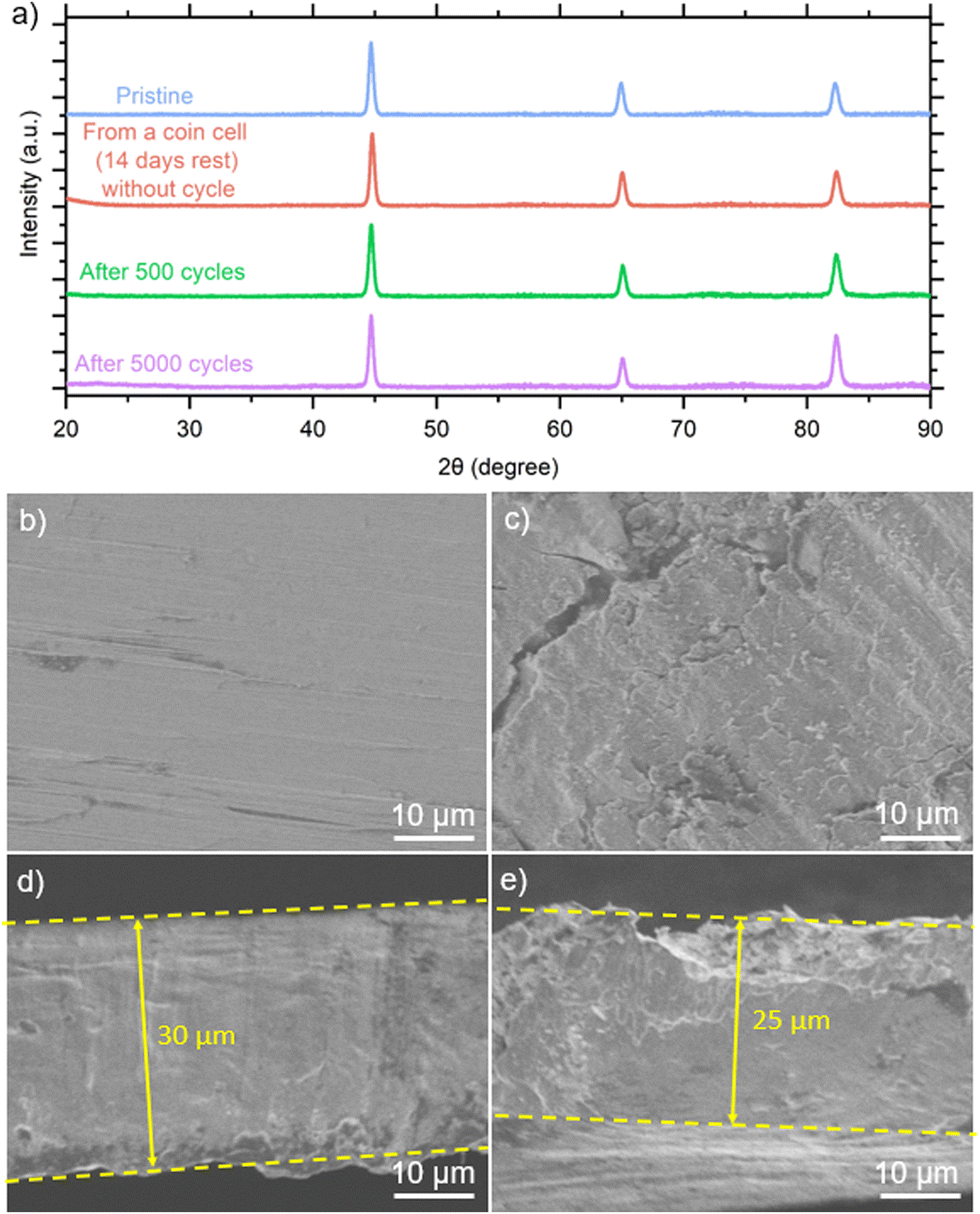 | ||
| Fig. 5 (a) X-ray diffraction data for pristine carbon steel, carbon steel anode in a coin cell without cycling after 14 days, and anode after 500 and 5000 cycles. SEM images of (b) surface of the pristine carbon steel, (c) surface of the carbon steel anode after 5000 cycles, (d) cross-section of the pristine carbon steel, and (e) cross section of the anode after 5000 cycles. | ||
We inspected the surface morphology of the carbon steel anode before cycling (Fig. 5(b)) and after 5000 cycles (Fig. 5(c)). Compared with the pristine carbon steel, the anode surface after 5000 cycles appeared rougher. According to the cross-section of the pristine carbon steel anode (Fig. 5(d)) compared to that after 5000 cycles (Fig. 5(e) and ESI,† Fig. S22), the thickness of the carbon steel anode after 5000 cycles was not uniform, appearing several microns thinner than the pristine anode as a result of Fe plating/stripping efficiencies lower than 100%.
We also examined the safety of our FIB by intentionally short-circuiting the cathode and the anode internally or externally at the beginning of discharge. The cathode and anode were made to contact after multiple cycles, as shown in Fig. 6(a)–(c) for electrode configurations before, during, and after the internal short-circuit test, respectively. The corresponding voltage profile is displayed in Fig. 6(d) as a function of time. The two electrodes were kept in contact for ∼25 min, and the voltage dropped to around 0 V. Surprisingly, after the two electrodes were detached, the cell was fully recovered, showing the normal voltage profile without having any noticeable fire hazard. Similarly, the external short-circuit test did not degrade the performance of the battery, besides demonstrating its safe operation (ESI,† Fig. S23). The lack of fire hazard is also advantageous in battery manufacturing, unlike Li-ion batteries, where the small misalignment of the electrodes often leads to an unpredictable explosion.
 | ||
| Fig. 6 Test setup with a cathode and an anode in the electrolyte (∼590 μL) with three different electrode configurations (a) before, (b) during, and (c) after the short circuit, and (d) corresponding voltage profile against time. | ||
The aforementioned experiments were carried out with conventional foil-type electrodes. In LIB, thin electrodes are wound or stacked to fabricate thick jelly rolls despite the significantly increased complexity in manufacturing because the sluggish ion transport in LIB limits the thickness of the electrodes. Here, the outstanding kinetics of our FIB, which was evidenced by its high C rates, allows a new electrode design with simple manufacturing. Our electrode design completely eliminated the slurry mixing, coating, and drying processes, which are time-consuming and capital-intensive processes, using the cylindrical 3D CNT (cathode) and a carbon steel wire (anode) wrapped by an electronically-insulating braided sleeve (separator), as schematically shown in Fig. 7(a).
 | ||
| Fig. 7 (a) Schematic of the cylindrical cell with a cylindrical PANI-deposited CNT sponge cathode, a carbon steel wire anode, a braided polyester sleeve separator, and an aqueous 1 M FeSO4 electrolyte. Graphite foil was used as a current collector because a transparent glass container was used for monitoring (the current collector is not required if a metal container is used). (b) Photograph of the cylindrical cell along with a dry cell (AA size) for comparison. The inset is a photograph of the CNT sponge used for the cathode. (c) Specific discharge capacity and Coulombic efficiency of the cylindrical cell at 1C (1C = 300 mA per g). The radar chart in the inset compares crucial battery aspects of our FIB with that of lead-acid and Li-ion batteries. | ||
The actual cylindrical battery is displayed in Fig. 7(b) with dimensions similar to an AA-size dry cell. The as-synthesized CNT sponge (diameter of 2.1 cm) was cut with an axial length of 1.5 cm. Then, PANI was electro-deposited into the CNT sponge using a DC power supply. A small hole was created at the center of the cylindrical CNT, and then the carbon steel wire with the sleeve was inserted into the hole. The CNT sponge was slightly pressurized while inserting it into a transparent glass vial, which was used to monitor the variations in the electrodes during charge/discharge. The current collector for the cathode was eliminated when the glass vial was replaced by a metal one. The discharge capacity of the cylindrical cell at a 1C rate is shown in Fig. 7(c), revealing negligible capacity fading for 3000 cycles. This impressive performance with the new electrode design underscores the promise of our FIB with a PANI cathode.
Our Fe-ion batteries are advantageous in numerous aspects including cycle life, cost per cycle, safety, non-toxicity, and power density (Fig. 7(c) inset, ESI,† Tables S5 and S6). The energy density of FIB is superior to that of lead-acid batteries (LAB), but not that of LIB. Carbon steel, used as an Fe anode, is not only inexpensive but also free from supply chain issues. The FeSO4·7H2O in the electrolyte costs a mere 0.03 $ per kg, and the cathode material PANI is priced at approximately 2.17 $ per kg.36 Furthermore, all the material costs are expected to be significantly lowered when mass-produced. Owing to its long cycle life and low-cost components, the cost per cycle ($ per kW h per cycle) of our battery surpasses that of LIB and LAB by an order of magnitude (see ESI,† Table S5). More importantly, nonflammability is vital for avoiding potentially catastrophic fires because energy storage stations often consist of thousands of batteries. However, a limitation of the aqueous electrolyte is its low voltage (<1.23 V), which makes it challenging for our FIB to surpass the energy density of LIB.
In summary, this work presents newly-emerged and low-cost Fe-ion batteries consisting of PANI-incorporated CNT cathodes and carbon steel anodes with two different configurations. The first configuration is to simply sandwich the active material in the cathode between sponge-like CNT layers and readily adjust the loading. The second design greatly simplified the manufacturing process by adopting as-synthesized cylindrical 3D CNT, thereby eliminating complicated multi-layered jelly rolls in the conventional battery designs. Despite the use of the low-cost carbon steel (Fe source) with a moderate Fe purity as the anode, our FIB delivered an impressive discharge specific capacity of 225 mA h g−1 at 15C, and it could be cycled 27000 times with the capacity retention of 82% at 15C. More importantly, our theoretical study unveiled the largely unexplored working mechanisms in the behaviors of the experimentally observed FIB including the underlying causes of the peak shifts in CV and voltage drops in the discharge process, which can be attributed to the formation energy of Fe on the empty N sites through our systematic approaches such as the convex hull and charge density diagrams, Fe-ion transfer behaviors, and structural deformations with the Fe attachment. Our investigations revealed that the nitrogen site, which was occupied by hydrogen initially, was gradually replaced by Fe depending on the state of discharge without having hydrogen due to the significant structural deformations. Concomitantly, the voltage drop during discharge is strongly related to the large changes in the average dihedral angles. This discovery emphasizes the necessity of understanding and managing the structural changes to enhance the electrochemical performance of PANI-based systems. Lastly, we demonstrated that our aqueous FIB are immune to performance degradation and fire due to short circuit, which is the main cause of fire and failure in LIB. This work substantiates that FIB can be cost-effective, long-lasting, safe, and manufacturing-friendly solutions, encouraging further research in the fields of renewable energy storage.
Method
Polyaniline (emeraldine base, molecular weight of ∼50000, Sigma Aldrich); aniline (>99%; Alfa Aesar), hydrochloric acid (HCl, 36.5–38.0%, Macron Fine Chemicals); FeSO4·7H2O (>99%, BeanTown Chemical); graphite foil (99.8% metals basis, thickness of 0.254 mm, Alfa Aesar); carbon steel shim (1008–1010 carbon steel, iron >99%, thickness of 0.001 or 0.005 inch, Precision Brand Products); carbon steel wire (1006–1008 carbon steel, diameter of 0.063 inch; purchased from McMaster); filter paper (pore size: 10 μm, thickness: 150 μm; VWR quantitative filter paper); braided expandable sleeve (polyester, diameter: 1/8 inches; purchased from McMaster.); deionized (DI) water (>18 MΩ cm−1).
Cathode preparation
Cylindrical 3D CNT sponges were synthesized using the procedure of our previous work.20,37 For the sandwich-type cathode, the CNT sponge was sliced with a thickness of ∼100–500 μm, and then punched to obtain circular electrodes with a diameter of 3/8 inch. PANI powder was sandwiched between two pieces of the circular CNT electrodes followed by cold-pressing (∼500 kPa). The PANI loadings were in the range of 0.2 to 1.4 mg with 2.2∼10 mg of CNT (area: ∼0.7 cm2), achieving 2% to 39 wt% active material loading in the entire cathode. It should be noted that our cathode is binder-free, with CNT serving as both the conductive additive and the current collector. In the cylindrical cell, the CNT sponge was cut to an axial length of 1.5 cm without any modification in its diameter (2.1 cm). PANI was electro-deposited into the CNT sponge by immersing it into an aniline solution (0.1 M in 1 M HCl) with 10 mA DC supply for 3 h. The typical PANI loading was ∼50 mg for the CNT with a mass of ∼500 mg.Coin and cylindrical cell fabrication
The coin cells (2025 size) were composed of a CNT cathode, carbon steel anode, filter paper separator with diameters of 3/8, 1/2, and 5/8 inch, respectively, together with 100 μL of 1 M aqueous FeSO4 electrolyte. The cylindrical cells were assembled with the CNT cathode, carbon steel wire anode, and sleeve separator. The carbon steel wire was inserted into the sleeve through the center of cathode, and then the whole piece was put in a 2-dram vial with 4 mL of 1 M aqueous FeSO4 electrolyte. The cylindrical cells were tightly sealed for testing.Battery and electrochemical test
The galvanostatic charge/discharge tests were carried out using a Landt battery test system (Model: CT3002A). For cyclic voltammetry, PANI was electro-deposited on the CNT sponge as the working electrode with a platinum wire as the counter electrode and a standard Ag/AgCl electrode as the reference electrode.Short circuit tests
The PANI-sandwiched CNT cathode was prepared, and then attached to graphite foil using carbon paint. After the carbon paint was fully dried, the cathode and a carbon steel anode were placed in a transparent container with dimensions of 5.4 cm × 5.4 cm × 5.4 cm and a wall thickness of 2 mm. The initial gap between the cathode and anode was ∼8 mm. Then, the electrolyte was poured in the container and the container was purged with argon. Then, the container was sealed using silicone and the electrodes were connected to the battery tester. For the internal short-circuit test, seven regular discharge/charge cycles and one additional charge cycle followed by 20 s of rest were carried out. Then, the anode was tilted towards the cathode to create the short circuit. The electrodes were kept in the short circuit configuration for ∼25 min, and then detached with an ∼8 mm gap followed by eight more cycles. The external short-circuit test was carried out similarly by connecting the cathode and anode externally.Computational details
All ab initio calculations were performed with the Vienna Ab Initio Simulation Package (VASP 5.4.4).38–41 We used the projector augmented wave (PAW) method42,43 with the generalized gradient approximation based on the Perdew–Burke–Ernzerhof (PBE)44 functional. A plane-wave cutoff energy of 500 eV was used. Lattice constants and internal atomic positions were fully optimized until the residual forces were less than 0.04 eV Å−1. To verify the accuracy of this energy convergence criterion value, we systematically compared the differences between the less strict setting of 0.04 eV Å−1 and the stricter setting of 0.01 eV Å−1 (Section S5 in the ESI†). The vacuum slab space of a unit cell in the z-direction was set to 15 Å to avoid interactions between layers. Spin polarization and dipole correction were also included to decouple the electrostatic interaction between periodically repeated systems. Further, to simplify the modeling of complex Fe-incorporated PANI structures, we used the CASM method, which is useful in the field of materials science, where the behavior of large assemblies of atoms or molecules is often dictated by localized interactions within smaller clusters. This method allows the exploration of different ratios of Fe doping by simulating only the most critical interactions within clusters of atoms. The valence orbital of the active sites was obtained by density of states (DOS), which was calculated by sampling the Brillouin zone with a 10 × 10 × 1 k-point grid.45 Analysis of Bader charges is a widely adopted method to study the charge distribution. In this work, the Bader charge analysis was performed using grid-based charge density decomposition, as developed by Henkelman et al.46Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- M. Tang, et al., Ultrafast rechargeable aqueous zinc-ion batteries based on stable radical chemistry, Adv. Funct. Mater., 2021, 31, 2102011 CrossRef CAS.
- W. Deng, et al., Zn2+ induced phase transformation of K2MnFe(CN)6 boosts highly stable zinc-ion storage, Adv. Energy Mater., 2021, 11, 2003639 CrossRef CAS.
- L. Ma, et al., Achieving both high voltage and high capacity in aqueous zinc-ion battery for record high energy density, Adv. Funct. Mater., 2019, 29, 1906142 CrossRef CAS.
- T. Sun, S. Zheng, Q. Nian and Z. Tao, Hydrogen bond shielding effect for high-performance aqueous zinc ion batteries, Small, 2022, 18, 2107115 CrossRef CAS PubMed.
- A. Hollas, et al., A biomimetic high-capacity phenazine-based anolyte for aqueous organic redox flow batteries, Nat. Energy, 2018, 3, 508–514 CrossRef CAS.
- Daily Metal Prices, https://www.dailymetalprice.com.
- X. Wu, et al., A rechargeable battery with an iron metal anode, Adv. Funct. Mater., 2019, 29, 1900911 CrossRef.
- S. K. Sandstrom and X. Ji, Unlocking the longevity of the iron metal anode, ACS Cent. Sci., 2022, 8, 686–688 CrossRef CAS PubMed.
- X. Wu, et al., Rechargeable iron–sulfur battery without polysulfide shuttling, Adv. Energy Mater., 2019, 9, 1902422 CrossRef CAS.
- Y. Xu, et al., Fe-ion bolted VOPO4·2H2O as an aqueous Fe-ion battery electrode, Adv. Mater., 2021, 33, 2105234 CrossRef CAS PubMed.
- Z. Luo, L. Liu, Q. Zhao, F. Li and J. Chen, An insoluble benzoquinone-based organic cathode for use in rechargeable lithium-ion batteries, Angew. Chem., Int. Ed., 2017, 129, 12735–12739 CrossRef.
- I. A. Rodríguez-Pérez, et al., A hydrocarbon cathode for dual-ion batteries, ACS Energy Lett., 2016, 1, 719–723 CrossRef.
- F. Wan, et al., Nanoeffects promote the electrochemical properties of organic Na2C8H4O4 as anode material for sodium-ion batteries, Nano Energy, 2015, 13, 450–457 CrossRef CAS.
- X. Wu, et al., Unraveling the storage mechanism in organic carbonyl electrodes for sodium-ion batteries, Sci. Adv., 2015, 1, e1500330 CrossRef PubMed.
- L. Fan, Q. Liu, Z. Xu and B. Lu, An organic cathode for potassium dual-ion full battery, ACS Energy Lett., 2017, 2, 1614–1620 CrossRef CAS.
- F. Wan, et al., An aqueous rechargeable zinc-organic battery with hybrid mechanism, Adv. Funct. Mater., 2018, 28, 1804975 CrossRef.
- B. Lee, et al., Exploiting biological systems: Toward eco-friendly and high-efficiency rechargeable batteries, Joule, 2018, 2, 61–75 CrossRef CAS.
- H. Lv, et al., Cross-linked polyaniline for production of long lifespan aqueous iron||organic batteries with electrochromic properties, Nat. Commun., 2023, 14, 3117 CrossRef CAS PubMed.
- G. Yang, et al., Bifunctional nano-sponges serving as non-precious metal catalysts and self-standing cathodes for high performance fuel cell applications, Nano Energy, 2016, 22, 607–614 CrossRef CAS.
- P. Wu, M. Dong, J. Tan, D. A. Kang and C. Yu, Revamping Lithium–Sulfur Batteries for High Cell-Level Energy Density by Synergistic Utilization of Polysulfide Additives and Artificial Solid-Electrolyte Interphase Layers, Adv. Mater., 2021, 33, 2104246 CrossRef CAS PubMed.
- G. Yang, W. Choi, X. Pu and C. Yu, Scalable synthesis of bi-functional high-performance carbon nanotube sponge catalysts and electrodes with optimum C–N–Fe coordination for oxygen reduction reaction, Energy Environ. Sci., 2015, 8, 1799–1807 RSC.
- S. L. Kim, H. T. Lin and C. Yu, Thermally chargeable solid-state supercapacitor, Adv. Energy Mater., 2016, 6, 1600546 CrossRef.
- N. A. Kumar, et al., Polyaniline-grafted reduced graphene oxide for efficient electrochemical supercapacitors, ACS Nano, 2012, 6, 1715–1723 CrossRef CAS PubMed.
- J. Noh, et al., Understanding of Lithium Insertion into 3D Porous Carbon Scaffolds with Hybridized Lithiophobic and Lithiophilic Surfaces by In-Operando Study, Nano Lett., 2020, 20, 3681–3687 CrossRef CAS PubMed.
- V. Augustyn, et al., High-rate electrochemical energy storage through Li+ intercalation pseudocapacitance, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- K. Zhang, et al., Urchin-like CoSe2 as a high-performance anode material for sodium-ion batteries, Adv. Funct. Mater., 2016, 26, 6728–6735 CrossRef CAS.
- Y. Y. Smolin, M. Soroush and K. K. S. Lau, Oxidative chemical vapor deposition of polyaniline thin films, Beilstein J. Nanotechnol., 2017, 8, 1266–1276 CrossRef CAS PubMed.
- C. Dhand et al., Polyaniline-based biosensors, Nanobiosensors in disease diagnosis, 2015, pp. 25–46 Search PubMed.
- C. Li, et al., Hybrid-ion strategy enables ultra-long life aqueous iron-organic batteries, J. Power Sources, 2023, 581, 233506 CrossRef CAS.
- K. Ouyang, et al., A New Insight into Ultrastable Zn Metal Batteries Enabled by In Situ Built Multifunctional Metallic Interphase, Adv. Funct. Mater., 2022, 32, 2109749 CrossRef CAS.
- L. Li, C. Yin, R. Han, F. Zhong and J. Hu, CNT Composite β-MnO2 with Fiber Cable Shape as Cathode Materials for Aqueous Zinc-Ion Batteries, Inorg. Chem., 2024, 63, 13100–13109 CrossRef CAS PubMed.
- G. Schimanke and M. Martin, In situ XRD study of the phase transition of nanocrystalline maghemite (γ-Fe2O3) to hematite (α-Fe2O3), Solid State Ionics, 2000, 136–137, 1235–1240 CrossRef CAS.
- S. K. Sharma, et al., Synthesis and ageing effect in FeO nanoparticles: Transformation to core–shell FeO/Fe3O4 and their magnetic characterization, J. Alloys Compd., 2011, 509, 6414–6417 CrossRef CAS.
- Y.-h Zheng, Y. Cheng, F. Bao and Y.-s Wang, Synthesis and magnetic properties of Fe3O4 nanoparticles, Mater. Res. Bull., 2006, 41, 525–529 CrossRef CAS.
- A. L. Mackay, β-Ferric Oxyhydroxide, Mineral. Mag. J. Mineral. Soc., 1960, 32, 545–557 CrossRef CAS.
- Y. Li, H. Wang, T. Yn, J. Li and J. Kan, Cheap and eco-friendly synthesis of polyaniline and performance of aqueous Zn-polyaniline battery, Int. J. Electrochem. Sci., 2020, 15, 5956–5965 CrossRef CAS.
- G. Yang, et al., Creating effective nanoreactors on carbon nanotubes with mechanochemical treatments for high-areal-capacity sulfur cathodes and lithium anodes, Adv. Funct. Mater., 2018, 28, 1800595 CrossRef.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for open-shell transition metals, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal--amorphous-semiconductor transition in germanium, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03350g |
| ‡ Equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2025 |