
Evolving mineralogy and reactivity of hematite-coated sands during reduction of 4-chloronitrobenzene by Fe(II) in flow-through reactors†
Celina M.
Harris
a,
Adel
Soroush
b,
Alanna M.
Hildebrandt
 a,
Kamilah Y.
Amen
a,
Louis G.
Corcoran
a,
Joshua M.
Feinberg
c,
William A.
Arnold
*b and
R. Lee
Penn
*a
a,
Kamilah Y.
Amen
a,
Louis G.
Corcoran
a,
Joshua M.
Feinberg
c,
William A.
Arnold
*b and
R. Lee
Penn
*a
aDepartment of Chemistry, University of Minnesota – Twin Cities, 207 Pleasant St SE, Minneapolis, MN 55455, USA. E-mail: rleepenn@umn.edu; Fax: +1 (612) 626 7541; Tel: +1 (612) 626 4680
bDepartment of Civil, Environmental, and Geo-Engineering, University of Minnesota – Twin Cities, 500 Pillsbury Drive SE, Minneapolis, Minnesota 55455, USA. E-mail: arnol032@umn.edu; Fax: +1 612 626 7750; Tel: +1 612 625 8582
cInstitute for Rock Magnetism and Department of Earth & Environmental Sciences, University of Minnesota, Minneapolis, Minnesota 55455, USA
First published on 12th November 2024
Abstract
Naturally-occurring iron oxide nanoparticles provide reactive surfaces for the reduction of nitroaromatic compounds, which are common groundwater pollutants, by Fe(II). In many natural aquifer systems, iron oxide minerals continuously react with groundwater pollutants and other chemical species. To closely emulate field conditions, continuous flow columns packed with hematite-coated sands were used to study the reduction of 4-chloronitrobenzene (4-ClNB) by Fe(II) associated with the iron oxide. Columns were packed with sands coated with either a high or low mass loading of hematite nanoparticles (0.19 or 0.43 mg hematite per gram of sand after flushing). Following 36 hours of reaction (200–225 pore volumes), the total mass of iron oxide present in the columns increased, resulting from the concurrent Fe(III) oxidative mineral growth. The greatest increase was observed at the bottom of the column packed with the higher hematite mass loading sand. Acicular particles were observed on the post-reaction materials of both the high and low hematite loading sands. The acicular morphology is characteristic of goethite nanoparticles, and the presence of goethite was detected by low temperature magnetometry. Similar to results obtained under batch reactor conditions, goethite crystals heterogeneously nucleated on hematite as a result of the reductive degradation of 4-ClNB by Fe(II). Results tracking the rates of reductive degradation of the 4-ClNB and evolution of mineralogy demonstrate that reactivity is determined by the accessible reactive surface area, which evolves as goethite is deposited on hematite over time.
Environmental significanceIron oxide minerals mediate pollutant degradation, but the reactivity may change over time as mineral surfaces grow and change as electrons are transferred and additional or new minerals are formed. Results demonstrate that when beginning with hematite nanoparticles, the iron oxide mineralogy evolves from hematite to goethite-coated hematite with increasing extent of reaction. The reactivity is initially limited by the hematite loading but ultimately determined by the added reactive mineral surface area as reaction proceeds. The results will help predict how mineral nanoparticles will drive contaminant transformation in groundwater remediation schemes driven by natural attenuation or engineering interventions. |
Introduction
Globally, groundwater sources meet one-half the drinking water needs and, in some regions, are the only source of drinking-water.1 Access to safe and clean drinking water was recognized as a basic human right by the United Nations in 2010 and recognized again as a United Nations Sustainable Development Goal (SDG) in 2015 as SDG 6, “ensure availability and sustainable management of water and sanitation for all”, in the 2030 Agenda for Sustainable Development.2,3 Groundwater sources are vulnerable to contamination by organic pollutants and hazardous metal wastes, which emphasizes the need to understand and identify the transport and transformation of pollutants in groundwater to optimize remediation processes.1,4,5 A common class of groundwater contaminants is nitroaromatic compounds (NACs), which are frequently used as herbicides, pesticides, dyes, explosives, and pharmaceuticals.6–10 Many NACs have been classified as toxic and/or priority pollutants by the United States Environmental Protection Agency,11 and some are recalcitrant, mutagenic, and/or carcinogenic.9,10The iron oxides, herein used to mean iron oxides, oxyhydroxides, and hydroxides, occur in environmental systems as naturally occurring nanoparticles, surface coatings on other mineral particles, components of rocks, or products of rock and mineral weathering.12–18 Synthetic nanoparticles may also be used in engineered (bio)remediation systems.19,20 In batch systems, the reaction of Fe(II)/goethite nanoparticles has been extensively studied with NACs ranging from explosives to pesticides.21–26 Hematite, the most thermodynamically stable iron oxide, also facilitates the reduction of NACs by Fe(II).27,28 Hematite is found in soils and aquifer materials, and its presence is known affect the transport and effect the transformation of contaminants. Hematite is, however, less studied than goethite with respect to contaminant transformations.29,30
During a reduction–oxidation reaction at the mineral surface in the presence of aqueous Fe(II), NACs are reduced to their corresponding anilines and concurrent oxidative mineral growth of the iron oxide mineral takes place via oxidation of Fe(II), with the iron oxide mineral facilitating electron transfer between the Fe(II) and NAC molecule (Scheme 1).22,24,29,31,32 In many laboratory settings, nitrobenzene-based molecules are used to model the behavior of NACs due to the ease of tracking and quantifying both the NAC molecules and corresponding anilines by routine analytical methods.24,31–38 In natural aquifer environments, however, the reaction occurs under more complex solution conditions and in the presence of multiple minerals, which may lead to pronounced differences in reactivity as compared to the well-controlled laboratory scale (often batch) reactors.37,38
 | ||
| Scheme 1 Reaction for the reduction of 4-chloronitrobenzene (4-ClNB) by ferrous iron to form 4-chloroaniline (4-ClAn) and goethite (α-FeOOH) via oxidative mineral growth. The reaction occurs for numerous substituted nitrobenzenes when Fe(II) is associated with an iron oxide surface. | ||
Key factors in reactivity are the available (reactive) surface area, extent of Fe(II) sorption (which is affected by pH, the presence of organic matter, and other solution conditions), and reduction potential of the NAC.36,39 The oxidative mineral growth of the goethite occurs on the {021} facets producing new goethite material as the reaction product, with particles generally growing longer, but not wider, with increasing extent of reaction,33–36 unless conditions lead to blockage of reactive sites.38
In contrast to goethite, the oxidative mineral growth that accompanies the reductive degradation of 4-chloronitrobenzene (4-ClNB) by Fe(II) in the presence of hematite is more complex. In addition to the production of hematite, heterogeneous nucleation of goethite on the hematite nanoparticles is often observed,34,35,40 and the ratio of goethite to hematite production depends on experimental conditions such as the ratio of initial hematite to Fe(II) concentration,34 solution pH,34 the hematite morphology,40 and the addition of other chemical species, like organic matter.35 In addition, previous work demonstrated that once goethite nucleates, the overall reactivity of the phases present becomes dominated by that of goethite, with subsequent 4-ClNB degradation accompanied by additional goethite growth. As a result, the observed reactivity was more similar to goethite even when the mass loading of hematite was higher than that of goethite.34
Past works have shown similarities or differences in reactivity between these two mineral phases. Hematite was found to lead to faster NAC reduction than goethite, even when normalizing for surface associated Fe(II)41,42 or surface area34 but a different study found hematite to have low Fe(II) sorption capacity, with slower NAC reduction observed than with goethite.31 Based on redox potentials of the minerals, similar reaction rates for a given NAC could be expected.39,43,44 Thus, surface area and extent of Fe(II) association with the surface (i.e., differences in reactive surface area) may be driving factors in NAC transformation rates, and, thus, it is important to understand the dynamics between these two minerals in systems where oxidative growth occurs. This is especially relevant when reaction of hematite leads to formation of goethite.34,40
Previous work using flow-through reactor columns to study reduction of model pollutants as a function of dissolved oxygen concentration, mineral phases present, the role of organic matter, and aqueous saturation conditions has improved understanding of mineral-mediated reactivity of NACs in more realistic models of environmental systems.45–47 Specifically, Soroush et al. demonstrated that goethite (α-FeOOH) can be reversibly attached to sand grains and used to enhance the reduction of 4-ClNB by Fe(II) under continuous flow conditions.47,48 Characterization by electron microscopy before and after reaction demonstrated that oxidative mineral growth led to crystal growth of the already existing goethite nanoparticles, that growth primarily occurred on the tips of the goethite nanoparticles, and that the amount of crystal growth was greatest at the column inlet and decreased with distance from the inlet.47 The application of electron microscopy enabled detailed study of mineral growth with changing reactive conditions in the flow-through reactors.47 Furthermore, changes in flow pathways strongly influenced mineral growth, with variations in particle growth observed along the length of the column.47
Here, we report results tracking the reactivity of hematite-coated sands packed into column reactors and compare them to results obtained with batch reactors. Changes in mineralogy and solution chemistry were monitored as a function of the extent of reduction of 4-ClNB by Fe(II). 4-ClNB was used as a model NAC pollutant to facilitate comparisons between previous work using both batch reactors and flow-through columns.33–38,47,48 Hematite was coated on sands via electrostatic attraction between the hematite particles and quartz sand grains. Based on previous work, we hypothesized that goethite would heterogeneously nucleate on the hematite nanoparticles and that the overall reactivity would become more similar to goethite-coated sand with time.34 Characterization of both pre- and post-reaction materials enables a quantitative description of how reactivity and mineralogy evolves with the extent of reaction. Finally, the similarity in morphology of post-reaction materials collected from the column reactors and from previous work with batch reactors highlights the relevance of batch reactors, despite increasing complexity of the model system.34,35
Materials and methods
Chemicals
Chemical sources and purities are provided in the ESI† (Section S1.1). Ultrapure water was obtained from a MilliPore Milli-Q Advantage A10 system (18.2 MΩ cm) and used for all experiments. For anoxic experiments, ultrapure water was sparged with ultra-high purity N2 (Airgas, 3 hours per liter) and allowed to equilibrate overnight in anaerobic conditions prior to use. Solution pH was adjusted using aqueous 1 M solutions of HCl (BDH Aristar, 12.1 M) and NaOH (Fisher Scientific, 19 M). Ottawa sand (50–60 mesh, US Silica, Berkeley Springs, West Virginia) was continuously mixed by inversion in 0.1 M HCl for approximately 21 hours, rinsed three times with ultrapure water, and air-dried prior to use.Coating of sand with hematite
Rhombohedral hematite particles, previously prepared and characterized by Voelz et al.,34 were used. These particles had dimensions of 43 ± 8 nm by 29 ± 6 nm and specific surface area, as determined using N2 adsorption, of 72 m2 g−1.34 For lower surface coverage, hematite particles were dried and added at a ratio of 1.5 mg of hematite per gram of sand to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of acid-washed sand and ultrapure water that had previously been adjusted to a pH of 6. The pH of the mixture dropped to approximately 4 after addition of sand to the pH adjusted water. For higher surface coverage, hematite particles were suspended in ultrapure water at a mass loading of 0.75 mg mL−1. A 1:2 mixture of the acid-washed sand and this suspension as prepared, and the entire mixture was adjusted to a pH of 6. For both samples, suspensions were continuously mixed by inversion overnight (18–21 hours) in high density polyethylene (Nalgene) bottles that were twice the volume capacity of the added suspension. Both sand samples became reddish in color, and hematite remaining in the suspension following coating was quantified using a modified procedure from Voelz et al. wherein 0.5 mL increments of supernatant were air-dried and weighed to determine hematite loading.34 In both cases, a nonzero quantity of hematite remained in suspension. The remaining suspension was decanted before the sand was rinsed three times with ultrapure water and air-dried prior to use.
:2 mixture of acid-washed sand and ultrapure water that had previously been adjusted to a pH of 6. The pH of the mixture dropped to approximately 4 after addition of sand to the pH adjusted water. For higher surface coverage, hematite particles were suspended in ultrapure water at a mass loading of 0.75 mg mL−1. A 1:2 mixture of the acid-washed sand and this suspension as prepared, and the entire mixture was adjusted to a pH of 6. For both samples, suspensions were continuously mixed by inversion overnight (18–21 hours) in high density polyethylene (Nalgene) bottles that were twice the volume capacity of the added suspension. Both sand samples became reddish in color, and hematite remaining in the suspension following coating was quantified using a modified procedure from Voelz et al. wherein 0.5 mL increments of supernatant were air-dried and weighed to determine hematite loading.34 In both cases, a nonzero quantity of hematite remained in suspension. The remaining suspension was decanted before the sand was rinsed three times with ultrapure water and air-dried prior to use.
Reactive column preparation
Column experiments were conducted in an anaerobic NextGen system glovebox (Vacuum Atmosphere Company) with a 100% N2 gas environment (Airgas). The oxygen level was maintained below 1 ppm by reaction with copper catalysts and moisture was removed by molecular sieves. Columns were designed based on the work of Soroush et al.47,48 Briefly, columns were prepared with an internal diameter of 2.5 cm and length of 3 cm to avoid wall effects, optimize characterization capacity of post-reaction samples, and ensure that the reaction occurred over the entire length of the column. Flow rates of 0.5 to 1 mL min−1 were maintained using high-pressure piston pumps (Chrom Tech, P-LSP10S). Flow rates were chosen to maintain experimental feasibility while simulating groundwater flow velocities.47Borosilicate glass columns (Kimble FLEX-Columns) were fitted with polypropylene end caps and wet-packed with 23 g of hematite-coated sand. A flow adapter (Kimble) was secured to the top end of the column to prevent sand loss from the column. The sand in the column was saturated with ultrapure water to prevent entrapment of air in the columns. A solution of 10 mM NaCl was used as a tracer with effluent collected over 45 minutes at various time points using a fraction collector (BIORAD 2110). Chloride ion concentration was measured using a chloride ion selective electrode (Cole-Parmer). For each column, breakthrough curves for the conservative tracer before and after reaction were used to determine Peclet number, porosity, and hydrodynamic dispersion coefficient. Details of the hydrodynamic characterization of the columns, including determination of porosity and dispersion coefficients are presented in the ESI† (S1.2). Following tracer measurements, the pH of the column was adjusted by pumping in 10 mM NaHCO3 buffer (pH 7.0) for 1 hour at 0.5 mL min−1, which corresponded to approximately 3 pore volumes for each column. Bicarbonate buffer was used to provide conditions representative of groundwater.
After these column conditioning and rinse steps, a solution of 1 mM Fe(II) in 10 mM NaHCO3 (pH 7.0) was pumped into the column for 1 hour at a flow rate of 0.5 mL min−1. Effluent was collected every 5 minutes. After an hour, the Fe(II) concentration in the effluent reached >90% of the influent Fe(II) concentration for all columns tested. Next, 0.1 mM 4-ClNB in 10 mM NaHCO3 (pH 7.0) was added using a second pump at a flow rate of 0.5 mL min−1, for a combined flow rate of 1 mL min−1. Effluent was collected every 9 minutes, and sample tubes of effluent slated for Fe(II), 4-ClNB, and 4-chloroaniline (4-ClAn) quantification were spiked with 50 μL of 1 M HCl to quench the reaction and prevent precipitation of any iron oxides.13,32 For all columns, the first 10 samples were analyzed, followed by every fifth sample, until hour 24, after which every tenth sample was analyzed. Total reaction durations for columns were 6, 21, or 36 hours (32–40, 113–139, and 194–238 pore volumes). Finally, ultrapure water was used to rinse excess chloride ion from the column for 1 hour at a flow rate of 0.5 mL min−1, and then a final tracer measurement using NaCl at time points matching those used with the pre-reaction measurements was performed.
Following final tracer measurements, columns were removed from the anaerobic chamber, and the solid material was removed from the column as an intact cylinder from the top end of the column. Material was sliced in three 1 cm zones (0–1, 1–2, and 2–3 cm from the influent end indicated as bottom, middle, and top, respectively) and air-dried in polystyrene weigh boats. Upon drying, solid material characterization was performed as outlined below.
Fe(II) sorption and quantification in column experiments
Sorption isotherm measurements of Fe(II) to hematite-coated sand were conducted in 35 mL serum bottles, 5.0 g of coated sand was suspended in 10 mM sodium bicarbonate buffer (pH 7). Fe(II) was added to the serum bottles to achieve concentrations ranging from 0.0 mM to 1.0 mM using an acidified, deoxygenated FeCl2·4H2O stock solution (175 mM Fe(II), 0.3 M HCl). Data were fit using a Langmuir isotherm, and further experimental details are in the ESI† (S1.3). Sorption of Fe(II) to the mineral surface is complex and may not be simple monolayer coverage, but the Langmuir model provides an estimate of iron uptake capacity for a given aqueous equilibrium concentration.49 For the column effluent samples, Fe(II) concentration was normalized to the influent Fe(II) concentration and plotted against pore volume. Total Fe(II) eluted was determined by integrating the area under the concentration (in mM) versus volume eluted (in L) curve. This area was subtracted from the feed quantity to identify the quantity of Fe(II) consumed during the reaction. Integration was performed using Origin Lab 2019 using the mathematical area function.Fe(II) concentration was quantified via the ferrozine assay50 using a UV-1601PC UV-Visible spectrometer (Shimadzu) and a solution of 5 mg mL−1 ferrozine. Standards ranging in concentration from 0.005 mM to 0.050 mM Fe(II) were prepared from ferrozine, water, and 0.15 mM FeCl2. Samples were prepared using 0.2 mL of ferrozine, 2.8 mL of water, and 0.1 mL of filtered sample (isotherms) or unfiltered effluent (columns) in 1 cm polypropylene cuvettes.
4-ClNB and 4-ClAn quantification
4-ClNB and 4-ClAn were quantified via high-performance liquid chromatography (HPLC; Agilent 1200 Infinity) using a Zorbax SB-C18 column (4.6 × 150 mm, 5 μm). The mobile phase was a 7:3 mixture of solvents A:B where A was acetonitrile and B was 12 mM ammonium acetate in a pH 7 solution of 10% vol/vol acetonitrile in water. The flow rate was 0.7 mL min−1 and the detection wavelength was 254 nm. Calibration curves were prepared for both 4-ClNB and 4-ClAn over a concentration range of 0 to 0.1 mM in methanol.
Approximately 1 mL of each effluent sample was filtered using 13 mm syringe filters with a 0.2 μm nylon membrane into amber borosilicate glass vials. To each vial, 10 μL of 1 M HCl was added to prevent precipitation of Fe(II)-bearing phases. The concentration of 4-ClNB and 4-ClAn were plotted against pore volume. Mass balances were determined by summing the 4-ClNB and 4-ClAn concentrations. 4-ClNB consumed during the reaction column was determined by integrating the area under the concentration (in mM) versus volume eluted (in L) and subtracting this area from the feed quantity. 4-ClAn produced from the reaction column was determined by integrating the area under the concentration (in mM) versus volume eluted (in L). Integration was performed using Origin Lab 2019 using the mathematical area function.
Post-reaction material characterization
To characterize the mineralogy of the iron oxides on the sand surfaces, powder X-ray diffraction was attempted, as in previous work.34–38 However, the mass loading of iron-bearing phases was below the detection limit. Attempts to detach the mineral phases from the sand surfaces for further characterization were unsuccessful. This procedure and further discussion are detailed in the ESI† (S1.4).Thus, iron oxide mineralogy was characterized using low temperature magnetometry.35,51–54 Measurements were collected using a Quantum Design magnetic properties measurement system with a sensitivity of 10−10 Am2 at the Institute for Rock Magnetism at the University of Minnesota. Field-cooled (FC)–zero-field-cooled (ZFC) measurements were performed by cooling samples from 300 K to 10 K in the presence or absence of a 2.5 T field. At 10 K, samples were exposed to a 2.5 T isothermal remanent magnetization. Data was collected on warming to 300 K in 5 K steps. Room temperature isothermal remanent magnetization (RTSIRM) measurements were performed by imparting a 2.5 T isothermal remanent magnetization at 300 K onto samples. After the field was turned off, remanence was measured in 5 K steps from 300 K to 10 K and back to 300 K. Fig. S1† (S1.5) presents the low temperature behaviors of iron oxide minerals during low temperature magnetometry.
Pre- and post-reaction sands were imaged using a JEOL 6500F scanning electron microscope (SEM). Approximately 50 mg of dried sand was placed on conductive carbon tape applied to an aluminum stub (Structure Probe, Inc.) and subsequently sputter-coated with 5 nm of platinum (Leica EM, ACE 600). Samples were analyzed in the SEM using secondary-electron imaging. The working voltage was 5.0 kV.
Total iron per gram sand was quantified for the pre-reaction, post-rinsing, and post-reaction materials. The iron oxides were dissolved by placing sand samples into concentrated HCl for 1 hour at a mass loading of 0.5 mg sand per mL of concentrated HCl. After digestion, the color of the sand matched that of the original Ottawa sand standard, and the supernatant was bright yellow. The supernatant was separated from the solid sand and diluted 100-fold using ultrapure water. A 1 mL aliquot of this diluted sample was further diluted with 10 mL of 1% nitric acid. Calibration standards with ferric ion concentrations ranging from 0.05 ppm to 2.5 ppm were prepared using Fe(NO3)3·9H2O in solutions of 0.105 M nitric acid and 0.015 M HCl to match the acid background from mineral digestion. Finally, iron concentration in digestion solution and standards were quantified using a Thermo Scientific iCAP 7400 Inductively Coupled Plasma-Optical Emission Spectrometer (ICP-OES) in radial mode. Peak areas were measured in quintuplicate for three elemental iron wavelengths: 259.940, 238.204, and 239.562 nm. Once the iron was quantified in the digested samples, mass of iron per gram of sand was determined and an estimate of the goethite surface area produced were calculated as described in the ESI† (S1.6).
Results and discussion
Characterization of hematite sand
Hematite was successfully attached to the quartz sand. After the column rinsing, the low iron sand (hereafter referred to as LFe sand) sample contained 0.13 ± 0.01 mg iron per gram of sand (0.19 ± 0.02 mg hematite per g sand), and the high iron sand (HFe sand) sample had 0.30 ± 0.02 mg iron per g sand (0.43 ± 0.03 mg hematite per g sand). Interestingly, the hematite on the HFe sand was more strongly associated with the sand grains than was the hematite on the LFe sand likely due to the difference in pH during the mixing/attachment process (6 for HFe, 4 for LFe).Both LFe and HFe sand had an orange to red appearance (Fig. 1a), and the SEM images show that the distribution of hematite nanoparticles on the sand surfaces was not uniform (Fig. 1c and d). For the HFe sand, the Fe(II) sorption capacity was determined at pH 7 and the data fit to a Langmuir sorption model as shown in Fig. S1.† The sorption capacity was 51 ± 1 μg Fe(II) per gram of hematite coated sand.
 | ||
| Fig. 1 (a) Optical image of (left to right) bare Ottawa sand, low-density surface coverage hematite on sand (LFe sand; 0.19 ± 0.02 mg hematite per g sand), and high-density surface coverage hematite on sand (HFe sand; 0.43 ± 0.03 mg hematite per g sand) demonstrating the increasingly intense reddish-orange color with increasing hematite coverage. Secondary electron SEM images of (b) quartz surface in bare sand (c) quartz surface decorated with <50 nm hematite in the LFe sand and (d) quartz surface decorated with <100 nm hematite in HFe sand. | ||
Reactivity of hematite-coated sands in column reactors
To understand the reactivity of the hematite-coated sands, the concentrations of 4-ClNB and 4-ClAn in the effluent, as a function of pore volume (Fig. 2), were compared. For the 36 hour LFe sand (LFe 36, Fig. 2a), from 0 to ∼15 pore volumes following 4-ClNB introduction to the influent, a gradual increase in the 4-ClNB concentration and mass balance was observed. Following the observation of 4-ClNB concentration approaching the feed concentration at ∼15 pore volumes, a period of minimal reactivity is seen from ∼15 to ∼65 pore volumes wherein low 4-ClAn concentrations were observed, despite a mass balance approximately equivalent to the feed concentration, indicating little to no conversion to 4-ClAn. After ∼65 pore volumes, a steady increase in 4-ClAn concentration and decrease in 4-ClNB concentration was seen until ∼120 pore volumes. During this period, the slightly low mass balance may be attributed to two intermediates, 4-chloronitrosobenzene and 4-chlorophenylhydroxylamine, that form during the reduction process.47,55 One intermediate, likely the 4-chlorophenylhydroxylamine, was visible in the HPLC chromatograms but was not quantified.55 In the final ∼40 pore volumes, the mass balance once again approaches the feed concentration, variations in 4-ClNB and 4-ClAn concentration level off, and the reactivity approaches a steady state. | ||
| Fig. 2 Concentrations of 4-chloronitrobenzene (4-ClNB) in the column reactors for the 36 hour reaction column on (a) LFe sand and (b) HFe sand. The influent level is show by the yellow line. The effluent concentrations of 4-ClNB and 4-chloroaniline (4-ClAn) are shown as red squares and blue circles, respectively. The summative mass balance of 4-ClNB and 4-ClAn is a black ×. In panel (a), the shading shows initial, minimal, increasing, and steady state transformation regions. | ||
For the HFe sand (HFe 36, Fig. 2b), the concentration of 4-ClNB increased until ∼9 pore volumes, as expected. The 4-ClNB concentration in the effluent never reaches that of the influent concentration, indicating that conversion started shortly after introduction. From ∼9 to ∼160 pore volumes, fluctuations in the 4-ClNB and 4-ClAn concentrations were observed. We hypothesize that oxidative mineral growth caused changes in flow, which led to these observed variations, as previously observed in goethite-coated sand columns.47,48 After ∼160 pore volumes, the concentrations of 4-ClNB and 4-ClAn reach a plateau, and reactivity approaches a steady state. The concentration of 4-ClNB at this point, relative to the feed concentration, is comparable to the steady state level approached by the LFe 36 column, as seen in Fig. S2.†
Additional column reactors were prepared using LFe sand to better characterize the post-reaction material as a function of extent of reaction. One column was sacrificed after 6 hours of reaction (LFe 6, ∼40 pore volumes), which was prior to the increase in 4-ClAn concentrations. Another column was sacrificed after 21 hours of reaction (LFe 21, ∼140 pore volumes), beyond both the minimal reactivity period (15–65 pore volumes) and period of increasing conversion (65–120 pore volumes) but before a steady state was reached (∼160 pore volumes). Results are shown in Fig. S3.† Plots showing the concentrations of Fe(II), 4-ClNB, and 4-ClAn in the effluents of the LFe 6, LFe 21, and LFe 36 sand columns demonstrate the overall reproducibility and similarity across the three LFe columns (Fig. S4†). Additionally, no conversion from 4-ClNB to 4-ClAn was observed in the control experiment using uncoated sand, indicating that reactivity arises from the hematite (Fig. S5†).
Both the 4-ClNB consumed and 4-ClAn produced increased with increasing reaction duration for the LFe sand (Table 1). Additionally, the quantities of 4-ClNB consumed and 4-ClAn produced were greater for HFe 36 than the LFe 36 experiment. The Peclet number decreased after reaction, indicating an increased contribution from diffusive transport relative to the advective flow (Table S1†). Changes in porosity and hydrodynamic dispersion coefficient varied, and details are in the ESI.†
| ID | Fe(II) consumed (mmol) | 4-ClNB consumed (mmol) | 4-ClAn produced (mmol) | Fe(II) cons./4-Cl-NB cons. | Fe(II) cons./4-Cl-An prod. |
|---|---|---|---|---|---|
| LFe 6 | 0.0111 | 0.0024 | 0.0002 | 4.6 | 55.5 |
| LFe 21 | 0.2243 | 0.0130 | 0.0084 | 17.2 | 26.7 |
| LFe 36 | 0.1000 | 0.0375 | 0.0210 | 2.7 | 4.8 |
| HFe 36 | 0.5495 | 0.0684 | 0.0628 | 8.0 | 8.7 |
In contrast, while the Fe(II) consumed followed a similar trend, there was one major deviation where LFe 21 consumed over double the amount of Fe(II) than LFe 36. LFe 36 had an initial porosity of 75% (Table S1†), nearly 10 percentage points greater than all other columns. The increased porosity likely means increased accessible surface area, which would increase the amount of Fe(II) sorbed. The limited adsorption of nitrobenzene species to the surfaces of iron oxides39 and sand would explain why this deviation from the trend was not observed for the 4-ClNB and 4-ClAn integrated results.
In all cases, consumed quantities of 4-ClNB were greater than the formed quantities of 4-ClAn, with HFe 36 showing the lowest disparity. Based on the stoichiometry of the reaction, it would be expected that 6 moles of iron would be oxidized per mole 4-ClNB converted to 4-ClAn (Scheme 1). In most cases, the numbers in Table 1 exceed the value of 6, which could be explained by adsorption of Fe(II) to newly formed iron oxide materials. For values less than 6, a likely explanation is the unquantified intermediates of the reduction of 4-ClNB, resulting in incomplete mass balances for LFe 6 and LFe 36. Instrumental/analytical errors also cannot be ruled out.
Finally, the apparent first-order rate constant, kobs, as a function of time (Fig. 3) was determined following the approach of Heijman et al.56 where kobs was defined as:
 | (1) |
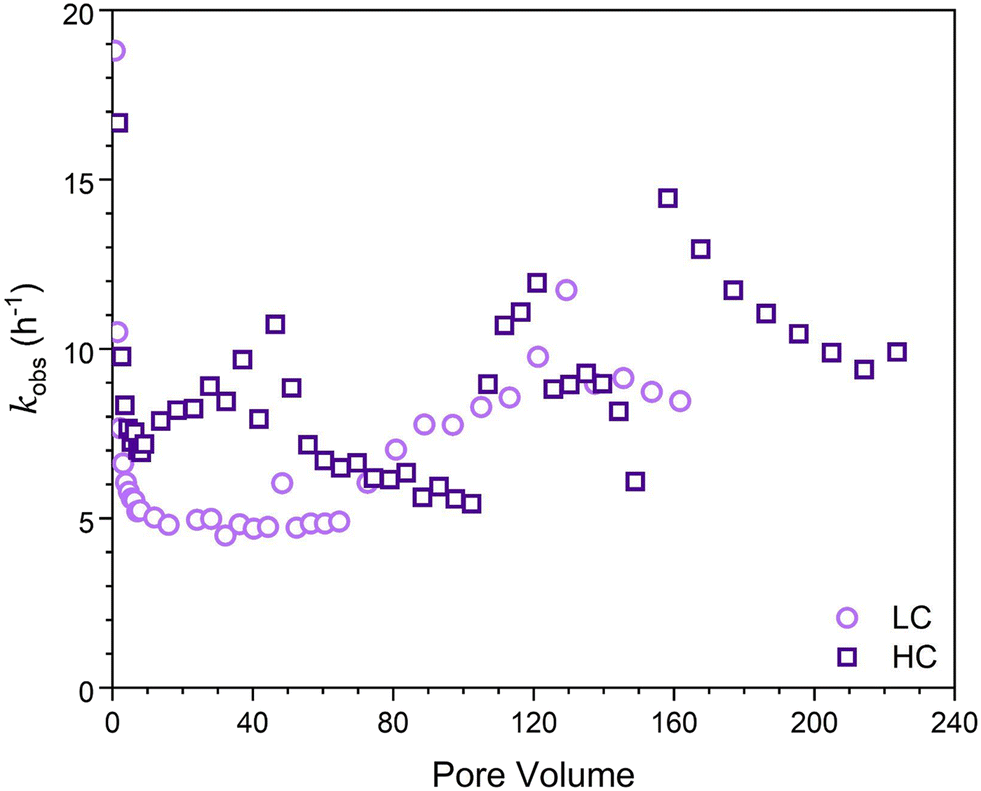 | ||
| Fig. 3 Apparent first-order rate constants (h−1) determined for the LFe (light purple circles) and HFe (dark purple squares) hematite coated sand columns after reacted for 36 hours using eqn (1). | ||
Post-reaction solid material characterization
The post-reaction materials from the different column zones of LFe 36 (Fig. 4) were compared to uncoated sand material and unreacted LFe sands (Fig. S7†). Readers are referred to the supplementary information to see examples of RTSIRM and FC/ZFC behavior for mineral standards of goethite, hematite, lepidocrocite, and magnetite (Fig. S1†). The RTSIRM and FC/ZFC measurements show inflections corresponding to the Verwey transition of magnetite at 120 K and the Besnus transition of pyrrhotite at 30 K, both of which likely occur as minor mineral inclusions within quartz grains for all samples. No evidence of the Morin transition, which would be indicative of the presence of hematite, was found in the RTSIRM data for uncoated or unreacted sands, likely due to the low volume concentration of hematite. For the top zone of LFe 36 (Fig. 4a), the RTSIRM data does show subtle evidence of the hematite's Morin transition (a slight inflection near 210 K, which is consistent with the Morin transition temperature observed in Voelz et al.35 No such evidence was detected for the middle and bottom of this column (Fig. 4b and c). Across the three zones, a consistent separation is observed in the FC/ZFC data, which is indicative of superparamagnetic goethite (Fig. 4d–f). While goethite does not have a signature magnetic transition, superparamagnetic grains of goethite can become thermally blocked at lower temperatures, which will make the magnetization of the goethite more susceptible to organization by a strong aligning field.35 This thermal unblocking is observed as decays in the FC magnetization data from 10 to 300 K. As such, the observed separation indicates the presence of goethite in all three LFe 36 column zones. The relative decrease of this separation along the length of the column also indicates that there is a greater amount of goethite at the bottom of the column, the inlet, relative to the top, the outlet. While we cannot completely rule out the presence of ferrihydrite or lepidocrocite, they are unlikely to be present based on the FC/ZFC data (see ESI†). | ||
| Fig. 4 RTSIRM (panels a–c) and FZ/ZFC (panels d–f) data collected from sand samples from the three zones of the LFe 36 reaction column (top: a and d; middle: b and e; bottom: c and f). In the RTSIRM data, the blue squares are for cooling, the red circles are for warming, and the lines are 9-point running averages. In the FC/ZFC plots, the blue squares are collected under FC conditions and the red circles under ZFC conditions. The red arrow in panel a indicates a subtle inflection in the running average at 210 K, which is consistent with the Morin transition in hematite observed in Voelz et al.35 Inflections at 120 K in both the RTSIRM and FC/ZFC data are associated with the Verwey transition in magnetite, where the mineral transforms from monoclinic symmetry below 120 K to cubic symmetry above 120 K. Golden lines in panels d–f indicate separation due to thermal unblocking of superparamagnetic goethite. | ||
SEM micrographs of samples from the three regions of each post-reaction column are shown in Fig. 5. In most cases, acicular particles were observed, consistent with the formation of acicular goethite crystals, which can form on hematite particles during reduction of nitrobenzenes.34,40 In the case of LFe 21 and LFe 36, the abundance of these particles was greatest at the bottom and decreased along the length of the column. For HFe 36, there is a dense collection of these particles throughout. In the case of LFe 6, no evidence of acicular goethite crystals was observed, and the morphology of the particles attached to the sand grain surfaces is consistent with pre-reacted LFe sand.
 | ||
| Fig. 5 SEM images of post-reaction hematite-coated sand grains for the LFe columns sacrificed at 6 hours (LFe 6), 21 hours (LFe 21), and 36 hours (LFe 36), and the HFe column sacrificed at 36 hours (HFe 36) at the three different regions of the column based on distance from the bottom inlet (bottom, middle, top). | ||
The total iron loading was highest near the inlet and lowest near the outlet for nearly all post-reaction column materials tested (Fig. 6). The amount of Fe found in each zone of the reactive columns was quantified and correlated with the extent of mineral growth. To account for variation in the starting Fe quantity on the sand surface, this quantity of Fe was normalized to the quantity of Fe obtained from the respective rinsed, unreacted hematite-coated sand material. While this method does not provide information about the mineral phase, owing to the digestion process required for ICP-OES analysis, it does facilitate the characterization of the extent of reaction as a function of position in the column. In general, the bottom zones of each column showed greater quantities of iron than the top zone, consistent with the higher concentration of reactants and thus higher reaction rates occurring at the bottom of the columns compared to the top. The results in Fig. 6 are consistent with the visual trends in Fig. 5.
 | ||
| Fig. 6 New iron mass (mg Fe per g sand) with respect to the iron mass of rinsed LFe or HFe hematite coated sand, from the three column zones for each reacted column. Y-Axis numbers indicate the length of time the column was exposed to 4-ClNB in hours. Error bars represent propagated error from the five measurements at each of the 3 iron wavelengths tested using ICP-OES. | ||
Compared to the other columns, LFe 6 shows a consistent decrease in Fe across all regions of the column. Given that 46% of the hematite rinsed off the LFe sand surfaces during the initial rinsing steps, this additional loss relative to the rinsed sand implies that additional hematite was lost during the six hours of flow. After six hours of flow, there is a slightly higher iron loading in the top zone as compared to the middle and bottom zones, which could have resulted from both loss from the top zone combined with entrapment of material dislodged from lower zones. Across the three zones, however, the loss (∼0.05 mg hematite per g sand) was within error of each other from ICP-OES measurement, supporting the hypothesis that the loss results from flow conditions removing poorly bound hematite that remained during the first 30 pore volumes even after rinsing the LFe sand during preparation.
For the other columns, the added mass of new Fe-bearing material on the sand surfaces decreased with increasing distance from the inlet of the column. For LFe 21, the Fe gained for the top and middle of the column was within error (0.229 ± 0.006 and 0.238 ± 0.006 mg Fe per g sand, respectively), whereas a greater amount of Fe was added in the bottom, 0.314 ± 0.006 mg Fe per g sand. For LFe 36 and HFe 36, the new Fe-bearing material added decreased substantially from the bottom to the top of the columns.
All regions of HFe 36 showed greater Fe mass gains relative to LFe 36. The Fe mass gained in the top zone of these two columns were within error of one another (0.336 ± 0.006 mg Fe per g sand for LFe 36 versus 0.38 ± 0.03 mg Fe per g sand). Whereas for the bottom zones of both columns, the Fe mass gain is twice as large for HFe 36 at 1.37 ± 0.03 mg Fe per g sand relative to 0.684 ± 0.006 mg Fe per g sand for LFe 36. Additionally, for HFe 36, the variation of relative Fe growth between the bottom and the top of the column is the greatest of all columns (>3× difference). This is likely due to the fact that no Fe was lost during rinsing for this column so more reactive sites were available at the bottom for Fe(II) consumption relative to the LFe sand. This aligns with the greater extent of 4-ClNB conversion in the HFe column.
Evolving mineralogy
As with previous studies investigating the reduction of 4-ClNB by Fe(II) in the presence of hematite,34,35 heterogeneous nucleation of goethite was observed. Based on the magnetometry data and past work,34,35 it is unlikely that other less stable, yet more reactive iron oxides contribute to reactivity, but they could be below detection limits. The extent of reaction is greatest near the inlet, where the highest concentrations of Fe(II) and 4-ClNB occur. The product of oxidative mineral growth at the bottom zone of the column appears to be dominated by goethite, where the Fe(II) to reactive surface area ratio is highest. With distance from the inlet, however, evidence for goethite and hematite growth can be observed in both the magnetometry data and SEM micrographs from the post-reaction material collected from the top zone of LFe 36. This is consistent with previous batch reactor results in which increasing the mass loading of hematite (reducing the Fe(II) to reactive surface area ratio) resulted in a lower goethite to hematite ratio in the material produced by oxidative mineral growth.34 Reduced splitting of the FC/ZFC curves with distance from the column inlet is consistent with a decreasing amount of goethite formed. The decreasing amount of goethite with distance from the column inlet, as detected, is expected based on the decreasing reactant concentrations as the fluid traverses the length of the column because higher concentrations of Fe(II) have been previously associated with greater mass fractions of goethite forming on hematite in batch reactors under similar reaction conditions.34 Given the evidence of nanophase hematite observed for the top of LFe 36 from the magnetometry data, it is likely that the formation of nanophase hematite occurs near the top of the column predominantly due to decreasing Fe(II) concentrations as along the column length (from bottom to top). An overall decreasing concentration of reactants as they are used along the column length results in a decreased concentration of Fe(II) at the top of the column, and subsequently, a lower mass fraction of goethite is expected at the top of the column based on results from Voelz et al.34 When correlated to the variation in Fe quantity along the column (Fig. 6), the decreasing Fe(II) concenration and decreased amount of observed oxidative growth of Fe(III) matches the observation of an overall lower net change in Fe% as distance from the inlet to the column increases.The magnetometry data are consistent with the SEM micrographs showing evidence of a greater amount of goethite at the bottom of the column and decreasing as a function of distance from the inlet. In addition, the SEM micrographs can help to further inform mineral growth behavior. For the bottom of this column (Fig. S8†), large swatches of the sand surface are covered by particles with the acicular morphology associated with goethite particles.47 Fewer and less densely packed acicular particles are observed on the post-reaction LFe 36 sands with increasing distance from the inlet (middle zone, Fig. S9;† top zone, Fig. S10†). In all three zones, the majority of observed goethite particles are found in dense clusters, consistent with the distribution of the hematite particles on the pre-reacted, coated sands. Thus, it is likely that the hematite serves as nucleation sites for goethite growth. Additionally, there is evidence of unaltered hematite particles for the top and middle of the LFe 36 post-reaction materials. From these micrographs, it also appears that goethite requires the initial nucleation site provided by the hematite particles as evident by the presence of regions where no particles are observed post-reaction, likely corresponding to uncoated sand surfaces.
No evidence of a Morin transition was observed in the RTSIRM data for uncoated or unreacted LFe and HFe sands. This indicates that the initial levels of hematite on the sand surfaces were below the detection limit. The Morin transition of hematite is observed in the top zone of LFe 36. This indicates that some quantity of hematite results as an oxidative growth product, which is consistent with previous results from batch reactor experiments, in which oxidative mineral growth results in the formation of both hematite and goethite growth.34 The Morin transition observed is at lower temperatures than typically reported for hematite, which is suggestive of a nanophase character to these particles. This is in line with previous work demonstrating a size dependence of the Morin transition when the hematite particle size is below 100 nm.53 Morin transitions at ∼200 K were previously observed following the reaction on hematite particles in batch reactors during the reduction of 4-ClNB by Fe(II) in the presence of organic matter.35
While it has been proposed that magnetite can precipitate from Fe(II) sorption on hematite surfaces,57,58 the evidence for the Verwey transition associated with magnetite observed in Fig. 4 does not indicate heterogeneous nucleation of magnetite surfaces because this transition was also observed in uncoated sands (Fig. S6†). This indicates that the transition likely arises from magnetite inclusions within the quartz sand itself. Additionally, the relatively consistent magnitude of the Verwey transition across all samples measured suggests that this is not a secondary mineral phase formed as a result of the 4-ClNB reduction or Fe(II) sorption to the mineral surfaces. Given that the ICP-OES data shows that Fe growth is directly correlated with distance from reactant inlets, oxidative growth products are expected to demonstrate a similar gradient across the zones of the post-reaction column material and no similar change in magnetite content was observed as a function of distance from the inlet. Additionally, in previous work studying hematite in batch reactors in the absence of quartz sand no evidence of magnetite or pyrrhotite formation was detected, which supports the conclusion that these are not secondary phases.35
Connecting evolution of reactivity to mineralogy
As was seen in Fig. 2, reaction dynamics were dependent on initial hematite coverage on the sand surfaces. Greater initial hematite coverage yielded a higher extent of conversion of 4-ClNB to 4-ClAn. This is attributed to the greater accessible surface area on which reactions can occur from the beginning of the experiment. This initial coverage led to an onset of significant conversion early in the reaction duration and overall greater conversion throughout the course of the reaction, irrespective of similar reaction lengths, as can be seen by comparing quantities of 4-ClAn produced from LFe 36 and HFe 36 (Table 1). This greater conversion by the HFe sand also corresponds to a greater mass of Fe, relative to the rinsed starting material, at the end of the reaction duration, as shown in Fig. 6.To allow for a comparison of reactivity to literature rates for batch reactors, the apparent-first order rate constants (Fig. 3) were normalized to the iron oxide mass. Given that the iron oxide mass loading varied over the course of the reaction, and that the reactive surface area also varied, it was necessary to normalize the apparent rates to surface area, using the estimated goethite surface areas calculated as described in the ESI.† As such, we focused on the rate constants obtained at steady state because at this point the surface areas could be estimated using the results from SEM imaging in combination with iron mass loadings from ICP-OES analysis. For LFe 36 and HFe 36, respectively, the approximate normalized rate constants at steady state were found to be 5 and 7 m−2 h−1. Interestingly, these rates are approximately the same even though the starting hematite coverage differed. Based on the dynamics of the observed kinetics and the formation of acicular goethite, it appears that once oxidative mineral growth occurred, goethite dominated the reactivity of the columns.
In comparison to literature results, these rates are nearly an order of magnitude greater than rates reported from batch reactors. Upon normalizing reported rate constants to surface area based on reported particle masses used in batch reactors, literature values for the reduction of 4-ClNB by Fe(II) in the presence of hematite34 and goethite38 are 0.72 m−2 h−1 and 0.18 m−2 h−1, respectively. A reason for the large discrepancy between these column reactors and batch reactors is not immediately clear. However, it has been suggestion by Chen et al. that reduction rates are dependent on the buffering capacity of sodium carbonate relative to the initial concentration of 4-ClNB.59 They suggested that the buffer capacity of 10 mM sodium bicarbonate was insufficient to buffer against the multiple protons generated by the reduction of 50 μM 4-ClNB.59 Previous work by Strehlau et al. has also shown that Fe(II) sorption on iron oxide surfaces is extremely pH dependent and very small decreases in pH can have significant impacts on rates due to the decreased Fe(II) sorption.38 Calculations based on Scheme 1 using Visual MINTEQ60 indicate that the complete reduction of the 4-ClNB and associated precipitation of goethite would lead to a drop in pH to 6.85. Because the 4-ClNB was not completely degraded, the pH change would be less. In these column experiments, the continued refreshment of the carbonate buffer along the column length could aid in pH stabilization and is potentially the reason that elevated rates are seen relative to batch reactors. We cannot rule out, however, that localized pH dynamics and changes in Fe(II) sorption affected the observed kinetics. The assumptions inherent in calculating the surface area used for the normalization could also be an issue.
The solids characterizations, however, provide evidence that the observed changes are a driving factor in the observed reactivity dynamics. From the four reaction periods defined for LFe 36, and subsequent post-reaction material characterization of columns sacrificed at different points in those periods, we can gain further insight into how the mineralogy evolves with the extent of reaction. Throughout the period of low reactivity, up to 65 pore volumes, minimal reaction and no clear morphological changes occur. Once the reaction begins after 65 pore volumes, indicated by conversion of 4-ClNB to 4-ClAn, we hypothesize that hematite and goethite formation are occurring in tandem. However, at the bottom of the column, goethite growth is likely more rapid due to the higher Fe(II) concentrations, which have been shown to favor the oxidative production of goethite.34 Based on the SEM micrographs for LFe 21 (Fig. 5) coupled with the net change in Fe mass loading, there is greater increase in Fe mass loading at the bottom of the LFe 21 column and more densely packed goethite particles. In contrast, the SEM micrographs of material collected from the top and middle zones of LFe 21 appear similar to the pre-reaction LFe sand and have similar net changes in Fe content. The lower Fe(II) concentrations at the top of the column likely favor the formation of hematite, however, which is supported by the observation of the Morin transition in material collected from the top of the column. Given that this transition was not observed for the pre-reaction material, this indicates that new hematite has formed and the hematite loading rose above the detection limit as a result of the reaction, but only for the region at the top of the column with the lowest Fe(II) concentrations.
The generally increasing trend of the rate constants with extent of 4-ClNB reduction indicates a change in mineralogy or additional reactive surface area formed over the course of the reaction. The redox potentials for Fe(II) bound to goethite and hematite are nearly identical43 and quantitative structure activity relationships for NAC reaction on these two minerals are similar.39,44 Thus, if surface area and extent of Fe(II) sorption were constant, it would be expected that the driving force for reaction and thus reactivity would be constant. This indicates that changes in reactivity over time are driven by changes in the amount of accessible mineral surface area, and this may be accompanied by changes in the Fe(II) sorption capacity/number of reactive sites. It is possible that surface area could increase and the capacity to sorb Fe(II) decrease depending the crystal facets that form. The combination of surface area, types of reactive sites,61 and Fe(II) sorption capacity will drive the reactivity in this system rather than the specific amount of goethite versus hematite. The role of organic matter on these dynamics, specifically the surface area of new mineral formed, needs to be further explored because organic matter can slow the reaction on mineral surface and affect the amount of goethite versus hematite produced.35,36,48
Given that adsorption of Fe(II) on goethite surfaces preferentially occurs on the {021} crystal face, which compromises a minor fraction of overall accessible surface area, rates are expected to decrease to reflect the change in accessible surface area as more goethite surfaces dominate over hematite surfaces. As flow pathways vary, hematite that was previously inaccessible, or not yet formed, could still react leading to variations in rate constant that cannot be easily normalized to accessible reactive surface area. Therefore, even though each subsequent spike in rate constant appears to be slightly higher, it is likely that if surface area could be quantified for these time points, the approximate surface area normalized rates would be similar to one another, if not slightly lower at later pore volumes as expected for the increased goethite content.
Conclusion
As with previously reported batch reactors,34,40 the heterogeneous growth of goethite on hematite was observed with the reduction of 4-ClNB by Fe(II). While our observed steady state rates were an order of magnitude faster than observed for batch reactors of goethite or hematite, the overall mineralogical outcome indicates that batch reactor data is environmentally relevant despite increasing complexity of subsequent model systems. The extent of goethite surface coverage during the reaction was dependent on the extent of reaction and reactant concentrations. This highlights how mineralogy is a moving target in aquifers with reactive species and must be continuously monitored to understand pollutant fate and transport. Because Fe(II) associated with goethite and hematite have similar redox potentials, the reactive surface area and extent of Fe(II) sorption will drive the observed reaction kinetics, rather than mineral identity.We found that a greater initial coverage resulted in greater overall conversion of 4-ClNB to 4-ClAn, irrespective of matching reaction length, indicating that the initial iron oxide content on sand grains has a significant impact on overall pollutant treatment potential in the short-term. In the long-term, however, our results suggest that properties will eventually become similar to one another irrespective of starting iron oxide mass loading. As such, the importance of understanding initial reaction conditions for predicting the fate of pollutant molecules environmentally is emphasized, as well as understanding how reactivity will change due to the evolving reactive surface areas as reaction duration progresses.
The results are important when iron oxide nanoparticles are mediating the reactions of oxidized contaminants. Relevant scenarios include groundwater systems contaminated with explosives or pesticides undergoing natural attenuation (where there is sufficient biologically produced ferrous iron) or engineered treatment (e.g., in situ chemical reduction to generate Fe(II)). Our work focused on coating an unreactive quartz surface with one reactive iron oxide mineral, hematite, and introducing such nanoparticles to contaminated systems lacking reactive capacity is also possible. The reactivity of these systems will be better understood in the context of evolving mineralogy based on the results herein. Overall, this work represents one step closer to mimicking field conditions, which feature a diversity of mineral particles, dissolved chemical species, spectator species, organic matter, and organisms. Incorporation of these variables will increase the environmental relevance of column studies and further our understanding of reactivity and mineralogy of natural systems and relevant engineering interventions. To further extend relevance to field conditions, variable flow rates could be studied to identify the impact that flow may have on kinetics and mineralogy of iron oxide minerals.
Data availability
The data supporting this article are available in the Data Repository for the University of Minnesota (https://hdl.handle.net/11299/166578) or have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the U.S. National Science Foundation grants ECS-1507496 and ECS-1904858. Additional support was provided by the University of Minnesota. Parts of this work were carried out in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program.References
- I. S. Zektser and L. G. Everett, Groundwater Resources of the World and Their Use, IHP-VI series on groundwater, UNESCO, Paris, France, 2004, vol. 6 Search PubMed.
- United Nations General Assembly, Resolution A/RES/64/292, UN, 2010, https://digitallibrary.un.org/record/687002 (accessed 2024-07-06) Search PubMed.
- Transforming our world: the 2030 Agenda for Sustainable Development | Department of Economic and Social Affairs, https://sdgs.un.org/2030agenda (accessed 2024-07-06).
- D. J. Lapworth, N. Baran, M. E. Stuart and R. S. Ward, Emerging Organic Contaminants in Groundwater: A Review of Sources, Fate and Occurrence, Environ. Pollut., 2012, 163, 287–303, DOI:10.1016/j.envpol.2011.12.034.
- A. Jurado, E. Pujades, M. Walther and M. S. Diaz-Cruz, Occurrence, Fate, and Risk of the Organic Pollutants of the Surface Water Watch List in European Groundwaters: A Review, Environ. Chem. Lett., 2022, 20, 3313–3333, DOI:10.1007/s10311-022-01441-w.
- P. Kovacic and R. Somanathan, Nitroaromatic Compounds: Environmental Toxicity, Carcinogenicity, Mutagenicity, Therapy and Mechanism, J. Appl. Toxicol., 2014, 34(8), 810–824, DOI:10.1002/jat.2980.
- K.-S. Ju and R. E. Parales, Nitroaromatic Compounds, from Synthesis to Biodegradation, Microbiol. Mol. Biol. Rev., 2010, 74(2), 250–272, DOI:10.1128/MMBR.00006-10.
- M. Kulkarni and A. Chaudhari, Microbial Remediation of Nitro-Aromatic Compounds: An Overview, J. Environ. Manage., 2007, 85(2), 496–512, DOI:10.1016/j.jenvman.2007.06.009.
- J. Tiwari, P. Tarale, S. Sivanesan and A. Bafana, Environmental Persistence, Hazard, and Mitigation Challenges of Nitroaromatic Compounds, Environ. Sci. Pollut. Res., 2019, 26(28), 28650–28667, DOI:10.1007/s11356-019-06043-8.
- C. M. Peres and S. N. Agathos, Biodegradation of Nitroaromatic Pollutants: From Pathways to Remediation, in Biotechnology Annual Review, Elsevier, 2000, vol. 6, pp. 197–220, DOI:10.1016/S1387-2656(00)06023-3.
- United States Environmental Protection Agency, Toxic and Priority Pollutants Under the Clean Water Act, 1972, vol. 33, USC § 1251, https://www.epa.gov/eg/toxic-and-priority-pollutants-under-clean-water-act (accessed 2022-04-25) Search PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, John Wiley & Sons, 2003 Search PubMed.
- N. S. Wigginton, K. L. Haus and M. F. Holchella, Jr, Aquatic Environmental Nanoparticles, J. Environ. Monit., 2007, 9(12), 1306–1316, 10.1039/B712709J.
- M. F. Hochella, There's Plenty of Room at the Bottom: Nanoscience in Geochemistry, Geochim. Cosmochim. Acta, 2002, 66(5), 735–743, DOI:10.1016/S0016-7037(01)00868-7.
- G. A. Waychunas, C. S. Kim and J. F. Banfield, Nanoparticulate Iron Oxide Minerals in Soils and Sediments: Unique Properties and Contaminant Scavenging Mechanisms, J. Nanopart. Res., 2005, 7(4), 409–433, DOI:10.1007/s11051-005-6931-x.
- R. L. Penn, C. Zhu, H. Xu and D. R. Veblen, Iron Oxide Coatings on Sand Grains from the Atlantic Coastal Plain: High-Resolution Transmission Electron Microscopy Characterization, Geology, 2001, 29(9), 843–846, DOI:10.1130/0091-7613(2001)029<0843:IOCOSG>2.0.CO;2.
- L. Carlson and U. Schwertmann, Natural Ferrihydrites in Surface Deposits from Finland and Their Association with Silica, Geochim. Cosmochim. Acta, 1981, 45(3), 421–429, DOI:10.1016/0016-7037(81)90250-7.
- D. J. Greenland and M. H. B. Hayes, The Chemistry of Soil Constituents, Wiley, Chichester, Eng., New York, 1978 Search PubMed.
- S. Mahanty, A. Sarkar, P. Chaudhuri and G. Krishna Darbha, Mycosynthesized Magnetic Iron-Oxide Nanoparticles for the Remediation of Heavy Metals – An Insight into the Mechanism of Adsorption, Process Optimization Using Algorithmic Approach and Its Application for the Treatment of Groundwater, Environ. Nanotechnol., Monit. Manage., 2023, 20, 100854, DOI:10.1016/j.enmm.2023.100854.
- K. Pandey, S. Sharma and S. Saha, Advances in Design and Synthesis of Stabilized Zero-Valent Iron Nanoparticles for Groundwater Remediation, J. Environ. Chem. Eng., 2022, 10(3), 107993, DOI:10.1016/j.jece.2022.107993.
- D. Colón, E. J. Weber and J. L. Anderson, Effect of Natural Organic Matter on the Reduction of Nitroaromatics by Fe(II) Species, Environ. Sci. Technol., 2008, 42(17), 6538–6543, DOI:10.1021/es8004249.
- T. B. Hofstetter, C. G. Heijman, S. B. Haderlein, C. Holliger and R. P. Schwarzenbach, Complete Reduction of TNT and Other (Poly)Nitroaromatic Compounds under Iron-Reducing Subsurface Conditions, Environ. Sci. Technol., 1999, 33(9), 1479–1487, DOI:10.1021/es9809760.
- T. P. Klupinski and Y. P. Chin, Abiotic Degradation of Trifluralin by Fe(II): Kinetics and Transformation Pathways, Environ. Sci. Technol., 2003, 37(7), 1311–1318, DOI:10.1021/es025673r.
- T. P. Klupinski, Y.-P. Chin and S. J. Traina, Abiotic Degradation of pentachloronitrobenzene by Fe(II): Reactions on Goethite and Iron Oxide Nanoparticles, Environ. Sci. Technol., 2004, 38(16), 4353–4360, DOI:10.1021/es035434j.
- K. B. Gregory, P. Larese-Casanova, G. F. Parkin and M. M. Scherer, Abiotic Transformation of Hexahydro-1,3,5-Trinitro-1,3,5-Triazine by FeII Bound to Magnetite, Environ. Sci. Technol., 2004, 38, 1408–1414, DOI:10.1021/es034588w.
- S. Wang and W. A. Arnold, Abiotic Reduction of Dinitroaniline Herbicides, Water Res., 2003, 37(17), 4191–4201, DOI:10.1016/S0043-1354(03)00340-3.
- X. Huang, X. Hou, X. Zhang, K. M. Rosso and L. Zhang, Facet-Dependent Contaminant Removal Properties of Hematite Nanocrystals and Their Environmental Implications, Environ. Sci.: Nano, 2018, 5(8), 1790–1806, 10.1039/C8EN00548F.
- P. A. Cárdenas-Hernández, K. A. Anderson, J. Murillo-Gelvez, D. M. Di Toro, H. E. Allen, R. F. Carbonaro and P. C. Chiu, Reduction of 3-Nitro-1,2,4-Triazol-5-One (NTO) by the Hematite-Aqueous Fe(II) Redox Couple, Environ. Sci. Technol., 2020, 54(19), 12191–12201, DOI:10.1021/acs.est.0c03872.
- M. J. Berens, T. B. Hofstetter, J. Bolotin and W. A. Arnold, Assessment of 2,4-Dinitroanisole Transformation Using Compound-Specific Isotope Analysis after In Situ Chemical Reduction of Iron Oxides, Environ. Sci. Technol., 2020, 54(9), 5520–5531, DOI:10.1021/acs.est.9b07616.
- T. M. Anggraini, S. An, S. H. Kim, M. J. Kwon, J. Chung and S. Lee, Influence of Iron (Hydr)Oxide Mineralogy and Contents in Aquifer Sediments on Dissolved Organic Carbon Attenuations during Aquifer Storage and Recovery, Chemosphere, 2024, 351, 141196, DOI:10.1016/j.chemosphere.2024.141196.
- F. Luan, L. Xie, J. Li and Q. Zhou, Abiotic Reduction of Nitroaromatic Compounds by Fe(II) Associated with Iron Oxides and Humic Acid, Chemosphere, 2013, 91(7), 1035–1041, DOI:10.1016/j.chemosphere.2013.01.070.
- J. Klausen, S. P. Troeber, S. B. Haderlein and R. P. Schwarzenbach, Reduction of Substituted Nitrobenzenes by Fe(II) in Aqueous Mineral Suspensions, Environ. Sci. Technol., 1995, 29(9), 2396–2404, DOI:10.1021/es00009a036.
- C. L. Chun, R. L. Penn and W. A. Arnold, Kinetic and Microscopic Studies of Reductive Transformations of Organic Contaminants on Goethite, Environ. Sci. Technol., 2006, 40(10), 3299–3304, DOI:10.1021/es0600983.
- J. L. Voelz, W. A. Arnold and R. L. Penn, Redox-Induced Nucleation and Growth of Goethite on Synthetic Hematite Nanoparticles, Am. Mineral., 2018, 103(7), 1021–1029, DOI:10.2138/am-2018-6342.
- J. L. Voelz, K. K. Hobart, K. A. Stahovich, H. E. Ziebol, N. A. Harper, J. M. Feinberg, W. A. Arnold and R. L. Penn, Organic Matter Inhibits Redox Activity and Impacts Heterogeneous Growth of Iron (Oxyhydr)Oxides on Nano-Hematite, ACS Earth Space Chem., 2022, 6(4), 847–860, DOI:10.1021/acsearthspacechem.1c00419.
- A. M. Vindedahl, M. S. Stemig, W. A. Arnold and R. L. Penn, Character of Humic Substances as a Predictor for Goethite Nanoparticle Reactivity and Aggregation, Environ. Sci. Technol., 2016, 50(3), 1200–1208, DOI:10.1021/acs.est.5b04136.
- J. H. Strehlau, J. D. Schultz, A. M. Vindedahl, W. A. Arnold and R. L. Penn, Effect of Nonreactive Kaolinite on 4-Chloronitrobenzene Reduction by Fe(II) in Goethite–Kaolinite Heterogeneous Suspensions, Environ. Sci.: Nano, 2017, 4(2), 325–334, 10.1039/C6EN00469E.
- J. H. Strehlau, M. S. Stemig, R. L. Penn and W. A. Arnold, Facet-Dependent Oxidative Goethite Growth As a Function of Aqueous Solution Conditions, Environ. Sci. Technol., 2016, 50(19), 10406–10412, DOI:10.1021/acs.est.6b02436.
- S. M. Stewart, T. B. Hofstetter, P. Joshi and C. A. Gorski, Linking Thermodynamics to Pollutant Reduction Kinetics by Fe2+ Bound to Iron Oxides, Environ. Sci. Technol., 2018, 52(10), 5600–5609, DOI:10.1021/acs.est.8b00481.
- P. Larese-Casanova, A. Kappler and S. B. Haderlein, Heterogeneous Oxidation of Fe(II) on Iron Oxides in Aqueous Systems: Identification and Controls of Fe(III) Product Formation, Geochim. Cosmochim. Acta, 2012, 91, 171–186, DOI:10.1016/j.gca.2012.05.031.
- D. Colón, E. J. Weber and J. L. Anderson, QSAR Study of the Reduction of Nitroaromatics by Fe(II) Species, Environ. Sci. Technol., 2006, 40(16), 4976–4982, DOI:10.1021/es052425x.
- D. Colón, E. J. Weber, J. L. Anderson, P. Winget and L. A. Suárez, Reduction of Nitrosobenzenes and N-Hydroxylanilines by Fe(II) Species: Elucidation of the Reaction Mechanism, Environ. Sci. Technol., 2006, 40(14), 4449–4454, DOI:10.1021/es0600429.
- C. A. Gorski, R. Edwards, M. Sander, T. B. Hofstetter and S. M. Stewart, Thermodynamic Characterization of Iron Oxide–Aqueous Fe2+ Redox Couples, Environ. Sci. Technol., 2016, 50(16), 8538–8547, DOI:10.1021/acs.est.6b02661.
- P. A. Cárdenas-Hernández, K. Hickey, D. M. Di Toro, H. E. Allen, R. F. Carbonaro and P. C. Chiu, Linear Free Energy Relationship for Predicting the Rate Constants of Munition Compound Reduction by the Fe(II)–Hematite and Fe(II)–Goethite Redox Couples, Environ. Sci. Technol., 2023, 57(36), 13646–13657, DOI:10.1021/acs.est.3c04714.
- J. Greskowiak, H. Prommer, G. Massmann, C. D. Johnston, G. Nützmann and A. Pekdeger, The Impact of Variably Saturated Conditions on Hydrogeochemical Changes during Artificial Recharge of Groundwater, Appl. Geochem., 2005, 20(7), 1409–1426, DOI:10.1016/j.apgeochem.2005.03.002.
- R. Simon, D. Colón, C. L. Tebes-Stevens and E. J. Weber, Effect of Redox Zonation on the Reductive Transformation of p-Cyanonitrobenzene in a Laboratory Sediment Column, Environ. Sci. Technol., 2000, 34(17), 3617–3622, DOI:10.1021/es000960l.
- A. Soroush, R. L. Penn and W. A. Arnold, Anisotropic Oxidative Growth of Goethite-Coated Sand Particles in Column Reactors during 4-Chloronitrobenzene Reduction by Fe(II)/Goethite, Environ. Sci.: Nano, 2022, 9(1), 275–288, 10.1039/d1en00788b.
- A. Soroush, C. M. Harris, A. M. Hildebrandt, M. Cruz-Reyes, R. L. Penn and W. A. Arnold, Interactions of Goethite and Natural Organic Matter: Fractionation and Impact on Contaminant Reduction, ACS Earth Space Chem., 2024, 8(2), 393–404, DOI:10.1021/acsearthspacechem.3c00334.
- P. Larese-Casanova and M. M. Scherer, Fe(II) Sorption on Hematite: New Insights Based on Spectroscopic Measurements, Environ. Sci. Technol., 2007, 41(2), 471–477, DOI:10.1021/es0617035.
- L. L. Stookey, Ferrozine---a New Spectrophotometric Reagent for Iron, Anal. Chem., 1970, 42(7), 779–781, DOI:10.1021/ac60289a016.
- A. R. Muxworthy and E. McClelland, Review of the Low-Temperature Magnetic Properties of Magnetite from a Rock Magnetic Perspective, Geophys. J. Int., 2000, 140(1), 101–114, DOI:10.1046/j.1365-246x.2000.00999.x.
- A. M. Hirt, L. Lanci, J. Dobson, P. Weidler and A. U. Gehring, Low-Temperature Magnetic Properties of Lepidocrocite, J. Geophys. Res.: Solid Earth, 2002, 107(B1), EPM 5, DOI:10.1029/2001JB000242.
- Ö. Özdemir, D. J. Dunlop and T. S. Berquó, Morin Transition in Hematite: Size Dependence and Thermal Hysteresis, Geochem., Geophys., Geosyst., 2008, 9(10) DOI:10.1029/2008GC002110.
- Q. Liu, Y. Yu, J. Torrent, A. P. Roberts, Y. Pan and R. Zhu, Characteristic Low-Temperature Magnetic Properties of Aluminous Goethite [α-(Fe, Al)OOH] Explained, J. Geophys. Res.: Solid Earth, 2006, 111(B12) DOI:10.1029/2006JB004560.
- C. A. Schultz and T. J. Grundl, pH Dependence on Reduction Rate of 4-Cl-Nitrobenzene by Fe(II)/Montmorillonite Systems, Environ. Sci. Technol., 2000, 34(17), 3641–3648, DOI:10.1021/es990931e.
- C. G. Heijman, E. Grieder, C. Holliger and R. P. Schwarzenbach, Reduction of Nitroaromatic Compounds Coupled to Microbial Iron Reduction in Laboratory Aquifer Columns, Environ. Sci. Technol., 1995, 29(3), 775–783, DOI:10.1021/es00003a027.
- B.-H. Jeon, B. A. Dempsey, W. D. Burgos and R. A. Royer, Reactions of Ferrous Iron with Hematite, Colloids Surf., A, 2001, 191(1), 41–55, DOI:10.1016/S0927-7757(01)00762-2.
- B.-H. Jeon, B. A. Dempsey, W. D. Burgos, R. A. Royer and E. E. Roden, Modeling the Sorption Kinetics of Divalent Metal Ions to Hematite, Water Res., 2004, 38(10), 2499–2504, DOI:10.1016/j.watres.2004.03.003.
- G. Chen, T. B. Hofstetter and C. A. Gorski, Role of Carbonate in Thermodynamic Relationships Describing Pollutant Reduction Kinetics by Iron Oxide-Bound Fe2+, Environ. Sci. Technol., 2020, 54(16), 10109–10117, DOI:10.1021/acs.est.0c02959.
- J. P. Gustafsson, Visual MINTEQ, 2023, https://vminteq.com/ (accessed 2024-10-07) Search PubMed.
- C. M. Johnston, Z. R. Wiethorn, R. A. Combs, M. Cruz-Reyes, S. Lodico, H. Nasri, W. A. Arnold and R. L. Penn, Reductive Degradation of 4-Chloronitrobenzene by Fe(II) on Aluminum-Substituted Goethite Nanoparticles, ACS Earth Space Chem., 2024, 8(3), 457–466, DOI:10.1021/acsearthspacechem.3c00126.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4en00602j |
| This journal is © The Royal Society of Chemistry 2025 |