
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencepH-responsive polymer-supported pyrene-based fluorescent dyes for CO2 detection in aqueous environments†
David A.
Londoño de la Cruz
 a,
David
Chappell
b,
Bilal
Rashid
b,
Blaine R.
Tookey
b,
Calum T. J.
Ferguson
a and
Rachel K.
O'Reilly
*a
a,
David
Chappell
b,
Bilal
Rashid
b,
Blaine R.
Tookey
b,
Calum T. J.
Ferguson
a and
Rachel K.
O'Reilly
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: r.oreilly@bham.ac.uk
bBP Exploration Operating Company Limited, Chertsey Road, Sunbury on Thames, Middlesex TW16 7BP, UK
First published on 28th January 2025
Abstract
Detecting fluctuations in carbon dioxide by switching ‘ON’ or ‘OFF’ fluorescence in aqueous environments has often been targeted for efficient monitoring. This switch needs to occur in a drastic and fast way that is visually observable to be effective. CO2 dissolves in water, leading to a reduction in pH, which can be used to trigger a response. Specifically, the modification of fluorescent dyes with pH-responsive units could create a dye that responds to CO2 and switches ‘off’ fluorescence. Here, we developed a water-soluble polymer functionalized with a CO2-responsive pyrene dye. This functional dye contains a tertiary amine conjugated to the aromatic pyrene, which can be protonated at pH ∼ 6.5 or lower. After only 15 s of CO2 bubbling, a significant fluorescence ‘OFF’ response was observed, with a drastic reduction in fluorescence at 480 nm as a consequence of disrupting non-covalent excimer bonds. Moreover, the exposure to atmospheric air results in a recovery of the excimer state and, therefore, the fluorescence, demonstrating its reversible nature.
Introduction
Carbon dioxide (CO2) is the primary greenhouse gas emitted by humans that lingers in the atmosphere for thousands of years. It accounts for 75% of all emissions, mainly from burning organic materials such as coal, oil, gas, wood, and solid waste.1–3 Detecting and monitoring CO2 is essential for a range of different applications, including climate research,4,5 biology,6 ecology,7 chemistry,8 agriculture,9 food10 and healthcare.11 Presently, a wide range of applications are used for detecting and monitoring CO2, from non-dispersive infrared sensors,12 positron emission tomography imaging,13 paper-based sensors,14,15 frameworks,16 and others.17,18 However, these methods can be expensive, non-reusable, time-consuming, have poor detection limits, or do not give a drastic response, so alternative strategies are needed. Chemical-based sensors, ranging from molecular-based to macromolecular sensors, have been investigated as an alternative strategy for detecting CO2. These sensors operate by undergoing a compositional change in their chemical structure upon exposure to CO2, which leads to a response.Polymer-based sensors offer a stable and easily applicable delivery system, typically comprising a responsive monomer and a dye molecule. Over the past decade, numerous CO2-responsive polymeric systems with fluorescence read-outs have been reported, commonly consisting of pH-responsive and polar-sensitive fluorescent units.19–28 They respond to CO2 by changing the conformation of the polymer, leading to a different microenvironment around the dye molecule, which changes its fluorescence. This response is due to CO2 forming carbonic acid in water, which can dissociate into HCO3−, CO32−, and H+, allowing protonation of amine-based monomers. However, the response rate to CO2 is often limited by conformational changes in the macromolecular structure.
Molecular-based CO2 sensors typically involve dye molecules that undergo a chemical transformation when exposed to CO2, resulting in a detectable signal change. For example, Morandi and coworkers developed a range of deactivated dye molecules that switched on in the presence of CO2. This highly sensitive and selective molecular sensor was capable of detecting CO2 at atmospheric levels (as low as eight ppm).29 Common dye molecules were modified to include an iminophosphorane group that, in the presence of CO2, undergoes a cascade aza-Wittig reaction that turns ‘ON’ its fluorescence. However, a significant limitation of this sensor is its non-reversibility, meaning it cannot be reused after detection, which poses challenges for continuous monitoring applications. Lee and coworkers developed an imidazole-based polymer sensor that can form N-heterocyclic carbenes, which undergo a photo-induced electron transfer (PET) to anthracene dye molecules and switch them ‘off’. The carbene reacts in the presence of CO2 switching on the anthracene dye by inhibiting the PET. This reaction could be reversed over multiple cycles of switching ‘ON’ and ‘OFF’ the dye. However, this exciting strategy requires heat to release the reacted CO2. Moreover, the detection limit of the material was not determined, and the visual response was not strong, potentially limiting the applicability.
Ideally, a combination of polymer- and molecular-based systems should be developed. Such a system would involve a reversibly CO2-responsive dye molecule that changes fluorescence due to direct fluctuation in CO2 levels. Incorporating this into a polymer delivery system would potentially increase its applicability and usability in aqueous environments. The desired characteristics for this system include reversibility, reusability, fast response, low detection limit, and activation in the presence of CO2 in water and deactivation upon the removal of CO2. Pyrene is a widely used fluorescent probe with outstanding features, including a large absorption coefficient, high fluorescence quantum yield, and excellent chemical stability.30 Pyrene-based fluorescence probes have been reported in macromolecular materials where the polymer responds to stimuli to alter the excimer formation of pyrene,31–40 changing fluorescent properties.
In this study, a modified pyrene-based CO2-sensing molecule was developed that could be readily incorporated into polymeric supports. Pyrene dye molecules were functionalized with pH-responsive amine functionality, which, upon protonation, turned ‘off’ fluorescence. This responsive dye-based sensor displayed visible changes in fluorescence, detecting changes in CO2 as a function of pH at parts per billion concentrations of dye. This pyrene-modified unit was incorporated into linear polymers and displayed a drastic response to changes in pH. Notably, this marks a significant advance as it represents an instance where the fluorescence of pyrene is extinguished by disrupting excimer bonds in the presence of CO2 within a polymeric system in aqueous solution. Furthermore, the quenching effect is entirely reversible upon exposure to atmospheric air.
Results and discussion
We aimed to modify the chemical structure of pyrene dye to be pH-responsive, creating drastic changes in fluorescence when interacting with CO2. To achieve this, we wanted to attach an amino pH-responsive unit to the dye that becomes protonated under acidic conditions. This protonation will directly affect its fluorescent properties by decreasing the excimer/unimer ratio due to electrostatic repulsion, drastically changing the dye's emission. However, due to its high hydrophobicity, testing its properties in water solution could be challenging. Consequently, we needed to incorporate this modified dye into a polymeric scaffold that enhances its water solubility.Our aim was to synthesize a water-soluble polymer integrating pyrene derivatives functionalized with N,N-dimethylamino and N,N-diethylamino groups, each offering distinct reactivity and responsiveness to pH and CO2 variations. The selection of these amines was guided by the pKa estimated (5.33 and 6.19, respectively) (Fig. S1 and S2†). We hypothesized that the diethylamino group would demonstrate optimal reactivity with CO2, due to its pKa value suggesting protonation would occur in the HCO3−/CO32− system. This enhanced reactivity was expected to result in a more pronounced change in the probe's fluorescence. The mechanism underlying these pH-responsive systems involves the protonation of amine groups upon a decrease in pH, which can be induced by CO2 as it partially dissolves in water to form an equilibrium with carbonic acid, and the generation of positive charges, leading to electrostatic repulsion between pyrene units. Pyrene dye molecules are only fluorescent when they have formed excimers, so they must be closely located next to each other. Creating a cationic charge on the pyrene induces electrostatic repulsion between pyrenes, breaking the excimers and turning off fluorescence. Here, we aimed to create a sensitive response to CO2, with the diethylamino group playing a crucial role in the system's reactivity and fluorescence changes.
For the synthesis of the pyrene-based monomers, we proposed a general synthetic route for the two designed monomers. We opted to modify position 1 of pyrene with a tertiary amine and a methacrylate functional group in position 6 (Fig. 1a). The most straightforward approach to accomplish this involved dialkylation of the commercially available 1-aminopyrene with the corresponding alkyl iodide, according to a protocol developed in the Wagenknecht laboratory.41 This was then followed by the addition of a bromo group at position 6 via radical bromination with N-bromosuccinimide (NBS). This enabled the subsequent Sonogashira cross-coupling reaction with 3-butyn-1-ol. Ultimately, esterification with EDC and methacrylic acid was used to obtain the methacrylate monomer diethylamino pyrene methacrylate (DEAPyMA) (Scheme S1†).
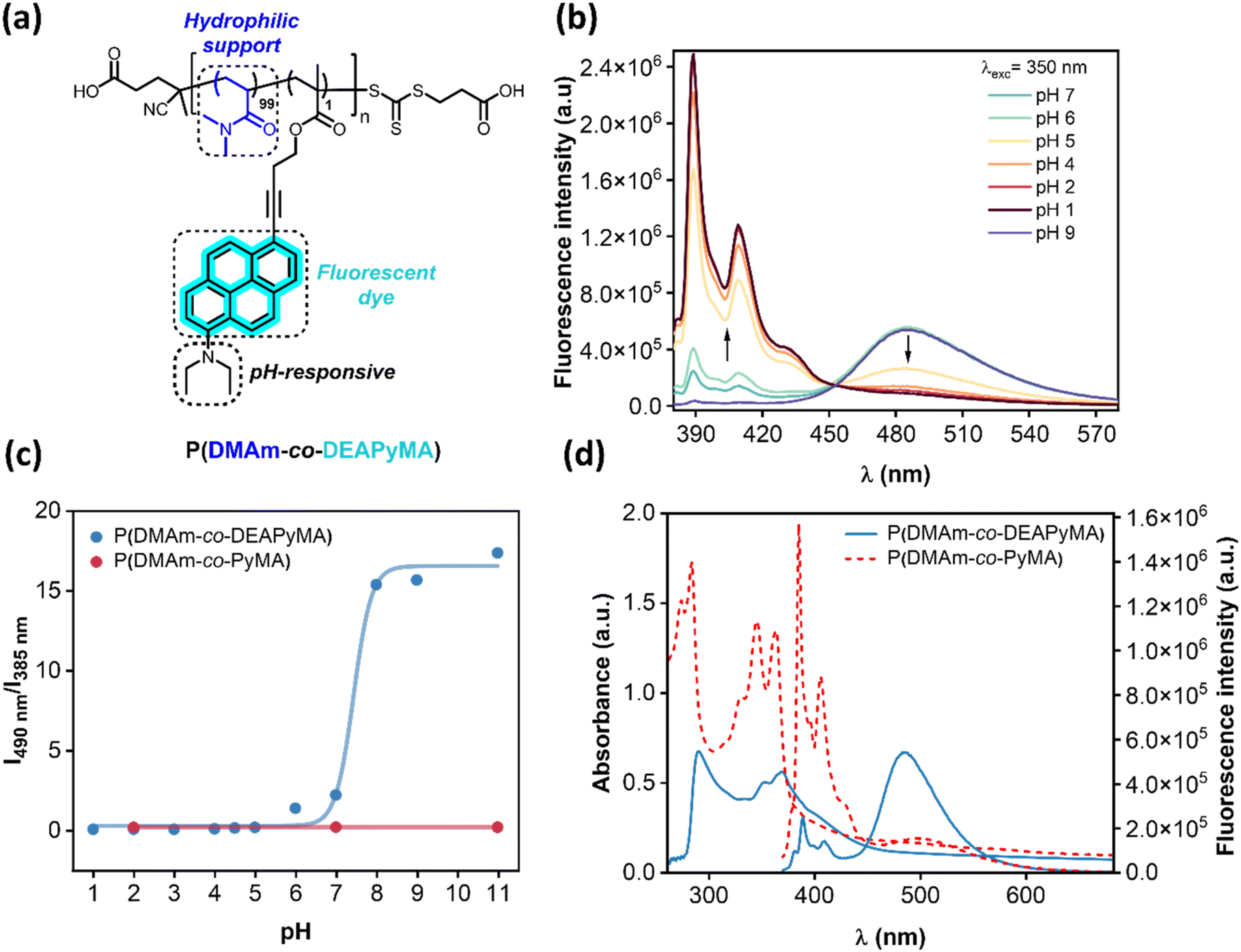 | ||
| Fig. 1 (a) Scheme of chemical structure of poly(dimethylacrylamide-co-diethylaminopyrenemethacrylate). (b) Fluorescence spectra of P(DMAm-co-DEAPyMA) at different pHs 7, 6, 5, 4, 3, 2 and 1. Concentration 0.1 mg mL−1. λexc = 350 nm. (c) Graphic representing change in intensity ratio I490nm/I385nm as effect of the pH for P(DMAm-co-DEAPyMA) and P(DMAm-co-PyMA). Concentration 0.1 mg mL−1. λexc = 350 nm. (d) UV-Vis and fluorescence spectra of both P(DMAm-co-DEAPyMA) (continuous line) and P(DMAm-co-PyMA) (discontinuous line) in water at pH 7. Concentration 1 mg mL−1. λexc = 350 nm. | ||
We hypothesized that because of the strong repulsion generated from the positive charge of protonated amines in the DEAPyMA moiety it would increase the distance between pyrene units and would consequently break the excimer state. This would be translated in switching the fluorescence from an ON to an OFF state. We empirically validated our hypothesis by examining the spectral properties of the different pyrene monomers in mixtures of H2O/MeCN (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) at various pH levels (Fig. S32†). As expected, the DEAPyMA dye exhibited distinctive emission peaks in the different mixtures, clearly transitioning from an ON state in basic pH to an OFF state in acidic conditions.
:5) at various pH levels (Fig. S32†). As expected, the DEAPyMA dye exhibited distinctive emission peaks in the different mixtures, clearly transitioning from an ON state in basic pH to an OFF state in acidic conditions.
Given the hydrophobic nature of the monomer, the subsequent objective involved evaluating its pH-sensitivity in an aqueous solution by polymerizing the fluorescent monomer within a water-soluble, non-pH-responsive system. An initial reaction was conducted at 80 °C by reacting 100 equivalents of N,N-dimethylacrylamide (DMAm) with 1 equivalent of DEAPyMA, 4-[[[(2-carboxyethyl)thio]thioxomethyl]thio]-4-cyanopentanoic acid as chain-transfer agent and 4,4′-azo-bis(4-cyanovaleric acid) (ACVA) as radical initiator for 16 hours. The obtained polymer (Fig. 1a) was purified by extensive dialysis using a 3 kDa membrane, in H2O/THF (1:1). The molecular weight of the polymer was calculated via end-group analysis, showing a 96% conversion for DMAm against a 76% conversion regarding DEAPyMA; this might be due to the different nature of the monomers as well as the bigger size of DEAPyMA in comparison to DMAm (approximately 4 times) resulting in steric hindrance, giving a value of (Mn,NMR = 10.1 kDa). We experimentally validated our hypothesis by investigating the optical characteristics of the polymer with a 1 mol% loading of DEAPyMA in an aqueous solution. The addition of the amine to the pyrene structure resulted in a noticeable redshift in both absorption and emission spectra compared to the non-pH-responsive PyMA (Fig. 1d).
The responsiveness of the polymer containing DEAPyMA was evaluated across the entire pH spectrum. As depicted in Fig. 1b, the dye exhibited two distinct fluorescent emission bands between 370–420 nm, corresponding to the pyrene unimer, and an additional band at 490 nm indicative of excimer species formation. We synthesized two other control polymers, one integrating a non-pH-responsive pyrene unit P(DMAm-co-PyMA) and the other with a dimethylamino moiety as a responsive unit, P(DMAm-co-DMPyMA). The first one showed a constant fluorescence emission through the whole pH spectrum (Fig. 1c), whereas the P(DMAm-co-DMPyMA) only showed a response at pH values below 3 (Fig. S11 and S12†). We prepared polymers with 0.1, 0.5 and 1 mol% of dimethylaminopyrenemethacrylate (DMAPyMA), and we could confirm that the fluorescence intensity corresponding to the excimer formation (490 nm) also depended on the concentration of the dye present in the solution (Fig. S14 and S15†). In contrast, with P(DMAm-co-DEAPyMA), a decrease in pH from basic to acidic conditions led to an increase in the intensity of the unimer pyrene emission peaks, coupled with a decrease in excimer peak intensity (Fig. 1b). In this case, the changes in the intensity of each peak were observed to be more pronounced than in the free molecule. We think this was due to a solubility issue resulting from the concentration of the dye, as this is lower in the polymer. This suggests that as the amine became protonated, electrostatic repulsion between positively charged amines likely caused the disruption of π–π interactions between excimers. This underscores the role of the pyrene unit in the observed pH-dependent fluorescence changes.
We confirmed our hypothesis by conducting fluorescence lifetime measurements on an aqueous solution of (DMAm-co-DEAPyMA) at pH 2, 4.5, and 7. As anticipated, we observed a decrease in fluorescence lifetime, indicative of excimer formation facilitating non-radiative pathways leading to a faster relaxation to the ground state compared to the unimers. Notably, we observed that the lifetimes corresponding to excimer peaks (490 nm) decreased to nearly half the values compared to those obtained at the same pHs at 385 nm (Fig. S25 and S26†).
Furthermore, we aimed to investigate the detection limit with respect to both the polymer and the dye. To achieve this, we prepared samples of P(DMAm-co-DEAPyMA) at five different concentrations and tested them over a pH range where a significant fluorescence change could be detected (Fig. 2b). The polymer exhibited a noteworthy pH-response with a distinct fluorescence ON/OFF transition in the parts per million (ppm) range (Fig. 2a). Interestingly, the visual response was even more pronounced in the parts per billion (ppb) range for the fluorescent dye, given its presence at around 1 mol% of charge within the polymer chains. This makes DEAPyMA potentially sensitive to CO2 in aqueous solution. Correspondingly, the fluorescence intensity ratio I490nm/I385nm of P(DMAm-co-DEAPyMA) gradually increases as the pH does. Following the work by Liu et al.,42 the pKa of DEAPyMA was calculated as 4.4 using the Henderson–Hasselbalch equation (Fig. S23†), suggesting its suitability for pH quantification in acidic to neutral environments and CO2 detection.
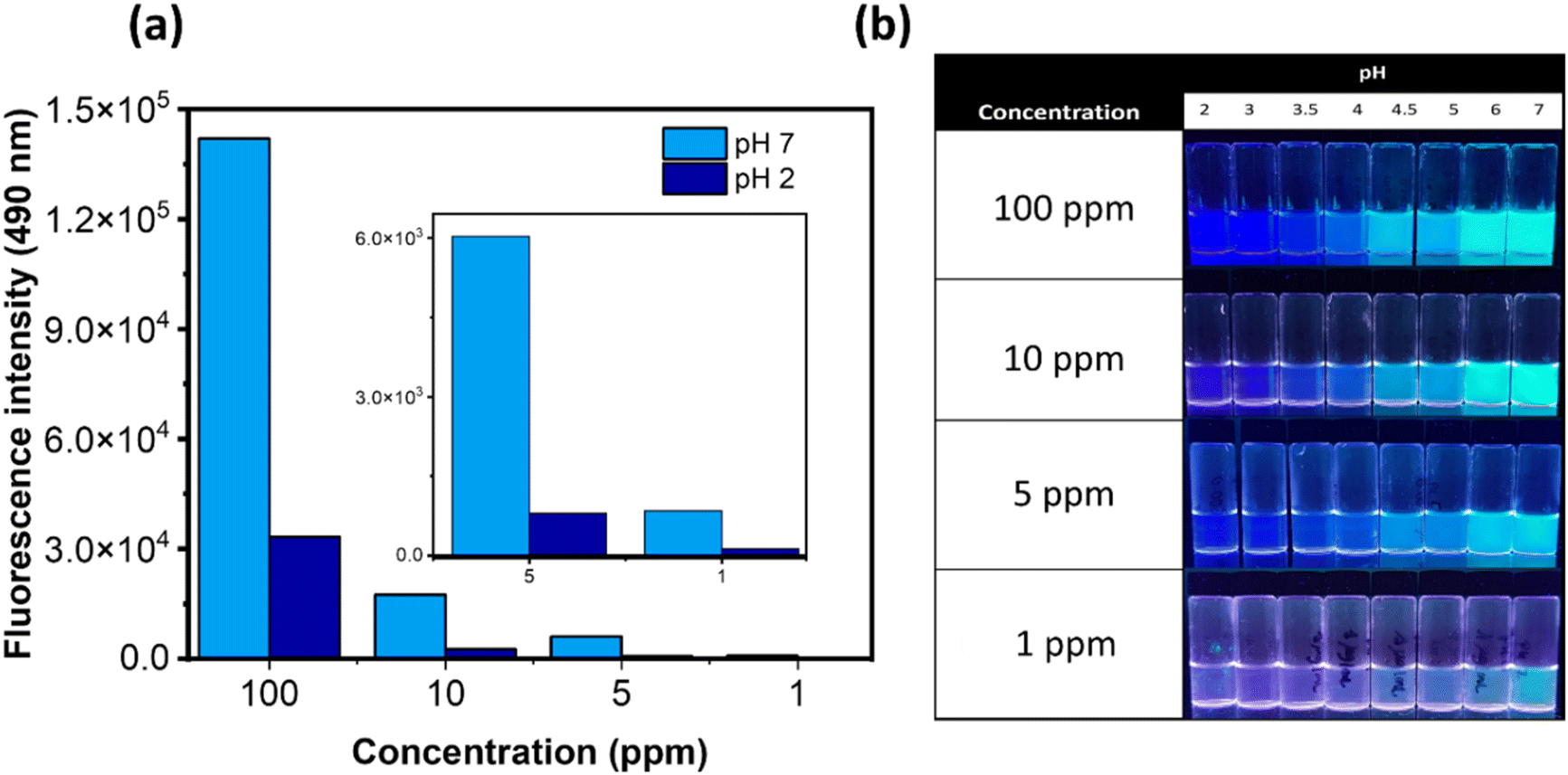 | ||
| Fig. 2 (a) Fluorescence intensity of P(DMAm-co-DEAPyMA) (concentrations: 100, 10, 5 and 1 ppm, pH 7 and 2, λexc = 350 nm, λem = 490 nm). (b) Photograph of P(DMAm-co-DEAPyMA) at different concentrations in aqueous solution and different pHs under 365 nm UV light. | ||
To evaluate the system's effectiveness in CO2 detection, we monitored the fluorescence emission of a 1 ppm sample of P(DMAm-co-DEAPyMA) in water before and after exposing it to a continuous flow and pressure of CO2. Within minutes, a notable change in visual fluorescence occurred. The reaction kinetics were further investigated by measuring the fluorescence ratio (I490nm/I385nm) at various time intervals with different CO2 volumes. Here, it can be seen that the response to CO2 depends on the flow rate of CO2 (Fig. S31, S40 and S41†).
Optical characteristics and pH assessments of the polymeric matrix were conducted before and after exposing it to a constant CO2 flow (50 cm3 min−1) and pressure (1 bar). After 15 seconds, an abrupt alteration in fluorescence intensity occurred, accompanied by a significant pH decrease from 7.4 to 5.6. This change in fluorescence corresponds to the exposure of the polymer to 12.5 cm3 of CO2. The fluorescence intensity ratio gradually declined from 1.8 to 0.07 as the CO2 volume reached 25 cm3, ultimately stabilizing (Fig. 3a and d). It is, therefore, clear that the system produced has a response to CO2 that saturates after exposure to around 20 cm3, which is likely because an equilibrium is reached, and the pH does not decrease any further.
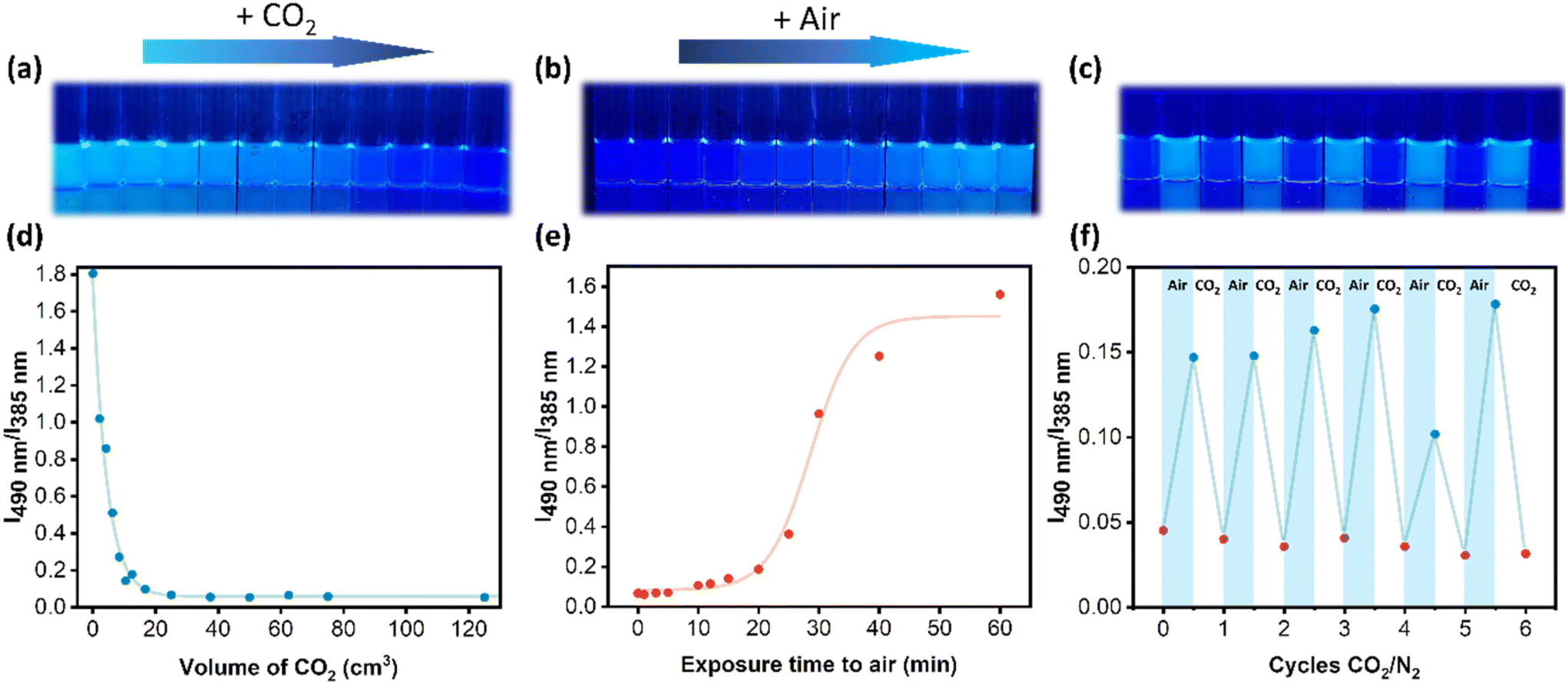 | ||
| Fig. 3 (a) Photograph of P(DMAm-co-DEAPyMA) samples of concentration 0.01 mg mL−1 after bubbling different CO2 volumes (OFF process) under 365 nm UV light. (b) Photograph of P(DMAm-co-DEAPyMA) samples of concentration 0.01 mg mL−1 after being exposed to atmospheric air for 1 h (ON process) under 365 nm UV light. (c) Photograph of P(DMAm-co-DEAPyMA) samples of concentration 0.01 mg mL−1 after undergoing several CO2/air cycles under 365 nm UV light. (d) Kinetics graphic representing a change in fluorescence intensity ratio I490nm/I385nm of a sample of concentration 0.01 mg mL−1 relative to the volume of CO2. (e) Kinetics graphic representing a change in fluorescence intensity ratio I490nm/I385nm of a sample of 0.01 mg mL−1 concentration when exposed to air for 1 h. (f) Graphic representing the change in fluorescence intensity ratio I490nm/I385nm of a sample of concentration 0.01 mg mL−1 when being exposed to CO2 for 5 min and air for 1 h for 6 cycles. | ||
As the protonation reaction of P(DMAm-co-DEAPyMA) was reversible, we assumed that the fluorescence could be recovered by removing CO2 by exposing the system to air. The system previously exposed to CO2 for 10 min was then exposed to atmospheric air, resulting in a gradual recovery of fluorescence (Fig. 3b). The fluorescence intensity ratio (I490m/I385nm) gradually increased from 0.07 to 1.56 (Fig. 3e). The fluorescence intensity at 490 nm exhibited an increase after being exposed to air, while the intensity at 385 nm underwent a sharp decrease. This reversal process took more time compared to the response to CO2, requiring exposure to air for 25 minutes to observe a substantial change in fluorescence, and 1 hour for complete recovery of the excimer state, as illustrated in Fig. 3e. Images of different samples were also taken at various times under 365 nm UV light (Fig. 3b), to visualize the reversible fluorescence change.
Subsequently, the system underwent multiple cycles of CO2 exposure for 5 minutes (OFF) and air exposure for 1 h (ON) to evaluate its reversibility. These results demonstrated that this system's CO2-responsive fluorescence property was stable and possessed excellent repeatability over at least 6 cycles, transitioning between an ON and OFF state, as shown in Fig. 3c and f. This transition from the off to on state as a function of atmosphere corresponds to changes in pH, as expected (Fig. S42†). Interestingly, after storing the sample in an aqueous environment for one week, a response to CO2 and air was still observed; however, increased variability was observed (Fig. S43†).
Conclusion
In summary, we have successfully synthesized a novel linear polymer through RAFT polymerization, incorporating a unique pH/CO2-responsive pyrene dye monomer. This water-soluble polymer not only displayed a fluorescence turn-off but also exhibited an exceptionally rapid response (15 s) and a high sensitivity in water in concentrations around ppb when exposed to CO2 bubbling. The interaction of CO2 with the amino response unit in the pyrene led to a meaningful change in the system's fluorescence. This change resulted from electrostatic repulsion between pyrene units, breaking the intermolecular excimer. Importantly, this process proved to be reversible, over at least six cycles under atmospheric conditions, indicating durability suitable for potential gas-sensing applications. The polymer exhibited promising features for real-time CO2 sensing and could be potentially applied in smart surfaces.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
Any additional relevant notes should be placed here.Acknowledgements
D.A.L.C. and R.O.R. acknowledge BP CO2 sensing project funding BPX/GPTL/JN/76937 (CW1905016). The authors would like to thank the BP Exploration Operating Company Ltd for funding D.A.L.C. The authors would also like to thank Mr Jake Barker for TD-DFT calculations and Dr Vasilios Stavros for useful discussion about excimer formation.References
- E. Ozturk, C. Ince, S. Derogar and R. Ball, Factors affecting the CO2 emissions, cost efficiency and eco-strength efficiency of concrete containing rice husk ash: A database study, Constr. Build. Mater., 2022, 326, 126905, DOI:10.1016/j.conbuildmat.2022.126905.
- H. F. Gholipour, A. Arjomandi and S. Yam, Green property finance and CO2 emissions in the building industry, Glob. Finance J., 2022, 51, 100696, DOI:10.1016/j.gfj.2021.100696.
- X. Lu, D. Jin, S. Wei, Z. Wang, C. An and W. Guo, Strategies to enhance CO2 capture and separation based on engineering absorbent materials, J. Mater. Chem. A, 2015, 3(23), 12118–12132, 10.1039/c4ta06829g.
- M. A. Clark, N. G. G. Domingo, K. Colgan, S. K. Thakrar, D. Tilman, J. Lynch, I. L. Azevedo and J. D. Hill, Global food system emissions could preclude achieving the 1.5° and 2 °C climate change targets, Science, 2020, 370(6517), 705–708, DOI:10.1126/science.aba7357.
- C. B. Field and K. J. Mach, Rightsizing carbon dioxide removal, Science, 2017, 356(6339), 706–707, DOI:10.1126/science.aam9726.
- E. P. Cummins, A. C. Selfridge, P. H. Sporn, J. I. Sznajder and C. T. Taylor, Carbon dioxide-sensing in organisms and its implications for human disease, Cell. Mol. Life Sci., 2014, 71(5), 831–845, DOI:10.1007/s00018-013-1470-6.
- E. Pennisi, Carbon dioxide increase may promote ‘insect apocalypse’, Science, 2020, 368(6490), 459–459, DOI:10.1126/science.368.6490.459.
- P. Gabrielli, M. Gazzani and M. Mazzotti, The Role of Carbon Capture and Utilization, Carbon Capture and Storage, and Biomass to Enable a Net-Zero-CO2 Emissions Chemical Industry, Ind. Eng. Chem. Res., 2020, 59(15), 7033–7045, DOI:10.1021/acs.iecr.9b06579.
- A. J. Bloom, M. Burger, J. S. R. Asensio and A. B. Cousins, Carbon Dioxide Enrichment Inhibits Nitrate Assimilation in Wheat, and Arabidopsis, Science, 2010, 328(5980), 899–903, DOI:10.1126/science.1186440.
- C. Agrimonti, M. Lauro and G. Visioli, Smart agriculture for food quality: facing climate change in the 21st century, Crit. Rev. Food Sci. Nutr., 2021, 61(6), 971–981, DOI:10.1080/10408398.2020.1749555.
- A. M. M. El-Betany, E. M. Behiry, M. Gumbleton and K. G. Harding, Humidified Warmed CO2 Treatment Therapy Strategies Can Save Lives with Mitigation and Suppression of SARS-CoV-2 Infection: An Evidence Review, Front. Med., 2020, 7, 594295, DOI:10.3389/fmed.2020.594295 , Hypothesis and Theory.
- R. K. Jha, Non-Dispersive Infrared Gas Sensing Technology: A Review, IEEE Sens. J., 2022, 22(1), 6–15, DOI:10.1109/jsen.2021.3130034.
- M. Hubeau, M. R. Thorpe, J. Mincke, J. Bloemen, I. Bauweraerts, P. E. H. Minchin, V. De Schepper, F. De Vos, C. Vanhove and S. Vandenberghe, et al., High-Resolution in vivo Imaging of Xylem-Transported CO2 in Leaves Based on Real-Time 11C-Tracing, Front. For. Glob. Change, 2019, 2, 00025, DOI:10.3389/ffgc.2019.00025 , Methods.
- H. Wang, S. I. Vagin, B. Rieger and A. Meldrum, An Ultrasensitive Fluorescent Paper-Based CO2 Sensor, ACS Appl. Mater. Interfaces, 2020, 12(18), 20507–20513, DOI:10.1021/acsami.0c03405.
- M. Chandramouli, V. K. Boraiah, R. P. Shivalingappa, V. Basavanna, S. Doddamani and S. Ningaiah, Paper-Based Carbon Dioxide Sensors: Past, Present, and Future Perspectives, Biointerface Res. Appl. Chem., 2022, 12(2), 2353–2360, DOI:10.33262/BRIAC122.23532360.
- M.-H. Xie, W. Cai, X. Chen, R.-F. Guan, L.-M. Wang, G.-H. Hou, X.-G. Xi, Q.-F. Zhang, X.-L. Yang and R. Shao, Novel CO2 Fluorescence Turn-On Quantification Based on a Dynamic AIE-Active Metal–Organic Framework, ACS Appl. Mater. Interfaces, 2018, 10(3), 2868–2873, DOI:10.1021/acsami.7b17793.
- H.-J. Li, S. K. Mellerup, X. Wang and S. Wang, D−π–A Triarylboranes as Reversible Fluorescent Probes for CO2 and Temperature, Org. Lett., 2019, 21(8), 2838–2842, DOI:10.1021/acs.orglett.9b00831.
- M. Y. Rezk, J. Sharma and M. R. Gartia, Nanomaterial-Based CO2 Sensors, Nanomaterials, 2020, 10(11), 2251, DOI:10.3390/nano10112251.
- A. B. Mabire, Q. Brouard, A. Pitto-Barry, R. J. Williams, H. Willcock, N. Kirby, E. Chapman and R. K. O'Reilly, CO2/pH-responsive particles with built-in fluorescence read-out, Polym. Chem., 2016, 7, 5943–5948 RSC . Royal Society of Chemistry.
- L. Q. Xu, B. Zhang, M. Sun, L. Hong, K. G. Neoh, E. T. Kang and G. D. Fu, CO2-triggered fluorescence “turn-on” response of perylene diimide-containing poly(N,N-dimethylaminoethyl methacrylate), J. Mater. Chem. A, 2013, 1, 1207–1212 RSC.
- D. Zhang, Y. Fan, H. Chen, S. Trépout and M. H. Li, CO2-Activated Reversible Transition between Polymersomes and Micelles with AIE Fluorescence, Angew. Chem., Int. Ed., 2019, 58, 10260–10265 CrossRef CAS PubMed.
- Q. Zhang and S. Zhu, Oxygen, and carbon dioxide dual responsive nanoaggregates of fluoro- and amino-containing copolymer, ACS Macro Lett., 2014, 3, 743–746 CrossRef CAS PubMed.
- Y. Yan, Y. Li, J. Wang, L. Li and F. Tang, A carbon dioxide responsive fluorescent system based on micellar transformation, Colloids Surf., A, 2022, 641, 128457, DOI:10.1016/j.colsurfa.2022.128457.
- J. Zhu, P. Jia, N. Li, S. Tan, J. Huang and L. Xu, Small-molecule fluorescent probes for the detection of carbon dioxide, Chin. Chem. Lett., 2018, 29(10), 1445–1450, DOI:10.1016/j.cclet.2018.09.002.
- J. Tian, B. Huang, C. Xiao and P. Vana, Intelligent CO2- and photo-dual-responsive polymer vesicles with tunable wall thickness, Polym. Chem., 2019, 10(13), 1610–1618, 10.1039/c8py01743c.
- Y. Wang, M. Huo, M. Zeng, L. Liu, Q. Q. Ye, X. Chen, D. Li, L. Peng and J. Y. Yuan, CO2-responsive Polymeric Fluorescent Sensor with Ultrafast Response, Chin. J. Polym. Sci., 2018, 36, 1321–1327 CrossRef CAS.
- L. Qiu, H. Zhang, B. Wang, Y. Zhan, C. Xing and C. Y. Pan, CO2-Responsive Nano-Objects with Assembly-Related Aggregation-Induced Emission and Tunable Morphologies, ACS Appl. Mater. Interfaces, 2020, 12, 1348–1358 CrossRef CAS PubMed.
- A. B. Mabire, Q. Brouard, A. Pitto-Barry, R. J. Williams, H. Willcock, N. Kirby, E. Chapman and R. K. O'Reilly, CO2/pH-responsive particles with built-in fluorescence read-out, Polym. Chem., 2016, 7(38), 5943–5948, 10.1039/C6PY01254J.
- O. Green, P. Finkelstein, M. A. Rivero-crespo, M. D. R. Lutz, M. K. Bogdos, M. Burger, J.-C. Leroux and B. Morandi, Activity-Based Approach for Selective Molecular CO 2 Sensing, J. Am. Chem. Soc., 2022, 144(19), 8717–8724, DOI:10.1021/jacs.2c02361.
- X. Song, M. Wang, W. Liu, H. Zheng, C. Redshaw, X. Feng, Z. Zhao and B. Z. Tang, A pyrene-based platform for studying the relationship of steric effects on optical properties, Dyes Pigm., 2023, 219, 111532, DOI:10.1016/j.dyepig.2023.111532.
- R. Rutkaite, L. Swanson, Y. Li and S. P. Armes, Fluorescence studies of pyrene-labelled, pH-responsive diblock copolymer micelles in aqueous solution, Polymer, 2008, 49(7), 1800–1811, DOI:10.1016/j.polymer.2008.02.007.
- X. He, S. Zhang, S. Qi, P. Xu, B. Dong and B. Song, Enhanced excimer fluorescence emission of pyrene derivatives: Applications in artificial light-harvesting systems, Dyes Pigm., 2023, 209, 110933, DOI:10.1016/j.dyepig.2022.110933.
- J. Zeng, X. Wang, X. Song, Y. Liu, B. Liao, J. Bai, C. Redshaw, Q. Chen and X. Feng, Steric influences on the photophysical properties of pyrene-based derivatives; mechanochromism and their pH-responsive ability, Dyes Pigm., 2022, 200, 110123, DOI:10.1016/j.dyepig.2022.110123.
- J. Cui, H. Nie, X. Liang, J. Bai and X. Zhang, A pyrene-derived ratiometric fluorescent probe for pH monitoring in cells and zebrafish based on monomer-excimer emission, J. Mol. Liq., 2022, 366, 120325, DOI:10.1016/j.molliq.2022.120325.
- T. Liu, Z. Huang, R. Feng, Z. Ou, S. Wang, L. Yang and L.-J. Ma, An intermolecular pyrene excimer-based ratiometric fluorescent probes for extremely acidic pH and its applications, Dyes Pigm., 2020, 174, 108102, DOI:10.1016/j.dyepig.2019.108102.
- K. Ayyavoo and P. Velusamy, Pyrene based materials as fluorescent probes in chemical and biological fields, New J. Chem., 2021, 45(25), 10997–11017, 10.1039/D1NJ00158B.
- Z. Gou, A. Wang, M. Tian and Y. Zuo, Pyrene-based monomer-excimer dual response organosilicon polymer for the selective detection of 2,4,6-trinitrotoluene (TNT) and 2,4,6-trinitrophenol (TNP), Mater. Chem. Front., 2022, 6(5), 607–612, 10.1039/D1QM01574E.
- M. Pose, K. M. Dillon, A. Denicola, B. Alvarez, J. B. Matson, M. N. Möller and E. Cuevasanta, Fluorescent detection of hydrogen sulfide (H2S) through the formation of pyrene excimers enhances H2S quantification in biochemical systems, J. Biol. Chem., 2022, 298(10), 102402, DOI:10.1016/j.jbc.2022.102402.
- S. Wang, Z. Zhang, Z. Huang, X. Lei, Y. Wang, L. Li, L. Yang, H. Liu, F. Sun and L.-J. Ma, A pyrene-based pH fluorescence probe with continuous multiple responses under acidic conditions and its application for environmental water systems and cells, J. Photochem. Photobiol., A, 2021, 418, 113438, DOI:10.1016/j.jphotochem.2021.113438.
- A. G. Crawford, Z. Liu, I. A. I. Mkhalid, M.-H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov and J. A. K. Howard, et al., Synthesis of 2- and 2,7-Functionalized Pyrene Derivatives: An Application of Selective C–H Borylation, Chem. – Eur. J., 2012, 18(16), 5022–5035, DOI:10.1002/chem.201103774.
- J. Frommer, B. Karg, K. Weisz and S. Müller, Preparation and characterization of pyrene modified uridine derivatives as potential electron donors in RNA, Org. Biomol. Chem., 2018, 16(41), 7663–7673, 10.1039/C8OB02246A.
- W. Luo, H. Jiang, X. Tang and W. Liu, A reversible ratiometric two-photon lysosome-targeted probe for real-time monitoring of pH changes in living cells, J. Mater. Chem. B, 2017, 5(24), 4768–4773, 10.1039/C7TB00838D.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figure and tables, detailed experimental procedures, and characterization data for all new compounds. See DOI: https://doi.org/10.1039/d4py01186d |
| This journal is © The Royal Society of Chemistry 2025 |