
Cooperative iridium and organocatalysis: a new frontier in asymmetric chemistry
Divakar
Chaudhary
and
Rakesh K.
Saunthwal
 *
*
Department of Chemistry, Indian Institute of Technology Roorkee, Roorkee, Uttarakhand 247667, India. E-mail: rakesh.saunthwal@cy.iitr.ac.in
First published on 28th January 2025
Abstract
The cooperative catalyst approach has become a significant and burgeoning area in synthetic organic chemistry, garnering substantial attention from the scientific community in recent years. Unlike single metal catalysts or organocatalysts, the cooperative catalysis strategy enables previously unattainable transformations to be accomplished in a stereocontrolled manner with high efficiency. In particular, cooperative iridium–organocatalysis has emerged as a valuable tool for producing biologically active chiral molecules from easily accessible starting materials. Iridium metal catalysts have been effectively combined with a diverse range of organocatalysts, including Brønsted acids, Lewis bases, N-heterocyclic carbene catalysts, and even phase transfer catalysts. This review provides a comprehensive overview of this evolving field, encompassing reaction advancements and mechanistic insights, to offer readers a better understanding of how metal–organic catalysts influence reaction mechanisms and the suitable starting materials for each catalytic process.
 Divakar Chaudhary | Divakar Chaudhary was born in 1999 in Basti (UP), India. He completed his B.Sc. (Hons.) degree in chemistry from Banaras Hindu University, Varanasi, India and pursued his Master's degree in chemistry from the Indian Institute of Technology Kanpur, India. At present, he is a doctoral student in Prof. Rakesh Kumar Saunthwal's group at the Department of Chemistry, Indian Institute of Technology, Roorkee, India. His current research primarily centers on Asymmetric C–C bond formation by metal–organo cooperative catalysis. |
 Rakesh K. Saunthwal | Rakesh K. Saunthwal was born in Rooppura Sikar (Rajasthan), India. He completed his master's at the University of Rajasthan, India. Then, he pursued his doctoral studies at the University of Delhi (India) under the guidance of Prof. Akhilesh K. Verma. Following his Ph.D., he held positions as a SERB Overseas postdoctoral fellow and Marie Curie Postdoctoral fellow in Prof. Jonathan Clayden's Lab at the University of Bristol, U.K. Subsequently, he relocated to Illinois to join Professor Scott E. Denmark's group at the University of Illinois at Urbana Champaign, USA (UIUC), as a Postdoctoral Research Associate. In 2023, he returned to India and became an Assistant Professor at IIT (BHU). Currently, he serves as an Assistant Professor at the Department of Chemistry, Indian Institute of Technology Roorkee, India. His research interests encompass asymmetric synthesis, catalysis, and reaction methodology. |
1. Introduction
The cooperative catalysis approach has advanced over the last two decades as an enabling strategy in chemical transformation.1 Transition metal and organocatalysts work together to activate inert coupling partners, leading to the creation of a new compound class.2,3 Recent advancements in this field highlight the use of dual catalysis,4 where two different catalysts act on distinct substrates to enable a reaction between two activated intermediates.5–7 Cooperative catalysis utilizes both single and dual catalytic cycles. Single-cycle activation involves two catalysts activating substrates A and B in one cycle to enhance the reaction rate. In the dual cycle, different catalysts simultaneously activate both substrates in separate catalytic cycles.8In the dual catalysis approach, both catalysts are placed in close proximity to substrates to form active sites containing electrophilic and nucleophilic centers in the reaction system.9,10 The collaboration of catalysts presents a perceived challenge in terms of kinetics. It might seem initially that the reaction between two catalytic intermediates present in low sub-stoichiometric concentrations would be inherently difficult. However, this assumption overlooks a crucial aspect of the rate equation – the rate constant k. In the case of the reaction between two activated intermediates, lowering the HOMO–LUMO gap in cooperative catalysis leads to a significant decrease in the activation energy. Consequently, this increase in the rate constant drives the desired pathway, favoring it over possible side reactions (Fig. 1).8,11,12
 | ||
| Fig. 1 Cooperative catalytic cycles. | ||
In the field of synthetic organic chemistry, organocatalysis has emerged as an innovative domain alongside transition metal catalysis for selectively building C–C and C–X bonds.13–15 When transition metals are combined with organocatalysts, the resulting catalytic system becomes more reactive, leading to the formation of new C–C and C–X bonds.16 In the past, transition metal–metal Lewis acids were used in the dual catalytic approach.17–20 However, the combination of transition metal and organocatalysts gained more popularity in asymmetric synthesis compared to the combination of two different metal catalysts. This preference is attributed to the redox compatibility of different metal catalysts and the balanced kinetics of the two catalytic cycles.2,21,22
Previously, many researchers have explored the use of various chiral organocatalysts,23 including Brønsted acids, Brønsted bases, Lewis bases, and phase transfer catalysts, in combination with different transition metals to collaboratively introduce a new chiral center at an achiral carbon. Cooperative catalysis offers several advantages over traditional catalytic approaches, such as improved selectivity, broader substrate applicability, cost reduction, and enhanced catalytic efficiency. As a result, it has been receiving increasing attention in the field.21,24
In the early stages, the iridium catalyst was utilized in collaboration with a Brønsted acid. Later on, its application was extended to include cooperation with Lewis bases such as amino catalysts, N-heterocyclic carbenes, and isothiourea catalysts, as well as phase transfer catalysts. Over the past two decades, it has become widely utilized as a photocatalyst also.25 For a successful cooperative catalytic cycle, iridium activates the [SM-A], and organocatalysts activate [SM-B] in a selective manner; otherwise, high reactivity and energy of the intermediate encourage the decomposition of the starting substrate or promote the side reaction, which diminishes the reaction efficacy and efficiency.26
In view of the significant role played by transition metal and organocatalysts in the context of synergistic catalysis, this review seeks to present an overview of the latest primary literature up until November 2024, focusing on the integration of synergistic dual catalysis and cooperative double activation, specifically involving iridium metal in combination with various organocatalysts. So far, all reviews have focused on either metal–metal cooperative catalysis or the use of specific organocatalysts alongside various metal catalysts for organic transformations. This review focuses specifically on one metal along with a selection of organocatalysts that remain to be explored. The emphasis will be on examining catalyst pairing, elucidating their proposed catalytic cycles, and highlighting notable instances of innovative transformation.
2. Combination of iridium and Brønsted acid
Brønsted acid catalysis has a rich history and remains one of the most widely utilized forms of catalysis in asymmetric organic transformations. It has been a cornerstone of the field for centuries, offering significant opportunities for advancing organic synthesis.27–30 Over the past two decades, researchers have increasingly combined Brønsted acids and transition metals to activate different substrates31 in organic synthesis. Notably, BINOL-derived Brønsted acid has been employed as a synergistic catalyst alongside metals such as Ag, Au, Pd, Rh, Ru, and Ir in asymmetric chemistry, yielding promising results in this field.32In the realm of catalysts, the pairing of iridium catalyst with Brønsted acids, specifically BINOL phosphoric acid (B*H-1 to B*H-3 and B*H-7), spirocyclic phosphoric acid (B*H-4 and B*H-5), and biphenyl phosphoric acid (B*H-6), has shown promise as a cooperative catalyst in driving asymmetric transformations (Fig. 2).
 | ||
| Fig. 2 Brønsted acid catalysts used with Ir-catalyst. | ||
In 2008, the Xiao group initially investigated the use of an iridium catalyst in conjunction with a BINOL-phosphate anion as a synergistic catalyst for the asymmetric hydrogenation of acyclic amine,33 by using chiral phosphate anion (1.0 mol%) and diamine-ligated iridium catalyst in the presence of 20 bar H2 pressure. By applying this protocol, a wide range of imines (1) can be efficiently hydrogenated, consistently yielding high product yields and impressive enantiomeric excess. In 2009, Xiao advanced the reductive amination approach by incorporating ketones (1′) and amines (2) as starting materials within a similar type of catalytic system.34 A broad range of substituted aromatic and aliphatic ketones were rapidly aminated in this reaction, exhibiting high yields and enantiomeric excess. Mechanistically, imines are generated in situ or can be isolated from ketones 1′ and amines 2 by a condensation reaction; the reduction of imines 1 is initiated by an ionic pathway using chiral phosphoric acid to form transient iminium cation 1-ii, which gives reduced amine product through enantioselective hydride transfer from iridium hydride complex by the facial attack. In 2013, the Xiao group built on the 2009 mechanistic hypothesis and revealed that combining an achiral [Ir]-catalyst with chiral phosphoric acid has produced a catalyst that enables highly enantioselective hydrogenation of both aliphatic and aromatic imines, achieving impressive enantiopurity.35 These findings indicated that chiral phosphoric acids have the potential to induce high enantioselectivity through hydride transfer from achiral organo-hydride donors to imines (Scheme 1).
 | ||
| Scheme 1 Asymmetric reductive amination. | ||
In a similar vein to Xiao's hypothesis, the Carreira group has developed an effective method for allylic etherification using a chiral ligand-enabled iridium complex and a Brønsted acid as a cooperative catalyst. This method demonstrates efficient coupling of two different alcohols in an enantioselective manner. By employing the widely available aliphatic alcohol 5 as the nucleophile and the branched allylic alcohol 4 as the electrophile, they achieved the asymmetric production of allylic ether 6 in high yield with up to 99% enantiomeric excess (Scheme 2).36
 | ||
| Scheme 2 Enantioselective allylic etherification. | ||
In 2014, Zhao's group explored the amination of alcohol by using iridium and chiral phosphoric acid catalysts. The amination of alcohols has been achieved by using the borrowing hydrogen methodology (also recognised as the hydrogen auto-transfer process). In this methodology, enantioselective production of secondary amines occurs by alcohol (7) and aniline substrate (2) in the presence of cooperative iridium and chiral phosphoric acid catalyst (B*H-1). Three mechanistic steps are involved in this approach: dehydrogenation of alcohol (7) to the corresponding ketone (1′), followed by the keto-amine condensation to give iminium ion (1-i); in the final step, imine hydrogenation and employing the alcohol as the H2 donor. This method demonstrates good tolerance to electron-donating and withdrawing groups on various alcohols and aniline substrates (Scheme 3).37
 | ||
| Scheme 3 Enantioselective amination of alcohols using borrowing hydrogen methodology. | ||
Zhao group utilized a synergistic dual catalyst to expand the concept of amination for α-branched alcohols 9 through dynamic kinetic asymmetric transformation (DYKAT). The enantioenriched amine product 10 was obtained from both enantiomers of racemic branched alcohol through a series of steps: oxidation of the alcohol (9-i), iminium ion formation (9-ii) via keto-amine condensation, followed by the enantioselective reduction of the iminium ion (Scheme 4).38
 | ||
| Scheme 4 Amination of α-branched alcohols by dynamic kinetic asymmetric transformation. | ||
In 2017, Zhao group further extended the borrowing hydrogen idea for enantioenriched tetrahydroquinolines 12 synthesis. In this procedure, chiral phosphoric acid (B*H-2) and Ir-complex are used as a cooperative catalyst to facilitate the enantioselective intramolecular cyclization of amino alcohol 11 in dimethyl carbonate solvent to afford the 2-substituted tetrahydroquinolines 12. The interaction between the Ir-catalyst and chiral phosphoric acid B*H-2 likely leads to the formation of complex I. This complex then reacts with the amino alcohol 11, resulting in the production of the amino ketone intermediate 11-i and iridium hydride III through a concerted transition state 11-i. In this reaction, alcohol functions as the H2 donor; this technique exploits three concurrent reactions: oxidation (11-i), condensation (11-ii), and reduction (12) without needing any additional reductant or oxidant. In this method, easily accessible racemic amino alcohols are transformed into tetrahydroquinolines in a redox-neutral manner, without requiring any stoichiometric reagents. Enantioselective hetero-cyclization reaction is well tolerated with electron-donating and electron-withdrawing groups with good yield and high enantioselectivity (Scheme 5).39
 | ||
| Scheme 5 Enantioselective tetrahydroquinolines synthesis via intramolecular amination of alcohols. | ||
Following the amination of alcohol by aniline derivatives, the Zhao group further developed the hydrogen borrowing method by incorporating aliphatic amines 14. This enantioenriched amination reaction demonstrated remarkable versatility with both alcohols and aliphatic primary amines. Alongside the previously suggested mechanistic pathway for the amination reaction, a new transition state mode, TS-1, has been proposed to account for the stereochemical outcome of the reduction step of the iminium ion intermediate (Scheme 6).40
 | ||
| Scheme 6 Enantioconvergent amination of alcohols using aliphatic amines. | ||
Upon investigating the potential of amines as a reactive nucleophile in conjunction with alcohols within the hydrogen borrowing technique, the Zhao group expanded this approach to enable the arylation of alcohols utilizing pyrrole 17 as a nucleophile. Similar to the amination process, the proposed mechanism commences with the dehydrogenation of alcohol 16, generating ketone 16-i and iridium hydride species. Subsequently, the acid-promoted nucleophilic addition of pyrrole results in the formation of the acid-bound tertiary alcohol 16-ii, which further transforms into species 16-iii through dehydration. Eventually, enantioselective hydride transfer from iridium hydride species produces the enantiopure arylated product 18. Zhao's group investigated a cost-effective and redox-neutral process using various substituted pyrroles (17). They found that secondary aryl–alkyl alcohols with ortho-, para-, and meta-positions containing electron-donating and withdrawing groups were well-tolerated. This resulted in the production of products with high enantioselectivity and good yield (Scheme 7).41
 | ||
| Scheme 7 Stereoconvergent heteroarylation of alcohols. | ||
In 2017, Zhong developed an extremely selective system for the allylic dearomatization of naphthol (19) by combining an Ir-catalyst and BINOL phosphoric acid (rac-BH-1). The system showed great tolerance towards aromatic groups with both electron-rich and electron-deficient substituents on the ortho-, meta-, or para-positions of the allyl alcohols, resulting in products with excellent enantioselectivities. The experimental results suggest that the chiral iridium complex and acid promoter rac-BH-1 react with allylic alcohol 4 to produce an Ir-π-allyl intermediate (4-i), while naphthol deprotonation and naphtholate react with the Ir-allyl intermediate to form the key intermediate 19-ii. Subsequently, the asymmetric allylic dearomatization proceeds through enantioselective nucleophilic addition of the allyl intermediate, ultimately yielding the desired products 20 (Scheme 8).42
 | ||
| Scheme 8 Asymmetric allylic dearomatization of naphthol derivatives. | ||
Over the past five years, researchers have not only employed chiral phosphoric acid as a cooperative catalyst with iridium but have also utilized achiral Brønsted acids in conjunction with iridium-catalysts for asymmetric synthesis. For instance, the Shi group has successfully used [Ir(cod)Cl]2 and TsOH·H2O Brønsted acid to achieve stereoselective reactions of vinyl benzoxazine 21 with azlactones 22. In this process, the iridium catalyst promotes the decarboxylation of vinyl benzoxazinanones 21, leading to the formation of π-allyl-Ir intermediate (21-i), while the Brønsted acid TsOH·H2O triggers the enolization of azlactones (22′) via hydrogen bonding interaction. The combination of these two active species (21-i and 22′) results in the production of the chiral 3,4-dihydroquinolin-2-one 23 through a [4 + 2] mode, regenerating the Ir-catalyst and TsOH acid in the process. This method has demonstrated high selectivity across a wide range of substituted azlactones and vinyl benzoxazinanones, yielding selectivity levels of up to 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dr (Scheme 9).43
:5 dr (Scheme 9).43
 | ||
| Scheme 9 Stereoselective reactions of vinyl benzoxazinones with azlactones. | ||
Following in the footsteps of Shi's work, Liu described a cooperative approach for producing enantioselective spiro-N,O-ketals by asymmetric [4 + 2] cycloaddition of 2-(1-hydroxyallyl) phenols and exocyclic enamides using [Ir(cod)Cl]2 and (PhO)2POOH. The formation of the Ir-π-allyl complex 24-i by dehydration is the first step in the reaction mechanism. Nucleophilic enamine 25 then attacks the Ir-π-allyl complex to generate an iminium ion intermediate (24-ii). Eventually, the enantioenriched product 26 was produced by the spirocyclic ring closure. This strategy is shown for a wide class of enamides that have the phenyl moiety substituted at C2, C3, C4, or C5 with groups that donate or withdraw electrons, as well as 2-(1-hydroxyallyl)phenols that have methoxy, bromo, and trifluoromethoxy groups at the C4 position of the aryl ring. This results in the production of distinct, well-yielding, high enantioenriched chromane-2,1′-isoindolins (Scheme 10).44
 | ||
| Scheme 10 Spiroketalization by cooperative catalysis. | ||
3. Combination of iridium and Lewis base
In recent decades, nucleophilic Lewis base organocatalysts have emerged as a promising and versatile tool in synthetic organic chemistry. N-heterocyclic carbene (NHC)45,46 and tertiary amine47 catalysts are commonly used examples, possessing a pair of nonbonding electrons.48,49 Initially, the Cordova group50 explored the use of a nucleophilic Lewis base catalyst in conjunction with palladium as a cooperative catalyst. Subsequently, various metals including Zn, Cu, Y, Au, In, Rh, Ru, and Ir have been employed with different Lewis bases.51–54 In recent reports, authors used Brønsted acid with Lewis base catalyst as an additive to activate allyl substrate in the reaction process, which aids in the production of the Ir-π-allyl complex and its counter ion also aids in stabilizing the Ir-π-allyl complex. This section provides a summary of the recent applications of amine6 and N-heterocyclic carbene (NHC) catalysts with iridium metal as a synergistic catalyst.In 2012, Jian Xiao developed a highly selective method for combining IrCl3 and a chiral diarylprolinol silyl ether catalyst A1 to perform intermolecular α-alkylation of aldehydes 27. This approach has proven effective for various bulky, linear aldehydes (27) and activated alcohols (7), consistently yielding high yield and excellent enantioselectivity (Scheme 11).55
 | ||
| Scheme 11 Stereoselective α-alkylation of aldehydes. | ||
Carreira group in 2014 devised a similar dual catalytic method for α-allylation of linear aldehydes by combining iridium catalysis with chiral diarylprolinol silyl ether A1, building on Xiao's stereoselective α-alkylation.55 By reacting aryl vinyl carbinol 4 and hexanal 27 with dimethyl hydrogen phosphate as the promoter and chiral iridium complex and diaryl silyl prolinol ether as cooperative catalysts, excellent enantio- and diastereoselective γ,δ-unsaturated aldehyde (29) was produced. Phosphoric acid activates allyl alcohol in the reaction process, which aids in the production of the Ir-π-allyl complex. Phosphoric acid's counter ion also aids in stabilizing the Ir-π-allyl complex, and prolinol ether combines with the carbonyl group in a different pathway to generate an enamine intermediate. Synthesis of (−)-paroxetine demonstrated the synthetic utility of this method (Scheme 12).56
 | ||
| Scheme 12 Stereodivergent α-allylation of linear aldehydes. | ||
The α-allylation process of linear aldehydes with alcohols evolved, and in 2015, the Carreira group expanded this method to include the synthesis of protected α-amino- and α-hydroxy-acetaldehydes (30 and 31). With this approach, it is also possible to achieve unsaturated product transformation in a stereodivergent way while maintaining the stereochemical integrity at the C-α location to show high selectivity. The reactivity of allylic alcohol 4 with electron-withdrawing substituents is aided by strong acid (trichloroacetic acid), producing compounds (30 and 31) with good diastereoselectivity and >99% ee (Scheme 13).57
 | ||
| Scheme 13 Stereodivergent α-allylation of protected α-amino- and α-hydroxyacetaldehydes. | ||
In continuation to Carreira's group, In 2021 Yang group employed Carreira's innovative stereodivergent Ir/amine dual catalysis method to couple pyrrole alcohol 32 and butanal 33, leading to the intermediates 34 for the bisdehydrotuberostemonine D and bisdedydrotuberostemonine E synthesis.58 Recently in 2024, the Yang group again incorporated the same method to develop a branched aldehyde intermediate 36 for (−)-daphenylline synthesis (Scheme 14).59
 | ||
| Scheme 14 Stereodivergent coupling of alcohol and aldehyde. | ||
Recent investigations conducted by Carreira's group have successfully demonstrated the compatibility between amino and iridium catalysts. Following this confirmation, the Jørgensen group introduced the pioneering regio- and stereoselective γ-allylation of cyclic α,β-unsaturated aldehydes, achieved through the integration of aminocatalysis and [Ir(cod)Cl]2 catalysis. This reaction proceeds via the formation of an enamine (37-i) and an Ir-π-allyl complex (38-i). Both electron-donating and electron-withdrawing groups having aldehyde and allylic alcohol exhibited favorable performance under standard reaction conditions (Scheme 15).60
 | ||
| Scheme 15 γ-Allylation of α,β-unsaturated aldehydes. | ||
For the exploration of enamine intermediate in a multi-component reaction (MCR), In 2016, Wenhao Hu revealed a three-component reaction of diazoacetates (40), indoles (41), and enals (42) by co-catalysis of iridium/chiral amine catalyst for the synthesis of the enantioselective 3-substituted indole derivative (43). This reaction involves the formation of an [Ir]-carbene intermediate (40-i), which then reacts with indole (41) to form a zwitterionic intermediate (41-i) or enolate (41-i′). Concurrently, 3,5-bis(trifluoromethyl)benzoic acid accelerates the formation of an iminium ion 42-i, capturing the zwitterionic intermediate (41-i) to produce the enamine 41-ii. Finally, acidic hydrolysis leads to the desired products (anti-43 and syn-43) with high diastereoselectivity (Scheme 16).61
 | ||
| Scheme 16 Asymmetric three-component reaction of diazoacetates with indoles and enals. | ||
Liu group successfully replaced the indole (36) nucleophile with alcohols (5) from Hu's work and developed a one-pot approach to the synthesis of enantioselective 1,2,5-triol (46) derivatives with vicinal chiral centers utilizing simple starting materials through the incorporation of a chiral secondary amine with an iridium(I) catalyst and a reducing agent. The reaction mechanism involves iridium-(I)-associated oxonium (5-i) and amine-activated enals intermediates (45-i) (Scheme 17).62
 | ||
| Scheme 17 Vicinal chiral centers having 1,2,5-triol derivatives synthesis. | ||
The Carreira group not only employed secondary amine catalysis but also made use of primary amine catalysts in tandem reactions. They successfully synthesized enantioselective γ,δ-unsaturated aldehydes (48) by combining allylic alcohol (4) and aldehyde (47) using primary amine A3 and a chiral iridium catalyst under acidic conditions. This approach involved the simultaneous activation of the aldehyde (47) and allylic alcohol (4) with primary amine and iridium catalysts, respectively, resulting in the formation of the enamine (47-i) and Ir-allyl-π complex (4-i) (Scheme 18).63
 | ||
| Scheme 18 α-Allylation of branched aldehydes by stereodivergent dual catalysis. | ||
A combination of diphenylmethanamine and iridium catalyst enables the [4 + 2] cycloadditions of vinyl amino alcohols (49) with aldehydes (47) to produce enantioselective dihydroquinolone derivatives 50′ in good yield. Ir-catalyst forms Ir-allyl-π intermediate (49-i) by reacting with vinyl amino alcohol (49). The carbonyl compound and amine catalyst then condense to produce an enamine intermediate (47-i). The hemiacetal 50 is created when intermediate 49-i and 47-i are coupled in a [4 + 2] manner through intermediate 49-ii. The Xiao group diversified the hemiaminal intermediate (50) into hydroquinolines (50a) by oxidation and tetrahydroquinolines (50b) by reduction in order to demonstrate the adaptability of the [4 + 2] cycloaddition reaction. Furthermore, the optically active 2-allyl (50c), 2-CN (50d), and 2-N3 (50e) substituted tetrahydroquinolines could be produced by substituting the hydroxyl group with other organosilicon reagents. Likewise, nucleophiles ending in –SPh (50f) and –OEt (50g) can take the place of the OH-group (Scheme 19).64
 | ||
| Scheme 19 Synergetic iridium/amine catalysis for the enantioconvergent [4 + 2] cycloadditions. | ||
The Xu group used traditional click chemistry to combine the squaramides and Ir-catalysts for the synthesis of axially chiral aryl triazoles. In order to carry out the atropo-enantioselective azide–alkyne cycloaddition (AAC) reaction, naturally occurring squaraine organocatalyst and Ir(I) are combined with ortho-hydroxy alkenyl naphthalene (51) and benzyl azide (52). Examining experimental data closely revealed that the metal/organic catalyst combined with the starting materials to create a cycloaddition adduct (Ts-51) and then vinylidene ortho-quinone methide (VQM) (51-i). The atrop-aryltriazoles 53 were then delivered by stereospecific [1,5]-H transfer (Scheme 20).65
 | ||
| Scheme 20 Asymmetric azide–alkyne cycloaddition reaction for the synthesis of atrop-aryltriazoles. | ||
Recently, the Yang group unveiled an innovative reaction that utilizes both Ir and Brønsted acid catalysis. This method effectively transforms 2-(1-hydroxyallyl)-phenols 55 in conjunction with isochroman ketals 54 into bis-benzannulated spiroketals 57, achieving remarkable efficiency and selectivity, with diastereomeric ratios reaching up to 17:1 and enantiomeric excess surpassing 99%. The process begins with the formation of exocyclic enol ethers (54-ii) from isochroman ketals 54, driven by Brønsted acid catalysis, which is then followed by an Ir-catalyzed enantioselective allylation and spiroketalization of the 2-(1-hydroxyallyl)-phenols 55 (Scheme 21).66
 | ||
| Scheme 21 Asymmetric synthesis of bisbenzannulated spiroketals and spiroaminals. | ||
Chen et al. combined achiral tertiary amine catalysts with iridium complexes to create a cooperative catalytic system in 2019. Stereoselective [4 + 3] annulated spiro compound (61) is formed by the chemoselective activation of Morita–Baylis–Hillman carbonate adduct (58) and carbamate-functionalized allylic carbonates (59) by the chiral iridium complex and DABCO, respectively. An essential development in stereoselective annulation chemistry is the independent activation by metal complexes and organocatalysts, leading to the emergence of two distinct dipole species. Apart from the [4 + 3] cycloaddition, Morita–Baylis–Hillman adducts (58) with vinyl aziridines (60) in the presence of iridium and tertiary amine catalysts A5 were also used to demonstrate outstanding regio-, chemo-, and stereoselective [3 + 3] annulation (Scheme 22).67
 | ||
| Scheme 22 Regio-, chemo-, and stereoselective [4 + 3] and [3 + 3] cycloaddition reactions. | ||
Enantioselective coupling of the α,β-unsaturated carbonyl-derived latent enolate, and metal-induced electrophile discovered by Mukherjee group in 2021 by combining cinchonidine Lewis base A6 with iridium metal catalyst. In this asymmetric procedure, an olefinic C(sp2)–H bond is enantioselectively alkylated by linear allylic carbonate (64). The reaction is initiated by the activation of the olefinic C(sp2)–H bond by the Iridium-Feringa's phosphoramidite ligand complex and coumalate ester (63) activated by the cinchonidine catalyst in a cooperative fashion. This method yields cinnamyl carbonates with a wide range of electrically biased substrate compatibility at various aryl ring locations. Without sacrificing enantioselectivity, a relatively reduced yield was seen by lengthening the ester's carbon chain. Enantioenriched α-allylic coumalate ester derivatives (65) can be functionalized as crucial synthetic building blocks for various reactions, including retro-cycloaddition, cascade reactions, and decarboxylation (Scheme 23).68
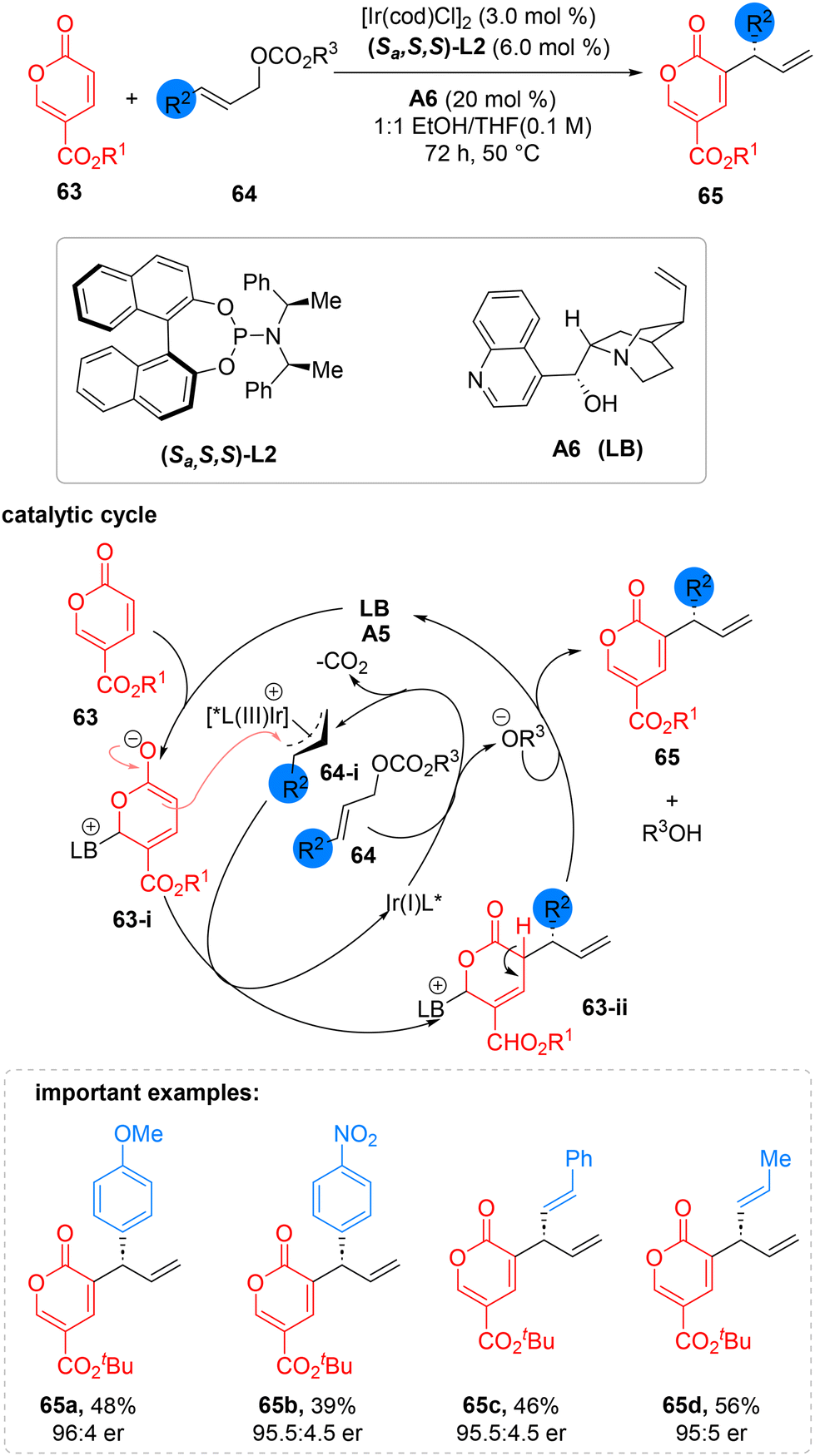 | ||
| Scheme 23 α-C(sp2)–H allylic alkylation of coumalates. | ||
A key obstacle in asymmetric catalysis is the generation of all possible stereoisomers of a given chiral molecule carrying multiple stereocenters using a straightforward and unified method. In 2017, Hartwig devised a protocol for stereodivergent substitution using allylic t-butyl carbonates (67) and aryl acetic acid ester (66) catalyzed cooperatively by iridium complex and benzotetramisole A7 in the presence of diisopropyl ethyl amine as a base. In this catalytic cycle, the iridium and Lewis base catalysts react with the allylic precursor (67) and the aryl acetic acid ester (66) cooperatively to produce Ir-allyl-π complex (67-ii) and C1-ammonium enolates (66-ii) respectively. Both intermediates react with high facial selectivity to yield stereo-divergent products (68). This dual catalytic cycle allows access to four stereoisomers by permutations of the enantiomers of the two chiral catalysts with high diastereo and enantioselectivity (Scheme 24).69
 | ||
| Scheme 24 Stereodivergent allylic substitutions of aryl acetic acid esters. | ||
Snaddon's team further investigated this chemical reaction to produce varied homoallylic amine compounds, using the allylic substitution protocol and sequential Hofmann rearrangement strategy to synthesize branched homoallylic amine 69 (Scheme 25).70
 | ||
| Scheme 25 Regio- and stereodivergent synthesis of branched homoallylic amines. | ||
Following in the footsteps of Hartwig and Snaddon, the Deng group revealed that iridium and isothiourea Lewis base catalyzed vinyl indoloxazolidones (70) and acid anhydrides (71) in a synergistic manner to produce Ir-π-allyl intermediate and C1 ammonium enolate for the enantioselective [3 + 2] cycloaddition chiral polycyclic indoles 72. The technique demonstrates a wide range of substrates, including substituted vinyl indoloxazolidones and anhydrides with various electron-donating and withdrawing groups at different positions. The result is the formation of asymmetric cyclic indoles 72 with high yield and excellent enantioselectivity (Scheme 26).71
 | ||
| Scheme 26 Asymmetric [3 + 2] cycloaddition reaction. | ||
The γ-lactam moiety plays a crucial role in various bioactive substances, serving as a fundamental framework in pharmaceuticals, natural products, and bioactive molecules. Recently, the Song group introduced a comprehensive and adaptable approach for synthesizing chiral γ-lactam compounds (74). This involved a chiral isothiourea A7 and [Ir(COD)Cl]2 cooperatively catalyzed [3 + 2] asymmetric annulation reaction among vinyl aziridines (73), pentafluorophenyl esters (66), and a base at room temperature. The cooperative catalytic framework resulted in the production of a wide range of optically active γ-lactams with excellent yield and high chiral induction. The catalytic cycle involved various steps, such as the reaction of isothiourea Lewis base with aryl acetic acid ester, leading to the formation of reactive C1-ammonium enolate. Additionally, co-catalytic cycles involving [Ir(I)]* complex resulted in the production of key intermediate (int-66) through intermolecular asymmetric allylic alkylation. Subsequently, the ring closure of the intermediate int-66 led to the final [3 + 2] ring expansion product 74 (Scheme 27).72
 | ||
| Scheme 27 Asymmetric [3 + 2] annulation for γ-lactam synthesis. | ||
In 2019, the Glorius group successfully developed a new method for synthesizing α,β-disubstituted γ-butyrolactones (77) using trans-cinnamaldehyde 75 and vinylethylene carbonate 76 through a dual catalysis approach involving iridium and N-heterocyclic carbene (NHC). This innovative process enables enantio- and diastereodivergent [3 + 2] annulation reactions. The catalytic cycle commences with the formation of Z-enol intermediate (75-ii) from α,β-unsaturated aldehyde via homoenolate formation, followed by facile β-protonation or base-mediated elimination of chloride in α-chloroaldehydes. In the co-catalytic cycle, an Ir-π-allyl intermediate (76-i) is formed from vinylethylene carbonate (76) via decarboxylation. The Sunoj group's73 DFT analogy demonstrates that the Si-face of both species 75-ii and 76-i come closer to form a new C–C bond (75-iii) and subsequently ring closer to produce the desired product 77 with high selectivity (Scheme 28).74
 | ||
| Scheme 28 Diastereodivergent [3 + 2] annulation reaction for α,β-disubstituted γ-butyrolactones synthesis. | ||
The remarkable effectiveness of switchable homoenolate and enolate intermediates derived from common enal precursors has been demonstrated through the use of the (NHC)/iridium catalyst system, enabling stereoselective and regiodivergent [3 + 2] and [3 + 3] annulation reactions. In 2021, the Wei-Ping Deng group published a study on a switchable annulation reaction of enals and 2-indolyl allyl carbonates 78 using NHC/iridium catalysis. The reaction, carried out in the presence of two different bases (triethyl amine and DBU), resulted in regiodivergent pyridine[1,2-a]indoles 80 and pyrrolo[1,2-a]indoles 79 derivatives with high diastereo- and enantioselectivity. A mechanistic study reveals that the NHC catalyst has the ability to activate cinnamaldehyde 75 or 75′, resulting in the formation of homoenolate intermediate 75-i. When weak base Et3N is present, homoenolate 75-i undergoes a quick proton transfer to yield enolate intermediate 75′-i. In the co-catalytic cycle, 2-indolyl π-allyl-iridium intermediate 78-i is formed from substrate 78. The enolate attacks the Re-face of the π-allyl-iridium intermediate. Ultimately, the catalytic cycle concludes with the lactamization of intermediate 75′-ii, producing product 79 with high selectivity. The use of DBU halts the proton transfer, leading to the predominance of homoenolate 75-i reactivity. As a result, the [3 + 3] annulation reaction proceeds through the Re–Si face attack of homoenolate 75-i to the π-allyl-iridium species 75-iii, ultimately leading to the synthesis of pyridine[1,2-a]indoles (80) (Scheme 29).75
 | ||
| Scheme 29 Regiodivergent [3 + 2] and [3 + 3] annulation reaction. | ||
Using an extremely efficient catalytic process, He group recently achieved a significant breakthrough with the first enantioselective creation of axially chiral enamides 82 with an N–C axis as a chiral component. With the aid of an iridacycle catalyst, they carried out an enantioselective allylation to form the C(sp2)–N link between 2-quinolinol 81 and cinnamyl carbonates 64. The final enamides product 82 was then created by in situ isomerizing the allylation product via a 1,3-H transfer that was made possible by the base diazabicycloundecene (DBU). This allowed the chirality to shift from central to axial. The corresponding axially chiral product is produced in a high yield with good enantiomeric excess by this reaction, which exhibits excellent functional group compatibility with both cinnamyl carbonates 64 and 2-quinolinol 81 analogs bearing electron-rich, electron-neutral, and electron-deficient substituents (Scheme 30).76
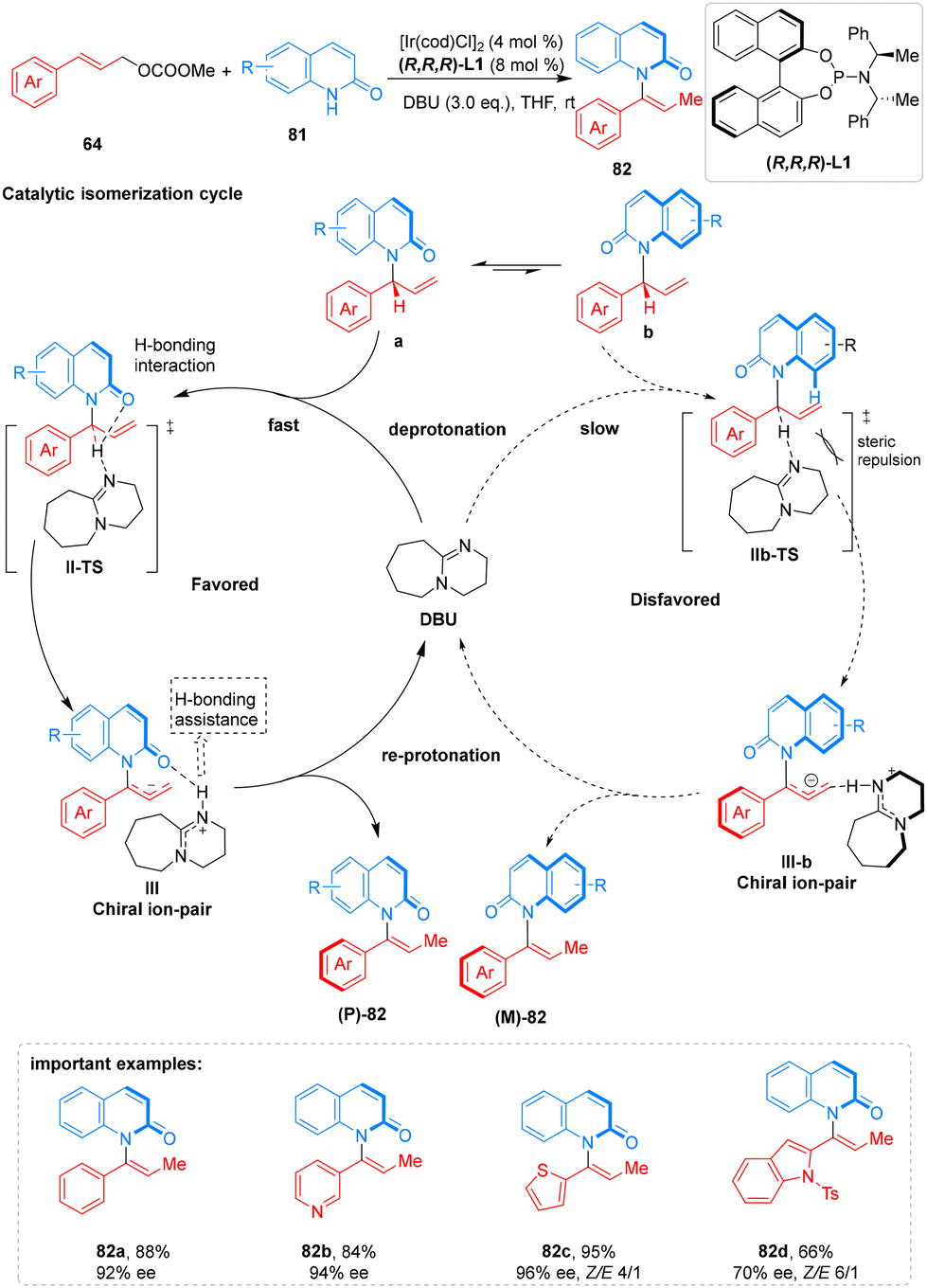 | ||
| Scheme 30 Enantioselective allylic substitution–isomerization of axially chiral enamides. | ||
4. Combination of iridium and phase-transfer catalyst
Over the past 30 years, asymmetric phase-transfer catalysis has proven to be a highly versatile method for organic synthesis.77,78 Recently, researchers have been exploring the combined use of phase-transfer catalysts and transition metal catalysts to activate normally inert C–C and C–H bonds. Notably, the Takemoto group was the first to demonstrate the synergistic effect of phase-transfer catalysis and palladium metal in facilitating asymmetric allylic alkylation of allylic acetates.79,80In 2017, Han and colleagues developed an innovative method to produce α-quaternary amino acid (85) with two vicinal stereocenters from α-imino esters 83 and allyl acetates 84. It was achieved by employing cooperative iridium and phase-transfer catalyst (PTC). During the process, the activation of α-imino esters by PTC (tetra butyl ammonium bromide) led to the formation of the 2-azallyl anion intermediate 83-ii, which then combined with the η3-π-allyliridium intermediate 84-i to generate the asymmetric final product 85 (Scheme 31).81
 | ||
| Scheme 31 α-Imino ester synthesis by Ir/PTC cooperative catalysis. | ||
Wang's 2019 publication introduces a practical and cost-effective approach for producing enantioselective homoallylic amines (87). This method involves the cooperative activation of substituted cinnamyl carbonate 64 and leucine-derived aldimine esters 86 by iridium, in combination with a phase-transfer catalyst (PTC), to create a diverse range of homoallylic amines 87. These amines exhibit outstanding enantioselectivity with yields ranging from moderate to good. The reaction is compatible with various allylic carbonates and aldimine esters featuring electron-donating, withdrawing, and neutral substituents (Scheme 32).82
 | ||
| Scheme 32 Asymmetric allylation/2-aza Cope rearrangement by Ir/PTC cooperative catalysis. | ||
5. Cooperative iridium photocatalysis
Over the past two decades, there has been notable progress in contemporary organic synthesis, largely attributed to the integration of visible light photocatalysis and organocatalysis.83,84 Visible light-induced cooperative iridium photocatalysis has emerged as a key tool in the cooperative catalysis regime for designing a stereoselective radical reaction to create site-selective carbon–carbon bonds under benign reaction conditions.85–87 Therefore, our goal in this section on cooperative photoredox is to present a particular perspective on current advancements through in-depth mechanistic analyses of cooperative organocatalyst/Ir-photocatalysis.Combination of iridium photocatalyst and amine catalyst
In the cooperative Iridium photocatalyst and amine catalyst reaction, the amino catalysts interacted with a carbonyl compound to produce a chiral enamine intermediate. Simultaneously, the Ir-photocatalyst engaged with an electrophile, leading to the formation of highly reactive electrophilic radical species. It's worth noting that both processes occurred in an overall redox-neutral manner.88The MacMillan research team developed a new method to achieve the first asymmetric α-trifluoromethylation and α-perfluoroalkylation of aldehydes 27 using a combination of cooperative iridium photocatalyst and imidazolidinone A8 organocatalyst under white light irradiation. This approach successfully allowed for a wide range of functional groups, including ethers, esters, amines, carbamates, and aromatic rings, to be compatible with the protocol. The resulting α-trifluoromethyl aldehyde 89 has the potential to be utilized in the synthesis of various organofluorine-containing compounds. Upon combination with the enamine 27a intermediate, the trifluoromethyl radical 88-i resulted in the α-trifluoromethylation of aldehyde 27. This method is applicable to aldehydes 27 with diverse functional groups like ethers, esters, amines, carbamates, and aromatic rings. Using α-trifluoromethyl aldehyde 89, a broad spectrum of valuable organofluorine-containing compounds can be synthesized. The MacMillan group expected89 that Ir*(ppy)2(dtb-bpy)+ would easily accept a single electron from enamine to create a potent reductant Ir(ppy)2(dtbbpy) (−1.51 V vs. SCE in CH3CN), with the electron-rich iridium system playing a crucial role in single electron transfer (SET) with trifluoromethyl iodide 88. This process generates the electrophilic radical 88-i and regenerates the photoredox catalyst (Scheme 33).90
 | ||
| Scheme 33 Enantioselective α-trifluoromethylation of aldehydes. | ||
In 2010, the MacMillan group further developed the α-trifluoromethylation mechanism to enable the enantioselective α-benzylation of aldehydes 27. This α-benzylation was achieved by employing an iridium photo-redox catalyst in combination with an imidazolidinone A8 organocatalyst under white light. The protocol proved effective for various substituted aldehydes containing functional groups like ethers, amines, and carbamates. Notably, the reaction process exhibited broad functional group resilience, even with benzyl bromide 90 substituents, consistently yielding the desired enantioselective product (Scheme 34).91
 | ||
| Scheme 34 Enantioselective α-benzylation of aldehydes. | ||
Combination of iridium photocatalyst and Brønsted acid catalyst
Over the past twenty years, Brønsted acid has been widely utilized as a co-catalyst alongside iridium photocatalyst in C–C bond formation reactions similar to the use of Lewis acids. It is suggested that Brønsted acid might catalysed the reaction through a proton-coupled electron transfer mechanism.10Knowles group successfully developed a groundbreaking enantioselective intramolecular aza-pinacol cyclization reaction in 2013. This innovative reaction, catalyzed by a Ir(ppy)2(dtbpy)PF6 photo-redox catalyst and aryl phosphoric acids, creates highly enantioselective products through a proton-coupled electron transfer (PCET) mechanism. Chiral phosphoric acid and an iridium photo redox catalyst facilitated this proton-coupled electron transfer (PCET) reaction in the presence of a blue LED, resulting in the intramolecular reductive coupling of ketones and hydrazones to produce a highly enantioselective product via the formation of ketyl radical intermediates (92). The process is suitable for aryl ketones with various substituents and consistently delivers syn-1,2-amino alcohol-derived molecules 93 with high yield and enantioselectivity (Scheme 35).92
 | ||
| Scheme 35 Enantioselective intramolecular aza-pinacol cyclization of keto-hydrazone. | ||
In 2018, Phipps and colleagues successfully developed a highly selective method for synthesizing α-heterocycle amines 96 utilizing a well-established minisci-type approach. The process involved the use of a chiral Brønsted acid catalyst (B*H-1 and B*H-2) along with an iridium photocatalyst to generate prochiral free radical 95-i from amino acid derivatives (95) using blue LED and [Ir]-photocatalyst. Radical 95-i was then added at the 2-position of a hydrogen-bonded pyridinium counterion (94-i) to form the aminium ion intermediate (94-ii). In the next step, the aminium ion undergoes deprotonation to form species (94-iii). This species could potentially yield the enantioenriched product 96 through rapid single-electron oxidation. In this reaction, the iridium photocatalyst facilitates the electron transfer process, while the chiral Brønsted acid plays a dual role by activating the substrate and inducing asymmetry. This approach demonstrates efficacy across a range of substituted amines, pyridines, and quinolines, consistently delivering highly enantioselective and regioselective α-heterocycle amines (Scheme 36).93
 | ||
| Scheme 36 Enantioselective Minisci-type addition to N-heterocycles. | ||
You et al. identified a fascinating example of the cooperatively catalyzed synthesis of an indoline derivative by using the umpolung chemistry of indole (97). Oxy-amine, tethered indoline derivatives (99) were produced using chiral phosphoric acid as a co-catalyst with iridium photocatalyst via an enantioselective dearomatization reaction of electrophilic indoles 97. This method is thoroughly investigated across a large range of substituted indoles 97, and it is discovered that introducing groups that withdraw electrons (–F, –Cl, –Br) into the indole moiety improves enantioselectivity when compared to groups that give electrons. The catalytic cycle begins with the activation of the Ir-photocatalyst using blue LED light. Once activated, the photoexcited [Ir]* assists in the initial SET oxidation of 97 to produce the indole radical cation intermediate (97-i). Following this, the [Ir]-photocatalyst is regenerated by molecular oxygen, and the indole radical cation intermediate is deprotonated by the oxygen anion radical. The deprotonated indole radical (97-iii) then undergoes ring closure through B*H-3 mediation, and the second SET oxidation takes place in the presence of [Ir]* to create the configurationally biased tertiary pyrroloindoline carbocation intermediate (97-vi), which is stabilized by electrostatic interactions with the chiral phosphate anion. Finally, the pyrroloindoline carbocation (97-vi) is trapped by a hydroxyl amine nucleophile to produce the optically enriched indoline derivatives 99 (Scheme 37).94
 | ||
| Scheme 37 Asymmetric photoredox dearomatization of indole derivatives. | ||
Yoon introduced a pioneering method for producing highly enantioselective pyridine-substituted cyclobutene 102 through a [2 + 2] cycloaddition reaction between vinyl pyridines 100 and a styrene derivative (101). This innovative approach involved the use of an iridium photocatalyst and chiral phosphoric acid (B*H-5) as a Brønsted acid catalyst. Yoon proposed that during a transient excited state, matched chiral catalyst pairs interact to generate an enantioenriched [2 + 2] photocycloaddition product 102, paving the way for the development of synergistic stereoinduction. Additionally, the authors noted that electron-rich styrenes (101) resulted in higher enantiomeric excess compared to electron-deficient styrene (Scheme 38).95
 | ||
| Scheme 38 Enantioselective [2 + 2] photo cycloaddition of vinylpyridines. | ||
An enantioselective three-component method for the synthesis of enantioenriched keto alkyl-functionalized 4-amino cyclopentenone was employed by the Cai group, using cooperative chiral Brønsted acid and iridium photocatalyst. In this process, the [Ir]-photocatalyst and chiral Brønsted acid catalyst (B*H) facilitated the creation of ketoalkyl radical (103-ii) from cycloalkyl silyl peroxide 103. The keto alkyl radical then attacked alkenyl furan 104, resulting in single-electron oxidation and the formation of a chiral-anion-stabilized keto-alkyl functionalized furan oxonium ion (103-iii). Subsequently, aryl amine 2 sequential nucleophilic attack on the furan ring led to ring opening and chiral anion-controlled asymmetric 4π-electrocyclization, producing the desired enantioenriched product 105. Extensive investigation revealed that the approach yielded highly enantioselective products in most cases, with moderate to good yield. This protocol enables the synthesis of various essential chiral cyclopentenone motifs with different functional groups (Scheme 39).96
 | ||
| Scheme 39 Asymmetric ketoalkylation/rearrangement of alkyenlfurans. | ||
In 2023, Wang's research group achieved the asymmetric synthesis of 3-substituted proline derivative (108) through a de novo approach, using a cascade radical addition/cyclization of racemic α-bromo γ-chloro ketone 106 and N-aryl glycine ester 107, facilitated by cooperative iridium photoredox/Chiral phosphoric acid catalysis. This reaction also demonstrated the broad applicability of the protocol, showing that both N-arylated glycinate methyl esters 107 and α-bromo γ-chloro aryl ketones 106, with electron-withdrawing and electron-donating substituents, yield exceptional enantio- and diastereoselectivity. However, this reaction does not work well with linear aliphatic dihalide ketones (Scheme 40).97
 | ||
| Scheme 40 Asymmetric de novo synthesis of 3-substituted proline derivatives. | ||
A spectacular demonstration of the enantioselective α-coupling between N-arylaminomethanes 110 and N-sulfonyl imines 109 was carried out in 2015 by Takashi Ooi and coworkers. Chiral arylaminophosphonium barfate (HBArF) and iridium metal photosensitizer were used in cooperative style with exposure to visible light to produce an enantioenriched coupled product between N-sulfonyl aldimines 109 and N-arylaminomethanes 110. The catalytic cycle commences with the excitation of the [Ir(ppy)2(bpy)]BArF complex under visible light irradiation, followed by the reductive quenching of *Ir(III) to form a neutral Ir(II) complex. This complex then transfers an electron to the imine to create an anion–radical. The anion–radical pairs with a positively charged Ir(III) complex (109-i) and then reacts with chiral arylamino phosphonium barfate to produce a crucial chiral ion pair (109-ii) while regenerating the parent photosensitizer. Simultaneously, an aminomethyl radical is formed through the deprotonation of the Ph2NMe 110 cation–radical species. Ultimately, the imine anion–radical and aminomethyl radicals are coupled to yield an enantioenriched 1,2-diamine derivative 111. A wide range of N-substituted aminomethane and aromatic N-sulfonyl imines (109) with electron-rich and electron-deficient substituents produced α-coupled products (111) with a high degree of enantiomeric excess (Scheme 41).98
 | ||
| Scheme 41 Redox neutral asymmetric α-coupling of N-arylaminomethanes with aldimines. | ||
Combination of iridium photocatalyst and Lewis acid catalyst
Synthesis of highly enantioenriched 1,2-amino tertiary alcohols (114) was reported by the Ryu group by employing chiral oxazaborolidinium ion organocatalyst and iridium photocatalyst. This protocol produces asymmetric amino tertiary alcohols (114) with high yield (up to 88%) and excellent enantioselectivity (up to 98%). This process involves the generation of α-amino alkyl radical (113-i) through visible light irradiation of α-silyl amine 113 using a single-electron transfer mechanism. The radical then undergoes an enantioselective attack on the COBI-activated aryl methyl ketone (TS). The reaction is concluded by a second single-electron transfer using α-silyl amine. The research also involved a thorough investigation of acetophenone derivatives with different substitution patterns on the phenyl ring, revealing that electron-donating substituents led to decreased selectivity and reactivity (Scheme 42).99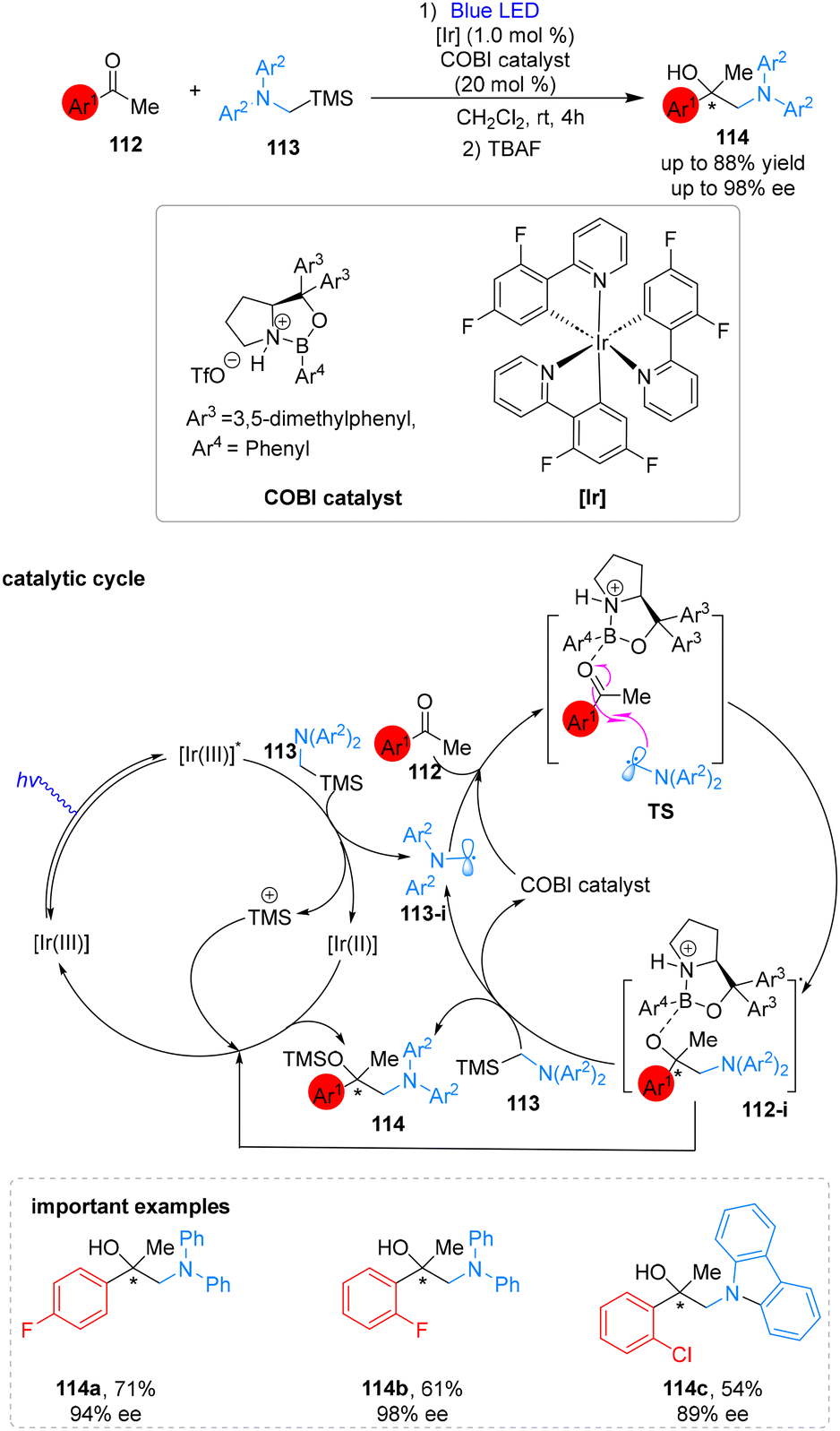 | ||
| Scheme 42 Enantioselective addition of α-aminoalkyl radicals to ketones. | ||
Combination of iridium photocatalyst and N-heterocyclic carbene
In 2022, Armido Studer discovered a method for synthesizing 2,3-difunctionalized dihydrobenzofurans (117) by utilizing a combination of N-heterocycle carbene (NHC) and iridium photoredox mediated reaction to form C–C and C–F bonds. This process involves the use of aroyl fluorides 115 as dual-purpose reagents in the presence of K2HPO4 base and 5 W blue LEDs, resulting in the conversion of benzofurans 116 into 3-aroyl-2-fluoro-2,3-dihydrobenzofurans 117 through dearomatizing fluoroaroylation.The mechanistic cycle commences with the oxidation of benzofuran 116 to its radical cation (116-i) by photoexcited Ir(III)*. Simultaneously, NHC generates acyl azonium ion (115-i) from acyl fluoride 115 in the co-catalytic cycle. The acyl azonium ion is then reduced by Ir(II) to regenerate Ir(III), leading to the formation of the persistent ketyl radical (115-ii). The subsequent merge of the benzofuran radical cation (116-i) with the ketyl radical (115-ii) results in the formation of the oxocarbenium ion (115-iii). Finally, the oxocarbenium ion is diastereoselectively trapped by F-anion in trans to the bulky alcoholate moiety, and NHC-regeneration produces product 117 in good yield. This photocatalytic protocol has been extensively studied for aroyl fluoride with para-position substituents that either donate or withdraw electrons from the aryl ring. Furthermore, benzofurans with various substituents at the aromatic ring have shown high tolerance, yielding acceptable yields with significant diastereoselectivity (Scheme 43).100a
 | ||
| Scheme 43 Photoredox dearomatizing fluoroaroylation of benzofurans. | ||
Recently, the Zhao group made an interesting breakthrough in the diastereoselective synthesis of oligopeptides by using a combination of synergistic photoredox, cobaloxime, and organophosphorus triple catalysis. This process creates a phosphine radical cation that engages in nucleophilic addition with a carboxylate, leading to the formation of the phosphoranyl radical (118-i). Further electron transfer between 118-i and a cobaloxime catalyst (CoIII) is expected to occur easily, leading to the formation of an oligopeptide 120via aminolysis, while also producing a phosphine oxide. Finally, the in situ reduction of PV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O by silanes and InBr3 completes the catalytic cycle (Scheme 44).100b
O by silanes and InBr3 completes the catalytic cycle (Scheme 44).100b
 | ||
| Scheme 44 Diastereoselective oligopeptide synthesis. | ||
Miscellaneous
Baoguo Zhao and his colleagues very recently released a study detailing how 1,8-diaza-fluoren-9-one (DFO) was able to activate the normally inactive α-C–H bond of an unprotected primary amine by increasing its acidity by an impressive 1044 times.By harnessing the excellent cooperative nature of Diazafluorenone with an Ir-catalyst, successfully crafted the chiral homoallylic amine 123 building block. This was achieved through an asymmetric allylic substitution, followed by in situ 2-aza-cope rearrangement using primary alkyl amine 121 and allylic carbonates 122 in the presence of ZnBr2 and DBU in THF/H2O solvent at room temperature. The reaction began with the condensation of alkyl amine 121 with diazafluorenone, resulting in the formation of imine 121-i. Following this, the delocalized carbanion 121-ii underwent an asymmetric addition to π-allyl-Ir species 122-i, generating a branched aza cope intermediate 121-iiiviaTS′. Species 122-i is generated from the iridium catalyst and allylic carbonate 122. Subsequently, compound 121-iii undergoes 2-aza-Cope rearrangement to form species 121-iv, followed by hydrolysis, leading to the production of α-C–H allylic alkylation product 123 and the regeneration of the diazafluorenone (Scheme 45).101
 | ||
| Scheme 45 Chiral homoallylic amine synthesis by keto-iridium dual catalysis. | ||
A straightforward and effective method for the asymmetric reduction of the C–C double bond to produce a highly enantioselective product in the presence of photocatalysis and enzymatic catalysis was established by Hartwig in 2018. The reaction is driven by a combination of photocatalysis and enzymatic catalysis, resulting in higher yield and enantiomeric excess. Utilizing blue light and a series of synergistic chemoenzymatic reactions, the reduction of the E/Z mixture of alkene 124 to the highly enantiopure product 125 is achieved. This process involves an iridium photocatalyst combined with an ene-reductase enzyme, resulting in the desired 125 products with high yield and high enantioselectivity (Scheme 46).102
 | ||
| Scheme 46 Asymmetric reduction by combining photocatalysis and enzymatic catalysis. | ||
Direct conversion of secondary amides 126 to chiral α-amino-nitriles 127 and α-aminophosphonates 128 can be accomplished through a one-pot process involving enantioselective reductive cyanation and phosphonylation. This approach utilizes a combination of iridium and chiral thiourea catalysis. During the catalytic cycle, the amides 126 undergo reduction in the iridium-catalyzed step, leading to the formation of imine intermediates. These intermediates are subsequently transformed into chiral α-functionalized amines through the action of the thiourea catalyst (Scheme 47).103
 | ||
| Scheme 47 Asymmetric reductive cyanation and phosphonylation of secondary amides. | ||
In 2022, Huang reported a novel method for the catalytic asymmetric reductive/deoxygenative alkynylation of secondary amides. This approach employs a multi-catalysis strategy, combining iridium and copper relay catalysis with cooperative catalysis from Cu and N-protected L-proline. The process involves three catalytic cycles: first, the Ir-catalyzed hydrosilylation of a secondary amide 129 using diethylsilane leads to the formation of an O-silyl hemi-aminal intermediate (129-i). This intermediate then undergoes a transformation by eliminating diethylsilanol, resulting in an imine intermediate (129-ii). Next, copper catalyzes the in situ formation of a nucleophilic Cu-alkynylide species (130-ii). Finally, the reaction proceeds with the N-Boc L-proline-catalyzed asymmetric alkynylation of the imine. Here, N-Boc L-proline acts both as a Brønsted acid, activating the relatively unreactive imine via hydrogen bonding, and as an asymmetric inducer, selectively blocking the Re-face approach. Consequently, the Cu-alkynylide (130-ii) preferentially adds to the reactive intermediate (TS) from the Si-face, yielding (R)-propargylamine 131 (Scheme 48).104
 | ||
| Scheme 48 Enantioselective reductive alkynylation of secondary amides. | ||
6. Conclusions
The development of a cooperative catalysis strategy using organocatalysts and transition metals in combination is heavily reliant on catalyst compatibility and the balanced kinetics of selective substrate activation in the reaction system. The coupling of metal and organocatalysis creates new reactivity and diverse applications for catalytic systems in the pharmaceutical, agrochemical, and fine chemical industries. Cooperative catalysis enables difficult chemical reactions that were not possible with either the organocatalyst or the metal catalyst alone.In the realm of asymmetric dual catalysis, chirality arises from either the ligand on the metal or from the chiral counter ion of an organic catalyst. Even though cooperative catalysis has become a highly effective approach for achieving high selective and complex functionality, however, it does have some synthetic limitations to consider. Issues often arise when iridium and organocatalysts disrupt the properties of each other. This incompatibility can compromise the effectiveness of the catalytic cycle, so it's essential to choose catalysts carefully to ensure a successful dual catalytic system. To make significant strides in cooperative catalysis, a deeper understanding of how catalysts interact both mechanistically and theoretically is crucial. Another notable limitation relates to organocatalysts, which have been used in conjunction with iridium metal. Specifically, various types of organocatalysts have been explored in cooperative catalysis along with iridium, including BINOL phosphoric acid-based catalysts, different Lewis bases like NHC, tertiary amines, and proline-based catalysts, as well as a few phase transfer catalysts. This dependency on delicate and costly organocatalysts underscores the importance of seeking alternatives that are more economical, readily accessible, and robust, in order to enhance the versatility of this process. The current obstacles in iridium–organo cooperative catalysis are sure to spark new innovations, paving the way for the development of novel synergistic catalytic systems.
This review offers a thorough examination of advanced strategies and foundational research in iridium–organo cooperative catalysis. Its goal is to provide a comprehensive insight into innovative techniques and potential pathways for synthesizing crucial pharmaceutical compounds, while also inspiring new discoveries and promoting the progress of novel cooperative catalytic processes.
Author contributions
D. C. and R. K. S. contributed to the writing and editing of the manuscript. All authors have given their approval for the final version.Data availability
No original research findings or new data were produced or examined in the course of this review.Conflicts of interest
There are no conflicts to report.Acknowledgements
We acknowledge financial support from the Science and Engineering Research Board (SERB), India (SRG/2023/000581), and the IIT Roorkee FIG-101034 grant. Divakar Chaudhary is thankful to the PMRF Foundation for the fellowship. We thank Sumit Kumar for his help in the literature search.References
- U. Bin Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S. G. Lee, Synergistic Dual Transition Metal Catalysis, Chem. Rev., 2020, 120, 13382–13433 CrossRef.
- (a) Z. Shao and H. Zhang, Combining transition metal catalysis and organocatalysis: A broad new concept for catalysis, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC; (b) B. A. Arndtsen and L. Z. Gong, Asymmetric Organocatalysis Combined with Metal Catalysis, Springer International Publishing, 2020 CrossRef.
- (a) D. S. Kim, W. J. Park and C. H. Jun, Metal-Organic Cooperative Catalysis in C-H and C-C Bond Activation, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS; (b) R. Peters, Cooperative Catalysis. Designing Efficient Catalysts for Synthesis, Wiley-VCH, Weinheim, 2015, pp. 1–427 CrossRef.
- J. Park and S. Hong, Cooperative bimetallic catalysis in asymmetric transformations, Chem. Soc. Rev., 2012, 41, 6931–6943 RSC.
- A. Sinibaldi, V. Nori, A. Baschieri, F. Fini, A. Arcadi and A. Carlone, Organocatalysis and beyond: Activating reactions with two catalytic species, Catalysts, 2019, 9, 928 CrossRef CAS.
- C. D. T. Nielsen, J. D. Linfoot, A. F. Williams and A. C. Spivey, Recent progress in asymmetric synergistic catalysis – the judicious combination of selected chiral aminocatalysts with achiral metal catalysts, Org. Biomol. Chem., 2022, 20, 2764–2778 RSC.
- X. Liang, T. Y. Zhang, X. Y. Zeng, Y. Zheng, K. Wei and Y. R. Yang, Ir-Catalyzed Asymmetric Total Synthesis of (−)-Communesin F, J. Am. Chem. Soc., 2017, 139, 3364–3367 CrossRef CAS.
- S. Afewerki and A. Córdova, Combinations of Aminocatalysts and Metal Catalysts: A Powerful Cooperative Approach in Selective Organic Synthesis, Chem. Rev., 2016, 116, 13512–13570 CrossRef CAS PubMed.
- Y. Kuang, J. Lai and J. P. Reid, Transferrable selectivity profiles enable prediction in synergistic catalyst space, Chem. Sci., 2023, 14, 1885–1895 RSC.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS.
- A. E. Allen and D. W. C. MacMillan, Synergistic catalysis: A powerful synthetic strategy for new reaction development, Chem. Sci., 2012, 3, 633–658 RSC.
- A. Del Vecchio, A. Sinibaldi, V. Nori, G. Giorgianni, G. Di Carmine and F. Pesciaioli, Synergistic Strategies in Aminocatalysis, Chem. – Eur. J., 2022, 28, e202200818 CrossRef CAS PubMed.
- Y. Qin, L. Zhu and S. Luo, Organocatalysis in Inert C-H Bond Functionalization, Chem. Rev., 2017, 117, 9433–9520 CrossRef CAS.
- R. Parella, S. Jakkampudi and J. C. G. Zhao, Recent Applications of Asymmetric Organocatalytic Methods in Total Synthesis, ChemistrySelect, 2021, 6, 2252–2280 CrossRef CAS.
- R. Mitra and J. Niemeyer, Dual Brønsted-acid Organocatalysis: Cooperative Asymmetric Catalysis with Combined Phosphoric and Carboxylic Acids, ChemCatChem, 2018, 10, 1221–1234 CrossRef CAS.
- D. F. Chen, Z. Y. Han, X. Le Zhou and L. Z. Gong, Asymmetric organocatalysis combined with metal catalysis: Concept, proof of concept, and beyond, Acc. Chem. Res., 2014, 47, 2365–2377 CrossRef CAS PubMed.
- J. Becica and G. E. Dobereiner, The roles of Lewis acidic additives in organotransition metal catalysis, Org. Biomol. Chem., 2019, 17, 2055–2069 RSC.
- X. Liang, S. Z. Jiang, K. Wei and Y. R. Yang, Enantioselective Total Synthesis of (-)-Alstoscholarisine A, J. Am. Chem. Soc., 2016, 138, 2560–2562 CrossRef CAS PubMed.
- S. Zhou, H. Chen, Y. Luo, W. Zhang and A. Li, Asymmetric total synthesis of mycoleptodiscin A, Angew. Chem., Int. Ed., 2015, 54, 6878–6882 CrossRef CAS.
- S. Z. Jiang, X. Y. Zeng, X. Liang, T. Lei, K. Wei and Y. R. Yang, Iridium-Catalyzed Enantioselective Indole Cyclization: Application to the Total Synthesis and Absolute Stereochemical Assignment of (-)-Aspidophylline A, Angew. Chem., Int. Ed., 2016, 55, 4044–4048 CrossRef CAS.
- C. C. Malakar, L. Dell'Amico and W. Zhang, Dual Catalysis in Organic Synthesis: Current Challenges and New Trends, Eur. J. Org. Chem., 2023, 26, e202201114 CrossRef CAS.
- N. Chakraborty, B. Das, K. K. Rajbongshi and B. K. Patel, Combined Power of Organo- and Transition Metal Catalysis in Organic Synthesis, Eur. J. Org. Chem., 2022, 2022, e202200273 CrossRef CAS.
- C. Zhong and X. Shi, When organocatalysis meets transition-metal catalysis, Eur. J. Org. Chem., 2010, 2999–3025 CrossRef CAS.
- J. M. Lee, Y. Na, H. Han and S. Chang, Cooperative multi-catalyst systems for one-pot organic transformations, Chem. Soc. Rev., 2004, 33, 302–312 RSC.
- S. H. Xiang and B. Tan, Advances in asymmetric organocatalysis over the last 10 years, Nat. Commun., 2020, 11, 3786 CrossRef CAS PubMed.
- E. Weis, Iridium-Catalyzed C–H Activation Methods for Late-Stage Functionalization of Pharmaceuticals, University, Stockholm, 2022 Search PubMed.
- C. Min and D. Seidel, Asymmetric Brønsted acid catalysis with chiral carboxylic acids, Chem. Soc. Rev., 2017, 46, 5889–5902 RSC.
- M. H. Aukland and B. List, Organocatalysis emerging as a technology, Pure Appl. Chem., 2021, 93, 1371–1381 CrossRef CAS.
- T. Akiyama, J. Itoh and K. Fuchibe, Recent progress in chiral Brønsted acid catalysis, Adv. Synth. Catal., 2006, 348, 999–1010 CrossRef CAS.
- A. Zamfir, S. Schenker, M. Freund and S. B. Tsogoeva, Chiral BINOL-derived phosphoric acids: Privileged Brønsted acid organocatalysts for C-C bond formation reactions, Org. Biomol. Chem., 2010, 8, 5262–5276 CAS.
- M. Rueping, R. M. Koenigs and I. Atodiresei, Unifying metal and Brønsted acid catalysis-concepts, mechanisms, and classifications, Chem. – Eur. J., 2010, 16, 9350–9365 CrossRef CAS PubMed.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS.
- C. Li, C. Wang, B. Villa-Marcos and J. Xiao, Chiral counteranion-aided asymmetric hydrogenation of acyclic imines, J. Am. Chem. Soc., 2008, 130, 14450–14451 CrossRef CAS PubMed.
- C. Li, B. Villa-Marcos and J. Xiao, Metal-bronsted acid cooperative catalysis for asymmetric reductive amination, J. Am. Chem. Soc., 2009, 131, 6967–6969 CrossRef CAS PubMed.
- W. Tang, S. Johnston, C. Li, J. A. Iggo, J. Bacsa and J. Xiao, Cooperative catalysis: Combining an achiral metal catalyst with a chiral Brønsted acid enables highly enantioselective hydrogenation of imines, Chem. – Eur. J., 2013, 19, 14187–14193 CrossRef CAS.
- M. Roggen and E. M. Carreira, Enantioselective allylic etherification: Selective coupling of two unactivated alcohols, Angew. Chem., Int. Ed., 2011, 50, 5568–5571 CrossRef CAS PubMed.
- Y. Zhang, C. Lim, D. S. B. Sim, H. Pan and Y. Zhao, Catalytic Enantioselective Amination of Alcohols by the Use of Borrowing Hydrogen Methodology: Cooperative Catalysis by Iridium and a Chiral Phosphoric Acid, Angew. Chem., Int. Ed., 2014, 53, 1399–1403 CrossRef CAS PubMed.
- Z. Q. Rong, Y. Zhang, R. H. B. Chua, H. J. Pan and Y. Zhao, Dynamic kinetic asymmetric amination of alcohols: From a mixture of four isomers to diastereo- and enantiopure α-branched amines, J. Am. Chem. Soc., 2015, 137, 4944–4947 CrossRef CAS.
- C. S. Lim, T. T. Quach and Y. Zhao, Enantioselective Synthesis of Tetrahydroquinolines by Borrowing Hydrogen Methodology: Cooperative Catalysis by an Achiral Iridacycle and a Chiral Phosphoric Acid, Angew. Chem., Int. Ed., 2017, 56, 7176–7180 CrossRef CAS.
- X. Q. Ng, C. S. Lim, M. W. Liaw, T. T. Quach, B. M. Yang, V. Isoni, J. Wu and Y. Zhao, Direct access to chiral aliphatic amines by catalytic enantioconvergent redox-neutral amination of alcohols, Nat. Synth., 2023, 2, 572–580 CrossRef CAS.
- Y. Liu, R. Tao, Z. K. Lin, G. Yang and Y. Zhao, Redox-enabled direct stereoconvergent heteroarylation of simple alcohols, Nat. Commun., 2021, 12, 5035 CrossRef CAS PubMed.
- D. Shen, Q. Chen, P. Yan, X. Zeng and G. Zhong, Enantioselective Dearomatization of Naphthol Derivatives with Allylic Alcohols by Cooperative Iridium and Brønsted Acid Catalysis, Angew. Chem., Int. Ed., 2017, 56, 3242–3246 CrossRef CAS.
- M. Sun, X. Wan, S. J. Zhou, G. J. Mei and F. Shi, Iridium and a Brønsted acid cooperatively catalyzed chemodivergent and stereoselective reactions of vinyl benzoxazinones with azlactones, Chem. Commun., 2019, 55, 1283–1286 RSC.
- X. Q. Xie, X. Li and P. N. Liu, Enantioselective synthesis of spiro-N,O-ketals via iridium and Brønsted acid co-catalyzed asymmetric formal [4 + 2] cycloaddition, Chem. Commun., 2024, 60, 1448–1451 RSC.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- C. De Risi, A. Brandolese, G. Di Carmine, D. Ragno, A. Massi and O. Bortolini, Oxidative N-Heterocyclic Carbene Catalysis, Chem. – Eur. J., 2023, 29, e202202467 CrossRef CAS PubMed.
- G. J. Knox, L. S. Hutchings-Goetz, C. M. Pearson and T. N. Snaddon, Tertiary Amine Lewis Base Catalysis in Combination with Transition Metal Catalysis, Springer International Publishing, 2020, vol. 378 Search PubMed.
- K. Liu, I. Khan, J. Cheng, Y. J. Hsueh and Y. J. Zhang, Asymmetric Decarboxylative Cycloaddition of Vinylethylene Carbonates with β-Nitroolefins by Cooperative Catalysis of Palladium Complex and Squaramide, ACS Catal., 2018, 8, 11600–11604 CrossRef CAS.
- S. E. Denmark and G. L. Beutner, Lewis base catalysis in organic synthesis, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS.
- I. Ibrahem and A. Córdova, Direct catalytic intermolecular α-allylic alkylation of aldehydes by combination of transition-metal and organocatalysis, Angew. Chem., Int. Ed., 2006, 45, 1952–1956 CrossRef CAS.
- M. H. Wang and K. A. Scheidt, Cooperative Catalysis and Activation with N-Heterocyclic Carbenes, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS.
- K. Nagao and H. Ohmiya, N-Heterocyclic Carbene (NHC)/Metal Cooperative Catalysis, Top. Curr. Chem., 2019, 377, 1–15 CrossRef CAS.
- Q. Jia, Y. Li, Y. Lin and Q. Ren, The combination of lewis acid with N-heterocyclic carbene (NHC) catalysis, Catalysts, 2019, 9, 863–886 CrossRef CAS.
- T. Kurihara, M. Kojima, T. Yoshino and S. Matsunaga, Achiral Cp*Rh(III)/Chiral Lewis Base Cooperative Catalysis for Enantioselective Cyclization via C-H Activation, J. Am. Chem. Soc., 2022, 144, 7058–7065 CrossRef CAS PubMed.
- J. Xiao, Merging organocatalysis with transition metal catalysis: Highly stereoselective α-alkylation of aldehydes, Org. Lett., 2012, 14, 1716–1719 CrossRef CAS PubMed.
- S. Krautwald, M. A. Schafroth, D. Sarlah and E. M. Carreira, Stereodivergent α-allylation of linear aldehydes with dual iridium and amine catalysis, J. Am. Chem. Soc., 2014, 136, 3020–3023 CrossRef CAS PubMed.
- T. Sandmeier, S. Krautwald, H. F. Zipfel and E. M. Carreira, Stereodivergent Dual Catalytic α–Allylation of Protected α–Amino– and α–Hydroxyacetaldehydes, Angew. Chem., Int. Ed., 2015, 54, 14363–14367 CrossRef CAS.
- Y. Deng, X. Liang, K. Wei and Y. R. Yang, Ir-Catalyzed Asymmetric Total Syntheses of Bisdehydrotuberostemonine D, Putative Bisdehydrotuberostemonine e and Structural Revision of the Latter, J. Am. Chem. Soc., 2021, 143, 20622–20627 CrossRef CAS.
- B. L. Wu, J. N. Yao, X. X. Long, Z. Q. Tan, X. Liang, L. Feng, K. Wei and Y. R. Yang, Enantioselective Total Synthesis of (−)-Daphenylline, J. Am. Chem. Soc., 2024, 146, 1262–1268 CrossRef CAS.
- L. Næsborg, K. S. Halskov, F. Tur, S. M. N. Mønsted and K. A. Jørgensen, Asymmetric γ-Allylation of α,β-Unsaturated Aldehydes by Combined Organocatalysis and Transition-Metal Catalysis, Angew. Chem., Int. Ed., 2015, 54, 10193–10197 CrossRef.
- M. Li, X. Guo, W. Jin, Q. Zheng, S. Liu and W. Hu, An enantioselective three-component reaction of diazoacetates with indoles and enals by iridium/iminium co-catalysis, Chem. Commun., 2016, 52, 2736–2739 RSC.
- M. Li, X. Guo, Q. Zheng, W. Hu and S. Liu, Enantioselective Multicomponent Reaction for Rapid Construction of 1,2,5-Triol Derivatives with Vicinal Chiral Centers, J. Org. Chem., 2017, 82, 5212–5221 CrossRef CAS.
- S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Enantio- and diastereodivergent dual catalysis: α-Allylation of branched aldehydes, Science, 2013, 340, 1065–1068 CrossRef CAS PubMed.
- M. M. Zhang, Y. N. Wang, B. C. Wang, X. W. Chen, L. Q. Lu and W. J. Xiao, Synergetic iridium and amine catalysis enables asymmetric [4 + 2] cycloadditions of vinyl aminoalcohols with carbonyls, Nat. Commun., 2019, 10, 2716 CrossRef PubMed.
- X. Zhang, S. Li, W. Yu, Y. Xie, C. H. Tung and Z. Xu, Asymmetric Azide-Alkyne Cycloaddition with Ir(I)/Squaramide Cooperative Catalysis: Atroposelective Synthesis of Axially Chiral Aryltriazoles, J. Am. Chem. Soc., 2022, 144, 6200–6207 CrossRef CAS.
- Y. Chen, H. Yan, H. Zheng, W. P. Deng, Z. Li and W. L. Yang, Ir/Brønsted acid dual-catalyzed asymmetric synthesis of bisbenzannulated spiroketals and spiroaminals from isochroman ketals, Org. Chem. Front., 2024, 11, 5831–5840 RSC.
- Z. Chen, Z. Chen, Z. Yang, L. Guo, W. Du and Y. Chen, Cooperative Tertiary Amine/Chiral Iridium Complex Catalyzed Asymmetric [4 + 3] and [3 + 3] Annulation Reactions, Angew. Chem., Int. Ed., 2019, 58, 15021–15025 CrossRef CAS.
- R. Sarkar and S. Mukherjee, Erratum: Iridium-catalyzed enantioselective olefinic C(sp2)-H allylic alkylation, Chem. Sci., 2021, 12, 3076 RSC.
- X. Jiang, J. J. Beiger and J. F. Hartwig, Stereodivergent allylic substitutions with aryl acetic acid esters by synergistic iridium and lewis base catalysis, J. Am. Chem. Soc., 2017, 139, 87–90 CrossRef CAS.
- C. M. Pearson, J. W. B. Fyfe and T. N. Snaddon, A Regio– and Stereodivergent Synthesis of Homoallylic Amines by a One–Pot Cooperative–Catalysis–Based Allylic Alkylation/Hofmann Rearrangement Strategy, Angew. Chem., Int. Ed., 2019, 58, 10521–10527 CrossRef CAS.
- F. Tian, W. L. Yang, T. Ni, J. Zhang and W. P. Deng, Catalytic asymmetric dipolar cycloadditions of indolyl delocalized metal-allyl species for the enantioselective synthesis of cyclopenta [b]indoles and pyrrolo[1,2-a]indoles, Sci. China: Chem., 2021, 64, 34–40 CrossRef CAS.
- Q. Wang, T. Fan and J. Song, Cooperative Isothiourea/Iridium-Catalyzed Asymmetric Annulation Reactions of Vinyl Aziridines with Pentafluorophenyl Esters, Org. Lett., 2023, 25, 1246–1251 CrossRef CAS PubMed.
- B. Bhaskararao and R. B. Sunoj, Origin of Stereodivergence in Cooperative Asymmetric Catalysis with Simultaneous Involvement of Two Chiral Catalysts, J. Am. Chem. Soc., 2015, 137, 15712–15722 CrossRef CAS PubMed.
- S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Diastereodivergent synthesis of enantioenriched α,β-disubstituted γ-butyrolactones via cooperative N-heterocyclic carbene and Ir catalysis, Nat. Catal., 2020, 3, 48–54 CrossRef CAS.
- J. Zhang, Y. S. Gao, B. M. Gu, W. L. Yang, B. X. Tian and W. P. Deng, Cooperative N-heterocyclic Carbene and Iridium Catalysis Enables Stereoselective and Regiodivergent [3 + 2] and [3 + 3] Annulation Reactions, ACS Catal., 2021, 11, 3810–3821 CrossRef CAS.
- C. Sun, X. Qi, X. L. Min, X. D. Bai, P. Liu and Y. He, Asymmetric allylic substitution-isomerization to axially chiral enamides: Via hydrogen-bonding assisted central-to-axial chirality transfer, Chem. Sci., 2020, 11, 10119–10126 RSC.
- T. Hashimoto and K. Maruoka, Recent development and application of chiral phase-transfer catalysts, Chem. Rev., 2007, 107, 5656–5682 CrossRef CAS.
- S. Shirakawa and K. Maruoka, Recent developments in asymmetric phase-transfer reactions, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS.
- M. Nakoji, T. Kanayama, T. Okino and Y. Takemoto, Chiral phosphine-free Pd-mediated asymmetric allylation of prochiral enolate with a chiral phase-transfer catalyst, Org. Lett., 2001, 3, 3329–3331 CrossRef CAS.
- M. Nakoji, T. Kanayama, T. Okino and Y. Takemoto, Pd-catalyzed asymmetric allylic alkylation of glycine imino ester using a chiral phase-transfer catalyst, J. Org. Chem., 2002, 67, 7418–7423 CrossRef CAS PubMed.
- Y. L. Su, Y. H. Li, Y. G. Chen and Z. Y. Han, Ir/PTC cooperatively catalyzed asymmetric umpolung allylation of α-imino ester enabled synthesis of α-quaternary amino acid derivatives bearing two vicinal stereocenters, Chem. Commun., 2017, 53, 1985–1988 RSC.
- L. Wei, L. Xiao, Z. F. Wang, H. Y. Tao and C. J. Wang, Ir/Phase-Transfer-Catalysis Cooperatively Catalyzed Asymmetric Cascade Allylation/2-aza-Cope Rearrangement: An Efficient Route to Homoallylic Amines from Aldimine Esters, Chin. J. Chem., 2020, 38, 82–86 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- J. H. Shen, M. Shi and Y. Wei, Synergistic Visible Light Photocatalysis with Organocatalysis, Chem. – Eur. J., 2023, 29, e202301157 CrossRef CAS.
- W. Yao, E. A. Bazan-Bergamino and M. Y. Ngai, Asymmetric Photocatalysis Enabled by Chiral Organocatalysts, ChemCatChem, 2022, 14, e202101292 CrossRef CAS PubMed.
- Y. Y. Liu, J. Liu, L. Q. Lu and W. J. Xiao, Organocatalysis Combined with Photocatalysis, Top. Curr. Chem., 2019, 377, 1–15 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, Photoredox Catalysis in Organic Chemistry, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, Efficient Yellow Electroluminescence from a Single Layer of a Cyclometalated Iridium Complex, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS PubMed.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, Enantioselective α-trifluoromethylation of aldehydes via photoredox organocatalysis, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed.
- H. W. Shih, M. N. Vander Wal, R. L. Grange and D. W. C. MacMillan, Enantioselective α-benzylation of aldehydes via photoredox organocatalysis, J. Am. Chem. Soc., 2010, 132, 13600–13603 CrossRef CAS.
- L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, Enantioselective photoredox catalysis enabled by proton-coupled electron transfer: Development of an asymmetric aza-pinacol cyclization, J. Am. Chem. Soc., 2013, 135, 17735–17738 CrossRef CAS PubMed.
- R. S. J. Proctor, H. J. Davis and R. J. Phipps, Catalytic enantioselective Minisci-type addition to heteroarenes, Science, 2018, 360, 419–422 CrossRef CAS PubMed.
- Y. Cheng, Q. Zhao, X. Zhang and S. You, Asymmetric Dearomatization of Indole Derivatives with N–Hydroxycarbamates Enabled by Photoredox Catalysis, Angew. Chem., Int. Ed, 2019, 58, 18069–18074 CrossRef CAS.
- S. J. Chapman, W. B. Swords, C. M. Le, I. A. Guzei, F. D. Toste and T. P. Yoon, Cooperative Stereoinduction in Asymmetric Photocatalysis, J. Am. Chem. Soc., 2022, 144, 4206–4213 CrossRef CAS PubMed.
- J. Wei, Y. Tang, Q. Yang, H. Li, D. He and Y. Cai, Asymmetric Ketoalkylation/Rearrangement of Alkyenlfurans via Synergistic Photoredox/Brønsted Acid Catalysis, Org. Lett., 2022, 24, 7928–7933 CrossRef CAS PubMed.
- C. Che, Y. N. Lu and C. J. Wang, Enantio- and Diastereoselective De Novo Synthesis of 3-Substituted Proline Derivatives via Cooperative Photoredox/Brønsted Acid Catalysis and Epimerization, J. Am. Chem. Soc., 2023, 145, 2779–2786 CrossRef CAS PubMed.
- D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines, J. Am. Chem. Soc., 2015, 137, 13768–13771 CrossRef CAS PubMed.
- S. M. Cho, J. Y. Kim, S. Han and D. H. Ryu, Visible Light-Mediated Enantioselective Addition of α-Aminoalkyl Radicals to Ketones Catalyzed by Chiral Oxazaborolidinium Ion, J. Org. Chem., 2022, 87, 11196–11203 CrossRef CAS.
- (a) X. Yu, Q. Y. Meng, C. G. Daniliuc and A. Studer, Aroyl Fluorides as Bifunctional Reagents for Dearomatizing Fluoroaroylation of Benzofurans, J. Am. Chem. Soc., 2022, 144, 7072–7079 CrossRef CAS; (b) J. Su, J.-N. Mo, G. Zhang, Z. Jiang and J. Zhao, A practical approach for oligopeptide synthesis via synergistic photoredox, cobaloxime and organophosphorus triple catalysis, Org. Chem. Front., 2023, 10, 4895–4904 RSC.
- J. Li, S. Gong, S. Gao, J. Chen, W. W. Chen and B. Zhao, Asymmetric α-C(sp3)–H allylic alkylation of primary alkylamines by synergistic Ir/ketone catalysis, Nat. Commun., 2024, 15, 939 CrossRef CAS.
- Z. C. Litman, Y. Wang, H. Zhao and J. F. Hartwig, Cooperative asymmetric reactions combining photocatalysis and enzymatic catalysis, Nature, 2018, 560, 355–359 CrossRef CAS.
- D. H. Chen, W. T. Sun, C. J. Zhu, G. S. Lu, D. P. Wu, A. E. Wang and P. Q. Huang, Enantioselective Reductive Cyanation and Phosphonylation of Secondary Amides by Iridium and Chiral Thiourea Sequential Catalysis, Angew. Chem., Int. Ed., 2021, 60, 8827–8831 CrossRef CAS PubMed.
- H. Chen, Z. Z. Wu, D. Y. Shao and P. Q. Huang, Multicatalysis protocol enables direct and versatile enantioselective reductive transformations of secondary amides, Sci. Adv., 2022, 8, eade3431 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2025 |