
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular analysis and design using generative artificial intelligence via multi-agent modeling
Isabella
Stewart
and
Markus J.
Buehler
 *
*
Laboratory for Atomistic and Molecular Mechanics (LAMM), Massachusetts Institute of Technology, 77 Mass. Ave 1-165, Cambridge, MA, USA. E-mail: mbuehler@MIT.EDU; Tel: +1 617 452 2759
First published on 24th January 2025
Abstract
We report the use of a multiagent generative artificial intelligence framework, the X-LoRA-Gemma large language model (LLM), to analyze, design and test molecular design. The X-LoRA-Gemma model, inspired by biological principles and featuring 7 billion parameters, dynamically reconfigures its structure through a dual-pass inference strategy to enhance its problem-solving abilities across diverse scientific domains. The model is used to first identify molecular engineering targets through a systematic human–AI and AI–AI self-driving multi-agent approach to elucidate key targets for molecular optimization to improve interactions between molecules. Next, a multi-agent generative design process is used that includes rational steps, reasoning and autonomous knowledge extraction. Target properties of the molecule are identified either using a principal component analysis (PCA) of key molecular properties or sampling from the distribution of known molecular properties. The model is then used to generate a large set of candidate molecules, which are analyzed via their molecular structure, charge distribution, and other features. We validate that as predicted, increased dipole moment and polarizability is indeed achieved in the designed molecules. We anticipate an increasing integration of these techniques into the molecular engineering workflow, ultimately enabling the development of innovative solutions to address a wide range of societal challenges. We conclude with a critical discussion of challenges and opportunities of the use of multi-agent generative AI for molecular engineering, analysis and design.
Design, System, ApplicationThis work presents a novel molecular design and optimization strategy that leverages multimodal generative artificial intelligence (AI) to accelerate the discovery of molecules with targeted properties. By integrating human–AI collaboration, multi-agent AI interactions, and inverse problem-solving techniques, our approach offers a powerful and flexible framework for tackling complex molecular design challenges. The incorporation of multiple modalities as well as full text sources of scientific papers offers a good level of traceability and human–AI interactions that helps with the incorporation into existing research or technology workflows. As a general utility, the desired functionality of the system is to generate molecules with enhanced properties, such as increased dipole moment and polarizability, while considering design constraints informed by chemical principles and the distribution of known molecular properties, as well as natural language reasoning over chemical, engineering and design principles. The immediate application potential of this work lies in its ability to streamline the molecular design process and generate novel molecules with tailored properties for various fields, such as sustainable materials, personalized medicine, and molecular sensors. In the future, we anticipate that this approach will be extended to address a wide range of challenges in molecular engineering, including the design of self-assembling materials, molecular robotics, catalysis, and energy storage and conversion, ultimately enabling the development of innovative solutions to pressing societal needs. |
1 Introduction
The discovery and design of new molecules are of paramount importance across a multitude of disciplines, including materials science, environmental technology and pharmacology. The ability to predict and tailor molecular properties can lead to breakthroughs in drug discovery, the development of new materials with novel properties, and solutions to sustainability challenges. Traditional experimental approaches in these domains are often time-consuming, expensive, and limited by the sheer complexity of possible molecular configurations to explore. As a result, computational techniques have increasingly become a cornerstone in the quest to expedite and refine molecular discovery processes.Recent advancements in computational power and algorithmic sophistication have enabled significant strides in this area. In particular, machine learning (ML) techniques have emerged as powerful tools to tackle complex problems in vast chemical spaces that were previously thought intractable. Among the plethora of ML techniques, generative methods stand out for their remarkable acuity in both forward problems, predicting the properties and behaviors of molecules from their structures, and inverse problems, designing molecules with desired characteristics. In particular, generative ML tools built on large language models (LLMs)1–12 can leverage vast datasets of textual descriptions to explore new molecular possibilities and handle complex tasks across diverse domains. Their ability to perform complex reasoning, integrating and generating knowledge across diverse types of data, opens new avenues for addressing multifaceted challenges in molecular science.
In recent years, deep learning models have received more attention due to their performance and potential applications in chemistry. New efforts are being made toward tackling chemistry problems with diverse knowledge by integrating language learning. Performance studies on artificial intelligence systems driven by off-the-shelf LLMs like GPT-4 and Llama have been explored.13–16 And additional fine-tuning of these LLMs with new datasets have been applied to create richer results on chemistry outputs. Fine-tuning for chemistry has been applied using instructional datasets for small molecules17–19 and are able to perform well on downstream tasks like property prediction for single applications14 and chemical design20 with diverse chemical tools for safety and reaction prediction.21
In this context, this paper introduces an application of a multiagent LLM, X-LoRA-Gemma, which builds upon the foundational work reported in an earlier paper detailing this novel architecture and its robustness in chemistry, physics, and biology tasks.22 X-LoRA-Gemma may serve as a step in the evolution of computational techniques for molecular design, leveraging the power of LLMs to navigate the intricate landscape of molecular properties and interactions. By synthesizing and reasoning across multi-textual data, including scientific text, structural information, and experimental data, X-LoRA-Gemma offers an integrated approach to the predictive design of molecules.
Fig. 1 shows the overall approach where a starting molecule is fed into the system, AI–AI and human–AI interactions advise on how to reach the set of target properties, and the AI system retorts by generating a set of plausible molecular designs.
 | ||
| Fig. 1 Overview of the approach discussed in this paper, using a series of generative AI tools to explore molecular design. We use both knowledge extraction and discovery via agentic modeling as well as targeted molecular design for specific properties. Created with https://BioRender.com. | ||
This paper aims to explore the capabilities of X-LoRA-Gemma in the domain of molecular design, focusing on its application to the QM9 dataset, a comprehensive collection of quantum mechanical (QM) properties of small molecules.23,24 Although limited to molecules containing H, C, N, O, F and up to nine heavy atoms, this dataset encompasses equilibrium structures and properties for 133![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 885 synthetically feasible organic molecules calculated from density functional theory (DFT) at the accuracy of B3LYP/6-31G(2df,p) level of theory. The homogeneity, purity, and lack of noise in QM9 has made it a classic benchmark for evaluating machine learning models in predicting molecular properties.25,26
885 synthetically feasible organic molecules calculated from density functional theory (DFT) at the accuracy of B3LYP/6-31G(2df,p) level of theory. The homogeneity, purity, and lack of noise in QM9 has made it a classic benchmark for evaluating machine learning models in predicting molecular properties.25,26
Through this exploration, we seek to demonstrate the potential of multiagent LLMs to improve molecular discovery while offering insights into their practical applications and laying the groundwork for future advancements in the field.
The X-LoRA model22 is a LLM inspired by biological principles. A key feature of this model is that it is trained to have the ability to dynamically rearrange its own structure before responding to a question or a task given. While conventional inference strategy processes a prompt through its entire architecture and generates a response in one shot, this model takes a more efficient approach wherein the modified inference strategy incorporates two forward passes. In the first pass, the model analyzes the question to identify the most relevant parts of its internal structure and determines a way by which it may reconfigure itself. In the second pass, the model responds to the question or task using the configuration identified. Refinement of the internal structure to prioritize modules specific to the prompt in the first pass suggests a form of ‘experiential learning’ that improves the responsiveness of the model during the second pass. It has been proposed that this realizes a simple implementation of ‘self-awareness’ and as a result even though the model has a relatively small parameter count of only around 7 billion parameters, it can reason across diverse scientific domains (biological materials, math, physics, chemistry, logic, mechanics, etc.), significantly enhancing its capacity for generating innovative solutions and solving specific chemical molecular design tasks.
The X-LoRA-Gemma model specifically is based on the Gemma-7B-it model27 and incorporates four expert adapters trained on mechanics and materials, protein mechanics, bioinspired materials, as well a quantum-mechanics based molecular properties QM9 dataset (see Table 1):28,29
| Label | Definition |
|---|---|
| Mu | Dipole moment: measures separation of charge within the molecule, affecting its interaction with electric fields and other molecules |
| Alpha | Polarizability: indicates how much the electron cloud around the molecule distorts in an external electric field, influencing the molecule's optical properties and interactions |
| HOMO | Highest occupied molecular orbital (HOMO) energy: related to the energy of the highest occupied electron orbital, important for understanding a molecule's chemical reactivity |
| LUMO | Lowest unoccupied molecular orbital (LUMO) energy: pertains to the energy of the lowest unoccupied electron orbital, also critical for reactivity and optical properties |
| Gap | HOMO–LUMO gap: the energy difference between HOMO and LUMO, significant for determining a molecule's chemical stability and reactivity |
| r 2 | Electronic spatial extent: this is a measure of the size of the electron cloud of a molecule, related to its electronic properties |
| z pve | Zero-point vibrational energy: the energy of a molecule at its lowest vibrational state, contributing to its stability and reactivity |
| c v | Heat capacity at constant volume: relates to the amount of heat required to change the temperature of a molecule by a certain amount, important for thermodynamics |
| u 0 | Internal energy at 0 K: the total energy of a molecule including electronic, vibrational, rotational, and translational contributions at absolute zero |
| u 298 | Internal energy at 298.15 K: similar to u0, but measured at room temperature (approximately 25 °C) |
| h 298 | Enthalpy at 298.15 K: the total heat content of a molecule at room temperature, including internal energy and the product of pressure and volume |
| g 298 | Free energy at 298.15 K: Gibbs free energy of the molecule at room temperature, indicating the maximum amount of work obtainable from a thermodynamic process at constant temperature and pressure |
1. Bioinspired materials.9
2. Mechanics and materials.6
3. Protein mechanics tasks (featuring generative sequence-to-property and inverse capabilities).30
4. Quantum-mechanics based molecular properties QM9 (featuring generative SMILES-to-property and inverse capabilities, see Table 1 for a definition and summary of all properties computed in that dataset).28,29
The fine-tuning of the model reported in earlier work improves task performance by efficiently repurposing the four pre-trained networks. The first two adapters (bioinspired and mechanics of materials) were trained using question–answer pairs, as described in ref. 22. The protein mechanics tasks were trained through instructions,22 with bidirectional forward and inverse instruction sets to predict protein mechanical properties and to design proteins to meet a set of mechanical properties. Using the QM9 dataset, two additional tasks, a forward and an inverse task, are developed to calculate molecular properties and to design molecules to meet a set of molecular properties. Sample tasks are:
CalculateMolecularProperties<CC(C)N1CC1C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O> O>
[0.098, 0.358, 0.581, 0.395, 0.309, 0.330, 0.570, 0.649, 0.519, 0.519, 0.519, 0.518] GenerateMolecularProperties <0.098, 0.358, 0.581, 0.395, 0.309, 0.330, 0.570, 0.649, 0.519, 0.519, 0.519, 0.518> [CC(C)N1CC1C |
Each molecule in the QM9 dataset has 12 computed QM properties that may be broadly categorized into 4 groups: geometric, energetic, electronic, and thermodynamic. An essential aspect of molecular geometry is the electronic spatial extent of a molecule, which reflects the average distribution of electrons. This distribution not only provides a basic understanding of a molecule's shape and size, but also directly influences its electronic features.31 Key thermodynamic properties such as the zero point vibrational energy, heat capacity at constant volume, internal energy (at 0 K and 298.15 K), enthalpy (at 298.15 K), and free energy (at 298.15 K) offer valuable insight into the dynamic behavior of a molecule under heat and pressure conditions.32 Electronic attributes such as the polarizability and dipole moment of a molecule inform how electrons are distributed within a molecule and how they respond to external factors. These are key criteria that can influence a molecule's reactions with other molecules and solvents. Energetic properties including the energy of the HOMO (highest occupied molecular orbital), energy of the LUMO (lowest unoccupied molecular orbital), and the corresponding HOMO–LUMO gap encompass a molecule's potential to participate in chemical reactions.33 The incorporation of these 12 properties and their relationships facilitates a systematic and comprehensive exploration of new structure–property landscapes in molecular design. Therefore, all 12 properties featured in the QM9 dataset are predicted or designed for, respectively (see, Table 1).
Since we will explore designing new molecules, it is important to understand the distribution of properties in the original QM9 dataset. Fig. 2 shows a pair plot of the molecular properties, focusing on the set of 12 properties within QM9. Correlations may be identified to understand molecular trends. When two properties are positively correlated, it means that as one increases, the other tends to increase as well. For instance, the HOMO–LUMO gap (gap) is strongly correlated with several thermodynamic variables, such as h298, g298, and u298 (indicating enthalpy, Gibbs free energy, and internal energy at different temperatures). A larger gap often corresponds to greater chemical stability, as molecules with a larger HOMO–LUMO gap are less reactive and less likely to engage in electronic transitions. This suggests that molecules with a larger gap are thermodynamically more stable, as reflected in their lower internal energies and enthalpies. The strong linear correlation between u298, h298, and g298 reflects the interconnected nature of internal energy, enthalpy, and Gibbs free energy in thermodynamic systems. This is expected since these quantities are all related to the thermal properties of the molecule, often differing by constant factors such as temperature and pressure. The relationship between the HOMO (HOMO) and LUMO (LUMO) energies shows some non-linear trends, possibly indicating that these frontier molecular orbitals do not scale in a simple linear fashion across different compounds. This can imply variability in the electronic structure of different molecules, particularly in their reactivity and optical properties. On the other hand, poor correlation is also observable in this set. Heat capacity (cv) shows little to no correlation with other variables like gap or frontier molecular orbital energies. This might suggest that heat capacity is largely independent of the electronic structure and more influenced by vibrational or rotational degrees of freedom. Molecules with similar HOMO–LUMO gaps may still exhibit a broad range of heat capacities, highlighting the role of molecular complexity and vibrational modes in determining the ability to store thermal energy.
 | ||
| Fig. 2 Pair plot of the molecular properties as defined in the QM9 dataset, focusing on a set of 12 quantum mechanical properties of molecules (dipole moment, mu; polarizability, alpha; highest occupied molecular orbital energy, HOMO; lowest unoccupied molecular orbital energy, LUMO; HOMO–LUMO gap: the energy difference between HOMO and LUMO, gap; electronic spatial extent; zero-point vibrational energy; heat capacity at constant volume; internal energy at 0 K; internal energy at 298.15 K; enthalpy at 298.15 K and free energy at 298.15 K). | ||
In the following sections, we will present a series of computational experiments that leverage the X-LoRA-Gemma model for several tasks, including question answering, knowledge discovery, and generation of research as well as design ideas. We will focus on one specific design idea in more detail to demonstrate the molecular generative capacity of the model.
1.1 Paper outline
We start with an exploration of interactive use cases, both human–AI collaboration to explore design strategies as well as multi-agent AI collaborations to autonomously explore topics and areas of interest.2 Results and discussion
The results are organized by the type of approach we take, leading from human–AI collaboration for planning, AI–AI collaboration through self-driving discovery, to molecular optimization and validation. First, we report a human–AI collaboration about a specific molecular design task that involves a combination of conversational AI with numerical predictions, inverse design, validation and multi-agentic analysis. Second, we report the results from another autonomous two-agent interaction to explore molecular design for organic electronic devices that also features in-context learning. We conclude with a detailed discussion of results to explore the multifaceted impacts this type of approach can have, and highlight rich insights for molecular design developed by these approaches.2.1 Identifying and planning molecular design via agentic modeling
We begin with a first experiment focused on human–AI collaboration. We ask X-LoRA-Gemma questions via chat interactions, in a human–AI collaboration. We begin the conversation by challenging the model to identify important biomolecules used in biological materials to make polymers with multifunctional qualities. The human user first asks for a list of important biomolecules used in creating polymers with multifunctional qualities. X-LoRA-Gemma explains that proteins, carbohydrates, polynucleotides (DNA and RNA), lipids, polysaccharides, peptides, and natural polymers like cellulose and chitin are crucial for developing polymers with a wide range of qualities including strength, elasticity, and biocompatibility. These biomolecules can be engineered to bind specific molecules, respond to environmental cues, store/transmit genetic information, act as catalysts for chemical reactions, serve as scaffolds for tissue engineering, and have specific mechanical properties. As the conversation progresses, context of the conversation is stored and fed to the model, which enables follow-up questions or clarifications. When context reaches the model's input limit, older parts of the conversation are summarized or truncated to feed new information. Truncation of older tokens starts from the beginning since recent context is usually more relevant for generating coherent responses.The user then inquires about how chitin forms a material on a molecular level and X-LoRA-Gemma describes the formation process of chitin involving the condensation of N-acetylglucosamine units linked by hydrogen bonds to form chitin chains. It is explained how these chains are arranged in a crystal structure stabilized by ionic bonds and van der Waals forces, leading to a strong and rigid material. X-LoRA-explains that the unique structure and arrangement of chitin give it high strength and rigidity. Then inspired by the insight developed through the conversation, the user seeks advice on enhancing hydrogen bond interactions between molecules with a specific SMILES code. The user lists several parameters that can be adjusted, reflecting those available via the X-LoRA-Gemma specific chemistry tasks.22 Following, X-LoRA-Gemma recommends focusing on increasing the dipole moment and polarizability of a specific molecule (COC(N)N1CCC1N, a starting point randomly selected from the QM9 test set) to strengthen hydrogen bond interactions. Several specific possible design ideas are shared, such as introducing more electronegative or polarizable atoms like nitrogen or oxygen into the molecule, enhancing the strength and number of hydrogen bonds, and potentially leading to stronger materials. The conversation is technical, focused on the biochemical and molecular engineering aspects of creating and improving polymers and materials. The full conversation is:
Chat between a user and X-LoRA-GemmaUser: you are an expert in biological molecular engineering. List the most important biomolecules used in biological materials to make polymers with multifunctional qualities.X-LoRA-Gemma: biological molecular engineering involves the use of various biomolecules to create polymers with multifunctional qualities. Some of the most important biomolecules used in this field include: 1. Proteins: proteins are essential biomolecules that are used to create polymers with specific properties, such as strength, elasticity, and biocompatibility. They can be engineered to have specific functions, such as the ability to bind to specific molecules or to respond to environmental cues. 2. Carbohydrates: carbohydrates are another important class of biomolecules used in biological molecular engineering. They can be used to create polymers with specific properties, such as the ability to bind to specific molecules or to have specific mechanical properties. 3. Polynucleotides (DNA and RNA): polynucleotides are used to create polymers with specific functions, such as the ability to store and transmit genetic information or to act as catalysts for chemical reactions. 4. Lipids: lipids are used to create polymers with specific properties, such as the ability to act as membranes or to store energy. 5. Polysaccharides: polysaccharides are used to create polymers with specific properties, such as the ability to act as scaffolds for tissue engineering or to have specific mechanical properties. 6. Peptides: peptides are used to create polymers with specific properties, such as the ability to bind to specific molecules or to act as catalysts for chemical reactions. 7. Proteins and polynucleotides: proteins and polynucleotides are used to create polymers with specific properties, such as the ability to bind to specific molecules or to act as catalysts for chemical reactions. 8. Natural polymers: natural polymers, such as cellulose and chitin, are used to create polymers with specific properties, such as the ability to act as scaffolds for tissue engineering or to have specific mechanical properties. Overall, these biomolecules are used in biological molecular engineering to create polymers with a wide range of multifunctional qualities, such as strength, elasticity, biocompatibility, and the ability to store and transmit genetic information. User: how does chitin form a material, specifically in terms of molecular interactions? X-LoRA-Gemma: chitin, a polysaccharide found in the exoskeletons of crustaceans, is formed through the condensation of N-acetylglucosamine units, which are linked by hydrogen bonds to form chitin chains. These chains are then stacked in a specific arrangement to create a crystal structure. The crystal structure is further stabilized by ionic bonds and van der Waals forces, leading to the formation of a strong and rigid material. This process is similar to the formation of other crystalline polymers, such as cellulose and chitin. However, chitin has a unique structure and arrangement of chains that gives it specific properties, such as its high strength and rigidity. User: thank you. Please tell me which properties I should change, in an organic molecule with SMILES code CCCCC, in order to facilitate H-bonding and van der Waals interactions with other molecules. I have access to tuning the following parameters: Dipole moment Polarizability Highest occupied molecular orbital (HOMO) energy Lowest unoccupied molecular orbital (LUMO) energy HOMO–LUMO gap: the energy difference between HOMO and LUMO Electronic spatial extent Zero-point vibrational energy Heat capacity at constant volume Internal energy at 0 K Internal energy at 298.15 K Enthalpy at 298.15 K Free energy at 298.15 K Discuss the most important properties I should change, and how. Brief answer. X-LoRA-Gemma: to facilitate H-bonding and van der Waals interactions with other molecules in an organic molecule with SMILES code CCCCC, you should consider changing the following properties: 1. Dipole moment: increase the dipole moment of the molecule to enhance its ability to interact with other molecules through H-bonding. 2. Polarizability: increase the polarizability of the molecule to enhance its ability to interact with other molecules through van der Waals interactions. 3. Highest occupied molecular orbital (HOMO) energy: decrease the HOMO energy of the molecule to enhance its ability to donate electrons to other molecules, which can facilitate H-bonding and van der Waals interactions. 4. Lowest unoccupied molecular orbital (LUMO) energy: increase the LUMO energy of the molecule to enhance its ability to accept electrons from other molecules, which can also facilitate H-bonding and van der Waals interactions. By changing these properties, you can increase the overall H-bonding and van der Waals interactions of the molecule, making it more likely to interact with other molecules and form a strong and rigid structure. |
Interestingly, the initial question from the user that prompts the AI agent to consider multi-functionality within biological materials leads the AI agent to mostly focus its response on material properties driven by molecular interactions. This focus stems from the internal knowledge-base of X-Lora-Gemma, prioritizing well-understood mechanisms. The AI response prompts the user to ask a new question, drawing on external knowledge, about chitin and its materiality in terms of its molecular interactions. This iterative exchange demonstrates the symbiotic advantages of AI–human collaboration, where the user's initial query steers the AI agent and the AI response enables the user to refine or change their questions based on the acquired knowledge.
The use of the LLM for interactive discussion and development of design ideas can alternatively be automated, by having two (or more) LLM serve as interacting agents. This can be implemented through the self-driving interaction between multiple generative models. Starting from an initial question asked by the first LLM agent (a question asker, defined as molecular designer here) the second LLM agent provides a response (an answering focused agent, here referred to as molecular engineer). The question asker is based on a general-purpose LLM, Zephyr-7b-β,34 and the answering agent uses X-LoRA-Gemma.22 Each is defined with specific instructions to behave in a particular way (details, see Materials and methods).
The question complements the last one in the human–AI conversation, but now forms the starting point for an independent multi-agent interaction that will unfold autonomously. The question is:
|
I have an organic molecule with SMILES code CCCCC.
Suggest key molecular design changes in order to enhance interactions with other molecules, in the context of bio-inspired design. Brief answer. |
The conversation provides a detailed discussion on enhancing interactions with other molecules for the organic molecule of interest. The molecular designer agent suggests key molecular design changes to improve interactions, while the molecular engineer proposes modifications to increase the polarity and reactivity of the molecule, or to have a longer carbon chain or a more branched structure. It is suggested that in order to ensure that the added functional groups or modified structure will not negatively impact the overall stability and biological activity of the molecule, factors such as chemical properties and reactivity should be considered. Computational methods and experimental techniques are proposed to be used to predict the specific functional groups or structural modifications that will enhance interactions while minimizing negative impacts. Balancing the need for enhanced interactions with specific target molecules against potential negative impacts on overall stability and biological activity is crucial, as well as considering the solubility of the molecule in different environments. Key takeaways to enhance interactions with other molecules for an organic molecule with SMILES code CCCCC are:
1. Key molecular design changes include adding functional groups, modifying the carbon chain length, or branching the structure.
2. Considerations for ensuring stability and biological activity, which include structural and chemical properties of the molecule and its reactivity, while reaching to meet the target specificity.
3. Computational methods and experimental techniques can be used to predict functional groups or structural modifications.
4. Balancing enhanced interactions with specific target molecules against potential negative impacts is crucial.
5. Considerations for solubility include the environment and the molecule's affinity for the solvent.
The full conversation is as follows:
Conversation between a molecular designer and a molecular engineerMolecular designer: I have an organic molecule with SMILES code CCCCC.Suggest key molecular design changes in order to enhance interactions with other molecules, in the context of bio-inspired design. Brief answer. Molecular engineer: the SMILES code for the organic molecule is CCCCC. To enhance interactions with other molecules, the molecular design changes could include adding functional groups to the molecule, such as amines or alcohols, to increase the polarity and reactivity of the molecule. Alternatively, the molecule could be modified to have a longer carbon chain or a more branched structure, which could increase its molecular weight and surface area, leading to improved interactions with other molecules. Molecular designer: how can we ensure that the added functional groups or modified structure will not negatively impact the overall stability and biological activity of the molecule? Molecular engineer: to ensure that the added functional groups or modified structure will not negatively impact the overall stability and biological activity of the molecule, it is important to consider the chemical properties and reactivity of the molecule. For example, if the molecule is used in a biological system, the added functional groups or modified structure should be biocompatible and not cause any adverse reactions. Additionally, the molecule should be stable under the conditions in which it will be used, such as temperature and pH, and should not undergo any significant structural changes that could affect its function. By carefully considering these factors, it is possible to enhance interactions with other molecules while maintaining the stability and biological activity of the molecule. Molecular designer: how can we predict the specific functional groups or structural modifications that will enhance interactions while minimizing negative impacts on stability and biological activity? Are there any computational methods or experimental techniques we can use to guide our design decisions? Molecular engineer: there are several computational methods and experimental techniques that can be used to predict the specific functional groups or structural modifications that will enhance interactions while minimizing negative impacts on stability and biological activity. One approach is to use molecular modeling software, such as AMBER or CHARMM, to simulate the interactions between the molecule and other molecules. These software programs can predict the strength and specificity of the interactions, such as hydrogen bonding, van der Waals forces, or electrostatic interactions. Additionally, experimental techniques such as X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy can be used to determine the molecular structure and interactions of the molecule. These techniques can provide valuable insights into the molecular properties and reactivity of the molecule, which can be used to guide the design of new molecules or modifications to existing molecules. By combining these computational and experimental methods, it is possible to optimize the design of the molecule for enhanced interactions with other molecules while maintaining its stability and biological activity. Molecular designer: how can we ensure that the added functional groups or modified structure will not negatively impact the overall selectivity of the molecule? In other words, how can we optimize interactions with specific target molecules while minimizing interactions with unwanted molecules? Molecular engineer: to ensure that the added functional groups or modified structure will not negatively impact the overall selectivity of the molecule, it is important to consider the specificity of the interactions between the molecule and other molecules. For example, if the molecule is used in a biological system, the added functional groups or modified structure should be selective and only interact with the target molecules, rather than binding to a wide range of molecules. Additionally, the molecule should have a high affinity for the target molecules and a low affinity for unwanted molecules. By carefully considering these factors, it is possible to enhance interactions with specific target molecules while minimizing interactions with unwanted molecules. For example, the molecule could be modified to have a specific binding site for the target molecules, or to have a higher concentration of functional groups that interact with the target molecules. By optimizing the design of the molecule, it is possible to enhance its selectivity and improve its overall performance. Molecular designer: how can we balance the need for enhanced interactions with specific target molecules against potential negative impacts on overall stability and biological activity when designing modifications to a molecule? Are there any trade-offs that we should be aware of, and how can we minimize these trade-offs? Molecular engineer: when designing modifications to a molecule to enhance interactions with specific target molecules, it is important to consider the potential negative impacts on overall stability and biological activity. For example, the added functional groups or modified structure could increase the molecular weight of the molecule, which could affect its stability and biological activity. Additionally, the molecule could undergo significant structural changes that could impact its function. By carefully considering these factors, it is possible to balance the need for enhanced interactions with specific target molecules against potential negative impacts on overall stability and biological activity. For example, the molecule could be modified to have a specific binding site for the target molecules, but the binding site should not be too crowded or too exposed, as this could affect the stability of the molecule. Additionally, the molecule could be modified to have a higher concentration of functional groups that interact with the target molecules, but the concentration should not be too high, as this could also affect the stability of the molecule. By carefully optimizing the design of the molecule, it is possible to enhance its interactions with specific target molecules while minimizing negative impacts on overall stability and biological activity. Molecular designer: how can we ensure that the added functional groups or modified structure will not negatively impact the overall solubility of the molecule? In other words, how can we optimize interactions with specific target molecules while maintaining adequate solubility in various environments? Molecular engineer: to ensure that the added functional groups or modified structure will not negatively impact the overall solubility of the molecule, it is important to consider the solubility of the molecule in different environments. For example, if the molecule is used in a biological system, the added functional groups or modified structure should not significantly affect the solubility of the molecule in water or other biological fluids. Additionally, the molecule should have adequate solubility in the solvent in which it will be used, such as water, ethanol, or chloroform. By carefully considering these factors, it is possible to enhance interactions with specific target molecules while maintaining adequate solubility in various environments. For example, the molecule could be modified to have a specific binding site for the target molecules, but the binding site should not be too crowded or too exposed, as this could affect the solubility of the molecule. Additionally, the molecule could be modified to have a higher concentration of functional groups that interact with the target molecules, but the concentration should not be too high, as this could also affect the solubility of the molecule. By carefully optimizing the design of the molecule, it is possible to enhance its interactions with specific target molecules while minimizing negative impacts on overall solubility. |
The AI–AI interaction discusses the “overall stability” of the molecule. However, it should be noted that stability encompasses a wide range of factors in the context of real-world applications, including thermodynamic stability, kinetic stability, and environmental stability (e.g., stability in water, air, under varying pH, temperature, or oxidative conditions). Each of these types of stability presents unique challenges in both prediction and assessment. Therefore, the AI–AI interaction could have been enriched by contextualizing “overall stability” in molecular design and how it might be experimentally validated. This highlights a scenario where human interjection into the conversation can be used for clarification.
The scenario can be expanded to explore other molecular designs, for instance considering a different starting molecule given as a reference in the process. In a second example, the question posed is:
|
How could I improve the interactions of an organic molecule with SMILES code COC(N)N1CCC1N in order to strengthen H-bond interactions between molecules.
I have access to tuning the following parameters: Dipole moment Polarizability Highest occupied molecular orbital (HOMO) energy Lowest unoccupied molecular orbital (LUMO) energy HOMO–LUMO gap: the energy difference between HOMO and LUMO Electronic spatial extent Zero-point vibrational energy Heat capacity at constant volume Internal energy at 0 K Internal energy at 298.15 K Enthalpy at 298.15 K Free energy at 298.15 K Discuss the top TWO properties I should change, and how. |
The conversation between the molecular designer (asking questions) and the molecular engineer (providing answers) proceeds to explore the optimal balance between enhancing H-bond interactions and maintaining overall stability, solubility, biological activity, selectivity, and pharmacokinetic properties for a specific organic molecule. Key points made include the importance of considering factors such as dipole moment, polarizability, binding affinity, specificity, and potential side effects when making changes to enhance H-bond interactions. Computational methods and experimental techniques can be employed to make informed decisions during the molecular design process. The optimal balance between enhancing H-bond interactions and maintaining conformational stability should also be considered.
It is pointed out that these factors can impact the molecule's overall activity and selectivity, so it is important to carefully consider the trade-offs between enhancing H-bond interactions and maintaining the molecule's stability and solubility.
The full conversation is:
Conversation between a molecular designer and a molecular engineerMolecular designer: how could I improve the interactions of an organic molecule with SMILES code COC(N)N1CCC1N in order to strengthen H-bond interactions between molecules.
I have access to tuning the following parameters: Dipole moment Polarizability Highest occupied molecular orbital (HOMO) energy Lowest unoccupied molecular orbital (LUMO) energy HOMO–LUMO gap: the energy difference between HOMO and LUMO Electronic spatial extent Zero-point vibrational energy Heat capacity at constant volume Internal energy at 0 K Internal energy at 298.15 K Enthalpy at 298.15 K Free energy at 298.15 K Discuss the top TWO properties I should change, and how. Molecular engineer: 1. **Dipole moment**: increase the dipole moment of the molecule to strengthen H-bond interactions. This can be achieved by introducing more polar groups or increasing the number of polar groups in the molecule. 2. **Polarizability**: increase the polarizability of the molecule to enhance H-bond interactions. Polarizability measures the ability of a molecule to interact with other molecules through dipole–dipole interactions. Increasing the polarizability of the molecule will make it more capable of forming H-bonds with other molecules. These changes can be made by manipulating the molecular structure and electronic properties of the organic molecule. By increasing the dipole moment and polarizability of the molecule, you can enhance H-bond interactions between molecules and improve their interactions with the SMILES code COC( Molecular designer: how do the changes in dipole moment and polarizability affect the overall stability and solubility of the organic molecule, and are there any trade-offs between enhancing H-bond interactions and maintaining these properties? Molecular engineer: the changes in dipole moment and polarizability can affect the overall stability and solubility of the organic molecule. Increasing the dipole moment can improve the molecule's ability to interact with other molecules through H-bond interactions, but it can also make the molecule more soluble in polar solvents. Increasing the polarizability of the molecule can enhance its H-bond interactions with other molecules, but it can also make the molecule less soluble in non-polar solvents. There may be trade-offs between enhancing H-bond interactions and maintaining the overall stability and solubility of the organic molecule. For example, increasing the dipole moment of the molecule can improve its ability to interact with other molecules through H-bond interactions, but it can also make the molecule more soluble in polar solvents. Similarly, increasing the polarizability of the molecule can enhance its H-bond interactions with other molecules, but it can also make the molecule less soluble in non-polar solvents. Therefore, the changes in dipole moment and polarizability should be carefully considered to achieve the desired balance between enhancing H-bond interactions and maintaining the overall stability and solubility of the organic molecule. Molecular designer: how can we ensure that the changes made to enhance H-bond interactions do not negatively impact the molecule's biological activity or selectivity? Consider factors such as binding affinity, specificity, and potential side effects. Provide a specific example of a molecule where this trade-off has been successfully navigated.
Molecular engineer: to ensure that the changes made to enhance H-bond interactions do not negatively impact the molecule's biological activity or selectivity, it is important to consider factors such as binding affinity, specificity, and potential side effects. For example, in the case of the organic molecule COC( One example of a molecule where this trade-off has been successfully navigated is the drug imatinib, which is used to treat chronic myeloid leukemia. Imatinib has a high binding affinity for the BCR–ABL protein, but it also has a low solubility in water. To improve the solubility of imatinib, researchers have developed various strategies, such as using surfactants or co-solvents. These strategies have enhanced the H-bond interactions between imatinib and the BCR–ABL protein, but they have also been found to have minimal impact on the molecule's biological activity or selectivity. In conclusion, when making changes to enhance H-bond interactions in an organic molecule, it is important to carefully consider factors such as binding affinity, specificity, and potential side effects. By taking a holistic approach that considers the molecule's biological activity and selectivity, it is possible to achieve the desired balance between enhancing H-bond interactions and maintaining the overall stability and solubility of the molecule. Molecular designer: how can we ensure that the changes made to enhance H-bond interactions do not negatively impact the molecule's pharmacokinetic properties, such as absorption, distribution, metabolism, and excretion? Provide specific examples of how these properties may be affected by altering dipole moment and polarizability. Molecular engineer: to ensure that the changes made to enhance H-bond interactions do not negatively impact the molecule's pharmacokinetic properties, such as absorption, distribution, metabolism, and excretion, it is important to consider factors such as solubility, bioavailability, and the presence of metabolic enzymes. For example, in the case of the organic molecule COC( To address these issues, researchers have developed various strategies, such as using surfactants or co-solvents to improve the solubility of the molecule in water. They have also studied the effects of changing the dipole moment and polarizability of the molecule on its metabolism and excretion. These studies have helped to identify the optimal balance between enhancing H-bond interactions and maintaining the molecule's pharmacokinetic properties. In conclusion, when making changes to enhance H-bond interactions in an organic molecule, it is important to consider factors such as solubility, bioavailability, and the presence of metabolic enzymes. By taking a holistic approach that considers the molecule's pharmacokinetic properties, it is possible to achieve the desired balance between enhancing H-bond interactions and maintaining the overall stability and solubility of the molecule. Molecular designer: how can we predict the optimal balance between enhancing H-bond interactions and maintaining overall stability, solubility, biological activity, selectivity, and pharmacokinetic properties for a specific organic molecule, given its unique structural features and target binding site? What computational methods or experimental techniques can be employed to make informed decisions during the molecular design process? Molecular engineer: to predict the optimal balance between enhancing H-bond interactions and maintaining overall stability, solubility, biological activity, selectivity, and pharmacokinetic properties for a specific organic molecule, it is important to consider factors such as dipole moment, polarizability, binding affinity, specificity, and potential side effects. Computational methods, such as molecular modeling and simulations, can be employed to predict the effects of changing the dipole moment and polarizability of the molecule on its H-bond interactions, overall stability, solubility, and other properties. Experimental techniques, such as X-ray crystallography and nuclear magnetic resonance (NMR), can also be used to determine the structure and properties of the molecule. By combining computational and experimental approaches, researchers can gain a better understanding of the relationships between the dipole moment, polarizability, and other structural features of the molecule and its biological activity, selectivity, and pharmacokinetic properties. This knowledge can be used to optimize the molecule's properties for specific applications, such as drug discovery or materials science. In conclusion, the optimal balance between enhancing H-bond interactions and maintaining overall stability, solubility, biological activity, selectivity, and pharmacokinetic properties for a specific organic molecule can be predicted by considering factors such as dipole moment, polarizability, binding affinity, specificity, and potential side effects. Computational methods and experimental techniques can be employed to make informed decisions during the molecular design process. Molecular designer: how can we ensure that the changes made to enhance H-bond interactions do not lead to unexpected conformational changes in the molecule, which could impact its overall activity and selectivity? Molecular engineer: to ensure that the changes made to enhance H-bond interactions do not lead to unexpected conformational changes in the molecule, it is important to consider factors such as the molecule's flexibility, rigidity, and the presence of any rigid or flexible regions. For example, in the case of the organic molecule COC( To address these issues, researchers have developed various strategies, such as using surfactants or co-solvents to improve the solubility of the molecule in water. They have also studied the effects of changing the dipole moment and polarizability of the molecule on its conformational flexibility and stability. These studies have helped to identify the optimal balance between enhancing H-bond interactions and maintaining the molecule's conformational stability. In conclusion, when making changes to enhance H-bond interactions in an organic molecule, it is important to consider factors such as the molecule's flexibility, rigidity, and the presence of any rigid or flexible regions. By taking a holistic approach that considers the molecule's conformational stability, it is possible to achieve the desired balance between enhancing H-bond interactions and maintaining the overall stability and solubility of the molecule. |
This interaction provides numerous useful considerations that can be utilized by a human user, specifically pointing out key issues for the design process that include:
• The optimal balance between enhancing H-bond interactions and maintaining overall stability, solubility, biological activity, selectivity, and pharmacokinetic properties for a specific organic molecule can be predicted by considering the interplay of factors such as dipole moment, polarizability, binding affinity, specificity, and potential medical side effects.
• Computational methods and experimental techniques can be employed to make informed decisions during the molecular design process.
• The optimal balance between enhancing H-bond interactions and maintaining conformational stability should also be considered.
• Factors such as the molecule's flexibility, rigidity, and the presence of any rigid or flexible regions can impact the molecule's overall activity and selectivity, so it would be important to carefully consider the trade-offs between enhancing H-bond interactions and maintaining the molecule's stability and solubility.
Providing a more detailed analysis, the conversation contains several concrete examples of good molecular design provided by the molecular engineer. The molecular engineer mentions imatinib, a drug known for its strong binding affinity to the cancer-causing protein BCR–ABL but poor water solubility. This case highlights various strategies used by researchers to navigate this trade-off including surfactants and co-solvents to enhance H-bond interactions and improve solubility, while preserving the target protein specificity. A deeper literature search by the authors points specifically to co-solvents such as toluene, n-propanol, and acetone for improving water solubility and its positive impact on the oral bioavailability of imatinib.35 Additionally, bioinspired design of co-crystals have been reported to enhance kinetic solubility as well as carriers that promote imatinib base solid dispersions.36,37
We note that as the AI–AI conversation progresses, it narrows its initial focus on biological activity and selectivity to optimizing pharmacokinetic activity within the field of drug design. The molecular engineer as the answering AI agent again mentions research on co-solvents and surfactants to control the effect of enhanced polarizability on a molecule's susceptibility to enzyme digestion. This is of particular importance in drug design as non-optimal adsorption, distribution, metabolism, and excretion (ADME) properties accounts for 40 percent of all drug failures. Late-stage failures account for significant loss in investment of time and money.38 As the answering AI agent mentions, drug elimination typically occurs in the liver and kidneys. However, contrary to the predicted response, enzymes typically act on hydrophobic drugs, bio-transforming them into polar substances for the body to excrete. More polar hydrophilic drugs, on the other hand, do not require metabolic changes to their molecular structures and are directly excreted.39,40 The answering AI agent is accurate in that molecules with enhanced polarizability and dipole moment are less likely to have a strong therapeutic effect. This presents a scenario where subsequent inquiry or clarification by the human-user may be addressed with literature research or by halting the AI–AI conversation and re-inserting the human user into conversation with the AI agent. This research methodology enables a fluid interchange of multi-agents configurable to the user's research goal or direction.
With respect to the improvement of ADME and biological effects, the answering AI agent proceeds to suggest a combination of computational and experimental techniques to refine predictions of molecular behavior. This section of the dialogue further supports the essential consideration of reaching target properties while maintaining others, and also provides basis for new inspiration for the human-user.
The AI–AI interaction is distinct and complimentary to the human–AI interaction. In the AI–AI interaction model, the question asking LLM follows a series of logic and reasoning based on the response of the answering LLM to guide its next query.
This type of conversation can inform the human (the user) on important considerations free of the user's biases, potentially leading to unexpected, diverse and relevant ideas. In the human–AI interaction, the human can be more flexible as the querying agent and can draw upon more sophisticated chemical intuition, including insights outside of the AI model's capabilities, or knowledge of real-world constraints to guide the next query even if contextually unrelated to the previous AI response. It is noted that with the use of more powerful AI models, some of these shortcomings can be addressed in the future. Another possibility is to provide one of the agents with access to scientific papers, to better inform their responses with new research or specific knowledge. We will explore this strategy in section 2.3.
2.2 Inverse problem solving to design molecules with specific target properties
We now take concrete steps towards molecular design. Based on the suggestion to increase the dipole moment and polarizability of the molecule, we explore ways of achieving this in an actual design process that optimizes the chemical makeup. The generative tasks of the X-LoRA-Gemma model (specifically: CalculateteMolecularProperties<…> and GenerateMolecularProperties<…>) allow us to predict molecular properties and to design a molecular structure to meet a certain target. The target set of properties we want to focus on can be informed based on a particular known design target, or be derived from the agent modeling described in the previous sections.Developing the specific distribution of the molecular properties we want to achieve could be a selection made by simply coming up with a set of 12 parameters. Alternatively we could use an existing molecule as the basis for the optimization, in order to generate alternative designs with a similar target. Another possibility is to use an existing molecule as a basis but change certain properties according to a design goal, such as increasing the dipole moment.
To better understand how molecular property changes are correlated, we conduct a principal component analysis (PCA) analysis with 11 total components based on the training data to determine which components may be suitable to determine a new set of properties that meet our design target, but also follow the initial data distribution of molecular structures. This can help us understand limitations that not all combinations of properties are chemically possible, and that changing the properties in a way that likely meets these considerations helps us discover more viable solutions to develop more realistic molecules.
Fig. 3 shows an analysis of the set of QM9 properties, including a heatmap of the correlation matrix, a heatmap of the PCA loadings. As can be seen 5th and 7th principal component have a quite strong sensitivity with respect to the dipole moment, mu and polarizability, alpha (with high loading of 0.97 and 0.95, respectively).
 | ||
| Fig. 3 Analysis of the set of QM9 properties, including a heatmap of the correlation matrix (a), a heatmap of the principal component analysis (PCA) loadings (b), and a PCA loadings plot for the 5th and 7th principal component that have strong sensitivity with respect to the dipole moment, mu and polarizability, alpha. The analysis in (b) reveals which components are best suited to use as coordinate to alter set of molecular properties. As seen in (c), for the dipole moment and polarizability, the 5th and 7th principal component stand out with high loading of 0.97 and 0.95, respectively. | ||
Further, Fig. 4 shows plots of various molecular properties over principal components, featuring the dipole moment, mu, polarizability, alpha, and the HOMO–LUMO gap, gap. Therein Fig. 4(a)–(c) show results over principal components 0 and 1, and Fig. 4(d)–(f) over principal components 5 and 7. These analyses provide a good overview of the distribution of properties and how our design process will expand these.
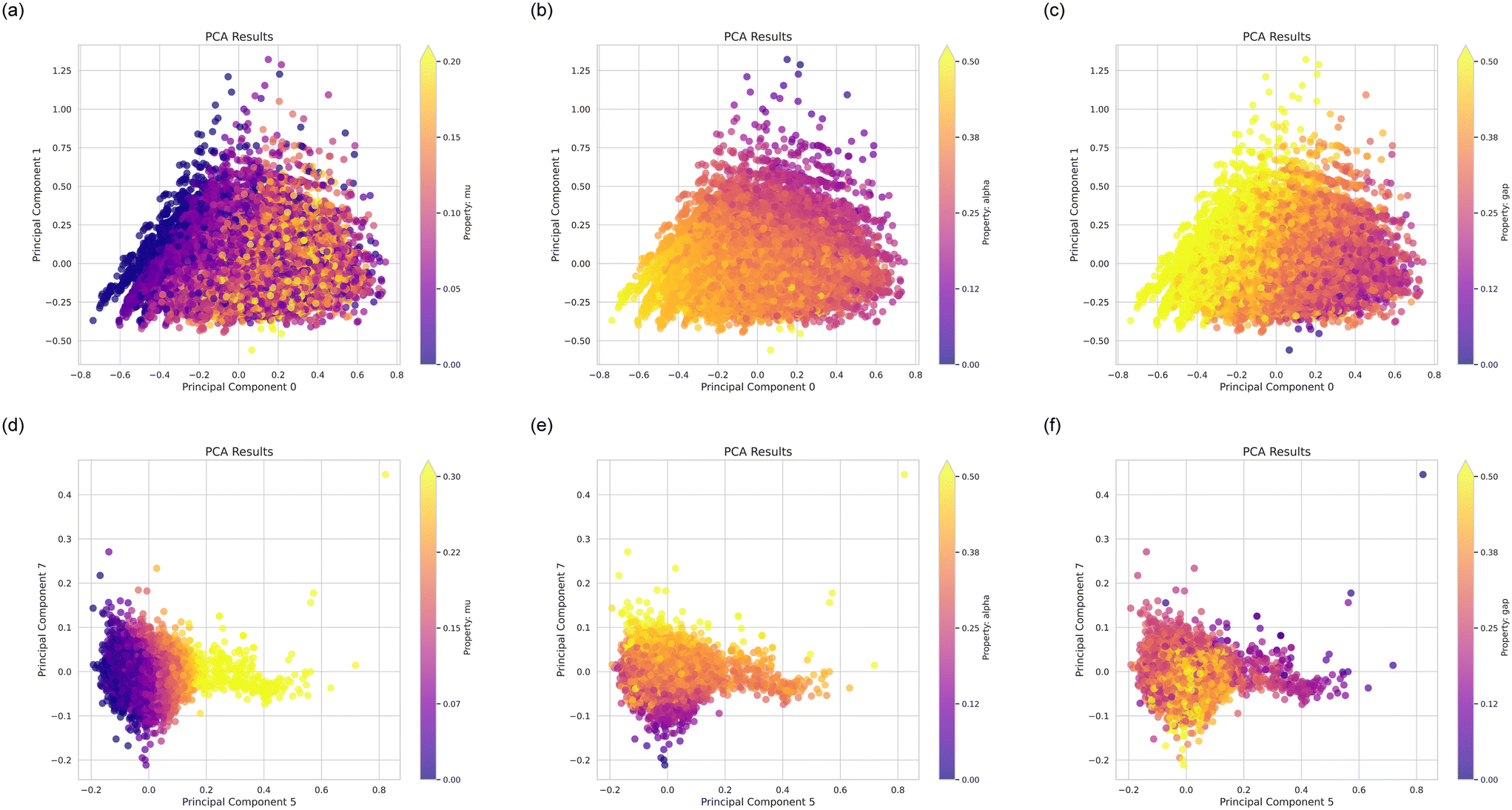 | ||
| Fig. 4 Plots of various molecular properties over principal components (PCs), featuring the dipole moment, mu, polarizability, alpha, and the HOMO–LUMO gap, gap. Panels (a)–(c) show results over PCs 0 and 1, and panels (d)–(f) over PCs 5 and 7. All values are normalized to non-dimensional measures as described in the methods section. Panels (a)–(c) provide evidence that these PCs capture the largest overall variance representing broad global trends in the dataset that may not strongly correlate with specific molecular properties. By contrast, panels (d)–(f) highlight the relevance of PCs 5 and 7 to properties mu and alpha with tighter clustering of datapoints. In this way, molecular designers can efficiently navigate the property space, focusing on changes that optimize properties like mu and alpha while minimizing irrelevant modifications. | ||
The design process will be implemented using the X-LoRA-Gemma model,22 and specifically the forward and inverse tasks for molecular property calculations. To show that the model's capabilities can successfully carry our forward predictions, we show a correlation analysis for a test set of QM9 predictions in Fig. 5. The plot shows results over all molecular properties in the test data and we find R2 = 0.93 that reveals excellent performance of the model for the forward task.
 | ||
| Fig. 5 A correlation analysis for a test set of QM9 predictions based on the X-LoRA-Gemma model.22 The plot shows results over all molecular properties in the test data. We find R2 = 0.93, showing excellent performance of the model for the forward task. | ||
Having established a solid understanding of the dataset and the capabilities of the X-LoRA-Gemma model to compute molecular properties, we now move on to the design task. Fig. 6 depicts two example molecular design process, including one where we start with a starting molecule CCCCC (Fig. 6(a)) and where the target properties obtained via increasing mu and alpha both by Δ = 0.1 (Fig. 6(b)) through changing the molecular properties via principal components 5 and 7. In the second design example (Fig. 6(c–e)), we use CCCC as the starting molecule but the design target is the distribution of properties of Cc1ccc(cn1)C#N.
 | ||
| Fig. 6 Molecular design process, including the starting molecule and the target properties obtained, for two design examples. Panel (a) shows the original molecule with SMILES code CCCCC. Panel (b) shows the property profile of the original molecule and the design target, showing an increase of mu and alpha as suggested by X-LoRA-Gemma, both by Δ = 0.1 (in this example, the error between target and current design is calculated only for the first two properties, mu and alpha). The panel shows that mu and alpha can be targeted for modification in this design process, while leaving other molecular properties unchanged. The lower example shows a second design objective, where we start from a molecule with SMILES code CCCCC in panel (c), and target the properties of an existing molecule with SMILES code Cc1ccc(cn1)C#N in panel (d). The entire set of 12 properties is used to compute the error shown as black arrows in panel (e). This showcases that the model can handle multi-property objectives, evolving toward more sophisticated target molecules. | ||
We conduct the design process via a multi-agent setup that iterates through these steps:
1. Generate new molecular design using the GenerateMolecularProperties<…> task, where the target properties reflect those seen in Fig. 6(b). To seed the design process with an initial molecule, we use in context learning and prepend the generation with the initial molecule: GenerateMolecularProperties<…>[CCCCC] (that is, we provide a design example that shows how a particular design objective is met, starting with the properties of the initial seed molecule, denoted by <…>).
2. Compute the molecular properties of the new design using the forward task CalculateMolecularProperties<…>.
3. Compute the error between the predicted molecular properties and the target, and store the information. This can be done for all 12 properties, or only a subset.
4. If the error between the predicted molecular properties and the target has decreased, the seed molecule for in-context learning is updated with the newly identified, better design and associated properties: GenerateMolecularProperties<PROPERTIES OF NEW MOLECULE>[SMILES OF NEW MOLECULE].
5. If error is below threshold or we have reached the maximum number of iterations, stop. If error is not below threshold, go back to step 1.
In this algorithm the generative agent produces new molecules and the calculation agent computes the properties and decides how well these match the desired target. The updating of the seed molecule supports the process by identifying increasingly more suitable designs while retaining a certain memory of the original cues designated by the starting point. Fig. 7 shows a set of generated molecules as a result of the multi-agent algorithm, with the best performing design on the left top, to the worst on the right lower part. In this algorithm, we use two agents – one to generate a design given the target properties and a second agent to assess the performance of the design. The error between the target and the prediction from the assessment is used to rank predicted solutions. The greater the error between the target and the prediction, the worse the ranking.
 | ||
| Fig. 7 Set of molecules design by the multi-agent algorithm for the design example defined in Fig. 6(a and b), with the best performing design on the left top, to the worst on the right lower part. In this algorithm, we use two agents – one to generate a design given the target properties and a second agent to assess the performance of the design. The error between the target and the prediction from the assessment is used to rank predicted solutions. | ||
Once the generative task is completed, we can further analyze the predicted designs. All designs have been generated using the target properties as generative condition. However, the resulting molecules show different levels of performance with respect to the desired target. The resulting distribution of error can be analyzed and used to select certain top performing molecules. Fig. 8 depicts a summary of the performance of the top designs generated by the multi-agent algorithm, where Fig. 8(a) shows a histogram of the MSE between the target and achieved properties and Fig. 8(b) shows the MSE over the top designs, with the best performing result on the right.
 | ||
| Fig. 8 Summary of the performance of the top designs generated by the multi-agent algorithm, for the design example depicted in Fig. 6(a and b). Panel (a) shows a histogram of the MSE between the target and achieved properties. Panel (b) depicts the MSE over the top designs, with the best performing result on the right. This summary of possible designs provides a rich set of possible solutions that can be explored using other techniques, such as experimental or computational analysis. Further computational steps, such as performing DFT runs for the highest performing options, would provide additional avenues to validate the predictions and add additional context. | ||
Using our earlier PCA analysis, we can explore how well the design has advanced towards our objective. Fig. 9 shows an overview of several measures to assess how the designed molecules move in the PCA space, here focused on the 5th and 7th principal component as they have the highest loadings with respect to the target properties. In the plot, the original design is depicted in blue, the target in red, and the best design per MSE measure is shown in dark blue, small circle. The results show that the design indeed moves closer towards the design target.
 | ||
| Fig. 9 Exploration of movement of the designed molecules in the space of the principal components, here 5th and 7th components, for the design example depicted in Fig. 6(a and b). The original design is depicted in blue, the target in red, and the best design per MSE measure is shown in dark blue, small circle. Visually one can see that the algorithm has indeed moved closer to the target. | ||
Further analysis can be used to validate the predictions using independent calculations. To this we conduct a DFT simulation using PySCF41 to predict the dipole moment. As a proxy measure for polarizability we compute the molar refractivity using RDKit.42,43 It is noted that molar refractivity can act as a proxy for polarizability under certain conditions, primarily because both properties are related to the ability of a molecule to distort its electron cloud in response to an external electromagnetic field. We find that both calculations confirm that the new molecular designs indeed meet the design target and show increases in both dimension. Table 2 shows a comparison with independent predictions, confirming the results. The results are further underscored via additional analyses of the molecular structure and specifically, charge distributions (Fig. 10). This figure shows a visualization of the original molecule, the overall best performing molecule, and the best performer with respect to mu and alpha, including the distribution of atom-level Mulliken charges computed from DFT calculations.41
| Compound | Context | SCF energy (a.u.) [ratio] | Dipole moment (Debye) [ratio] | Molar refractivity (cm3 mol−1) [ratio] |
|---|---|---|---|---|
| CCCCC | Original molecule | −197.786 [1.000] | 0.084 [1.000] | 25.199 [1.000] |
| C1C(CC(C1)N)C |
Best design (example 1) | −288.796 [1.460] | 1.797 [21.393] | 30.644 [1.216] |
| CCCC | Original molecule | −158.470 [1.000] | 3.336 × 10−7 [1.000] | 20.582 [1.000] |
| Cc1cc(ccn1)C | Best design (example 2) | −326.941 [2.063] | 2.259 [6.771 × 106] | 33.711 [1.638] |
 | ||
| Fig. 10 Analysis of charges and molecular structure for three different molecules. Panel (a): visualization of the original molecule used in the design example defined in Fig. 6(a and b). Panel (b): overall best performing molecule for the design example defined in Fig. 6(a and b). Panel (c): analysis of the design example defined in Fig. 6(c–e) (starting molecule is not shown here, as it is quite similar to the case in panel (a)). For each case, the top row shows a 2D projection, the center row DFT-based Mulliken charges, and the lower row a 3D model with color indicating element type (including a mesh representation, whereby the colors of the mesh show the identity of the atom at the molecule's surface at that position). In the designed molecules, pronounced shifts in electron density, particularly around electronegative atoms or polarizable regions, are accommodated by structural changes and directly correlate with the targeted property changes selected by the model. | ||
This model demonstrates great performance in moving toward the target. For real-world applicability of these molecules, computational tools like SYLVIA may be used to obtain the synthetic accessibility (SA) score of each generated molecule to enable chemists to weigh the trade-offs between error to target properties and synthetic feasibility.
The second design example, defined in Fig. 6(c–e), conducts a similar analysis but the target is identified from an existing molecule with SMILES code Cc1ccc(cn1)C#N. The entire set of 12 properties is used to compute the error. The best performing molecule is depicted in Fig. 10(c).
Finally, Fig. 11 offers a summary of the performance, focused on the best design for each of the design examples defined in Fig. 6. Both distributions show good agreement with the target, in spite of both molecules being novel, that is, not included in the QM9 dataset. It is noted that in Fig. 11 the error is only calculated for mu and alpha, reflecting the particular design objective for that case. The other molecular properties change in ways to reflect chemical principles by which molecular properties are distributed, and hence, identified in the generative task.
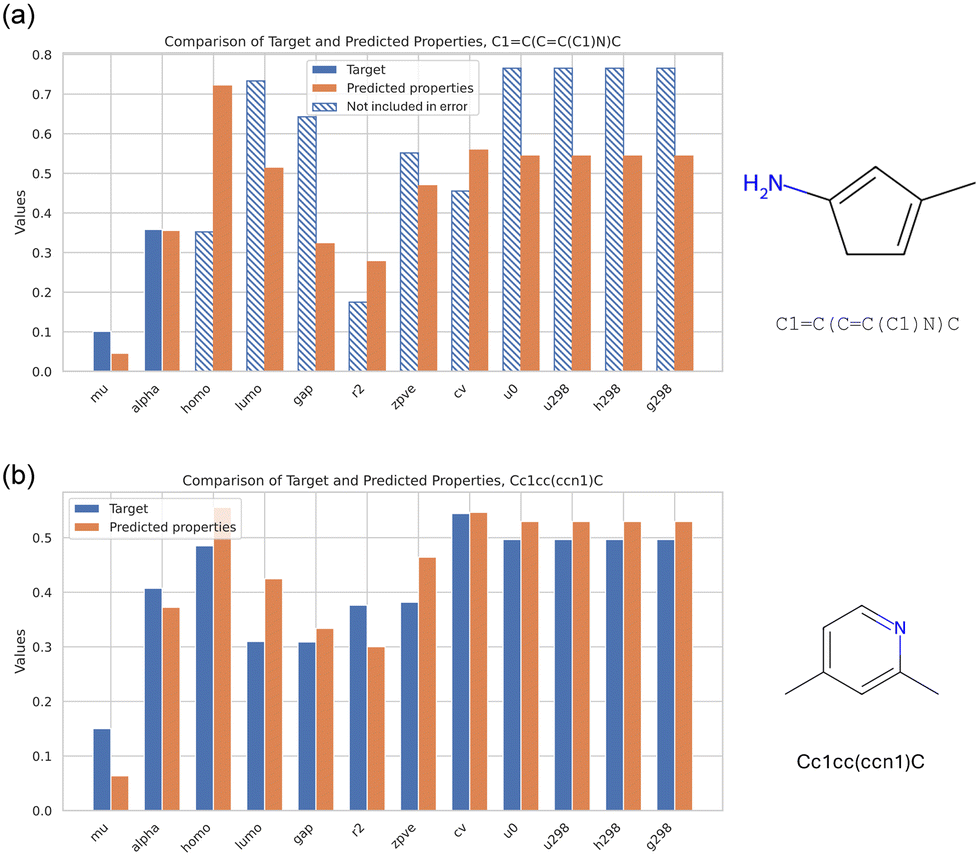 | ||
| Fig. 11 Summary of the performance of the two designs, focused on the best design per MSE for the two properties of interest, mu and alpha (a) (for the first design example defined in Fig. 6(a and b)). In panel (b), the results for the second design objective is shown, this time targeting the entire set of all 12 properties (for the second design example defined in Fig. 6(c–e)). Both cases show good agreement with the target, in spite of both molecules being novel. Note, for the case shown in (a) the error is only calculated for mu and alpha, reflecting the particular design objective. The other molecular properties change in ways to reflect chemical principles by which molecular properties are distributed. | ||
These two design examples demonstrate two conditional generation methods by which this model can reach design objectives. The first design example enables targeted fine-tuning of specific properties, such as dipole moment (mu) and polarizability (alpha). This is particularly useful for applications where small adjustments to certain molecular characteristics are needed to optimize performance for a specific task (e.g., fine-tuning solubility, reactivity, or polarity). The method enables a controlled approach to adjusting molecular properties, which can be crucial when working with molecules where preserving the core structure is important. The method of the second design example is beneficial for achieving significant transformations in molecular properties, aiming to emulate the characteristics of a more complex target molecule. This allows for starting from simpler molecules and evolving them to exhibit properties of more complex structures, which can be useful in de novo design or discovery of new functional molecules.
2.3 Molecular research design via autonomous agentic modeling
As a last example for the proposed approach we expand on the earlier discussion and provide an example of developing a specific research idea that utilizes H-bonds to integrate silk proteins in order to make organic electronic devices. This is done using the same multi-agent setup as before, but we now give one of the agents access to the source text of four scientific papers.In terms of topical choice, the concept of using silk proteins, a naturally abundant and biocompatible material, in the realm of organic electronics may lead to a paradigm shift toward more sustainable and environmentally friendly technology. Silk proteins, with their complex structures and robust mechanical properties,44–46 offer a unique substrate for electronic components that are not only high-performing but also biodegradable. We aim to particularly explore the manipulation of H-bonds as driver of assembly and performance of organic electronic devices.
We provide the following research papers as a foundational baseline for in-context learning about contemporary insights into the multifaceted role of H-bonds in both biological systems and organic electronics:
1. Herschlag et al., hydrogen bonds: simple after all?47
2. Głowacki et al., hydrogen-bonds in molecular solids – from biological systems to organic electronics.48
3. Fang et al., electron transport through hydrogen bonded single molecule junctions.49
4. Zhang et al., repurposing DNA-binding agents as H-bonded organic semiconductors.50
The question posed is
| Develop a specific research idea that utilizes H-bonds to integrate silk proteins in order to make organic electronic devices. |
The conversation then unfolds and discusses the development of H-bonded organic materials derived from silk proteins for use in organic electronic devices. The use of silk proteins in these devices has the potential to enhance their biocompatibility and effectiveness, as well as open up a wide range of new biomedical applications. The key points made in the conversation include the use of specific silk protein structures, such as silk fibroin,51,52 to improve the flexibility and stretchability of organic electronic devices. Additionally, the use of stabilizing agents and optimization of processing conditions can help to minimize protein degradation and maintain the biocompatibility of the materials over time. However, key challenges that need to be addressed are identified, such as the durability of the H-bonds and the potential for protein degradation.
The full conversation is as follows:
Conversation between a molecular designer and a molecular engineerMolecular designer: develop a specific research idea that utilizes H-bonds to integrate silk proteins in order to make organic electronic devices.Molecular engineer: sure, I'd be happy to help! Here's a specific research idea that utilizes H-bonds to integrate silk proteins in order to make organic electronic devices: Objective: The objective is to develop a method to integrate silk proteins into organic electronic devices using H-bonds. This will allow for the creation of bio-based electronics with improved properties. Methodology: The research will focus on the synthesis and characterization of H-bonded organic materials derived from silk proteins. The materials will be designed to have specific electrical and ionic conduction properties, as well as semiconducting properties. The H-bonds will be used to control the ordering of the organic molecules, creating a more efficient and stable device. Chemistry: The research will involve the use of silk proteins as the H-bond donor and acceptor. The silk proteins will be modified to enhance their electrical and ionic conduction properties. The H-bonds will be used to control the ordering of the organic molecules, creating a more efficient and stable device. Mechanics: The research will involve the development of new fabrication techniques to integrate the H-bonded organic materials into organic electronic devices. The devices will be tested for their electrical and ionic conduction properties, as well as their semiconducting properties. Conclusion: The development of H-bonded organic materials derived from silk proteins has the potential to revolutionize the field of bio-based electronics. By integrating silk proteins into organic electronic devices, the research aims to create a more efficient and stable device that can be used in a wide range of applications. Molecular designer: how will the use of silk proteins in organic electronic devices impact their biocompatibility and potential for biomedical applications? Consider factors such as protein degradation, toxicity, and cellular response to silk-based materials. Provide specific examples of potential biomedical applications that could benefit from this technology. Molecular engineer: the use of silk proteins in organic electronic devices could have a significant impact on their biocompatibility and potential for biomedical applications. Silk proteins are biocompatible and biodegradable, making them an attractive option for biomedical applications. The H-bonds in silk proteins can be used to control the ordering of organic molecules, creating a more efficient and stable device. This could lead to a wide range of biomedical applications, such as biosensors, analytical/diagnostic devices, drug delivery systems, and interfaces with neural tissues. However, there are still some challenges that need to be addressed. For example, the stability of the H-bonds and the potential for protein degradation are important factors to consider. Additionally, the toxicity and cellular response to silk-based materials must be carefully evaluated before they can be used in biomedical applications. Overall, the development of H-bonded organic materials derived from silk proteins has the potential to revolutionize the field of bio-based electronics and open up a wide range of new biomedical applications. Molecular designer: how will the use of silk proteins in organic electronic devices impact their flexibility and stretchability, and how can this be optimized for potential wearable technology applications? Molecular engineer: the use of silk proteins in organic electronic devices could have a significant impact on their flexibility and stretchability, which are important factors to consider for potential wearable technology applications. Silk proteins can be designed to have specific mechanical properties, such as elasticity and toughness, which can be optimized for wearable technology applications. Additionally, the H-bonds in silk proteins can be used to control the ordering of organic molecules, creating a more flexible and stretchable device. However, there are still some challenges that need to be addressed. For example, the durability of the H-bonds and the potential for protein degradation are important factors to consider. Additionally, the toxicity and cellular response to silk-based materials must be carefully evaluated before they can be used in wearable technology applications. Overall, the development of H-bonded organic materials derived from silk proteins has the potential to revolutionize the field of wearable technology and open up a wide range of new applications. Molecular designer: how can the stability of the H-bonds in silk proteins be enhanced to ensure their longevity and reliability in organic electronic devices, especially under varying environmental conditions? Additionally, what strategies can be employed to minimize protein degradation and maintain the biocompatibility of these materials over time? Molecular engineer: the stability of the H-bonds in silk proteins can be enhanced by using a combination of factors, such as the design of the silk proteins to minimize protein degradation, the use of stabilizing agents, and the optimization of the processing conditions. Additionally, the use of biodegradable and non-toxic materials, such as silk proteins, can help to minimize protein degradation and maintain the biocompatibility of the materials over time. Overall, the development of H-bonded organic materials derived from silk proteins has the potential to revolutionize the field of organic electronics and open up a wide range of new applications. By addressing the challenges of stability and protein degradation, the use of silk proteins in organic electronic devices could have a significant impact on their biocompatibility and potential for biomedical applications. Molecular designer: how can the toxicity of silk-based materials be minimized while maintaining their biocompatibility and effectiveness in organic electronic devices for potential biomedical applications? Provide specific examples of strategies that have been successful in reducing toxicity without compromising performance. Molecular engineer: the toxicity of silk-based materials can be minimized by using a combination of factors, such as the design of the silk proteins to minimize protein degradation, the use of stabilizing agents, and the optimization of the processing conditions. Additionally, the use of biodegradable and non-toxic materials, such as silk proteins, can help to minimize protein degradation and maintain the biocompatibility of the materials over time. Overall, the development of H-bonded organic materials derived from silk proteins has the potential to revolutionize the field of bio-based electronics and open up a wide range of new biomedical applications. By addressing the challenges of stability and protein degradation, the use of silk proteins in organic electronic devices could have a significant impact on their biocompatibility and potential for biomedical applications. Molecular designer: what specific silk protein structures have been successfully used in organic electronic devices, and how do these structures contribute to their biocompatibility and effectiveness? Can you provide examples of successful applications in wearable technology or biomedical fields? Molecular engineer: the use of specific silk protein structures in organic electronic devices has been successful in enhancing their biocompatibility and effectiveness. For example, the use of silk fibroin, a protein derived from silk, has been shown to improve the flexibility and stretchability of organic electronic devices. Additionally, the use of silk proteins with specific sequences and structures can help to minimize protein degradation and maintain the biocompatibility of the materials over time. Overall, the development of H-bonded organic materials derived from silk proteins has the potential to revolutionize the field of organic electronics and open up a wide range of new applications. By addressing the challenges of stability and protein degradation, the use of silk proteins in organic electronic devices could have a significant impact on their biocompatibility and potential for biomedical applications. |
The key takeaway from the conversation is the potential of H-bonded organic materials derived from silk proteins to impact the field of organic electronics and open up a wide range of new biomedical applications. By addressing the challenges of stability and protein degradation, the use of silk proteins in organic electronic devices could have a significant impact on their biocompatibility and potential for biomedical applications. Specific points made are:
• Specific silk protein structures, such as silk fibroin, can improve the flexibility and stretchability of organic electronic devices.
• Stabilizing agents and optimization of processing conditions can help to minimize protein degradation and maintain the biocompatibility of the materials over time.
• The durability of the H-bonds and the potential for protein degradation are important factors to consider.
• The use of biodegradable and non-toxic materials, such as silk proteins, can help to minimize protein degradation and maintain the biocompatibility of the materials over time.
• The development of H-bonded organic materials derived from silk proteins has the potential to revolutionize the field of organic electronics and open up a wide range of new applications.
• The use of silk proteins in organic electronic devices can enhance their biocompatibility and effectiveness, as well as open up a wide range of new biomedical applications.
Table 3 presents a summary of the papers and pages within the papers used for the six turns in the conversation, providing traceability. Among the four papers provided, only two were used, specially the paper by Głowacki et al.48 focused on hydrogen bonds in molecular solids (from biological systems to organic electronics) as well as Zhang et al.'s paper focused on repurposing DNA-binding agents as H-bonded organic semiconductors.50
| Conversation turn | References and page number within each of the papers |
|---|---|
| 1 | Głowacki et al., page 11 |
| Głowacki et al.,48 page 2 | |
| 2 | Zhang et al.,50 page 10 |
| Głowacki et al., page 3 | |
| 3 | Zhang et al., page 10 |
| Głowacki et al., page 11 | |
| 4 | Głowacki et al., page 11 |
| Głowacki et al., page 2 | |
| 5 | Głowacki et al., page 3 |
| Zhang et al., page 10 | |
| 6 | Zhang et al., page 10 |
| Głowacki et al., page 3 |
This conversation provides specific research ideas that could be followed up on, including with more detailed, further probing questions or computational or experimental studies. We view the initial result of the autonomous agentic interaction as a starting point to develop additional questions, either generated by human users or by extending the conversation. Analyzing the conversation, we identify several important new ideas that the model has developed:
• Bio-based electronics: H-bonded organic materials through the use of modified silk proteins can have enhanced ionic and electronic conduction properties. Specifically, the silk protein can serve as the H-bond donor/acceptor and the ordering of H-bonds can create more stable and efficient devices.
– Biomedical applications: bio-based electronics geared toward biomedical applications can use silk for its biocompatible and biodegradable properties. The model mentions a variety of possible technologies that could benefit from this approach including biosensors, analytical/diagnostic devices, drug delivery systems, and interfaces with neural tissues.
– Wearable technology: bio-based electronics geared toward wearable technology can use silk for its flexible and stretchable qualities. The manipulation of silk's mechanical properties, such as elasticity and toughness, can be optimized for wearable technology applications.
• Optimizing silk-material usage: the model identifies strategies for leveraging silk as a material for bioelectronics.
– Enhancing bio-compatibility and longevity: the design of non-toxic silk proteins, the use of stabilizing agents, and the optimization of the processing conditions can influence bio-compatibility and longevity of silk-based bio-electronics.
– Minimizing toxicity: the extraction of silk proteins from silk offers the possibility of maintaining biocompatibility and effectiveness if challenges like protein degradation and stability are properly addressed.
3 Conclusions
This paper explored the use of a multi-agent LLM, X-LoRA-Gemma, to develop reasoning, discovery and generative capabilities to address molecular design tasks. The workflow used here first identified targets for molecular optimization and then used it to drive a multi-agent generative design process. We showed that a set of target properties of the molecule can be identified using a principal component analysis (PCA) of key molecular properties or by targeting a known distribution of properties derived from an existing molecule. We used the model to generate a large set of candidate molecules. We validate that as predicted, increased dipole moment and polarizability is likely to be achieved in the designed molecules, suggesting that the workflow is reasonable and can be used for other applications. We also presented the use of autonomous agents to explore question development and answering as a system to go deeper into topical areas of molecular design. We demonstrated this both for the development of molecular interactions and a second example focused on protein design, specifically the utilization of H-bonds to integrate silk proteins for novel organic electronic devices. Key takeaways include:• Human–AI collaborations offer powerful strategies, specifically given the adaptability since human can inject a broader breadth of knowledge into the conversation with more nuanced background and context of design objectives.
• AI–AI collaborations typically result in a deep-dive into a focused topic as the querying AI agent logically refines its questions based on the previous responses from the answering AI agent.
• The interchange of these multi-agents facilitates the human user to identify novel ideas and methods.
• The use of principal component analysis (PCA) in AI molecular design, or the development of design objectives based on known molecular property distributions, enables the development of more realistic molecules.
• This model can be used creatively not only to answer questions based on existing research, but to form new research ideas and protocols as exemplified by the silk-based bioelectronics conversation.
The representativeness of responses to chemical queries reflects the selection of training datasets, which encompass geometric, electronic, and thermodynamic properties to ensure comprehensive chemical property coverage. We opted for a low sampling temperature to ensure consistent responses in all design tasks, but this can be easily tuned to endow the model with greater creativity in its responses. Further, a key strength of this model in molecular design tasks is its improved robustness, achieved through a dual-pass inference strategy. This approach dynamically reconfigures the model's internal structure based on the prompt context before generating responses. In this adaptive mechanism, the model's scaling head can leverage the inherent strengths of the underlying LLM and optimally combine adapters to deliver accurate answers. Such a design presents a significant advantage for future LLM-based approaches in chemical discovery, as the framework is compatible with any model without necessitating changes to its internal logic. Notably, it can be seamlessly integrated into the vast resources of the Hugging Face ecosystem, further enhancing its applicability. The model's performance is thus also highly dependent on the training approach, particularly how the scaling head learns to optimally mix adapters. For this reason, future training should include complex question–answer samples or conversations, enabling the model to refine its combinatorial methods. Exploring alternative techniques for layer-wise scaling optimization is also of interest, such as leveraging system messages or other strategies to provide targeted prompts that guide the scaling head toward better domain understanding. Future work could address limitations of this work, such as the limited range of molecular properties (for small molecules defined by the original QM9 dataset23), and the possibility to include other molecular features.
To start, expanding the predictive and generative capabilities can be achieved by adding to the X-LoRA-Gemma model, for instance by including additional training data to improve prediction to target distance and forward/inverse tasks. In this case, selection of the inverse design space is not limited to PCA analysis and the model is highly adaptable to other advanced variable models. We opted for PCA analysis in this work as it simplifies complex datasets, identifies key correlated properties, and aligns changes with chemically feasible directions.
While the importance of small molecule design should not be overlooked especially with respect to property optimization to improve the efficacy and safety of potential drugs and accessibility via chemical synthesis, future work may involve expanding the system to consider molecular interactions and chemical reactions as a bottom-up materials innovation strategy. Tuning on this multiagent system may be used to integrate existing tools like RXN Predict for reaction chemistry prediction, safety assessments, explosive checks, and SYNTH Plan for synthesis planning, which has previously been shown for off-the-shelf LLMs like GPT-4.21 Furthermore, an exciting future direction of this predictive multi-agent model may be in integrating automation capabilities as another agent to enable chemical hardware to execute in synthesizing the generated molecular designs that meet the user's desired target properties.53 Human interjection in a human–AI interaction will be crucial in this context for guiding complex decision-making, interpreting the results, and providing insights that the AI might miss such as subtle considerations of synthesis routes, stability under certain environmental stresses, or other experimental constraints. In such cases, human expertise can override algorithmic recommendations to ensure that the designs are practical and relevant, playing a key role in refining the algorithm's performance. We have previously demonstrated human–AI interactions in such multi-agent contexts where human feedback is used to align the model with human preferences and improve AI safety.9 In this study, it becomes evident that the flow and development of conversations differ significantly between human–AI and AI–AI collaboration, each presenting unique advantages. In human–AI collaboration, the human guides the interaction by posing queries. The AI, in turn, provides tailored responses that include explanations, insights, or solutions, fostering an iterative, interactive exchange where the AI adapts to the user's objectives and context. In contrast, AI–AI collaboration within the X-LoRA-Gemma framework operates autonomously, with pre-defined roles for each AI agent. These interactions are much more structured, task-oriented exchanges, where one AI generates hypotheses or design suggestions, and the other evaluates and refines them. The conversation evolves through logical progression, with minimal redundancy and a focus on efficient decision-making, leveraging the shared expertise of the systems. Unlike human–AI collaboration, AI–AI exchanges are not as restricted by human biases and contextual interruptions or deviations from the established context driven by human curiosity or intuition. This distinction underscores the complementary strengths of both collaboration types in advancing molecular design.
The usability of the model can be further extended to include other text-based representations of chemical structures. In this model, the QM9 dataset was utilized with the SMILES representation, enabling molecules to be extracted from the model as strings of characters. SMILES is a compact and widely supported format that integrates seamlessly with cheminformatics tools and libraries such as RDKit, Open Babel, and ChemAxon.54 For future work, SELFIES presents a robust alternative to address the issue of invalid representations.55,56 Unlike SMILES, every SELFIES string guarantees a valid chemical structure by encoding molecules through molecular substructures (such as atoms, bonds, and functional groups) and linking them with a predefined grammar. This grammar ensures chemical validity.
Although SELFIES employs a more complex syntax that may require additional preprocessing, the representation eliminates syntactic ambiguity, thereby reducing dependence on tokenization. In contrast, SMILES is sensitive to tokenization, which can lead to variations in molecular representations and predictions.56
Although this sensitivity might pose limitations, it can also introduce diverse representations of the same molecule and enables data set augmentation, improving the robustness of the model.56,57 A comparative analysis of SMILES and SELFIES could highlight their respective strengths in achieving accurate and flexible molecular representations. However, the transition to SELFIES may require the adaptation or replacement of workflows and tools built around SMILES, which could constrain validation efforts. Other text-based representations, particularly those relying on detailed natural language descriptions, offer alternative approaches and can be integrated with general LLMs trained on diverse textual datasets. However, these representations risk ambiguity and, therefore, necessitate rigorous standardization.
Other research could focus on experimental validation and more sophisticated quantum mechanical validation beyond what was done in this study.
The ability to combine question asking, reasoning and numerical predictions via multi-agent LLMs as done in this work opens many other possibilities and shows a powerful use case of such models for molecular discovery, especially in a multi-agent setup.
Sometimes, the self-driving multi-agent interaction provides limited insights. For example, the model might reveal the obvious that a molecule should have a modified binding site or a high concentration of functional groups that attract the target molecule to enhance its selectivity. This type of response may be rudimentary for expert chemists but can still serve to lower the barrier for non-experts. In other instances, a domain expert might be required to test different questions to obtain a useful answer. With more powerful AI models (going beyond relatively small 7 billion parameter models, as well as with increased context length) we can likely address these issues. Other possibilities include the use of retrieval-augmented strategies by which we can inject specific or new knowledge into the conversation, providing also referable results that link back to sources used in the development of answers or ideas, as shown in section 2.3. This can also help with improving accuracy and to limit hallucinations, as well as to achieve more consistent results.
The unique benefit of X-LoRA-Gemma is that these future functionalities toward greater chemical discovery can be easily handled owing to its flexible and fine-grained combination of low-rank adapter experts. We have demonstrated how each adapter can be fine-tuned for specific tasks or domains, such as chemistry, mechanics, or protein design, and mixed dynamically during inference to leverage the most relevant expertise. This token-level gating and dynamic scaling approach enable the model to integrate diverse capabilities seamlessly across different tasks, offering a more granular and task-specific response than traditional static LLM mixtures. Furthermore, modularity is enabled by this open-source framework, which encourages transparency and community-driven enhancements. The use of low-rank adapters allows efficient fine-tuning and retraining, significantly reducing computational costs compared to training full models while preserving the ability to specialize in different domains.
Additionally, we find that the use of generative AI can serve as an impactful platform to complement conventional approaches. The availability of multi-agent tools in particular can be an interesting way to connect data-driven and physics-driven modeling and design, such as done in recent work on mechanics58 and proteins.59 Using such approaches, this methodology could be adapted to the design of materials with specific mechanical, thermal, or optical properties, opening up new possibilities for the development of advanced functional materials. This model could address current limitations in reasoning about interconnected material science principles. For instance, this approach may prove particularly useful for processing the multimodal data common in material science literature, while enabling resource-efficient fine-tuning at pace with growing material science knowledge – a current bottleneck in existing LLMs for material science.60–63
Possible ideas for application include utilizing the framework to design novel catalysts and optimize reaction pathways for improved efficiency, selectivity, or sustainability. This could be achieved by including reaction mechanism data and catalyst properties into the design objectives. Thereby, the generative AI approach could enable the discovery of new catalytic systems and the optimization of industrial chemical processes. The method may also be useful for the development of advanced energy storage and conversion technologies, such as high-capacity battery electrodes, efficient solar cells, or novel fuel cell membranes. In other applications, building on the ideas proposed for silk-based electronics and the other conversations, we may target the design of molecules that can self-assemble into complex, hierarchical structures with specific functional properties, such as stimuli-responsiveness, self-healing, or adaptive behavior.
Furthermore, the ability to combine question-asking, reasoning, and numerical predictions via multi-agent LLMs could be extended to a broad range of fields such as drug discovery, catalysis, and renewable energy, where the rapid generation and screening of molecular candidates could accelerate the identification of promising compounds. Future work could also include experimental validation of the designed molecules' properties and performance, which is essential to fully assess the effectiveness of the approach in real-world molecular engineering applications. Other directions could include further assessment of the diversity of generated molecules, for instance, along with additional measures to assess synthesizability, for example.
4 Materials and methods
Detailed methods used for the work presented here are outlined in this section.4.1 Generative AI models
We use the X-LoRA-Gemma model as proposed in ref. 22. The XLoRA-Gemma code and model weights are available at https://huggingface.co/lamm-mit/x-lora-gemma-7b.4.2 Multi-agent AI model
For agentic modeling, we develop an autonomous agent interface that consists of multiple AI agents or AI–human agent combinations. The code is available at https://github.com/lamm-mit/GraphReasoning/tree/main/GraphReasoning.This self-driving discussion is automated by having two (or more) LLMs serve as interacting agents to form a self-driving interaction. The process starts with an initial question (a question asker, defined as molecular designer here), the second LLM agent provides a response (an answering agent, defined as molecular engineer). The question asker is based on a general-purpose LLM, Zephyr-7b-β,34 and the answering agent uses X-LoRA-Gemma.22 Each agent defined with specific instructions to behave in a particular way.
The molecular designer is instructed as follows:
|
You are molecular designer, and taking part in a discussion and ask difficult questions that span a broad spectrum from materials science to manufacturing.
Always challenge statements in a provocative way. As a creative individual, you inject ideas from other fields and push the boundaries. |
The molecular engineer is instructed as follows:
|
You are molecular engineer, a creative engineer with knowledge in biology, chemistry and materials science.
You are taking part in a discussion. Keep your answers brief. You come up with excellent ideas and new directions of thought, always logical. |
The AI agent is set up so that it can have optionally have access to additional data, in our case research papers. We use vector index embedding https://huggingface.co/BAAI/bge-large-en implemented in Llama-Index,64 to realize a retrieval-augmented chat interactions (i.e., responses to the chat are provided with context chunks retrieved from the embedding vector). This enables the agents to draw upon new information via in-context learning, and also provides a level of traceability and sourcing as we can track which paper, and chunk within the paper, was used to answer a particular query in a chat turn.
The conversation starts with the initial question posted. Once the second AI agent answers, a follow-up question is generated. This can be done either in one-shot generation where the agent is given a copy of the entire (or part of) previous conversational exchange, and prompted to develop a follow-up question. Alternatively, the second agent is first given the entire (or part of) previous conversational exchange and then tasked to write a concise summary. Then, it is tasked to develop a follow-up question. An example is (where […] is a copy of the previous conversation between the two agents):
Development of summary and follow-up questionAlgorithm: read this conversation between molecular designer and molecular engineer:“‘[…]”’ ### Instruction: summarize the conversation, with details. Include logic and reasoning, and think step by step. Question developer: the conversation between the molecular designer and molecular engineer revolves around developing a research idea to integrate silk proteins into organic electronic devices using H-bonds. The objective is to create bio-based electronics with improved properties. The research will focus on synthesizing and characterizing H-bonded organic materials derived from silk proteins, which will have specific electrical, ionic conduction, and semiconducting properties. The H-bonds will be used to control the ordering of the organic molecules, creating a more efficient and stable device. New fabrication techniques will be developed to integrate these materials into organic electronic devices, which will be tested for their electrical, ionic conduction, and semiconducting properties. The use of silk proteins in organic electronic devices could have a significant impact on their biocompatibility and potential for biomedical applications. Silk proteins are biocompatible and biodegradable, making them an attractive option for biomedical applications. The H-bonds in silk proteins can be used to control the ordering of organic molecules, creating a more efficient and stable device. This could lead to a wide range of biomedical applications such as biosensors, analytical/diagnostic devices, drug delivery systems, and interfaces with neural tissues. However, there are still some challenges that need to be addressed, such as the stability of H-bonds, protein degradation, toxicity, and cellular response to silk-based materials. These factors must be carefully evaluated before using silk-based materials in biomedical applications. In summary, the research aims to develop a method to integrate silk proteins into organic electronic devices using H-bonds, which could lead to bio-based electronics with improved properties and a wide range of potential biomedical applications. However, careful consideration must be given to factors such as protein degradation, toxicity, and cellular response to silk-based materials before these materials can be used in biomedical applications. Algorithm: please generate a thoughtful and challenging follow-up question. ### You provide only the question, NOT the answer. Only respond with a SINGLE probing, concise question. The question is: Question developer: how will the use of silk proteins in organic electronic devices impact their flexibility and stretchability, and how can this be optimized for potential wearable technology applications? |
4.3 Calculation of molecular properties using physics-based approaches for validation
We use PySCF for density functional theory (DFT) simulations to compute energies and the dipole moment.41 We compute an estimate for the polarizability using RDKit65via the molar refractivity of the molecule. We use Crippen.MolMR(…) within RDKit42 that deals with calculating various physicochemical properties based on fragment contributions.43 The correlation between the RDKit predictions and the ground truth values proves sufficient as a surrogate to the DFT protocol used in QM9 as a computationally cost-efficient approach. The molar refractivity is a measure related to the volume occupied by a mole of a substance and its refractive index, and it is calculated from contributions of various atom types and fragments within the molecule. Molar refractivity can act as a proxy for polarizability under certain conditions, primarily because both properties are related to the ability of a molecule to distort its electron cloud in response to an external electromagnetic field. Here we choose it as a way to validate the generative predictions, as possible quantum mechanical models would be more challenging to conduct.4.4 Chemical analysis and visualization
RDKit42 is used to work with simplified molecular-input line-entry system (SMILES) representations of molecular structures,66,67 visualization and some property calculation. We use PyMOL68 to visualize and analyze molecular structures.A generated molecule is identified as new by checking against all molecules included in the QM9 dataset.
Data availability
Trained weights for the models utilized in this study can be found at https://huggingface.co/lamm-mit/x-lora-gemma-7b, along with codes/notebooks and datasets. The Zephyr-7b-β model is available at https://huggingface.co/HuggingFaceH4/zephyr-7b-beta. Further tools and datasets are provided viahttps://github.com/lamm-mit/GraphReasoning (multi-agent LLM platform) and https://huggingface.co/lamm-mit (general codes and model weights). Training codes and other tools for X-LoRA are available at: https://github.com/EricLBuehler/xlora.Author contributions
MJB designed the research, conducted the model development and inference. IS and MJB carried out simulations and data analysis. MJB and IS wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from Google, the MIT Generative AI Initiative, DOE-SERDP (WP22-S1-3475), the Army Research Office (79058LSCSB, W911NF-22-2-0213 and W911NF2120130), the USDA (2021-69012-35978), and the National Institutes of Health (NIH) (U01EB014976 and R01AR077793).References
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. Kaiser and I. Polosukhin, Advances in Neural Information Processing Systems, 2017, pp. 5999–6009 Search PubMed.
- H. Touvron, L. Martin, K. Stone, P. Albert, A. Almahairi, Y. Babaei, N. Bashlykov, S. Batra, P. Bhargava, S. Bhosale, D. Bikel, L. Blecher, C. C. Ferrer, M. Chen, G. Cucurull, D. Esiobu, J. Fernandes, J. Fu, W. Fu, B. Fuller, C. Gao, V. Goswami, N. Goyal, A. Hartshorn, S. Hosseini, R. Hou, H. Inan, M. Kardas, V. Kerkez, M. Khabsa, I. Kloumann, A. Korenev, S. Koura, M.-A. Lachaux, T. Lavril, J. Lee, D. Liskovich, Y. Lu, Y. Mao, X. Martinet, T. Mihaylov, P. Mishra, I. Molybog, Y. Nie, A. Poulton, J. Reizenstein, R. Rungta, K. Saladi, A. Schelten, R. Silva, E. Michael, S. Ranjan, S. Xiaoqing, E. Tan, B. Tang, R. Taylor, A. Williams, J. X. Kuan, P. Xu, Z. Yan, I. Zarov, Y. Zhang, A. Fan, M. Kambadur, S. Narang, A. Rodriguez, R. Stojnic, S. Edunov and T. Scialom, Llama 2: Open Foundation and Fine-Tuned Chat Models, arXiv, 2023, preprint, arXiv:2307.09288, DOI:10.48550/arXiv.2307.09288.
- A. Q. Jiang, A. Sablayrolles, A. Mensch, C. Bamford, D. S. Chaplot, D. d. l. Casas, F. Bressand, G. Lengyel, G. Lample, L. Saulnier, L. R. Lavaud, M.-A. Lachaux, P. Stock, T. L. Scao, T. Lavril, T. Wang, T. Lacroix and W. E. Sayed, Mistral 7B, arXiv, 2023, preprint, arXiv:2310.06825, DOI:10.48550/arXiv.2310.06825.
- S. Gunasekar, Y. Zhang, J. Aneja, C. César, T. Mendes, A. D. Giorno, S. Gopi, M. Javaheripi, P. Kauffmann, G. De, R. Olli, S. Adil, S. Shital, S. Harkirat, S. Behl, X. Wang, S. Bubeck, R. Eldan, A. Tauman, K. Yin, T. Lee and Y. Li, , Textbooks Are All You Need, arXiv, 2023, preprint, arXiv:2306.11644, DOI:10.48550/arXiv.2306.11644.
- S. Bubeck, V. Chandrasekaran, R. Eldan, J. Gehrke, E. Horvitz, E. Kamar, P. Lee, Y. T. Lee, Y. Li, S. Lundberg, H. Nori, H. Palangi, M. T. Ribeiro and Y. Zhang, Sparks of Artificial General Intelligence: Early experiments with GPT-4, arXiv, 2023, preprint, arXiv:2303.12712, DOI:10.48550/arXiv.2303.12712.
- M. J. Buehler, Appl. Mech. Rev., 2024, 76, 021001 CrossRef.
- M. Nejjar, Z. Luca, F. Stiehle and I. Weber, LLMs for Science: Usage for Code Generation and Data Analysis, arXiv, 2023, preprint, arXiv:2311.16733, DOI:10.48550/arXiv.2311.16733.
- M. J. Buehler, ACS Eng. Au, 2024, 4, 241–277 CrossRef CAS PubMed.
- R. K. Luu and M. J. Buehler, Adv. Sci., 2024, 11, 2306724 CrossRef PubMed.
- R. K. Luu, M. Wysokowski and M. J. Buehler, Appl. Phys. Lett., 2023, 122, 234103 CrossRef CAS.
- M. J. Buehler, J. Mech. Phys. Solids, 2023, 181, 105454 CrossRef.
- Y. Ge, W. Hua, K. Mei, J. Ji, J. Tan, S. Xu, Z. Li and Y. Zhang, OpenAGI: When LLM Meets Domain Experts, arXiv, 2023, preprint, arXiv:2304.04370, DOI:10.48550/arXiv.2304.04370.
- A. D. White, G. M. Hocky, H. A. Gandhi, M. Ansari, S. Cox, G. P. Wellawatte, S. Sasmal, Z. Yang, K. Liu, Y. Singh and W. J. Pena-Ccoa, Digital Discovery, 2023, 2, 368–376 RSC.
- K. Jablonka, P. Schwaller and A. Ortega-Guerrero, Nat. Mach. Intell., 2024, 6, 161–169 CrossRef.
- S. Liu, J. Wang, Y. Yang, C. Wang, L. Liu, H. Guo and C. Xiao, ChatGPT-powered Conversational Drug Editing Using Retrieval and Domain Feedback, arXiv, 2023, preprint, arXiv:2305.18090, DOI:10.48550/arXiv.2305.18090.
- M. C. Ramos, C. J. Collison and A. D. White, A Review of Large Language Models and Autonomous Agents in Chemistry, arXiv, 2024, preprint, arXiv:2407.01603, DOI:10.48550/arXiv.2407.01603.
- Y. Fang, X. Liang, N. Zhang, K. Liu, R. Huang, Z. Chen, X. Fan and H. Chen, Mol-Instructions: A Large-Scale Biomolecular Instruction Dataset for Large Language Models, arXiv, 2023, preprint, DOI:10.48550/ARXIV.2306.08018.
- Y. Liang, R. Zhang, L. Zhang and P. Xie, DrugChat: Towards Enabling ChatGPT-Like Capabilities on Drug Molecule Graphs, arXiv, 2023, preprint, DOI:10.48550/ARXIV.2309.03907.
- B. Yu, F. N. Baker, Z. Chen, X. Ning and H. Sun, LlaSMol: Advancing Large Language Models for Chemistry with a Large-Scale, Comprehensive, High-Quality Instruction Tuning Dataset, arXiv, 2024, preprint, DOI:10.48550/ARXIV.2402.09391.
- G. X. Gu, C. T. Chen and M. J. Buehler, Extreme Mech. Lett., 2018, 18, 19–28 CrossRef.
- A. M. Bran, S. Cox, O. Schilter, O. Baldassari, A. D. White and P. Schwaller, Nat. Mach. Intell., 2024, 6, 525–535 CrossRef.
- E. L. Buehler and M. J. Buehler, X-LoRA: Mixture of Low-Rank Adapter Experts, a Flexible Framework for Large Language Models with Applications in Protein Mechanics and Design, arXiv, 2024, preprint, arXiv:2402.07148, DOI:10.48550/arXiv.2402.07148.
- Z. Qin, Q. Yu and M. J. Buehler, Machine learning model for fast prediction of the natural frequencies of protein molecules, RSC Adv., 2020, 10, 16607–16615 RSC.
- P. Y. Bruice, Organic Chemistry, 2016, p. 1340 Search PubMed.
- M. Glavatskikh, J. Leguy, G. Hunault, T. Cauchy and B. Da Mota, J. Cheminf., 2019, 11, 69 Search PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. Von Lilienfeld, Sci. Data, 2014, 1(1), 1–7 Search PubMed.
- google/gemma-7b-it·Hugging Face, https://huggingface.co/google/gemma-7b-it.
- L. Ruddigkeit, R. Van Deursen, L. C. Blum and J. L. Reymond, J. Chem. Inf. Model., 2012, 52, 2864–2875 CrossRef CAS.
- R. Ramakrishnan, M. Hartmann, E. Tapavicza and O. A. Von Lilienfeld, J. Chem. Phys., 2015, 143, 84111 CrossRef.
- B. Ni, D. L. Kaplan and M. J. Buehler, Sci. Adv., 2024, 10(6), eadl4000 CrossRef CAS.
- G. Falk von Rudorff, Molecular shape as a (useful) bias in chemistry, arXiv, 2019, preprint, arXiv:1904.07035, DOI:10.48550/arXiv.1904.07035.
- Y. Cengel and M. Boles, Thermodynamics: An Engineering Approach, McGraw-Hill, 5th edn, 2006 Search PubMed.
- J. Clayden, N. Greeves and S. Warren, Thermodynamics: An Engineering Approach, Oxford University Press, 2nd edn, 2012 Search PubMed.
- HuggingFaceH4/zephyr-7b-beta·Hugging Face, https://huggingface.co/HuggingFaceH4/zephyr-7b-beta.
- Z. Yang, D. Shao and G. Zhou, J. Chem. Thermodyn., 2020, 144, 106031 CrossRef CAS.
- M. Gutmann, S. H. Zottnick, P. Piechon, I. Dix, K. Müller-Buschbaum, U. Holzgrabe, L. Meinel and B. Galli, Eur. J. Pharm. Biopharm., 2018, 128, 290–299 CrossRef.
- B. Karolewicz, M. Gajda and A. Górniak, J. Therm. Anal. Calorim., 2017, 130, 383–390 CrossRef CAS.
- N. A. Durán-Iturbide, B. L. Díaz Erufacio and J. L. Medina-Franco, ACS Omega, 2020, 5, 16076–16084 CrossRef PubMed.
- S. Phang-Lyn and V. A. Llerena, Biochemistry, Biotransformation, StatPearls Publishing, 2023 Search PubMed.
- A. Z. Garza, S. B. Park and R. Kocz, Drug Elimination, StatPearls Publishing, 2023 Search PubMed.
- Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova, S. Sharma, S. Wouters and G. K. L. Chan, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1340 Search PubMed.
- RDKit, RDKit: Open-source cheminformatics, https://www.rdkit.org.
- S. A. Wildman and G. M. Crippen, J. Chem. Inf. Comput. Sci., 1999, 39, 868–873 CrossRef CAS.
- F. G. Omenetto and D. L. Kaplan, Nat. Photonics, 2008, 2, 641–643 CrossRef CAS.
- D. Ebrahimi, O. Tokareva, N. Rim, J. Wong, D. Kaplan and M. Buehler, ACS Biomater. Sci. Eng., 2015, 1, 23739878 Search PubMed.
- A. Motta, M. Floren and C. Migliaresi, in Biomaterials from Nature for Advanced Devices and Therapies, ed. N. M. Neves and R. L. Reis, Wiley, 1st edn, 2016, pp. 127–141 Search PubMed.
- D. Herschlag and M. M. Pinney, Biochemistry, 2018, 57, 3338–3352 CrossRef CAS PubMed.
- E. D. Głowacki, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, J. Mater. Chem. B, 2013, 1, 3742–3753 RSC.
- J. H. Fang, Z. H. Zhao, A. X. Li and L. Wang, Chin. J. Chem., 2023, 41, 3433–3446 CrossRef CAS.
- F. Zhang, V. Lemaur, W. Choi, P. Kafle, S. Seki, J. Cornil, D. Beljonne and Y. Diao, Nat. Commun., 2019, 10(1), 1–11 CrossRef.
- J. Wu, L. Cao, Y. Liu, A. Zheng, D. Jiao, D. Zeng, X. Wang, D. L. Kaplan and X. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 8878–8895 CrossRef CAS PubMed.
- S. Ling, Z. Qin, W. Huang, S. Cao, D. Kaplan and M. Buehler, Sci. Adv., 2017, 3, 23752548 Search PubMed.
- D. A. Boiko, R. Macknight, B. Kline and G. Gomes, Nature, 2023, 624, 570–578 CrossRef CAS.
- V. Chandrasekhar, N. Sharma, J. Schaub, C. Steinbeck and K. Rajan, J. Cheminf., 2023, 15, 98 Search PubMed.
- M. Leon, Y. Perezhohin, F. Peres, A. PopoviÄ and M. Castelli, Sci. Rep., 2024, 14, 25016 CrossRef CAS.
- M. Krenn, Q. Ai, S. Barthel, N. Carson, A. Frei, N. C. Frey, P. Friederich, T. Gaudin, A. A. Gayle, K. M. Jablonka, R. F. Lameiro, D. Lemm, A. Lo, S. M. Moosavi, J. M. Nápoles-Duarte, A. Nigam, R. Pollice, K. Rajan, U. Schatzschneider, P. Schwaller, M. Skreta, B. Smit, F. Strieth-Kalthoff, C. Sun, G. Tom, G. Falk von Rudorff, A. Wang, A. D. White, A. Young, R. Yu and A. Aspuru-Guzik, Patterns, 2022, 3, 100588 CrossRef CAS PubMed.
- H. Jang, Y. Jang, J. Kim and S. Ahn, Can LLMs Generate Diverse Molecules? Towards Alignment with Structural Diversity, arXiv, 2024, preprint, arXiv:2410.03138, DOI:10.48550/arXiv.2410.03138.
- B. Ni and M. J. Buehler, MechAgents: Large language model multi-agent collaborations can solve mechanics problems, generate new data, and integrate knowledge, Extreme Mech. Lett., 2024, 67, 102131 CrossRef.
- A. Ghafarollahi and M. J. Buehler, ProtAgents: protein discovery via large language model multi-agent collaborations combining physics and machine learning, Digital Discovery, 2024, 3, 1389–1409 RSC.
- S. Miret and N. M. A. Krishnan, Are LLMs Ready for Real-World Materials Discovery?, arXiv, 2024, preprint, arXiv:2402.05200, DOI:10.48550/arXiv.2402.05200.
- N. Gruver, A. Sriram, A. Madotto, A. G. Wilson, C. L. Zitnick and Z. Ulissi, Fine-Tuned Language Models Generate Stable Inorganic Materials as Text, arXiv, 2024, preprint, arXiv:2402.04379, DOI:10.48550/arXiv.2402.04379.
- L. M. Antunes, K. T. Butler and R. Grau-Crespo, Nat. Commun., 2024, 15, 10570 CrossRef CAS PubMed.
- Q. Ding, S. Miret and B. Liu, MatExpert: Decomposing Materials Discovery by Mimicking Human Experts, arXiv, 2024, preprint, arXiv:2410.21317, DOI:10.48550/arXiv.2410.21317.
- run-llama/llama_index: LlamaIndex (formerly GPT Index) is a data framework for your LLM applications, https://github.com/run-llama/llama_index.
- G. Bradski, Dr. Dobb's Journal of Software Tools, 2000 Search PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- D. Weininger, A. Weininger and J. L. Weininger, J. Chem. Inf. Comput. Sci., 1989, 29, 97–101 CrossRef CAS.
- L. L. C. Schrödinger, The PyMOL molecular graphics system, version 1.8, 2015.
| This journal is © The Royal Society of Chemistry 2025 |