
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Harnessing borane-potassium cooperativity for sulfurated ring-opening copolymerisation†
Bhargav R.
Manjunatha
 a,
Kailey
Sun Marcus
b,
Rosa M.
Gomila
c,
Antonio
Frontera
c and
Alex J.
Plajer
*ad
a,
Kailey
Sun Marcus
b,
Rosa M.
Gomila
c,
Antonio
Frontera
c and
Alex J.
Plajer
*ad
aMakromolekulare Chemie, Universität Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: alex.plajer@uni-bayreuth.de
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34-36, Berlin 14195, Germany
cDepartament de Química, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma, Baleares, Spain
dBayrisches Polymer Institut (BPI), Universität Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany
First published on 26th February 2025
Abstract
Sulfur-containing polymers, such as thioesters and thiocarbonates, can exhibit improved thermal properties and degradability compared to their all-oxygen analogues, yet their synthesis remains challenging. In this respect, ring-opening copolymerization (ROCOP) offers access to sulfur-containing polymers; however, the catalysts used for this process often rely on toxic, expensive or synthetically complex components. Here, we demonstrate that combining commercial borane Lewis acids with easily accessible potassium acetate crown ether complexes highly selectively mediates the ring-opening copolymerization of oxetanes with a wide range of sulfur-containing monomers. Mechanistic investigations clearly indicate a cooperative mode of action between boron and potassium, yielding high-melting, semicrystalline materials that exhibit improved thermal stability compared to those generated via chromium catalysis. Our study establishes new concepts in cooperative catalysis to produce sustainable materials that are otherwise difficult to access.
 Alex J. Plajer | Alex Plajer is a Junior Professor at the University of Bayreuth. Born in Mannheim, Germany, he studied chemistry at Heidelberg University graduating top of his class. He then completed an M.Phil. and Ph.D. at the University of Cambridge, supported by a Vice-Chancellor's Scholarship. Following his doctorate, he was awarded a prestigious 1851 Fellowship at the University of Oxford. He then secured a Liebig Fellowship at Freie Universität Berlin, establishing his independent research. In 2024, he joined Bayreuth as a tenure-track professor, continuing his work on sustainable polymer materials focusing on how sulfur (and soon other more inorganic elements) shape catalysis and properties. |
Green foundation1. We set a precedent for sustainable catalysis, providing access to more degradable and recyclable alternatives to current commodity materials.2. We achieve the synthesis of sulfur-containing polymers using catalysts based on inexpensive, abundant, and colorless elements, replacing the previously required expensive, toxic, and colored chromium-based compounds. 3. Our catalysis still relies on air-sensitive boranes, necessitating specialized and more energy-intensive setups compared to standard benchtop conditions. Additionally, monomer synthesis yields require further optimization. |
Introduction
Incorporating sulfur atoms into polymer main chains can enhance material properties, for example, by improving semi-crystallinity compared to all-oxygen analogues, while also providing access to oxidative degradation pathways unavailable in current commodity materials.1–13 Sulfur-containing polymers, such as polythioesters and polythiocarbonates, are typically synthesized via polycondensation or ring-opening polymerization (ROP), but these methods allow access to only a limited range of polymer structures.14–16 In contrast, ring-opening copolymerization (ROCOP) of strained heterocycles with heteroallenes or (thio)anhydrides has emerged as a popular approach for synthesizing sulfur-containing copolymers, for example yielding polythioesters from the ROCOP of cyclic thioanhydrides with oxygenated heterocycles, and polythiocarbonates from carbon disulfide.17–23 Although alternating enchainment of the two monomers, resulting in perfect microstructures, can be achieved, transesterification side reactions often occur, leading to a scrambled polymer microstructure—commonly referred to as O/S scrambling.24–32 This scrambling process negatively impacts material properties, particularly by reducing or altogether eliminating semi-crystallinity, resulting in disordered polymers.Addressing these issues, a series of selective sulfurated ROCOPs involving oxetane have recently been reported, mediated by heterobimetallic catalysts.33–36 For example, ROCOP of phthalic thioanhydride (PTA) with oxetane yields alternating poly(trimethylene-ester-alt-thioesters), ROCOP of CS2 with oxetane yields poly(trimethylene dithiocarbonates), and ROCOP of PhNCS with oxetane yields the corresponding poly(monothioimidocarbonates). In these the improved nucleophilicity of sulfurated intermediates after PTA, CS2 or PhNCS insertion achieves efficient ring opening of the four membered oxetane ring that typically requires activation by strong Lewis acids.36b,c Unfortunately, this process relies on toxic chromium(III) coordinated within a non-commercial, complex-to-synthesize ligand scaffold, limiting its utility in other research laboratories. Furthermore, the polymers obtained are often heavily discolored, indicating residual catalyst contamination even after multiple purification steps. Residual traces of toxic catalysts not only render the materials unsuitable for consumer applications but can also negatively impact thermal properties.37 To address this issue, borane catalysis, a highly effective methodology in oxygenated ROCOP, presents a potential solution.38,39 Indeed, BEt3-based borane catalysis has demonstrated selective sulfurated ROCOP, although Ph3PNPPh3Cl (PPNCl) cocatalyst was required.40 Again, PPNCl is expensive and incompatible with larger-scale reactor setups, while Phosphazene residues in polymers are also known to by cytotoxic.41 Therefore, viable alternatives are needed.
Addressing this problem, we report that the combination of BEt3 with KOAc complexed by 18-crown-6 acts as a fast and highly selective catalyst for the copolymerization of PTA with oxetane, yielding perfectly alternating poly(ester-alt-thioester) as well as enabling a range of other ROCOPs to access a wide variety of sulfur-containing polymers. The high degree of alternation results in semi-crystalline materials with melting points up to 180 °C, while the facile deactivation of the catalyst improves the thermal stability of the polymers by up to 100 °C compared to previous reports.
Results and discussion
Attempting to achieve chromium-free PTA/OX ROCOP, we first employed KOAc (Table 1, run #1) at a loading of 1 eq. Cat.: 300 eq. PTA: 600 eq. OX at 80 °C, as alkali metal acetates had previously proven to be viable ROCOP catalysts.42–44 However, in this case, no polymerization occurred. Similarly, BEt3 alone showed no catalytic activity. Interestingly, when KOAc and BEt3 were combined (Table 1, run #5), a poly(ester-alt-thioester) was formed with quantitative polymer selectivity achieving 78% PTA conversion after 24 hours. In this system KOAc represents the cocatalyst, effectively replacing PPNCl which is usually referred to as the cocatalyst, and BEt3 represents the catalyst. To the best of our knowledge, this catalyst pair has not been successfully employed in the past in such polymerisation reactions.| No. | Catalyst BEt3 (1) + X | Temp. (°C) | Time (h) | PTA conv.a (%) | TOF (h−1) | M n (Đ)b (kg mol−1) |
|---|---|---|---|---|---|---|
| ROCOPs conducted with 1 eq. Cat. + coCat.: 300 eq. PTA: 600 eq. OX.a Relative integral of aromatic resonances from residual PTA versus polymer in the normalised 1H NMR (CDCl3, 400 MHz) spectrum of crude mixture.b Determined by GPC (gel permeation chromatography) measurements in THF calibrated against a narrow polystyrene standard. L = (S,S)-N,N′-Bis(3,5-di-tert-butylsalicyliden)-1,2-cyclohexandiamin also known as SalCy. TOF – turnover frequency calculated w.r.t to equivalents of converted PTA per equivalent of catalyst. | ||||||
| 1 | KOAc | 80 | 24 | 18 | 2.3 | — |
| 2 | 1 | 80 | 24 | 0 | 0.0 | — |
| 3 | 1 + LiOAc | 80 | 24 | 20 | 2.5 | 14.2 (1.2) |
| 4 | 1 + NaOAc | 80 | 24 | 66 | 8.3 | 33.9 (1.1) |
| 5 | 1 + KOAc | 80 | 24 | 78 | 9.8 | 25.1 (1.2) |
| 6 | 1 + RbOAc | 80 | 24 | 64 | 8.0 | 24.6 (1.4) |
| 7 | 1 + Mg(OAc)2 | 80 | 24 | 16 | 2.0 | 9.4 (1.3) |
| 8 | 1 + KPF6 | 80 | 24 | 0 | 0.0 | — |
| 9 | 1 + KOAc@18c6 | 80 | 1 | 98 | 294.0 | 31.4 (1.2) |
| 10 | BPh3 + KOAc@18c6 | 80 | 6.3 | 80 | 38.1 | 23.8 (1.2) |
| 11 | (C6F5)3B + KOAc@18c6 | 80 | 6 | 0 | 0.0 | — |
| 12 | LAlCl + KOAc@18c6 | 80 | 18 | 98 | 16.3 | 21.4 (1.5) |
| 13 | 1 + KOAc@18c6 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
80 | 0.66 | 96 | 436.0 | 27.0 (1.5) |
| 14 | 1 + KCl@18c6 | 80 | 10 | 74 | 22.2 | 12.6 (1.9) |
| 15 | 1 + KOAc@18c6 | 60 | 3 | 100 | 100.0 | 33.2 (1.4) |
| 16 | 1 + KOAc@18c6 | 100 | 0.5 | 88 | 528.0 | 23.4 (1.2) |
| 17 | 1 + KOAc@18c6 | 120 | 1.6 | 74 | 138.8 | 18.9 (1.4) |
| 18 | 1 + KOAc@18c6 | 140 | 0.8 | 60 | 225.0 | 13.7 (1.5) |
The polymer can be easily isolated via precipitation with methanol. Gel permeation chromatography (GPC) reveals a molecular weight of Mn = 25.1 kg mol−1 and a polydispersity of Đ = 1.2. The 1H-NMR spectrum of the polymer shows resonances at 7.51, 7.58, and 7.82 ppm, corresponding to an ester-thioester-substituted phthalate unit, and at 2.12, 3.18, and 4.39 ppm, corresponding to the three CH2 groups from ring-opened oxetane, which in reference to previous studies, supports a perfectly alternating microstructure without ether linkages from oxetane homopropagation or linkages from O/S scrambling. 13C-NMR spectroscopy confirms this exhibiting sharp quarternary ester and thioester resonances at 166.59 and 193.57 ppm respectively (see ESI Fig. S4†). The effect of the metal cation on the ROCOP was investigated by employing different alkali and alkaline earth metal acetates as co-catalyst in combination with BEt3. Rubidium acetate (Table 1, run #6) resulted in similar molecular weights and lower turnover frequency (TOF) compared to potassium. Lithium and magnesium acetate (Table 1, run #3, #7) showed much slower rates and molecular weights. Sodium acetate showed lower TOF and somewhat improved molecular weight. Combined these results clearly indicate that the metal cation represents a real catalyst rather than just a mere spectator to the propagating chain end. Investigating the role of the acetate anion, a potassium salt with a weakly coordinating and non-nucleophilic anion KPF6 was employed as a cocatalyst but did not result in polymer formation (see ESI Fig. S14†). This suggests that acetate represents the initiator of the polymerisation. Accordingly, we performed ROCOP at a reduced monomer loading (see ESI section S2†) and could indeed identify methyl ester resonances in the 1H and 13C NMR spectra formed from acetate initiation which could further be confirmed by MALDI-TOF mass spectrometry (see ESI Fig. S29†). Furthermore, signals corresponding to propane diol initiated chains could be identified indicating co-initiated chain from alcohol impurities in the oxetane monomer. This produces a mixture of α-OAc,ω-OH and α,ω-OH terminated chains also explaining the bimodality of molecular weight distributions as reported for the previous chromium base catalysts.33–35
As the crown ether 18-crown-6, represents an ideal size fit for the potassium cation, we hypothesized that this could potentially enhance catalytic activity by breaking up aggregates of potassium salts and increase nucleophilicity of the anion. Indeed, employing the 18-crown-6 complex of potassium acetate (KOAc@18c6) with BEt3 (Table 1, run #9) substantially accelerated the polymerization, resulting in near-quantitative PTA conversion within 1 hour and yielding perfectly alternating poly(ester-alt-thioester) with Mn = 31.4 kg mol−1 (Đ = 1.2) (Fig. 1). Having demonstrated that the coordinative environment of potassium influences ROCOP, we then turned our attention to the effect of the Lewis acid, with the goal of investigating the potentially cooperative mode of action between boron and potassium.
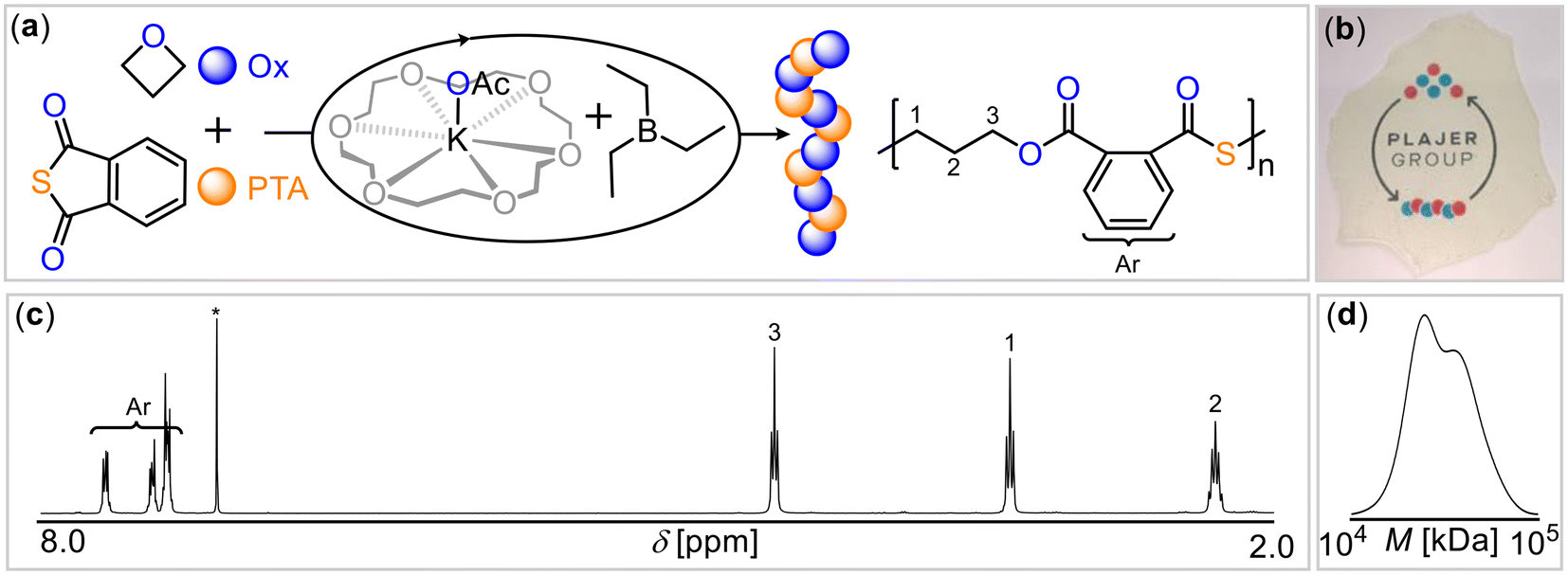 | ||
| Fig. 1 (a) PTA/OX ROCOP scheme. (b) Photograph of polymer film. (c) 1H spectrum (CDCl3*, 400 MHz) as well as (d) gel-permeation chromatogram (GPC) of copolymer corresponding to Table 1 run #9. | ||
Replacing BEt3 with its more Lewis acidic aromatic variant, BPh3 (Table 1, run #10), slowed down the polymerization, requiring six times longer and resulting in a slightly decreased PTA conversion of 80%, yielding a polymer with Mn = 23.8 kg mol−1 and a good polydispersity of Đ = 1.2. Moving to (C6F5)3B resulted in no catalytic activity (Table 1, run #11), consistent with the observation that increased borane Lewis acidity negatively correlates with catalyst activity. When the commercial (S,S)-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamino aluminum chloride complex (also known as Salcy) was used in place of BEt3 (Table 1, run #12), the ROCOP was much slower and 98% conversion was achieved in 18 h while the Mn was 21.4 kg mol−1 (Đ = 1.5). Increasing the stoichiometry of the Lewis acid, BEt3, relative to KOAc@18c6 led to slightly faster ROCOP; however, this came at the expense of polydispersity, yielding a polymer with a broader molecular weight distribution of Đ = 1.5. Similarly, moving away from the acetate coligand, which likely acts as an initiator, negatively impacted catalysis. Replacing acetate with chloride, i.e., KCl@18c6 (Table 1, run #14), slowed down ROCOP by more than an order of magnitude, achieving 74% conversion in 10 hours and resulting in decreased molecular weights and much broader polydispersity of Đ = 1.9. This suggests not only slower initiation by the chloride coligand but also more side reactions, such as chain-end coupling involving the alkyl chloride chain ends.45
To study the effect of reaction temperature, we performed polymerisation runs analogous to run #9 at temperatures ranging from 60 °C to 140 °C (Table 1, run #15 to #18). Reaction times were adapted in a way that excessive continuation of the polymerisation past full monomer consumption is avoided as we previously showed this to negatively affect sequence selectivity due to intermolecular scrambling reactions which can also lead to polymer degradation.36a As expected, decreasing the reaction temperature slowed down ROCOP, taking three times as long to achieve complete PTA conversion. Increasing the temperature to 100 °C resulted in faster polymerisation, requiring 30 minutes to reach 88% PTA conversion, with good polydispersities, although with a lower molecular weight of Mn = 23.4 kg mol−1 than anticipated based on conversion compared to run #9. This trend continued at 120 °C and 140 °C, with no significant rate increase observed beyond 80 °C, suggesting catalyst degradation at these higher temperatures. Remarkably, across all runs presented in Table 1, perfectly alternating poly(ester-alt-thioester) was consistently produced, demonstrating the catalyst system's ability to suppress O/S scrambling reactions under a range of conditions.
The clear dependence of catalyst performance on both the borane Lewis acid and the potassium coordination environment indicates that both components act cooperatively to achieve copolymerization. In the PTA/OX system, the first step after initiation involves oxetane ring-opening by the acetate initiator, generating an alkoxide chain end. This is followed by propagation via PTA insertion, forming a thiocarboxylate chain end, which subsequently inserts oxetane to regenerate the alkoxide. To investigate this further, we studied the polymerization kinetics via in situ IR spectroscopy. First, ROCOP was conducted in the presence of excess oxetane, monitoring the decrease in PTA concentration. A constant linear decline was observed (R2 = 0.98 by linear fitting), indicating that the reaction rate is zero-order (or constant) with respect to the PTA concentration in the reaction mixture. In contrast, when polymerization was conducted with excess PTA, monitoring oxetane consumption showed a clear concentration dependence, although the precise reaction order could not be determined. Nevertheless, this indicates that oxetane ring opening is the rate-determining step of the ROCOP.
To understand how the two catalyst components facilitate this step, we performed density functional theory (DFT) calculations at the BP86-D4/def2TZVP level of theory. We considered three scenarios for the oxetane ring-opening step, in which the sulfur atoms remain uncoordinated, allowing for nucleophilic attack (see Fig. 2(a)). To assess whether potassium actively participates or merely serves as a counterbalancing cation, we calculated the energy barriers for the three transition states. For these calculations, the thiocarboxylate chain-end was simplified to a thiobenzoate for computational efficiency.
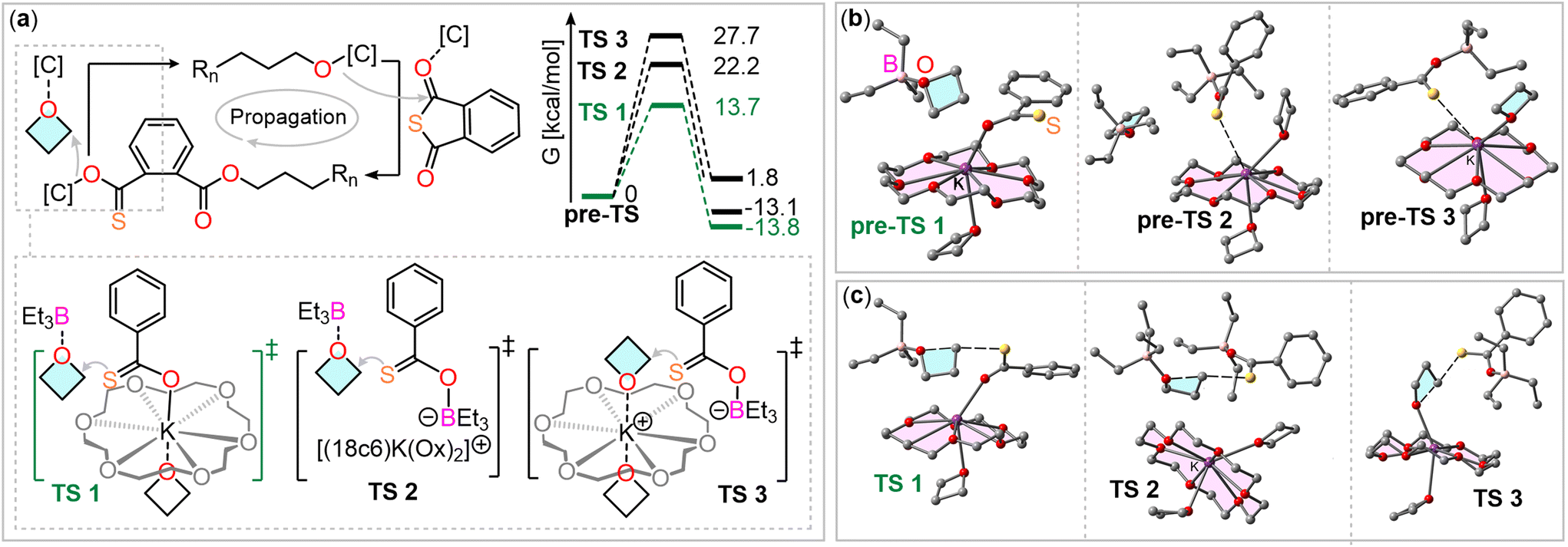 | ||
| Fig. 2 (a) Comparison of different transitions state in the oxetane ring-opening step of PTA/OX ROCOP. Fully optimized (b) “pre-transition state (pre-TS)” and (c) TS structures for the three routes analyzed. H-atoms omitted for clarity. Level of theory BP86-D4/def2TZVP. | ||
The first investigated transition state (TS1, Fig. 2(a)) involves coordination of the inserted oxetane with BEt3, while the thiocarboxylate is coordinated to potassium. In the second scenario (TS2), both reaction partners are coordinated to two different BEt3 molecules. The third scenario (TS3) involves coordination of the inserted oxetane to potassium and the thiocarboxylate to BEt3. In all cases, the coordination sphere of potassium is completed by an equivalent of oxetane in axial position to the crown ether plane.
Fig. 2(b) presents the optimized structures prior to oxetane insertion for the three scenarios, serving as starting points for examining the oxetane ring-opening mechanism. These structures are referred to as “pre-transition states” (pre-TS). In route 1 (pre-TS1), oxetane is positioned near the thiobenzoate ring. In route 2, the interaction between the thiobenzoate-BEt3 adduct and potassium (from K@18c6) occurs at a distance of 3.29 Å, which is shorter than the combined van der Waals radii (3.40 Å), suggesting a noncovalent interaction. This likely reduces the sulfur atom's nucleophilicity. In route 3, a similar sulfur-potassium interaction is observed, facilitating the proximity of the nucleophile and electrophile. However, this interaction also reduces the nucleophilicity of the sulfur atom and the electrophilicity of the carbon atom, as the interaction between the oxetane oxygen and potassium weakens.
We then investigated the transition states for all three routes, with the corresponding energy profiles depicted in Fig. 2(a). Notably, the energy barrier for route 1 is significantly lower (13.7 kcal mol−1) compared to the others (22.2 kcal mol−1 for TS2 and 27.7 kcal mol−1 for TS3). This suggests that the propagation mechanism most likely involves coordination of the thiobenzoate chain end to a K@18c6 complex via the oxygen atom, with the oxetane ring being activated through coordination with the BEt3 Lewis acid. Additionally, the process is exergonic for routes 1 and 2 (approximately −13 kcal mol−1), but slightly endergonic for route 3 (1.8 kcal mol−1). These results contrast with what is commonly observed in borane-catalyzed ROCOP.38,46 Typically, pathways involving activation of both the propagating chain end and the ring-opened heterocycle, coordinated to boranes, are the most favorable. However, in the BEt3/KOAc@18c6 system, we infer that the potassium cation, which balances the charge of the anionic chain end, actively participates in the oxetane ring-opening step. Fig. 2(c) presents the optimized geometries of the transition states, all demonstrating a typical SN2 mechanism characterized by an almost linear S⋯C⋯O angle (>174°). Notably, the noncovalent S⋯K interaction, seen in the initial pre-TS2 and pre-TS3 structures, is absent in the TS2 and TS3 geometries. This absence likely accounts for the higher energy barriers observed for these transition states. In the transition states, the O⋯C distances range from 1.85 to 1.99 Å, and the S⋯C distances span 2.40 to 2.55 Å. These measurements corroborate the SN2 mechanism, characterized by simultaneous bond formation and breaking.
Having demonstrated the cooperative mode of action between the borane and potassium centers, we applied this strategy to other monomer combinations (Fig. 3). Based on the results above, the BEt3/KOAc@18c6 catalyst at 80 °C showed the overall best performance, and we continued our study under these conditions. Replacing PTA with an aliphatic anhydride, succinic thioanhydride (STA, Table 2, run #2), resulted in the formation of an aliphatic polythioester. However, the polymer had a substantially reduced molecular weight and broad polydispersity with some scrambled links occurring due to O/S exchange. We also explored the use of 3,3′-disubstituted oxetanes (Table 2, run #3 to #6) as comonomers with PTA for ROCOP. Specifically, we used 3,3′-dimethyloxetane (OXMe) as well as various 3-methyl,3′-methylenetheroxetanes with ethyl (OXOEt), benzyl (OXOBn), and allyl (OXOAll) groups. All substituted oxetanes required higher temperatures (120 to 140 °C) and longer reaction times for ROCOP, resulting in materials with Mn = 4.0–9.6 kDa and Đ = 1.2–1.9. For all the polymers, minor resonances, in addition to the symmetric sets of resonances corresponding to links from alternating copolymerization, were observed in the 1H and 13C NMR spectra (ESI Fig. S58 to S77†). These minor resonances correspond to some disordered links, arising from O/S scrambling side reactions.24
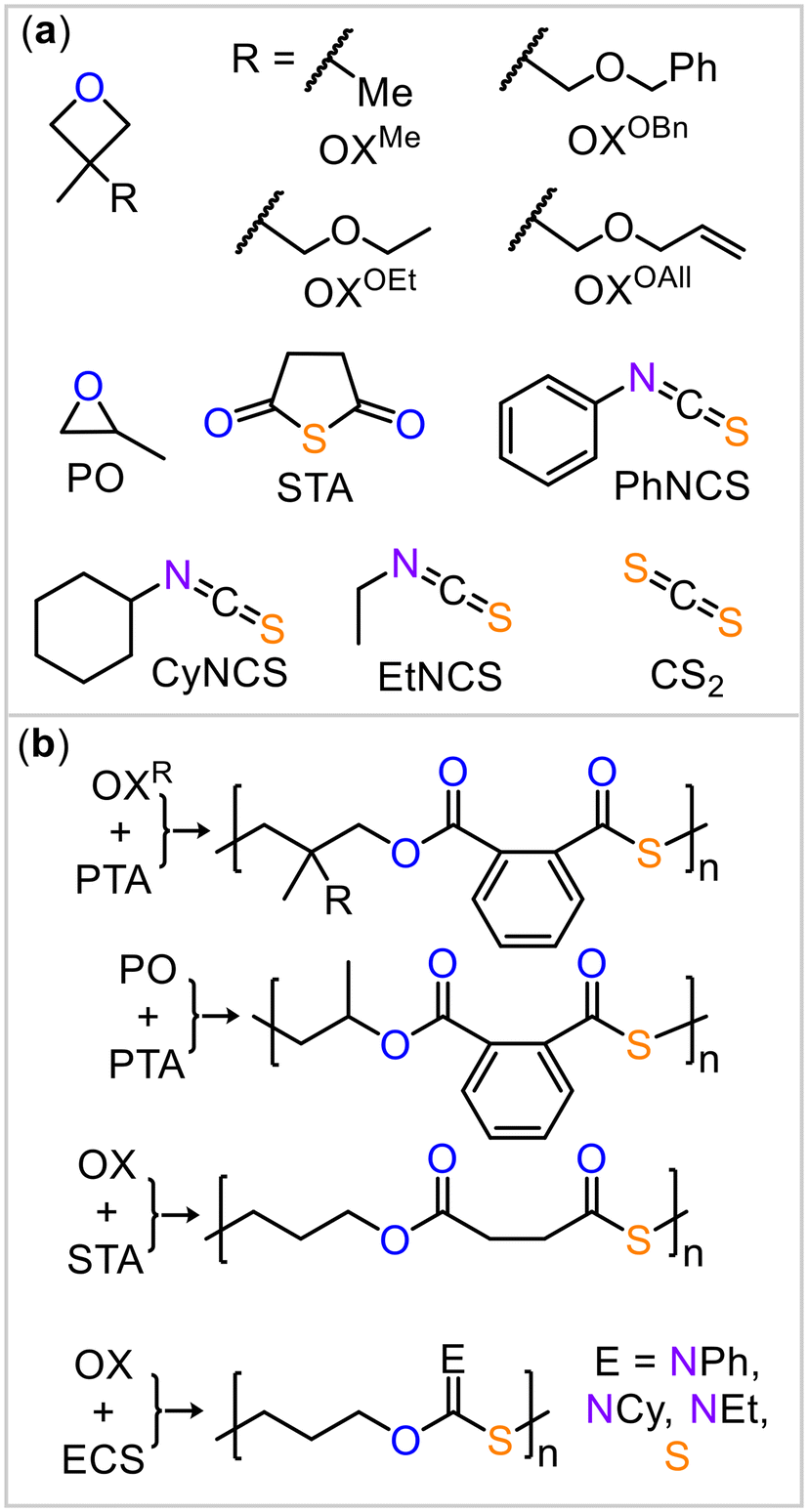 | ||
| Fig. 3 (a) Monomer scope explored in this study. (b) Ideal polymer structure resulting from the employed monomers. | ||
:1)
| No. | Monomers (X, Y) | Temp. (°C) | Time (h) | Conversion of Xa (%) | Pol.b (%) | M n(Đ)c (kg mol−1) |
T
d,5%d (°C) |
T
g, Tme (°C) |
|---|---|---|---|---|---|---|---|---|
ROCOPs conducted with 1 eq. Cat. + coCat.: 300 eq. PTA: 600 eq. OX.a Relative integral of aromatic resonances from residual monomer X versus polymer in the normalised 1H NMR (CDCl3, 400 MHz) spectrum of crude mixture.b Relative integral in the normalised 1H NMR spectrum of deconvoluted resonances from –O–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPh)–S– (ONS) repeat units versus other signals.c Determined by GPC (gel permeation chromatography) measurements in THF versus a narrow polystyrene standard.d Degradation temperature Td,5% determined by thermogravimetric analysis.e Melting point Tm determined from first heating scan of the as-synthesised polymer; glass transition temperature Tg determined from the second cycle by differential scanning calorimetry.f Conversion/selectivity not determined due to overlapping signals in 1H NMR spectrum.g Due to insolubility polymer purification by precipitation could not be performed potentially explaining the decrease in Td. NPh)–S– (ONS) repeat units versus other signals.c Determined by GPC (gel permeation chromatography) measurements in THF versus a narrow polystyrene standard.d Degradation temperature Td,5% determined by thermogravimetric analysis.e Melting point Tm determined from first heating scan of the as-synthesised polymer; glass transition temperature Tg determined from the second cycle by differential scanning calorimetry.f Conversion/selectivity not determined due to overlapping signals in 1H NMR spectrum.g Due to insolubility polymer purification by precipitation could not be performed potentially explaining the decrease in Td. |
||||||||
| 1 | PTA, OX | 80 | 1 | 98 | >99 | 31.4 (1.2) | 312.3 | T g = 28.3 |
| T m = 83.5 | ||||||||
| 2 | STA, OX | 80 | 72 | —f | >99 | 2.2 (3.0) | 268.6 | T g = −33.5 |
| 3 | PTA, OX-Me2 | 80 | 144 | 90 | >99 | 4.0 (1.2) | 332.6 | T g = 42.2 |
| 4 | PTA, OX-OEt | 120 | 25 | 68 | >99 | 8.6 (1.4) | 310.6 | T g = 15.5 |
| 5 | PTA, OX-OBn | 140 | 88 | >99 | >99 | 4.8 (1.5) | 301.0 | T g = 17.5 |
| 6 | PTA, OX-OAll | 120 | 72 | 48 | >99 | 9.6 (1.9) | 308.3 | T g = 18.2 |
| 7 | PTA, PO | 50 | 9 | 84 | 99 | 25.3 (1.7) | 243.1 | T g = 63.0 |
| 8 | PhNCS, OX | 80 | 0.33 | —f | >99 | 28.9 (1.2) | 228.0 | T g = 30.5 |
| T m = 180.1 | ||||||||
| 9 | CyNCS, OX | 80 | 17.5 | 100 | >99 | 14.9 (1.2) | 244.3 | T g = 5.8 |
| 10 | EtNCS, OX | 80 | 17.5 | 63 | >99 | 5.7 (1.8) | 180.2 | T g = −36.8 |
| 11 | CS2, OX | 80 | 0.5 | >99 | >85 | 19.2 (1.8) | 232.5 | T g = −22.2 |
| T m = 85.5 | ||||||||
| 12 | CS2, OX | 60 | 1.5 | 90 | >99 | 32.2 (1.8) | 121.5g | T g = −18.8 |
| T m = 82.0 | ||||||||
In contrast, the ROCOP of PTA with industrially relevant propylene oxide (Table 2, run #7) occurs readily at 80 °C, resulting in 84% PTA conversion after 9 hours. A perfectly alternating poly(ester-alt-thioester) with >95% selectivity (see ESI Fig. S42†) is formed with Mn = 25.3 kg mol−1 and Đ = 1.7 in quantitative polymer selectivity. This is evident in the 13C NMR spectrum, which shows two well-defined carbonyl resonances corresponding to the ester (δ(13C) = 165.78 ppm) and thioester (δ(13C) = 192.80 ppm) groups. Previous catalysts achieving such sequence selectivity were based on metal complexes with complex ligand frameworks that required multistep synthesis.47,48 In contrast, our catalytic system is easily assembled from commercial building blocks and achieves equally high sequence selectivity.
Transitioning from PTA to the ROCOP of sulfurated heteroallenes with oxetane, similarly resulted in good to excellent linkage selectivities. Specifically, the PhNCS/oxetane ROCOP (Table 2, run #8) produced a perfectly alternating poly(thioimidocarbonate) featuring –O–C(NPh)–S– (ONS) linkages, with Mn = 28.9 kg mol−1 and Đ = 1.2. The aliphatic variant, cyclohexylisothiocyanate (Table 2, run #9), also underwent selective ROCOP, with ONS linkage selectivity >98% (see ESI Fig. S51 and S52†) and yielding a polymer with a narrow polydispersity of Đ = 1.2 and a somewhat reduced molecular weight of Mn = 14.9 kg mol−1. The ethyl variant (Table 2, run #10) resulted in broadened dispersities and decreased molecular weights with due scrambling. Carbon disulfide (CS2) also underwent fast ROCOP, achieving full conversion after 30 minutes, thus exceeding the speed of the PTA/oxetane ROCOP under the same conditions. This reaction produced a polymer with Mn = 19.2 kg mol−1 and Đ = 1.8, with 92% polymer selectivity versus cyclic six-membered trithiocarbonate.13C NMR spectroscopy revealed good linkage selectivity for the alternating dithiocarbonate –O–(CS)–S– links (δ(Cq) = 214.22 ppm), with 85% selectivity based on integration of the 1H NMR spectrum compared to links from scrambling processes. Interestingly, these predominantly comprise monothiocarbonate –O–(CO)–S–, indicating oxygen enrichment of the polymer and the deployment of the lost sulfur atom in the trithiocarbonate byproduct. Decreasing the temperature to 60 °C (Table 2, run #12) resulted in >99% polymer selectivity vs. cyclic byproducts with 97% links (see ESI Fig. S32†) being dithiocarbonate –O–(CS)–S– alongside a significant increase in Mnto 31.2 kg mol−1.
With a range of materials in hand, we assessed their thermal properties. Polymers obtained from the ROCOP of substituted oxetanes, epoxides, and aliphatic isothiocyanates resulted in amorphous materials with glass transition temperatures ranging from −33.5 °C to 63 °C (see ESI section S4†). In contrast, polymers derived from unsubstituted oxetane were found to be semicrystalline. For example, the poly(ester-alt-thioester) obtained from PTA/oxetane ROCOP (Table 1, run #9) exhibited a complex, multimodal endotherm in the differential scanning calorimetry (DSC) at a heating rate of 10 °C min−1, associated with the melting of crystals when heating the sample for the first time. The most pronounced peak was observed at Tm = 83.5 °C. However, the sample did not crystallize upon cooling the melt in the calorimeter. The subsequent cooling and second heating only displayed glass transitions. Thus, while the as-synthesized polymer is semi-crystalline, its crystallization from the melt is slow.
In an attempt to achieve crystallization, we employed a self-nucleation strategy (see ESI section S5†), where heating was halted at different domains of the melting peak at a temperature Ts to leave crystallization nuclei in the sample, aiding crystallization upon cooling.34 Despite this strategy, crystallization during cooling was not achieved. However, it was possible to anneal the sample during partial melting at Ts = 83 °C. In the second heating scan, a sharpened endothermic peak at a higher Tm = 89.3 °C was observed, (Fig. 4(a)) indicating the melting of annealed crystals and surpassing the previously achieved melting points for the PTA/oxetane copolymers.
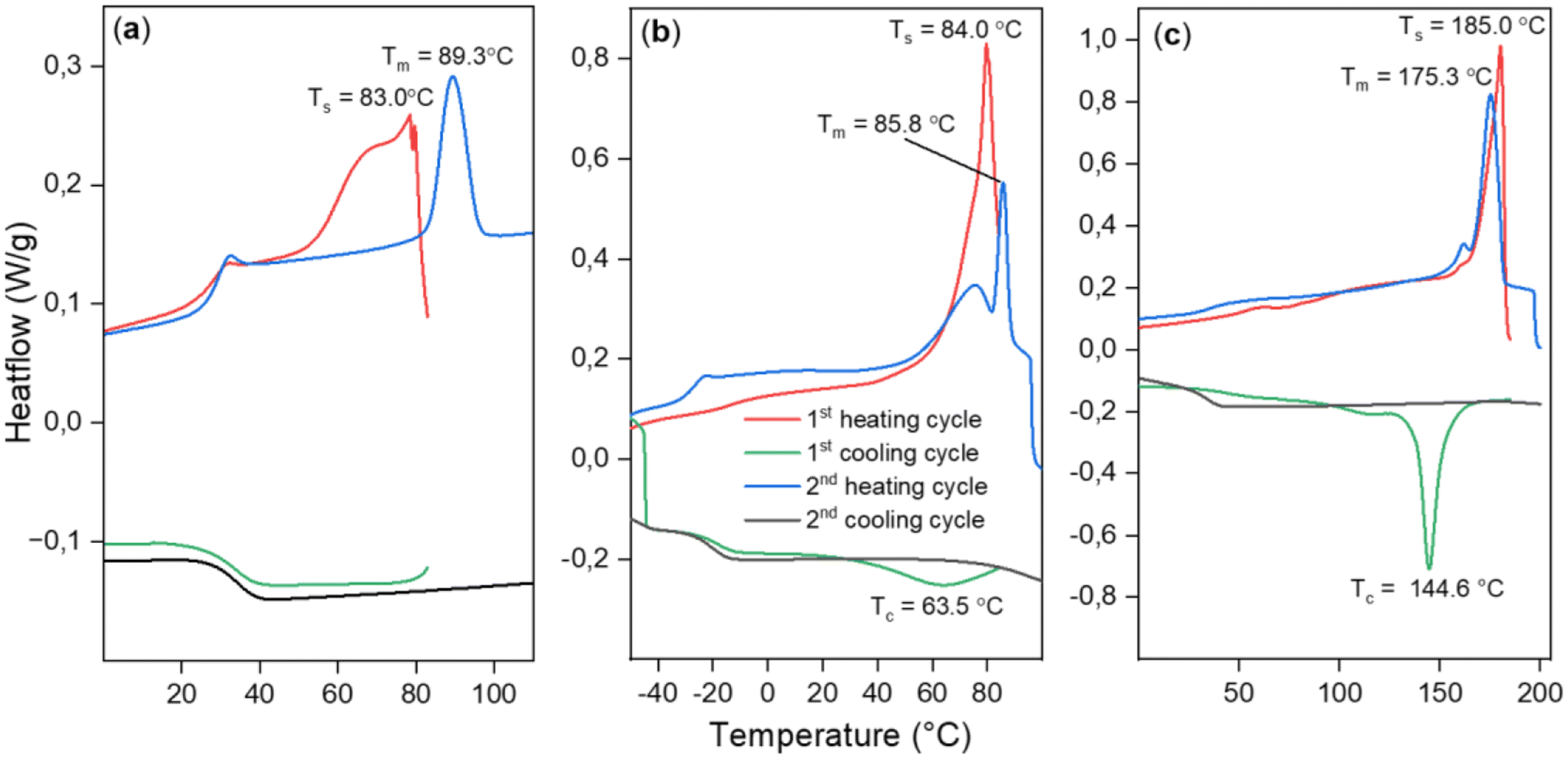 | ||
| Fig. 4 DSC data showing results of self-nucleation experiments for (a) PTA/OX copolymer (Table 2, run #1), (b) CS2/OX copolymer (Table 2, run #12), (c) PhNCS/OX copolymer (Table 2, run #8). | ||
Regarding thermal stability, thermogravimetric analysis shows a Td,5% = 312 °C. Compared to PTA/oxetane copolymers of similar molecular weights synthesized using Cr(III) catalysis, this represents an increase in Td,5% by more than 50 °C.35 The difference is even more pronounced when considering the onset temperature of degradation, which improves by more than 100 °C. We attribute this improvement to the complete removal (or at least deactivation by decomposition) of the borane catalyst during the polymerization work-up. In contrast, sulfur-containing polymers are known to absorb transition metals, which may explain why Cr(III) catalyst residues have been challenging to remove, leading to decreased thermal stability and catalysing degradation during heating.49,50
The scrambled CS2/OX copolymer (Table 2, run #11) shows a similar melting behaviour such as that the first heating scan shows a complex multimodal endotherm due to the melting of crystals with the most pronounced peak at Tm = 85.5 °C; no crystallisation is observed in the cooling scan due to slow crystallization. The less scrambled material showed the melting peak at Tm = 82 °C. This material could be crystallised during the first cooling cycle of the DSC when it was heated to a Ts = 84 °C with a broad recrystallization peak at Tc = 63.5 °C. But going to temperatures closer to the end of the melting peak (90 °C) resulted in the erasure of thermal history and no crystallization was seen.
The PhNCS/OX copolymer (Table 2, run #8) exhibits a melting peak at Tm = 180.1 °C in the first heating cycle, followed by again no crystallisation upon cooling. A broad crystallization peak was observed when the sample was incompletely melted when Ts = 180 °C. At Ts = 185 °C, the semi-crystalline polymer melts almost completely yet still leaving a few nuclei for crystallization. After heating to this temperature, a sharp crystallization peak was observed during the cooling cycle at Tc = 144.6 °C. Furthermore, an improved Td,5% = 228 °C, which is ca. 50 °C higher than for the PhNCS/OX copolymer obtained from chromium catalysis, is obtained clearly highlighting the benefits of the new catalytic system.
Conclusion
We have developed a transition metal-free catalytic system using simple and inexpensive components based on colorless and abundant main group elements with reduced toxicity for the ring-opening copolymerization (ROCOP) of sulfur-containing monomers with oxetanes and epoxides. This catalyst achieves polymers with perfect sequence selectivity through a cooperative mechanism between the borane and potassium centers, resulting in semi-crystalline materials with melting points up to 180 °C. Additionally, the polymers can demonstrate enhanced thermal stability compared to those synthesized with Cr(III) catalysts, likely due to the effective removal or deactivation of the borane catalyst. Our study highlights the potential of cooperative catalysis exclusively based on main group elements for sustainable polymerization catalysis in general.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest.Acknowledgements
We thank the “Verband der chemischen Industrie” and the “Daimler and Benz Foundation” for financial support (personal scholarships for A. J. P).References
- T.-J. Yue, W.-M. Ren and X.-B. Lu, Chem. Rev., 2023, 123, 14038–14083 CrossRef CAS PubMed.
- Y. Xia, X. Yue, Y. Sun, C. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202219251 CrossRef CAS PubMed.
- Y.-L. Su, L. Yue, H. Tran, M. Xu, A. Engler, R. Ramprasad, H. J. Qi and W. R. Gutekunst, J. Am. Chem. Soc., 2023, 145, 13950–13956 CrossRef CAS PubMed.
- C. Hardy, G. Kociok-Köhn and A. Buchard, Chem. Commun., 2022, 58, 5463–5466 RSC.
- T. Lee, P. T. Dirlam, J. T. Njardarson, R. S. Glass and J. Pyun, J. Am. Chem. Soc., 2022, 144, 5–22 Search PubMed.
- P. Yuan, Y. Sun, X. Xu, Y. Luo and M. Hong, Nat. Chem., 2022, 14, 294–303 CrossRef CAS PubMed.
- N. M. Bingham, Z. Abousalman-Rezvani, K. Collins and P. J. Roth, Polym. Chem., 2022, 13, 2880–2901 RSC.
- Y. Zhu, M. Li, Y. Wang, X. Wang and Y. Tao, Angew. Chem., Int. Ed., 2023, 62, e202302898 CrossRef CAS PubMed.
- X.-F. Zhu, G.-W. Yang, R. Xie and G.-P. Wu, Angew. Chem., Int. Ed., 2022, 61, e202115189 CrossRef CAS PubMed.
- H. Huang, S. Zheng, J. Luo, L. Gao, Y. Fang, Z. Zhang, J. Dong and N. Hadjichristidis, Angew. Chem., Int. Ed., 2024, 63, e202318919 Search PubMed.
- J. Jia, J. Liu, Z.-Q. Wang, T. Liu, P. Yan, X.-Q. Gong, C. Zhao, L. Chen, C. Miao, W. Zhao, S. (Diana) Cai, X.-C. Wang, A. I. Cooper, X. Wu, T. Hasell and Z.-J. Quan, Nat. Chem., 2022, 14, 1249–1257 Search PubMed.
- S. Wang, Z.-Y. Tian and H. Lu, Angew. Chem., Int. Ed., 2024, 63, e202411630 Search PubMed.
- C. Shi, M. L. McGraw, Z.-C. Li, L. Cavallo, L. Falivene and E. Y.-X. Chen, Sci. Adv., 2020, 6, eabc0495 Search PubMed.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 Search PubMed.
- T. Sehn, B. Huber, J. Fanelli and H. Mutlu, Polym. Chem., 2022, 13, 5965–5973 RSC.
- A. W. Woodhouse, A. Kocaarslan, J. A. Garden and H. Mutlu, Macromol. Rapid Commun., 2024, 45, 2400260 CrossRef CAS PubMed.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 Search PubMed.
- L.-Y. Wang, G.-G. Gu, B.-H. Ren, T.-J. Yue, X.-B. Lu and W.-M. Ren, ACS Catal., 2020, 10, 6635–6644 CrossRef CAS.
- T.-J. Yue, M.-C. Zhang, G.-G. Gu, L.-Y. Wang, W.-M. Ren and X.-B. Lu, Angew. Chem., 2019, 131, 628–633 CrossRef.
- D. K. Tran, A. N. Braaksma, A. M. Andras, S. K. Boopathi, D. J. Darensbourg and K. L. Wooley, J. Am. Chem. Soc., 2023, 145, 18560–18567 Search PubMed.
- K. Nakano, G. Tatsumi and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 15116–15117 Search PubMed.
- M. Sengoden, G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 2535–2541 Search PubMed.
- E. F. Clark, G. Kociok-Köhn, M. G. Davidson and A. Buchard, Polym. Chem., 2023, 14, 2838–2847 Search PubMed.
- Y. Sun, C. Zhang and X. Zhang, Chem. – Eur. J., 2024, 30, e202401684 CrossRef CAS PubMed.
- J. Stephan, M. R. Stühler, S. M. Rupf, S. Neale and A. J. Plajer, Cell Rep. Phys. Sci., 2023, 101510 Search PubMed.
- C. Gallizioli, D. Battke, H. Schlaad, P. Deglmann and A. J. Plajer, Angew. Chem., Int. Ed., 2024, 63, e202319810 CrossRef CAS PubMed.
- S. Rupf, P. Pröhm and A. J. Plajer, Chem. Sci., 2022, 13, 6355–6365 RSC.
- P. Deglmann, S. Machleit, C. Gallizioli, S. M. Rupf and A. J. Plajer, Catal. Sci. Technol., 2023, 13, 2937–2945 RSC.
- D. Silbernagl, H. Sturm and A. J. Plajer, Polym. Chem., 2022, 13, 3981–3985 Search PubMed.
- L.-Y. Wang, G.-G. Gu, T.-J. Yue, W.-M. Ren and X.-B. Lu, Macromolecules, 2019, 52, 2439–2445 Search PubMed.
- T.-J. Yue, W.-M. Ren, L. Chen, G.-G. Gu, Y. Liu and X.-B. Lu, Angew. Chem., 2018, 130, 12852–12856 CrossRef.
- T. M. McGuire and A. Buchard, Polym. Chem., 2021, 12, 4253–4261 RSC.
- C. Fornacon-Wood, B. R. Manjunatha, M. R. Stühler, C. Gallizioli, C. Müller, P. Pröhm and A. J. Plajer, Nat. Commun., 2023, 14, 4525 CrossRef CAS PubMed.
- J. Stephan, J. L. Olmedo-Martínez, C. Fornacon-Wood, M. Stühler, M. Dimde, D. Braatz, R. Langer, A. J. Müller, H. Schmalz and A. J. Plajer, Angew. Chem., Int. Ed., 2024, 63, e202405047 CrossRef CAS PubMed.
- C. Fornacon-Wood, M. R. Stühler, C. Gallizioli, B. R. Manjunatha, V. Wachtendorf, B. Schartel and A. J. Plajer, Chem. Commun., 2023, 59, 11353–11356 RSC.
- (a) M. R. Stühler, M. Kreische, C. Fornacon-Wood, S. M. Rupf, R. Langer and A. J. Plajer, Chem. Sci., 2024, 15, 19029–19036 RSC; (b) C.-J. Zhang, S.-Q. Wu, S. Boopathi, X.-H. Zhang, X. Hong, Y. Gnanou and X.-S. Feng, ACS Sustainable Chem. Eng., 2020, 34, 13056–13063 CrossRef; (c) M. Alves, B. Grignard, A. Boyaval, R. Méreau, J. De Winter, P. Gerbaux, C. Detrembleur, T. Tassaing and C. Jérôme, ChemSusChem, 2017, 10, 1128–1138 CrossRef CAS PubMed.
- K. V. Khopade, S. H. Chikkali and N. Barsu, Cell Rep. Phys. Sci., 2023, 4, 101341 CrossRef CAS.
- (a) S. Naumann, Polym. Chem., 2023, 14, 1834–1862 RSC; (b) Y.-Y. Zhang, G.-W. Yang, C. Lu, X.-F. Zhu, Y. Wang and G.-P. Wu, Chem. Soc. Rev., 2024, 53, 3384–3456 RSC; (c) C. Zhang, X. Geng, X. Zhang, Y. Gnanou and X. Feng, Prog. Polym. Sci., 2023, 136, 101644 Search PubMed.
- G.-W. Yang, Y.-Y. Zhang and G.-P. Wu, Acc. Chem. Res., 2021, 54, 4434–4448 CrossRef CAS PubMed.
- G. Feng, X. Feng, X. Liu, W. Guo, C. Zhang and X. Zhang, Macromolecules, 2023, 56, 6798–6805 CrossRef CAS.
- Y. Xia, J. Shen, H. Alamri, N. Hadjichristidis, J. Zhao, Y. Wang and G. Zhang, Biomacromolecules, 2017, 18, 3233–3237 CrossRef CAS PubMed.
- X. Xia, R. Suzuki, K. Takojima, D.-H. Jiang, T. Isono and T. Satoh, ACS Catal., 2021, 11, 5999–6009 CrossRef CAS.
- X. Xia, R. Suzuki, T. Gao, T. Isono and T. Satoh, Nat. Commun., 2022, 13, 163 CrossRef CAS PubMed.
- (a) X. Xia, T. Gao, F. Li, R. Suzuki, T. Isono and T. Satoh, J. Am. Chem. Soc., 2022, 144, 17905–17915 CrossRef CAS PubMed; (b) X. Dou, X.-H. Liu, B. Wang and Y.-S. Li, Chin. J. Chem., 2023, 41, 83–92 CrossRef CAS.
- J. Xu, P. Zhang, Y. Yuan and N. Hadjichristidis, Angew. Chem., Int. Ed., 2023, 62, e202218891 CrossRef CAS PubMed.
- A. Sirin-Sariaslan and S. Naumann, Chem. Sci., 2022, 13, 10939–10943 RSC.
- L.-Y. Wang, G.-G. Gu, B.-H. Ren, T.-J. Yue, X.-B. Lu and W.-M. Ren, ACS Catal., 2020, 10, 6635–6644 CrossRef CAS.
- X.-L. Chen, B. Wang, D.-P. Song, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 1153–1164 Search PubMed.
- T. Tian, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 6156–6163 CrossRef CAS PubMed.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05665e |
| This journal is © The Royal Society of Chemistry 2025 |