
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Configurational entropy-tailored NASICON cathode redox chemistry for capacity-dense and ultralong cyclability†
Wei Zhang‡
 *ab,
Liang He‡c,
Jiantao Lid,
Ruohan Yue,
Zhenming Xuf,
Yulun Wuc,
Haotian Qub,
Qi Zhangb,
Jianwei Lia,
Xian Wub,
Qingjin Fub,
Yanqing Lai*c,
Guangmin Zhou*b,
Guanjie He*a and
Ivan P. Parkina
*ab,
Liang He‡c,
Jiantao Lid,
Ruohan Yue,
Zhenming Xuf,
Yulun Wuc,
Haotian Qub,
Qi Zhangb,
Jianwei Lia,
Xian Wub,
Qingjin Fub,
Yanqing Lai*c,
Guangmin Zhou*b,
Guanjie He*a and
Ivan P. Parkina
aChristopher Ingold Laboratory, Department of Chemistry, University College London, London, WC1H 0AJ, UK. E-mail: wei.zhang.21@ucl.ac.uk; g.he@ucl.ac.uk
bTsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518000, P. R. China. E-mail: guangminzhou@sz.tsinghua.edu.cn
cSchool of Metallurgy and Environment, Central South University, Changsha, 410083, P. R. China. E-mail: laiyanqing@csu.edu.cn
dDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, Illinois, 60208, USA
eWuhan University of Technology, The Sanya Science and Education Innovation Park, Sanya, 572000, P. R. China
fJiangsu Key Laboratory of Electrochemical Energy Storage Technologies, College of Materials Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing, 210016, P. R. China
First published on 6th June 2025
Abstract
Sodium-ion batteries (SIBs) are a promising solution for large-scale energy storage, but their development is hindered by the limited performance of cathode materials. NASICON (Na superionic conductor)-type compounds offer fast Na+ diffusion and structural robustness, yet still suffer from low specific capacities (<120 mA h g−1), poor cycling stability, and large volume changes – even after conventional doping. High-entropy (HE) engineering, a strategy that enhances structural and functional stability via multiple equimolar dopants, has shown great promise in layered oxides, but its application in NASICONs has been fundamentally challenging due to rigid polyanionic frameworks, dopant incompatibility, and redox mismatch. Here we report an entropy-stabilized NASICON cathode, Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 (HE-V), synthesized by compositionally complex doping at the 12c Wyckoff site. The HE strategy enables the development of a single-phase material with enhanced redox flexibility and suppressed lattice strain. HE-V delivers a high reversible capacity of 170 mA h g−1, enabled by multi-electron V5+/V4+/V3+/V2+ redox reactions, and demonstrates exceptional rate performance and cycling stability—maintaining performance over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 50C. Operando and ex situ characterization reveals a nearly zero-strain structure, low defect formation, and enhanced local bonding. Theoretical calculations further confirm bandgap narrowing and improved charge transport. This work establishes a new class of high-entropy polyanionic cathodes, overcoming long-standing limitations in NASICON chemistry and offering a viable path toward durable, high-energy SIBs.
000 cycles at 50C. Operando and ex situ characterization reveals a nearly zero-strain structure, low defect formation, and enhanced local bonding. Theoretical calculations further confirm bandgap narrowing and improved charge transport. This work establishes a new class of high-entropy polyanionic cathodes, overcoming long-standing limitations in NASICON chemistry and offering a viable path toward durable, high-energy SIBs.
Broader contextThe shift toward sustainable energy requires advanced, cost-effective, and scalable battery technologies. While lithium-ion batteries (LIBs) dominate the market, concerns over lithium's supply constraints and high costs have driven interest in alternatives like sodium-ion batteries (SIBs), which benefit from sodium's abundance and lower cost. NASICON-type materials are promising SIB cathodes due to their fast Na+ diffusion and structural stability. However, challenges such as limited energy density and capacity fading hinder their commercialization. High-entropy (HE) engineering, originally developed in metallurgy, offers a novel strategy to enhance battery materials by incorporating multiple principal elements, thus improving stability and electrochemical performance. While HE strategies have succeeded in layered oxide cathodes, their application in NASICON materials remains underexplored. This study leverages HE engineering to improve NASICON cathodes, addressing key performance limitations and paving the way for long-lasting, energy-dense SIBs suitable for grid storage and electric vehicles. |
Introduction
The development of high-performance, cost-effective energy storage systems is essential to meet global decarbonization goals.1–7 While lithium-ion batteries (LIBs) dominate today's market, concerns over critical material scarcity and rising costs are driving a rapid pivot toward alternatives such as sodium-ion batteries (SIBs).8,9 SIBs offer compelling advantages—low raw material cost, wide geographic availability of sodium, and compatibility with existing LIB manufacturing infrastructure.10,11 However, unlocking their full potential requires breakthroughs in cathode materials that combine high energy density with long-term cycling stability under fast charge/discharge conditions.12,13Among candidate SIB cathodes, sodium superionic conductor (NASICON)-type materials (NaxMeMe′(PO4)3, where Me/Me′ are transition metals) have received tremendous interest by virtue of the fast Na+ conductivity, three-dimensional open framework, and tunable voltage/capacity profiles enabled by compositional diversity.14,15 The [MeO6]/[Me′O6] octahedra within NASICON structures are key to their electrochemical performance, influencing charging compensation during cycling and stabilizing the framework. In this regard, one can anticipate that rational compositional design in Me/Me′ sites (i.e., the redox center) is capable of achieving multi-electron redox reactions, structure modulation and output voltage. Early studies on unitary NASICON, such as Na3V2(PO4)3 (NVP), demonstrated a capacity of 108.5 mA h g−1 and a moderate voltage platform of 3.4 V (versus Na+/Na) from the V3+/V4+ redox couple, despite a significant volumetric change of 8.26%.16
To address concerns about cost-effectiveness and energy density, Goodenough et al. in 2016 reported binary NASICON materials like Na4MnV(PO4)3 by substituting vanadium with manganese, a lower-cost element.17 This cathode delivered a comparable capacity of ∼110 mA h g−1 and successfully activated a higher voltage plateau at 3.6 V based on a Mn2+/Mn3+ redox couple, thereby improving the energy density. Since then, a plethora of binary NASICON materials have been reported, such as Na3FeV(PO4)3,17 Na3MnTi(PO4)3,18 Na4MnCr(PO4)3,19–21 Na3MnZr(PO4)3,22 Na2TiV(PO4)3,23 Na3Cr0.5V1.5(PO4)3,24 Na4MnAl(PO4)3,25 etc., very much enriching the NASICON cathode family with diverse voltage plateaus, electrochemical properties, and structural evolution. Subsequently, Hu et al. presented a pioneering report on a ternary NASICON, Na4VMn0.5Fe0.5(PO4)3, which combined the advantages of Na4MnV(PO4)3 and Na4FeV(PO4)3 with good battery performance.26 Despite these advances, challenges still remain, including unsatisfactory long-term cycling at high current rates, structural instability, limited capacities (e.g., <120 mA h g−1), and low electronic conductivity. These limitations are symptomatic of a deeper design constraint: the intrinsic trade-off between structural stability and redox activity in polyanionic systems. Therefore, it is a must but challenging to identify an efficient method to increase the competence of NASICONs (e.g., thousands of cycles without noticeable capacity fading, fast-charging capability within the minute time level, and high capacities of >160 mA h g−1) compared with the LiFePO4 counterpart in LIBs.
To overcome this, a radically different approach is needed—one that can introduce new functional complexity without destabilizing the structure. High-entropy (HE) engineering, originally developed in the field of metallic alloys, has recently emerged as a promising approach to enhance the properties of materials by incorporating multi-principal elements within a single phase in catalysis,27 batteries,28–30 capacitors,31 etc. This strategy increases the configurational entropy, improving the solubility of different components and optimizing target properties.28 As far as practicality is concerned, configurational entropy engineering would alleviate the reliance on any single metal source (e.g., Co and Ni) for corresponding industries if incorporating more abundant metal elements, which could reduce the manufacturing cost.28 This concept was initially applied in SIB layered oxide cathodes by Hu et al. in 2020, which enhanced phase transition (O3 to P3) reversibility and improved cycling stability.32 The same group in 2022 further developed a new HE oxide –NaNi0.25Mg0.05Cu0.1Fe0.2Mn0.2Ti0.1Sn0.1O2 – with suppressed Jahn–Teller distortion, lattice parameter changes, and vacancy/Na+ ordering, thereby enhancing electrochemical performance and thermal safety.33 Very recently, they pointed out that the structural stability of the high-entropy oxides is significantly influenced by how well the constituent elements within the transition metal layers are compatible with each other; even one different dopant (from Sn4+ to Ti4+) contributes to much suppressed planar strain and thus to the inhibited elemental segregation and crack formation upon battery operation.34 In general, most studies of the HE strategy in SIBs are mainly in the field of layered oxide cathodes and achieved fascinating results.32,33,35,36
Despite this success, extending the HE strategy to NASICON-type cathodes has remained elusive, and for good reason. The NASICON structure is governed by a rigid polyanionic lattice, where PO43− tetrahedra and MeO6 octahedra form a tightly interconnected framework. Unlike layered oxides that tolerate cation mixing and planar disorder, NASICONs require precise site occupancy and local symmetry to maintain Na+ mobility and structural integrity. Introducing multiple transition metals into the 12c Wyckoff site37,38 creates a high risk of phase separation, cation site distortion, or redox incompatibility, any of which can degrade capacity or induce irreversible structural evolution.
Additionally, the very premise of entropy stabilization – disorder – runs counter to the ordered framework required for efficient Na+ conduction in NASICONs. There is also a common pitfall: many HE cathodes suffer from low capacities due to the inclusion of redox-inactive elements, which dilute the electrochemically active centers. As such, the challenge is not only how to stabilize a NASICON phase with multiple cations, but also how to do so while preserving – or even enhancing – its energy density and rate capability.
Herein, in response to the problems of NASICON-type materials with an undesirable lattice change, an inferior capacity retention (especially at high current rates), and a low capacity from the inactive V4+/V5+ redox couple (Fig. 1a), we applied a configurational entropy engineering strategy via compositionally complex doping at the 12c Wyckoff site37 in improving the NASICON (i.e., Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3) material (HE-V) that demonstrated high energy-density Na+ storage capabilities and “low-strain” properties. Thanks to this configurational entropy tuning method, HE-V is able to present a record-breaking cycling performance to stably run 10000 cycles even at an ultrahigh current rate of 50C with negligible capacity fading, which remarkably promotes the application of NASICONs. Also, it delivered a much-improved specific capacity of 170 mA h g−1 (close to the theoretical value) compared to the above-mentioned normal value (<120 mA h g−1) of NASICONs, based on a successive multi-electron reaction from the V5+/V4+, V4+/V3+, and V3+/V2+ redox couples identified by synchrotron-based X-ray absorption spectroscopy and X-ray photoelectron spectroscopy.
 | ||
| Fig. 1 Composition design principle and theoretical studies of physicochemical properties. (a) Prototype composition design principle. (b) Na+ ion diffusion pathways (blue isosurface) in HE-V from the BV calculation. (c) Integral ICOHP values of all V–O bonds in HE-V and NVP. (d) Illustrations of crystalline structures and transition metal compositions in HE-V. (e) Calculated voltage profile. | ||
Structural evolution was disclosed to have a nearly zero volumetric change throughout a wide electrochemical window by operando X-ray diffraction, bringing about significantly decreased lattice defects and local strain-induced cracks by the aberration-corrected transmission electron microscope and the finite element method simulation. Density functional theory studies showed a reduced forbidden-band gap and a strengthened neighbor chemical bonding around the redox center, which benefits an excellent rate performance up to 50C.
This work demonstrates that configurational entropy can be harnessed not just to stabilize complex NASICON structures, but also to unlock redox richness and rate robustness previously unattainable in this class of cathodes. It offers a new blueprint for polyanionic cathode design, bridging the gap between high structural order and high compositional complexity.
Results and discussion
Material design and theoretical studies
One big challenge to apply the HE concepts in NASICONs lies in how to select suitable metal dopant elements and to constrain their concentration and the design principal needs to be elaborately proposed. Furthermore, HE compounds are always composed of inactive redox centers (usually >5),34,39 which definitely reduce the specific capacity to some extent. In order to demonstrate the effectiveness of the HE concept, the NASICON cathode material was rationally designed as shown in Fig. 1a: (i) the pristine Na3V2(PO4)3 (denoted as NVP) serves as the baseline sample; (ii) considering the availability, economic concerns, and lattice match constraints, Mg, Al, Ti, V, Cr, Fe, Ni, Cu, Zn, and Mn from the periodic table are selected by Wu et al.;30 furthermore, the high-voltage platform of the V5+/V4+ redox couple in Na3V2(PO4)3, according to previous reports, can be electrochemically activated by specific activators/dopants such as Cr3+,40 Mn2+,41 Fe3+,42 Al3+,43 and Mg2+,44 which is conducive to a “three-electron reaction” with a much-boosted specific capacity (e.g., from 108.5 mA h g−1 in NVP16 to 163.2 mA h g−1 in Na3V1.5Cr0.5(PO4)345) and thus improves the overall energy density; (iii) goodenough et al.,40 Okada et al.,44 and Liang et al.46 have shown that the doped NASICON Na3V2(PO4)3 with a ratio of V to dopants (1.5:0.5 per formula unit) exhibited an optimal performance and a pure phase without compromising capacities significantly, and an equal ratio of all dopants can help enhance the configurational entropy of the compound39 to further stabilize the crystal structure and thus each dopant shares 0.1 per formula unit; (iv) the content of Na is controlled by the charge balance mechanism (i.e., V3+, Cr3+, Mn2+, Fe3+, Al3+, Mg2+, and PO43−). Therefore, the doped Na3V2(PO4)3 was designed as Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 (denoted as HE-V), in which the equal molar dopants of Cr, Mn, Fe, Al, and Mg are anticipated to share the same Wyckoff 12c sites with V (Fig. 1d and Fig. S1, ESI†).
Firstly, density functional theory (DFT) calculations were performed to provide additional theoretical insights into the intrinsic physicochemical features. Calculations of bond valence (BV) and nudged elastic band (NEB) were used to theoretically investigate the behaviors of Na+ diffusion within Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3. An empirical method that is widely used to identify Na+ diffusion channels is the BV method (the isosurface value is 0.5 e Å−3).47 Since the lowest energy locations in HE-V are well-connected,46,48 3D Na+ diffusion channels may be found in Fig. 1b and Fig. S2 (ESI†) (NVP). Twelve routes for Na+ diffusion from the Na+ local habitats in HE-V were found by the NEB investigation. Low energy barriers of 0.162–0.425 eV could be detected, which are comparable to/less than NVP (Fig. S3, ESI†) and other NASICON materials,20,23,40,45 as illustrated in Fig. S4 (ESI†). As seen in Fig. S5 (ESI†), galvanostatic intermittent titration technique (GITT) measurements were conducted to evaluate Na+ diffusion coefficients across the voltage range. The HE-V sample exhibited consistently higher diffusion coefficients compared to NVP, indicating enhanced ionic conductivity and more efficient Na+ transport kinetics. Cyclic voltammetry (CV) curves of NVP and HE-V in Fig. S6 (ESI†) at a scan rate of 0.1 mV s−1 were analyzed to evaluate redox kinetics and Na+ diffusion behavior. HE-V exhibits a markedly higher response current, a larger peak area, and a narrower potential gap between cathodic and anodic peaks, indicating superior redox kinetics and enhanced Na+ diffusion compared to NVP.
Furthermore, we employed a theoretical framework to analyze the local bonding environment around vanadium atoms. Crystal orbital Hamilton population (COHP) analysis, increasingly recognized as a reliable tool for visualizing chemical bonding in battery materials,49–51 was used to probe the electronic interactions. By projecting the Kohn–Sham states onto atomic orbitals, we examined the bonding characteristics through orbital overlap populations. To obtain a statistically robust and physically meaningful assessment, we re-evaluated the V–O coordination by considering all vanadium-centered octahedra within the HE-V unit cell, rather than relying on a representative subset. Given the presence of three crystallographically distinct V sites (Fig. S7, ESI†), we performed integrated COHP (ICOHP) calculations for each site, enabling a comprehensive evaluation of entropy-induced lattice distortions and their effect on local bonding.
Fig. 1c and Fig. S8–S13 and Tables S1–S6 (ESI†) display the COHP and ICOHP (integrated COHP) results based on the six O atoms that surround a particular V atom. According to reports, the modulus of COHP relates to the covalency degree between O and V; the zero point of COHP indicates a pure ionic bond or non-interacting bonds, and the negative value of COHP of V–O implies constructive (i.e., bonding) interference of atomic orbitals. The true ICOHP value is found at the junction of the ICOHP curve and Fermi level in each pattern shown in Fig. S8–S13 (ESI†). According to Fig. 1c, HE-V has an accumulated ICOHP value of 6.8 eV, which is greater than NVP's 6 eV (all V–O pairings, including spin-down and spin-up orientations). This finding suggests that, in the local scope, this configurational entropy engineering strategy increases the V–O bonds’ overall chemical stability, which is favorable for stabilizing structures and thus prolonging cycling stability.21
To evaluate the capacity contribution of the vanadium redox couples in HE-V, we calculated the theoretical capacity based on the full utilization of the V5+/V4+, V4+/V3+, and V3+/V2+ redox transitions, assuming a three-electron transfer per formula unit. This yields a theoretical capacity of 176.2 mA h g−1. As shown in Fig. 1e, the experimental capacity obtained from GITT measurements reaches approximately 170 mA h g−1, which is in excellent agreement with the theoretical prediction. This close match confirms the effective activation of all three vanadium redox couples during cycling. Additionally, the observed voltage plateaus in the GITT profile closely align with the calculated redox potentials, indicating low polarization and high reversibility. The minimal discrepancy between theoretical and experimental values suggests that capacity loss from electrochemically inactive dopants or structural limitations is negligible, underscoring the success of the high-entropy design in preserving redox activity while enhancing structural robustness.
Fig. S14 (ESI†) depicting the structural evolution during cycling was created using a convex-hull phase diagram in terms of formation energies and various Na+ concentrations in NaxV1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3.47 The most thermodynamically stable structure is Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3, and the metastable phases of Na0V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 to Na4V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 can be achieved electrochemically, as shown by negative formation energy values that support the multi-electron redox reactions.
From a kinetics standpoint, it offers compelling evidence for the good Na+ diffusion capability. In addition, density of state (DOS) diagrams showing the presence of hybridized orbitals in the valence bands of all elements are shown in Fig. S15 (ESI†). Compared to Na3V2(PO4)3, a significantly smaller forbidden band gap can be achieved, leading to a better electronic conductivity.45 This echoes the EIS results in Fig. S16 (ESI†). Thus, one can anticipate an improved rate performance of Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3.
Materials characterization
In this section, the designed materials were prepared by a sol–gel method and systematically characterized. In Fig. S17a (ESI†), X-ray diffraction (XRD) determined that HE-V and NVP were indexed to the pure rhombohedral phase (JCPDS#96-222-5133; space group: R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c). Both the structural details and the comprehensive atomic occupation were obtained by Rietveld refinement (Fig. 2a and Fig. S17b and Tables S7 and S8, ESI†). In Fig. 2a, HE-V featured a NASICON structure by a low Rwp value of 3.41% (atomic occupation and lattice properties are presented in Table S7, ESI†). The fundamental “lantern” [V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3] units are made up of [PO4] tetrahedra and [MeO6] octahedra through corner-sharing, as illustrated in Fig. 1d. V/Cr/Mn/Fe/Al/Mg atoms occupy the same 12c positions at random. As a result, a 3D open-framework structure with high Na+ conductivity was created, which is advantageous for high-rate performances. Because of the lower bonding force between Na2 cations and O,17 it is easier to extract Na2 with eight coordination or insert Na+.17 With a high theoretical capacity of 176.2 mA h g−1, which exceeds that of other NASICONs, one can expect that three sodium ions can insert/take up to/from Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 hinged on V5+/V4+, V4+/V3+ and V3+/V2+ redox processes.15
c). Both the structural details and the comprehensive atomic occupation were obtained by Rietveld refinement (Fig. 2a and Fig. S17b and Tables S7 and S8, ESI†). In Fig. 2a, HE-V featured a NASICON structure by a low Rwp value of 3.41% (atomic occupation and lattice properties are presented in Table S7, ESI†). The fundamental “lantern” [V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3] units are made up of [PO4] tetrahedra and [MeO6] octahedra through corner-sharing, as illustrated in Fig. 1d. V/Cr/Mn/Fe/Al/Mg atoms occupy the same 12c positions at random. As a result, a 3D open-framework structure with high Na+ conductivity was created, which is advantageous for high-rate performances. Because of the lower bonding force between Na2 cations and O,17 it is easier to extract Na2 with eight coordination or insert Na+.17 With a high theoretical capacity of 176.2 mA h g−1, which exceeds that of other NASICONs, one can expect that three sodium ions can insert/take up to/from Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 hinged on V5+/V4+, V4+/V3+ and V3+/V2+ redox processes.15
 | ||
Fig. 2 Material characterization of HE-V. (a) XRD Rietveld refinement results. (b) and (c) TEM and HRTEM images. (d)–(h) Aberration-corrected STEM images viewed from the (d) [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10], (e) [2 10], (e) [2![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 1], (g) [4], and (h) [1] crystallographic directions. (f) EDS mapping images (scale bar: 50 nm). 1], (g) [4], and (h) [1] crystallographic directions. (f) EDS mapping images (scale bar: 50 nm). | ||
The site occupancy of the dopant elements in the HE-V framework was carefully considered to understand their role in structural stability and electrochemical behavior. All five dopants (Cr3+,40 Mn2+,41 Fe3+,42 Al3+,43 and Mg2+ (ref. 44)) were intentionally introduced to substitute vanadium at the 12c Wyckoff position, which corresponds to the octahedrally coordinated transition metal site in the NASICON structure. Rietveld refinement of the XRD data was performed using a structural model that assigns all dopants to the 12c site. The refined model achieved excellent agreement with the experimental pattern (Rwp = 3.41%), and no significant residuals or misfits were observed, indicating that the assumption of exclusive dopant occupation at the 12c site is well supported. Furthermore, the ionic radii and coordination preferences of the dopants are consistent with the six-fold octahedral environment of the 12c site; substitution at non-transition-metal sites (e.g., Na or P positions) would likely lead to phase instability or symmetry disruption, which was not observed. This assignment is further supported by previous reports on individually doped NVP systems, in which Cr3+,40 Mn2+,41 Fe3+,42 Al3+,43 and Mg2+(ref. 44) have all been experimentally verified to occupy the vanadium site (12c). Taken together, these findings support the conclusion that the dopants are incorporated exclusively at the 12c transition metal site in the HE-V structure.
To further observe the morphologies, transmission electron microscopy (TEM) was used. Both samples shown in Fig. 2b and c and Fig. S18a (ESI†) had a thin film of amorphous carbon covering them (5 nm in thickness). This is explained by the pyrolysis of organic molecules during the calcination process, which is advantageous to improve electronic conductivities. Additionally, a lattice fringe of 0.38 nm is indexed to the HE-V lattice plane (23) (Fig. 2c). To understand the sodium storage mechanism, aberration-corrected STEM, in Fig. 2d–h, was used to acquire a direct image of the atomic-scale arrangement in the crystal structure of HE-V. Since the atomic displacements in a single direction are readily missed and the separated columns of Na, P, O, and V/Cr/Mn/Fe/Al/Mg ions are aligned in various crystal directions, several projections were used for observation. V and Cr/Mn/Fe/Al/Mg are almost indistinguishable in STEM images because they have the same location in HE-V. Since the atomic number affects the intensity of STEM images, heavy elements (Cr/Mn/Fe/Al/Mg and P) dominate the image contrast. Due to the comparatively low atomic number, the contrast between Na and O in STEM is much weaker and not very visible in most projections. As presented in Fig. 2d–h, it is evident that the framework structure has void space for Na+ migration along various directions in the atomic alignment. Na+ transportation channels are clearly visible in all four directions, and because the channel's dimension is greater than the Na+ ion's diameter, Na+ ions can move through these channels quickly, which enables rapid rate performance and is in good agreement with NASICON's characteristics. Fig. 2f (HE-V) and Fig. S18b (ESI†) (NVP) show the homogeneous elemental distribution of V (Cr, Mn, Fe, Al, and Mg) Na, O, C, and P in both samples, according to energy dispersive spectrometry (EDS) mapping results. The chemical formula was further determined by ICP-OES findings. The atomic ratios were determined to be Na(3.154), V(1.453), Cr(0.103), Mn(0.101), Fe(0.099), Al(0.098), Mg(0.100), and P(2.960) for HE-V, as shown in Fig. S20 (ESI†), which is very consistent with the designed ratio of Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3.
The thermal gravimetric analysis (TGA) results indicate that the amount of carbon was calculated to be 9.6% for HE-V and 9.4% for NVP, as shown in Fig. S21 (ESI†). The results from X-ray photoelectron spectroscopy (XPS) show that V3+52 (Cr3+,53 Mn2+,54 Fe2+/Fe3+,55,56 Al3+,57 and Mg2+ (ref. 58)) and C–C/C–O/O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O are present in HE-V (Fig. S22, ESI†) and NVP (Fig. S23, ESI†). Furthermore, the virtually similar Fourier transform infrared (FT-IR) spectra are presented in Fig. S24 (ESI†). At approximately 620 and 1000 cm−1, symmetric and asymmetric stretching/bending vibration signals of [MeO6] octahedra and [PO4] tetrahedra were detected.46,59,60 All evidence demonstrates the successful preparation of both samples.
O are present in HE-V (Fig. S22, ESI†) and NVP (Fig. S23, ESI†). Furthermore, the virtually similar Fourier transform infrared (FT-IR) spectra are presented in Fig. S24 (ESI†). At approximately 620 and 1000 cm−1, symmetric and asymmetric stretching/bending vibration signals of [MeO6] octahedra and [PO4] tetrahedra were detected.46,59,60 All evidence demonstrates the successful preparation of both samples.
Electrochemical performance
HE-V/NVP was then coupled with Na anodes to assemble the CR2032 coin cell for performance measurements. Three apparent voltage platforms were shown by HE-V in Fig. 3a and b: 1.67 V (V3+/V2+), 3.43 V (V4+/V3+), and 4.03 V (V5+/V4+). As a result, at 0.2C, a high capacity of 170 mA h g−1 (almost the theoretical value) was produced, surpassing that of the majority of NASICON cathodes.16–18,22,26,41,61,62 Furthermore, this verifies that HE-V's three-electron redox reactions were triggered. Three apparent reduction/oxidation peaks of HE-V are identified by the dQ/dV curves in Fig. 3b, indicating good kinetics and consecutive transformations of Na1.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 ↔ Na2.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 ↔ Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 ↔ Na4.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3, which will be described in depth later. In addition, the HE-V's enhanced electronic conductivity and short Na+ diffusion length enhanced the redox kinetics, which resulted in more symmetric dQ/dV profiles (Fig. 3b). On the other hand, a lower capacity of 155.2 mA h g−1 with greater potential differences of dQ/dV peaks was produced in NVP due to the absence of the high-voltage platform of V5+/V4+ (Fig. 3a and b). Moreover, capacities within different voltage areas in Fig. 3c revealed that mid (3–3.6 V) to high (>3.6 V) voltage ranges showed higher capacities and larger capacity fractions in total for HE-V in contrast to NVP. It verified higher output voltage and increased capacity resulting from this strategy's activation of the V5+/V4+ redox pair. Fig. 3d also shows that HE-V has a higher energy density than other polyanionic compounds.16–18,22,26,41,61,62 | ||
| Fig. 3 Electrochemical performances. (a) Charge/discharge curves at 0.2C (1C = 100 mA g−1); (b) corresponding dQ/dV curves and (c) the summary of capacities within different voltage ranges. (d) A comparison among HE-V and other reported materials from the literature. (e) Rate properties. (f) Radar plot of the comparison in terms of specific capacity, voltage, CE, energy density, cycling performance, cost-effectiveness, and rate capability. Cycling capabilities at (g) 0.2C and (h) 50C, respectively. (i) Voltage curves of the HC anode and the HE-V cathode. (j) Cycling stability of the HC‖HE-V full cell at 1C. | ||
As shown in Fig. 3e–g and Fig. S26 (ESI†), HE-V demonstrates noticeably improved rate capability (up to 50C) and cycling performance (at 0.2C). Remarkably, as demonstrated in Fig. 3h, HE-V obtained record-breaking cycling stability at an ultrahigh current density of 50C: a high capacity retention of 95.2% over the first 5000 cycles and a capacity retention of 93.3% for the latter 5000 cycles (i.e., 88.8% throughout 10000 cycles), surpassing the earlier works (Fig. S27, ESI†).17,21–24,41,43,60 For example, notably, in our previous contribution,24 our group employed a hybrid strategy by Cr3+ doping (from Na3V2(PO4)3 to Na3Cr0.5V1.5(PO4)3) and reduced graphene oxide coating to modify the V-based NASICON with improved battery performance, but it only realized a limited cycling capability (∼62% capacity retention after 1000 cycles at 20C). This demonstrated once more how effective this configurational entropy engineering strategy is in making these materials (Fig. 3f).
To assess the practical potential of HE-V, the HE-V cathode was coupled with a commercial hard carbon (HC) anode to assemble full cells (HC‖HE-V). The HC electrodes were initially pre-cycled against Na metal anodes at 25 mA g−1 (Fig. S28, ESI†) to prevent the cathode's Na+ loss from the SEI layer formation, consistent with previous studies.20,63,64 The pre-sodiated HC was disassembled at the discharge state following five cycles. With respect to Na+/Na, HC delivered a reversible capacity of ∼300 mA h g−1 and a low voltage platform of ∼0.1 V (Fig. S28, ESI†). Fig. S29a (ESI†) displays the galvanostatic charge–discharge patterns of HC‖HE-V at 0.2C within the voltage window of 1.5–4.1 V. As a result, there were three unique, characteristic voltage platforms with a high initial capacity of 170.8 mA h g−1. Consequently, a high energy density of 485.9 W h kg−1 was reached. Furthermore, good rate capabilities from 0.2C to 5C and an exceptional cycling stability at 1C were obtained (Fig. 3j and Fig. S29b, ESI†). The above described full-cell properties provided additional evidence for the superiority of this technique.
Structural evolution and charge compensation analysis
Operando XRD was carried out at 0.1C to further elucidate Na+ storage mechanisms and structural alterations (left side of Fig. 4a and b; Fig. 4c). The robust structure and highly reversible electrochemical performance are demonstrated by the main reflections of (10), (104), (20), (23), (006), (20![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) ), (31), (26), and (108), all of which had extremely reversible evolution during cycling. At first, a rhombohedral Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 phase with the space group Rc was used to index all peaks. All reflections progressively shifted positively after the initial charge, indicating a solid-solution reaction and structural contraction, with the exception of (20), (31), (26), and (108), which disappeared after additional charging. A biphasic reaction was determined by the peak vanishment, per earlier research.17,25,60,63 Similar behavior was observed in the symmetrical discharge procedure that follows. Lower potentials caused all reflections moving to smaller angles and the (26) peak reappearance, consistent with a coupled biphasic and monophasic process. The good structural reversibility was confirmed by the reflections shifting back to their original positions (Fig. 4c). As a result, these findings showed that HE-V cycles through mixed biphasic and monophasic (solid-solution) mechanisms; solid-solution reactions seem to control the entire process,45,65 which is advantageous for the quick diffusion of Na+ and good electrochemical reversibility.
), (31), (26), and (108), all of which had extremely reversible evolution during cycling. At first, a rhombohedral Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 phase with the space group Rc was used to index all peaks. All reflections progressively shifted positively after the initial charge, indicating a solid-solution reaction and structural contraction, with the exception of (20), (31), (26), and (108), which disappeared after additional charging. A biphasic reaction was determined by the peak vanishment, per earlier research.17,25,60,63 Similar behavior was observed in the symmetrical discharge procedure that follows. Lower potentials caused all reflections moving to smaller angles and the (26) peak reappearance, consistent with a coupled biphasic and monophasic process. The good structural reversibility was confirmed by the reflections shifting back to their original positions (Fig. 4c). As a result, these findings showed that HE-V cycles through mixed biphasic and monophasic (solid-solution) mechanisms; solid-solution reactions seem to control the entire process,45,65 which is advantageous for the quick diffusion of Na+ and good electrochemical reversibility.
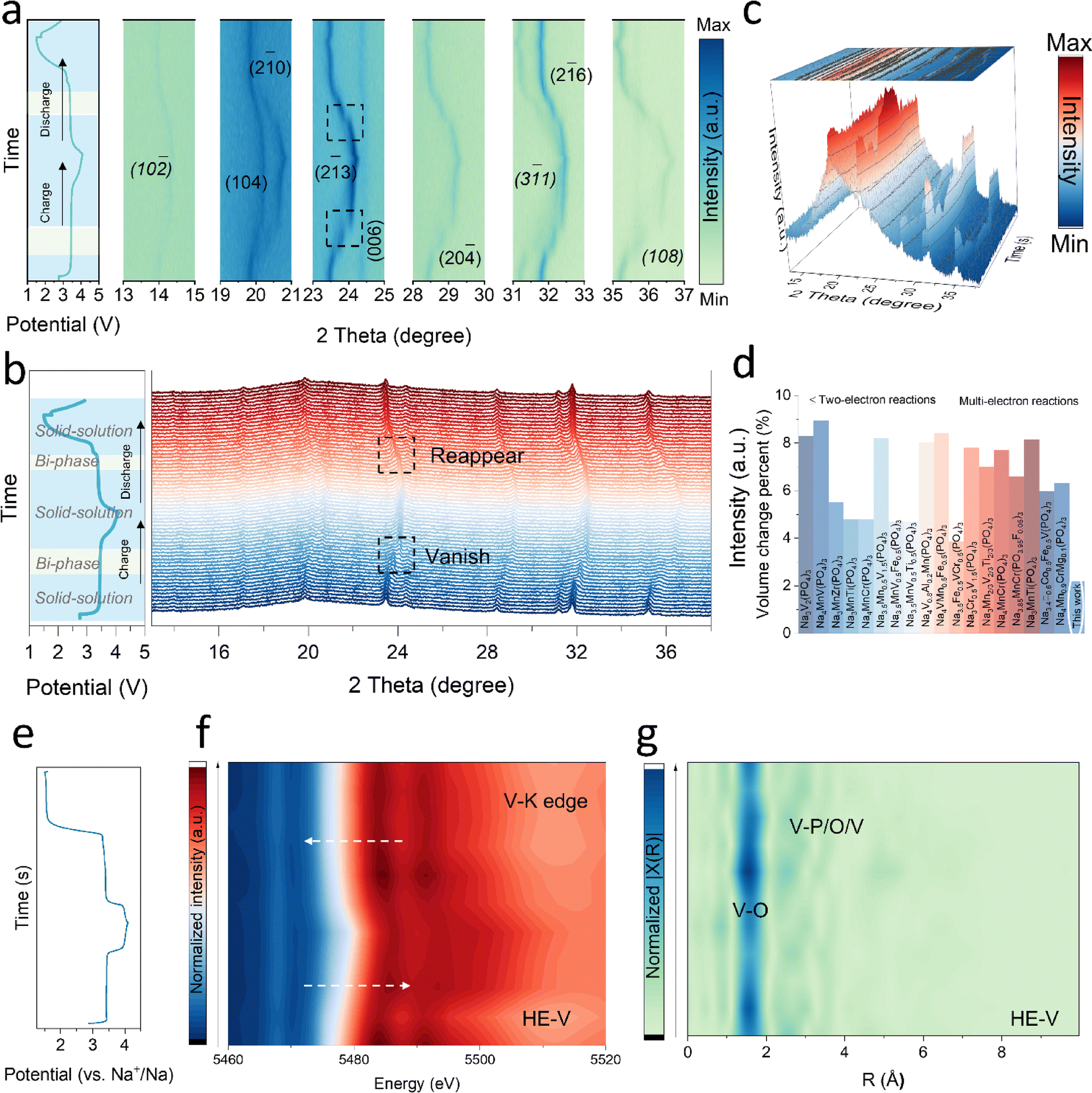 | ||
| Fig. 4 Structural evolution and charge compensation analysis. (a)–(c) In situ XRD patterns of HE-V. (d) A volumetric change comparison of HE-V and reported NASICON cathodes upon battery operation. (e) The galvanostatic charge–discharge curve and (f) XANES spectra at different charging states as a function of the V K-edge of the HE-V electrode (2D contour plot) and (g) corresponding k2-weighted Fourier transform magnitudes of HE-V. | ||
To further aid in understanding the structure alterations, the changes in lattice parameters were gathered and are presented in Fig. S30 (ESI†). It is evident that throughout the first charging process, the a (=b)-axis and the c-axis decreased, leading to a declining trend in v (volume). The a-axis grew when discharging, but the c-axis barely changed. As a result, the c-axis showed an increasing tendency that was similar to the a-axis. Consequently, HE-V showed a minor volume change of 2.2%, which is significantly less than those of common NASICON materials for SIBs (Fig. 4d).62,63 The robust NASICON framework structure is one of the reasons for HE-V's exceptional cyclabilities. The structural evolution of HE-V can be summed up as follows based on the discussions above: the initial two Na+ ions were extracted, and the pristine state changed to Na1.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3; three Na+ ions were inserted during the next cycling to create Na4.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3, which is consistent with the multi-electron electrochemical characteristics.
Next, XPS was used to investigate the HE-V's charge compensation mechanisms at different voltages (Fig. S31, ESI†). It is assumed that the changes in valence states of V occur with the transitions of Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 → Na1.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 and Na1.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 ↔ Na2.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 ↔ Na3.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3 ↔ Na4.2V1.5Cr0.1Mn0.1Fe0.1Al0.1Mg0.1(PO4)3. The initial peak of V 2p3/2 was at 516.8 eV (V3+), as shown in Fig. S31b (ESI†).52 It had a positive shift to 517.08 eV (V4+) when charged to 3.72 V.23,66 Subsequently, the peak of V 2p3/2 developed to 517.40 eV (V5+) when charged to 4.1 V.67 The valence states of V shift to +2 when discharged to 1.5 V, with the peak of V 2p3/2 appearing at 516.50 eV.68 In Fig. S32 (ESI†), the XPS spectra demonstrate that the oxidation states of Cr, Al, Fe, and Mg remain essentially unchanged during cycling, confirming their electrochemical inactivity throughout the charge/discharge process. A slight shift in the Mn signal suggests a potential minor oxidation from Mn2+ to Mn3+/Mn4+; however, given the low doping concentration, its contribution to capacity is negligible. These results support the conclusion that the dopants primarily serve to enhance the structural robustness and configurational entropy of the NASICON framework rather than acting as active redox centers. This finding further reinforces the entropy-engineering strategy by confirming that capacity is predominantly delivered by vanadium redox reactions.
Furthermore, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) investigations were carried out to look more closely at the charge compensation mechanisms of HE-V. Fig. 4f and Fig. S33 (ESI†) display the XANES spectra in the V K-edge region. During de-sodiation, the V K-edge spectra shift toward the high-energy side, indicating an increase in the oxidation state of V. Peak intensities in the V K-edge region shrank toward the end of charge, which might be attributed to both the shortening of V–O bonds and electron loss in the VO6 octahedra.45 As a result, during the subsequent discharge operation, the V K-edge spectra returned to the lower energy area. The strong V–O hybridization is suggested by the pre-edge region's apparent pre-peaks. The discharge capacity is significantly more than the charge capacity, as indicated by the galvanostatic charge–discharge curve in the left of Fig. 4e. As a result, more Na+ ions are intercalated into the structure upon discharge, resulting in V2+/V3+ redox reactions.40,45 The reduction of V3+ to V2+ and the additional Na+ ions may cause VO6 octahedra to be severely distorted, or they may even transform the octahedral symmetry into a tetrahedral symmetry that is distinguished by strong V–O hybridization.
The V K-edge EXAFS spectra's Fourier-transformed (FT) magnitude plot was also acquired. With radii of approximately 1.4 and 2.5 Å, two prominent peaks are shown in the right of Fig. 4g, which correspond to the V–O bond and the V–P/O/V bond, respectively. It should be mentioned that because these FT spectra were not phase-corrected, these distances do not correspond to the real bond lengths. The absence of a noticeable alteration in the V–O bond throughout cycling suggests that the octahedral symmetry was effectively preserved in the initial cycle. Still, there has been a noticeable change in the peak intensities. Due to the shorter atom distance and enhanced electrostatic repulsion during deep sodium extraction, the peak strengths of the V–O bond and the V–P/O/V bond rose at the final part of the charge process. Furthermore, even with deep sodium insertion, there is essentially no V local structural change, as evidenced by the excellent overlap between the EXAFS spectra after one cycle and that of the pristine sample. This agrees with the operando XRD findings that were previously presented.
COMSOL simulation and postmortem analysis
Based on the variation in the ionic diffusion coefficient and the diffusion barrier and volumetric change upon cycling, a multi-physical simulation was run to visualize the Na+ ion concentration and the associated stress field for both electrodes. Even at a highly desodiated state (90%), partial Na+ ions were still retained on the cathode surface, exhibiting a clearly heterogeneous concentration distribution in NVP (Fig. 5a). This could be explained by the limited Na+-ion mass transfer resulting from the intrinsic inferior ionic diffusion coefficient. However, the enhanced diffusion kinetics, particularly in 90% SOC, nearly non-disparity inside particles, and the surface allowed for HE-V to obtain a uniform Na+ ion concentration (Fig. 5a). As seen in Fig. 5b, the homogenous Na+-ion concentration field would balance the stress field's distribution and reduce strain formation. We then performed postmortem analysis on the cycled HE-V cathode. | ||
| Fig. 5 COMSOL simulation and postmortem analysis. (a) Na+ distribution and (b) stress field analysis at different depths of discharge (DOD) states based on the COMSOL platform. Postmortem analysis of the cycled HE-V: (c) TEM, (d) HRTEM, (e) FFT, and (f) aberration-corrected STEM image. (g) EDS mapping images. | ||
The TEM image in Fig. 5c shows that HE-V has maintained its intact morphology after cycling and Fig. 5d confirms a thin film of amorphous carbon covering it with a thickness of 5 nm. Additionally, a lattice fringe of 0.37 nm is indexed to the HE-V lattice plane (23). (42), (20), and (29) planes can be clearly identified in Fig. 5e as well. Atomic scale HRTEM images in Fig. 5f further confirmed the good crystallinities of HE-V even after cycling. Fig. 5g shows the homogeneous elemental distribution of V, Cr, Mn, Fe, Al, Mg, Na, O, C, and P in HE-V, according to EDS mapping results. In sharp contrast, after cycling, many cracks and nonuniform elemental distribution can be clearly found in the NVP particles (Fig. S34, ESI†) due to the large local lattice strain and lattice defects. The above results further confirm the merits of this configurational entropy engineering strategy for NASICONs.
Conclusions
In summary, in response to the issues of NASICONs with low capacity, poor capacity retention at high current densities, and noticeable volumetric variation, this work proposed a configurational entropy engineering strategy by introducing five other dopants at the 12c site for improving the NASICON material (HE-V) that demonstrated high energy-density Na+ storage capabilities and “low-strain” characteristics. It delivered a specific capacity of 170 mA h g−1, based on a multi-electron reaction from V5+/V4+, V4+/V3+, and V3+/V2+ redox couples. Benefiting from the synergistical effect of all dopants, an almost zero volumetric change was revealed with the dominant solid-solution reaction mechanism and a strengthened neighbor chemical bonding around the redox center was also identified, which led to reduced lattice defects and local strain-induced cracks. A significantly reduced energy gap contributed to an improved electronic conductivity. Consequently, HE-V could stably run 10000 cycles at 50C, which greatly facilitates the application of NASICONs. This work provides a novel strategy for boosting the structural stability of NASICON cathodes for energy-dense and long-lasting sodium-ion batteries.
Author contributions
Conceptualization: W. Z; methodology: W. Z. and L. H; software: Z. X., H. Q., and G. Z; validation: W. Z., L. H., J. L., R. Y., Z. X., Q. Z., and J. L; formal analysis: W. Z., L. H., J. L., and R. Y; investigation: W. Z., L. H., J. L., R. Y., and Y. W; resources: Z. X., Y. L., G. Z., G. H., and I. P. P; data curation: W. Z., L. H., J. L., R. Y., and Z. X; writing – original draft: W. Z; writing – reviewing and editing: W. Z., L. H., J. L., R. Y., Z. X., Q. Z., Y. L., G. Z., G. H., and I. P. P; visualization: R. Y., Z. X., and H. Q; supervision: Y. L., G. Z., G. H., and I. P. P; project administration: W. Z. and G. H; and funding acquisition: W. Z., J. L., R. Y., Y. L., G. Z., and G. H.Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors would like to acknowledge the Engineering and Physical Sciences Research Council, United Kingdom (EPSRC, EP/L015862/1, and EP/V027433/3), the UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding guarantee (101077226; EP/Y008707/1), EPSRC Centre for Doctoral Training in Molecular Modelling and Materials Science (EP/L015862/1), Open Foundation of the State Key Laboratory of Silicate Materials for Architectures at WUT (No. SYSJJ2020-04), the Royal Society (RGS/R1/211080; IEC/NSFC/201261), and STFC Batteries Network (ST/R006873/1) for funding support. This work forms part of the NMS battery project: Metrology and Standards for the UK Battery Value Chain of the National Measurement System of the UK Department of Business, Energy and Industrial strategy. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by the Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This work was supported by the Fundamental Research Funds for the Central Universities (WUT: 2019III012GX and 2020III002GX). This work is partially supported by the High Performance Computing Platform of Nanjing University of Aeronautics and Astronautics. This S/TEM work was performed at the Nanostructure Research Center (NRC), which was supported by the State Key Laboratory of Advanced Technology for Materials Synthesis and Processing and the State Key Laboratory of Silicate Materials for Architectures (all of the laboratories are at the Wuhan University of Technology).References
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- W. Zhang, Y. Dai, R. Chen, Z. Xu, J. Li, W. Zong, H. Li, Z. Li, Z. Zhang, J. Zhu, F. Guo, X. Gao, Z. Du, J. Chen, T. Wang, G. He and I. Parkin, Angew. Chem., Int. Ed., 2023, 62, e202212695 CrossRef CAS PubMed.
- Y. Dai, C. Zhang, W. Zhang, L. Cui, C. Ye, X. Hong, J. Li, R. Chen, W. Zong, X. Gao, J. Zhu, P. Jiang, Q. An, D. J. L. Brett, I. P. Parkin, G. He and L. Mai, Angew. Chem., Int. Ed., 2023, 62, e202301192 CrossRef CAS PubMed.
- R. Chen, W. Zhang, C. Guan, Y. Zhou, I. Gilmore, H. Tang, Z. Zhang, H. Dong, Y. Dai, Z. Du, X. Gao, W. Zong, Y. Xu, P. Jiang, J. Liu, F. Zhao, J. Li, X. Wang and G. He, Angew. Chem., Int. Ed., 2024, 63, e202401987 CrossRef CAS PubMed.
- Q. Fu, W. Zhang, X. Liu, Y. Liu, Z. Lei, M. Zhang, H. Qu, X. Xiao, X. Zhong, Z. Liu, P. Qin, J. Yang and G. Zhou, J. Am. Chem. Soc., 2024, 146, 34950–34961 CrossRef CAS PubMed.
- X. Wu, W. Zhang, H. Qu, C. Guan, C. Li, G. Lu, C. Chang, Z. Lao, Y. Zhu, L. Nie and G. Zhou, Energy Environ. Sci., 2025, 18, 1835–1846 RSC.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- H. Li, W. Zhang, K. Sun, J. Guo, K. Yuan, J. Fu, T. Zhang, X. Zhang, H. Long, Z. Zhang, Y. Lai and H. Sun, Adv. Energy Mater., 2021, 11, 2100867 CrossRef CAS.
- J.-M. Tarascon, Joule, 2020, 4, 1616–1620 CrossRef.
- A. Rudola, R. Sayers, C. J. Wright and J. Barker, Nat. Energy, 2023, 8, 215–218 CrossRef.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- S. Xu, H. Dong, D. Yang, C. Wu, Y. Yao, X. Rui, S. Chou and Y. Yu, ACS Cent. Sci., 2023, 9, 2012–2035 CrossRef CAS PubMed.
- Y. Liu, X. Rong, R. Bai, R. Xiao, C. Xu, C. Zhang, J. Xu, W. Yin, Q. Zhang, X. Liang, Y. Lu, J. Zhao, L. Chen and Y.-S. Hu, Nat. Energy, 2023, 8, 1088–1096 CrossRef CAS.
- Y. Liu, X. Rong, F. Xie, Y. Lu, J. Zhao, L. Chen and Y.-S. Hu, Mater. Futures, 2023, 2, 023502 CrossRef CAS.
- Z. Jian, W. Han, X. Lu, H. Yang, Y.-S. Hu, J. Zhou, Z. Zhou, J. Li, W. Chen, D. Chen and L. Chen, Adv. Energy Mater., 2013, 3, 156–160 CrossRef CAS.
- W. Zhou, L. Xue, X. Lü, H. Gao, Y. Li, S. Xin, G. Fu, Z. Cui, Y. Zhu and J. B. Goodenough, Nano Lett., 2016, 16, 7836–7841 CrossRef CAS PubMed.
- H. Gao, Y. Li, K. Park and J. B. Goodenough, Chem. Mater., 2016, 28, 6553–6559 CrossRef CAS.
- J. Wang, Y. Wang, D. H. Seo, T. Shi, S. Chen, Y. Tian, H. Kim and G. Ceder, Adv. Energy Mater., 2020, 10, 1903968 CrossRef CAS.
- W. Zhang, H. Li, Z. Zhang, M. Xu, Y. Lai and S.-L. Chou, Small, 2020, 16, 2001524 CrossRef CAS PubMed.
- W. Zhang, Y. Wu, Y. Dai, Z. Xu, L. He, Z. Li, S. Li, R. Chen, X. Gao, W. Zong, F. Guo, J. Zhu, H. Dong, J. Li, C. Ye, S. Li, F. Wu, Z. Zhang, G. He, Y. Lai and I. P. Parkin, Chem. Sci., 2023, 14, 8662–8671 RSC.
- H. Gao, I. D. Seymour, S. Xin, L. Xue, G. Henkelman and J. B. Goodenough, J. Am. Chem. Soc., 2018, 140, 18192–18199 CrossRef CAS PubMed.
- W. Zhang, Z. Xu, H. Li, M. Xu, S. Wang, Z. Li, A. Wang, L. Zhang, L. He, S. Li, B. Zhu, Z. Zhang and Y. Lai, Chem. Eng. J., 2022, 433, 133542 CrossRef CAS.
- W. Zhang, Y. Wu, Z. Xu, H. Li, M. Xu, J. Li, Y. Dai, W. Zong, R. Chen, L. He, Z. Zhang, D. J. L. Brett, G. He, Y. Lai and I. P. Parkin, Adv. Energy Mater., 2022, 12, 2201065 CrossRef CAS.
- Q. Wang, C. Ling, J. Li, H. Gao, Z. Wang and H. Jin, Chem. Eng. J., 2021, 425, 130680 CrossRef CAS.
- C. Xu, J. Zhao, E. Wang, X. Liu, X. Shen, X. Rong, Q. Zheng, G. Ren, N. Zhang, X. Liu, X. Guo, C. Yang, H. Liu, B. Zhong and Y. S. Hu, Adv. Energy Mater., 2021, 11, 2100729 CrossRef CAS.
- Y. Yao, Q. Dong, A. Brozena, J. Luo, J. Miao, M. Chi, C. Wang, I. G. Kevrekidis, Z. J. Ren, J. Greeley, G. Wang, A. Anapolsky and L. Hu, Science, 2022, 376, eabn3103 CrossRef CAS PubMed.
- Z. Lun, B. Ouyang, D. H. Kwon, Y. Ha, E. E. Foley, T. Y. Huang, Z. Cai, H. Kim, M. Balasubramanian, Y. Sun, J. Huang, Y. Tian, H. Kim, B. D. McCloskey, W. Yang, R. J. Clement, H. Ji and G. Ceder, Nat. Mater., 2021, 20, 214–221 CrossRef CAS PubMed.
- R. Zhang, C. Wang, P. Zou, R. Lin, L. Ma, T. Li, I.-h Hwang, W. Xu, C. Sun, S. Trask and H. L. Xin, Nat. Energy, 2023, 8, 695–702 CrossRef CAS.
- W. Zeng, F. Xia, J. Wang, J. Yang, H. Peng, W. Shu, Q. Li, H. Wang, G. Wang, S. Mu and J. Wu, Nat. Commun., 2024, 15, 7371 CrossRef CAS PubMed.
- B. Yang, Y. Zhang, H. Pan, W. Si, Q. Zhang, Z. Shen, Y. Yu, S. Lan, F. Meng, Y. Liu, H. Huang, J. He, L. Gu, S. Zhang, L.-Q. Chen, J. Zhu, C.-W. Nan and Y.-H. Lin, Nat. Mater., 2022, 21, 1074–1080 CrossRef CAS PubMed.
- C. Zhao, F. Ding, Y. Lu, L. Chen and Y. S. Hu, Angew. Chem., Int. Ed., 2020, 59, 264–269 CrossRef CAS PubMed.
- F. Ding, C. Zhao, D. Xiao, X. Rong, H. Wang, Y. Li, Y. Yang, Y. Lu and Y. S. Hu, J. Am. Chem. Soc., 2022, 144, 8286–8295 CrossRef CAS PubMed.
- F. Ding, P. Ji, Z. Han, X. Hou, Y. Yang, Z. Hu, Y. Niu, Y. Liu, J. Zhang, X. Rong, Y. Lu, H. Mao, D. Su, L. Chen and Y.-S. Hu, Nat. Energy, 2024, 9, 1529–1539 CrossRef CAS.
- F. Ding, H. Wang, Q. Zhang, L. Zheng, H. Guo, P. Yu, N. Zhang, Q. Guo, F. Xie, R. Dang, X. Rong, Y. Lu, R. Xiao, L. Chen and Y. S. Hu, J. Am. Chem. Soc., 2023, 145, 13592–13602 CrossRef CAS PubMed.
- H. Wang, X. Gao, S. Zhang, Y. Mei, L. Ni, J. Gao, H. Liu, N. Hong, B. Zhang, F. Zhu, W. Deng, G. Zou, H. Hou, X. Y. Cao, H. Chen and X. Ji, ACS Nano, 2023, 17, 12530–12543 CrossRef CAS PubMed.
- P. Senguttuvan, G. Rousse, M. E. Arroyo y de Dompablo, H. Vezin, J. M. Tarascon and M. R. Palacin, J. Am. Chem. Soc., 2013, 135, 3897–3903 CrossRef CAS PubMed.
- S. Park, J.-N. Chotard, D. Carlier, F. Fauth, A. Iadecola, C. Masquelier and L. Croguennec, Chem. Mater., 2023, 35, 3181–3195 CrossRef CAS.
- B. Ouyang and Y. Zeng, Nat. Commun., 2024, 15, 973 CrossRef CAS PubMed.
- Y. Zhao, X. Gao, H. Gao, H. Jin and J. B. Goodenough, Adv. Funct. Mater., 2020, 30, 1908680 CrossRef CAS.
- J. Zhang, X. Zhao, Y. Song, Q. Li, Y. Liu, J. Chen and X. Xing, Energy Storage Mater., 2019, 23, 25–34 CrossRef.
- C. Xu, J. Zhao, Y. A. Wang, W. Hua, Q. fu, X. Liang, X. Rong, Q. Zhang, X. Guo, C. Yang, H. Liu, B. Zhong and Y. S. Hu, Adv. Energy Mater., 2022, 12, 2200966 CrossRef CAS.
- F. Lalère, V. Seznec, M. Courty, R. David, J. N. Chotard and C. Masquelier, J. Mater. Chem. A, 2015, 3, 16198–16205 RSC.
- A. Inoishi, Y. Yoshioka, L. Zhao, A. Kitajou and S. Okada, ChemElectroChem, 2017, 4, 2755–2759 CrossRef CAS.
- M. Chen, W. Hua, J. Xiao, J. Zhang, V. W. Lau, M. Park, G. H. Lee, S. Lee, W. Wang, J. Peng, L. Fang, L. Zhou, C. K. Chang, Y. Yamauchi, S. Chou and Y. M. Kang, J. Am. Chem. Soc., 2021, 143, 18091–18102 CrossRef CAS PubMed.
- Y. Zhou, G. Xu, J. Lin, Y. Zhang, G. Fang, J. Zhou, X. Cao and S. Liang, Adv. Mater., 2023, 35, 2304428 CrossRef CAS PubMed.
- M. Chen, W. Hua, J. Xiao, D. Cortie, X. Guo, E. Wang, Q. Gu, Z. Hu, S. Indris, X. Wang, S. Chou and S. Dou, Angew. Chem., Int. Ed., 2020, 132, 2470–2477 CrossRef.
- J. Zhang, Y. Yan, X. Wang, Y. Cui, Z. Zhang, S. Wang, Z. Xie, P. Yan and W. Chen, Nat. Commun., 2023, 14, 3701 CrossRef CAS PubMed.
- R. Dronskowski and P. E. Bloechl, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- Y. You, S. Xin, H. Y. Asl, W. Li, P.-F. Wang, Y.-G. Guo and A. Manthiram, Chem, 2018, 4, 2124–2139 CAS.
- C. Wang, L. Liu, S. Zhao, Y. Liu, Y. Yang, H. Yu, S. Lee, G. H. Lee, Y. M. Kang, R. Liu, F. Li and J. Chen, Nat. Commun., 2021, 12, 2256 CrossRef CAS PubMed.
- F. E. M. N. K. Nag, J. Catal., 1990, 124, 127–132 CrossRef.
- R. M. L. T. P. Moffat and R. R. Ruf, Electrochim. Acta, 1995, 40, 1723–1734 CrossRef.
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, J. Am. Chem. Soc., 1991, 113, 855–861 CrossRef CAS.
- J. Lian, Y. He, N. Li, P. Liu, Z. Liu and Q. Liu, Inorg. Chem., 2021, 60, 1893–1901 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- K. Arata and M. Hino, Appl. Catal., 1990, 59, 197–204 CrossRef CAS.
- H. Seyama and M. Soma, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 237–248 RSC.
- D. Wang, Z. Wei, Y. Lin, N. Chen, Y. Gao, G. Chen, L. Song and F. Du, J. Mater. Chem. A, 2019, 7, 20604–20613 RSC.
- H. Li, M. Xu, C. Gao, W. Zhang, Z. Zhang, Y. Lai and L. Jiao, Energy Storage Mater., 2020, 26, 325–333 CrossRef.
- X. Shen, Y. Su, S. He, Y. Li, L. Xu, N. Yang, Y. Liao, M. Wang and F. Wu, J. Mater. Chem. A, 2023, 11, 16860–16870 RSC.
- C. Xu, R. Xiao, J. Zhao, F. Ding, Y. Yang, X. Rong, X. Guo, C. Yang, H. Liu, B. Zhong and Y.-S. Hu, ACS Energy Lett., 2022, 7, 97–107 CrossRef CAS.
- J. Zhang, Y. Liu, X. Zhao, L. He, H. Liu, Y. Song, S. Sun, Q. Li, X. Xing and J. Chen, Adv. Mater., 2020, 32, 1906348 CrossRef CAS PubMed.
- Q. Shi, R. Qi, X. Feng, J. Wang, Y. Li, Z. Yao, X. Wang, Q. Li, X. Lu, J. Zhang and Y. Zhao, Nat. Commun., 2022, 13, 3205 CrossRef PubMed.
- T. Zhu, P. Hu, X. Wang, Z. Liu, W. Luo, K. A. Owusu, W. Cao, C. Shi, J. Li, L. Zhou and L. Mai, Adv. Energy Mater., 2019, 9, 1803436 CrossRef.
- A. E. E. Bailón-García, F. Ribeiro, C. Henriques, A. F. Pérez-Cadenas, F. Carrasco-Marín and F. J. Maldonado-Hódar, Carbon, 2020, 156, 194–204 CrossRef.
- S. F. Geoffrey and C. Bond, Appl. Catal., 1989, 46, 89–102 CrossRef.
- J. G. Chen, C. M. Kirn, B. Frühberger, B. D. DeVries and M. S. Touvelle, Surf. Sci., 1994, 321, 145–155 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00877h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |