
Insight into the role of tunable nitrogen vacancies in transition metal nitrides for ammonia synthesis†
Shiqi
Yu
a,
Ziyu
Mei
b,
Luyuan
Wang
b,
Yuping
Ren
b,
Wei
Wu
a,
Mao
Liu
*a,
Tianyi
Wang
*b and
Chuangwei
Liu
 *bc
*bc
aSchool of Metallurgy, Northeastern University, Shenyang 110819, China. E-mail: liumao@mail.neu.edu.cn
bSchool of Material Science and Engineering, Northeastern University, Shenyang 110819, China. E-mail: tianyiwang@mail.neu.edu.cn
cDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: cwliu@dicp.ac.cn
First published on 9th April 2025
Abstract
The thermally catalyzed nitrogen reduction reaction (NRR) is significant in the fertilizer industry and basic catalytic science. This study employs density-functional theory (DFT) calculations to explore the performance of 12 metal nitrides in thermocatalytic NRR by focusing on them. The surface incompleteness in the catalytic environment is simulated by constructing nitrogen vacancies on the metal nitride surfaces. Then the catalytic activity of these surfaces is evaluated in the thermally catalyzed ammonia synthesis process under specific experimental conditions (temperatures of 573, 673, and 773 K, and a pressure of 1 bar), as well as the effect on the activation of N2 and H2 molecules. It was found that the NRR performance can be optimized by considering the relationship between the N coordination structure on the catalyst surface and the NRR activity, thus identifying LaN(110)–V(N) and NbN(110) as two highly promising catalysts with notable stability and kinetic activity. It is also found that for metal nitride catalysts, a surface with lattice nitrogen as tetra-coordinated exhibits better NRR activity with lower reaction energy, and the distal pathway is more favorable for all catalyst surfaces studied. The present results provide new ideas for developing efficient metal nitride catalysts for ammonia synthesis and enrich the basic knowledge of metal nitride-catalyzed NRR.
Introduction
Ammonia is considered a potential green energy carrier and an essential foundation for chemicals and fertilizer products.1–3 The most critical and rate-limiting step in the ammonia synthesis process is the dissociation of N2, which requires significant energy input to overcome the inert triple bond.4 To reduce energy consumption, efficient ammonia synthesis under mild conditions is an urgent target of current scientific research.5,6 At present, electrochemical and photocatalytic methods receive more attention in investigating ammonia synthesis under mild conditions, but their ammonia production efficiency and scale are far from meeting industrial requirements.7 Although the Haber–Bosch method needs high temperature and pressure to achieve the desired ammonia yield, the traditional process dominates industrial ammonia synthesis.8,9 Therefore, large-scale industrial production of NH3 still relies on traditional thermocatalytic processes.10,11In recent years, researchers have been searching for more efficient and environmentally friendly methods for NH3 synthesis under harsh conditions, and some novel catalysts have been developed.12–14 Compared to traditional Fe-based catalysts, supported Ru-based catalysts exhibit good NH3 synthesis performance under mild reaction conditions, and their performance improves further when alkali metals and alkaline earth metals are used as promoters, making the NH3 synthesis process more efficient.15–17 In addition, Co-based catalysts also exhibit remarkable catalytic activity under mild conditions.1,18 For example, a Ba–Co/C catalyst achieved an NH3 production rate of 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 235 μmol g−1 h−1 at 400 °C and 9 MPa, in which Ba, as a promoter, significantly enhanced the catalytic activity by suppressing the undesirable methane formation on the carbon matrix, even at low loadings.11,15,19,20
235 μmol g−1 h−1 at 400 °C and 9 MPa, in which Ba, as a promoter, significantly enhanced the catalytic activity by suppressing the undesirable methane formation on the carbon matrix, even at low loadings.11,15,19,20
Some ternary nitrides have also attracted increasing attention for their NH3 synthesis performance.21 For instance, a study of Co3Mo3N surfaces explored whether nitrogen vacancies could adsorb and activate N2.22 It was found that within the temperature range for NH3 synthesis, the (111) surface with nitrogen vacancies, surrounded by a triple hollow structure, exhibited an unexpectedly high concentration of nitrogen vacancies (1.6 × 1016 to 3.7 × 1016 cm−2), which could effectively adsorb and activate N2.23 The Co3Mo3N catalyst promoted by Cs exhibited high activity (900 μmol g−1 h−1) at 400 °C and 0.1 MPa, comparable to that of Fe-based and Co-based catalysts under the same conditions.24 Other nitrides such as Co3ZnN, Ni3ZnN, Co3InN, and Ni3InN have also been found to be ineffective as catalysts, though they decomposed to produce ammonia, indicating their potential in chemical cycles.25 Furthermore, NH3 production through the reduction of nitrides, including Cu3N, Ni3N, Re3N, Zn3N2 and Ta3N5, has been reported, but the regeneration of nitrogen gas for chemical looping applications has yet to be realized.26,27
Some metal nitrides have attracted increasing attention due to their high activity in ammonia synthesis reactions.28,29 Metal nitrides can provide abundant lattice nitrogen for NH3 formation and abundant nitrogen vacancies to activate N2.30–32 The reaction conditions and catalytic rates of various nitride-based catalysts have been reported and are summarized in Fig. 1. In 2018, a systematic study of manganese nitride materials in NH3 synthesis was conducted, and the reactivity of lattice nitrogen was assessed. For Mn3N2, limited reactivity was observed, with only 3.1% of the available lattice nitrogen reacting with hydrogen to form NH3, while most lattice nitrogen was lost as N2. In contrast, the addition of a co-metal to manganese nitride altered the pathway of lattice nitrogen reactivity, and it was found that when lithium was used as a co-metal, the nitrogen transfer properties were significantly enhanced, yielding 1306 ± 44 μmol g−1 NH3 during the first 5 h of the reaction and a total of 1571 ± 53 μmol g−1 NH3 after 15 h.33 The amount of NH3 yielded at the end of the reaction corresponded to 18% of the total usable lattice nitrogen for the reaction. The higher reactivity of Li–Mn–N may be attributed to the dihydrogen activation of lithium, in which case Li–Mn–N behaves more as a nitrogen-transferring reagent than a catalyst.34,35
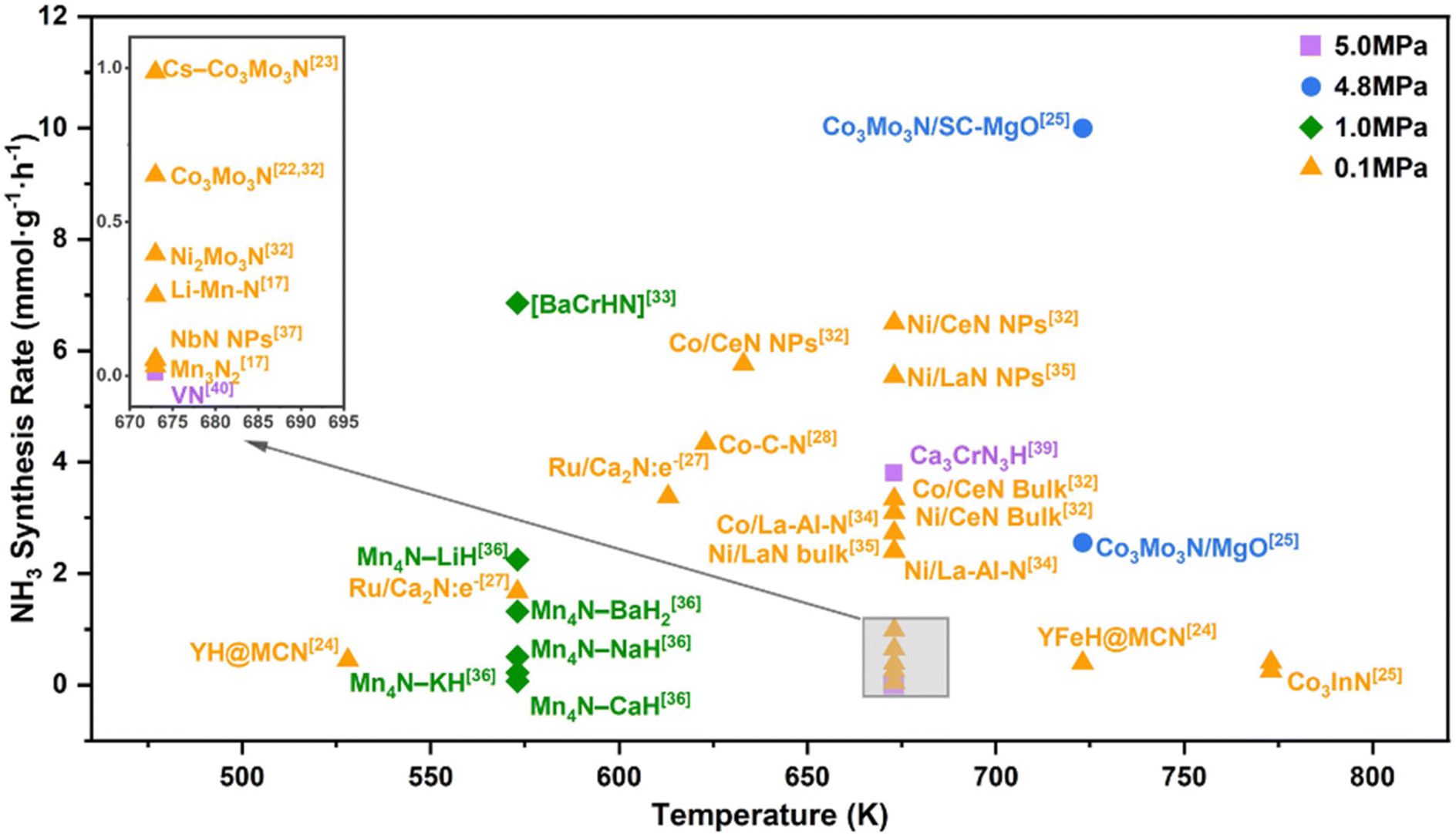 | ||
| Fig. 1 Experimental activities for ammonia synthesis. Overview of the most recent and promising metal nitride catalysts reported for ammonia synthesis. The data points (filled symbols) are extracted from original references based on the following condition: H2/N2 = 3. | ||
In addition, nitrogen vacancies are generated during the reduction process, and Li doping also greatly enhances the formation of nitrogen vacancies and reduces the energy demand of N removal.36 Fe and Ni doping in the Mn4N lattice facilitated the diffusion of N, and Ni doping also reduces the overall flexibility, leading to good H adsorption. Ye et al. have compared the NH3 synthesis from rare-earth metal nitrides such as CeN, NbN, and LaN.35,37 For CeN, the catalytic activities of CeN NPs at 0.1 and 0.9 MPa (673 K) were 250 and 1450 μmol g−1 h−1, respectively.32 They also investigated the associative and dissociative synergistic mechanisms for ammonia synthesis on Ni/CeN catalysts. In the Ni/CeN catalytic system, the activation of H2 and N2 occurred on Ni and surface nitrogen vacancies, where N2 molecules were adsorbed and activated on the nitrogen vacancies, continuously reacting with H* to achieve a stable catalytic cycle.38 In contrast, for pure CeN, VN sites can activate both H2 and N2 simultaneously for stable ammonia production. The formation of nitrogen vacancies in Co/CeN leads to a lower figure of merit due to the strong electron-donating capacity of CeN, which facilitates efficient N2 cleavage on Co. The strong nitrogen affinity of the nitrogen vacancies on the CeN surface leads to promotion of N2 adsorption and activation at these sites. Due to this unique mechanism, the catalytic activity of Co/CeN is much higher than that of other reported Co-based and Ni-based catalysts, even surpassing that of the traditional Ru-based catalysts. For the Ni/LaN system, the surface nitrogen vacancy formation energy is only 1.6 eV, and the nitrogen vacancy sites on the surface weaken the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonding, an effect enhanced by the dissociation of H on Ni metal supported on the substrate.39 This effect is enhanced by the dissociation of H on the Ni metal loaded on the carrier. Researchers have also proposed a new high-chemical-stability lanthanide-based catalyst: Ni- and Co-loaded La–Al–N. In NH3 synthesis, the La3AlN anti-perovskite structure transformed into the distorted rock salt structure of La–Al–N, with Al atoms doping into the LaN lattice.40 Even in this structure, the original functionality of LaN was retained, and the La–Al bonding protected the catalyst from oxidation. After Al doping and H2O adsorption, OH dissociation was hindered, providing the surface with better chemical stability, significant stability, and catalytic activity. The use of non-precious metals such as Ni and Co made TM/La–Al–N stand out as a promising catalyst in various NH3 synthesis processes.34
N bonding, an effect enhanced by the dissociation of H on Ni metal supported on the substrate.39 This effect is enhanced by the dissociation of H on the Ni metal loaded on the carrier. Researchers have also proposed a new high-chemical-stability lanthanide-based catalyst: Ni- and Co-loaded La–Al–N. In NH3 synthesis, the La3AlN anti-perovskite structure transformed into the distorted rock salt structure of La–Al–N, with Al atoms doping into the LaN lattice.40 Even in this structure, the original functionality of LaN was retained, and the La–Al bonding protected the catalyst from oxidation. After Al doping and H2O adsorption, OH dissociation was hindered, providing the surface with better chemical stability, significant stability, and catalytic activity. The use of non-precious metals such as Ni and Co made TM/La–Al–N stand out as a promising catalyst in various NH3 synthesis processes.34
In future catalyst development for NH3 synthesis, evaluating stable and effective catalysts based on nitrogen vacancy formation energy is a promising approach. Building on this premise, twelve metal nitrides—CeN, Co3Mo3N, CrN, Fe3Mo3N, La3AlN, LaN, Mn4N, MoN, NbN, ScN, TiN, and YN—are investigated as potential catalysts. Their three low-index surfaces are selected as candidate active sites, and the catalytic mechanism of the nitrogen reduction reaction (NRR) is explored by introducing surface defects, with a particular focus on the role of surface nitrogen vacancies in enhancing NH3 synthesis activity. LaN(110) and NbN(110) are promising candidates, with the LaN(110)–V(N) model showing excellent stability and activity. This study proposes a rational design principle for NRR catalysts based on four-coordinate lattice nitrogen to enhance catalytic performance through surface engineering.
Computational methods
All structural optimization and electronic structure analyses were conducted using the Vienna Ab Initio Simulation Package (6.3.2 VASP) through density functional theory (DFT). In the DFT computational flow, we applied the revised Perdew–Burke–Ernzerhof (RPBE) generalization under the generalized gradient approximation (GGA), and the description of the core electrons was accurately treated with the help of the projected augmented wave (PAW) method.41,42 In this work, a 500 eV energy cutoff was used to create a plane-wave basis set, and the Brillouin zone was sampled at 3 × 3 × 1 k-points using a Monkhorst–Pack scheme grid, with geometrical optimization until the force becomes less than −0.02 eV m−2 and the energy difference was less than 10−5 eV.43,44 The calculation structures hold 50–60 atoms and two periodic layers. Then three low-index surfaces were selected for the calculations based on the experimentally and theoretically derived X-ray diffraction patterns of the selected metal nitrides. A vacuum space of more than 18 Å was used to avoid interactions between the two periodic cells. In the structural optimization, the bottom three layers of atoms of the surface model were kept fixed to maintain the characteristics of the real surface, while the first layer of atoms of the unit cell was allowed to be fully relaxed during the geometrical optimization for further VN formation and thermal and zero-point-energy (ZPE) correction calculations.45 The geometries of the transition states were determined using the creeping image nudging elastic band (CI-NEB) method. The vibrational frequencies of all species were calculated to analyze the thermodynamic contribution to the free energy.46 For each step, the Gibbs free energy change is calculated by the following equation:| ΔG = ΔE + ΔZPE − ΔTS |
The nitrogen vacancy formation energy (ENV) at the surface is defined as
| ENV = E(VN/slab) − E(slab) + 1/2E(N2) |
The proton-coupled electron transfer steps involved in the NRR can be carefully divided into two main pathways: the distant pathway and the alternating pathway.
Results and discussion
Exploration of potentially active surfaces
In this study, we aim to explore and identify the most suitable metal nitride catalysts for the NRR. To this end, 12 structures were chosen as initial candidates, including single-transition-metal nitrides and double-transition-metal nitrides. To better understand the mechanistic details behind the unique catalytic properties of metal nitrides, DFT calculations were employed.Based on the XRD patterns of the metal nitrides (Fig. S1†), three low-index surfaces were selected from the original unit cell to explore the catalytic active surfaces. To investigate the effect of surface defects on catalytic performance, nitrogen vacancies (denoted as V(Nx)) were introduced into the original metal surfaces, systematically constructing a series of potential defective surfaces. Several typical surface structures and vacancy positions are shown in Fig. 2a and Fig. S2.† The introduction of nitrogen vacancies aims to simulate surface imperfections in real catalytic environments, and subsequently to evaluate their potential impact on catalytic activity. After constructing the defected surfaces with nitrogen vacancies, we further evaluated the thermodynamic stability of these surfaces in the catalytic ammonia synthesis process (Haber–Bosch process). Specifically, we calculated the free formation energies at three key reaction temperatures (T = 573 K, 673 K, 773 K) and at standard pressure (P = 1 bar), as shown in Fig. 2b and Fig. S3–S5.† It was easy to find that double-metal nitrides Co3Mo3N((100), (110), (211)), Fe3Mo3N((100), (110), (211)), and La3AlN((100), (110), (111)) have positive Gibbs free energies of V(N) and V(N2) indicating their poor stability. Any vacancy formation on these surfaces requires significant energy. For the single-metal nitrides CrN((100), (110)), Mn4N((100), (110)), NbN((100), (110)), LaN((100), (110)), and TiN(111), the surfaces are thermodynamically neutral or favorable for vacancy formation. Therefore, these relatively stable surfaces were further studied for their effect on the activation of N2 and H2 molecules.
 | ||
| Fig. 2 (a) Top view of the optimized pristine (100), (110), and (111) surfaces. (b) Relative formation free energies of N vacancies on the above three pristine surfaces at experimental temperature (from 298 K to 773 K) and at 1 bar. The exact locations of the N vacancies created on these three potential surfaces are marked by the numbers in (a). | ||
NRR activity of metal nitrides
The optimized configurations of N2 adsorption on selected CrN((100), (110)), Mn4N((100), (110)), NbN((100), (110)), LaN((100), (110)) and TiN(111) pristine and defective surfaces are shown in Fig. S6 and S7.† The activation degree of N2 was analyzed through adsorption energy and N–N bond length. For the CrN(100) surface, it can be seen that N2 molecules cannot be adsorbed on the pristine and V(N) surfaces, which is manifested by positive adsorption energy. On the V(N2) surface, N2 is adsorbed in an end-on mode with an adsorption energy of −0.66 eV, suggesting that nitrogen vacancy formation on the surface favors N2 adsorption. For the CrN(110) surface, both the pristine and defective surfaces adsorb N2 efficiently, and the adsorption energy decreases sequentially with the increase of nitrogen vacancies, indicating that the adsorption of N2 becomes easier and easier. For Mn4N, N2 could not be adsorbed on the intact (100) surface, while it was adsorbed more strongly on the V(N) surface with increased N–N bond lengths and reduced adsorption energies, and the activation of N2 on the defective surface is not significantly enhanced compared to the pristine surface for the (110) surface. For the NbN(100) surface, for which N2 is adsorbed in an end-on mode on the pristine surface, the activation of N2 is gradually enhanced with the increase of nitrogen vacancy concentration, which is manifested by the growth of N–N and the increase of Bader charge of the N2 adsorbed state. As for the (110) surface, the defective surface is more effective than the pristine surface for N2 activation. N2 cannot be effectively activated on either pristine or defective surfaces of LaN(100) and (110). For surfaces with small adsorption energy but difficult N2 dissociation, H-mediated activation of N2 is considered.In summary, for surfaces with positive N2 adsorption energy on the intact surface, the adsorption energy becomes more and more negative with the increase of surface nitrogen vacancies. For surfaces with negative N2 adsorption energy on the intact surface, the increase of surface nitrogen vacancies did not have a significant promotion effect on N2 adsorption. Previous studies have shown that the formation energy of oxygen vacancies on metal oxides is a reliable descriptor for evaluating their catalytic performance. Here, we tested the descriptor of Nvac formation energy with the aim of expressing the energetic intermediate information of the NRR on different metal nitrides. It is noteworthy that a larger Evac is favorable for N2 adsorption at the metal site. Quantitatively, the adsorption energy (Eads) on the defect surface becomes more negative when the nitrogen vacancy formation energy (Evac) exceeds 1.2 eV, which implies that the nitrogen vacancies (Nvac) have stronger nitrogen (N2) adsorption and activation capabilities for metal nitrides possessing higher Evac. Therefore, an increase in Evac not only contributes to the activation and hydrogenation of N2 but also weakens the hydrogen binding at the surface Nlat sites; thus Evac is an effective performance descriptor. A lower vacancy formation energy (Evac) indicates that vacancies are more easily formed on the catalyst surface. Nitrogen vacancies, as active sites, can lower the reaction energy barrier (ΔG) by stabilizing the reaction intermediates or lowering the transition state energy, which is consistent with the expectation that the presence of nitrogen vacancies can improve the catalytic activity. The adsorption energy of nitrogen (Eads) reflects the reactant adsorption strength, and its weak correlation with Evac indicates that the adsorption process is not fully coupled with the vacancy formation mechanism. Vacancies in the system may indirectly affect the local electronic structure of the adsorption sites, but are not directly involved in the adsorption process.
For the NRR, a catalyst surface with Eads < 0.5 eV of N2 was selected for the next catalytic hydrogenation reaction. Two reaction mechanism pathways for the NRR have been discussed in the literature: (I) dissociation mechanism, in which the NN triple bond is completely dissociated prior to hydrogenation, and (II) association mechanism, in which N2 is hydrogenated to produce N2Hxvia a distal or alternating pathway prior to NN bond breakage. The relevant data for DFT calculations for all surfaces are listed in Table S1.† Considering both the N–N bond length and adsorption energy, we studied the 15 potential surfaces mentioned above, including CrN(110), CrN(110)–V(N), CrN(110)–V(N2), Mn4N(100)–V(N), Mn4N(100)–V(N2), NbN(100)–V(N), NbN(100)–V(N2), NbN(100)–V(N3), NbN(110), NbN(110)–V(N), LaN (110), LaN(110)–V(N), TiN(110), TiN(111)–V(N3), and TiN(111)–V(N4) surfaces. To further analyze the NRR activity of the selected nitride structures, the NRR paths under experimental conditions (T = 573, 673, 773 K, P = 1 bar) were calculated by DFT (Fig. S8–S27†). The complete free energy diagrams of the surface distal and alternate reaction paths were plotted, along with the optimized intermediate structures.
The complete distal pathway free energy diagram of the better-performing LaN(110)–V(N) surface and the optimized intermediate structure are shown in Fig. 3c and d. The distal pathway has the lowest thermodynamic energy span (0.59 eV) on the better-performing LaN(110)–V(N) surface, which is significantly lower than that of the other surfaces and the alternating pathway (e.g., the alternating pathway has an energy span of 1.57 eV on the same surface). The alternating pathway was not selected as the primary pathway because it had a higher energy span and did not show a clear advantage. The distal pathway is able to achieve efficient electron and proton transfer through the active centers on the LaN(110)–V(N) surface. The distal N atoms are preferentially hydrogenated to form NH3 and released, and the remaining N atoms continue to hydrogenate to form a second NH3. The transition state energy barriers of this pathway (e.g., TS3–4 and TS5–6) are much milder in the experimental temperature range (573 K). The alternating pathway, on the other hand, leads to overall poorer kinetics due to the strong adsorption of intermediates (e.g., NxHy) and higher energy barriers. For another surface with better properties (NbN(110)), the step diagram shows that the process N2* → NNH* requires overcoming a large thermodynamic barrier, and therefore the step of N2 hydrogenation to produce NNH also determines the rate of this reaction. The N–N bond length in the NNH* intermediate on its defective surface is 1.36 Å, which is significantly longer than that on the intact surface, which is 1.33 Å. The significant extension of the bond length suggests that the modulation of the electronic structure induced by the surface defects weakened the bond strength of the N–N bond, which proves the optimization of the kinetics of the NRR by the surface defects. To investigate the correlation between electron transfer and NRR activity of nitride catalysts, Fig. 3a shows ΔGmaxNRR as a function of Bader charge. Bader charge analysis shows that the NRR performance (red line) increases as the transferred Bader charge increases. The d-band center and ΔGNRRmax show an overall negative correlation, ΔGNRRmax gradually decreasing as the d-band center becomes larger (blue line in Fig. 3b). For CeN, since Ce is a rare earth metal, its NRR catalytic performance may be related to the f orbital, and the d-band center is not suitable for use as a descriptor. In Fig. 3c, N2 is first spontaneously adsorbed at the catalytic center (ΔG = −0.45 eV) with NN distance of 1.18 Å. Comparing the difference in the performance of the two NRR catalysts LaN and NbN, DFT calculations show that the N–N bond length in the N2 molecule adsorbed on the surface of NbN is 1.20 Å, which is significantly longer than that of 1.18 Å in the LaN system. The Bader charge analysis shows that N2 acquires 1.50|e|, suggesting that N2 is activated by chemisorption. Second, NNH2* and NHNH* are produced upward in the subsequent protonation step at energies of −0.14 and 0.15 eV, respectively, suggesting that the distal mechanism is the energetically favorable pathway. Thus, the complete energy profiles of the six calculated transition states (TS) in the distal mechanism are shown in Fig. 3c. Bader charge analysis further reveals the electronic structure differences between NbN and LaN in the N2 activation mechanism. For the adsorbed-state N2 molecule, the net charge transfer of N2 in the NbN system is 0.81|e|, which is slightly higher than that of 0.65|e| in the LaN system, suggesting that the nitrogen vacancies on the NbN surface inject more electrons into the N2 antibonding orbitals (π*), which leads to an increased degree of polarization of the NN bond.
 | ||
| Fig. 3 (a) Relation between ΔGNRRmax and Bader charge (δ, in units of e). (b) Relation between ΔGNRRmax and d-band center. (c) Free energy diagram of the distal reaction pathway on the V(N) surface of LaN(110) at 573 K. (d) Structural diagram of the intermediates of the NRR process on the V(N) surface of LaN(110). | ||
Reaction mechanism and electronic structure
To further understand the intrinsic electronic properties of the intermediate-activated La atoms, the bonding properties involved in species were investigated. The densities of states (PDOS) of N2* and NNH* on the LaN(110)–V(N) and NbN(110) surfaces are illustrated in Fig. 4a and b. The PDOS of N2* exhibits significant variations, especially near the Fermi energy level. The d-orbital of La overlaps significantly with the p-orbital state of N2* near the Fermi energy level. This orbital hybridization is attributed to the involvement of the antibonding π* orbital of the N2 molecule, suggesting that the catalyst partially fills the π* orbitals by electron transfer, thereby weakening the triple bond within the N2 molecule and promoting N2 activation. | ||
| Fig. 4 The projected density of states (PDOS) of the N2* and NNH* intermediate states on the (a) LaN(110)–V(N) and (b) NbN(110) surfaces. (c) The –COHP analysis of LaN(110) and LaN(110)–V(N) surface N2*. (d) Fluctuation of temperature and energy versus time (10 ps) in AIMD simulation. | ||
For the NNH* adsorbed state, the orbital interactions are further enhanced. The d orbital density of states of La exhibits a more pronounced distribution near the Fermi energy level and strongly couples to the p orbitals of NNH*. The broad density of state peaks near the Fermi energy level indicates that the catalyst injects more electrons into the antibonding orbitals of the NNH* molecule (including π* and part of σ*). This electron rearrangement weakens the NN bond and enhances N–H bond formation, suggesting that the La atom further activates the adsorbed species. Therefore, the d orbitals of La play a dominant role in the N2 chemisorption. COHP is a visual indicator of bonding to investigate the chemical bonding and antibonding of atomic pairs. Therefore, COHP analysis was performed for N2* as shown in Fig. 4c. Compared with the LaN(110) intact surface, the antibonding orbitals of N–N bonds are stronger on the surface with N defects, and the ICOHP result, −8.37 (>−10.96), quantitatively explains the degree of activation of the catalyst for N2. In addition to high activity and selectivity, AIMD simulations and phonon spectroscopy (see Fig. 4d) further confirmed that the LaN(110)–V(N) catalyst is well stabilized. The slight distortion of the geometrical image shows that the catalyst has significant thermodynamic stability at 773 K. AIMD simulations were performed to analyze the temperature and energy of the structure over 10 ps which indicated that the vibrations in both results are close to the equilibrium state.
Conclusions
Through DFT calculations, 12 different metal nitride catalysts were proposed for the catalytic conversion of N2 to NH3 under experimental conditions (T = 573, 673, and 773 K, 1 bar). By analyzing the reaction selectivity and activity, LaN(110) and NbN(110) were identified as promising catalysts. By considering the relationship between the catalyst surface nitrogen coordination structure and NRR activity, this study proposes a rational design principle for NRR catalysts based on the coordination number of lattice nitrogen. The four-coordinate lattice nitrogen is more favorable for NRR activity, with lower reaction energies in the distal pathway. These predictions and findings enrich the fundamental understanding of metal nitrides in NRR catalysis, suggesting that surface engineering with fine local structures of lattice nitrogen on metal nitrides can improve catalytic performance and provide new insights for the rational design and improvement of metal nitride NH3 synthesis catalysts. In addition, this study breaks through and reveals the unique bifunctional mechanism of metal-free NbN catalysts in the ammonia synthesis reaction. Based on the efficient activation ability of Nb sites for H2 and the selective adsorption property of nitrogen vacancies for N2 in precursor materials, NbN exhibits the spatial synergistic effect of multiphase-like catalysis in the homogeneous catalytic system, which provides promising ideas for the design of a new type of nonprecious-metal catalytic system for ammonia synthesis.Data availability
The data supporting this article have been included as part of the ESI.† All conventional cells are from Materials Project: https://next-gen.materialsproject.org/.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
C. L. greatly appreciates financial support provided by the National Natural Science Foundation of China (grant no. 22473108) and the Foundation of National Key Research and Development Program of China (grant no. 2023YFB3810601).References
- A. Cao, V. J. Bukas, V. Shadravan, Z. Wang, H. Li, J. Kibsgaard, I. Chorkendorff and J. K. Nørskov, Nat. Commun., 2022, 13, 2382 CrossRef CAS PubMed.
- F. Chang, Y. Guan, X. Chang, J. Guo, P. Wang, W. Gao, G. Wu, J. Zheng, X. Li and P. Chen, J. Am. Chem. Soc., 2018, 140, 14799–14806 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- C. Liu, Q. Li, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Mater. Chem. A, 2019, 7, 4771–4776 RSC.
- Z. Zeng, X. Shen, R. Cheng, O. Perez, N. Ouyang, Z. Fan, P. Lemoine, B. Raveau, E. Guilmeau and Y. Chen, Nat. Commun., 2024, 15, 3007 CrossRef CAS PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- X. Peng, M. Zhang, T. Zhang, Y. Zhou, J. Ni, X. Wang and L. Jiang, Chem. Sci., 2024, 15, 5897–5915 RSC.
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- B. A. MacKay and M. D. Fryzuk, Chem. Rev., 2004, 104, 385–402 CrossRef CAS PubMed.
- Y. Guan, C. Liu, Q. Wang, W. Gao, H. A. Hansen, J. Guo, T. Vegge and P. Chen, Angew. Chem., Int. Ed., 2022, 61, e202205805 CrossRef CAS PubMed.
- Q. Wang, J. Pan, J. Guo, H. A. Hansen, H. Xie, L. Jiang, L. Hua, H. Li, Y. Guan, P. Wang, W. Gao, L. Liu, H. Cao, Z. Xiong, T. Vegge and P. Chen, Nat. Catal., 2021, 4, 959–967 CrossRef CAS.
- C. Liu, H. Zheng, T. Wang, Z. Guo, F. Zhu, H. Xie, G. Qin, H. Li and S. Li, J. Mater. Sci. Technol., 2023, 159, 244–250 CrossRef CAS.
- T. Wang, Z. Guo, H. Oka, A. Kumatani, C. Liu and H. Li, J. Mater. Chem. A, 2024, 12, 8438–8446 RSC.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- C. Liu, Q. Wang, J. Guo, T. Vegge, P. Chen and H. A. Hansen, ACS Catal., 2022, 12, 4194–4202 CrossRef CAS.
- S. Liu, Y. Wang, S. Wang, M. You, S. Hong, T.-S. Wu, Y.-L. Soo, Z. Zhao, G. Jiang, Q. Jieshan, B. Wang and Z. Sun, ACS Sustainable Chem. Eng., 2019, 7, 6813–6820 CrossRef CAS.
- S. Laassiri, C. D. Zeinalipour-Yazdi, C. R. A. Catlow and J. S. J. Hargreaves, Appl. Catal., B, 2018, 223, 60–66 CrossRef CAS.
- K. Zhang, A. Cao, L. H. Wandall, J. Vernieres, J. Kibsgaard, J. K. Nørskov and I. Chorkendorff, Science, 2024, 383, 1357–1363 CrossRef CAS PubMed.
- N. Morlanés, S. Sayas, G. Shterk, S. P. Katikaneni, A. Harale, B. Solami and J. Gascon, Catal. Sci. Technol., 2021, 11, 3014–3024 RSC.
- K. Sato, S.-i. Miyahara, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura, K. Inazu, H. Mohri, T. Iwasa, T. Taketsugu and K. Nagaoka, ACS Catal., 2021, 11, 13050–13061 CrossRef CAS.
- S. Laassiri, C. D. Zeinalipour-Yazdi, N. Bion, C. R. A. Catlow and J. S. J. Hargreaves, Faraday Discuss., 2021, 229, 281–296 RSC.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2018, 122, 6078–6082 CrossRef CAS.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- P. K. Roy and S. Kumar, Int. J. Hydrogen Energy, 2024, 64, 497–506 CrossRef CAS.
- R. K. Rai, R. Maligal-Ganesh, W. A. Maksoud, S.-J. Kim, Y. Vaishnav, C. T. Yavuz and Y. Kobayashi, ChemCatChem, 2024, 16, e202301579 CrossRef CAS.
- A. M. Alexander, J. S. J. Hargreaves and C. Mitchell, Top. Catal., 2012, 55, 1046–1053 CrossRef CAS.
- M. Kitano, Y. Inoue, H. Ishikawa, K. Yamagata, T. Nakao, T. Tada, S. Matsuishi, T. Yokoyama, M. Hara and H. Hosono, Chem. Sci., 2016, 7, 4036–4043 RSC.
- X. Wang, X. Peng, W. Chen, G. Liu, A. Zheng, L. Zheng, J. Ni, C.-t. Au and L. Jiang, Nat. Commun., 2020, 11, 653 CrossRef CAS PubMed.
- L. Ye, Chem, 2021, 7, 3198–3199 CAS.
- W. Gao, Y. Wang, Q. Wang, Z. Sun, J. Guo and P. Chen, EES Catal., 2024, 2, 780–788 RSC.
- J. O. Conway and T. J. Prior, J. Alloys Compd., 2019, 774, 69–74 CrossRef CAS.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2020, 142, 14374–14383 CrossRef CAS PubMed.
- Y. Guan, W. Zhang, Q. Wang, C. Weidenthaler, A. Wu, W. Gao, Q. Pei, H. Yan, J. Cui, H. Wu, S. Feng, R. Wang, H. Cao, X. Ju, L. Liu, T. He, J. Guo and P. Chen, Chem. Catal., 2021, 1, 1042–1054 CrossRef CAS.
- Y. Lu, T.-N. Ye, J. Li, Z. Li, H. Guan, M. Sasase, Y. Niwa, H. Abe, Q. Li, F. Pan, M. Kitano and H. Hosono, Angew. Chem., Int. Ed., 2022, 61, e202211759 CrossRef CAS PubMed.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano, T. Tada and H. Hosono, Nature, 2020, 583, 391–395 CrossRef CAS PubMed.
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- H. Kumagai, K. Takanabe, J. Kubota and K. Domen, Bull. Chem. Soc. Jpn., 2015, 88, 584–590 CrossRef CAS.
- Q. Li, C. Liu, S. Qiu, F. Zhou, L. He, X. Zhang and C. Sun, J. Mater. Chem. A, 2019, 7, 21507–21513 RSC.
- Y. Cao, M. A. Kirsanova, M. Ochi, W. Al Maksoud, T. Zhu, R. Rai, S. Gao, T. Tsumori, S. Kobayashi, S. Kawaguchi, E. Abou-Hamad, K. Kuroki, C. Tassel, A. M. Abakumov, Y. Kobayashi and H. Kageyama, Angew. Chem., Int. Ed., 2022, 61, e202209187 CrossRef PubMed.
- Y. Cao, A. Saito, Y. Kobayashi, H. Ubukata, Y. Tang and H. Kageyama, ChemCatChem, 2021, 13, 191–195 CrossRef CAS.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- N. Ouyang, C. Wang and Y. Chen, Int. J. Heat Mass Transfer, 2022, 192, 122859 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- N. Ouyang, C. Wang, Z. Zeng and Y. Chen, Appl. Phys. Lett., 2021, 119, 061902 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00282f |
| This journal is © The Royal Society of Chemistry 2025 |