
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall upconversion-ruthenium nanohybrids for cancer theranostics†
Anne
Nsubuga
*a,
Nour
Fayad
a,
Federico
Pini
ab,
Marta M.
Natile
 b and
Niko
Hildebrandt
*c
b and
Niko
Hildebrandt
*c
aUniv Rouen, CNRS, INSA Rouen, Normandie Université, Laboratoire COBRA, 76000 Rouen, France. E-mail: anne.nsubuga@univ-rouen.fr
bIstituto di Chimica della Materia Condensata e Tecnologie per l'Energia (ICMATE), Consiglio Nazionale delle Ricerche (CNR), Dipartimento di Scienze Chimiche, Università di Padova, 35131 Padova, Italy
cMcMaster University, Department of Engineering Physics, Hamilton, ON M8S 4K1, Canada. E-mail: hildebrandt@mcmaster.ca
First published on 27th December 2024
Abstract
Photoresponsive drug delivery systems have great potential for improved cancer therapy. However, most of the currently available drug-delivery nanosystems are relatively large and require light excitation with low tissue penetration. Here, we designed a near infrared responsive drug delivery system by loading [Ru(terpyridine)(dipyridophenazine)(H2O)]2+ (Ru(tpy)DPPZ) in azobenzene-modified mesoporous silica coated NaGdF4:Nd0.01/Yb0.2/Tm0.01 upconversion nanoparticles (azo-mSiO2-UCNPs). Upon 808 nm excitation, the generated ultraviolet and blue upconversion luminescence induced a reversible cis–trans isomerization of azobenzene for on-demand release of Ru(tpy)DPPZ. Imaging of both the UCNPs and Ru(tpy)DPPZ revealed targeted drug delivery to the nucleus of MCF-7 breast cancer cells, inducing DNA damage and concomitant cell destruction. Considering that cell nuclei are the core of cellular transcription and the main site of action for multiple chemotherapeutic drugs, our NIR-excitable and small (10 nm diameter) nanohybrids can potentially become highly versatile tools for targeted cancer theranostics.
Introduction
Transition metal complexes have been extensively investigated for different medicinal and diagnostic applications against various types of cancer.1 Metal-based anticancer complexes inhibit cell proliferation by irreversibly creating intrastrand DNA cross-links, provoking cellular DNA damage and ultimately tumor cell death.2 Cisplatin is currently employed as the first step in treating cancer, including metastatic breast and ovarian tumors.3,4 However, the full exploitation of cisplatin anticancer agents is still limited due to development of drug-resistance, adverse side effects, and low solubility.5Efforts have been made to develop alternative metal-based chemotherapeutics with improved treatment and diagnosis outcome.6–8 Ruthenium polypyridyl complexes have emerged as promising anticancer therapeutics and DNA imaging probes due to their tunable electrochemical/photophysical properties and high coordination number.9 Thus, a wide variety of photoactive ruthenium polypyridyl molecules have been considered as interesting alternatives to current treatments.10 They have been shown to bind avidly to DNA with potential applications for cell imaging, in particular, for nuclear staining of high-density DNA and for phototherapeutics.11,12 Complexes featuring the dipyridophenazine (DPPZ) moiety have the ability to intercalate between the base pairs of double-stranded DNA. Interestingly, ruthenium complexes featuring a DPPZ moiety exhibit a unique property known as ‘DNA light switch’. In aqueous solutions, these complexes display negligible photoluminescence (PL); however, upon intercalation with DNA, they show significantly enhanced PL intensities originating from to a 3MLCT (triplet metal-to-ligand charge transfer) excited state.13–16
In vivo applications of ruthenium polypyridyl complexes have raised concerns regarding their stability, cell transport, and targeted delivery.17,18 To address this, recent strategies have involved the use of polymers,19,20 liposomes,21,22 gold nanoparticles,23,24 and silica nanoparticles25,26 as carriers, to ensure effective delivery of specific biological targets. For example, Stoddart and coworkers developed a nanocarrier platform by grafting a benzonitrile ligand, covalently linked with ruthenium polypyridyl complexes, onto the surface of mesoporous SiO2 nanoparticles. Upon irradiation with visible light, the photolabile benzonitrile ligand was cleaved, resulting in the release of [Ru(tpy)(DPPZ)(H2O)]2+ (for simplicity abbreviated as Ru(tpy)DPPZ throughout the article) from the nanoparticle surface. Despite an anticancer effect in cell culture, the substantial size of these nanocarriers (∼150 nm) limited their efficient cellular uptake.27 Photoactivation of ruthenium polypyridyl complexes with high energy photons (<500 nm) is another disadvantage because such light can be phototoxic and has limited penetration depth into tissues.
To reduce phototoxicity associated with blue and UV light and to increase the depth of tissue penetration, lanthanide doped upconversion nanoparticles (UCNPs) have emerged as nano transducers that generate visible or UV emission upon NIR excitation.28 Other key advantages of UCNPs include narrow emission bands, no photobleaching, and low cytotoxicity.28–30 Thus, UCNPs have become promising candidates for applications in bioimaging,31 photodynamic therapy,32–34 and biosensing.35–37 The most frequently applied UCNPs have Yb3+ sensitizer ions for excitation around 980 nm and Er3+ activator ions for emission in the green and red visible range.38,39 However, it is also possible to use Nd3+ sensitizers for excitation around 800 nm and Tm3+ activators for emission in the UV, blue, red, and NIR spectral range.40 Due to lower water absorption, the use of 800 nm causes significantly less heating than 980 nm excitation.41 On the emission side, the UV and blue upconversion luminescence (UCL) can be used for local energy transfer to ruthenium complexes.
For example, Natile, Bonnet et al. developed Yb/Tm-doped UCNPs with ruthenium-encapsulated lipid coatings which could generate reactive oxygen species in aqueous media upon 980 nm excitation.42 To avoid heating effects, the authors extended their research to Nd/Yb/Tm-doped UCNPs with surface-attached photocleavable bis(thioether) coordinated to a ruthenium(II) bipyridyl complex. However, despite 6 h of irradiation with a 796 nm laser (50 W cm−2), only one of the two thioether groups was photocleaved, resulting in relatively inefficient ruthenium release.43 In addition, the UCNPs of both studies were still quite large with more than 40 nm in diameter. Cerruti and coworkers recently developed Yb/Tm-doped UCNPs that required only 2 h of irradiation at 980 nm with relatively low power densities (∼1 W cm−2) to very efficiently release 44% of 7-dehydrocholesterol from a relatively thin (∼10 nm) mesoporous photoactivatable silica coating.44 This study also demonstrated delivery of the UCNPs into HeLa cells but the intracellular drug release could unfortunately not be monitored. With more than 50 nm in diameter, these nanocarriers were also relatively large.
Theoretically, the perfect nanocarrier should be very small to yield efficient cellular internalization and present high surface-to-volume ratios for carrying a large surface load per volume. However, smaller UCNPs are significantly dimmer than larger ones, which can be disadvantageous to photoactivate the UCNP coating for drug release and may require high excitation power densities. That said, the distance between the photoactive molecules and the UCNP would be shorter for a more efficient energy transfer that would be beneficial for photoactivation. The thickness of the coating is also important. A thick shell could potentially carry a large amount of drugs but an efficient release would be more difficult and energy transfer from UCNP to the entire coating would also be limited. Considering those pre-thoughts about the “anatomy” of an ideal UCNP-based nanocarrier and keeping in mind that ∼800 nm NIR excitation (Nd3+ sensitizers) for lower heating and deeper tissue penetration as well as UV/blue emission (Tm3+ activators) for efficient photoactivated release of ruthenium polypyridyl complexes would also need to be on the list of desired properties, we wanted to find out if such UCNP drug carriers can actually be produced and applied for killing cancer cells. An additional feature of ruthenium-releasing Nd/Yb/Tm-based UCNPs is the ability of being able to independently study the emission of the UCNPs and the ruthenium complexes. Thus, not only the nanocarrier internalization into cells but also the intracellular drug release and drug delivery to the nucleus can be monitored, thereby providing the nanohybrids with a very useful theranostic functionality.
With the aim of developing such nuclear targeting nanotheranostic agents, we loaded Ru(tpy)DPPZ in the interior pores of azobenzene-modified mesoporous silica coated NaGdF4:Nd3+/Yb3+/Tm3+ UCNPs (azo-mSiO2-UCNPs) excitable at 808 nm. Whereas photocleavage of covalent bonds generally demands high energy and extended irradiation times, azobenzene (azo) molecules offer a unique advantage. Azo molecules can reversibly switch between their trans and cis forms when exposed to visible or UV light, making them ideal as photo-responsive molecular machines for controlled drug release.45–48 The trans–cis photo-isomerization is induced by UV light, while cis–trans isomerization proceeds either thermally in the dark or can be accelerated by exposure to illumination with longer wavelengths, typically in the blue/green range. Upon absorption of NIR light (808 or 980 nm), the UCL in the UV-visible region can be used for non-radiative (i.e., FRET) or radiative (i.e., reabsorption) energy transfer to the photoresponsive azo molecules in the porous network of the silica layer (Fig. 1). The reversible photoisomerization via UV and visible light simultaneously emitted by the UCNPs can create a continuous back and forth movement (molecular motor) that results in controlled Ru(tpy)DPPZ release. Notably, once the drug is released from the azo-mSiO2-UCNPs, it can generate fluorescence on its own by binding to DNA, which would provide experimental evidence for targeting DNA in the cell nuclei.
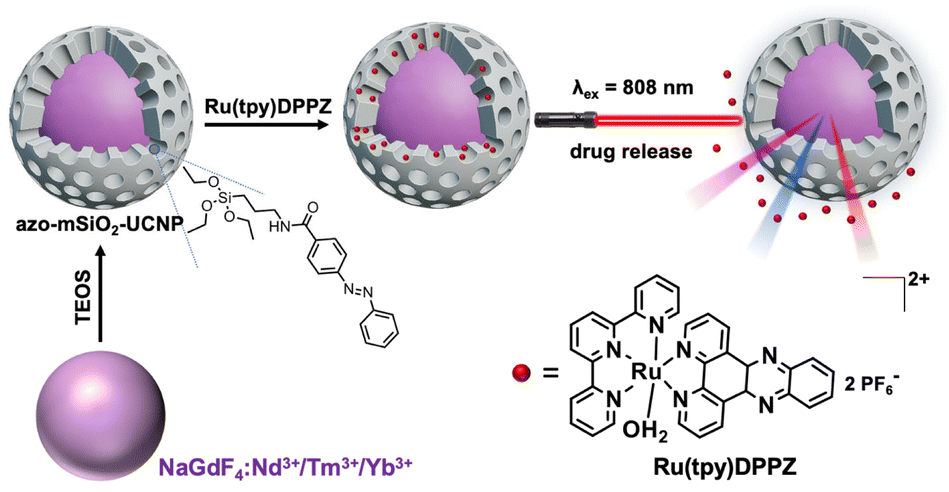 | ||
| Fig. 1 Preparation of Ru(tpy)DPPZ loaded azo-mSiO2-UCNP nanohybrids and NIR-photoactivated drug release. TEOS = tetraethyl orthosilicate; tpy = 2,2′:6′,2′′-terpyridine; DPPZ = dipyrido[3,2-a:2′,3′-c] phenazine. | ||
Results and discussion
Ru(tpy)DPPZ was synthesized following established protocols (see ESI including ESI Fig. S1–S4† for details).27,49–51 Characterization results were consistent with previous reports about this complex. The UV-visible absorption spectrum of Ru(tpy)DPPZ showed the MLCT band of the Ru(II) center at 487 nm (ε = 1671 cm−1 M−1) while the band centered at 273 nm π–π* (ε = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 737 cm−1 M−1) was assigned to the intraligand transitions in the Ru complex (ESI Fig. S5†). NaGdF4:Nd3+/Yb3+/Tm3+ UCNPs were synthesized using a co-precipitation method (see ESI†). The as-synthesized UCNPs exhibited a spherical morphology with an average size of circa 8.2 ± 1.6 nm as evidenced by TEM (ESI Fig. S6†). The UCNPs displayed the characteristic diffractions of the hexagonal β-phase and exhibited phase purity (ESI Fig. S7†). Upon excitation at 808 nm (0.65 W cm−2), the Nd3+ ions transfered energy to Yb3+ and Tm3+, resulting in characteristic UCL bands centered around 346 nm (3P0 → 3F4), 362 nm (1D2 → 3H6), 452 nm (1D2 → 3F4), 476 nm (1G4 → 3H6), 647 nm (1G4 → 3F4), and 692 and 740 nm (3F2,3 → 3H6) (Fig. 2A).
737 cm−1 M−1) was assigned to the intraligand transitions in the Ru complex (ESI Fig. S5†). NaGdF4:Nd3+/Yb3+/Tm3+ UCNPs were synthesized using a co-precipitation method (see ESI†). The as-synthesized UCNPs exhibited a spherical morphology with an average size of circa 8.2 ± 1.6 nm as evidenced by TEM (ESI Fig. S6†). The UCNPs displayed the characteristic diffractions of the hexagonal β-phase and exhibited phase purity (ESI Fig. S7†). Upon excitation at 808 nm (0.65 W cm−2), the Nd3+ ions transfered energy to Yb3+ and Tm3+, resulting in characteristic UCL bands centered around 346 nm (3P0 → 3F4), 362 nm (1D2 → 3H6), 452 nm (1D2 → 3F4), 476 nm (1G4 → 3H6), 647 nm (1G4 → 3F4), and 692 and 740 nm (3F2,3 → 3H6) (Fig. 2A).
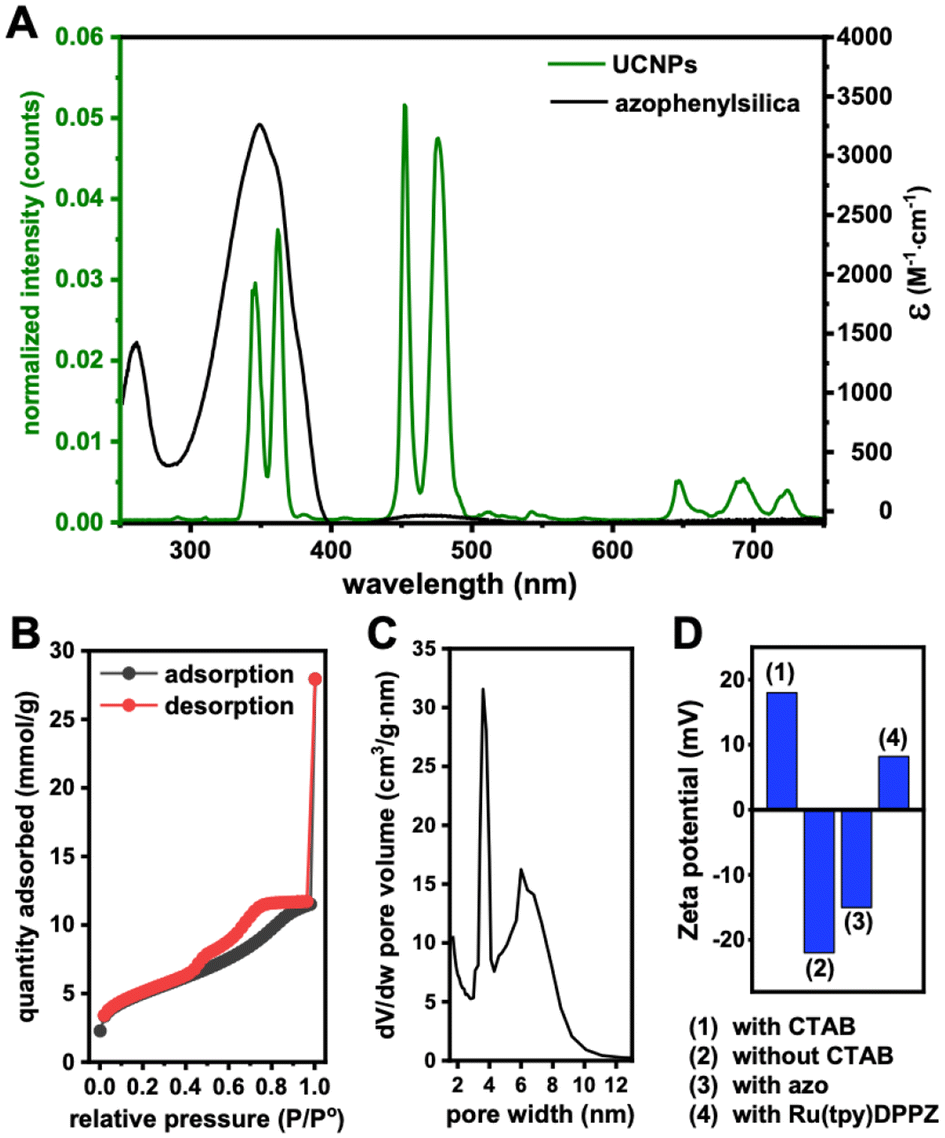 | ||
| Fig. 2 (A) Absorption spectrum of azophenylsilica (black) and UCL spectrum of OA-capped UCNPs (green, 3 mg mL−1) upon 808 nm laser excitation (0.65 W cm−2). (B) Nitrogen adsorption/desorption isotherm of mSiO2-UCNPs. (C) Pore size distribution of mSiO2-UCNPs. (D) Zeta potentials of mSiO2-UCNPs before (1) and after (2) CTAB extraction, azo-mSiO2 (3), and Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs (4). | ||
Oleic acid (OA)-capped UCNPs were functionalized with a mesoporous silica shell using tetraethyl orthosilicate (TEOS) hydrolysis via a template-based mechanism using cetyltrimethyl ammonium bromide (CTAB) as surfactant. To characterize the growth of the shell on the (OA)-capped UCNPs, we performed dynamic light scattering (DLS) analysis (ESI Fig. S8†). The hydrodynamic diameter (Dh) of the (OA)-capped UCNPs (circa 10 nm) showed the expected increase compared to the hard-sphere diameter of 8.2 nm found via TEM (vide supra). Whereas the Dh of the mSiO2-UCNPs increased only slightly to approximately 11 nm, the functionalized mesoporous shells revealed a significant size increase to circa 27 nm for both azo-mSiO2-UCNPs and Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs. The surface area and pore size distribution of mSiO2-UCNPs were analyzed by the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively.52,53 The hysteretic loop of the N2 adsorption/desorption isotherm (Fig. 2B) indicated that the mSiO2-UCNP can be classified as type IV according to the IUPAC classification for mesoporous materials. The pore size distribution was calculated using the BJH method and resulted in pore widths mainly between 3 and 4 nm (Fig. 2C), which indicated a mesoporous structure. Calculations based on BET analysis revealed a surface area of 390 m2 g−1, suggesting that the mesoporous silica shell had a large surface-to-volume ratio, facilitating efficient loading of payloads. As a result, high concentrations of cargo can potentially be delivered into cells while maintaining a lower overall concentration of UCNPs. The resulting mSiO2-UCNPs were functionalized with N-(3-triethoxysilyl)-propyl-4-phenylazobenzamide, which underwent conformational changes from trans to cis upon UV/blue light irradiation and concomitant payload release from the mesopores.54
To confirm successful derivatization, azo-mSiO2-UCNPs were characterized using Fourier transform infrared spectroscopy (FTIR) and zeta potential analysis. As illustrated in the FTIR spectra (ESI Fig. S9†), the mSiO2-UCNPs exhibited strong transmission bands attributed to the stretching vibrations of Si–O–Si at 1084 and 806 cm−1 and Si–OH at 950 cm−1. In the spectra of the azo-mSiO2-UCNPs, new bands corresponding to the symmetric and asymmetric C–H stretching vibrations at 2852 cm−1 and 2922 cm−1, respectively, as well as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic elongation, confirmed the successful grafting of 3-aminopropyltriethoxysilane onto the mSiO2-UCNPs. The surface charge of the mSiO2-UCNPs and azo-mSiO2-UCNPs decreased to circa −22 and −15 mV, respectively, indicating the removal of the cationic CTAB surfactant and the subsequent generation of negatively charged silanol groups on the surface (Fig. 2D). Ru(tpy)DPPZ complexes were encapsulated within the pore channels of the azo-mSiO2-UCNPs nanocarriers by overnight incubation, facilitating diffusion. Excess Ru(tpy)DPPZ that did not enter the pores was removed through filtration, centrifugation, and washing with methanol. Following Ru(tpy)DPPZ loading, the surface charge of the azo-mSiO2-UCNPs increased to ∼8 mV (Fig. 2D), suggesting strong electrostatic interactions between the ruthenium complex and the surface silanol groups within the mesopores. To quantify the concentration of Ru(tpy)DPPZ within the azo-mSiO2-UCNPs nanohybrids, the absorbance of Ru(tpy)DPPZ at 480 nm was measured, and a calibration curve was established (ESI Fig. S10†). Using this curve, the Ru(tpy)DPPZ loading efficiency was calculated to be approximately 17 wt%.
C aromatic elongation, confirmed the successful grafting of 3-aminopropyltriethoxysilane onto the mSiO2-UCNPs. The surface charge of the mSiO2-UCNPs and azo-mSiO2-UCNPs decreased to circa −22 and −15 mV, respectively, indicating the removal of the cationic CTAB surfactant and the subsequent generation of negatively charged silanol groups on the surface (Fig. 2D). Ru(tpy)DPPZ complexes were encapsulated within the pore channels of the azo-mSiO2-UCNPs nanocarriers by overnight incubation, facilitating diffusion. Excess Ru(tpy)DPPZ that did not enter the pores was removed through filtration, centrifugation, and washing with methanol. Following Ru(tpy)DPPZ loading, the surface charge of the azo-mSiO2-UCNPs increased to ∼8 mV (Fig. 2D), suggesting strong electrostatic interactions between the ruthenium complex and the surface silanol groups within the mesopores. To quantify the concentration of Ru(tpy)DPPZ within the azo-mSiO2-UCNPs nanohybrids, the absorbance of Ru(tpy)DPPZ at 480 nm was measured, and a calibration curve was established (ESI Fig. S10†). Using this curve, the Ru(tpy)DPPZ loading efficiency was calculated to be approximately 17 wt%.
Energy transfer from the UCNPs to the conjugated azobenzene molecules was confirmed by PL spectroscopy (Fig. 3A). Upon excitation at 808 nm, UCL spectra of mSiO2-UCNP and azo-mSiO2-UCNP showed that the peaks centered at 368 and 353 nm were the principal contributors of energy transfer from the UCNPs to the azobenzene molecules. These emission bands overlapped strongest with the π–π* transition (absorption band) of the trans-azo-moieties and were therefore quenched via energy transfer (most likely Förster resonance energy transfer – FRET). The overall averaged FRET efficiency (E(avg)), which includes all Tm3+ donors and all azo acceptors independent of their distance and if they actually participate in FRET, was estimated to be 24% using eqn (1):
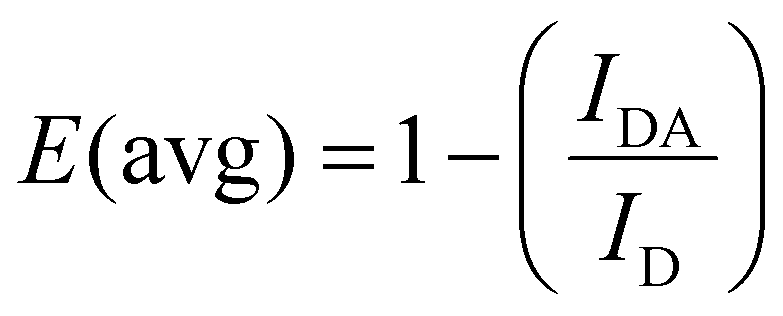 | (1) |
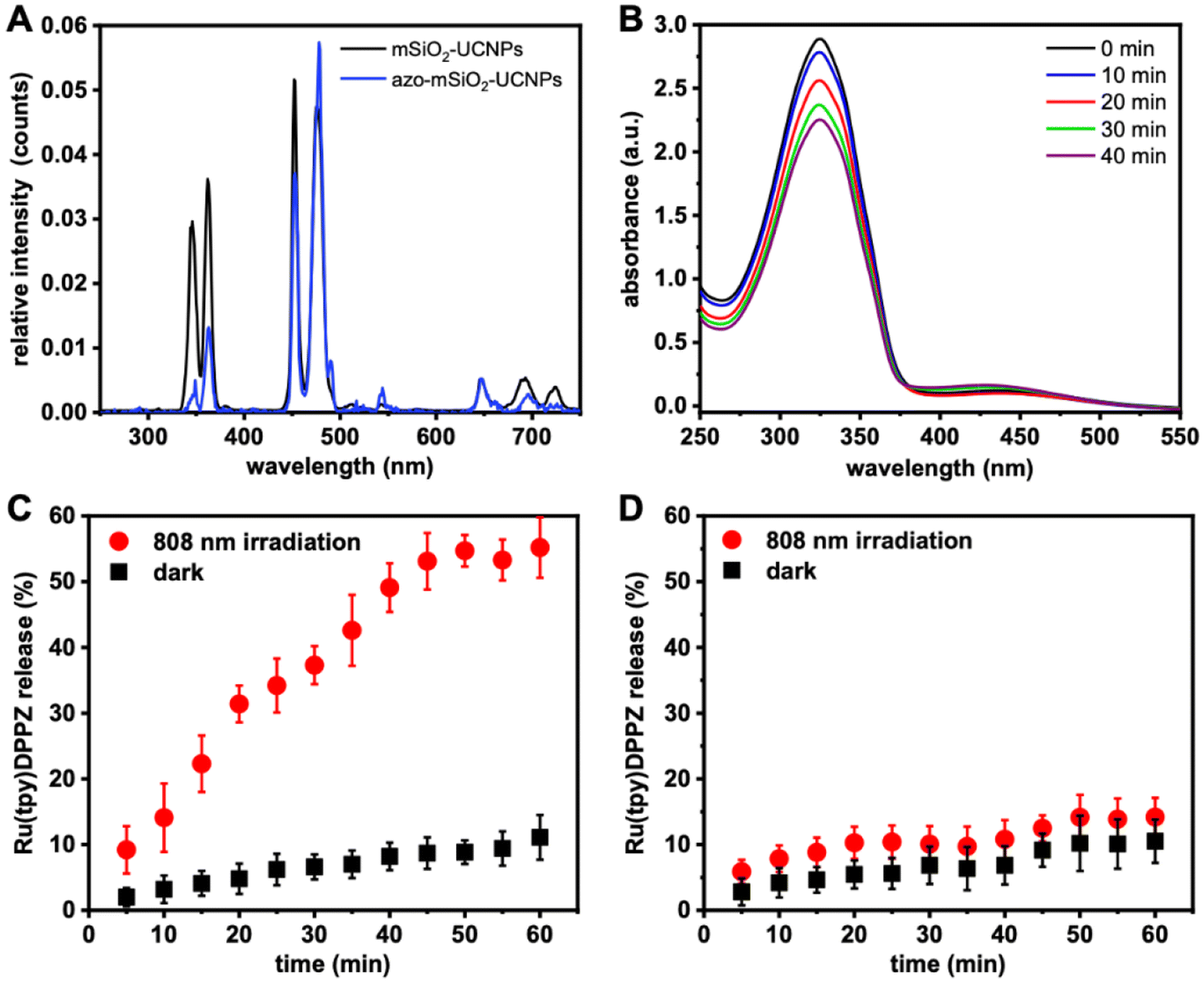 | ||
| Fig. 3 (A) UCL spectra of mSiO2-UCNPs (black) and azo-mSiO2-UCNPs (blue) dispersed in H2O upon 808 nm NIR excitation. (B) Absorption spectra of azo-mSiO2-UCNPs before and after irradiation with 808 nm NIR light for up to 40 min. Release profiles of Ru(tpy)DPPZ from azo-mSiO2-UCNPs (C) and mSiO2-UCNPs (D) with and without NIR irradiation. Standard deviations are shown as error bars for triplicate experiments. | ||
A dispersion of Ru(tpy)DPPZ-loaded azo-mSiO2 UCNPs was irradiated with a 808 nm laser in 5 min intervals for a total of 40 min. Upon exposure to NIR light (808 nm), the UV emissions from Tm3+ ions facilitated the conversion of the trans to the cis isomer of azo-mSiO2. Consequently, the absorption band of the azo molecules at 334 nm, associated with the π–π* transition of the trans isomer, decreased, while the absorption around 435 nm, corresponding to the n–π* transition of the cis isomer, increased. Simultaneously, the visible emissions of Tm3+ ions at 475 and 450 nm caused a reversal of the cis isomer back to the trans isomer, resulting in relatively moderate but still clearly visible absorption intensity changes (Fig. 3B).
To investigate if the NIR-induced trans–cis conversion actually leads to drug release, a dispersion of Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs was irradiated with an 808 nm laser in 5 min intervals for one hour. After each 5 min irradiation period, the samples were centrifuged at 13500g for 20 min and the supernatant was analyzed by UV-visible absorption spectroscopy to determine the concentration of the released anticancer drug (Fig. 3C). In the absence of NIR irradiation, only minimal amounts of Ru(tpy)DPPZ were released (<10%), suggesting that the large majority of azo molecules remained in the trans conformation within the pores, thereby preventing Ru(tpy)DPPZ leakage. In the presence of NIR irradiation, progressive release of Ru(tpy)DPPZ was observed, with approximately 37% of the payload released within 30 min and about 55% released within 1 h of irradiation. This accelerated release following NIR excitation was attributed to the isomerization of azo molecules triggered by UV and blue UCL. A control experiment was conducted using Ru(tpy)DPPZ-loaded mSiO2-UCNPs (without azo). Both the absence and the presence of NIR irradiation resulted in less than 15% drug release even after 1 h of irradiation (Fig. 3D). This result suggests that without the photoactivated azo molecular motor the majority of the Ru(tpy)DPPZ complexes remain confined within the mSiO2-UCNPs. In this case, the H2O ligand likely forms hydrogen bonds with the silica coating, stabilizing the complex and preventing significant release under NIR irradiation. In contrast, with azo moieties present, the trans–cis photoisomerization activates a “molecular machine” mechanism, enabling controlled release. Thus, only the complete azo-mSiO2-UCNPs were capable of photocontrolled drug release.
Before conducting in vitro studies, we assessed the stability of azo-mSiO2-UCNPs and Ru(tpy)DPPZ loaded azo-mSiO2-UCNPs by measuring their hydrodynamic diameters after incubation in DMEM supplemented with 10% fetal bovine serum at 37 °C to simulate biological conditions. No signs of aggregation were observed for at least 7 days. The DLS measurements confirmed that the NPs remained stable throughout the incubation period, demonstrating their suitability for biological applications (ESI Table S1†). The cytotoxicities of free Ru(tpy)DPPZ, azo-mSiO2-UCNPs, and Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs were evaluated on MCF-7 breast carcinoma cells both in the dark and after NIR irradiation using a standard methyl thiazolyl tetrazolium (MTT) assay. As an additional control, the toxicity of cisplatin was also determined on the same cell line (ESI Fig. S11†). Interestingly, the half-maximum inhibitory concentration of Ru(tpy)DPPZ (IC50 = 17.1 ± 0.9 μM) was similar to the one of cisplatin (IC50 = 12.1 ± 1.6 μM). A 24 h incubation with azo-mSiO2-UCNPs and Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs in the dark resulted in concentration-dependent cytotoxicity, which was almost negligible for the azo-mSiO2-UCNPs and relatively weak for the Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs (blue bars in Fig. 4). Incubation for approximately 12 h, followed by NIR irradiation (808 nm, 0.65 W cm−2) for 20 min and another 12 h of incubation (orange bars in Fig. 4), resulted in minimal toxicity for azo-mSiO2-UCNPs (Fig. 4A) and significant toxicity for Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs (Fig. 4B). These findings demonstrated the biocompatibility and negligible toxicity of the free azo-mSiO2-UCNPs, the relatively weak leakage of Ru(tpy)DPPZ from UCNPs without NIR irradiation, and the strong and concentration-dependent NIR photoactivatable cytotoxicity of the Ru(tpy)DPPZ-loaded azo-mSiO2-UCNP nanohybrids. The significant cell death, reaching up to 90%, provided strong evidence for a phototriggered drug release followed by the accumulation and intracellular action of the drug.
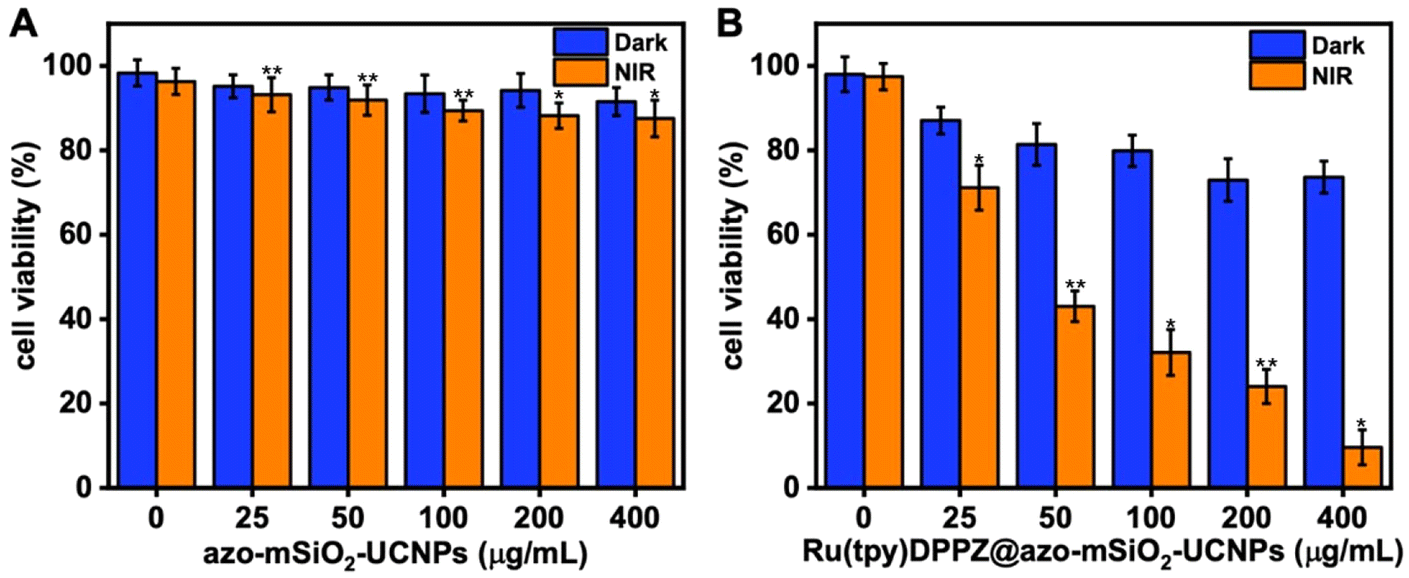 | ||
| Fig. 4 In vitro cell viabilities of MCF-7 cells incubated with different concentrations (0, 25, 50, 100, 200, 400 μg mL−1) of (A) azo-mSiO2-UCNPs and (B) Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs for 24 h. Blue bars represent incubation in the dark for 24 h. Orange bars represent circa 12 h incubation in the dark, followed by 20 min NIR irradiation (808 nm, 0.65 W cm−2), and an additional 12 h of incubation in the dark. Error bars represent the relative standard deviation from triplicate measurements. A statistically significant difference between non-irradiated and irradiated samples is indicated by * at P < 0.02 and ** at P < 0.0006, obtained using a two-tailed Student's t-test, for which P < 0.05 is considered statistically significant. | ||
To show that the drug release actually happened within the cells, i.e., cellular uptake of the Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs and drug release within the cells (as opposed to drug release outside the cells followed by cellular uptake of the drug), we monitored the nanohybrid-incubated cells via PL microscopy. First, MCF-7 cells were incubated in the dark with Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs (100 μg mL−1) for 1, 2, and 4 h, respectively. Following incubation, the cells were stained with the nucleus DNA-staining dye Hoechst 33342 and then irradiated at 808 nm (0.65 W cm−2) for 25 min to initiate drug release. Overlapping the UCNP and Hoechst 33342 emission with differential interference contrast (DIC) transmission images clearly showed that the nanocarriers were mainly located within the cytoplasm of the cells (Fig. 5). Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs underwent rapid uptake with intracellular PL signals already visible after 1 h of incubation. The images after 2 h and 4 h incubation were similar but the cellular contrast clearly decreased over time, an indicator of the drug starting to already decompose the cells. The drug release was clearly confirmed upon excitation of Ru(tpy)DPPZ at 480 ± 25 nm which resulted in bright MLCT phosphorescence at 607 ± 8 nm. Ru(tpy)DPPZ emission strongly overlapped with the nuclear DNA stain (Hoechst 33342), confirming that the drug specifically targeted the cell nuclei and interacted with the nuclear DNA. Despite their small size, the Ru(tpy)DPPZ-loaded azo-mSiO2-UCNP nanohybrids clearly demonstrated their strong potential for theranostics, i.e., real-time imaging of nanocarrier uptake and intracellular drug release followed by drug action.
 | ||
| Fig. 5 Widefield microscopy images of Ru(tpy)DPPZ-loaded azo-mSiO2-UCNP cellular uptake, photo-triggered Ru(tpy)DPPZ release, and Ru(tpy)DPPZ interaction with nuclear DNA in MCF-7 cells. For each panel (1 h, 2 h, and 4 h incubation), the images from left to right show DIC transmission, UCL (ex: 808 nm, em: 450 ± 30 nm, green), Ru(tpy)DPPZ phosphorescence (ex: 480 ± 25 nm, em: 607 ± 8 nm, red), and an overlay of the four images. Scale bars: 20 μm. | ||
Control experiments with and without NIR irradiation after 4 h of incubation confirmed the previous experiments of phototriggered intracellular Ru(tpy)DPPZ release (Fig. 6 top) and also showed minimal passive leakage (without NIR irradiation) of Ru(tpy)DPPZ despite the presence of Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs in the cytoplasm of the cells (Fig. 6 bottom).
 | ||
| Fig. 6 Widefield microscopy images of MCF-7 cells incubated with 100 μg mL−1 Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs for 4 h, with (top) and without (bottom) NIR irradiation (808 nm, 0.65 W cm−2, 25 min). For each panel, the images from left to right show DIC transmission, UCL (ex: 808 nm, em: 450 ± 30 nm, green), Ru(tpy)DPPZ phosphorescence (ex: 480 ± 25 nm, em: 607 ± 8 nm, red), and an overlay of the four images. Scale bars: 20 μm. | ||
Conclusions
Considering the state-of-the-art concerning NIR-activatable drug delivery nanosystems, we aimed at developing and applying small nanoparticles capable of carrying drugs into cells and delivering them to the nucleus upon NIR excitation. This approach allowed us to monitor nanocarrier entry into cells, track drug delivery to the nucleus, and ensure efficient tumor cell destruction. While the unique property of UCNPs to transform NIR excitation into UV and visible emission has been used to photoactivate surface molecules to deliver drugs, a particular challenge has been the design of small nanocarriers in the range of 10 nm in diameter and the use of moderate NIR irradiation power densities. The smaller the nano-delivery systems, the higher the surface-to-volume ratio and the more efficient the cell penetration that is required to deliver the drugs to the cell nucleus. However, small UCNPs contain a lower amount of lanthanide ions which resultor requires high excit/or requires high excitation power densities.Here, we designed sub-10 nm UCNPs that could be excited at 808 nm (due to Nd3+ sensitizers) and emitted in the UV and visible spectral range (due to Tm3+ activators) for energy transfer to photo-switchable azobenzene molecules. These azo molecules were functionalized to a thin mesoporous silica shell on the UCNP. Ru(tpy)DPPZ drug molecules could be captured by the azo-mSiO2-UCNPs and only released via NIR excitation due to the reversible photoactivation of trans–cis conformation in azobenzene. NIR excitation at power densities below 1 W cm−2 for a short duration was sufficient to release a significant amount of Ru(tpy)DPPZ, with approximately 37% of the payload released within 30 min and about 55% within 1 h of irradiation. A 20 min NIR irradiation at 0.65 W cm−2 was enough to kill over 90% of MCF-7 breast cancer cells that had been incubated with 400 μg mL−1 of Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs. PL imaging demonstrated that incubating MCF-7 cells with 100 μg mL−1 of Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs for a few hours, followed by NIR excitation (808 nm, 0.65 W cm−2) for 25 min, led to the intracellular release of Ru(tpy)DPPZ and its delivery into cell nuclei, where it intercalated with DNA, inducing DNA damage and subsequent cell death. Importantly, azo-mSiO2-UCNPs were biocompatible and did not induce any significant cell damage. Moreover, only small amounts of Ru(tpy)DPPZ were released from non-irradiated azo-mSiO2-UCNPs, causing relatively low dark damage to cells.
Overall, our theranostic nano delivery platform integrates all the desirable properties: small size, thin coating, efficient cell penetration, NIR-activatable drug release, low excitation power densities, short irradiation times, high biocompatibility, and effective tumor cell destruction, along with the capability to monitor each delivery and activation step via PL microscopy. These findings highlight the significant potential of our nanohybrids for photoactivatable deep tissue drug delivery. Future research will focus on functionalizing the Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs with tumor cell-binding molecules to add a targeting function, enabling the delivery of nanohybrids to specific cells for targeted cancer therapy.
Materials and methods
All reagents and chemicals used were purchased from Sigma-Aldrich and used without further purification unless specified.General methods
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AVANCE III 300. Chemical shifts are given in ppm relative to the residual 1H resonance of the deuterated solvent and coupling constants J in Hz. UCL spectra were recorded on a Fluorolog fluorescence spectrometer (Horiba) equipped with external 980 and 808 nm lasers (MDL-III-980/808 nm, CNI Laser). Samples had a concentration of 1 mg mL−1 and were measured in the 350–750 nm spectral range with a power density of 60 W cm−2, an emission slit width of 3 nm, and a step size of 1 nm. Powder X-ray diffraction (XRD) patterns were obtained on a Bruker D8 Advance diffractometer equipped with a Cu Kα source (λ = 1.54056 Å) operating at a power of 40 kV and 40 mA. The 2θ scan range was set from 10–70° with a scanning step size of 0.01° and integration time of 2 s. UV-visible absorption spectra were recorded on a Cary 5000 series UV-Vis-NIR spectrophotometer (Agilent) in a range of 190–900 nm. The samples vibrational modes were characterized using a Nicolet iS50 FTIR spectrometer (Thermo Scientific). The instrument was used in its attenuated total reflectance (ATR) mode. The dried sample was placed against a diamond ATR crystal and the spectra were acquired from 500 to 4000 cm−1. DLS measurements of the particles (1 mg mL−1) were performed with the Malvern Zetasizer Nano ZS. Hydrodynamic diameters were determined with the corresponding error as standard deviations from three measurements. Surface zeta potential of the UCNPs (0.2 mg mL−1) in Milli-Q H2O was determined with a Malvern Zetasizer Nano ZS. TEM was used to characterize nanoparticle size and morphology. TEM analysis of the oleate-capped NaGdF4:Nd3+/Yb3+/Tm3+ UCNPs was performed using a FEI Tecnai G2 instrument operating at 100 kV, equipped with an Olympus Veleta camera. Prior to analysis, a 1 wt% sample was dispersed in n-hexane. A drop of the resulting solution was evaporated on a formvar/carbon film supported 400 mesh copper grid. All obtained micrographs were analyzed with ImageJ. BET analysis was used to characterize samples via a N2 adsorption–desorption isotherm at 77 K using a ASAP 2020 Plus instrument from Micromeritics. The sample was degassed under vacuum conditions (P ≈ 10−5 bar) at a temperature of 130 °C for 24 h before measurement. Specific surface areas were calculated using standard expressions for BET isotherms. Nitrogen adsorption at relative pressures of P/P0 = 0.02–1.0 was used. The total pore volume was determined from the adsorbed volume at the highest P/P0 point on the isotherm. The BJH adsorption model was used to calculate the pore volume in the BJH desorption graphs.53Synthesis of dipyrido[3,2-a:2′,3′-c]phenazine (DPPZ – Scheme 1)
DPPZ was synthesized following a previously reported protocol with slight modifications.49 In this procedure, 1.2 mmol of phendione and 1.2 mmol of o-phenylenediamine were dissolved in 10 mL of absolute ethanol. The resulting solution was refluxed for 30 min. After cooling to room temperature (RT), the product was filtered, washed with methanol, recrystallized and dried under vacuum, yielding 216 mg (77%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 7.97 (dd, 2H, J = 8.1, 4.4 Hz); 8.09 (m, 2H); 8.42 (m, 2H); 9.24 (dd, 2H, J = 4.3, 1.8 Hz); 9.56 (m, 2H).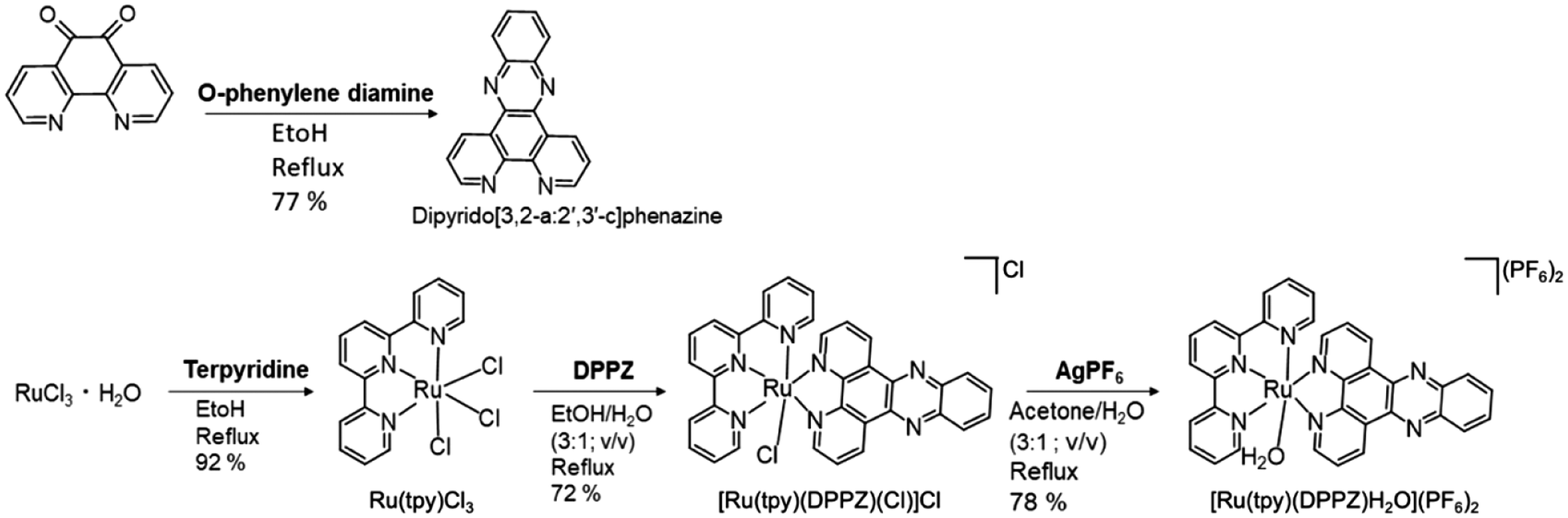 | ||
| Scheme 1 Synthetic routes of DPPZ (top) and Ru(tpy)DPPZ (bottom). | ||
Synthesis of RuCl3(terpyridine)
Ruthenium terpyridine chloride was prepared following a reported method with slight modifications.55 0.3265 mg (1.25 mmol) of RuCl3·3H2O and 0.2915 g (1.25 mmol, 1 equiv.) of 2,2′:6′,2′′-terpyridine were combined in 50 mL of absolute ethanol. The resulting solution was heated at reflux under an argon atmosphere for 3 h and then allowed to cool to RT. Subsequently, the solution was further cooled in an ice bath. The brown solid obtained from the reddish-yellow solution was collected through vacuum filtration, washed with cold methanol until the filtrate became colorless, and further washed four times with Et2O before being air-dried. A total of 0.505 g of product (1.15 mmol) was obtained, with a yield of 92%, and was utilized without additional purification.Synthesis of [Ru(tpy)(DPPZ)(Cl)](Cl−)
A mixture of Ru(tpy)Cl3 (100.6 mg, 0.23 mmol), dipyrido[3,2-a:2′,3′-c]phenazin (74.6 mg, 0.23 mmol), and Et3N (55 μL) was dissolved in 10 mL of EtOH/H2O (3:1, v/v). The solution was degassed at RT and then heated to reflux for 2 h. Following this, the brownish solution was hot-filtered, and the volume was reduced to 2 mL under reduced pressure. The resulting solid was obtained by filtration, thoroughly rinsed with DI–H2O, and air-dried, yielding 72.3%.
1H NMR (CD3OD): 10.40 (1H, d), 9.91 (1H), 9.32–9.30 (1H), 8.89 (1H), 8.81 (2H, d), 8.66 (2H, d), 8.60–8.53 (2H, m), 8.44–8.35 (1H), 8.33 (1H), 8.02 (2H, t), 7.91–7.87 (3H, m), 7.56 (1H, t), 7.36 (2H, t).
Synthesis of [Ru(tpy)(DPPZ)(H2O)](PF6)2 (Ru(tpy)DPPZ – Scheme 1)
[Ru(tpy)(DPPZ)(Cl)] (Cl−) (50 mg, 0.07 mmol) and AgPF6 (17.7 mg, 0.07 mmol) were dissolved in a mixture of acetone and H2O (3:1, 8 mL). The solution underwent degassing and was heated to reflux under argon atmosphere for 2 h. After cooling, the solution was filtered. The filtrate was then reduced to 1 mL under reduced pressure and precipitated by the addition of KPF6 (aq. Sat.). The resulting precipitate was filtered, washed with DI–H2O, recovered with acetone, and dried under vacuum, yielding a dark red solid (78% yield).
1H NMR (CD3COCD3, 500 MHz): δ = 10.06 (1H, dd, J = 5.0 Hz, 1.0 Hz), 9.92 (1H, dd, J = 8.5 Hz, 1.0 Hz), 9.29 (1H, dd, J = 8.5 Hz, 1.0 Hz), 8.77 (2H, d, J = 8.0), 8.61 (1H, dd, J = 8.5 Hz, 5.0 Hz), 8.60 (2H, d), 8.52 (1H, dd), 8.39 (1H, dd), 8.33 (1H), 8.17–8.10 (2H, m), 7.98 (2H, td), 7.94 (1H, dd), 7.87 (2H, d), 7.57 (1H, dd), 7.27 (2H, td).
13C NMR (CD3COCD3, 125 MHz): δ = 159.8, 159.6, 156.2, 155.6, 154.2, 153.9, 153.1, 152.3, 150.6, 143.5, 143.1, 141.4, 140.8, 139.6, 137.3, 133.9, 133.7, 133.5, 132.6, 131.1, 130.5, 130.3, 128.5, 128.3, 127.2, 125.1, 124.1.
Preparation of N-(3-triethoxysilyl)propyl-4-phenyl azobenzene (amide)
4-Phenylazobenzoyl (260 mg; 1.07 mmol) was mixed in 10 mL absolute ethanol. (3-Aminopropyl)triethoxysilane (0.275 mL, 1.17 mmol), and triethylamine (163 μL, 1.17 mmol) were added into the reaction mixture and the reaction was allowed to stir overnight at RT under argon. Next, the solids were filtered off and the filtrate was concentrated under rotary evaporator. The obtained solid was placed under reduced pressure over night.Synthesis of NaGdF4 UCNPs
25%Yb3+,0.5%Tm3+,1%Nd3+: nanoparticles were synthesized at a 1 mmol Ln3+ scale a high-temperature coprecipitation method utilizing a non-coordinating solvent 1-octadecene in combination with oleic acid to direct nanoparticle growth. 1 mmol total of hydrated lanthanide chlorides were combined with 15 mL 1-octadecene and 6 mL oleic acid in a 50 mL round-bottom flask. The reaction mixture was degassed at RT for 10 min and then heated to 160 °C under vacuum for 30 min to form a clear solution. Next, the reaction mixture was cooled to 65 °C, NH4F (4 mmol) and Na-oleate (2.5 mmol) were added simultaneously. The suspension was kept at 120 °C for 30 min under argon flow and then the temperature was increased to 325 °C at a mean rate of ∼15 °C min−1. The reaction temperature was maintained at 325 °C for 25 min. The resulting UCNPs were precipitated through the addition of excess ethanol and collected via centrifugation at 5000 rpm for 5 min. The obtained white pellet was re-dispersed, absolute ethanol (40 mL) was added to induce precipitation. This process was repeated twice more. Finally, the purified oleate-coated UCNPs were dispersed in 10 mL of n-hexane and stored at 4 °C.Synthesis of mSiO2-UCNPs
OA-capped UCNPs were rendered hydrophilic by using CTAB. Typically, CTAB (35 mg) was added to 12 mL ethanol/water (5:1 v/v) and the mixture was stirred and sonicated until a clear solution was obtained. Next, 2 mL of oleate-capped UCNPs dispersed in n-hexane (5 mg mL−1) were added dropwise. Then, 150 μL of an aqueous ammonia solution (32%) was added and TEOS (75 μL) was slowly added dropwise. The reaction solution was stirred overnight at RT. The silica-coated UCNPs were precipitated by adding acetone (40 mL) and collected via centrifugation (6000 rpm for 20 min). Three cycles of re-dispersion and centrifugation were performed to wash the pellet with a methanol/water mixture (1:1 v/v). To obtain the mesoporous silica shell, the CTAB surfactant was extracted. Typically, the as-synthesized silica-coated NPs were mixed with 30 mL EtOH/H2O (1:1, v/v) and the pH was adjusted to 1.5 by adding HCl (2 M). The mixture was heated at 75 °C for 4 h. The mSiO2 UCNPs were precipitated by adding acetone (40 mL) and purified the same way as for the silica capped UCNPs.
Preparation of azobenzene-functionalized mSiO2-UCNPs (azo-mSiO2-UCNPs)
mSiO2 UCNPs (25 mg) were added into a solution containing methanol/H2O (1:1, v/v) and N-(3-triethoxysilyl)propyl-4-phenylazobenzamide (30 mg). The suspension was stirred at 60 °C for 3 h. Afterward, the particles were collected by centrifugation at 12500 rpm for 15 minutes. The obtained orange pellet was re-dispersed in deionized water (DI–H2O), and acetone (25 mL) was added to induce precipitation. This process was repeated twice more. Finally, the particles were dispersed in DI–H2O for subsequent measurements.
Ru(tpy)DPPZ loading into azo-mSiO2-UCNPs
Ru(tpy)DPPZ was incorporated into the mesoporous silica through physical adsorption and electrostatic interactions. Initially, 1 mg of azo-mSiO2-UCNP nanocomposites was dispersed in 5 mL of deionized water, followed by the addition of 10 μmol Ru(tpy)DPPZ. The resulting mixture was stirred in the dark for 12 h. To remove excess ruthenium(II) complex molecules, centrifugation (13500g for 20 min) and methanol washing were employed. UV-visible absorption spectra of the obtained ruthenium-loaded nanocarriers were measured to determine the loading concentrations of Ru(tpy)DPPZ. The quantity of Ru(tpy)DPPZ loaded into azo-mSiO2-UCNPs was then calculated as the difference between the initially added amount of Ru(tpy)DPPZ and the excess amount of Ru(tpy)DPPZ.
NIR triggered release of Ru(tpy)DPPZ
A dispersion of Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs (5 mg in 2 mL H2O) was irradiated with an 808 nm laser for periods of 5 min. A control experiment was done without NIR irradiation. After 5 min of irradiation the sample was centrifuged at 13500g for 20 min, the supernatant was measured by UV-visible absorption spectroscopy to obtain the concentration of released Ru(tpy)DPPZ using a standard calibration curve. The pellet was re-dispersed in distilled H2O (1 mL) and exposed again to NIR light for 5 min.
Cell culture
MCF-7 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Sigma–Aldrich) supplemented with 10% fetal bovine serum (FBS; Sigma–Aldrich), 1% penicillin/streptomycin (10000 U mL−1 penicillin and 10 mg mL−1 streptomycin; Sigma Aldrich), and 1% L-glutamine (Sigma–Aldrich). The cells were maintained at 37 °C in a humidified incubator with 5% CO2. Subsequent passages were performed using trypsin–EDTA (0.05%).Cytotoxicity assays
The viability of MCF-7 cells was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. MCF-7 cells were seeded at a density of 104 cells per well in a 48-well cell culture plate, Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs, azo-mSiO2-UCNPs, Ru(tpy)DPPZ, and cisplatin in culture medium were added to each well. Cells alone were used as controls. A control for the effect of dimethyl sulfoxide (DMSO) was also performed by treating the cells with DMSO. The cells were incubated for 24 h at 37 °C in a 5% CO2 atmosphere. Following incubation, the medium containing particles was aspirated, and the wells were washed with Hank's Balanced Salt Solution (Thermo Fisher Scientific – HBSS, 10×, calcium, magnesium, no phenol red) to eliminate non-uptaken particles. Subsequently, the cells were washed twice with HBSS (100 μL). Fresh medium was added to each well. MTT (3-[4,5-dimethylthiozol-2-yl]-2,5-diphenyltetrazoliumbromide) reagent (0.1 mg mL−1 in cell culture medium) was added to each well and incubated for 4 h at 37 °C in a 5% CO2 atmosphere. The precipitated formazan crystals were dissolved by the addition of DMSO (200 μL). Finally, the absorption of the dissolved formazan crystals in each well were measured at 590 nm using a SPARK (Tecan) microplate reader. All samples were prepared in triplicates. Cell viabilities were expressed as percentages relative to the absorption of the control group (cells incubated with fresh media only).Phototoxicity assays
MCF-7 cells were incubated with Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs at concentrations between 25 and 400 μg mL−1 for 4 h. Excess azo-mSiO2-UCNPs were subsequently removed by washing with HBSS. Following this, an 808 nm laser (0.65 W cm−2) was used to irradiate the cells for 25 min. The culture medium was then replaced with fresh medium, and the cells were further cultivated for 20 h at 37 °C. This experimental setup aimed to assess the effects of 808 nm laser irradiation and laser induced Ru(tpy)DPPZ release on cell viability. All the samples were prepared in triplicates. Cytotoxicity/phototoxicity was expressed as the percentage of cell viability compared to untreated control cells.Live cell imaging
MCF-7 cells were seeded in 35 mm glass-bottom dishes (CELLview Petri dishes, 35 × 10 mm) at a density of 1 × 104 cells and allowed to adhere overnight at 37 °C in a 5% CO2 atmosphere. The following day, cells were washed with HBSS. The Ru(tpy)DPPZ-loaded azo-mSiO2-UCNPs were diluted with Opti-MEM to achieve a final concentration of 100 μg mL−1 and incubated with cells for 4 h at 37 °C in a 5% CO2 environment. After incubation, cells were washed twice with HBSS to remove any free conjugates, and 1.5 mL of Opti-MEM was added to the cells. Subsequently, the cells were irradiated with an 808 nm laser. Following laser exposure, the cells were stained with Hoechst 33342 (1 μM) for 15 min, and the wells were washed with HBSS three times. Nanocarrier uptake was demonstrated through live-cell imaging under 808 nm excitation. The cells were imaged using an Olympus IX83 microscope equipped with a 60× oil immersion objective, a wide-field fluorescence microscope using excitation light source (X-Cite) and an EMCCD camera (Andor iXon 888 Ultra) for detection. The lamp was set to provide a power of 50 W. Images were taken with a 5 ms acquisition time and an electron-multiplying gain set at 200. All data were processed using ImageJ, free software courtesy of The National Institutes of Health.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Région Normandie and the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 101034329 (WINNINGNormandy Fellowship Program), Labex SynOrg (ANR-11-LABX-0029), University of Rouen Normandy, Normandy University, INSA Rouen Normandy, the Centre National de la Recherche Scientifique (CNRS), the European Regional Development Fund (ERDF), Institut Carnot I2C, XL-Chem graduate school (ANR-18-EURE-0020 XL CHEM), the Canada Excellence Research Chairs Program (CERC in Nano-Optical Biosensing and Molecular Diagnostics, CERC-2022–00072), Istituto di Chimica della Materia Condensata e Tecnologie per l'Energia, Consiglio Nazionale delle Ricerche, Dipartimento di Scienze Chimiche, Università di Padova, and the Italian Ministry of University and Research (project PRIN-Lancia – 2022PNR4ET).References
- L. Gourdon, K. Cariou and G. Gasser, Phototherapeutic Anticancer Strategies with First-Row Transition Metal Complexes: A Critical Review, Chem. Soc. Rev., 2022, 51(3), 1167–1195, 10.1039/D1CS00609F.
- F. Heinemann, J. Karges and G. Gasser, Critical Overview of the Use of Ru(II) Polypyridyl Complexes as Photosensitizers in One-Photon and Two-Photon Photodynamic Therapy, Acc. Chem. Res., 2017, 50(11), 2727–2736, DOI:10.1021/acs.accounts.7b00180.
- D. García Sar, M. Montes-Bayón, E. Blanco González, L. M. Sierra Zapico and A. Sanz-Medel, Reduction of Cisplatin-Induced Nephrotoxicity in Vivo by Selenomethionine: The Effect on Cisplatin–DNA Adducts, Chem. Res. Toxicol., 2011, 24(6), 896–904, DOI:10.1021/tx200085n.
- B. Ding, S. Shao, F. Jiang, P. Dang, C. Sun, S. Huang, P. Ma, D. Jin, A. A. Al Kheraif and J. Lin, MnO 2 -Disguised Upconversion Hybrid Nanocomposite: An Ideal Architecture for Tumor Microenvironment-Triggered UCL/MR Bioimaging and Enhanced Chemodynamic Therapy, Chem. Mater., 2019, 31(7), 2651–2660, DOI:10.1021/acs.chemmater.9b00893.
- Y. Liu, Y. Wang, X. Guan, Q. Wu, M. Zhang, P. Cui, C. Wang, X. Chen, X. Meng and T. Ma, Reversal of Cisplatin Resistance in Ovarian Cancer by the Multitargeted Nanodrug Delivery System Tf-Mn-MOF@Nira@CDDP, ACS Appl. Mater. Interfaces, 2023, 15(22), 26484–26495, DOI:10.1021/acsami.3c05374.
- M. G. Ferraro, M. Piccolo, G. Misso, R. Santamaria and C. Irace, Bioactivity and Development of Small Non-Platinum Metal-Based Chemotherapeutics, Pharmaceutics, 2022, 14(5), 954, DOI:10.3390/pharmaceutics14050954.
- P. C. Bruijnincx and P. J. Sadler, New Trends for Metal Complexes with Anticancer Activity, Curr. Opin. Chem. Biol., 2008, 12(2), 197–206, DOI:10.1016/j.cbpa.2007.11.013.
- S. Bonnet, Why Develop Photoactivated Chemotherapy?, Dalton Trans., 2018, 47(31), 10330–10343, 10.1039/C8DT01585F.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, The Development of Ruthenium(II) Polypyridyl Complexes and Conjugates for in Vitro Cellular and in Vivo Applications, Chem. Soc. Rev., 2017, 46(24), 7706–7756, 10.1039/C7CS00680B.
- S. Bonnet, Ruthenium-Based Photoactivated Chemotherapy, J. Am. Chem. Soc., 2023, 145(43), 23397–23415, DOI:10.1021/jacs.3c01135.
- E. Rüba, J. R. Hart and J. K. Barton, [Ru(Bpy) 2 (L)]Cl 2 : Luminescent Metal Complexes That Bind DNA Base Mismatches, Inorg. Chem., 2004, 43(15), 4570–4578, DOI:10.1021/ic0499291.
- M. R. Gill and J. A. Thomas, Ruthenium(II) Polypyridyl Complexes and DNA—from Structural Probes to Cellular Imaging and Therapeutics, Chem. Soc. Rev., 2012, 41(8), 3179–3192, 10.1039/c2cs15299a.
- C. E. Elgar, N. A. Yusoh, P. R. Tiley, N. Kolozsvári, L. G. Bennett, A. Gamble, E. V. Péan, M. L. Davies, C. J. Staples, H. Ahmad and M. R. Gill, Ruthenium(II) Polypyridyl Complexes as FRET Donors: Structure- and Sequence-Selective DNA-Binding and Anticancer Properties, J. Am. Chem. Soc., 2023, 145(2), 1236–1246, DOI:10.1021/jacs.2c11111.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, A Molecular Light Switch for DNA: Ru(Bpy)2(Dppz)2+, J. Am. Chem. Soc., 1990, 112(12), 4960–4962, DOI:10.1021/ja00168a052.
- D. Ossipov, S. Gohil and J. Chattopadhyaya, Synthesis of the DNA–[Ru(Tpy)(Dppz)(CH 3 CN)] 2+ Conjugates and Their Photo Cross-Linking Studies with the Complementary DNA Strand, J. Am. Chem. Soc., 2002, 124(45), 13416–13433, DOI:10.1021/ja0269486.
- M. Frasconi, Z. Liu, J. Lei, Y. Wu, E. Strekalova, D. Malin, M. W. Ambrogio, X. Chen, Y. Y. Botros, V. L. Cryns, J.-P. Sauvage and J. F. Stoddart, Photoexpulsion of Surface-Grafted Ruthenium Complexes and Subsequent Release of Cytotoxic Cargos to Cancer Cells from Mesoporous Silica Nanoparticles, J. Am. Chem. Soc., 2013, 135(31), 11603–11613, DOI:10.1021/ja405058y.
- J. Karges, J. Li, L. Zeng, H. Chao and G. Gasser, Polymeric Encapsulation of a Ruthenium Polypyridine Complex for Tumor Targeted One- and Two-Photon Photodynamic Therapy, ACS Appl. Mater. Interfaces, 2020, 12(49), 54433–54444, DOI:10.1021/acsami.0c16119.
- N. Soliman, G. Gasser and C. M. Thomas, Incorporation of Ru(II) Polypyridyl Complexes into Nanomaterials for Cancer Therapy and Diagnosis, Adv. Mater., 2020, 32(47), 2003294, DOI:10.1002/adma.202003294.
- L. Mascheroni, M. V. Dozzi, E. Ranucci, P. Ferruti, V. Francia, A. Salvati and D. Maggioni, Tuning Polyamidoamine Design To Increase Uptake and Efficacy of Ruthenium Complexes for Photodynamic Therapy, Inorg. Chem., 2019, 58(21), 14586–14599, DOI:10.1021/acs.inorgchem.9b02245.
- J. P. M. António, A. Gandioso, F. Nemati, N. Soliman, R. Vinck, F. Sun, C. Robert, P. Burckel, D. Decaudin, C. M. Thomas and G. Gasser, Polymeric Encapsulation of a Ruthenium(II) Polypyridyl Complex: From Synthesis to in Vivo Studies against High-Grade Epithelial Ovarian Cancer, Chem. Sci., 2023, 14(2), 362–371, 10.1039/D2SC05693C.
- J. Shen, H.-C. Kim, J. Wolfram, C. Mu, W. Zhang, H. Liu, Y. Xie, J. Mai, H. Zhang, Z. Li, M. Guevara, Z.-W. Mao and H. Shen, A Liposome Encapsulated Ruthenium Polypyridine Complex as a Theranostic Platform for Triple-Negative Breast Cancer, Nano Lett., 2017, 17(5), 2913–2920, DOI:10.1021/acs.nanolett.7b00132.
- S. Askes, M. Meijer, T. Bouwens, I. Landman and S. Bonnet, Red Light Activation of Ru(II) Polypyridyl Prodrugs via Triplet-Triplet Annihilation Upconversion: Feasibility in Air and through Meat, Molecules, 2016, 21(11), 1460, DOI:10.3390/molecules21111460.
- S. Estalayo-Adrián, G. J. McManus, H. L. Dalton, A. J. Savyasachi, J. M. Kelly and T. Gunnlaugsson, Functionalisation of Gold Nanoparticles with Ruthenium(II) Polypyridyl Complexes for Their Application in Cellular Imaging, Dalton Trans., 2020, 49(40), 14158–14168, 10.1039/D0DT02754E.
- R. B. P. Elmes, K. N. Orange, S. M. Cloonan, D. C. Williams and T. Gunnlaugsson, Luminescent Ruthenium(II) Polypyridyl Functionalized Gold Nanoparticles; Their DNA Binding Abilities and Application As Cellular Imaging Agents, J. Am. Chem. Soc., 2011, 133(40), 15862–15865, DOI:10.1021/ja2061159.
- L. He, Y. Huang, H. Zhu, G. Pang, W. Zheng, Y. Wong and T. Chen, Cancer–Targeted Monodisperse Mesoporous Silica Nanoparticles as Carrier of Ruthenium Polypyridyl Complexes to Enhance Theranostic Effects, Adv. Funct. Mater., 2014, 24(19), 2754–2763, DOI:10.1002/adfm.201303533.
- Y. Ellahioui, M. Patra, C. Mari, R. Kaabi, J. Karges, G. Gasser and S. Gómez-Ruiz, Mesoporous Silica Nanoparticles Functionalised with a Photoactive Ruthenium(II) Complex: Exploring the Formulation of a Metal-Based Photodynamic Therapy Photosensitiser, Dalton Trans., 2019, 48(18), 5940–5951, 10.1039/C8DT02392A.
- M. Frasconi, Z. Liu, J. Lei, Y. Wu, E. Strekalova, D. Malin, M. W. Ambrogio, X. Chen, Y. Y. Botros, V. L. Cryns, J.-P. Sauvage and J. F. Stoddart, Photoexpulsion of Surface-Grafted Ruthenium Complexes and Subsequent Release of Cytotoxic Cargos to Cancer Cells from Mesoporous Silica Nanoparticles, J. Am. Chem. Soc., 2013, 135(31), 11603–11613, DOI:10.1021/ja405058y.
- F. Auzel, Upconversion and Anti-Stokes Processes with f and d Ions in Solids, Chem. Rev., 2004, 104(1), 139–174, DOI:10.1021/cr020357g.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Upconversion Nanoparticles: Design, Nanochemistry, and Applications in Theranostics, Chem. Rev., 2014, 114(10), 5161–5214, DOI:10.1021/cr400425h.
- X. Liu, C.-H. Yan and J. A. Capobianco, Photon Upconversion Nanomaterials, Chem. Soc. Rev., 2015, 44(6), 1299–1301, 10.1039/C5CS90009C.
- X. Qiu, J. Xu, M. Cardoso Dos Santos and N. Hildebrandt, Multiplexed Biosensing and Bioimaging Using Lanthanide-Based Time-Gated Förster Resonance Energy Transfer, Acc. Chem. Res., 2022, 55(4), 551–564, DOI:10.1021/acs.accounts.1c00691.
- C. A. del Valle, T. Hirsch and M. J. Marín, Recent Advances in near Infrared Upconverting Nanomaterials for Targeted Photodynamic Therapy of Cancer, Methods Appl. Fluoresc., 2022, 10(3), 034003, DOI:10.1088/2050-6120/ac6937.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Upconversion Nanoparticles: Design, Nanochemistry, and Applications in Theranostics, Chem. Rev., 2014, 114(10), 5161–5214, DOI:10.1021/cr400425h.
- A. Nsubuga, K. Morice, N. Fayad, F. Pini, V. Josserand, X. Le Guével, A. Alhabi, M. Henry, D. Puchán Sánchez, N. Plassais, P. Josse, J. Boixel, P. Blanchard, C. Cabanetos and N. Hildebrandt, Sub 20 Nm Upconversion Photosensitizers for Near–Infrared Photodynamic Theranostics, Adv. Funct. Mater., 2024, 2410077, DOI:10.1002/adfm.202410077.
- F. Pini, L. Francés-Soriano, V. Andrigo, M. M. Natile and N. Hildebrandt, Optimizing Upconversion Nanoparticles for FRET Biosensing, ACS Nano, 2023, 17(5), 4971–4984, DOI:10.1021/acsnano.2c12523.
- L. Francés-Soriano, N. Estebanez, J. Pérez-Prieto and N. Hildebrandt, DNA–Coated Upconversion Nanoparticles for Sensitive Nucleic Acid FRET Biosensing, Adv. Funct. Mater., 2022, 32(37), 2201541, DOI:10.1002/adfm.202201541.
- C. Corbella Bagot, E. Rappeport, A. Das, T. Ba Tis and W. Park, True FRET–Based Sensing of PH via Separation of FRET and Photon Reabsorption, Adv. Opt. Mater., 2022, 10(15), 2200242, DOI:10.1002/adom.202200242.
- K. Malhotra, D. Hrovat, B. Kumar, G. Qu, J. Van Houten, R. Ahmed, P. A. E. Piunno, P. T. Gunning and U. J. Krull, Lanthanide-Doped Upconversion Nanoparticles: Exploring A Treasure Trove of NIR-Mediated Emerging Applications, ACS Appl. Mater. Interfaces, 2023, 15(2), 2499–2528, DOI:10.1021/acsami.2c12370.
- R. Lv, M. Raab, Y. Wang, J. Tian, J. Lin and P. N. Prasad, Nanochemistry Advancing Photon Conversion in Rare-Earth Nanostructures for Theranostics, Coord. Chem. Rev., 2022, 460, 214486, DOI:10.1016/j.ccr.2022.214486.
- U. Kostiv, M. M. Natile, D. Jirák, D. Půlpánová, K. Jiráková, M. Vosmanská and D. Horák, PEG-Neridronate-Modified NaYF4:Gd3+,Yb3+,Tm3+/NaGdF4 Core–Shell Upconverting Nanoparticles for Bimodal Magnetic Resonance/Optical Luminescence Imaging, ACS Omega, 2021, 6(22), 14420–14429, DOI:10.1021/acsomega.1c01313.
- C. T. Xu, N. Svensson, J. Axelsson, P. Svenmarker, G. Somesfalean, G. Chen, H. Liang, H. Liu, Z. Zhang and S. Andersson-Engels, Autofluorescence Insensitive Imaging Using Upconverting Nanocrystals in Scattering Media, Appl. Phys. Lett., 2008, 93(17), 171103, DOI:10.1063/1.3005588.
- M. S. Meijer, V. S. Talens, M. F. Hilbers, R. E. Kieltyka, A. M. Brouwer, M. M. Natile and S. Bonnet, NIR-Light-Driven Generation of Reactive Oxygen Species Using Ru(II)-Decorated Lipid-Encapsulated Upconverting Nanoparticles, Langmuir, 2019, 35(37), 12079–12090, DOI:10.1021/acs.langmuir.9b01318.
- M. S. Meijer, M. M. Natile and S. Bonnet, 796 Nm Activation of a Photocleavable Ruthenium(II) Complex Conjugated to an Upconverting Nanoparticle through Two Phosphonate Groups, Inorg. Chem., 2020, 59(20), 14807–14818, DOI:10.1021/acs.inorgchem.0c00043.
- V. Tam, P. Picchetti, Y. Liu, A. Skripka, M. Carofiglio, G. Tamboia, A. Bresci, F. Manetti, G. Cerullo, D. Polli, L. De Cola, F. Vetrone and M. Cerruti, Upconverting Nanoparticles Coated with Light-Breakable Mesoporous Silica for NIR-Triggered Release of Hydrophobic Molecules, ACS Appl. Mater. Interfaces, 2024, 16(22), 29029–29041, DOI:10.1021/acsami.4c03444.
- Y. Zhang, Y. Zhang, G. Song, Y. He, X. Zhang, Y. Liu and H. Ju, A DNA–Azobenzene Nanopump Fueled by Upconversion Luminescence for Controllable Intracellular Drug Release, Angew. Chem., Int. Ed., 2019, 58(50), 18207–18211, DOI:10.1002/anie.201909870.
- J. Park, L. Sun, Y. Chen, Z. Perry and H. Zhou, Azobenzene–Functionalized Metal–Organic Polyhedra for the Optically Responsive Capture and Release of Guest Molecules, Angew. Chem., 2014, 126(23), 5952–5956, DOI:10.1002/ange.201310211.
- H. M. D. Bandara and S. C. Burdette, Photoisomerization in Different Classes of Azobenzene, Chem. Soc. Rev., 2012, 41(5), 1809–1825, 10.1039/C1CS15179G.
- G. A. Mandl, P. A. Rojas-Gutierrez and J. A. Capobianco, A NIR-Responsive Azobenzene-Based Supramolecular Hydrogel Using Upconverting Nanoparticles, Chem. Commun., 2018, 54(46), 5847–5850, 10.1039/C8CC03101K.
- A. Savić, A. M. Kaczmarek, R. Van Deun and K. Van Hecke, DNA Intercalating Near-Infrared Luminescent Lanthanide Complexes Containing Dipyrido[3,2-a:2′,3′-c]Phenazine (Dppz) Ligands: Synthesis, Crystal Structures, Stability, Luminescence Properties and CT-DNA Interaction, Molecules, 2020, 25(22), 5309, DOI:10.3390/molecules25225309.
- N. Gupta, N. Grover, G. A. Neyhart, W. Liang, P. Singh and H. H. Thorp, [RuO(Dppz)(Tpy)] 2+ : A DNA Cleavage Agent with High DNA Affinity, Angew. Chem., Int. Ed. Engl., 1992, 31(8), 1048–1050, DOI:10.1002/anie.199210481.
- A. K. F. Mårtensson and P. Lincoln, Binding of Ru(Terpyridine)(Pyridine)Dipyridophenazine to DNA Studied with Polarized Spectroscopy and Calorimetry, Dalton Trans., 2015, 44(8), 3604–3613, 10.1039/C4DT02642J.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60(2), 309–319, DOI:10.1021/ja01269a023.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms, J. Am. Chem. Soc., 1951, 73(1), 373–380, DOI:10.1021/ja01145a126.
- Z. Zhang, M. K. G. Jayakumar, X. Zheng, S. Shikha, Y. Zhang, A. Bansal, D. J. J. Poon, P. L. Chu, E. L. L. Yeo, M. L. K. Chua, S. K. Chee and Y. Zhang, Upconversion Superballs for Programmable Photoactivation of Therapeutics, Nat. Commun., 2019, 10(1), 4586, DOI:10.1038/s41467-019-12506-w.
- B. P. Sullivan, J. M. Calvert and T. J. Meyer, Cis-Trans Isomerism in (Trpy)(PPh3)RuC12. Comparisons between the Chemical and Physical Properties of a Cis-Trans Isomeric Pair, Inorg. Chem., 1980, 19(5), 1404–1407, DOI:10.1021/ic50207a066.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra (Fig. S1–S3), 13C NMR spectra (Fig. S4), UV-visible spectrum (Fig. S5), TEM micrograph (Fig. S6), XRD diffraction pattern (Fig. S7), DLS spectra (Fig. S8), FTIR spectra (Fig. S9), calibration curve for Ru(tpy)DPPZ quantification (Fig. S10), cell viability curves for Ru(tpy)DPPZ and cisplatin (Fig. S11) and DLS particle size distributions (Table S1). See DOI: https://doi.org/10.1039/d4nr04210g |
| This journal is © The Royal Society of Chemistry 2025 |