
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of large language models and autonomous agents in chemistry
Mayk Caldas
Ramos
 ab,
Christopher J.
Collison
c and
Andrew D.
White
*ab
ab,
Christopher J.
Collison
c and
Andrew D.
White
*ab
aFutureHouse Inc., San Francisco, CA, USA. E-mail: andrew@futurehouse.org
bDepartment of Chemical Engineering, University of Rochester, Rochester, NY, USA. E-mail: mcaldasr@ur.rochester.edu
cSchool of Chemistry and Materials Science, Rochester Institute of Technology, Rochester, NY, USA. E-mail: cjcscha@rit.edu
First published on 9th December 2024
Abstract
Large language models (LLMs) have emerged as powerful tools in chemistry, significantly impacting molecule design, property prediction, and synthesis optimization. This review highlights LLM capabilities in these domains and their potential to accelerate scientific discovery through automation. We also review LLM-based autonomous agents: LLMs with a broader set of tools to interact with their surrounding environment. These agents perform diverse tasks such as paper scraping, interfacing with automated laboratories, and synthesis planning. As agents are an emerging topic, we extend the scope of our review of agents beyond chemistry and discuss across any scientific domains. This review covers the recent history, current capabilities, and design of LLMs and autonomous agents, addressing specific challenges, opportunities, and future directions in chemistry. Key challenges include data quality and integration, model interpretability, and the need for standard benchmarks, while future directions point towards more sophisticated multi-modal agents and enhanced collaboration between agents and experimental methods. Due to the quick pace of this field, a repository has been built to keep track of the latest studies: https://github.com/ur-whitelab/LLMs-in-science.
1 Introduction
The integration of Machine Learning (ML) and Artificial Intelligence (AI) into chemistry has spanned several decades.1–10 Although applications of computational methods in quantum chemistry and molecular modeling from the 1950s–1970s were not considered AI, they laid the groundwork. Subsequently in the 1980s expert systems like DENDRAL11,12 were expanded to infer molecular structures from mass spectrometry data.13 At the same time, Quantitative Structure–Activity Relationship (QSAR) Models were developed5 that would use statistical methods to predict the effects of chemical structure on activity.14–17 In the 1990s, neural networks, and associated Kohonen Self-Organizing Maps were introduced to domains such as drug design,18,19 as summarized well by Yang et al.5 and Goldman and Walters,20 although they were limited by the computational resources of the time. With an explosion of data from High-Throughput Screening (HTS),21,22 models then started to benefit from vast datasets of molecular structures and their biological activities. Furthermore, ML algorithms such as Support Vector Machines and Random Forests became popular for classification and regression tasks in cheminformatics,1 offering improved performance over traditional statistical methods.23Deep learning transformed the landscape of ML in chemistry and materials science in the 2010s.24 Recurrent Neural Networks (RNNs),25–29 Convolutional Neural Networks (CNNs)30–32 and later, Graph Neural Networks (GNNs),33–38 made great gains in their application to molecular property prediction, drug discovery,39 and synthesis prediction.40 Such methods were able to capture complex patterns in data, and therefore enabled the identification of novel materials for high-impact needs such as energy storage and conversion.41,42
In this review, we explore the next phase of AI in chemistry, namely the use of Large Language Models (LLMs) and autonomous agents. Inspired by successes in natural language processing (NLP), LLMs were adapted for chemical language (e.g., Simplified Molecular Input Line Entry System (SMILES)43) to tackle tasks from synthesis prediction to molecule generation.44–46 We will then explore the integration of LLMs into autonomous agents as illustrated by M. Bran et al.47 and Boiko et al.,48 which may be used for data interpretation or, for example, to experiment with robotic systems. We are at a crossroads where AI enables chemists to solve major global problems faster and streamline routine lab tasks. This enables, for instance, the development of larger, consistent experimental datasets and shorter lead times for drug and material commercialization. As such, language has been the preferred mechanism for describing and disseminating research results and protocols in chemistry for hundreds of years.49
1.1 Challenges in chemistry
We categorize some key challenges that can be addressed by AI in chemistry as: property prediction, property-directed molecule generation, and synthesis prediction. These categories, as illustrated in Fig. 1 can be connected to a fourth challenge in automation. The first task is to predict a property for a given compound to decide if it should be synthesized for a specific application, such as an indicator,50 light harvester,51 or catalyst.52 To achieve better models for property prediction, high-quality data is crucial. We discuss the caveats and issues with the current datasets in Section 3.1 and illustrate state-of-the-art findings in Section 3.2.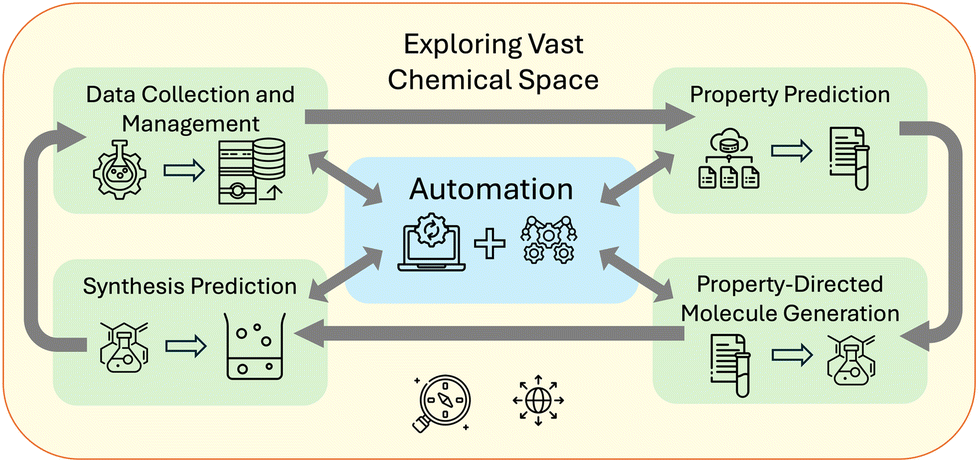 | ||
| Fig. 1 AI-powered LLMs accelerate chemical discovery with models that address key challenges in property prediction, property directed molecule generation, and synthesis prediction. Autonomous agents connect these models and additional tools thereby enabling rapid exploration of vast chemical spaces. | ||
The second task is to generate novel chemical structures that meet desired chemical profiles or exhibit properties.53 Success in this area would accelerate progress in various chemical applications, but reliable reverse engineering (inverse design)54 is not yet feasible over the vast chemical space.55 For instance, inverse design, when coupled with automatic selection of novel structures (de novo molecular design) could lead to the development of drugs targeting specific proteins while retaining properties like solubility, toxicity, and blood–brain barrier permeability.56 The complexity of connecting de novo design with property prediction is high and we show how state-of-the-art models currently perform in Section 3.3.
Once a potential target molecule has been identified, the next challenge is predicting its optimal synthesis using inexpensive, readily available, and non-toxic starting materials. In a vast chemical space, there will always be an alternative molecule “B” that has similar properties to molecule “A” but is easier to synthesize. Exploring this space to find a new molecule with the right properties and a high-yield synthesis route brings together these challenges. The number of possible stable chemicals is estimated to be up to 10180.57–60 Exploring this vast space requires significant acceleration beyond current methods.61 As Restrepo57 emphasizes, cataloguing failed syntheses is essential to building a comprehensive dataset of chemical features. Autonomous chemical resources can accelerate database growth and tackle this challenge. Thus, automation is considered a fourth major task in chemistry.62–65 The following discussion explores how LLMs and autonomous agents can provide the most value. Relevant papers are discussed in Section 3.4.
This review is organized within the context of these categories. The structure of the review is as follows. Section 2 provides an introduction to transformers, including a brief description of encoder-only, decoder-only and encoder–decoder architectures. Section 3 provides a detailed survey of work with LLMs, where we connect each transformer architecture to the areas of chemistry that it is best suited to support. We then progress into a description of autonomous agents in Section 4, and a survey of how such LLM-based agents are finding application in chemistry-centered scientific research, Section 5. After providing some perspective on future challenges and opportunities in Section 6, and we conclude in Section 7. We distinguish between “text-based” and “mol-based” inputs and outputs, with “text” referring to natural language and “mol” referring to the chemical syntax for material structures, as introduced by Zhang et al.66
2 Large language models
The prior state-of-the-art for sequence-to-sequence (seq2seq) tasks had been the Recurrent Neural Network (RNN),67 typically as implemented by Hochreiter and Schmidhuber.68 In a seq2seq task, an input sequence, such as a paragraph in English, is processed to generate a corresponding output sequence, such as a translation into French. The RNN retains “memory” of previous steps in a sequence to predict later parts. However, as sequence length increases, gradients can become vanishingly small or explosively large,69,70 preventing effective use of earlier information in long sequences. Due to these limitations, RNNs have thus fallen behind Large Language Models (LLMs), which primarily implement transformer architectures, introduced by Vaswani et al.71 LLMs are deep neural networks (NN) characterized by their vast number of parameters and, though transformers dominate, other architectures for handling longer input sequences are being actively explored.72–75 A detailed discussion of more generally applied LLMs can be found elsewhere.76 Since transformers are well-developed in chemistry and are the dominant paradigm behind nearly all state-of-the-art sequence modeling results, they are a focus in this review.2.1 The transformer
The transformer was introduced in, “Attention is all you need” by Vaswani et al.71 in 2017. A careful line-by-line review of the model can be found in “The Annotated Transformer”.77 The transformer was the first seq2seq model based entirely on attention mechanisms, although attention had been a feature for RNNs some years prior.78 The concept of “attention” is a focus applied to certain words of the input, which would convey the most importance, or the context of the passage, and thereby would allow for better decision-making and greater accuracy. However, in a practical sense, “attention” is implemented simply as the dot-product between token embeddings and a learned non-linear function, which will be described further below.The transformer architecture is built around two key modules: the encoder and the decoder. Fig. 2a provides a simplified diagram of the general encoder–decoder transformer architecture. The input is The input is tokenized, from the model's vocabulary,81–85 embedded and positionally encoded, as described above. The encoder consists of multiple stacked layers (six layers in the original model),71 with each layer building on the outputs of the previous one. Each token is represented as a vector, that gets passed through these layers. At each encoder layer, a self-attention mechanism is applied, which calculates the attention between tokens, as discussed earlier. Afterward, the model uses normalization and adds the output back to the input through what's called a residual connection. Residual connection is represented in Fig. 2a by the “by-passing” arrow. This bypass helps prevent issues with vanishing gradients,69,70 ensuring that information flows smoothly through the model. The final step in each encoder layer is a feed-forward neural network with an activation function (such as ReLU,88 SwiGLU,89 GELU,90etc.) that further refines the representation of the input.
 | ||
| Fig. 2 (a) The generalized encoder–decoder transformer: the encoder on the left converts an input into a vector, while the decoder on the right predicts the next token in a sequence. (b) Encoder–decoder transformers are traditionally used for translation tasks and, in chemistry, for reaction prediction, translating reactants into products. (c) Encoder-only transformers provide a vector output and are typically used for sentiment analysis. In chemistry, they are used for property prediction or classification tasks. (d) Decoder-only transformers generate likely next tokens in a sequence. In chemistry, they are used to generate new molecules given an instruction and description of molecules. | ||
The decoder works similarly to the encoder but with key differences. It starts with an initial input token – usually a special start token – embedded into a numerical vector. This token initiates the output sequence generation. Positional encodings are applied to preserve the token order. The decoder is composed of stacked layers, each containing a masked self-attention mechanism that ensures the model only attends to the current and previous tokens, preventing access to future tokens. Additionally, an encoder–decoder attention mechanism aligns the decoder's output with relevant encoder inputs, as depicted by the connecting arrows in Fig. 2a. This alignment helps the model focus on the most critical information from the input sequence. Each layer also employs normalization, residual connections, and a feed-forward network. The final layer applies a softmax function, converting the scores into a probability density over the vocabulary of tokens. The decoder generates the sequence autoregressively, predicting each token based on prior outputs until an end token signals termination.
2.2 Model training
The common lifetime of an LLM consists of being first pretrained using self-supervised techniques, generating what is called a base model. Effective prompt engineering may lead to successful task completion but this base model is often fine-tuned for specific applications using supervised techniques and this creates the “instruct model”. It is called the “instruct model” because the fine-tuning is usually done for it to follow arbitrary instructions, removing the need to specialize fine-tuning for each downstream task.91 Finally, the instruct model can be further tuned with reward models to improve human preference or some other non-differentiable and sparse desired character.92 These concepts are expanded on below.One important technique for LLM alignment is instruction tuning. This method refines the model by training it on datasets that contain specific instructions and examples of preferred responses. By doing so, the model learns to generalize from these examples and follow user instructions more effectively, leading to outputs that are more relevant and safer for real-world applications.96,97 Instruction tuning establishes a baseline alignment, which can then be further improved in the next phase using reinforcement learning (RL).98
In RL-based alignment, the model generates tokens as actions and receives rewards based on the quality of the output, guiding the model to optimize its behavior over time. Unlike post-hoc human evaluations, RL actively integrates preference feedback during training, refining the model to maximize cumulative rewards. This approach eliminates the need for token-by-token supervised fine-tuning by focusing on complete outputs, which better capture human preferences.99–101
The text generation process in RL is typically modeled as a Markov Decision Process (MDP), where actions are tokens, and rewards reflect how well the final output aligns with human intent.102 A popular method, Reinforcement Learning with Human Feedback (RLHF),103 leverages human input to shape the reward system, ensuring alignment with user preferences. Variants such as reinforcement learning with synthetic feedback (RLSF),104 Proximal Policy Optimization (PPO),105 and REINFORCE106 offer alternative strategies for assigning rewards and refining model policies.99,102,107,108 A broader exploration of RL's potential in fine-tuning LLMs is available in works by Cao et al.109 and Shen et al.95
There are ways to reformulate the RLHF process into a direct optimization problem with a different loss. This is known as reward-free methods. Among the main examples of reward-free methods, we have the direct preference optimization (DPO),110 Rank Responses to align Human Feedback (RRHF),111 and Preference Ranking Optimization (PRO).112 These models are popular competitors to PPO and other reward-based methods due to its simplicity. It overcomes the lack of token-by-token loss signal by comparing two completions at a time. The discussions about which technique is superior remain very active in the literature.113
Finally, the alignment may not be to human preferences but to downstream tasks that do not provide token-by-token rewards. For example, Bou et al.114 and Hayes et al.115 both use RL on a language model for improving its outputs on a downstream scientific task.
2.3 Model types
While the Vaswani Transformer71 employed an encoder–decoder structure for sequence-to-sequence tasks, the encoder and decoder were ultimately seen as independent models, leading to “encoder-only”, and “decoder-only” models described below.Examples of how such models can be used are provided in Fig. 2b–d. Fig. 2b illustrates the encoder–decoder model's capability to transform sequences, such as translating from English to Spanish or predicting reaction products by mapping atoms from reactants (amino acids) to product positions (a dipeptide and water). This architecture has large potential on sequence-to-sequence transformations.116,117Fig. 2c highlights the strengths of an encoder-only model in extracting properties or insights directly from input sequences. For example, in text analysis, it can assign sentiment scores or labels, such as tagging the phrase “Chemistry is great” with a positive sentiment. In chemistry, it can predict molecular properties, like hydrophobicity or pKa, from amino acid representations, demonstrating its applications in material science and cheminformatics.118–120 Finally, Fig. 2d depicts a decoder-only architecture, ideal for tasks requiring sequence generation or completion. This model excels at inferring new outputs from input prompts. For instance, given that “chemistry is great,” it can propose broader implications or solutions. It can also generate new peptide sequences from smaller amino acid fragments, showcasing its ability to create novel compounds. This generative capacity is particularly valuable in drug design, where the goal is to discover new molecules or expand chemical libraries.44,121–123
Subsequent evolutions of BERT include, for example, RoBERTa, (Robustly optimized BERT approach), described in 2019 by Liu et al.125 RoBERTa was trained on a larger corpus, for more iterations, with larger mini-batches, and longer sequences, improving model understanding and generalization. By removing the NSP task and focusing on the MLM task, performance improved. RoBERTa dynamically changed masked positions during training and used different hyperparameters. Evolutions of BERT also include domain-specific pretraining and creating specialist LLMs for fields like chemistry, as described below (see Section 3).
GPT employed positional encodings to maintain word order in its predictions. Its self-attention mechanism prevented tokens from attending to future tokens, ensuring each word prediction depended only on preceding words. Hence a decoder-only architecture represents a so-called causal language model, one that generates each item in a sequence based on the previous items. This approach is also referred to as “autoregressive”, meaning that each new word is predicted based on the previously generated words, with no influence from future words. The generation of each subsequent output is causally linked to the history of generated outputs and nothing ahead of the current word affects its generation.
Multi-task models, such as the Text-to-Text Transfer Transformer (T5) developed by Raffel et al.128 demonstrate that various tasks can be reframed into a text-to-text format, allowing the same model architecture and training procedure to be applied universally. By doing so, the model can be used for diverse tasks, but all with the same set of weights. This reduces the need for task-specific models and increases the model's adaptability to new problems. The relevance of this approach is particularly significant as it enables researchers to tackle multiple tasks without needing to retrain separate models, saving both computational resources and time. For instance, Flan-T5 (ref. 129) used instruction fine-tuning with chain-of-thought prompts, enabling it to generalize to unseen tasks, such as generating rationales before answering. This fine-tuning expands the model's ability to tackle more complex problems. More advanced approaches have since been proposed to build robust multi-task models that can flexibly switch between tasks at inference time.130–133
Additionally, LLMs have been extended to process different input modalities, such as image and sound, even though they initially only processed text. For example, Fuyu134 uses linear projection to adapt image representations into the token space of an LLM, allowing a decoder-only model to generate captions for figures. Expanding on this, next-GPT135 was developed as an “any-to-any” model, capable of processing multiple modalities, such as text, audio, image, and video, through modality-specific encoders. The encoded representation is projected into a decoder-only token space, and the LLM's output is processed by a domain-specific diffusion model to generate each modality's output. Multitask or multimodel methods are further described below as these methods start to connect LLMs with autonomous agents.
3 LLMs for chemistry and biochemistry
The integration of large language models (LLMs) into chemistry and biochemistry is opening new frontiers in molecular design, property prediction, and synthesis. As these models evolve, they increasingly align with specific chemical tasks, capitalizing on the strengths of their architectures. Specifically, encoder-only models excel at property prediction,118 decoder-only models are suited for inverse design,136 and encoder–decoder models are applied to synthesis prediction.137 However, with the development improvement of decoder-only models138 and the suggestion that regression tasks can be reformulated as a text completion task,139 decoder-only models started being also applied for property prediction.140–143 This section surveys key LLMs that interpret chemical languages like SMILES and InChI, as well as those that process natural language descriptions relevant to chemistry.We provide a chronological perspective on the evolution of LLMs in this field (Fig. 4), presenting broadly on the design, functionality, and value of each model. Our approach primarily centers on models that use chemical representations like SMILES strings as inputs, but we also examine how natural language models extract valuable data from scientific literature to enhance chemical research.
Ultimately, this discussion underscores the potential for mol-based and text-based LLMs to work together, addressing the growing opportunity for automation in chemistry. This sets the stage for a broader application of autonomous agents in scientific discovery. Fig. 3 illustrates the capabilities of different LLMs available currently, while Fig. 4 presents a chronological map of LLM development in chemistry and biology.
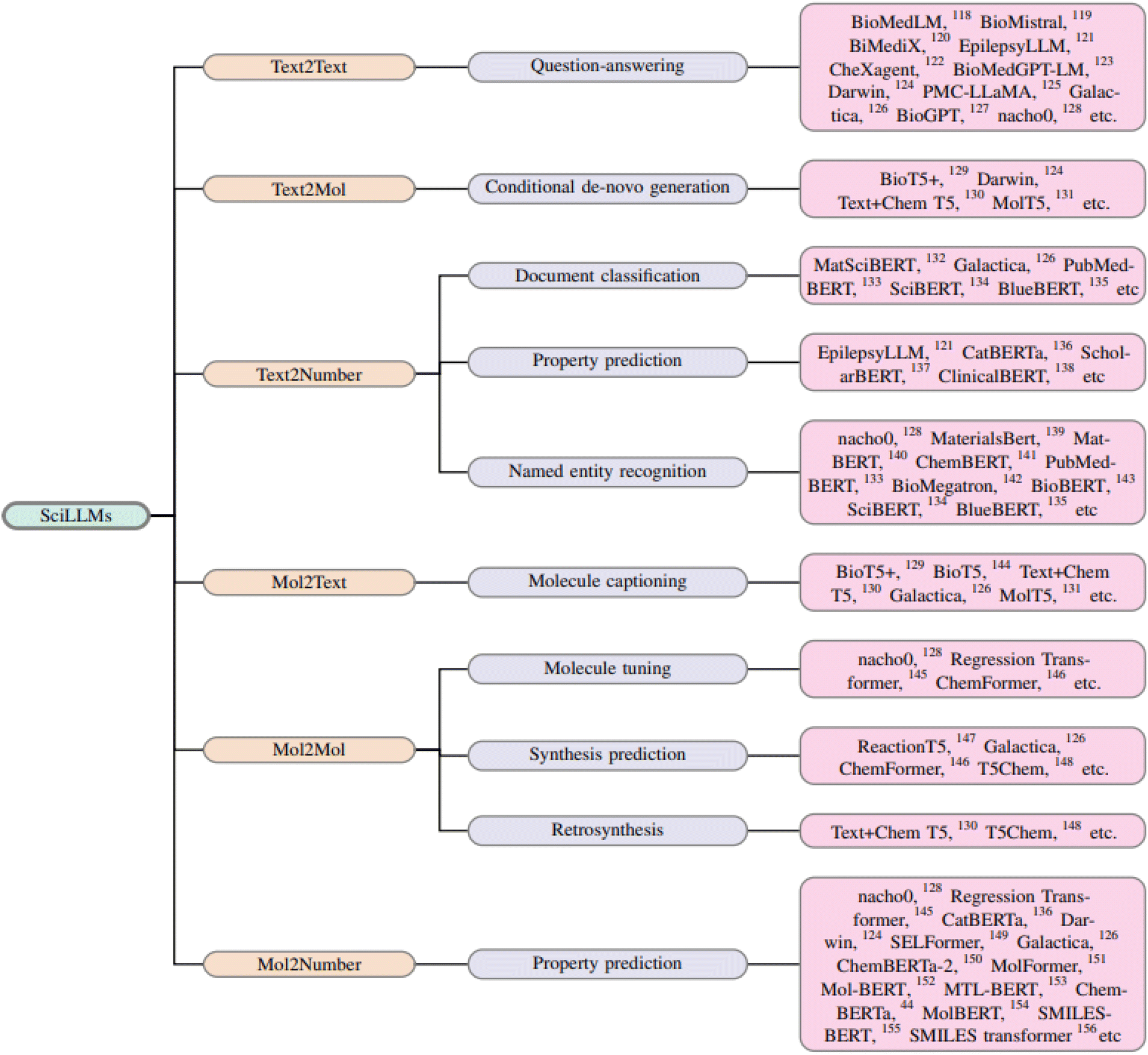 | ||
| Fig. 3 Classification of LLMs in chemistry and biochemistry according to their application. | ||
 | ||
| Fig. 4 Illustration of how Large Language Models (LLMs) evolved chronologically. The dates display the first publication of each model. | ||
Of critical importance, this section starts by emphasizing the role of trustworthy datasets and robust benchmarks. Without well-curated, diverse datasets, models may fail to generalize across real-world applications. Benchmarks that are too narrowly focused can limit the model's applicability, preventing a true measure of its potential. While natural language models take up a smaller fraction of this section, these models will be increasingly used to curate these datasets, ensuring data quality becomes a key part of advancing LLM capabilities in chemistry.
3.1 Molecular representations, datasets, and benchmarks
Molecules can be described in a variety of ways, ranging from two-dimensional structural formulas to more complex three-dimensional models that capture electrostatic potentials. Additionally, molecules can be characterized through properties such as solubility, reactivity, or spectral data from techniques like NMR or mass spectrometry. However, to leverage these descriptions in machine learning, they must be converted into a numerical form that a computer can process. Given the diversity of data in chemistry-based machine learning, multiple methods exist for representing molecules,144–149 highlighting this heterogeneity. Common representations include molecular graphs,150–152 3D point clouds,153–156 and quantitative feature descriptors.145,157–160 In this review, we focus specifically on string-based representations of molecules, given the interest in language models. Among the known string representations, we can cite IUPAC names, SMILES,43 DeepSMILES,161 SELFIES,162 and InChI,163 as recently reviewed by Das et al.164Regarding datasets, there are two types of data used for training LLMs, namely training data and evaluation data. Training data should be grounded in real molecular structures to ensure the model develops an accurate representation of what constitutes a valid molecule. This is similar to how natural language training data, such as that used in models like GPT-4, must be based on real sentences or code to avoid generating nonsensical outputs. Fig. 5 shows a comparison of the number of tokens in common chemistry datasets with those used to train LLaMA2, based on literature data.165–169 With this in mind, we note the largest chemical training corpus, which largely comprises hypothetical chemical structures, amounts to billions of tokens, almost two orders of magnitude fewer than the trillions of tokens used to train LLaMA2. When excluding hypothetical structures from datasets like ZINC,165 (Fig. 5), the number of tokens associated with verifiably synthesized compounds is over five orders of magnitude lower than that of LLaMA2's training data. To address this gap, efforts such as the Mol-instructions dataset, curated by Fang et al.,170 prioritize quality over quantity, providing ∼2M biomolecular and protein-related instructions. Mol-instructions170 was selectively built from multiple data sources,56,171–180 with rigorous quality control. Given the success of literature-based LLMs, one may naturally assume that large datasets are of paramount importance for chemistry. However, it is crucial not to overlook the importance of data quality. Segler et al.181 demonstrated that even using the Reaxys dataset, a very small, human-curated collection of chemical reactions, was sufficient to achieve state-of-the-art results in retrosynthesis. Therefore, the issue is not merely a lack of data, but rather a lack of high-quality data that may be the pivotal factor holding back the development of better scientific LLMs. Ultimately, the focus must shift from sheer quantity to the curation of higher-quality datasets to advance these models.
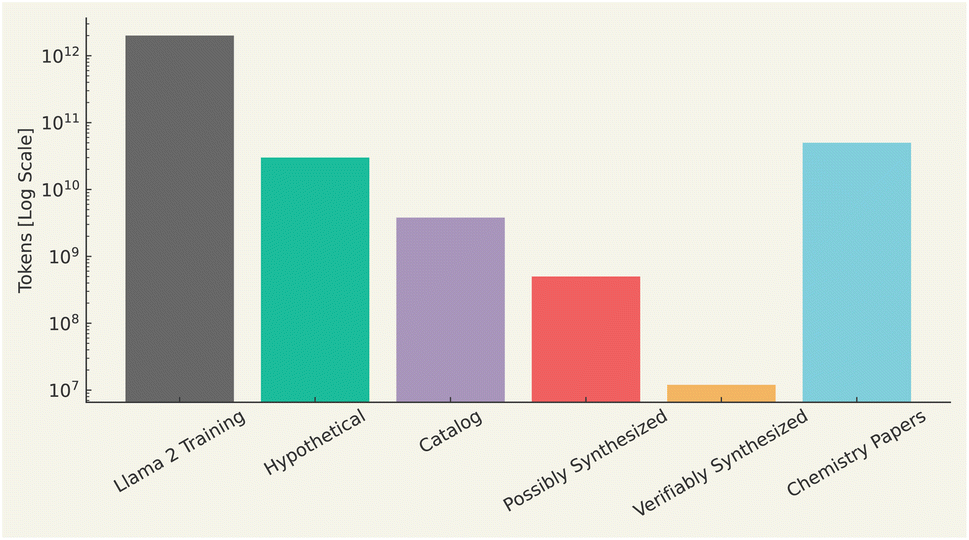 | ||
| Fig. 5 Number of training tokens (on log scale) available from various chemical sources compared with typical LLM training runs. The numbers are drawn from ZINC,165 PubChem,166 Touvron et al.,167 ChEMBL,168 and Kinney et al.169 | ||
To evaluate the accuracy of these models, we compare their performance against well-established benchmarks. However, if the benchmarks are not truly representative of the broader chemistry field, it becomes difficult to gauge the expected impact of these models. Numerous datasets, curated by the scientific community, are available for this benchmarking.182,183 Among them, MoleculeNet,56 first published in 2017, is the most commonly used labeled dataset for chemistry. However, MoleculeNet has several limitations: it is small, contains errors and inconsistencies, and lacks relevance to a larger number of real-world chemistry problems.184–187 Pat Walters, a leader in ML for drug discovery, has emphasized, “I think the best way to make progress on applications of machine learning to drug discovery is to fund a large public effort that will generate high-quality data and make this data available to the community”.188
Walters provides several constructive critiques noting, for example, that the QM7, QM8, and QM9 datasets, intended for predicting quantum properties from 3D structures, are often misused with predictions based incorrectly on their 1D SMILES strings, which inadequately represent 3D molecular conformations. He also suggests more relevant benchmarks and also datasets with more valid entries. For example, he points to the Absorption, Distribution, Metabolism, and Excretion (ADME) data curated by Fang et al.,189 as well as the Therapeutic Data Commons (TDC)190,191 and TDC-2.192 These datasets contain measurements of real compounds, making them grounded in reality. Moreover, ADME is crucial for determining a drug candidate's success, while therapeutic results in diverse modalities align with metrics used in drug development.
Here, we hypothesize that the lack of easily accessible, high-quality data in the correct format for training foundational chemical language models is a major bottleneck to the development of the highly desired “super-human” AI-powered digital chemist. A more optimistic view is presented by Rich and Birnbaum193 They argue that we do not need to wait for the creation of new benchmarks. Instead, they suggest that even the currently available, messy public data can be carefully curated to create benchmarks that approximate real-world applications. In addition, we argue that extracting data from scientific chemistry papers might be an interesting commitment to generating data of high quality, grounded to the truth, and on a large scale.194 Some work has been done in using LLMs for data extraction.195,196 Recently, a few benchmarks following these ideas were created for evaluating LLMs' performance in biology (LAB-Bench197) and material science (MatText,198 MatSci-NLP199 and MaScQA200).
3.2 Property prediction and encoder-only Mol-LLMs
Encoder-only transformer architectures are primarily composed of an encoder, making them well-suited for chemistry tasks that require extracting meaningful information from input sequences, such as classification and property prediction. Since encoder-only architectures are mostly applied to capturing the underlying structure–property relationships, we describe here the relative importance of the property prediction task. Sultan et al.201 also discussed the high importance of this task, the knowledge obtained in the last years, and the remaining challenges regarding molecular property prediction using LLMs. A table of encoder-only scientific LLMs is shown in Table 1.| LLM | Model sizea | Training data | Architecture | Application | Release date |
|---|---|---|---|---|---|
| a “Model Size” is reported as the number of parameters. b The authors report they not used as many encoder layers as it was used in the original BERT paper. But the total number of parameters was not reported. | |||||
| CatBERTa202 | 355M | OpenCatalyst2020 (OC20) | RoBERTa | Property prediction | 2023.09 (2023.11) |
| SELFormer203 | ∼86M | ∼2M compounds from ChEMBL | RoBERTa | Property prediction | 2023.04 (2023.06) |
| ChemBERTa-2 (ref. 122) | 5–46M | 77M SMILES from PubChem | RoBERTa | Property prediction | 2022.09 |
| MaterialsBERT204 | 110M | 2.4M material science abstracts + 750 annotated abstract for NER | BERT | NER and property extraction | 2022.09 (2023.04) |
| SolvBERT205 | 1M SMILES of solute–solvent pairs from CombiSolv-QM and LogS from Boobier et al.206 | BERT | Property prediction | 2022.07 (2023.01) | |
| ScholarBERT207 | 340M, 770M | Public.Resource.Org, Inc. | BERT | Property prediction | 2022.05 (2023.05) |
| BatteryBERT208 | ∼110M | ∼400k papers from RSC, Elsevier and Springer | BERT | Document classification | 2022.05 |
| MatBERT209 | 110M | Abstracts from solid state articles and abstracts and methods from gold nanoparticle articles | BERT | NER | 2022.04 |
| MatSciBERT210 | 110M | ∼150k material science paper downloaded from Elsevier | BERT | NER and text classification | 2021.09 (2022.05) |
| Mol-BERT118 | 110M | ∼4B SMILES from ZINC15 and ChEMBL27 | BERT | Property prediction | 2021.09 |
| MolFormer211 | PubChem and ZINC | BERT | Property prediction | 2021.06 (2022.12) | |
| ChemBERT212 | 110M | ∼200k extracted using ChemDataExtractor | BERT | NER | 2021.06 |
| MolBERT213 | ∼85M | ChemBench | BERT | Property prediction | 2020.11 |
| ChemBERTa44 | 10M SMILES from PubChem | RoBERTa | Property prediction | 2020.10 | |
| BioMegatron214 | 345M, 800M, 1.2B | Wikipedia, CC-Stories, Real-News, and OpenWebtext | Megatron-LM | NER and QA | 2020-10 |
| PubMedBERT215 | 110M | 14M abstracts from PubMed | BERT | NER, QA, and document classification | 2020.07 (2021.10) |
| Molecule attention transformer216 | ZINC15 | Encoder with GCN features | Property prediction | 2020.02 | |
| SMILES-BERT217 | ∼18M SMILES from ZINC | BERT | Property prediction | 2019.09 | |
| BlueBERT218 | 110M | PubMed and MIMIC-III | BERT | NER, and document classification | 2019.06 |
| ClinicalBERT219 | 110M | MIMIC-III | BERT | Patient readmission probability | 2019.04 |
| SciBERT220 | 110M | 1.14M papers from Semantic Scholar | BERT | NER and sentence classification | 2019.03 (2019.11) |
| BioBERT221 | 110M | PubMed and PMC | BERT | NER and QA | 2019.01 (2019.09) |
While encoder-only LLMs are predominantly used for property prediction, they are also applicable for synthesis classification. Schwaller et al.244 used a BERT model to more accurately classify complex synthesis reactions by generating reaction fingerprints from raw SMILES strings, without the need to separate reactants from reagents in the input data, thereby simplifying data preparation. The BERT model achieved higher accuracy (98.2%) compared to the encoder–decoder model (95.2%) for classifying reactions. Accurate classification aids in understanding reaction mechanisms, vital for reaction design, optimization, and retrosynthesis. Toniato et al.245 also used a BERT architecture to classify reaction types for downstream retrosynthesis tasks that would enable the manufacture of any molecular target. Further examples of BERT use include self-supervised reaction atom-to-atom mapping.246,247 These chemical classifications would accelerate research and development in organic synthesis, described further below.
Beyond synthesis classification, encoder-only models like BERT have shown great promise for molecular property prediction, especially when labeled data is limited. Recognizing this, Wang et al. introduced a semi-supervised SMILES-BERT model, which was pretrained on a large unlabeled dataset with a Masked SMILES Recovery task.248 The model was then fine-tuned for various molecular property prediction tasks, outperforming state-of-the-art methods in 2019 on three chosen datasets varying in size and property. This marked a shift from using BERT for reaction classification towards property prediction and drug discovery. Maziarka et al.216 also claimed state-of-the-art performance in property prediction after self-supervised pretraining in their Molecule Attention Transformer (MAT), which adapted BERT to chemical molecules by augmenting the self-attention with inter-atomic distances and molecular graph structure.
Zhang et al.249 also tackled the issue of limited property-labeled data and the lack of correlation between any two datasets labeled for different properties, hindering generalizability. They introduced multitask learning BERT (MTL-BERT), which used large-scale pretraining and multitask learning with unlabeled SMILES strings from ChEMBL,168 which is a widely-used database containing bioactive molecules with drug-like properties, designed to aid drug discovery. The MTL-BERT approach mined contextual information and extracted key patterns from complex SMILES strings, improving model interpretability. The model was fine-tuned for relevant downstream tasks, achieving better performance than state-of-the-art methods in 2022 on 60 molecular datasets from ADMETlab250 and MoleculeNet.56
In 2021, Li and Jiang118 introduced Mol-BERT, pretrained on four million unlabeled drug SMILES from the ZINC15 (ref. 251) and ChEMBL27 (ref. 168) databases to capture molecular substructure information for property prediction. Their work leveraged the underutilized potential of large unlabeled datasets like ZINC, which contains over 230 million commercially available compounds, and is designed for virtual screening and drug discovery. Mol-BERT consisted of three components: a PretrainingExtractor, Pretraining Mol-BERT, and Fine-Tuning Mol-BERT. It treated Morgan fingerprint fragments as “words” and complete molecular compounds as “sentences,” using RDKit and the Morgan algorithm for canonicalization and substructure identification. This approach generated comprehensive molecular fingerprints from SMILES strings, used in a Masked Language Model (MLM) task for pretraining. Mol-BERT was fine-tuned on labeled samples, providing outputs as binary values or continuous scores for classification or regression, and it outperformed existing sequence and graph-based methods by at least 2% in ROC-AUC scores on Tox21, SIDER, and ClinTox benchmark datasets.56
Ross et al.252 introduced MoLFormer, a large-scale self-supervised BERT model, with the intention to provide molecular property predictions with competitive accuracy and speed when compared to density functional theory calculations or wet-lab experiments. They trained MoLFormer with rotary positional embeddings on SMILES sequences of 1.1 billion unlabeled molecules from ZINC,251 and PubChem,166 another database of chemical properties and activities of millions of small molecules, widely used in drug discovery and chemical research. The rotary positional encoding captures token positions more effectively than traditional methods,71 improving modeling of sequence relationships. MoLFormer outperformed state-of-the-art GNNs on several classification and regression tasks from ten MoleculeNet56 datasets, while performing competitively on two others. It effectively learned spatial relationships between atoms, predicting various molecular properties, including quantum-chemical properties. Additionally, the authors stated how MoLFormer represents an efficient and environment-friendly use of computational resources, claiming a reduced GPU usage in training by a factor of 60 (16 GPUs instead of 1000).
With ChemBERTa, Chithrananda et al.44 explored the impact of pretraining dataset size, tokenization strategy, and the use of SMILES or SELFIES, distinguishing their work from other BERT studies. They used HuggingFace's RoBERTa transformer,253 and referenced a DeepChem56 tutorial for accessibility. Their results showed improved performance on downstream tasks (BBBP, ClinTox, HIV, Tox21 from MoleculeNet56) as the pretraining dataset size increased from 100k to 10M. Although ChemBERTa did not surpass state-of-the-art GNN-based baselines like Chemprop (which used 2048-bit Morgan Fingerprints from RDKit),254 the authors suggested that with expansion to larger datasets they would eventually beat those baselines. The authors compared Byte-Pair Encoder (BPE) with a custom SmilesTokenizer and its regular expression developed by255 while exploring tokenization strategies. They found the SmilesTokenizer slightly outperformed BPE, suggesting more relevant sub-word tokenization is beneficial. No difference was found between SMILES and SELFIES, but the paper highlighted how attention heads in transformers could be visualized with BertViz,256 showing certain neurons selective for functional groups. This study underscored the importance of appropriate benchmarking and addresses the carbon footprint of AI in molecular property prediction.
In ChemBERTa-2, Ahmad et al.122 aimed to create a foundational model applicable across various tasks. They addressed a criticism that LLMs were not so generalizable because the training data was biased or non-representative. They addressed this criticism by training on 77M samples and adding a Multi-Task Regression component to the pretraining. ChemBERTa-2 matched state-of-the-art architectures on MoleculeNet.56 As with ChemBERTa, the work was valuable because of additional exploration, in this case into how pretraining improvements affected certain downstream tasks more than others, depending on the type of fine-tuning task, the structural features of the molecules in the fine-tuning task data set, or the size of that fine-tuning dataset. The result was that pretraining the encoder-only model is important, but gains could be made by considering the chemical application itself, and the associated fine-tuning dataset.
In June 2023, Yuksel et al.203 introduced SELFormer, building on ideas from ChemBERTa2 (ref. 122) and using SELFIES for large data input. Yuksel et al.203 argue that SMILES strings have validity and robustness issues, hindering effective chemical interpretation of the data, although this perspective is not universally held.257 SELFormer uses SELFIES and is pretrained on two million drug-like compounds, fine-tuned for diverse molecular property prediction tasks (BBBP, SIDER, Tox21, HIV, BACE, FreeSolv, ESOL, PDBbind from MoleculeNet).56 SELFormer outperformed all competing methods for some tasks and produced comparable results for the rest. It could also discriminate molecules with different structural properties. The paper suggests future directions in multimodal models combining structural data with other types of molecular information, including text-based annotations. We will discuss such multimodal models below.
In 2022, Yu et al.205 published SolvBERT, a multi-task BERT-based regression model that could predict both solvation free energy and solubility from the SMILES notations of solute–solvent complexes. It was trained on the CombiSolv-QM dataset,258 a curation of experimental solvent free energy data called CombiSolv-Exp-8780,259–262 and the solubility dataset from Boobier et al.206 SolvBERT's performance was benchmarked against advanced graph-based models263,264 This work is powerful because there is an expectation that solvation free energy depends on 3-dimensional conformational properties of the molecules, or at least 2D properties that would be well characterized by graph-based molecular representations. It shows an overachieving utility of using SMILES strings in property prediction, and aligns with other work by Winter et al.,265 regarding activity coefficients. SolvBERT showed comparable performance to a Directed Message Passing Neural Network (DMPNN) in predicting solvation free energy, largely due to its effective clustering feature in the pretraining phase as shown by TMAP (Tree Map of All Possible) visualizations. Furthermore, SolvBERT outperformed Graph Representation Of Molecular Data with Self-supervision (GROVER)264 in predicting experimentally evaluated solubility data for new solute–solvent combinations. This underscores the significance of SolvBERT's ability to capture the dynamic and spatial complexities of solvation interactions in a text-based model.
While models like SolvBERT have achieved impressive results in solvation free energy prediction, challenges such as limited labeled data continue to restrict the broader application of transformer models in chemistry. Recognizing this issue, Jiang et al. introduced INTransformer in 2024,266 a method designed to enhance property prediction by capturing global molecular information more effectively, even when data is scarce. By incorporating perturbing noise and using contrastive learning to artificially augment smaller datasets, INTransformer delivered improved performance on several tasks. Ongoing work continues to explore various transformer strategies for smaller datasets. Again using contrastive learning, which maximizes the difference between representations of similar and dissimilar data points, but in a different context, MoleculeSTM267 uses LLM encoders to create representations for SMILES and for descriptions of molecules extracted from PubChem.268 Similar work was performed by Xu et al.269 The authors curated a dataset with descriptions of proteins. Subsequently, to train ProtST, a protein language model (PLM) was used to encode amino acid sequences and LLMs to encode the descriptions.
In this section, we outlined the advancements of encoder-only models like BERT and their evolution for property prediction and synthesis classification. Chemists traditionally hypothesize molecular properties, but these models, ranging from Mol-BERT to SolvBERT, showcase the growing efficiency of machine learning in property prediction. Approaches such as multitask learning and contrastive learning, as seen in INTransformer, offer solutions to challenges posed by limited labeled data.
3.3 Property directed inverse design and decoder-only mol-LLMs
Decoder-only GPT-like architectures offer significant value for property-directed molecule generation and de novo chemistry applications because they excel at generating novel molecular structures by learning from vast datasets of chemical compounds. These models can capture intricate patterns and relationships within molecular sequences, proposing viable new compounds that adhere to desired chemical properties and constraints. This enables rapid exploration and innovation within an almost infinite chemical space. Moreover, such large general-purpose models can be fine-tuned with small amounts of domain-specific scientific data,142,270 allowing them to support specific applications efficiently. In this section, we first describe property-directed inverse design from a chemistry perspective and then examine how decoder-only LLMs have propelled inverse design forward. A table of decoder-only scientific LLMs is shown in Table 2.| LLM | Model sizea | Training data | Architecture | Application | Release date |
|---|---|---|---|---|---|
| a “Model Size” is reported as the number of parameters. “PubMed” refer to the PubMed abstracts dataset, while PMC (PubMed Corpus) refers to the full-text corpus dataset. b The total number of parameters was not reported. | |||||
| Tx-LLM271 | TDC datasets | PaLM-2 | Property prediction and retrosynthesis | 2024.06 | |
| BioMedLM272 | 2.7B | PubMed abstracts and full articles | GPT | QA | 2024.03 |
| LlasMol273 | ∼7B | SMolInstruct | Galactica, LLaMa, Mistral | Property prediction, molecule captioning, molecule generation, retrosynthesis, name conversion | 2024.02 (2024.08) |
| BioMistral274 | 7B | PubMed Central (PMC) | Mistral | QA | 2024.02 (2024.08) |
| BiMediX275 | 8× 7B | 1.3M Arabic–English instructions (BiMed) | Mixtral | QA | 2024.02 |
| EpilepsyLLM276 | 7B | Data from the Japan Epilepsy Association, Epilepsy Information Center, and Tenkan Net | LLaMa | QA | 2024.01 |
| CheXagent277 | 7B | 28 Publicly available datasets, including PMC, MIMIC, wikipedia, PadChest, and BIMCV-COVID-19 | Mistral | QA, image understanding | 2024.01 |
| ChemSpaceAL278 | ChEMBL 33, GuacaMol v1, MOSES, and BindingDB 08-2023 | GPT | Molecule generation | 2023.09 (2024.02) | |
| BioMedGPT-LM279 | 7B and 10B | 5.5M Bbiomedical papers from S2ORC | LLaMA2 | QA | 2023.08 |
| Darwin280 | 7B | SciQ and web of science | LLaMA | QA, property prediction, NER, and molecule generation | 2023.08 |
| cMolGPT46 | MOSES | GPT | Molecule generation | 2023.05 | |
| PMC-LLaMA281 | 7B and 13B | MedC-k and MedC-I | LLaMA | QA | 2023.04 (2024.04) |
| GPTChem142 | 175B | Curation of multiple classification and regression benchmarks | GPT-3 | Property prediction and inverse design | 2023.02 (2024.02) |
| Galactica123 | 125M, 1.3B, 6.7B, 30B, 120B | The galactica corpus, a curation with 62B scientific documents | Decoder-only | QA, NER, document summarization, property prediction | 2022.11 |
| BioGPT282 | 355M | 15M of title and abstract from PubMed | GPT-2 | QA, NER, and document classification | 2022-09 (2023.04) |
| SMILES-to-properties-transformer265 | 6.5M | Synthetic data generated with the thermodynamic model COSMO-RS | GPT-3 | Property prediction | 2022.06 (2022.09) |
| ChemGPT283 | ∼1B | 10M molecules from PubChem | GPT-neo | Molecule generation | 2022.05 (2023.11) |
| Regression transformer139 | ∼27M | ChEMBL, MoleculeNet, USPTO, etc. | XLNet | Property prediction, molecule tuning, molecule generation | 2022.02 (2023.04) |
| MolGPT284 | 6M | MOSES and GuacaMol | GPT | Molecule generation | 2021.10 |
| Adilov2021 (ref. 285) | 13.4M | 5M SMILES from ChemBERTa's PubChem-10M | GPT-2 | Property prediction and molecule generation | 2021.09 |
While natural products have been a valuable starting point, we are not confined to their derivatives. AI, particularly generative LLMs, allows us to go beyond nature and explore a much larger chemical space. In silico molecular design enables rapid modification, akin to random mutation,289 where only valid, synthesizable molecules that meet predefined property criteria remain in the generated set.242,290 This approach allows us to test modifications in silico, expanding exploration beyond the boundaries of natural products.
The true innovation of AI-driven molecular design, however, lies in its ability to directly generate candidate molecules based on desired properties, without the need for iterative stepwise modifications.291 This “inverse design” capability allows us to start with a target property and directly generate candidate molecules that meet the predefined property requirements. Generative LLMs applied to sequences of atoms and functional groups offer a powerful opportunity for out-of-the-box exploration, tapping into the vast chemical space that extends far beyond the confines of nature. This accelerates the path from concept to viable therapeutic agents, aligning seamlessly with decoder-only LLM architectures.
A key advancement then came with MolGPT,284 a 6-million-parameter decoder-only model designed for molecular generation. MolGPT introduced masked self-attention, enabling the learning of long-range dependencies in SMILES strings. The model ensured chemically valid SMILES representations, respecting structural rules like valency and ring closures. It also utilized salience measures for interpretability, aiding in predicting SMILES tokens and understanding which parts of the molecule were most influential in the model's predictions. MolGPT outperformed many existing Variational Auto-Encoder (VAE)-based approaches,293–300 in predicting novel molecules with specified properties, being trained on datasets like MOSES301 and GuacaMol.302
While MolGPT's computational demands may be higher than traditional VAEs, its ability to generate high-quality, novel molecules justifies this trade-off. MolGPT demonstrated strong performance on key metrics such as validity, which measures the percentage of generated molecules that are chemically valid according to bonding rules; uniqueness, the proportion of generated molecules that are distinct from one another; Frechet ChemNet Distance (FCD),303 which compares the distribution of generated molecules to that of real molecules in the training set, indicating how closely the generated molecules resemble real-world compounds; and KL divergence,302 a measure of how the probability distribution of generated molecules deviates from the true distribution of the training data. These metrics illustrate MolGPT's ability to generate high-quality, novel molecules while maintaining a balance between diversity and similarity to known chemical spaces. A brief summary of advancements in transformer-based models for de novo molecule generation from 2023 and 2024 follows, which continue to refine and expand upon the foundational work laid by models like MolGPT.
Haroon et al.304 further developed a GPT-based model with relative attention for de novo drug design, showing improved validity, uniqueness, and novelty. This work was followed by Frey et al.,283 who introduced ChemGPT to explore hyperparameter tuning and dataset scaling in new domains. ChemGPT's contribution lies in refining generative models to better fit specific chemical domains, advancing the understanding of how data scale impacts generative performance. Both Wang et al.305 and Mao et al.306 presented work that surpassed MolGPT. Furthermore, Mao et al.140 showed that decoder-only models could generate novel compounds using IUPAC names directly.
This marked a departure from typical SMILES-based molecular representations, as IUPAC names offer a standardized, human-readable format that aligns with how chemists conceptualize molecular structures. By integrating these chemical semantics into the model, iupacGPT140 bridges the gap between computational predictions and real-world chemical applications. The IUPAC name outputs are easier to understand, validate, and apply, facilitating smoother integration into workflows like regulatory filings, chemical databases, and drug design. Focusing on pretraining with a vast dataset of IUPAC names and fine-tuning with lightweight networks, iupacGPT excels in molecule generation, classification, and regression tasks, providing an intuitive interface for chemists in both drug discovery and material science.
In a similar vein, Zhang et al.307 proposed including target 3D structural information in molecular generative models, even though their approach is not LLM-based. However, it serves as a noteworthy contribution to the field of structure-based drug design. Integrating biological data, such as 3D protein structures, can significantly improve the relevance and specificity of generated molecules, making this method valuable for future LLM-based drug design. Similarly, Wang et al.308 discussed PETrans, a deep learning method that generates target-specific ligands using protein-specific encoding and transfer learning. This study further emphasizes the importance of using transformer models for generating molecules with high binding affinity to specific protein targets. The significance of these works lies in their demonstration that integrating both human-readable formats (like IUPAC names) and biological context (such as protein structures) into generative models can lead to more relevant, interpretable, and target-specific drug candidates. This reflects a broader trend in AI-driven chemistry to combine multiple data sources for more precise molecular generation, accelerating the drug discovery process.
In 2024, Yoshikai et al.309 discussed the limitations of transformer architectures in recognizing chirality from SMILES representations, which impacts the prediction accuracy of molecular properties. To address this, they coupled a transformer with a VAE. Using contrastive learning from NLP to generate new molecules with multiple SMILES representations, enhancing molecular novelty and validity. Kyro et al.278 presented ChemSpaceAL, an active learning method for protein-specific molecular generation, efficiently identifying molecules with desired characteristics without prior knowledge of inhibitors. Yan et al.310 proposed the GMIA framework, which improves prediction accuracy and interpretability in drug–drug interactions through a graph mutual interaction attention decoder. These innovations represent significant strides in addressing key challenges in molecular generation, such as chirality recognition, molecular novelty, and drug–drug interaction prediction. By integrating new techniques like VAEs, contrastive learning, and active learning into transformer-based models, they have improved both the accuracy and interpretability of molecular design.
Building on these developments, Shen et al.311 reported on AutoMolDesigner, an open-source tool for small-molecule antibiotic design, further emphasizing the role of automation in molecular generation. This work serves as a precursor to more complex models, such as Taiga101 and cMolGPT,46 which employ advanced methods like autoregressive mechanisms and reinforcement learning for molecular generation and property optimization.
For a deeper dive into decoder-only transformer architecture in chemistry, we highlight the May 2023 “Taiga” model by Mazuz et al.,101 and cMolGPT by Wang et al.46 Taiga first learns to map SMILES strings to a vector space, and then refines that space using a smaller, labeled dataset to generate molecules with targeted attributes. It uses an autoregressive mechanism, predicting each SMILES character in sequence based on the preceding ones. For property optimization, Taiga employs the REINFORCE algorithm,106 which helps refine molecules to enhance specific features. While this reinforcement learning (RL) approach may slightly reduce molecular validity, it significantly improves the practical applicability of the generated compounds. Initially evaluated using the Quantitative Estimate of Drug-likeness (QED) metric,312 Taiga has also demonstrated promising results in targeting IC50 values,168 the BACE protein,313 and anti-cancer activities they collected from a variety of sources. This work underscores the importance of using new models to address applications that require a higher level of chemical sophistication, to illustrate how such models could ultimately be applied outside of the available benchmark datasets. It also builds on the necessary use of standardized datasets and train-validation-test splitting, to demonstrate progress, as explained by Wu et al.56 Yet, even the MoleculeNet benchmarks56 are flawed, and we point the reader here to a more detailed discussion on benchmarking,188 given that a significant portion of molecules in the BACE dataset have undefined stereo centers, which, at a deeper level, complicates the modeling and prediction accuracy.
While models like Taiga demonstrate the power of autoregressive learning and reinforcement strategies to generate molecules with optimized properties, the next step in molecular design incorporates deeper chemical domain knowledge. This approach is exemplified by Wang et al.46 They introduced cMolGPT, a conditional generative model that brings a more targeted focus to drug discovery by integrating specific protein–ligand interactions, which underscores the importance of incorporating chemical domain knowledge to effectively navigate the vast landscape of drug-like molecules. Using self-supervised learning and an auto-regressive approach, cMolGPT generates SMILES guided by predefined conditions based on target proteins and binding molecules. Initially trained on the MOSES dataset301 without target information, the model is fine-tuned with embeddings of protein-binder pairs, focusing on generating compound libraries and target-specific molecules for the EGFR, HTR1A, and S1PR1 protein datasets.314–317
Their approach employs a QSAR model5 to predict the activity of generated compounds, achieving a Pearson correlation coefficient over 0.75. However, despite the strong predictive capabilities, this reliance on a QSAR model, with its own inherent limitations, highlights the need for more extensive experimental datasets. cMolGPT46 tends to generate molecules within the sub-chemical space represented in the original dataset, successfully identifying potential binders but struggling to broadly explore the chemical space for novel solutions. This underscores the challenge of generating diverse molecules with varying structural characteristics while maintaining high binding affinity to specific targets. While cMolGPT advances the integration of biological data and fine-tuned embeddings for more precise molecular generation, models like Taiga and cMolGPT differ in their approach. Taiga101 employs reinforcement learning to optimize generative models for molecule generation, while cMolGPT uses target-specific embeddings to guide the design process. Both highlight the strengths of decoder-only models but emphasize distinct strategies; Taiga optimizes molecular properties through autoregressive learning, and cMolGPT focuses on conditional generation based on protein–ligand interactions.
In contrast, Yu et al.273 follow a different approach with LlaSMol,273 which utilizes pretrained models (for instance Galactica, LlaMa2, and Mistral) and performs parameter efficient fine-tuning (PEFT) techniques318,319 such as LoRa.320 PEFT enables fine-tuning large language models with fewer parameters, making the process more resource-efficient while maintaining high performance. LlaSMol demonstrated its potential by achieving state-of-the-art performance in property prediction tasks, particularly when fine-tuned on benchmark datasets like MoleculeNet.56
There continue to be significant advancements being made in using transformer-based models to tackle chemical prediction tasks with optimized computational resources, including more generalist models, such as Tx-LLM,271 designed to streamline the complex process of drug discovery. For additional insights on how these models are shaping the field, we refer the reader to several excellent reviews,164,321–323 with Goel et al.324 highlighting the efficiency of modern machine learning methods in sampling drug-like chemical space for virtual screening and molecular design. Goel et al.324 discussed the effectiveness of generative models, including large language models (LLMs), in approximating the vast chemical space, particularly when conditioned on specific properties or receptor structures.
We provide a segue from this section by introducing the work by Jablonka et al.,142 which showcases a decoder-only GPT model that, despite its training on natural language rather than specialized chemical languages, competes effectively with decoder-only LLMs tailored to chemical languages. The authors finetuned GPT-3 to predict properties and conditionally generate molecules and, therefore, highlight its potential as a foundational tool in the field. This work sets the stage for integrating natural language decoder-only LLMs, like GPT, into chemical research, where they could serve as central hubs for knowledge discovery.
Looking ahead, this integration foreshadows future developments that pair LLMs with specialized tools to enhance their capabilities, paving the way for the creation of autonomous agents that leverage deep language understanding in scientific domains. Decoder-only models have already significantly advanced inverse molecular design, from improving property prediction to enabling target-specific molecular generation. Their adaptability to various chemical tasks demonstrates their value in optimizing drug discovery processes and beyond. As models like LlaSMol and cMolGPT continue to evolve, integrating chemical domain knowledge and biological data, they offer exciting opportunities for more precise molecular generation. The growing potential for combining large language models like GPT-4 with specialized chemical tools signals a future where AI-driven autonomous agents could revolutionize chemical research, making these models indispensable to scientific discovery.
3.4 Synthesis prediction and encoder–decoder Mol-LLMs
The encoder–decoder architecture is designed for tasks involving the translation of one sequence into another, making it ideal for predicting chemical reaction outcomes or generating synthesis pathways from given reactants. We begin with a background on optimal synthesis prediction and describe how earlier machine learning has approached this challenge. Following that, we explain how LLMs have enhanced chemical synthesis prediction and optimization. Although, our context below is aptly chosen to be synthesis prediction, other applications exist. For example, SMILES Transformer (ST)325 is worth a mention, historically, because it explored the benefits of self-supervised pretraining to produce continuous, data-driven molecular fingerprints from large SMILES-based datasets. A list of encoder-decoder scientific LLMs is shown in Table 3.| LLM | Model sizea | Training data | Architecture | Application | Release date |
|---|---|---|---|---|---|
| a “Model Size” is reported as the number of parameters. b The total number of parameters was not reported. | |||||
| BioT5+117 | 252M | ZINC20, UniRef50, 33M PubMed articles, 339k mol-text pairs from PubChem, 569k FASTA-text pairs from Swiss-prot | T5 | Molecule captioning, molecule generation, property prediction | 2024.02 (2024.08) |
| nach0 (ref. 187) | 250M | MoleculeNet, USPTO, ZINC | T5 | Property prediction, molecule generation, question answering, NER | 2023.11 (2024.05) |
| ReactionT5 (ref. 326) | 220M | ZINC and ORD | T5 | Property prediction and reaction prediction | 2023.11 |
| BioT5 (ref. 116) | 252M | ZINC20, UniRef50, full-articles from BioRxiv and PubMed, mol-text-IUPAC information from PubChem | T5 | Molecule captioning, property prediction | 2023-10 (2024.12) |
| MOLGEN327 | ZINC15 | BART | Molecule generation | 2023.01 (2024.03) | |
| Text+Chem T5 (ref. 328) | 60M, 220M | 11.5M or 33.5M samples curated from Vaucher et al.,329 Toniato et al.,245 and CheBI-20 | T5 | Molecule captioning, product prediction, retrosynthesis, molecule generation | 2023.01 (2023.06) |
| MolT5 (ref. 330) | 60M, 770M | C4 dataset | T5 | Molecule captioning and molecule generation | 2022.04 (2022.12) |
| T5Chem179 | 220M | USPTO | T5 | Product prediction, retrosynthesis, property prediction | 2022.03 |
| Text2Mol331 | CheBI-20 | SciBERT w/decoder | Molecule captioning and conditional molecule generation | 2021.11 | |
| ChemFormer185 | 45M, 230M | 100M SMILES from ZINC-15 | BART | Product prediction, property prediction, molecular generation | 2021.07 (2022.01) |
| SMILES transformer325 | ChEMBL24 | Transformer | Property prediction | 2019.11 | |
| Molecular transformer255 | 12M | USPTO | Transformer | Product prediction | 2018.11 (2019.08) |
Building on RNNs and GRUs, the field advanced with the introduction of template-based models. In parallel with the development of the Chematica tool339,340 for synthesis mapping, Segler and Waller341 highlighted that traditional rule-based systems often failed by neglecting molecular context, leading to “reactivity conflicts”. Their approach emphasized transformation rules that capture atomic and bond changes, applied in reverse for retrosynthesis. Trained on 3.5 million reactions, their model achieved 95% top-10 accuracy in retrosynthesis and 97% for reaction prediction on a validation set of nearly 1 million reactions from the Reaxys database (1771–2015). Although not transformer-based, this work laid the foundation for large language models (LLMs) in synthesis. However, template-based models depend on explicit reaction templates from known reactions, limiting their ability to predict novel reactions and requiring manual updates to incorporate new data.
Semi-template-based models offered a balance between rigid template-based methods and flexible template-free approaches. They used interpolation or extrapolation within template-defined spaces to predict a wider range of reactions and to adjust based on new data. In 2021, Somnath et al.342 introduced a graph-based approach recognizing that precursor molecule topology is largely unchanged during reactions. Their model broke the product molecule into “synthons” and added relevant leaving groups, making results more interpretable.343 Training on the USPTO-50k dataset,338 they achieved a top-1 accuracy of 53.7%, outperforming previous methods.
However, the template-free approaches align well with transformer-based learning approaches because they learn retrosynthetic rules from raw training data. This provides significant flexibility and generalizability across various types of chemistry. Template-free models are not constrained by template libraries and so can uncover novel synthetic routes that are undocumented or not obvious from existing reaction templates. To pave the way for transformer use in synthesis, Cadeddu et al.344 drew an analogy between fragments in a compound and words in a sentence due to their similar rank distributions. Schwaller et al.345 further advanced this with an LSTM network augmented by an attention-mechanism-based encoder–decoder architecture, using the USPTO dataset.338 They introduced a new “regular expression” (or regex) for tokenizing molecules, framing synthesis (or retrosynthesis) predictions as translation problems with a data-driven, template-free sequence-to-sequence model. They tracked which starting materials were actual reactants, distinguishing them from other reagents like solvents or catalysts, and used the regular expression to uniquely tokenize recurring reagents, as their atoms were not mapped to products in the core reaction. This regex for tokenizing molecules is commonly used today in all mol-based LLMs.
In 2019, going beyond the “neural machine” work of Nam and Kim,336 Schwaller et al.255 first applied a transformer for synthesis prediction, framing the task as translating reactants and reagents into the final product. Their model inferred correlations between chemical motifs in reactants, reagents, and products in the dataset (USPTO-MIT,346 USPTO-LEF,347 USPTO-STEREO345). It required no handcrafted rules and accurately predicted subtle chemical transformations, outperforming all prior algorithms on a common benchmark dataset. The model handled inputs without a reactant-reagent split, following their previous work,345 and accounted for stereochemistry, making it valuable for universal application. Then, in 2020, for automated retrosynthesis, Schwaller et al.348 developed an advanced Molecular Transformer model with a hyper-graph exploration strategy. The model set a standard for predicting reactants and other entities, evaluated using four new metrics. “Coverage” measured how comprehensively the model could predict across the chemical space, while “class diversity” assessed the variety of chemical types the model could generate, ensuring it was not limited to narrow subsets of reactions. “Round-trip accuracy” checked whether the retrosynthetically predicted reactants could regenerate the original products, ensuring consistency in both directions. “Jensen–Shannon divergence” compared the predicted outcomes to actual real-world distributions, indicating how closely the model's predictions matched reality. Constructed dynamically, the hypergraph allowed for efficient expansion based on Bayesian-like probability scores, showing high performance despite training data limitations. Notably, accuracy improved when the re-synthesis of the target product from the generated precursors was factored in, a concept also employed by Chen and Jung349 and Westerlund et al.350 Also in 2020, Zheng et al.351 developed a “template-free self-corrected retrosynthesis predictor” (SCROP) using transformer networks and a neural network-based syntax corrector, achieving 59.0% accuracy on a benchmark dataset.338,352 This approach outperformed other deep learning methods by over 2% and template-based methods by over 6%.
We now highlight advancements in synthesis prediction using the BART encoder–decoder architecture, starting with Chemformer by Irwin et al.185 This paper emphasized the computational expense of training transformers on SMILES and the importance of pretraining for efficiency. It showed that models pretrained on task-specific datasets or using only the encoder stack were limited for sequence-to-sequence tasks. After transfer learning, Chemformer achieved state-of-the-art results in both sequence-to-sequence synthesis tasks and discriminative tasks, such as optimizing molecular structures for specific properties. They studied the effects of small changes on molecular properties using pairs of molecules from the ChEMBL database168 with a single structural modification. Chemformer's performance was tested on the ESOL, Lipophilicity, and Free Solvation datasets.56 Irwin et al.185 also described their use of an in-house property prediction model, but when models train on calculated data for ease of access and uniformity, they abstract away from real-world chemical properties. We again emphasize the importance of incorporating experimentally derived data into Chemistry LLM research to create more robust and relevant models. Continuously curating new, relevant datasets that better represent real-world chemical complexities will enhance the applicability and transferability of these models.
In 2023, Toniato et al.245 also applied LLMs to single-step retrosynthesis as a translation problem, but increased retrosynthesis prediction diversity by adding classification tokens, or “prompt tokens,” to the target molecule's language representation, guiding the model towards different disconnection strategies. Increased prediction diversity has high value by providing out-of-the-box synthetic strategies to complement the human chemist's work. To measure retrosynthesis accuracy, Li et al.353 introduced Retro-BLEU, a metric adapted from the BLEU (Bilingual Evaluation Understudy) score used in machine translation.354 Despite progress in computer-assisted synthesis planning (CASP), not all generated routes are chemically feasible due to steps like protection and deprotection needed for product formation. Widely accepted NLP metrics like BLEU354 and ROUGE355 focus on precision and recall by computing n-gram overlaps between generated and reference texts. Similarly, in retrosynthesis, reactant–product pairs can be treated as overlapping bigrams. Retro-BLEU uses a modified BLEU score, emphasizing precision over recall, as there is no absolute best route for retrosynthesis. Although not yet applied to LLM-based predictions, this approach has value by allowing future performance comparison with a single standard.
Finally, by expanding the use of encoder–decoder architectures outside synthesis prediction into molecular generation, Fang et al.327 introduced MOLGEN, a BART-based pretrained molecular language model, in a 2023 preprint updated in 2024. MOLGEN addressed three key challenges: generating valid SMILES strings, avoiding an observed bias that existed against natural product-like molecules, and preventing hallucinations of molecules that didn't retain the intended properties. Pretrained on 100 million molecules using SELFIES162 and a masked language model approach, MOLGEN predicts missing tokens to internalize chemical grammar. An additional highlight of this work is how MOLGEN uses “domain-agnostic molecular prefix tuning”. This technique integrates domain knowledge directly into the model's attention mechanisms by adding molecule-specific prefixes, trained simultaneously with the main model across various molecular domains. The model's parameters would thus be adjusted to better capture the complexities and diversities of molecular structures, and domain-specific insights would be seamlessly integrated. To prevent molecular hallucinations, MOLGEN employs a chemical feedback mechanism, to autonomously evaluate generated molecules for appropriate properties, to guide learning and optimization. Such feedback foreshadows a core aspect of autonomous agents, which is their capacity for reflection. We will explore this further below.
The advancements in synthesis prediction and molecular generation using encoder–decoder architectures have revolutionized the field, moving from rigid, template-based models to more flexible, template-free approaches. Early work with LSTMs and GRUs laid the foundation, while transformer-based models like Molecular Transformer and Chemformer set new benchmarks in accuracy and versatility. New metrics, such as Retro-BLEU, and domain-aware techniques, like MOLGEN's prefix tuning, have further refined predictions and molecular design. These innovations, coupled with self-correcting mechanisms, point to a future of autonomous molecular design, where AI agents can predict, evaluate, and optimize synthetic pathways and molecular properties, accelerating chemical discovery.
3.5 Multi-modal LLMs
We have demonstrated the impact of LLMs on chemistry through their ability to process textual representations of molecules and reactions. However, LLMs can also handle diverse input modalities, representing molecular and chemical data in various formats.356–358 In chemistry, data can be represented in various forms, each providing unique insights and information (see Section 3.1). Chemical representations can be broadly classified into 1D, 2D, and 3D categories, depending on how much structural detail they convey.148,149 1D representations include basic numerical descriptors, such as molecular features and fingerprints, as well as textual representations like SMILES,43 SELFIES,162 and IUPAC names. These descriptors vary in the amount of chemical information they carry.359 2D representations involve graph-based structures and visual formats, which can be extended with geometric information to produce 3D representations. Examples of 3D representations include molecular graphs enriched with spatial data, molecular point clouds, molecular grids, and 3D geometry files.360Some of these representations can be input into models in different ways. For instance, a point cloud can be expressed either as a vector of coordinates (numerical input) or as a text-based PDB file. However, due to the distinct nature of the information conveyed, we treat textual descriptions of different molecular representations as separate modalities, even though both are technically strings. Additionally, molecule images have been utilized to train transformer-based models.361 However, spectral data—such as Nuclear Magnetic Resonance (NMR), Infrared (IR) spectroscopy, and mass spectrometry, remain underexplored as inputs for LLM-based applications.
Multi-modal LLMs leverage and integrate these diverse data types to enhance their predictive and analytical capabilities. This integration improves the accuracy of molecular property predictions and facilitates the generation of novel compounds with desired properties. A key example is Text2Mol proposed by Edwards et al.331 in 2021, which integrates natural language descriptions with molecular representations, addressing the cross-lingual challenges of retrieving molecules using text queries. The researchers created a paired dataset linking molecules with corresponding text descriptions and developed a unified semantic embedding space to facilitate efficient retrieval across both modalities. This was further enhanced with a cross-modal attention-based model for explainability and reranking. One stated aim was to improve retrieval metrics, which would further advance the ability for machines to learn from chemical literature.
In their 2022 follow-up, MolT5, Edwards et al.330 expanded on earlier work by utilizing both SMILES string representations and textual descriptions to address two tasks: generating molecular captions from SMILES and predicting molecular structures from textual descriptions of desired properties. However, several key challenges remain. Molecules can be described from various perspectives, such as their therapeutic effects, applications (e.g., aspirin for pain relief or heart attack prevention), chemical structure (an ester and a carboxylic acid connected to a benzene ring in ortho geometry), or degradation pathways (e.g., breaking down into salicylic acid and ethanoic acid in moisture).362 This complexity demands expertise across different chemistry domains, unlike typical image captioning tasks involving everyday objects (e.g., cats and dogs), which require minimal specialized knowledge. Consequently, building large, high-quality datasets pairing chemical representations with textual descriptions is a challenging task.
Moreover, standard metrics like BLEU, effective in traditional NLP, are insufficient for evaluating molecule-text tasks. To address these challenges, Edwards et al.330 employed a denoising objective, training the model to reconstruct corrupted input data, thereby learning the structure of both text and molecules. Fine-tuning on gold-standard annotations further improved the model's performance, enhancing previous Text2Mol metrics331 and enabling MolT5 to generate accurate molecular structures and their corresponding captions.
Other multimodal approaches similarly target the fusion of chemical and linguistic data to advance applications in molecular design. Seidl et al.363 developed CLAMP, which combines separate chemical and language modules to predict biochemical activity, while Xu et al.364 presented BioTranslator, a tool that translates text descriptions into non-text biological data to explore novel cell types, protein function, and drug targets. These examples highlight the growing trend of using language-based interfaces to enhance molecular exploration. The potential of multimodal LLMs extends beyond chemistry into more interactive and accessible tools. ChatDrug, by Liu et al.,365 integrates multimodal capabilities through a prompt module, a retrieval and domain feedback module, and a conversation module for systematic drug editing. It identifies and manipulates molecular structures for better interpretability in pharmaceutical research. Similarly, Christofidellis et al.328 introduced a multi-domain, multi-task language model capable of handling tasks across both chemical and natural language domains without requiring task-specific pretraining. Describe Joint Multi-domain Pre-training (JMP), which operates on the hypothesis that pre-training across diverse chemical domains, showed improved generalization for a foundational model. In this context, Liu et al.366 developed MolXPT, introduced MolXPT, which further demonstrated the strength of multimodal learning by achieving robust zero-shot molecular generation.
Finally, models that integrate even more diverse data types, such as GIT-Mol,367 which combines graphs, images, and text, and MolTC,368 which integrates graphical information for molecular interaction predictions illustrate how multimodal data improves accuracy and generalizability. Moreover, multimodal fusion models like PremuNet369 and 3M-Diffusion, Zhu et al.370 which use molecular graphs and natural language for molecule generation, represent a significant leap forward in the creation of novel compounds. Gao et al.371 advanced targeted molecule generation with DockingGA, combining transformer neural networks with genetic algorithms and docking simulations for optimal molecule generation, utilizing Self-referencing Chemical Structure Strings to represent and optimize molecules. Zhou et al.372 developed TSMMG, a teacher-student LLM designed for multi-constraint molecular generation, leveraging a large set of text–molecule pairs to generate molecules that satisfy complex property requirements. Gong et al.373 introduced TGM-DLM, a diffusion model for text-guided molecule generation that overcomes limitations of autoregressive models in generating precise molecules from textual descriptions. These advances culminate in works like MULTIMODAL-MOLFORMER by Soares et al.,374 which integrates chemical language and physicochemical features with molecular embeddings from MOLFORMER,211 significantly enhancing prediction accuracy for complex tasks like biodegradability and PFAS toxicity.
Overall, the shift to multimodal LLMs represents a robust approach to molecular design. By integrating diverse data sources, these models significantly enhance accuracy, interpretability, and scalability, opening new avenues for drug discovery, material design, and molecular property prediction. Combining linguistic, chemical, and graphical data into unified frameworks enables AI-driven models to make more informed predictions and generate innovative molecular structures.
3.6 Textual scientific LLMs
LLMs are large neural networks known for their performance across various machine learning tasks, with the main advantage of not requiring well-structured data like molecular descriptors.375 Their true power lies in their ability to handle more challenging tasks, such as extracting insights from less structured data sources like scientific texts or natural language descriptions. In chemistry, this opens doors to new methods of data extraction, classification, and generation, although it depends heavily on the availability of high-quality and diverse datasets (as discussed in Section 3.1). Unfortunately, many datasets are locked behind paywalls or are not machine-readable, limiting the full potential of LLMs in scientific applications. Encouraging open data initiatives and standardization of formats will play a vital role in expanding LLM applications in chemistry and related fields.Guo et al.212 argue that manually curating structured datasets is a sub-optimal, time-consuming, and labor-intensive task. Therefore, they automated data extraction and annotation from scientific papers using ChemDataExtractor379 and their in-house annotation tool.380 Text extraction tasks, like NER, can be formulated as multi-label classification tasks, which motivates using NER-like approaches and LLMs to extract structured data directly from unstructured text. LLMs developed for data mining include the work of Zhang et al.381 and Chen et al.382
Text extraction tasks, like NER, can be formulated as multi-label classification tasks, which motivates using NER-like approaches and LLMs to extract structured data directly from unstructured text. LLMs developed for data mining include the work of Zhang et al.381 and Chen et al.382 Building upon this, Wang et al.383 conducted a study comparing GPT-4 and ChemDataExtractor379 for extracting band gap information from materials science literature. They found that GPT-4 achieved a higher level of accuracy (correctness 87.95% vs. 51.08%) without the need for training data, demonstrating the potential of generative LLMs in domain-specific information extraction tasks. Additionally, LLMs with support for image inputs have been shown to enable accurate data extraction directly from images of tables.196 A detailed discussion can be found in the study by Schilling-Wilhelmi et al.384
In contrast to broad domain models, some LLMs focus on narrow, specialized fields to improve performance. ChemBERT212 was pretrained using a BERT model to encode chemical reaction information, followed by fine-tuning a NER head. ChemBERT outperformed other models such as BERT87 and BioBERT221 in the product extraction task, presenting an improvement of ∼6% in precision. For product role labeling, that is by identifying the role an extracted compound plays in a reaction, ChemBERT showed a ∼5% improvement in precision. This suggests that training on narrower datasets enables models to learn specific patterns in the data more effectively.
This trend continued with MatSciBERT,210 and MaterialsBERT.385 With MatSciBERT, Gupta et al.210 fine-tuned SciBERT220 on the Material Science Corpus (MSC), a curated dataset of materials extracted from Elsevier's scientific papers and improved article subject classification accuracy by 3% compared to SciBERT. In a similar vein, with MaterialsBERT, Shetty et al.385 fine-tuned PubMedBERT215 on 2.4 million abstracts, showing incremental precision improvements in NER tasks. BatteryBERT208 also followed this strategy, outperforming baseline BERT models in battery-related tasks.
Considerable effort has also been devoted to developing LLMs for biology tasks, following a similar trend of training models on large corpora such as Wikipedia, scientific databases, and textbooks, and then fine-tuning them for specific downstream tasks. Shin et al.214 pretrained various sizes of Megatron-LM,386 another BERT-like LLM, to create the BioMegatron family of models. These models, which had 345M, 800M, and 1.2B parameters and vocabularies of either 30k or 50k tokens, were pretrained using abstracts from the PubMed dataset and full-text scientific articles from PubMed Central (PMC), similar to BioBERT.221
Surprisingly, the largest 1.2B model did not perform better than the smaller ones, with the 345M parameter model using the 50k tokens vocabulary consistently outperforming others in tasks like Named Entity Recognition (NER) and Relation Extraction (RE). NER identifies specific entities, such as chemicals or diseases, while RE determines the relationships between them—both crucial for structuring knowledge from unstructured data. These processes streamline research by converting raw textual information into structured, useable formats for further analysis. This suggests that, for certain tasks, increasing model size does not necessarily lead to better performance. The relevance of model size was more apparent in the SQuAD387 dataset, suggesting that LLMs trained on smaller, domain-specific datasets may face limitations in broader generalization.
BioBERT221 pretrained using data from Wikipedia, textbooks, PubMed abstracts, and the PMC full-text corpus, outperformed the original BERT in all tested benchmarks, and in some cases even achieved state-of-the-art (SOTA) performance in benchmarks such as NCBI disease, 2010 i2b2/VA, BC5CDR, BC4CHEMD, BC2GM, JNLPBA, LINNAEUS, and Species-800. Peng et al.388 developed BlueBERT, a multi-task BERT model, which was evaluated on the Biomedical Language Understanding Evaluation (BLUE) benchmark.218 BlueBERT was pretrained on PubMed abstracts and MIMIC-III,376 and fine-tuned on various BLUE tasks, showing performance similar to BioBERT across multiple benchmarks.
PubMedBERT,215 following the approach adopted in SciBERT, created a domain-specific vocabulary using 14M abstracts from PubChem for pretraining. In addition to pretraining, the team curated and grouped biomedical datasets to develop BLURB, a comprehensive benchmark for biomedical natural language processing (NLP) tasks, including NER, sentence similarity, document classification, and question-answering. Gu et al.215 demonstrated that PubMedBERT significantly outperformed other LLMs in the BLURB benchmark, particularly in the PubMedQA and BioQSA datasets. The second-best model in these datasets was BioBERT, emphasizing the importance of domain-specific training for high-performance LLMs in biomedical applications.
Text classification using LLMs, particularly in biomedicine and materials science, has demonstrated that domain-specific pretraining is most effective for enhancing model performance. Models like BioBERT, BlueBERT, and PubMedBERT highlight how focusing on specialized datasets, such as PubMed and MIMIC-III, improves accuracy in tasks like NER, RE, and document classification. These advances illustrate how narrowing the training scope to relevant data enables more effective extraction of structured information from unstructured scientific texts.
In the broader context of this work, text classification serves as a key element that allows AI models to interface with chemical, biological, and medical literature, thereby accelerating progress in drug design, materials discovery, and other research fields. This ability to classify and extract relevant information from scientific texts directly impacts the efficiency and precision of data interpretation, facilitating real-world applications across multiple domains.
The Darwin model, as outlined by Xie et al.,280 is one such example. It fine-tunes LLaMA-7B on FAIR, a general QA dataset, followed by specific scientific QA datasets. Instructions for scientific QA were sourced from SciQ389 and generated using the Scientific Instruction Generation (SIG) model, a tool fine-tuned from Vicuna-7B that converts full-text scientific papers into question–answer pairs. This multi-step training process significantly improved Darwin's performance on regression and classification benchmarks. Notably, LLaMA-7B fine-tuned only on FAIR achieved nearly the same results as the fully fine-tuned model on six out of nine benchmarks, indicating that the integration of domain-specific datasets may not always require extensive fine-tuning for performance gains.
Similarly, Song et al.390 created HoneyBee by fine-tuning LLaMA-7B and LLaMa-13B on MatSci-Instruct, a dataset with ∼52k instructions curated by the authors. HoneyBee outperformed other models, including MatBERT, MatSciBERT, GPT, LLaMa, and Claude, within its specialized dataset. However, Zhang et al.391 showed that HoneyBee did not generalize well to other benchmarks, such as MaScQA200 and ScQA,392 highlighting the limitations of models trained on narrow domains in terms of broader applicability.
In biology, BioGPT282 pretrained a GPT-2 model architecture using 15M abstracts from PubChem corpus. BioGPT was evaluated across four tasks and five benchmarks, including end-to-end relation extraction on BC5CDR, KD-DTI, and DDI, question-answering on PubMedQA, document classification on HoC, and text generation on all these benchmarks. After fine-tuning on these tasks (excluding text generation), BioGPT consistently outperformed encoder-only models like BioBERT and PubMedBERT, particularly in relation extraction and document classification. Focusing specifically on text generation, the authors compared BioGPT's outputs to those of GPT-2, concluding that BioGPT was superior, although no quantitative metric was provided for this comparison.
Building on these ideas, Wu et al.281 pretrained LLaMA2 with the MedC-k dataset, which included 4.8M academic papers and 30k textbooks. This model was further refined through instruction tuning using the MedC-I dataset, a collection of medical QA problems. PMC-LLaMA281 outperformed both LLaMa-2 and ChatGPT on multiple biomedical QA benchmarks, even though it was ∼10 times smaller in size. Notably, the model's performance on MedQA,393 MedMCQA,394 and PubMedQA121 benchmarks improved progressively as additional knowledge was incorporated, the model size increased, and more specific instructions were introduced during tuning.
Text generation through decoder models has significantly expanded the applications of LLMs in scientific fields by enabling the generation of contextual answers and labels from scientific data. Unlike encoder-only models that rely on predefined classifications, decoder models such as Darwin, HoneyBee, and BioGPT can produce outputs tailored to domain-specific needs. This capability is important in fields like biomedicine, where accurate question-answering and document generation are highly valued. By leveraging multi-step pretraining and fine-tuning on specialized datasets, decoder models offer greater flexibility in handling both general and domain-specific tasks.
In the broader context of this work, text generation marks a key methodological advance that complements other LLM tasks, such as classification and extraction. The ability to generate structured responses and create new text from scientific literature accelerates research and discovery across chemistry, biology, and medicine. This generative capacity bridges the gap between raw data and meaningful scientific insights, equipping AI-driven models with a more comprehensive toolkit for addressing complex research challenges.
3.7 The use of ChatGPT in chemistry
With the rise of ChatGPT, we review here how many researchers have wanted to test the capability of such an accessible decoder-only LLM. Castro Nascimento and Pimentel395 wrote the first notable paper on ChatGPT's impact on Chemistry. The authors emphasize that LLMs, trained on extensive, uncurated datasets potentially containing errors or secondary sources, may include inaccuracies limiting their ability to predict chemical properties or trends. The paper highlighted that while LLMs could generate seemingly valid responses, they lacked true reasoning or comprehension abilities and would perpetuate existing errors from their training data. However, the authors suggested that these limitations could be addressed in the future. The work serves as a benchmark to qualitatively assess improvements in generative pretrained transformers. For example, five tasks were given to ChatGPT (GPT-3). The accuracy for converting compound names to SMILES representations and vice versa was about 27%, with issues in differentiating alkanes and alkenes, benzene and cyclohexene, or cis and trans isomers. ChatGPT found reasonable octanol–water partition coefficients with a 31% mean relative error, and a 58% hit rate for coordination compounds' structural information. It had a 100% hit rate for polymer water solubility and a 60% hit rate for molecular point groups. Understandably, the best accuracies were achieved with widely recognized topics. The authors concluded that neither experimental nor computational chemists should fear the development of LLMs or task automation; instead, they advocated for enhancing AI tools tailored to specific problems and integrating them into research as valuable facilitators.The use of ChatGPT in chemistry remains somewhat limited. Studies by Humphry and Fuller,396 Emenike and Emenike,397 and Fergus et al.398 focus on its role in chemical education. Some research also explores ChatGPT's application in specific areas, such as the synthesis and functional optimization of Metal–Organic Frameworks (MOFs), where computational modeling is integrated with empirical chemistry research.399–402 Deb et al.403 offer a detailed yet subjective evaluation of ChatGPT's capabilities in computational materials science. They demonstrate how ChatGPT assisted with tasks like identifying crystal space groups, generating simulation inputs, refining analyses, and finding relevant resources. Notably, the authors emphasize ChatGPT's potential to write code that optimizes processes and its usefulness for non-experts, particularly in catalyst development for CO2 capture.
Three key points emerge regarding the use of ChatGPT alone. First, reliable outputs depend on precise and detailed input, as Deb et al.403 found when ChatGPT struggled to predict or mine crystal structures. Second, standardized methods for reproducing and evaluating GPT-based work remain underdeveloped. Third, achieving complex reasoning likely requires additional chemical tools or agents, aligning with Bloom's Taxonomy.404,405 Bloom's Taxonomy organizes educational objectives into hierarchical levels: remembering, understanding, applying, analyzing, evaluating, and creating. These range from recalling facts to constructing new concepts from diverse elements. While LLMs and autonomous agents can support lower-level tasks, they currently fall short of replicating higher-order cognitive skills comparable to human expertise.
Currently, LLMs and autonomous agents are limited in replicating higher-level thinking compared to human understanding. To better assess LLMs' capabilities in this domain, we propose using Bloom's Taxonomy as a quality metric.404,405 This framework offers a structured approach for evaluating the sophistication of LLMs and autonomous agents, especially when addressing complex chemical challenges. It can help quantify their ability to engage in higher-level reasoning and problem-solving.
This increased productivity may be the only possible approach to exploring the vastness of all chemical space. To fully leverage AI in property prediction, inverse design, and synthesis prediction, it must be integrated with automated synthesis, purification, and testing. This automation should be high-throughput and driven by AI-based autonomous decision-making (sometimes called “lights-out” automation). Janet et al.411 highlighted challenges in multi-step reactions with intermediate purifications, quantifying uncertainty, and the need for standardized recipe formats. They also stated the limitations of automated decision-making. Organa417 addresses some of these challenges. It can significantly reduce physical workload and improve users' lab experience by automating diverse common lab routine tasks such as solubility assessment, pH measurement, and recrystallization. Organa interacts with the user through text and audio. The commands are converted into a detailed LLM prompt and used to map the goal to the robot's instructions. Interestingly, Organa is also capable of reasoning over the instructions, giving feedback about the experiments, and producing a written report with the results.
Other limitations exist, like a machine being restricted to pre-defined instructions, its inability to originate new materials, and the lower likelihood of lucky discoveries. Yet, when dedicated tools can be connected to address each step of an automated chemical design, these limitations can be systematically addressed through advancements in LLMs and autonomous agents, discussed in the next section.
4 LLM-based autonomous agents
The term “agent” originates in philosophy, referring to entities capable of making decisions.418 Hence, in artificial intelligence, an “agent” is a system that can perceive its environment, make decisions, and act upon them in response to external stimuli.419 Language has enabled humans to decide and act to make progress in response to the environment and its stimuli, and so LLMs are naturally ideal for serving as the core of autonomous agents. Thus, in agreement with Gao et al.,420 we define a “language agent” as a model or program (typically based on LLMs) that receives an observation from its environment and executes an action in this environment. Here, environment means a set of tools and a task. Hence, “LLM-based autonomous agents” refer to language agents whose core is based on an LLM model. Comprehensive analyses of these agents are available in the literature,419–421 but this section highlights key aspects to prepare the reader for future discussions.There is no agreed definition of the nomenclature to be used to discuss agents. For instance, Gao et al.420 created a classification scheme that aims to group agents by their autonomy in biological research. This means a level 0 agent has no autonomy and can only be used as a tool, while a level 3 agent can independently create hypotheses, design experiments, and reason.
Following this perspective, Wang et al.421 categorizes agent components into four modules: profiling, memory, planning, and action. In contrast, Weng422 also identifies four elements — memory, planning, action, and tools — but with a different emphasis. Meanwhile, Xi et al.419 proposes a division into three components: brain, perception, and action, integrating profiling, memory, and planning within the brain component, where the brain is typically an LLM. Recently, Sumers et al.423 proposed Cognitive Architectures for Language Agents (CoALA), a conceptual framework to generalize and ease the design of general-purpose cognitive language agents. In their framework, a larger cognitive architecture composed of modules and processes is defined. CoALA defines a memory, decision-making, and core processing module, in addition to an action space composed of both internal and external tools. While internal tools mainly interact with the memory to support decision-making, external tools make up the environment, as illustrated in Fig. 6. Given a task that initiates the environment, the “decision process” runs continuously in a loop, receiving observations and executing actions until the task is completed. For more details, read Sumers et al.423
 | ||
| Fig. 6 Agent's architecture as defined in this review. According to our definition, an agent is composed of a central program (typically an LLM and the code to implement the agent's dynamic behavior) and the agent modules. The agent continuously receives observations from the environment and decides which action should be executed to complete the task given to it. Here, we define the agent as the set of elements whose decision is trainable, that is, the LLM, the agent code, the decision process, and the agent modules. Given a task, the agent uses the agent modules (memory, reasoning, planning, profiling) and the LLM to decide which action should be executed. This action is executed by calling a tool from the environment. After the action is executed, an observation is produced and fed back to the agent. The agent can use perception to receive inputs in different modalities from the environment. (A) Description of agent modules, (B) illustration of the agent architecture, (C) illustration of the environment components, (D) description of tools elements present in the environment. | ||
In this review, we define an autonomous agent system as a model (typically an LLM) that continuously receives observations from the environment and executes actions to complete a provided task, as described by Gao et al.420 Nevertheless, in contrast to CoALA,420 we will rename “internal tools” as “agent modules” and “external tools” simply as “tools”, for clarity. The agent consists of trainable decision-making components such as the LLM itself, policy, memory, and reasoning scheme. In contrast, the environment comprises non-trainable elements like the task to be completed, Application Programming Interface (API) access, interfaces with self-driving labs, dataset access, and execution of external code. By referring to decision-making components as agent modules, we emphasize their inclusion as parts of the agent. By referring to non-trainable elements as tools, we highlight their role as part of the environment. We discuss six main types of actions. As shown in Fig. 6, four of the six, memory, planning, reasoning, and profiling are agent modules. The remaining two actions (or tools) and perception are part of the environment. Since the perception is how the agent interacts with the environment and is not a trainable decision, we therefore included it as part of the environment.
4.1 Memory module
The role of the memory module is to store and recall information from past interactions and experiences to inform future decisions and actions. There are multiple types of memory in agents, namely sensory memory, short-term memory, and long-term memory. A major challenge in using agents is the limited context window, which restricts the amount of in-context information and can lead to information loss, thereby impacting the effectiveness of short-term and long-term memory. Solutions involve summarizing memory content,424 compressing memories into vectors,425–427 and utilizing vector databases428 or combinations thereof,429 with various databases available such as ChromaDB, FAISS, Pinecone, Weaviate, Annoy, and ScaNN.430 Addressing these challenges to enhance agent memory continues to be a significant area of research.431 Sensory, or procedural memory is knowledge embedded into the model's parameters during pretraining and/or in heuristics implemented into the agent's code. Short-term, or working, memory includes the agent's finite knowledge during a task, incorporating interaction history and techniques like in-context learning93 (ICL), which leverages the limited input's context length for information retention. Long-term memory involves storing information externally, typically through an embedded vector representation in an external database. In the original CoALA420 paper, long-term memory is further categorized as episodic, which registers previous experiences, and semantic, which stores general information about the world.4.2 Planning and reasoning modules
The planning and reasoning module is made of two components. Planning involves identifying a sequence of actions required to achieve a specified goal. In the context of language agents, this means generating steps or strategies that the model can follow to solve a problem or answer a question, which can be enhanced with retrieval from previous experiences,432 and from feedback from post-execution reasoning.433,434 We note that Retrieval-Augmented Generation (RAG) enhances the planning phase by enabling models to access external knowledge bases, integrating retrieved information into the generation process. This approach improves accuracy and relevance, especially when handling complex or knowledge-intensive tasks. Reasoning refers to the process of drawing conclusions or making decisions based on available information and logical steps. For example, there are studies that demonstrate the benefits of LLM reasoning for question answering, where new context tokens can be integrated in a step-by-step way to guide the model towards more accurate answers.435–440 One popular reasoning strategy is Chain-of-Thought (CoT),107,437,441–444 a reasoning strategy which substantially boosts QA performance by generating intermediate reasoning steps in a sequential manner. CoT involves breaking down complex problems into smaller, manageable steps, allowing the model to work through reasoning one step at a time rather than attempting to solve the entire problem at once. CoT thereby reduces hallucinations and enhances interpretability, as demonstrated by improved results in models like PaLM445 and GPT-3 with benchmarks like GSM8K,446 SVAMPs,447 and MAWPS.448In advanced reasoning, final tasks are often decomposed into intermediary ones using a cascading approach, similar to Zero-shot-CoT436 and RePrompt.433 However, while CoT is considered as single-path reasoning, CoT extensions like Tree-of-Thoughts,439 Graph-of-Thoughts,449 Self-consistent CoT,438 and Algorithm-of-Thoughts450 offer multi-path reasoning. Furthermore, other models have pitted multiple agents against each other to debate or discuss various reasoning paths,451–453 while others use external planners to create plans.454,455 A feedback step during the execution of the plan was a further extension of the CoT ideas; this enables agents to refine their actions based on environmental responses adaptively, which is crucial for complex tasks.456,457
Another interesting reasoning scheme is the Chain-of-Verification(CoVe),434 where once an answer is generated, another LLM is prompted to generate a set of verification questions to check for agreement between the original answer and the answers to the verification questions such that the final answer can be refined. The ReAct439 – Reason + Act – model proposes adding an observation step after acting. This means the LLM first reasons about the task and determines the necessary step for its execution, it performs the action and then observes the action's result. Reasoning on that result, it can subsequently perform the following step. Similarly, Reflexion107 also implements a reasoning step after executing an action. However, Reflexion implements an evaluator and self-reflection LLMs to not only reason about each step but also to evaluate the current trajectory the agent is following using a long-term memory module. As the context increases, it may become challenging for agents to deal with the long prompt. Aiming to solve this issue, the Chain-of-Agents (CoA)80 extends reasoning schemes that leverage multi-agent collaboration to reason over long contexts. This framework employs workers and manager agents to process and synthesize information to generate the final response. CoA demonstrated improvements of up to 10% when compared against an RAG baseline.
ReAct and Reflexion are closed-ended approaches where the agent starts with all the tools and must determine which to use. To address more open-world challenges, Wang et al.458 introduced the Describe, Explain, Plan, and Select (DEPS) method, which extends this approach. Lastly, human inputs can also be used to provide feedback to the agent. Providing feedback using a human-in-the-loop approach is particularly interesting in fields where safety is a main concern.
4.3 Profiling module
LLMs can be configured to perform in specific roles, such as coders, professors, students, and domain experts, through a process known as profiling. Language agents can thus incorporate the profile through the LLM or through the agent code. The profiling approach involves inputting psychological characteristics to the agent, significantly impacting its decision-making process.459–462 Profiling enables the creation of multi-agent systems that simulate societal interactions, with each agent embodying a unique persona within the group.432,463 The most prevalent technique for profiling, called “handcrafting”, requires manually defining the agent's profile, often through prompts or system messages.464,465 While profiling can also be automated with LLMs,466 that automation method may only be suited for generating large numbers of agents since it offers less control over their overall behavior. An interesting application of profiling is the development of agent sets that reflect demographic distributions.4674.4 Perception
Perception is an analog to the human sensory system, which interprets multimodal information such as text, images, or auditory data, transforming it into a format comprehensible by LLMs, as demonstrated by SAM,468 GPT4-V,469 LLaVa,470 Fuyu8B,134 and BuboGPT.471 In our proposed architecture, the perception is responsible for converting the task and the observations to a data representation that can be understood by the agent. Moreover, advancements in LLMs have led to the development of even more versatile models, such as the any-to-any Next-GPT135 and the any-to-text Macaw-LLM.472 Employing such multimodal LLMs in decision-making processes can simplify perception tasks for agents, with several studies exploring their use in autonomous systems.473,4744.5 Tools
In our proposed definition (see Fig. 6b), tools or actions are part of the environment. The agent can interact with this environment by deciding which action to execute through the decision-making process. The set of all possible actions that can be selected is also known as the “action space”.The decision process is composed of three main steps: proposal, evaluation, and selection. During the proposal, one or more action candidates are selected using reasoning,439 code structures,432,475 or simply by selecting every tool available.435,438,476,477 The evaluation process consists of evaluating each selected action according to some metric to predict which action would bring more value to the agent. Lastly, the action is selected and executed.
Given that pretrained parameters (sensory memory) are limited, the model must use tools for complex tasks in order to provide reliable answers. However, LLMs need to learn how to interact with the action space and how and when to use those tools most accurately.478 LLMs can be pretrained or fine-tuned with examples of tool use, enabling them to operate tools and directly retrieve tool calls from sensory memory during a zero-shot generation.479 Recent studies investigate this approach, particularly focusing on open-source LLMs.480–482
As foundational AI models become more advanced, their abilities can be expanded. It was shown that general-purpose foundation models can reason and select tools even with no fine-tuning. For example, MRKL483 implements an extendable set of specialized tools known as neuro-symbolic modules and a smart “router” system to retrieve the best module based on the textual input. Specifically, this router smartly parses the agent's output and selects which neuro-symbolic module is more suitable to perform the task following some heuristic. These neuro-symbolic modules are designed to handle specific tasks or types of information and are equipped with built-in capabilities and task-relevant knowledge. This pre-specialization allows the model to perform domain-specific tasks without needing a separate, domain-specific dataset. This design addresses the problem of LLMs lacking domain-specific knowledge and eliminates the need for the costly and time-consuming LLM fine-tuning step, using specialized data annotation.484 The router can receive support from a reasoning strategy to help select the tools484 or follow a previously created plan.458 Recent advances have shown that LLMs can develop new tools of their own,485–487 enabling agents to operate, as needed, in dynamic and unpredictable “open-worlds”, on unseen problems as illustrated by Voyager.475 This capability allows agents to evolve and improve continually.
5 LLM-based autonomous agents in scientific research
The previous section introduced key concepts relevant to any description of the development of autonomous agents. Here, we now focus on which agents were developed for scientific purposes, and ultimately for chemistry. Previous sections of this review have discussed how LLMs could be powerful in addressing challenges in molecular property prediction, inverse design, and synthesis prediction. When we consider the value of agents in chemistry and the ability to combine tools that, for example, search the internet for established synthetic procedures, look up experimental properties, and control robotic synthesis and characterization systems, we can see how autonomous agents powerfully align with the broader theme of automation, which will lead to an acceleration of chemical research and application (Table 4). 488 and multi-agent systems with the icon
488 and multi-agent systems with the icon  .
.  ,
,  , and
, and  489 mean the agent bases his behavior on sensory, short, and long memory components, respectively. Besides the textual capabilities of LLM-based agents,
489 mean the agent bases his behavior on sensory, short, and long memory components, respectively. Besides the textual capabilities of LLM-based agents,  490 and
490 and  491 mean the agent has additional audio and visual perception, respectively. The release date column displays the date of the first publication for each paper. When available, the publication date of the last updated version is displayed between parentheses
491 mean the agent has additional audio and visual perception, respectively. The release date column displays the date of the first publication for each paper. When available, the publication date of the last updated version is displayed between parentheses
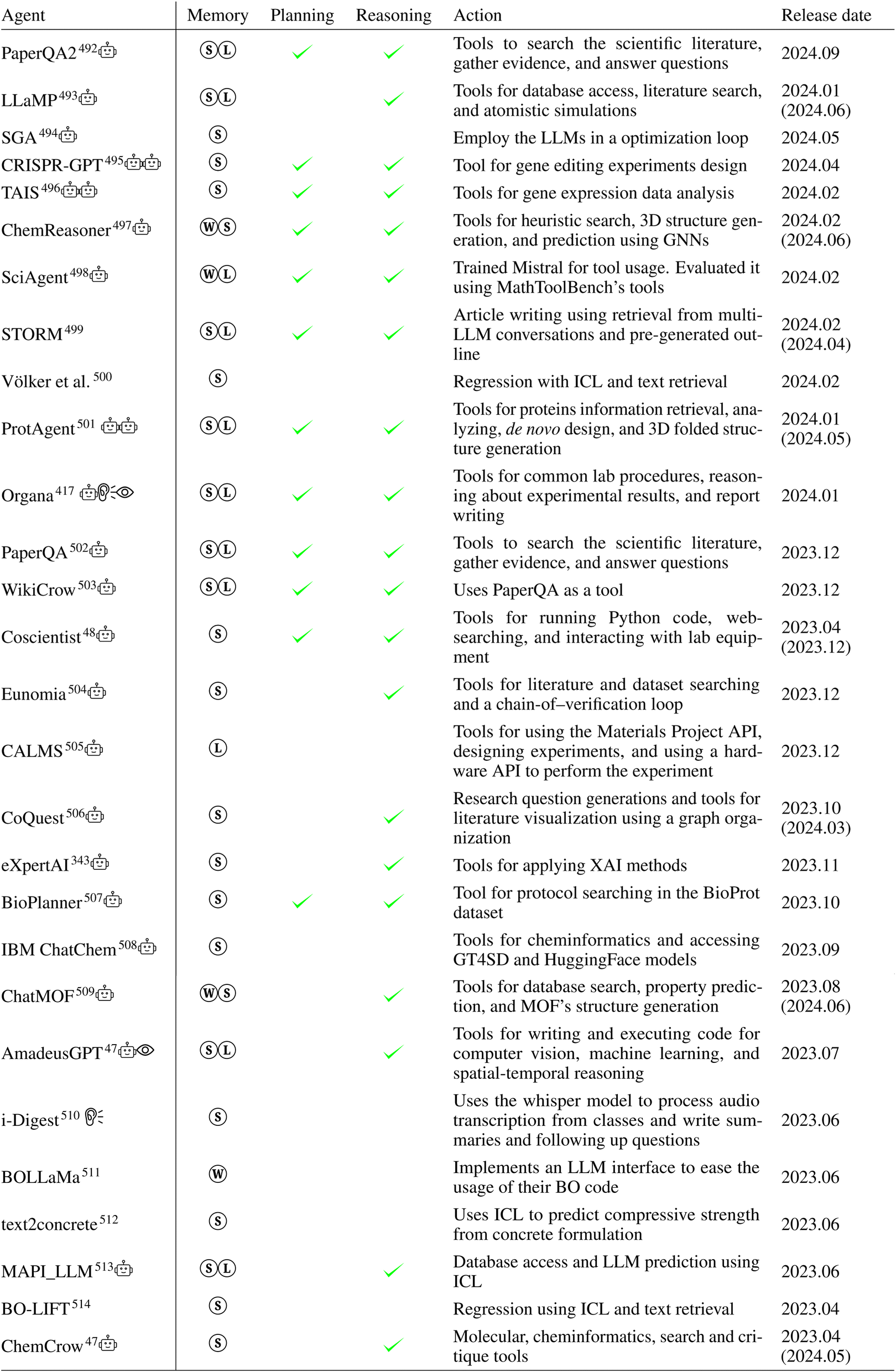
|
It was Hocky and White515 who discussed the early stages of models that could automate programming and, hence, the expected impacts in chemistry. Then, early work by White et al.516 applied LLMs that could generate code to a benchmark set of chemical problems. In that case, not only were LLMs demonstrated to possess a notable understanding of chemistry, based on accurate question answering, but White et al.516 imagined a potential to use them as base models to control knowledge augmentation and a variety of other tools. Thus, these LLMs could be used to execute routine tasks, optimize procedures, and enhance the retrieval of information from scientific literature across a range of scientific domains. To the best of our knowledge, this is the first review of autonomous agents in chemistry that have evolved since these two visionary conceptual perspectives. A deeper exploration follows below. One driving motivation for the need to augment LLMs with a more pertinent and dedicated knowledge base is the need to circumvent problems of a limited context prompt window, and the restriction that once an LLM is trained, any new information is beyond it’s reach since it necessarily has fallen outside its corpus of training data. Furthermore, LLMs are also known to hallucinate. Their predictions are probabilistic and, in science, if experimental evidence is available, then there is great value in building from known domain-specific information. Some improved prompt engineering can aid in the generation of results that are more likely to be accurate, but the use of autonomous agents may solve such problems completely in this next phase of AI in chemistry. In fact, even adding one or two components when building an agent, as opposed to a whole suite, has shown some significant gains.
Building on this foundation, Ramos et al.514 illustrated that LLMs could directly predict experimental outcomes from natural language descriptions, incorporating this ability into a Bayesian optimization (BO) algorithm to streamline chemical processes. Using in-context learning (ICL), where a model learns from examples provided during inference without requiring retraining, their approach avoided additional model training or fine-tuning, simplifying the optimization process. In a similar vein, Kristiadi et al.517 demonstrated similar results with a smaller, domain-specific model, using parameter-efficient fine-tuning (PEFT) rather than ICL. Ranković and Schwaller518 also explored BO using natural language. They used an LLM to encode chemical reaction procedures, described using natural language, and then trained a Gaussian process (GP) head to predict the reaction yield from the latent encoded representation of the procedure. By keeping the LLM frozen and only updating the multilayer perceptron (MLP) head, this approach minimized training time. Völker et al.500 extended these ideas by sampling multiple model completions and adding a verifier model to select the next best step in the BO algorithm. They also used ICL and a short-term memory component to optimize alkali-activated concrete mix design. These examples demonstrate how agent-based systems can execute complex optimization algorithms step by step, directly contributing to automation and more efficient experimental design.
To better promote new ideas regarding AI in scientific research, Jablonka et al.519 organized a one-day hackathon in March 2023 where participants developed 14 innovative projects addressing chemical problems centered on predictive modeling, automation, knowledge extraction, and education. Several agent-based approaches emerged from this hackathon. First, MAPI_LLM513 is an agent with access to the Materials Project API (MAPI) database that receives a query asking for a property of a material and then retrieves the relevant information from the dataset. If the material is not available on MAPI, the agent can search for similar materials and use in-context learning (ICL) to provide a prediction of the requested property. Additionally, MAPI_LLM also has a reaction module for synthesis proposal. Second, Rankovic et al.511 used LLMs to make BO algorithms more accessible to a broader group of scientists; BOLLaMa implements a natural language interface to easily interact with BO software developed by their group.520 Third, and similar to Ramos et al.514 and Ranković and Schwaller518 who employed LLMs in BO, Weiser et al.521 focused on genetic algorithms (GA), a different optimization algorithm. In GA, pieces of information are stochastically combined and evaluated to guide the algorithm during the optimization. For chemistry, these pieces are often molecular fragments that are combined to compose a final whole molecular structure. Thus, Weiser et al.521 used LLMs to implement common GA operators under the hypothesis that LLMs can generate new combined molecules better than random cross-over due to their sensory memory. Fourth, InsightGraph522 can draw general relationships between materials and their properties from JSON files. Circi and Badhwar522 showed that LLMs can understand the structured data from a JSON format and reorganize the information in a knowledge graph. Further refinement of this tool could automate the process of describing relationships between materials across various scientific reports, a task that remains labor-intensive today. Fifth, Kruschwitz et al.512 used ICL and LLMs to accurately predict the compressive strength of concrete formulations; Text2Concrete achieved predictive accuracy comparable with a Gaussian process regression (GPR) model, with the advantage that design principles can be easily added as context. This model was successfully applied in a BO algorithm following the Ramos et al.514 approach.500 For education purposes, multiple authors have raised the discussion about how LLMs can be used to support educators' and instructors' daily work.523–527 Finally, in this direction, Mouriño et al.510 developed i-Digest, an agent whose perception module can understand audio tracks and video recordings. These audio recordings are transcribed to text using the Whisper138 model, and therefore, i-Digest is a digital tutor that generates questions to help students test their knowledge about the course material. These are just a few examples to showcase the capabilities of AI systems to innovate and generate solutions rapidly.
More recently, Ma et al.498 showed that agents can be trained to use tools. SciAgent498 was developed under the premise that finetuning LLMs for domain-specific applications is often impractical. Nevertheless, the agent can be fine-tuned with a set of tools that will enable them to perform well in a domain-specific task. These tools, typically Python functions, enable SciAgent to plan, retrieve, and use these tools to facilitate reasoning and answer domain-related questions effectively. The benchmark developed for SciAgent, known as SciToolBench, includes five distinct domains, each equipped with a set of questions and corresponding tools. The development of its retrieval and planning modules involved finetuning different LLMs on the MathFunc benchmark, resulting in a notable performance improvement of approximately ∼20% across all domains in SciToolBench compared to other LLMs.
These examples demonstrate the rapidly growing potential of autonomous agents to drive innovation and automation across scientific tasks, from optimizing experiments and materials discovery to enhancing education. As these tools advance, they streamline processes, generate new insights, and empower researchers to tackle complex challenges. By combining reasoning, optimization, and tool usage in real time, agents mark a significant leap in AI-driven research. In the next section, we focus on how agents are transforming literature review processes, a critical aspect of scientific discovery.
5.1 Agents for literature review
Another fantastic opportunity for automation in the sciences is associated with high-quality literature review, a pivotal aspect of scientific research that requires reading and selecting relevant information from large numbers of papers, and thereby distilling the current state of knowledge relevant to a particular research direction. This extremely time-consuming task is being revolutionized by advanced AI tools designed to automate and enhance such analysis and summarization.PaperQA introduces a robust model that significantly reduces misinformation while improving the efficiency of information retrieval. This agent retrieves papers from online scientific databases, reasons about their content, and performs question-answering (QA) tasks. Its mechanism involves three primary components—“search”, “gather_evidence”, and “answer_question” and the authors adapted the Retrieval-Augmented Generation (RAG)528 algorithm to include inner loops on each step. For instance, PaperQA can perform multiple rounds of search and gather_evidence if, upon reflection, not have enough evidence has been acquired to successfully answer_question.
To further validate its capabilities, the authors developed a new benchmark called LitQA, specifically designed to evaluate the performance of models like PaperQA in solving complex, real-world scientific questions. LitQA focuses on tasks that mimic the intricacy of scientific inquiry, comprising 50 multiple-choice questions derived from biomedical papers published post-September 2021, ensuring that these papers were not included in the training data of LLMs. In this challenging setting, PaperQA not only meets but exceeds human performance, achieving a precision rate of 87.9% and an accuracy score of 69.5%, compared to the human baseline of 66.8%.502 By applying the RAG technique to full-text scientific papers, PaperQA sets a new standard in QA capabilities, achieving human-like performance in curated datasets without hallucination or selecting irrelevant citations.502
Building on top of PaperQA, WikiCrow exemplifies the practical application of AI in generating concise and relevant Wikipedia-style summaries. The authors show that while 16% of a human-created Wikipedia article comprises irrelevant statements, WikiCrow displays irrelevant information only 3% of the time. Their system also added 5% more correct citations when compared with original articles. Moreover, thanks to its foundation in the PaperQA framework,502 WikiCrow achieves remarkable cost-efficiency. The authors estimate that WikiCrow can accomplish in a few days what would take humans approximately 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours, or about 6.8 years, thereby underscoring its ability to rapidly produce extensive scientific content. This efficiency exemplifies the reliability and transformative potential of AI in content creation.503
000 hours, or about 6.8 years, thereby underscoring its ability to rapidly produce extensive scientific content. This efficiency exemplifies the reliability and transformative potential of AI in content creation.503
Following a different approach, the STORM model also addressed the problem of writing Wikipedia-like summaries, where the STORM acronym represents the Synthesis of Topic Outlines through Retrieval and Multi-perspective questions.499 This approach implements a two-step procedure. First, STORM retrieves multiple articles on a topic and uses an LLM to integrate various perspectives into a cohesive outline. Second, this outline is used to write each section of the Wikipedia-like summary individually. To create the outline, multiple articles discussing the topic of interest are retrieved by an “expert” LLM, which processes each one to create N perspectives. Each perspective is then fed to a “writer” LLM, and a conversation is initiated between writer and expert. Finally, the N conversations are used to design the final outline. The outline and the set of references, accessed by RAG, are given to the writer LLM. The writer LLM is prompted to use these inputs to generate each section of the article sequentially. Following this, all sections are merged and refined to eliminate redundancies and enhance coherence. Upon human evaluation, STORM is reported to be ∼25% more organized and present ∼10% better coverage when compared to a pure RAG approach. However, it was also less informative than human-written Wikipedia pages, and STORM presented a transfer of internet-borne biases, producing emotional articles, which is a major concern.
5.2 Agents for chemical innovation
Transitioning from literature synthesis to practical chemistry applications, we next explore how LLM-based agents have proven their capabilities to revolutionize routine chemical tasks toward an acceleration of molecular discovery and scientific research. Agents are flexible entities capable of developing prompt-specific workflows and executing a plan toward accomplishing a specific task. ChemCrow47 introduced a significant shift in how LLMs would be applied in chemistry, given that LLMs alone do not access information outside of their training data nor can they directly perform chemistry-related tasks.By augmenting LLMs with common chemical tools, computational or robotic, ChemCrow automates a broad spectrum of routine chemical tasks, demonstrating a significant leap in LLM applicability. Under human evaluation, ChemCrow consistently outperformed GPT-4, achieving an accuracy score of 9.24/10 compared to 4.79/10.47 The developers of ChemCrow have also considered the ethical implications and potential risks associated with its capabilities. ChemCrow's high potential could be misused and exploited for malicious objectives, and therefore the authors have implemented safety checks and guidelines to prevent such misuse, or “dual usage”. Additionally, they acknowledge that ChemCrow, relying on an LLM, may not always provide completely accurate answers due to gaps in its chemical knowledge. As such, they recommend careful and responsible use of the tool, along with thorough scrutiny of its outputs. In summary, while ChemCrow presents a powerful new chemical assistant,47 oversight of its use is required, and this agent's access to tools has been deliberately limited to enhance security and avoid misuse.
Similarly to ChemCrow,47 Chemist-X529 uses RAG to get up-to-date literature information and use it to reliably solve a user's questions. Nevertheless, Chemist-X focuses on designing chemical reactions to achieve a given molecule. It works in three phases: (1) first, the agent searches molecule databases for similar molecules to the given molecule, then (2) it searches online literature searching for chemical reactions capable of converting the list of similar molecules in the target. Lastly, (3) machine learning models are used to propose the reaction conditions. To validate their agent, the authors used Chemist-X to design a High-Throughput Screening (HTS) experiment aiming to produce 6-(1-methyl-1H-indazol-4-yl), resulting in a maximum yield of 98.6%.
Another system called Coscientist48 system exemplifies the integration of semi-autonomous robots in planning, conceiving, and performing chemical reactions with minimal human intervention. At its core, the system features a main module named ‘PLANNER’, which is supported by four submodules. These submodules, or tools, are responsible for performing actions such as searching the web for organic synthesis, executing Python code, searching the hardware documentation, and performing a reaction in an automated lab.48 Utilizing this framework, the Coscientist successfully conducted two types of chemical coupling reactions, Suzuki–Miyaura and Sonogashira, in a semi-automated fashion, with manual handling of initial reagents and solvents. Additionally, Coscientist was also used to optimize reaction conditions. In contrast to Ramos et al.,514 who used LLMs within a Bayesian Optimization (BO) algorithm as a surrogate model, Boiko et al.48 approached the optimization task as a strategic “game” aimed at maximizing reaction yield by selecting optimal reaction conditions. This demonstrates the ability of GPT-4 to effectively reason about popular chemical reactions – possibly via comprehensive coverage in pretraining. The authors have indicated that the code for their agent will be released following changes in U.S. regulations on AI and its scientific applications. At the time of writing, the code remains unreleased, but a simple example that calculates the square roots of random numbers has been provided to illustrate their approach.48 These examples underscore the transformative role of LLMs in enhancing and automating chemical processes, which will likely accelerate chemical discovery.
Automated workflows in protein research have also been explored. ProtAgent501 is a multi-agent system designed to automate and optimize protein design with minimal human intervention. This system comprises three primary agents: Planner, Assistant, and Critic. The Planner is tasked with devising a strategy to address the given problem, the Assistant executes the plan using specialized tools and API calls, and the Critic supervises the entire process, providing feedback and analyzing outcomes. These agents collaborate through a dynamic group chat managed by a fourth agent, the Chat Manager. Tasks executed by this team include protein retrieval and analysis, de novo protein design, and conditioned protein design using Chroma530 and OmegaFold.531
Similarly to ProtAgent, Liu et al.496 created a team of AI-made scientists (TAIS) to conduct scientific discovery without human intervention. However, their agents have roles analogous to human roles, such as project manager, data engineer, code reviewer, statistician, and domain expert. While in ProtAgent501 agents interact through the Chat Manager only, TAIS496 enables AI scientists to interact between themselves directly using pre-defined collaboration pipelines. To evaluate TAIS, the authors curated the Genetic Question Exploration (GenQEX) benchmark, which consists of 457 selected genetic data questions. As a case study, the authors show TAIS's answer to the prompt “What genes are associated with Pancreatic Cancer when considering conditions related to Vitamin D Levels?”. The system identified 20+ genes with a prediction accuracy of 80%.
Innovation can also be achieved by looking into data from a different point-of-view to get new insights. Automating querying databases was investigated by Ramos et al.513 with a ReAct agent with access to the MAPI dataset. This concept was extended by Chiang et al.493 using LLaMP,493 which is a RAG-based ReAct agent that can interact with MAPI, arXiv, Wikipedia, and has access to atomistic simulation tools. The authors showed that grounding the responses on high-fidelity information (a well-known dataset) enabled the agent to perform inferences without fine-tuning.
The agents in chemistry, as exemplified by ChemCrow47 and Coscientist,48 highlight a significant shift towards automation and enhanced efficiency in molecular discovery and scientific research. These systems demonstrate the potential of integrating LLMs with chemical tools and automation frameworks, achieving impressive accuracy and effectiveness in tasks ranging from routine chemical operations to complex reaction optimizations. Similarly, ProtAgent501 and TAIS496 systems showcase the versatility of multi-agent frameworks in automating protein design and genetic research, pushing the boundaries of what AI-driven scientific discovery can achieve. These studies collectively showcase the incredible potential of agents in chemical and biological research, promising automation of routine tasks, easing the application of advanced techniques and analyses, and accelerating discoveries. However, they also underscore the necessity for meticulous oversight and responsible development to harness their full potential while mitigating risks.532
5.3 Agents for experiments planning
Building on the capabilities of ChemCrow and Coscientist in automating chemistry-related tasks, recent advances have focused on bridging the gap between virtual agents and physical laboratory environments. For example, Context-Aware Language Models for Science (CALMS),505 BioPlanner,507 and CRISPR-GPT495 focus on giving support to researchers with wet-lab experimental design and data analysis.CALMS505 focuses on improving laboratory efficiency through the operation of instruments and management of complex experiments, employing conversational LLMs to interact with scientists during experiments. In addition, this agent can perform actions using lab equipment after lab equipment APIs have been provided to the agent as tools. CALMS was designed to enhance instrument usability and speed up scientific discovery, providing on-the-spot assistance for complex experimental setups, such as tomography scans, and enabling fully automated experiments. For instance, its capability was showcased through the operation of a real-world diffractometer. Although CALMS excelled in several tasks, a comparison between GPT-3.5 and Vicuna 1.5 revealed Vicuna's limitations in handling tools.
In contrast, BioPlanner507 significantly improves the efficiency of scientific experimentation by creating pseudocode representations of experimental procedures, showcasing AI's capacity to streamline scientific workflows. Therefore, Rather than interacting directly with lab equipment through APIs, BioPlanner creates innovative experimental protocols that can be expanded upon within a laboratory setting. The initial step in BioPlanner's process involves assessing the capability of LLMs to produce structured pseudocode based on detailed natural language descriptions of experimental procedures.
In testing, BioPlanner successfully generated correct pseudocode for 59 out of 100 procedures using GPT-4, although the most common errors involved omitted units. Afterward, the authors used BioPlanned to generate a procedure for culturing an E. coli bacteria colony and storing it with cryopreservation, which ran successfully.
Focusing on gene editing experiments, CRISPR-GPT495 is an agent developed to design experiments iteratively with constant human feedback. CRISPR-GPT495 aims to bridge the gap for non-experts by simplifying this process into manageable steps solvable by an LLM with access to useful tools. This agent operates in three modes based on user prompts: “Meta mode” provides predefined pipelines for common gene-editing scenarios; “Auto mode” uses the LLM to plan a sequence of tasks; and “Q&A mode” answers general questions about the experimental design. The authors demonstrate that based on human evaluations, CRISPR-GPT outperforms GPT-3.5 and GPT-4 in accuracy, reasoning, completeness, and conciseness. Additionally, they applied CRISPR-GPT to design real-world experiments for knocking out TGFBR1, SNAI1, BAX, and BCL2L1 in the human A375 cell line, achieving an editing efficiency of approximately 70% for each gene.
Following the ideas of developing agents for automating experimental protocol generation, Ruan et al.533 created a multi-agents system composed of 6 agents: Literature Scouter, Experiment Designer, Hardware Executor, Spectrum Analyzer, Separation Instructor, and Result Interpreter. The Large Language Models-based Reaction Development Framework (LLM-RDF)533 automates every step of the synthesis workflow. While other studies focus on the literature review,381,534,535 HTS,529 and reaction optimization,48,536 LLM-RDF can support researchers from literature search until the product purification. Using this system, the authors showed they could design a copper/TEMPO catalyzed alcohol oxidation reaction, optimize reaction conditions, engineer a scale-up, and purify the products, obtaining a yield of 86% and a purity >98% while producing 1 gram of product.
Interestingly, despite covering different fields and having diverse goals, all of these studies, from the fully automated systems like CALMS,505 and LLM-RDF,533 to human-driven protocols in BioPlanner507 and CRISPR-GPT,495 share a “human-in-the-loop” approach. This ensures the researcher remains integral to the development process, enhancing reliability and mitigating potential agent limitations, such as errors or hallucinations. Moreover, this approach addresses risks and dual-use concerns, as humans can assess whether the agents' suggestions are safe.421,537 On a slightly different track, Organa417 fully automates the laboratory workload while providing feedback to the researcher and producing reports with the results, as discussed on Section 3.7.1.
Autonomous agents significantly enhance productivity and efficiency in scientific research, but human creativity and decision-making remain vital to ensure quality and safety. In the next section, we explore agents designed to automate cheminformatics tasks, continuing our focus on how AI systems are reshaping the chemical sciences.
5.4 Agents for automating cheminformatics tasks
Cheminformatics consists of applying information technology techniques to convert physicochemical information into knowledge. The process of solving cheminformatics problems commonly involves retrieving, processing, and analyzing chemical data.538 Getting inspiration from ChemCrow47 ideas, Chemistry Agent Connecting Tool Usage to Science (CACTUS)539 focused on assisting scientists by automating cheminformatics tasks. CACTUS automates the applications of multiple cheminformatics tools, such as property prediction and calculation, while maintaining the human-in-the-loop for molecular discovery. The authors investigated the performance of a diverse set of open-source LLMs, where Gemma-7B and Mistral-7B demonstrated superior performance against LLaMA-7B and Falcon-7B. In addition, the authors reported that adding domain-specific information in the prompt to align the agent to chemistry problems considerably increases a model's performance. For instance, predicting drug-likeness with a Gemma-7B agent improves the accuracy of ∼60% when aligning the agent in this way, and prompt alignment improved the prediction of all properties they studied.Further illustrating the versatility of AI in scientific research and domain-specific tools usage is ChatMOF,509 which focuses on the prediction and generation of Metal–Organic Frameworks (MOFs). ChatMOF integrates MOF databases with its MOFTransformer540 predictor module, thereby showcasing the innovative use of genetic algorithms in guiding generative tasks from associated predictions. The authors showed that ChatMOF achieved an accuracy of ∼90% in search and prediction tasks while generative tasks have an accuracy of ∼70%. The genetic algorithm used by ChatMOF allows for the generation of a diverse array of MOF structures, which can be further refined based on specific properties requested by users. For instance, when prompted to, “generate structures with the largest surface area”, the system initially generated a broad distribution of structures with surface area centered in 3784 m2 g−1, and the GA evolves it to a narrower distribution with a peak at 5554 m2 g−1 after only three generations. It is important to note that even though ChatMOF has access to a dataset of experimental values for MOFs, language model predictions guide their GA, and no further validation has been made. Lastly, Ansari and Moosavi504 developed Eunomia, another domain-specific autonomous AI agent that leverages existing knowledge to answer questions about materials. Eunomia504 can use chemistry tools to access a variety of datasets, scientific papers and unstructured texts to extract and reason about material science information. The authors implemented a CoVe434 (Consistency Verification) scheme to evaluate the model's answer and minimize hallucination. The authors showed that including CoVe increased the model's precision by ∼20% when compared to previous methods such as an agent using ReAct only.439
Promoting molecular discovery is a topic with great attention in the literature devoted to it and, as described extensively above, LLMs have leveraged a large amount of unstructured data to accelerate that discovery. Janakarajan et al.508 discuss the advantages of using LLMs in fields such as de novo drug design, reaction chemistry, and property prediction, but they augment the LLM in IBM ChemChat, a chatbot with the capability of using common APIs and python packages commonly used daily by a cheminformatics researcher to access molecular information. ChemChat has access to tools such as Generative Toolkit for Scientific Discovery (GT4SD),541 a package with dozens of trained models generative models for science, rxn4chemistry,542 a package for computing chemistry reactions tasks, HuggingMolecules,543 a package developed to aggregate molecular property prediction LMs, and RDKit,544 a package to manipulating molecules. Since ChemChat implements an agent in a chat-like environment, users can interactively refine design ideas. Despite being developed to target de novo drug design, ChemChat nonetheless is a multi-purpose platform that can be more broadly used for molecular discovery.
In addition to the capabilities described above, LLM-based agents can empower users to tackle tasks that typically require extensive technical knowledge. In previous work, Wellawatte and Schwaller343 and Gandhi and White290 showed that including natural language explanations (NLE) in explainable AI (XAI) analysis can improve user understanding. More recently, Wellawatte and Schwaller343 developed XpertAI343 to seamlessly integrate XAI techniques with LLMs to interpret and explain raw chemical data autonomously. Applying XAI techniques is usually restricted to technical experts but by integrating such techniques with an LLM-based agent to automate the workflow, the authors made XAI accessible to a wider audience.
Their system receives raw data with labels for physicochemical properties. The raw data is used to compute human-interpretable descriptors and then calculate SHAP (or SHapley Addictive exPlanations) values or Z-scores for Local Interpretable Model-agnostic Explanations (LIME). By calculating SHAP values, a value can be assigned to each feature, indicating its contribution to a model's output. LIME interprets a model by making a local approximation, around a particular prediction, to indicate what factors have contributed to that prediction in the model. It may use, for example, a surrogate local linear regression fit to recognized features.290 In addition to XAI tools, XpertAI can search and leverage scientific literature to provide accessible natural language explanations (NLEs). While ChatGPT provides scientific justifications with similar accuracy, its explanation is often too broad. On the other hand, XpertAI provides data-specific explanations and visual XAI plots to support its explanations.343 With a similar goal, Zheng et al.545 prompted the LLM to generate explanatory rules from data.
These developments signify a growing trend in the integration of tools and LLMs in autonomous AI within scientific research. By automating routine tasks, enhancing information retrieval and analysis, and facilitating experimentation, AI is expanding the capabilities of researchers and accelerating the pace of scientific discovery. This review underscores the transformative impact of AI across various scientific domains, heralding a new era of innovation and efficiency in chemical research.
5.5 Agents for hypothesis creation
Following the agent's classification proposed by Gao et al.,420 the studies we have discussed previously lie mainly in level 1, i.e. AI agents as a research assistant. Therefore, such agents can support researchers in executing predefined tasks, but they lack the autonomy to propose, test, and refine new scientific hypotheses. New research has been focusing on making agents able to refine scientists' initial hypotheses collaboratively, which is a required skill to achieve level 2 in the Gao et al.420 classification.The idea of an “AI scientist” who can generate new, relevant research questions (RQ) has been pursued by groups such as Wang et al.,546 who developed a framework called Scientific Inspiration Machines Optimized for Novelty (SciMON). SciMON uses LLMs to produce new scientific ideas grounded in existing literature. It retrieves inspirations from past papers and iteratively refines generated ideas to optimize novelty by comparing them with prior work. Extending these ideas, Gu and Krenn547 used LLMs to search over a knowledge graph for inspiration to propose new personalized research ideas. Aligned with this vision, Liu et al.506 developed CoQuest, partially automating the brainstorming for new the RQ process. This system uses a human-computing interface (HCI) to allow the agent to create new RQs that can be further enhanced by human feedback. They developed two strategies for RQ generation: breadth-first, where the agent generates multiple RQs simultaneously following the original user's prompt, and depth-first, where multiple RQs are created sequentially, building on the top of the previously generated RQ. For each RQ generation, the agent implements a ReAct439 framework with tools for literature discovery, hypothesis proposition, refinement, and evaluation. Upon evaluation of 20 HCI doctoral researchers by a post-interaction survey, the breadth-first approach was preferred by 60% of the evaluators. Interestingly, despite the evaluators' report that the breadth-first approach gave them more control and resulted in more trustworthy RQs, the depth-first had better scores for novelty and surprise. This difference might be caused by the fact that the depth-first uses its own RQ to iterate. This process can introduce new keywords that users have not considered.
Focusing on generating and testing hypotheses, ChemReasoner497 uses a domain-specific reward function and computational chemistry feedback to validate agent responses. The authors combined a Monte Carlo thought search548 for catalysis with a reward function from atomistic GNNs trained to predict adsorption energy or reaction energy barriers. While the search is responsible for exploiting literature information and allowing the model to propose new materials, the hypothetic material is further tested by the GNN. This framework was applied to suggest materials for adsorbates, biofuel catalysts, and catalysts for CO2 to methanol conversion. The LLM generated the top five catalysts for each task, with ChemReasoner significantly outperforming GPT-4 based on the reward score.
Similarly, Ma et al.494 developed Scientific Generative Agent (SGA) to generate hypotheses and iteratively refine them through computational simulations. Initially, the LLM generates a hypothesis. In the use cases considered, it can be a code snippet or a molecule. In the sequence, a search algorithm is used to find a better initial hypothesis for solving the initial query. Finally, this hypothesis—code or molecule—is optimized using a gradient-based algorithm. Lastly, the optimization output serves as feedback to the LLM to iterate. In their molecule design task, the goal was to generate a molecule with a specified HOMO–LUMO gap. The hypothesis is a molecule, that is, a SMILES string and a set of atomic coordinates. The gap is predicted by employing UniMol.549 They showed that SGA could generate molecules based on quantum mechanical properties, but the results were not validated.
6 Challenges and opportunities
LLMs hold great potential in chemistry due to their ability to both predict properties, new molecules, and their syntheses and to orchestrate existing computational and experimental tools. These capabilities enhance the accuracy and efficiency of chemical research and open up new avenues for discovery and innovation. By encapsulating AI models, data analysis software, and laboratory equipment within agent-based frameworks, researchers can harness these sophisticated tools through a unified interface. This approach not only simplifies the interaction with complex systems but also democratizes the immense capabilities of modern computational tools, thereby maximizing their utility in advancing chemical research and development. Other publications and reviews also shared their opinions on the challenges for the future of LLMs and LLM-based agents in chemistry.76,194,423,550–552 Nonetheless, some important challenges and opportunities for progress remain, which we summarize here.6.1 Data quality and availability
Quality and availability of data are critical factors that influence the efficacy of LLMs. Indeed, scaling both the model size and the amount of training data used has proven to improve capabilities.553 However, current AI models are not trained on large amounts of chemical data, which limits their capabilities to reason about advanced chemical concepts.183There are two types of datasets commonly used to train LLMs: unlabeled and labeled datasets. Unlabeled datasets, or pretraining data, are used during the semi-supervised training, which focuses on creating a “prior belief” about a molecule. Currently, we have huge datasets composed of hypothetical and/or theoretical data. When a model is trained on data that is not grounded in real chemical information, this might cause the model to learn a wrong prior belief.554
Labeled datasets, often used in benchmarks, also suffer from their inclusion of hypothetical and calculated data. Benchmarks are necessary for quantifying improvements in AI modeling and prediction within a competitive field. However, dominant benchmarks like MoleculeNet,56 have significant limitations that may restrict the generalizability and applicability of evolving models. In his blog, Walters188 brings to light numerous errors and inconsistencies within the MoleculeNet data, which substantially impact model performance and reliability.184–187 Walters also argues that the properties present in these benchmarks do not directly correlate with real chemistry improvement. As such, new benchmarks need to translate to practical chemistry problems directly. For instance, increasing accuracy in predicting LogP is not necessarily mapped to drugs with greater bioavailability. Some promising work has come from the Therapeutic Common Data (TDC),190,191 which includes data from actual therapeutic essays, providing a more practical foundation for model training.
The community continues to work to organize and curate datasets to prepare data for LLM training and evaluation. Scientific benchmarks,218,502,555 repositories with curated datasets,182 and packages for model evaluation183 have been developed. However, the challenges concerning grounded truth and consistent datasets remain. With advancements in scientific document processing,379 there is now the opportunity to obtain new datasets from peer-reviewed scientific papers.197–200 Due to the multi-modal capabilities of such AI models, these new benchmarks can comprise multiple data types, potentially enhancing the applicability and transferability of these models. The continual curation of new, relevant datasets that represent the complexities of real-world chemical problems will further enhance the robustness and relevance of LLMs in chemistry.
6.2 Model interpretability
Model interpretability is a significant challenge for LLMs due to their “black-box” nature, which obscures the understanding of how predictions are made. However, innovative approaches are being developed to enhance LLMs' interpretability. For instance, Schwaller et al.556 and Schilter et al.557 used information from the different multi-attention heads. While Schwaller et al.556 connected atoms from reactants to atoms in the products, Schilter et al.557 assigned H-NMR peaks to specific hydrogens in a molecule to indicate how spectra were comprehended, or structures deduced. Additionally, since the LLMs use language, which is intrinsically interpretable, LLMs may be incrementally modified to explain their reasoning processes directly, exemplified with tools like eXpertAI343 and or simply adjusting prompting.437,484 These methods address the critical need for transparency in the mechanism of understanding for a good prediction beyond the good prediction itself.6.3 Integration with domain knowledge and cross-disciplinary applications
While LLMs excel at pattern recognition, integrating explicit chemical rules and domain knowledge into these systems remains challenging. This integration is essential to make predictions that are not only statistically valid but also chemically reasonable. It was shown by Beltagy et al.220 and Gu et al.215 that better performance on common NLP tasks can be achieved by developing a vocabulary and pretraining on a domain-specific training corpus. While pretraining with domain-specific datasets that include chemical properties, reaction mechanisms, and experimental results may better capture the nuances of chemistry, but using AI to foster multi-disciplinary research remains a significant challenge. The Galactica LLM123 also used special tokens for delineating chemical information, to relatively good success on chemistry tasks. Aryal et al.558 also progress by creating an ensemble of specialist agents with different domains of knowledge, allowing them to interact to better answer the user query. Specifically, Aryal et al.558 used agents with chemistry, physics, electrochemistry, and materials knowledge.6.4 Tool development
The effectiveness of a combined LLM/autonomous agent approach hinges significantly on the availability and quality of the tools, as well as on the complexity and diversity of the chemical tasks at hand. Some emphasis should be placed on refining standalone tools, with the confidence that overarching frameworks, like a GPT-4-type wrapper, or “assistant”, will eventually integrate these tools seamlessly. Developers should stay informed about existing tools and design their tools to interface effectively with such a wrapper. This ensures that each tool is ready to contribute its unique capabilities to a cohesive agent system.6.5 Reinforcement learning
RL has been successfully used in LLMs,103,559,560 with a few applications also proposed for use in agents.561,562 The next frontier is applying RL to agents directly, to improve their ability on specific tasks. Bou et al.114 provided a recent framework and example for generative molecular design when viewed as an RL problem (similar to RLHF) and some early success has been seen in applying the RLHF algorithm directly to protein language models where the reward model comes from scientific tasks.115 Neither of these are direct RL on language model agents, but are a step towards this goal.6.6 Agent evaluation
Comparing different agent systems is challenging due to the lack of robust benchmarks and evaluation schemes. Consequently, it is difficult to define what constitutes a “superhuman” digital chemist and reach a consensus on the criteria for success.563,564 This issue is similar to the ongoing discussions about defining artificial general intelligence (AGI) and the expected capabilities of cognitive architectures.565,566 Once a reliable metric for evaluating such AI systems is established, it is crucial for the AI scientific community to set clear guidelines for conducting research. Currently, assessing success is challenging because the goals are not well defined. Building on this, we propose using Bloom's taxonomy404,405 as a reference point for developing a metric to evaluate more complex reasoning and tool use in autonomous agents. This educational framework categorizes cognitive skills in a hierarchical manner, from basic recall to creative construction, providing a structured approach to assess higher-order thinking and reasoning capabilities in these systems. This adaptation could significantly enhance the evaluation of LLMs and autonomous agents, especially when tackling complex chemical challenges.6.7 Ethical and safety concerns
As with all AI technologies, deploying LLMs involves ethical considerations, such as biases in predictions and the potential misuse of AI-generated chemical knowledge. Ruan et al.567 and Tang et al.532 highlight the need for multi-level regulation, noting that current alignment methods may be insufficient for ensuring safety and that human evaluation alone is not scalable.The absence of specialized models for risk control and reliable safety evaluations poses a significant challenge to ensuring the safety of tool-using LLMs. This highlights the urgent need to automate red-teaming strategies to reinforce AI safety protocols. Additionally, the development of safe AI systems should prioritize minimizing harmful hallucinations. While managing dual-use risks is a human responsibility and should be controlled through safety assessments at publication or indirect regulation by the scientific community.
6.8 Human–AI collaboration in chemical research
LLMs are poised to transform fields such as drug discovery, materials science, and environmental chemistry due to their ability to predict chemical properties and reactions with remarkable accuracy. Models based on architectures like BERT have demonstrated their capability to achieve state-of-the-art performance in various property prediction tasks.45,248 Furthermore, studies by Jablonka et al.142 and Born and Manica139 have showcased the predictive power of LLMs by reformulating traditional regression and classification tasks as generative tasks, opening up new avenues for chemical modeling. However, as emphasized by Weng,422 maintaining the reliability of LLM outputs is essential, as inaccuracies in formatting, logical reasoning, or content can significantly impede their practical utility. Hallucination is also an intrinsic issue with LLMs.568 Though agents can deal with hallucinations to some extent by implementing sanity-checking tools, it does not make the response hallucination-proof. A possible approach to address this issue is to use a human-in-the-loop approach, where steps of human–agent interaction are added to the workflow to check if the agent is in the correct pathway to solve the request.569–571The potential of LLMs to design novel molecules and materials was highlighted by the AI-powered robotic lab assistant, A-Lab, which synthesized 41 new materials within just 17 days.572 Nonetheless, this achievement has sparked debates about the experimental methods and the actual integration of atoms into new crystalline materials, raising questions about the authenticity of the synthesized structures.573 These controversies underline the necessity for rigorous standards and the critical role of human expertise in validating AI-generated results. Again, the integration of advanced AI tools with the oversight of seasoned chemists is crucial, suggesting that a hybrid approach could significantly enhance both the innovation and integrity of materials science research.
In parallel, we have seen how LLM-based agents are increasingly capable of automating routine tasks in chemical research, which traditionally consume significant time and resources. These models excel in real-time data processing, managing vast datasets, and even conducting comprehensive literature reviews with minimal human intervention. Advances in AI technology now allow agents not only to perform predefined tasks but also to adapt and develop new tools for automating additional processes. For instance, tasks such as data analysis, literature review, and elements of experimental design are now being automated.47,48,343,502,505,507,574,575 This automation liberates chemists to focus on more innovative and intellectually engaging aspects of their work, and the opportunity is to expand productivity and creativity in their science.
6.9 Promotion of impactful discoveries
AI technologies offer experimental chemists significant opportunities to streamline repetitive tasks like data collection and analysis, freeing up time for innovation.417 AI-powered tools can suggest novel experiments and chemical pathways,533 but the black-box nature of many models raises concerns about trust and transparency. Human expertise remains essential to validate AI-generated results, especially in critical experiments.A key challenge is translating AI predictions into real-world experiments, where factors like reagent quality and equipment limitations must be considered. To integrate AI effectively in the lab, stronger collaboration between computational and experimental chemists is essential, ensuring AI tools are practical and aligned with lab conditions.576 Clear communication will help identify the most impactful AI advancements, ensuring tools address the real needs of experimentalists. AI's ability to explore new chemical spaces also offers exciting opportunities for discovery, allowing chemists to harness these insights while maintaining oversight for accuracy and reliability.
6.10 AI in everyday chemistry
In the near future, AI tools will become integral to the daily workflow of chemists, transforming how routine challenges are approached and resolved. Today's chemist may soon find themselves interacting directly with AI-driven systems, leveraging advanced simulations, literature analyses, and predictive models to accelerate discovery. While this may sound like a glimpse into the future, the reality is that such tools are already emerging, and their widespread adoption has likely already begun. To illustrate this transformation, we propose the following scenario, based largely on prior experience.577A chemist working on synthesizing a challenging target molecule encounters suboptimal yields and an unexpected side product. Despite verifying solvent purity, reaction conditions, and ruling out possible causes such as steric hindrance, or leaving group viability, the issue remains unresolved. The chemist plans a comprehensive systematic study, varying the leaving group and adjusting the length of a bulky alkyl chain in one of the secondary amines.577 This would require weeks of repeated testing and data analysis, creating two lengthy projects for PhD students, diverting 2–3 months of effort. Nonetheless, the starting materials are ordered, and are expected to arrive within a fortnight.
In contrast, another chemist, equipped with methods described here, approaches the problem differently. Through in silico studies, they evaluate the chemical properties of reactants and intermediates using a selection from the chemistry-specific LLMs described above. This strategy allows for rapid hypothesis testing and simulation of reaction conditions. With a human-in-the-loop workflow, the chemist refines the predictions, dismissing implausible pathways and focusing on a promising hypothesis. They use tools like PaperQA2 (ref. 492) to verify the reaction mechanism against existing literature, ensuring a solid foundation in prior knowledge. This AI-driven workflow enables the chemist to design three targeted experiments, each validating a critical model prediction, thus bypassing the need for a larger methodological studies. Using an automated ChemCrow system,47 the required starting materials are synthesized overnight. The following day, a PhD chemist performs the reactions, swiftly confirming the AI-derived hypothesis and achieving the desired product within 24 hours. The entire process, from problem identification to successful synthesis, concludes in just one week. Meanwhile, the first group of PhD students continues their extensive exploration of reaction conditions, gaining methodological insights but without directly achieving their original goal.
This comparison underscores how creativity and efficiency in research may benefit from a hybrid approach where there is some computational heavy lifting, along with a team of virtual chemistry experts to help hone and test ideas.
7 Conclusions
Since this review is targeted in part to an audience of chemists, who may not have yet embraced AI technology, we consider it valuable to point out our perspective that AI in chemistry is definitely here to stay. We predict that its use will only grow as a necessary tool that will inevitably lead to more jobs and greater progress. We hope to facilitate the change by connecting the technology to the chemical problems that our readership is already addressing through more traditional methods.Large Language Models (LLMs) have demonstrated remarkable potential in reshaping chemical research and development workflows. These models have facilitated significant advancements in molecular simulation, reaction prediction, and materials discovery. In this review, we discussed the evolution of LLMs in chemistry and biochemistry. Successful cases where LLMs have proven their potential in promoting scientific discovery were shown with caveats of such models.
Adopting LLM-based autonomous agents in chemistry has enhanced the accuracy and efficiency of traditional research methodologies and introduced innovative approaches to solving complex chemical problems. Looking forward, the continued integration of LLMs promises to accelerate the field's evolution further, driving forward the frontiers of scientific discovery and technological innovation in chemistry. We have shown how agents have been used in chemistry and proposed a framework for thinking about agents as a central LLM followed by interchangeable components.
However, despite the community's astonishing advances in this field, many challenges still require solutions. We identified the main challenges and opportunities that need to be addressed to promote the further development of agents in chemistry. Addressing the challenges related to model transparency, data biases, and computational demands will be crucial for maximizing their utility and ensuring their responsible use in future scientific endeavors.
While there are significant challenges to be addressed, the opportunities presented by LLMs in chemistry are vast and have the potential to fundamentally alter how chemical research and development are conducted. Effectively addressing these challenges will be crucial for realizing the full potential of LLMs in this exciting field. To keep pace with the ever-growing number of relevant publications, we will maintain a repository with an organized structure listing new studies regarding LLMs and LLM-based agents focused on scientific purposes. The repository can be found in https://github.com/ur-whitelab/LLMs-in-science.
Data availability
All data discussed and referenced within the paper are available in the original studies cited. For further information, please refer to the respective publications mentioned in the references section.Author contributions
All authors contributed to writing this review article.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
M. C. R. and A. D. W. acknowledge the U.S. Department of Energy, Grant No. DE-SC0023354, and C. J. C. gratefully acknowledges the Jane King Harris Endowed Professorship at Rochester Institute of Technology for the support provided for this publication. We are grateful for feedback from early drafts of this review from the following colleagues: Kevin Jablonka, Philippe Schwaller, Michael Pieler, Ryan-Rhys Griffiths, Geemi Wellawatte, and Mario Krenn.References
- P. Willett, Chemoinformatics: a history, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1(1), 46–56 Search PubMed.
- E. J. Griffen, A. G. Dossetter and A. G. Leach, Chemists: AI Is Here; Unite To Get the Benefits, J. Med. Chem., 2020, 63(16), 8695–8704, DOI:10.1021/acs.jmedchem.0c00163.
- Z. J. Baum, Yu Xiang, P. Y. Ayala, Y. Zhao, S. P. Watkins and Q. Zhou, Artificial Intelligence in Chemistry: Current Trends and Future Directions, J. Chem. Inf. Model., 2021, 61(7), 3197–3212, DOI:10.1021/acs.jcim.1c00619.
- L. B. Ayres, F. J. V. Gomez, J. R. Linton, M. F. Silva and C. D. Garcia, Taking the leap between analytical chemistry and artificial intelligence: A tutorial review, Anal. Chim. Acta, 2021, 1161, 338403, DOI:10.1016/j.aca.2021.338403.
- X. Yang, Y. Wang, R. Byrne, G. Schneider and S. Yang, Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery, Chem. Rev., 2019, 119(18), 10520–10594, DOI:10.1021/acs.chemrev.8b00728.
- C. M. Adam and M. L. Coote, Deep learning in chemistry, J. Chem. Inf. Model., 2019, 59(6), 2545–2559 CrossRef PubMed.
- Y.-F. Shi, Z.-X. Yang, S. Ma, P.-L. Kang, C. Shang, P. Hu and Z.-P. Liu, Machine learning for chemistry: basics and applications, Engineering, 2023, 27, 70–83 CrossRef.
- J. A. Keith, V. Vassilev-Galindo, B. Cheng, S. Chmiela, M. Gastegger, K.-R. Muller and A. Tkatchenko, Combining machine learning and computational chemistry for predictive insights into chemical systems, Chem. Rev., 2021, 121(16), 9816–9872 CrossRef PubMed.
- D. Kuntz and A. K. Wilson, Machine learning, artificial intelligence and chemistry: How smart algorithms are reshaping simulation and the laboratory, Pure Appl. Chem., 2022, 94(8), 1019–1054 CrossRef CAS.
- M. Meuwly, Machine learning for chemical reactions, Chem. Rev., 2021, 121(16), 10218–10239 CrossRef CAS PubMed.
- J. Lederberg, G. L. Sutherland, B. G. Buchanan, E. A. Feigenbaum, A. V. Robertson, A. M. Duffield and C. Djerassi, Applications of artificial intelligence for chemical inference. i. number of possible organic compounds. acyclic structures containing carbon, hydrogen, oxygen and nitrogen, J. Am. Chem. Soc., 1969, 91(11), 2973–2976 CrossRef CAS.
- R. K. Lindsay, B. G. Buchanan, E. A. Feigenbaum and J. Lederberg, Dendral: a case study of the first expert system for scientific hypothesis formation, Artif. Intell., 1993, 61(2), 209–261 CrossRef.
- B. G. Buchanan, D. H. Smith, W. C. White, R. J. Gritter, E. A. Feigenbaum, J. Lederberg and C. Djerassi, Applications of artificial intelligence for chemical inference. 22. Automatic rule formation in mass spectrometry by means of the meta-DENDRAL program, J. Am. Chem. Soc., 1976, 98(20), 6168–6178, DOI:10.1021/ja00436a017.
- C. Hansch, P. P. Maloney, T. Fujita and R. M. Muir, Correlation of biological activity of phenoxyacetic acids with Hammett substituent constants and partition coefficients, Nature, 1962, 194(4824), 178–180 CrossRef CAS.
- C. Hansch and T. Fujita, p-σ-π analysis. a method for the correlation of biological activity and chemical structure, J. Am. Chem. Soc., 1964, 86(8), 1616–1626 CrossRef CAS.
- C. Hansch and A. Leo, Exploring QSAR.: Fundamentals and applications in chemistry and biology, American Chemical Society, 1995, vol. 1 Search PubMed.
- J. G. Topliss and R. J. Costello, Chance correlations in structure-activity studies using multiple regression analysis, J. Med. Chem., 1972, 15(10), 1066–1068 CrossRef CAS.
- J. N. Weinstein, K. W. Kohn, M. R. Grever, V. N. Viswanadhan, L. V. Rubinstein, A. P. Monks, D. A. Scudiero, L. Welch, A. D. Koutsoukos, A. J. Chiausa and K. D. Paull, Neural Computing in Cancer Drug Development: Predicting Mechanism of Action, Science, 1992, 258(5081), 447–451, DOI:10.1126/science.1411538.
- W. W. Van Osdol, T. G. Myers, K. D. Paull, K. W. Kohn and J. N. Weinstein, Use of the kohonen self-organizing map to study the mechanisms of action of chemotherapeutic agents, J. Natl. Cancer Inst., 1994, 86(24), 1853–1859 CrossRef CAS PubMed.
- B. B. Goldman and W. P. Walters, Machine learning in computational chemistry, Annu. Rep. Comput. Chem., 2006, 2, 127–140 CAS.
- D. A. Pereira and J. A. Williams, Origin and evolution of high throughput screening, Br. J. Pharmacol., 2007, 152(1), 53–61 CrossRef CAS.
- J. L. Medina-Franco, M. A. Giulianotti, G. S. Welmaker and R. A. Houghten, Shifting from the single to the multitarget paradigm in drug discovery, Drug Discovery Today, 2013, 18(9), 495–501, DOI:10.1016/j.drudis.2013.01.008.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Machine learning for molecular and materials science, Nature, 2018, 559(7715), 547–555, DOI:10.1038/s41586-018-0337-2.
- M. Rupp, A. Tkatchenko, K.-R. Müller and O. Anatole von Lilienfeld, Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning, Phys. Rev. Lett., 2012, 108(5), 058301, DOI:10.1103/PhysRevLett.108.058301.
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, Molecular de-novo design through deep reinforcement learning, J. Cheminf., 2017, 9(1), 48, DOI:10.1186/s13321-017-0235-x.
- M. H. S. Segler, T. Kogej, C. Tyrchan and M. P. Waller, Generating focused molecule libraries for drug discovery with recurrent neural networks, ACS Cent. Sci., 2018, 4(1), 120–131, DOI:10.1021/acscentsci.7b00512.
- M. H. S. Segler, T. Kogej, C. Tyrchan and M. P. Waller, Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks, ACS Cent. Sci., 2018, 4(1), 120–131, DOI:10.1021/acscentsci.7b00512.
- A. Gupta, A. T. Muller, B. J. H. Huisman, J. A. Fuchs, P. Schneider and G. Schneider, Generative recurrent networks for de novo drug design, Mol. Inf., 2018, 37(1–2), 1700111, DOI:10.1002/minf.201700111.
- P. Karpov, G. Godin and I. V. Tetko, Transformer-CNN: Swiss knife for QSAR modeling and interpretation, J. Cheminf., 2020, 12(1), 17, DOI:10.1186/s13321-020-00423-w.
- K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko and K.-R. Müller, SchNet – A deep learning architecture for molecules and materials, J. Chem. Phys., 2018, 148(24), 241722, DOI:10.1063/1.5019779.
- M. Hirohara, Y. Saito, Y. Koda, K. Sato and Y. Sakakibara, Convolutional neural network based on SMILES representation of compounds for detecting chemical motif, BMC Bioinf., 2018, 19(Suppl 19), 526, DOI:10.1186/s12859-018-2523-5.
- W. C. Connor, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, A graph-convolutional neural network model for the prediction of chemical reactivity, Chem. Sci., 2019, 10(2), 370–377, 10.1039/c8sc04228d.
- V. Prakash Dwivedi, C. K. Joshi, A. Tuan Luu, T. Laurent, Y. Bengio and X. Bresson, Benchmarking graph neural networks, J. Mach. Learn. Res., 2023, 24(43), 1–48 Search PubMed.
- B. Sanchez-Lengeling, E. Reif, A. Pearce and A. B. Wiltschko, A gentle introduction to graph neural networks, Distill, 2021, 6(9), e33 Search PubMed.
- M. M. Bronstein, J. Bruna, Y. LeCun, A. Szlam and P. Vandergheynst, Geometric deep learning: going beyond euclidean data, IEEE Signal Process. Mag., 2017, 34(4), 18–42 Search PubMed.
- Z. Wu, S. Pan, F. Chen, G. Long, C. Zhang and S. Yu Philip, A comprehensive survey on graph neural networks, IEEE Transact. Neural Networks Learn. Syst., 2020, 32(1), 4–24 Search PubMed.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and E. D. George, Neural message passing for quantum chemistry, in International conference on machine learning, PMLR, 2017, pp. 1263–1272 Search PubMed.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules, ACS Cent. Sci., 2018, 4(2), 268–276, DOI:10.1021/acscentsci.7b00572.
- T. Gaudelet, B. Day, A. R. Jamasb, J. Soman, C. Regep, G. Liu, J. B. R. Hayter, R. Vickers, C. Roberts and J. Tang, et al., Utilizing graph machine learning within drug discovery and development, Briefings Bioinf., 2021, 22(6), bbab159 CrossRef PubMed.
- K. Choudhary, B. DeCost, C. Chen, A. Jain, F. Tavazza, R. Cohn, C. W. Park, A. Choudhary, A. Agrawal, S. J. L. Billinge, E. Holm, S. P. Ong and C. Wolverton, Recent advances and applications of deep learning methods in materials science, npj Comput. Mater., 2022, 8(1), 1–26, DOI:10.1038/s41524-022-00734-6.
- V. Fung, J. Zhang, E. Juarez and B. G. Sumpter, Benchmarking graph neural networks for materials chemistry, npj Comput. Mater., 2021, 7(1), 1–8, DOI:10.1038/s41524-021-00554-0.
- P. Reiser, M. Neubert, A. Eberhard, L. Torresi, C. Zhou, S. Chen, H. Metni, C. van Hoesel, H. Schopmans, T. Sommer and P. Friederich, Graph neural networks for materials science and chemistry, Commun. Mater., 2022, 3(1), 93, DOI:10.1038/s43246-022-00315-6.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36, DOI:10.1021/ci00057a005.
- S. Chithrananda, G. Grand and B. Ramsundar, ChemBERTa: Large-scale self-supervised pretraining for molecular property prediction, arXiv, 2020, preprint, arXiv:2010.09885, DOI:10.48550/arXiv.2010.09885, http://arxiv.org/abs/2010.09885.
- J. Li and X. Jiang, Mol-BERT: An Effective Molecular Representation with BERT for Molecular Property Prediction, Wireless Commun. Mobile Comput., 2021, 2021, 1–7, DOI:10.1155/2021/7181815.
- Y. Wang, H. Zhao, S. Sciabola and W. Wang, cMolGPT: A conditional generative Pre-Trained transformer for Target-Specific de novo molecular generation, Molecules, 2023, 28(11), 4430, DOI:10.3390/molecules28114430.
- A. M. Bran, S. Cox, O. Schilter, C. Baldassari, A. D. White and P. Schwaller, Augmenting large language models with chemistry tools, Nat. Mach. Intell., 2024, 1–11, DOI:10.1038/s42256-024-00832-8.
- D. A. Boiko, R. MacKnight, B. Kline and G. Gomes, Autonomous chemical research with large language models, Nature, 2023, 624(7992), 570–578, DOI:10.1038/s41586-023-06792-0.
- A. D. White, The future of chemistry is language, Nat. Rev. Chem, 2023, 7(7), 457–458, DOI:10.1038/s41570-023-00502-0.
- C. J. Collison, M. J. O'Donnell and J. L. Alexander, Complexation between Rhodamine 101 and Single-Walled Carbon Nanotubes Indicative of Solvent-Nanotube Interaction Strength, J. Phys. Chem. C, 2008, 112(39), 15144–15150, DOI:10.1021/jp804359j.
- J. W. Tyler, J. S. Sandoval, J. A. Cody, D. W. McCamant and C. J. Collison, Directional Exciton Diffusion, Measured by Subpicosecond Transient Absorption as an Explanation for Squaraine Solar Cell Performance, J. Phys. Chem. C, 2024, 128(11), 4616–4630, DOI:10.1021/acs.jpcc.3c06361.
- R. Ahmadov, S. S. Michtavy and M. D. Porosoff, Dual Functional Materials: At the Interface of Catalysis and Separations, Langmuir, 2024, 40(19), 9833–9841, DOI:10.1021/acs.langmuir.3c03888.
- T. Fischer, S. Gazzola and R. Riedl, Approaching Target Selectivity by De Novo Drug Design, Expet Opin. Drug Discov., 2019, 14(8), 791–803, DOI:10.1080/17460441.2019.1615435.
- Z. Wang, Z. Sun, H. Yin, X. Liu, J. Wang, H. Zhao, C. H. Pang, T. Wu, S. Li, Z. Yin and X.-F. Yu, Data-Driven Materials Innovation and Applications, Adv. Mater., 2022, 34(36), 2104113, DOI:10.1002/adma.202104113.
- B. Sridharan, M. Goel and U. Deva Priyakumar, Modern machine learning for tackling inverse problems in chemistry: molecular design to realization, Chem. Commun., 2022, 58(35), 5316–5331, 10.1039/D1CC07035E.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, MoleculeNet: a benchmark for molecular machine learning, Chem. Sci., 2018, 9(2), 513–530, 10.1039/C7SC02664A.
- G. Restrepo, Chemical space: limits, evolution and modelling of an object bigger than our universal library, Digital Discovery, 2022, 1(5), 568–585, 10.1039/D2DD00030J.
- P. Kirkpatrick and C. Ellis, Chemical space, Nature, 2004, 432(7019), 823–824 CrossRef CAS.
- A. Mullard, et al., The drug-maker’s guide to the galaxy, Nature, 2017, 549(7673), 445–447 CrossRef PubMed.
- E. J. Llanos, W. Leal, D. H. Luu, J. Jost, P. F. Stadler and G. Restrepo, Exploration of the chemical space and its three historical regimes, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(26), 12660–12665 CrossRef CAS.
- J. Schrier, A. J. Norquist, T. Buonassisi and J. Brgoch, In Pursuit of the Exceptional: Research Directions for Machine Learning in Chemical and Materials Science, J. Am. Chem. Soc., 2023, 145(40), 21699–21716, DOI:10.1021/jacs.3c04783.
- P. S. Gromski, A. B. Henson, J. M. Granda and L. Cronin, How to explore chemical space using algorithms and automation, Nat. Rev. Chem, 2019, 3(2), 119–128 CrossRef.
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, J. M. Granda, G. Keenan, T. Hinkley, G. Aragon-Camarasa, P. J. Kitson and D. Angelone, et al., Organic synthesis in a modular robotic system driven by a chemical programming language, Science, 2019, 363(6423), eaav2211 CrossRef CAS PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li and R. Clowes, et al., A mobile robotic chemist, Nature, 2020, 583(7815), 237–241 CrossRef CAS.
- B. P. MacLeod, F. G. L. Parlane, T. D. Morrissey, F. Häse, L. M. Roch, K. E. Dettelbach, R. Moreira, L. P. E. Yunker, M. B. Rooney and J. R. Deeth, et al., Self-driving laboratory for accelerated discovery of thin-film materials, Sci. Adv., 2020, 6(20), eaaz8867 CrossRef CAS.
- Q. Zhang, K. Ding, T. Lyv, X. Wang, Q. Yin, Y. Zhang, J. Yu, Y. Wang, X. Li, Z. Xiang, Z. Xiang, Z. Wang, M. Qin, M. Zhang, J. Zhang, J. Cui, R. Xu, H. Chen, X. Fan, H. Xing and H. Chen, Scientific large language models: A survey on biological & chemical domains, arXiv, 2024, preprint, arXiv:2401.14656, DOI:10.48550/arXiv.2401.14656, http://arxiv.org/abs/2401.14656.
- D. E. Rumelhart, G. E. Hinton and R. J. Williams, Learning internal representations by error propagation, Parallel distributed processing, explorations in the microstructure of cognition, ed. D. E. Rumelhart and J. Mcclelland, Biometrika, 1986, vol. 1, pp. 599–607 Search PubMed.
- S. Hochreiter and J. Schmidhuber, Long short-term memory, Neural Comput., 1997, 9(8), 1735–1780 CrossRef CAS.
- A. H. Ribeiro, K. Tiels, L. A. Aguirre and T. B. Schön, Beyond exploding and vanishing gradients: analysing RNN training using attractors and smoothness, arXiv, 2020, preprint, arXiv:1906.08482 [cs, math, stat], DOI:10.48550/arXiv.1906.08482, http://arxiv.org/abs/1906.08482.
- Dr Barak Or, The Exploding and Vanishing Gradients Problem in Time Series, 2023, https://medium.com/metaor-artificial-intelligence/the-exploding-and-vanishing-gradients-problem-in-time-series-6b87d558d22.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. Kaiser and I. Polosukhin, Attention is all you need, arXiv, June 2017, preprint, arXiv:1706.03762, DOI:10.48550/arXiv.1706.03762, http://arxiv.org/abs/1706.03762.
- A. Gu and T. D. Mamba, Linear-time sequence modeling with selective state spaces, arXiv, 2023, preprint, arXiv:2312.00752, DOI:10.48550/arXiv.2312.00752, http://arxiv.org/abs/2312.00752.
- S. Jelassi, D. Brandfonbrener, S. M. Kakade and E. Malach, Repeat after me: Transformers are better than state space models at copying, arXiv, 2024, preprint, arXiv:2402.01032, DOI:10.48550/arXiv.2402.01032, http://arxiv.org/abs/2402.01032.
- B. Peng, E. Alcaide, Q. Anthony, A. Albalak, S. Arcadinho, H. Cao, X. Cheng, M. Chung, M. Grella, K. G. V. Kranthi, et al., Rwkv: Reinventing rnns for the transformer era, arXiv, 2023, preprint, arXiv:2305.13048, DOI:10.48550/arXiv.2305.13048.
- M. Beck, K. Pöppel, M. Spanring, A. Auer, O. Prudnikova, M. Kopp, G. Klambauer, J. Brandstetter and S. Hochreiter, xLSTM: Extended Long Short-Term Memory, arXiv, 2024, preprint, arXiv:2405.04517 [cs, stat], DOI:10.48550/arXiv.2405.04517, http://arxiv.org/abs/2405.04517.
- S. Minaee, T. Mikolov, N. Nikzad, M. Chenaghlu, R. Socher, X. Amatriain and J. Gao, Large language models: A survey, arXiv, 2024, preprint, arXiv:2402.06196, DOI:10.48550/arXiv.2402.06196, http://arxiv.org/abs/2402.06196.
- The Annotated Transformer, 2022, https://nlp.seas.harvard.edu/annotated-transformer/.
- D. Bahdanau, K. Cho and Y. Bengio, Neural Machine Translation by Jointly Learning to Align and Translate, arXiv, 2016, preprint, arXiv:1409.0473 [cs, stat], DOI:10.48550/arXiv.1409.0473, http://arxiv.org/abs/1409.0473.
- T. Li, G. Zhang, Q. D. Do, Y. Xiang and W. Chen, Long-context LLMs struggle with long in-context learning, arXiv, 2024, preprint, arXiv:2404.02060, DOI:10.48550/arXiv.2404.02060, http://arxiv.org/abs/2404.02060.
- Y. Zhang, R. Sun, Y. Chen, T. Pfister, R. Zhang and S. Ö. Arik, Chain of agents: Large language models collaborating on long-context tasks, arXiv, 2024, preprint, arXiv:2406.02818, DOI:10.48550/arXiv.2406.02818, http://arxiv.org/abs/2406.02818.
- T. Kudo, Subword regularization: Improving neural network translation models with multiple subword candidates, arXiv, 2018, preprint, arXiv:1804.10959, DOI:10.48550/arXiv.1804.10959, http://arxiv.org/abs/1804.10959.
- T. Kudo and J. Richardson, SentencePiece: A simple and language independent subword tokenizer and detokenizer for neural text processing, arXiv, 2018, preprint, arXiv:1808.06226, DOI:10.48550/arXiv.1808.06226, http://arxiv.org/abs/1808.06226.
- X. Song, A. Salcianu, Y. Song, D. Dopson and D. Zhou, Fast WordPiece tokenization, arXiv, 2020, preprint, arXiv:2012.15524, DOI:10.48550/arXiv.2012.15524, http://arxiv.org/abs/2012.15524.
- P. Rust, J. Pfeiffer, I. Vulić, S. Ruder and I. Gurevych, How good is your tokenizer? on the monolingual performance of multilingual language models, arXiv, 2020, preprint, arXiv:2012.15613, DOI:10.48550/arXiv.2012.15613, http://arxiv.org/abs/2012.15613.
- M. Berglund and B. van der Merwe, Formalizing BPE tokenization, arXiv, 2023, preprint, arXiv:2309.08715, DOI:10.48550/arXiv.2309.08715, http://arxiv.org/abs/2309.08715.
- J. Gehring, M. Auli, D. Grangier, D. Yarats and Y. N. Dauphin, Convolutional sequence to sequence learning, arXiv, 2017, preprint, arXiv:1705.03122, DOI:10.48550/arXiv.1705.03122.
- J. Devlin, M.-W. Chang, K. Lee and K. Toutanova, BERT: Pre-training of deep bidirectional Transformers for language understanding, arXiv, 2018, preprint, arXiv:1810.04805, DOI:10.48550/arXiv.1810.04805, http://arxiv.org/abs/1810.04805.
- V. Nair and G. E. Hinton, Rectified linear units improve restricted boltzmann machines, in Proceedings of the 27th international conference on machine learning, ICML-10, 2010, pp. 807–814 Search PubMed.
- N. Shazeer, GLU Variants Improve Transformer, arXiv, 2020, preprint, arXiv:2002.05202 [cs, stat], DOI:10.48550/arXiv.2002.05202, http://arxiv.org/abs/2002.05202.
- D. Hendrycks and K. Gimpel, Gaussian Error Linear Units (GELUs), arXiv, 2023, preprint, arXiv:1606.08415 [cs], DOI:10.48550/arXiv.1606.08415, http://arxiv.org/abs/1606.08415.
- S. Ouyang, Z. Zhang, B. Yan, X. Liu, J. Han and L. Qin, Structured chemistry reasoning with large language models, arXiv, 2023, preprint, arXiv:2311.09656, DOI:10.48550/arXiv.2311.09656, http://arxiv.org/abs/2311.09656.
- N. Stiennon, L. Ouyang, J. Wu, D. Ziegler, R. Lowe, C. Voss, A. Radford, D. Amodei and P. F. Christiano, Learning to summarize with human feedback, Adv. Neural Inf. Process. Syst., 2020, 33, 3008–3021 Search PubMed.
- T. B. Brown, B. Mann, N. Ryder, M. Subbiah, J. Kaplan, P. Dhariwal, A. Neelakantan, P. Shyam, G. Sastry, A. Askell, S. Agarwal, A. Herbert-Voss, G. Krueger, T. Henighan, R. Child, A. Ramesh, D. M. Ziegler, J. Wu, C. Winter, C. Hesse, M. Chen, E. Sigler, M. Litwin, S. Gray, B. Chess, J. Clark, C. Berner, S. McCandlish, A. Radford, I. Sutskever and D. Amodei, Language Models are Few-Shot Learners, arXiv, 2020, preprint, arXiv:2005.14165, DOI:10.48550/arXiv.2005.14165, http://arxiv.org/abs/2005.14165.
- F. Errica, G. Siracusano, D. Sanvito and R. Bifulco, What did I do wrong? quantifying LLMs' sensitivity and consistency to prompt engineering, arXiv, 2024, preprint, arXiv:2406.12334, DOI:10.48550/arXiv.2406.12334, http://arxiv.org/abs/2406.12334.
- T. Shen, R. Jin, Y. Huang, C. Liu, W. Dong, Z. Guo, X. Wu, Y. Liu and D. Xiong, Large language model alignment: A survey, arXiv, 2023, preprint, arXiv:2309.15025, DOI:10.48550/arXiv.2309.15025, http://arxiv.org/abs/2309.15025.
- C. Lee, J. Han, S. Ye, S. J. Choi, H. Lee and K. Bae, Instruction matters, a simple yet effective task selection approach in instruction tuning for specific tasks, arXiv, 2024, preprint, arXiv:2404.16418, DOI:10.48550/arXiv.2404.16418, http://arxiv.org/abs/2404.16418.
- J. Hewitt, N. F. Liu, P. Liang and C. D. Manning, Instruction following without instruction tuning, arXiv, 2024, preprint, arXiv:2409.14254, DOI:10.48550/arXiv.2409.14254, http://arxiv.org/abs/2409.14254.
- S. Zhang, L. Dong, X. Li, S. Zhang, X. Sun, S. Wang, J. Li, R. Hu, T. Zhang, F. Wu and G. Wang, Instruction tuning for large language models: A survey, arXiv, 2023, preprint, arXiv:2308.10792, DOI:10.48550/arXiv.2308.10792, http://arxiv.org/abs/2308.10792.
- Y. Duan, J. Schulman, X. Chen, P. L. Bartlett, I. Sutskever and P. Abbeel, RL2: Fast reinforcement learning via slow reinforcement learning, arXiv, 2016, preprint, arXiv:1611.02779, DOI:10.48550/arXiv.1611.02779, http://arxiv.org/abs/1611.02779.
- D. M. Ziegler, N. Stiennon, J. Wu, T. B. Brown, A. Radford, D. Amodei, P. Christiano and G. Irving, Fine-tuning language models from human preferences, arXiv, 2019, preprint, arXiv:1909.08593, DOI:10.48550/arXiv.1909.08593, http://arxiv.org/abs/1909.08593.
- E. Mazuz, G. Shtar, B. Shapira and L. Rokach, Molecule generation using transformers and policy gradient reinforcement learning, Sci. Rep., 2023, 13(1), 8799, DOI:10.1038/s41598-023-35648-w.
- M. Laskin, L. Wang, J. Oh, E. Parisotto, S. Spencer, R. Steigerwald, D. J. Strouse, S. Hansen, A. Filos, E. Brooks, M. Gazeau, H. Sahni, S. Singh and V. Mnih, In-context reinforcement learning with algorithm distillation, arXiv, 2022, preprint, arXiv:2210.14215, DOI:10.48550/arXiv.2210.14215, http://arxiv.org/abs/2210.14215.
- Aligning language models to follow instructions, 2022, https://openai.com/index/instruction-following, accessed: 2024-5-1.
- S. Kim, S. Bae, J. Shin, S. Kang, D. Kwak, K. M. Yoo and M. Seo, Aligning large language models through synthetic feedback, arXiv, 2023, preprint, arXiv:2305.13735, DOI:10.48550/arXiv.2305.13735, http://arxiv.org/abs/2305.13735.
- J. Schulman, F. Wolski, P. Dhariwal, A. Radford and O. Klimov, Proximal policy optimization algorithms, arXiv, 2017, preprint, arXiv:1707.06347, DOI:10.48550/arXiv.1707.06347, http://arxiv.org/abs/1707.06347.
- J. Zhang, J. Kim, B. O'Donoghue and S. Boyd, Sample efficient reinforcement learning with REINFORCE, arXiv, 2020, preprint, arXiv:2010.11364, DOI:10.48550/arXiv.2010.11364, http://arxiv.org/abs/2010.11364.
- N. Shinn, F. Cassano, E. Berman, A. Gopinath, K. Narasimhan and S. Yao, Reflexion: Language agents with verbal reinforcement learning, arXiv, 2023, preprint, arXiv:2303.11366, DOI:10.48550/arXiv.2303.11366, http://arxiv.org/abs/2303.11366.
- A. Feyza Akyurek, E. Akyurek, A. Madaan, A. Kalyan, P. Clark, D. Wijaya and N. Tandon, RL4F: Generating natural language feedback with reinforcement learning for repairing model outputs, arXiv, 2023, preprint, arXiv:2305.08844, DOI:10.48550/arXiv.2305.08844, http://arxiv.org/abs/2305.08844.
- Y. Cao, H. Zhao, Y. Cheng, T. Shu, G. Liu, G. Liang, J. Zhao and Y. Li, Survey on large language model-enhanced reinforcement learning: Concept, taxonomy and methods, arXiv, 2024, preprint, arXiv:2404.00282, DOI:10.48550/arXiv.2404.00282, http://arxiv.org/abs/2404.00282.
- R. Rafailov, A. Sharma, E. Mitchell, S. Ermon, C. D. Manning and C. Finn, Direct preference optimization: Your language model is secretly a reward model, arXiv, 2023, preprint, arXiv:2305.18290, DOI:10.48550/arXiv.2305.18290, http://arxiv.org/abs/2305.18290.
- Y. Zheng, H. Yuan, C. Tan, W. Wang, S. Huang and F. Huang, RRHF: Rank responses to align language models with human feedback without tears, arXiv, 2023, preprint, arXiv:2304.05302, DOI:10.48550/arXiv.2304.05302, http://arxiv.org/abs/2304.05302.
- F. Song, B. Yu, M. Li, H. Yu, F. Huang, Y. Li and H. Wang, Preference ranking optimization for human alignment, arXiv, 2023, preprint, arXiv:2306.17492, DOI:10.48550/arXiv.2306.17492, http://arxiv.org/abs/2306.17492.
- S. Xu, W. Fu, J. Gao, W. Ye, W. Liu, Z. Mei, G. Wang, C. Yu and Y. Wu, Is DPO superior to PPO for LLM alignment? a comprehensive study, arXiv, 2024, preprint, arXiv:2404.10719, DOI:10.48550/arXiv.2404.10719, http://arxiv.org/abs/2404.10719.
- A. Bou, M. Thomas, S. Dittert, C. Navarro Ramírez, M. Majewski, Y. Wang, S. Patel, G. Tresadern, M. Ahmad, M. Vincent, et al., Acegen: Reinforcement learning of generative chemical agents for drug discovery, arXiv, 2024, preprint, arXiv:2405.04657, DOI:10.48550/arXiv.2405.04657.
- T. Hayes, R. Rao, H. Akin, N. J. Sofroniew, D. Oktay, Z. Lin, R. Verkuil, V. Q. Tran, J. Deaton, M. Wiggert, R. Badkundri, I. Shafkat, J. Gong, A. Derry, R. S. Molina, N. Thomas, Y. Khan, C. Mishra, C. Kim, L. J. Bartie, M. Nemeth, P. D. Hsu, T. Sercu, S. Candido and A. Rives, bioRxiv, 2024, preprint, DOI:10.1101/2024.07.01.600583v2, https://www.biorxiv.org/content/10.1101/2024.07.01.600583v2.
- Q. Pei, W. Zhang, J. Zhu, K. Wu, K. Gao, L. Wu, Y. Xia and R. Yan, BioT5: Enriching cross-modal integration in biology with chemical knowledge and natural language associations, in Proceedings of the 2023 Conference on Empirical Methods in Natural Language Processing, ed. H. Bouamor, J. Pino and K. Bali, Association for Computational Linguistics, Stroudsburg, PA, USA, 2023, pp. 1102–1123, DOI:10.18653/v1/2023.emnlp-main.70.
- Q. Pei, L. Wu, K. Gao, X. Liang, Y. Fang, J. Zhu, S. Xie, T. Qin and R. Yan, BioT5+: Towards generalized biological understanding with IUPAC integration and multi-task tuning, arXiv, 2024, preprint, arXiv:2402.17810, DOI:10.48550/arXiv.2402.17810, http://arxiv.org/abs/2402.17810.
- J. Li and X. Jiang, Mol-BERT: An effective molecular representation with BERT for molecular property prediction, Proc. Int. Wirel. Commun. Mob. Comput. Conf., 2021, 2021, 1530–8669, DOI:10.1155/2021/7181815.
- C. Qian, H. Tang, Z. Yang, H. Liang and Y. Liu, Can large language models empower molecular property prediction?, arXiv, 2023, preprint, arXiv:2307.07443, DOI:10.48550/arXiv.2307.07443, http://arxiv.org/abs/2307.07443.
- T.-H. Nguyen-Vo, Q. H. Trinh, L. Nguyen, T. T. T. Do, M. C. H. Chua and B. P. Nguyen, Predicting Antimalarial Activity in Natural Products Using Pretrained Bidirectional Encoder Representations from Transformers, J. Chem. Inf. Model., 2022, 62(21), 5050–5058, DOI:10.1021/acs.jcim.1c00584.
- Q. Jin, B. Dhingra, Z. Liu, W. W. Cohen and X. Lu, PubMedQA: A dataset for biomedical research question answering, arXiv, 2019, preprint, arXiv:1909.06146, DOI:10.48550/arXiv.1909.06146, http://arxiv.org/abs/1909.06146.
- W. Ahmad, E. Simon, S. Chithrananda, G. Grand and B. Ramsundar, ChemBERTa-2: Towards Chemical Foundation Models, arXiv, 2022, preprint, arXiv:2209.01712 [cs, q-bio], DOI:10.48550/arXiv.2209.01712, http://arxiv.org/abs/2209.01712.
- R. Taylor, M. Kardas, G. Cucurull, T. Scialom, A. Hartshorn, E. Saravia, A. Poulton, V. Kerkez and R. Stojnic, Galactica: A large language model for science, arXiv, 2022, preprint, arXiv:2211.09085, DOI:10.48550/arXiv.2211.09085, http://arxiv.org/abs/2211.09085.
- W. L. Taylor, “Cloze Procedure”: A New Tool for Measuring Readability, Journal. Q., 1953, 30(4), 415–433, DOI:10.1177/107769905303000401.
- Y. Liu, M. Ott, N. Goyal, J. Du, M. Joshi, D. Chen, O. Levy, M. Lewis, L. Zettlemoyer and V. Stoyanov, RoBERTa: A Robustly Optimized BERT Pretraining Approach, arXiv, 2019, preprint, arXiv:1907.11692 [cs], DOI:10.48550/arXiv.1907.11692, http://arxiv.org/abs/1907.11692.
- A. Radford, K. Narasimhan, T. Salimans and I. Sutskever, Improving language understanding with unsupervised learning, 2018 Search PubMed.
- M. Lewis, Y. Liu, N. Goyal, M. Ghazvininejad, A. Mohamed, O. Levy, V. Stoyanov and L. Zettlemoyer. BART: Denoising Sequence-to-Sequence Pre-training for Natural Language Generation, Translation and Comprehension, arXiv, 2019, preprint, arXiv:1910.13461 [cs, stat], DOI:10.48550/arXiv.1910.13461, http://arxiv.org/abs/1910.13461.
- C. Raffel, N. Shazeer, A. Roberts, K. Lee, S. Narang, M. Matena, Y. Zhou, W. Li and P. J. Liu, Exploring the limits of transfer learning with a unified text-to-text transformer, arXiv, 2019, preprint, arXiv:1910.10683, DOI:10.48550/arXiv.1910.10683, http://arxiv.org/abs/1910.10683.
- H. W. Chung, L. Hou, S. Longpre, B. Zoph, Y. Tay, W. Fedus, Y. Li, X. Wang, M. Dehghani, S. Brahma, A. Webson, S. S. Gu, Z. Dai, M. Suzgun, X. Chen, A. Chowdhery, A. Castro-Ros, M. Pellat, K. Robinson, D. Valter, S. Narang, G. Mishra, Y. Adams, Z. Vincent, Y. Huang, A. Dai, H. Yu, S. Petrov, H. Chi, J. Dean, J. Devlin, A. Roberts, D. Zhou, V. L. Quoc and J. Wei, Scaling instruction-finetuned language models, arXiv, 2022, preprint, arXiv:2210.11416, DOI:10.48550/arXiv.2210.11416, http://arxiv.org/abs/2210.11416.
- B. Tan, Y. Zhu, L. Liu, E. Xing, Z. Hu and J. Chen, Cappy: Outperforming and boosting large multi-task LMs with a small scorer, arXiv, 2023, preprint, arXiv:2311.06720, DOI:10.48550/arXiv.2311.06720.
- J. Shen, N. Tenenholtz, J. Brian Hall, D. Alvarez-Melis and N. Fusi, Tag-LLM: Repurposing general-purpose LLMs for specialized domains, arXiv, 2024, preprint, arXiv:2402.05140, DOI:10.48550/arXiv.2402.05140, http://arxiv.org/abs/2402.05140.
- G. Son, S. Baek, S. Nam, I. Jeong and S. Kim, Multi-task inference: Can large language models follow multiple instructions at once?, arXiv, 2024, preprint, arXiv:2402.11597, DOI:10.48550/arXiv.2402.11597, http://arxiv.org/abs/2402.11597.
- W. Feng, H. Chuzhan, Y. Zhang, Y. Han and H. Wang, Mixture-of-LoRAs: An efficient multitask tuning for large language models, arXiv, 2024, preprint, arXiv:2403.03432, DOI:10.48550/arXiv.2403.03432, http://arxiv.org/abs/2403.03432.
- Fuyu-8B: A multimodal architecture for AI agents, 2023, https://www.adept.ai/blog/fuyu-8b, accessed: 2023-11-8.
- S. Wu, H. Fei, L. Qu, W. Ji and T.-S. Chua, NExT-GPT: Any-to-any multimodal LLM, arXiv, 2023, preprint, arXiv:2309.05519, DOI:10.48550/arXiv.2309.05519, http://arxiv.org/abs/2309.05519.
- D. Bhattacharya, H. Cassady, M. Hickner and W. Reinhart, Large language models as molecular design engines, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-n0l8q.
- M. Vaškevičius, J. Kapočiūtė-Dzikienė and L. Šlepikas, Generative LLMs in organic chemistry: Transforming esterification reactions into natural language procedures, Appl. Sci., 2023, 13(24), 13140, DOI:10.3390/app132413140.
- A. Radford, J. W. Kim, T. Xu, G. Brockman, C. McLeavey and I. Sutskever, Robust speech recognition via large-scale weak supervision, arXiv, 2022, preprint, arXiv:2212.04356, DOI:10.48550/arXiv.2212.04356, http://arxiv.org/abs/2212.04356.
- J. Born and M. Manica, Regression Transformer enables concurrent sequence regression and generation for molecular language modelling, Nat. Mach. Intell., 2023, 5(4), 432–444, DOI:10.1038/s42256-023-00639-z.
- J. Mao, J. Wang, K.-H. Cho and K. Tai, No. iupacGPT: IUPAC-based large-scale molecular pre-trained model for property prediction and molecule generation, ChemRxiv, 2023, preprint, pp. 1–13, DOI:10.26434/chemrxiv-2023-5kjvh.
- N. Shoghi, A. Kolluru, J. R. Kitchin, Z. W. Ulissi, C. Lawrence Zitnick and B. M. Wood, From Molecules to Materials: Pre-training Large Generalizable Models for Atomic Property Prediction, arXiv, 2024, preprint, arXiv:2310.16802 [cs], DOI:10.48550/arXiv.2310.16802, http://arxiv.org/abs/2310.16802.
- K. M. Jablonka, P. Schwaller, A. Ortega-Guerrero and B. Smit, Leveraging large language models for predictive chemistry, Nat. Mach. Intell., 2024, 6(2), 161–169, DOI:10.1038/s42256-023-00788-1.
- R. Jacobs, M. P. Polak, L. E. Schultz, H. Mahdavi, V. Honavar and D. Morgan, Regression with large language models for materials and molecular property prediction, arXiv, 2024, preprint, arXiv:2409.06080, DOI:10.48550/arXiv.2409.06080, http://arxiv.org/abs/2409.06080.
- Yu-C. Lo, S. E. Rensi, T. Wen and R. B. Altman, Machine learning in chemoinformatics and drug discovery, Drug discovery today, 2018, 23(8), 1538–1546 CrossRef CAS.
- L. David, A. Thakkar, R. Mercado and O. Engkvist, Molecular representations in AI-driven drug discovery: a review and practical guide, J. Cheminf., 2020, 12(1), 56, DOI:10.1186/s13321-020-00460-5.
- K. Atz, F. Grisoni and G. Schneider, Geometric deep learning on molecular representations, arXiv, 2021, preprint, arXiv:2107.12375, DOI:10.48550/arXiv.2107.12375.
- W. P. Walters and R. Barzilay, Applications of deep learning in molecule generation and molecular property prediction, Acc. Chem. Res., 2021, 54(2), 263–270, DOI:10.1021/acs.accounts.0c00699.
- A. Karthikeyan and U. Deva Priyakumar, Artificial intelligence: machine learning for chemical sciences, J. Chem. Sci., 2022, 134(1), 2, DOI:10.1007/s12039-021-01995-2.
- Z. Li, M. Jiang, S. Wang and S. Zhang, Deep learning methods for molecular representation and property prediction, Drug Discov. Today, 2022, 27(12), 103373, DOI:10.1016/j.drudis.2022.103373.
- C. Chen, W. Ye, Y. Zuo, C. Zheng and S. P. Ong, Graph networks as a universal machine learning framework for molecules and crystals, Chem. Mater., 2019, 31(9), 3564–3572 CrossRef.
- W. Hu, M. Fey, M. Zitnik, Y. Dong, H. Ren, B. Liu, M. Catasta and J. Leskovec, Open graph benchmark: Datasets for machine learning on graphs, Adv. Neural Inf. Process. Syst., 2020, 33, 22118–22133 Search PubMed.
- S. Kearnes, K. McCloskey, M. Berndl, V. Pande and P. Riley, Molecular graph convolutions: moving beyond fingerprints, J. Comput. Aided Mol. Des., 2016, 30, 595–608 CrossRef PubMed.
- Y. Wang, S. Wu, Y. Duan and Y. Huang, A point cloud-based deep learning strategy for protein–ligand binding affinity prediction, Briefings Bioinf., 2022, 23(1), bbab474 CrossRef PubMed.
- N. Thomas, T. Smidt, S. Kearnes, L. Yang, L. Li, K. Kohlhoff and P. Riley, Tensor field networks: Rotation-and translation-equivariant neural networks for 3d point clouds, arXiv, 2018, preprint, arXiv:1802.08219, DOI:10.48550/arXiv.1802.08219.
- L. Wang, Z. Lu, X. Liu, J. Fu and A. Zhang, Seppcnet: deeping learning on a 3d surface electrostatic potential point cloud for enhanced toxicity classification and its application to suspected environmental estrogens, Environ. Sci. Technol., 2021, 55(14), 9958–9967 CrossRef PubMed.
- S. Ahmadi, M. Amin Ghanavati and S. Rohani, Machine learning-guided prediction of cocrystals using point cloud-based molecular representation, Chem. Mater., 2024, 36, 1153–1161, DOI:10.1021/acs.chemmater.3c01437.
- S. Singh and R. B. Sunoj, Molecular Machine Learning for Chemical Catalysis: Prospects and Challenges, Acc. Chem. Res., 2023, 56(3), 402–412, DOI:10.1021/acs.accounts.2c00801.
- M. B. Andres and P. Schwaller, Transformers and large language models for chemistry and drug discovery, arXiv, 2023, preprint, arXiv:2310.06083, DOI:10.48550/arXiv.2310.06083, http://arxiv.org/abs/2310.06083.
- S. Shilpa, G. Kashyap and R. B. Sunoj, Recent Applications of Machine Learning in Molecular Property and Chemical Reaction Outcome Predictions, J. Phys. Chem. A, 2023, 127(40), 8253–8271, DOI:10.1021/acs.jpca.3c04779.
- D. S. Wigh, J. M. Goodman and A. A. Lapkin, A review of molecular representation in the age of machine learning, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12(5) DOI:10.1002/wcms.1603.
- N. O'Boyle and A. Dalke, DeepSMILES: An Adaptation of SMILES for Use in Machine-Learning of Chemical Structures, ChemRxiv, 2018, preprint Search PubMed.
- M. Krenn, F. Häse, A. K. Nigam, P. Friederich and A. Aspuru-Guzik, Self-Referencing Embedded Strings (SELFIES): A 100% robust molecular string representation, Mach. Learn.: Sci. Technol., 2020, 1(4), 045024, DOI:10.1088/2632-2153/aba947.
- S. R. Heller, A. McNaught, I. Pletnev, S. Stein and D. Tchekhovskoi, InChI, the IUPAC International Chemical Identifier, J. Cheminf., 2015, 7(1), 23, DOI:10.1186/s13321-015-0068-4.
- M. Das, A. Ghosh and R. B. Sunoj, Advances in machine learning with chemical language models in molecular property and reaction outcome predictions, J. Comput. Chem., 2024, 45(14), 1160–1176, DOI:10.1002/jcc.27315.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, Zinc: a free tool to discover chemistry for biology, J. Chem. Inf. Model., 2012, 52(7), 1757–1768 CrossRef CAS.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He and B. A. Shoemaker, et al., Pubchem substance and compound databases, Nucleic Acids Res., 2016, 44(D1), D1202–D1213 CrossRef CAS PubMed.
- H. Touvron, L. Martin, K. Stone, P. Albert, A. Almahairi, Y. Babaei, N. Bashlykov, S. Batra, P. Bhargava, S. Bhosale, et al., Llama 2: Open foundation and fine-tuned chat models, arXiv, 2023, preprint, arXiv:2307.09288, DOI:10.48550/arXiv.2307.09288.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, ChEMBL: a large-scale bioactivity database for drug discovery, Nucleic Acids Res., 2012, 40(Database issue), D1100–D1107, DOI:10.1093/nar/gkr777.
- R. Kinney, C. Anastasiades, R. Authur, I. Beltagy, J. Bragg, A. Buraczynski, I. Cachola, S. Candra, Y. Chandrasekhar, A. Cohan, M. Crawford, D. Downey, J. Dunkelberger, O. Etzioni, R. Evans, S. Feldman, J. Gorney, D. Graham, F. Hu, R. Huff, D. King, S. Kohlmeier, K. Bailey, M. Langan, D. Lin, H. Liu, K. Lo, J. Lochner, K. MacMillan, M. Tyler, C. Newell, S. Rao, S. Rohatgi, P. Sayre, Z. Shen, A. Singh, L. Soldaini, S. Subramanian, A. Tanaka, A. D. Wade, L. Wagner, L. L. Wang, C. Wilhelm, C. Wu, J. Yang, A. Zamarron, M. V. Zuylen and D. S. Weld, The semantic scholar open data platform, arXiv, 2023, preprint, arXiv:2301.10140, DOI:10.48550/arXiv.2301.10140, http://arxiv.org/abs/2301.10140.
- Y. Fang, X. Liang, N. Zhang, K. Liu, R. Huang, Z. Chen, X. Fan and H. Chen, Mol-instructions: A large-scale biomolecular instruction dataset for large language models, arXiv, 2023, preprint, arXiv:2306.08018, DOI:10.48550/arXiv.2306.08018, http://arxiv.org/abs/2306.08018.
- W. Jin-Mao, X.-J. Yuan, Q.-H. Hu and S.-Q. Wang, A novel measure for evaluating classifiers, Expert Syst. Appl., 2010, 37(5), 3799–3809, DOI:10.1016/j.eswa.2009.11.040.
- M. Krallinger, O. Rabal, F. Leitner, M. Vazquez, D. Salgado, Z. Lu, R. Leaman, Y. Lu, D. Ji, D. M. Lowe, R. A. Sayle, R. T. Batista-Navarro, R. Rak, T. Huber, T. Rocktäschel, S. Matos, D. Campos, B. Tang, H. Xu, T. Munkhdalai, K. H. Ryu, S. V. Ramanan, S. Nathan, S. Žitnik, M. Bajec, L. Weber, M. Irmer, S. A. Akhondi, J. A. Kors, S. Xu, X. An, U. K. Sikdar, A. Ekbal, M. Yoshioka, T. M. Dieb, M. Choi, K. Verspoor, M. Khabsa, C. L. Giles, H. Liu, K. E. Ravikumar, A. Lamurias, F. M. Couto, H.-J. Dai, R. T.-H. Tsai, C. Ata, T. Can, A. Usié, R. Alves, I. Segura-Bedmar, P. Martínez, J. Oyarzabal and A. Valencia, The CHEMDNER corpus of chemicals and drugs and its annotation principles, J. Cheminf., 2015, 7(S2), 1758–2946, DOI:10.1186/1758-2946-7-S1-S2.
- L. Jiao, Y. Sun, R. J. Johnson, D. Sciaky, C.-H. Wei, R. Leaman, A. P. Davis, C. J. Mattingly, T. C. Wiegers and Z. Lu, BioCreative V CDR task corpus: a resource for chemical disease relation extraction, Database, 2016, baw068, DOI:10.1093/database/baw068.
- R. Islamaj Dogan, S. Kim, A. Chatr-Aryamontri, C.-H. Wei, D. C. Comeau, R. Antunes, S. Matos, Q. Chen, A. Elangovan, N. C. Panyam, K. Verspoor, H. Liu, Y. Wang, Z. Liu, B. Altinel, Z. M. Hüsünbeyi, A. Özgür, A. Fergadis, C.-K. Wang, H.-J. Dai, T. Tran, R. Kavuluru, L. Luo, A. Steppi, J. Zhang, J. Qu and Z. Lu, Overview of the BioCreative VI precision medicine track: mining protein interactions and mutations for precision medicine, Database, 2019, bay147, DOI:10.1093/database/bay147.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Gene ontology: tool for the unification of biology. the gene ontology consortium, Nat. Genet., 2000, 25(1), 25–29, DOI:10.1038/75556.
- D. Hendrycks, C. Burns, S. Basart, A. Zou, M. Mazeika, D. Song and J. Steinhardt, Measuring massive multitask language understanding, in International Conference on Learning Representations, 2020, https://openreview.net/pdf?id=d7KBjmI3GmQ Search PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, PubChem in 2021: new data content and improved web interfaces, Nucleic Acids Res., 2021, 49(D1), D1388–D1395, DOI:10.1093/nar/gkaa971.
- A. Pal, L. Kumar Umapathi and M. Sankarasubbu, MedMCQA: A large-scale multi-subject multi-choice dataset for medical domain question answering, in Conference on Health, Inference and Learning, PMLR, 2022, pp. 248–260 Search PubMed.
- J. Lu and Y. Zhang, Unified Deep Learning Model for Multitask Reaction Predictions with Explanation, J. Chem. Inf. Model., 2022, 62(6), 1376–1387, DOI:10.1021/acs.jcim.1c01467.
- UniProt Consortium, UniProt: The universal protein knowledgebase in 2023, Nucleic Acids Res., 2023, 51(D1), D523–D531, DOI:10.1093/nar/gkac1052.
- M. H. S. Segler, M. Preuss and M. P. Waller, Planning chemical syntheses with deep neural networks and symbolic ai, Nature, 2018, 555(7698), 604–610 CrossRef CAS PubMed.
- Awesome-chemistry-datasets/code_of_conduct.md at main · kjappelbaum/awesome-chemistry-datasets, 2023, https://github.com/kjappelbaum/awesome-chemistry-datasets/blob/main/code_of_conduct.md.
- A. Mirza, N. Alampara, S. Kunchapu, B. Emoekabu, A. Krishnan, M. Wilhelmi, M. Okereke, J. Eberhardt, A. M. Elahi, M. Greiner, C. T. Holick, T. Gupta, M. Asgari, C. Glaubitz, L. C. Klepsch, Y. Koster, J. Meyer, S. Miret, T. Hoffmann, F. A. Kreth, M. Ringleb, N. Roesner, U. S. Schubert, L. M. Stafast, D. Wonanke, M. Pieler, P. Schwaller and K. Maik Jablonka, Are large language models superhuman chemists?, arXiv, 2024, preprint, arXiv:2404.01475, DOI:10.48550/arXiv.2404.01475, http://arxiv.org/abs/2404.01475.
- D. E. Gloriam, Bigger is better in virtual drug screens, Nature, 2019, 566(7743), 193–194, DOI:10.1038/d41586-019-00145-6.
- R. Irwin, S. Dimitriadis, J. He and E. J. Bjerrum, Chemformer: a pre-trained transformer for computational chemistry, Mach. Learn.: Sci. Technol., 2022, 3(1), 015022, DOI:10.1088/2632-2153/ac3ffb.
- S. Liu, W. Nie, C. Wang, J. Lu, Z. Qiao, L. Liu, J. Tang, C. Xiao and A. Anandkumar, Multi-modal molecule structure–text model for text-based retrieval and editing, Nat. Mach. Intell., 2023, 5(12), 1447–1457, DOI:10.1038/s42256-023-00759-6.
- M. Livne, Z. Miftahutdinov, E. Tutubalina, M. Kuznetsov, D. Polykovskiy, A. Brundyn, A. Jhunjhunwala, A. Costa, A. Aliper, A. Aspuru-Guzik and A. Zhavoronkov, Nach0: Multimodal natural and chemical languages foundation model, arXiv, 2023, preprint, arXiv:2311.12410, DOI:10.48550/arXiv.2311.12410, http://arxiv.org/abs/2311.12410.
- P. Walters, We need better benchmarks for machine learning in drug discovery, 2023, http://practicalcheminformatics.blogspot.com/2023/08/we-need-better-benchmarks-for-machine.html.
- C. Fang, Y. Wang, R. Grater, S. Kapadnis, C. Black, P. Trapa and S. Sciabola, Prospective validation of machine learning algorithms for absorption, distribution, metabolism and excretion prediction: An industrial perspective, J. Chem. Inf. Model., 2023, 63(11), 3263–3274, DOI:10.1021/acs.jcim.3c00160.
- Therapeutics data commons, https://tdcommons.ai/, accessed: 2024-6-13.
- K. Huang, T. Fu, W. Gao, Z. Yue, Y. Roohani, J. Leskovec, C. W. Coley, C. Xiao, J. Sun and M. Zitnik, Therapeutics data commons: Machine learning datasets and tasks for drug discovery and development, arXiv, 2021, preprint, arXiv:2102.09548, DOI:10.48550/arXiv.2102.09548, http://arxiv.org/abs/2102.09548.
- A. Velez-Arce, K. Huang, M. M. Li, X. Lin, W. Gao, T. Fu, M. Kellis, B. L. Pentelute and M. Zitnik, TDC-2: Multimodal foundation for therapeutic science, bioRxiv, 2024, preprint, DOI:10.1101/2024.06.12.598655.
- A. Rich and B. Birnbaum, Building adme benchmark datasets that drive impact, 2023, https://www.inductive.bio/blog/building-better-benchmarks-for-adme-optimization.
- K. Hira, M. Zaki, D. Sheth, Mausam and N. M. A. Krishnan, Reconstructing the materials tetrahedron: challenges in materials information extraction, Digital Discovery, 2024, 3(5), 1021–1037, 10.1039/d4dd00032c.
- J. Dagdelen, A. Dunn, S. Lee, N. Walker, A. S. Rosen, G. Ceder, K. A. Persson and A. Jain, Structured information extraction from scientific text with large language models, Nat. Commun., 2024, 15(1), 1418, DOI:10.1038/s41467-024-45563-x.
- D. Circi, G. Khalighinejad, A. Chen, B. Dhingra and L. C. Brinson, How well do large language models understand tables in materials science?, Integr. Mater. Manuf. Innov., 2024, 13(3), 669–687, DOI:10.1007/s40192-024-00362-6.
- J. M. Laurent, J. D. Janizek, M. Ruzo, M. M. Hinks, M. J. Hammerling, S. Narayanan, M. Ponnapati, A. D. White and S. G. Rodriques, LAB-bench: Measuring capabilities of language models for biology research, arXiv, 2024, preprint, arXiv:2407.10362, DOI:10.48550/arXiv.2407.10362, http://arxiv.org/abs/2407.10362.
- N. Alampara, S. Miret and K. M. Jablonka, MatText: Do language models need more than text & scale for materials modeling?, arXiv, 2024, preprint, arXiv:2406.17295, DOI:10.48550/arXiv.2406.17295, http://arxiv.org/abs/2406.17295.
- Y. Song, S. Miret and B. Liu, MatSci-NLP: Evaluating scientific language models on materials science language tasks using text-to-schema modeling, arXiv, 2023, preprint, arXiv:2305.08264, DOI:10.48550/arXiv.2305.08264, http://arxiv.org/abs/2305.08264.
- M. Zaki, M. Jayadeva and N. M. Anoop Krishnan, MaScQA: investigating materials science knowledge of large language models, Digital Discovery, 2024, 3(2), 313–327, 10.1039/D3DD00188A.
- A. Sultan, J. Sieg, M. Mathea and A. Volkamer, Transformers for molecular property prediction: Lessons learned from the past five years, arXiv, 2024, preprint, arXiv:2404.03969, DOI:10.48550/arXiv.2404.03969, http://arxiv.org/abs/2404.03969.
- J. Ock, C. Guntuboina and A. B. Farimani, Catalyst Energy Prediction with CatBERTa: Unveiling Feature Exploration Strategies through Large Language Models, ACS Catal., 2023, 13(24), 16032–16044, DOI:10.1021/acscatal.3c04956.
- A. Yuksel, E. Ulusoy, A. Ünlü and T. Doğan, SELFormer: Molecular representation learning via SELFIES language models, arXiv, 2023, preprint, arXiv:2304.04662, DOI:10.48550/arXiv.2304.04662, http://arxiv.org/abs/2304.04662.
- M. Yoshitake, F. Sato, H. Kawano and H. Teraoka, MaterialBERT for natural language processing of materials science texts, Sci. Technol. Adv. Mater., 2022, 2(1), 372–380, DOI:10.1080/27660400.2022.2124831.
- J. Yu, C. Zhang, Y. Cheng, Y.-F. Yang, Y.-B. She, F. Liu, W. Su and A. Su, SolvBERT for solvation free energy and solubility prediction: a demonstration of an NLP model for predicting the properties of molecular complexes, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-0hl5p.
- S. Boobier, D. R. J. Hose, A. J. Blacker and B. N. Nguyen, Machine learning with physicochemical relationships: solubility prediction in organic solvents and water, Nat. Commun., 2020, 11(1), 5753 Search PubMed.
- Z. Hong, A. Ajith, G. Pauloski, E. Duede, K. Chard and I. Foster, The diminishing returns of masked language models to science, arXiv, 2022, preprint, arXiv:2205.11342, DOI:10.48550/arXiv.2205.11342, http://arxiv.org/abs/2205.11342.
- S. Huang and J. M. Cole, BatteryBERT: A Pretrained Language Model for Battery Database Enhancement, J. Chem. Inf. Model., 2022, 62(24), 6365–6377, DOI:10.1021/acs.jcim.2c00035.
- A. Trewartha, N. Walker, H. Huo, S. Lee, K. Cruse, J. Dagdelen, A. Dunn, K. A. Persson, G. Ceder and A. Jain, Quantifying the advantage of domain-specific pre-training on named entity recognition tasks in materials science, Patterns, 2022, 3(4), 100488, DOI:10.1016/j.patter.2022.100488.
- T. Gupta, M. Zaki, N. M. A. Krishnan and Mausam, MatSciBERT: A materials domain language model for text mining and information extraction, npj Comput. Mater., 2022, 8(1), 1–11, DOI:10.1038/s41524-022-00784-w.
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Large-scale chemical language representations capture molecular structure and properties, Nat. Mach. Intell., 2022, 4(12), 1256–1264, DOI:10.1038/s42256-022-00580-7.
- J. Guo, A. Santiago Ibanez-Lopez, H. Gao, V. Quach, C. W. Coley, K. F. Jensen and R. Barzilay, Automated chemical reaction extraction from scientific literature, J. Chem. Inf. Model., 2022, 62(9), 2035–2045, DOI:10.1021/acs.jcim.1c00284.
- B. Fabian, T. Edlich, H. Gaspar, M. Segler, J. Meyers, M. Fiscato and M. Ahmed. Molecular representation learning with language models and domain-relevant auxiliary tasks, arXiv, 2020, preprint, arXiv:2011.13230, DOI:10.48550/arXiv.2011.13230, http://arxiv.org/abs/2011.13230.
- H.-C. Shin, Y. Zhang, E. Bakhturina, R. Puri, M. Patwary, M. Shoeybi and R. Mani, BioMegatron: Larger biomedical domain language model, arXiv, 2020, preprint, arXiv:2010.06060, DOI:10.48550/arXiv.2010.06060, http://arxiv.org/abs/2010.06060.
- Y. Gu, R. Tinn, H. Cheng, M. Lucas, N. Usuyama, X. Liu, T. Naumann, J. Gao and H. Poon, Domain-Specific language model pretraining for biomedical natural language processing, ACM Trans. Comput. Healthc., 2021, 3(1), 1–23, DOI:10.1145/3458754.
- L. Maziarka, T. Danel, S. Mucha, K. Rataj, J. Tabor and S. Jastrzębski, Molecule attention transformer, arXiv, 2020, preprint, arXiv:2002.08264, DOI:10.48550/arXiv.2002.08264, http://arxiv.org/abs/2002.08264.
- S. Wang, Y. Guo, Y. Wang, H. Sun and J. Huang, SMILES-BERT: Large Scale Unsupervised Pre-Training for Molecular Property Prediction, in Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, BCB ’19, Association for Computing Machinery, Niagara Falls NY USA, 2019, pp. 429–436, DOI:10.1145/3307339.3342186.
- Y. Peng, S. Yan and Z. Lu, Transfer learning in biomedical natural language processing: An evaluation of BERT and ELMo on ten benchmarking datasets, arXiv, 2019, preprint, arXiv:1906.05474, DOI:10.48550/arXiv.1906.05474, http://arxiv.org/abs/1906.05474.
- K. Huang, J. Altosaar and R. Ranganath, ClinicalBERT: Modeling clinical notes and predicting hospital readmission, arXiv, 2019, preprint, arXiv:1904.05342, DOI:10.48550/arXiv.1904.05342, http://arxiv.org/abs/1904.05342.
- I. Beltagy, K. Lo and A. Cohan, SciBERT: A pretrained language model for scientific text, arXiv, 2019, preprint, arXiv:1903.10676, DOI:10.48550/arXiv.1903.10676, http://arxiv.org/abs/1903.10676.
- J. Lee, W. Yoon, S. Kim, D. Kim, S. Kim, C. H. So and J. Kang, BioBERT: a pre-trained biomedical language representation model for biomedical text mining, Bioinformatics, 2020, 36(4), 1234–1240, DOI:10.1093/bioinformatics/btz682.
- D. Wu, Q. Chen, X. Chen, F. Han, Z. Chen and Y. Wang, The blood–brain barrier: structure, regulation and drug delivery, Signal Transduct. Targeted Ther., 2023, 8(1), 1–27, DOI:10.1038/s41392-023-01481-w.
- C. Bissantz, B. Kuhn and M. Stahl, A medicinal chemist's guide to molecular interactions, J. Med. Chem., 2010, 53(14), 5061–5084 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, The medicinal chemist's toolbox: an analysis of reactions used in the pursuit of drug candidates, J. Med. Chem., 2011, 54(10), 3451–3479 Search PubMed.
- I. Doytchinova, Drug Design—Past, Present, Future, Molecules, 2022, 27(5), 1496, DOI:10.3390/molecules27051496.
- L. H. Sperling, Introduction to physical polymer science, Wiley-Blackwell, Chichester, England, 4th edn, 2005 Search PubMed.
- D. J. Newman and G. M. Cragg, Natural Products as Sources of New Drugs from 1981 to 2014, J. Nat. Prod., 2016, 79(3), 629–661, DOI:10.1021/acs.jnatprod.5b01055.
- P. M. P. Ferreira, D. D. R. Arcanjo and A. P. Peron, Drug development, Brazilian biodiversity and political choices: Where are we heading?, J. Toxicol. Environ. Health, Part B, 2023, 26(5), 257–274, DOI:10.1080/10937404.2023.2193762.
- H. C. Kolb and K. B. Sharpless, The growing impact of click chemistry on drug discovery, Drug Discovery Today, 2003, 8(24), 1128–1137, DOI:10.1016/S1359-6446(03)02933-7.
- N. J. Castellino, A. P. Montgomery, J. J. Danon and M. Kassiou, Late-stage Functionalization for Improving Drug-like Molecular Properties, Chem. Rev., 2023, 123(13), 8127–8153, DOI:10.1021/acs.chemrev.2c00797.
- K. Sharma, K. K. Sharma, A. Sharma and R. Jain, Peptide-based drug discovery: Current status and recent advances, Drug Discovery Today, 2023, 28(2), 103464, DOI:10.1016/j.drudis.2022.103464.
- B. J. Reizman and K. F. Jensen, Feedback in Flow for Accelerated Reaction Development, Acc. Chem. Res., 2016, 49(9), 1786–1796, DOI:10.1021/acs.accounts.6b00261.
- J. A. DiMasi, H. G. Grabowski and R. W. Hansen, Innovation in the pharmaceutical industry: New estimates of R&D costs, J. Health Econ., 2016, 47, 20–33, DOI:10.1016/j.jhealeco.2016.01.012.
- E. G. Lewars, Computational chemistry: Introduction to the theory and applications of molecular and quantum mechanics, Springer International Publishing, Cham, Switzerland, 4th edn, 2024, DOI:10.1007/978-3-031-51443-2.
- X. Bidault and S. Chaudhuri, How Accurate Can Crystal Structure Predictions Be for High-Energy Molecular Crystals?, Molecules, 2023, 28(11), 4471, DOI:10.3390/molecules28114471.
- E. O. Pyzer-Knapp, L. Chen, G. M. Day and A. I. Cooper, Accelerating computational discovery of porous solids through improved navigation of energy-structure-function maps, Sci. Adv., 2021, 7(33), eabi4763, DOI:10.1126/sciadv.abi4763.
- S. Fredericks, K. Parrish, D. Sayre and Q. Zhu, PyXtal: A Python library for crystal structure generation and symmetry analysis, Comput. Phys. Commun., 2021, 261, 107810, DOI:10.1016/j.cpc.2020.107810.
- D. H. Case, J. E. Campbell, P. J. Bygrave and G. M. Day, Convergence Properties of Crystal Structure Prediction by Quasi-Random Sampling, J. Chem. Theory Comput., 2016, 12(2), 910–924, DOI:10.1021/acs.jctc.5b01112.
- A. V. Kazantsev, P. G. Karamertzanis, C. S. Adjiman and C. C. Pantelides, Efficient Handling of Molecular Flexibility in Lattice Energy Minimization of Organic Crystals, J. Chem. Theory Comput., 2011, 7(6), 1998–2016, DOI:10.1021/ct100597e.
- G. Huang, Y. Guo, Y. Chen and Z. Nie, Application of Machine Learning in Material Synthesis and Property Prediction, Materials, 2023, 16(17), 5977, DOI:10.3390/ma16175977.
- K. Martinez-Mayorga, J. G. Rosas-Jiménez, K. Gonzalez-Ponce, E. López-López, A. Neme and J. L. Medina-Franco, The pursuit of accurate predictive models of the bioactivity of small molecules, Chem. Sci., 2024, 15(6), 1938–1952, 10.1039/D3SC05534E.
- P. W. Geemi, H. A. Gandhi, A. Seshadri and A. D. White, A Perspective On Explanations Of Molecular Prediction Models, J. Chem. Theory Comput., 2023, 19(8), 2149–2160, DOI:10.1021/acs.jctc.2c01235.
- D. Xiang, V. Bashlovkina, F. Han, S. Baumgartner and M. Bendersky, LLMs to the Moon? Reddit Market Sentiment Analysis with Large Language Models, in Companion Proceedings of the ACM Web Conference 2023, WWW ’23 Companion, Association for Computing Machinery, New York, NY, USA, 2023, pp. 1014–1019, DOI:10.1145/3543873.3587605.
- P. Schwaller, D. Probst, A. Vaucher, V. H. Nair, D. Kreutter, T. Laino and J. Reymond, Mapping the space of chemical reactions using attention-based neural networks, Nat. Mach. Intell., 2020, 3(2), 144–152, DOI:10.1038/s42256-020-00284-w.
- A. Toniato, A. C. Vaucher, P. Schwaller and T. Laino, Enhancing diversity in language based models for single-step retrosynthesis, Digital Discovery, 2023, 2(2), 489–501, 10.1039/D2DD00110A.
- P. Schwaller, B. Hoover, J.-L. Reymond, H. Strobelt and T. Laino, Extraction of organic chemistry grammar from unsupervised learning of chemical reactions, Sci. Adv., 2021, 7(15), eabe4166, DOI:10.1126/sciadv.abe4166.
- P. Schwaller, A. C. Vaucher, T. Laino and J.-L. Reymond, Prediction of chemical reaction yields using deep learning, Mach. Learn. Sci. Technol., 2021, 2(1), 015016, DOI:10.1088/2632-2153/abc81d.
- S. Wang, Y. Guo, Y. Wang, H. Sun and J. Huang, SMILES-BERT: Large Scale Unsupervised Pre-Training for Molecular Property Prediction, in Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, BCB ’19, Association for Computing Machinery, New York, NY, USA, 2019, pp. 429–436, DOI:10.1145/3307339.3342186.
- X.-C. Zhang, C.-K. Wu, J.-C. Yi, X.-X. Zeng, C.-Q. Yang, A.-P. Lu, T.-J. Hou, T.-J. Hou and D.-S. Cao, Pushing the Boundaries of Molecular Property Prediction for Drug Discovery with Multitask Learning BERT Enhanced by SMILES Enumeration, Research, 2022, 0004, DOI:10.34133/research.0004.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties, Nucleic Acids Res., 2021, 49(W1), W5–W14, DOI:10.1093/nar/gkab255.
- T. Sterling and J. John, Irwin. ZINC 15 – Ligand Discovery for Everyone, J. Chem. Inf. Model., 2015, 55(11), 2324–2337, DOI:10.1021/acs.jcim.5b00559.
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Large-scale chemical language representations capture molecular structure and properties, Nat. Mach. Intell., 2022, 4(12), 1256–1264, DOI:10.1038/s42256-022-00580-7.
- T. Wolf, L. Debut, V. Sanh, J. Chaumond, C. Delangue, A. Moi, P. Cistac, T. Rault, R. Louf, M. Funtowicz, J. Davison, S. Shleifer, P. von Platen, C. Ma, Y. Jernite, J. Plu, C. Xu, T. Le Scao, S. Gugger, M. Drame, Q. Lhoest and A. M. Rush, Huggingface's transformers: State-of-the-art natural language processing, 2020 Search PubMed.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, Analyzing Learned Molecular Representations for Property Prediction, J. Chem. Inf. Model., 2019, 59(8), 3370–3388, DOI:10.1021/acs.jcim.9b00237.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction, ACS Cent. Sci., 2019, 5(9), 1572–1583, DOI:10.1021/acscentsci.9b00576.
- J. Vig, Bertviz: A tool for visualizing multihead self-attention in the bert model, in ICLR workshop: Debugging machine learning models, 2019, vol. 3 Search PubMed.
- M. A. Skinnider, Invalid SMILES are beneficial rather than detrimental to chemical language models, Nat. Mach. Intell., 2024, 6(4), 437–448, DOI:10.1038/s42256-024-00821-x.
- F. H. Vermeire and W. H. Green, Transfer learning for solvation free energies: From quantum chemistry to experiments, Chem. Eng. J., 2021, 418, 129307 Search PubMed.
- D. L. Mobley and J. P. Guthrie, FreeSolv: a database of experimental and calculated hydration free energies, with input files, J. Comput. Aided Mol. Des., 2014, 28(7), 711–720, DOI:10.1007/s10822-014-9747-x.
- A. V. Marenich, C. P. Kelly, J. D. Thompson, G. D. Hawkins, C. C. Chambers, D. J. Giesen, P. Winget, C. J. Cramer and D. G. Truhlar, Minnesota solvation database (mnsol) version 2012, 2020.
- E. Moine, R. Privat, B. Sirjean and J.-N. Jaubert, Estimation of solvation quantities from experimental thermodynamic data: Development of the comprehensive compsol databank for pure and mixed solutes, J. Phys. Chem. Ref. Data, 2017, 46(3), 033102, DOI:10.1063/1.5000910.
- L. M. Grubbs, M. Saifullah, E. Nohelli, S. Ye, S. S. Achi, W. E. Acree Jr and M. H. Abraham, Mathematical correlations for describing solute transfer into functionalized alkane solvents containing hydroxyl, ether, ester or ketone solvents, Fluid Phase Equilib., 2010, 298(1), 48–53 CrossRef CAS.
- K. Yang, K. Swanson, W. Jin, C. Coley, H. Gao, A. Guzman-Perez, T. Hopper, B. P. Kelley, A. Palmer, V. Settels, et al., Are learned molecular representations ready for prime time?, ChemRxiv, 2019, preprint, DOI:10.26434/chemrxiv.7940594.v2.
- Y. Rong, Y. Bian, T. Xu, W.-Y. Xie, Y. Wei, W.-B. Huang and J. Huang, Self-Supervised Graph Transformer on Large-Scale Molecular Data, Adv. Neural Inf. Process. Syst., 2020, 33, 12559–12571 Search PubMed.
- B. Winter, C. Winter, J. Schilling and A. Bardow, A smile is all you need: predicting limiting activity coefficients from SMILES with natural language processing, Digital Discovery, 2022, 1(6), 859–869, 10.1039/D2DD00058J.
- J. Jiang, Y. Li, R. Zhang and Y. Liu, INTransformer: Data augmentation-based contrastive learning by injecting noise into transformer for molecular property prediction, J. Mol. Graphics Modell., 2024, 128, 108703, DOI:10.1016/j.jmgm.2024.108703.
- S. Liu, W. Nie, C. Wang, J. Lu, Z. Qiao, L. Liu, J. Tang, C. Xiao and A. Anandkumar, Multi-modal molecule structure-text model for text-based retrieval and editing, arXiv, 2022, preprint, arXiv:2212.10789, DOI:10.48550/arXiv.2212.10789, http://arxiv.org/abs/2212.10789.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, PubChem 2023 update, Nucleic Acids Res., 2023, 51(D1), D1373–D1380, DOI:10.1093/nar/gkac956.
- M. Xu, X. Yuan, S. Miret and J. Tang, ProtST: Multi-modality learning of protein sequences and biomedical texts, in International Conference on Machine Learning, PMLR, 2023, pp. 38749–38767 Search PubMed.
- G. M. Hocky, Connecting molecular properties with plain language, Nat. Mach. Intell., 2024, 6(3), 249–250, DOI:10.1038/s42256-024-00812-y.
- J. M. Z. Chaves, E. Wang, T. Tu, E. Dhaval Vaishnav, B. Lee, S. Sara Mahdavi, C. Semturs, D. Fleet, V. Natarajan and S. Azizi, Tx-LLM: A large language model for therapeutics, arXiv, 2024, preprint, arXiv:2406.06316, DOI:10.48550/arXiv.2406.06316, http://arxiv.org/abs/2406.06316.
- E. Bolton, A. Venigalla, M. Yasunaga, D. Hall, B. Xiong, T. Lee, R. Daneshjou, J. Frankle, P. Liang, M. Carbin and C. D. Manning, BioMedLM: A 2.7B parameter language model trained on biomedical text, arXiv, 2024, preprint, arXiv:2403.18421, DOI:10.48550/arXiv.2403.18421, http://arxiv.org/abs/2403.18421.
- B. Yu, F. N. Baker, Z. Chen, X. Ning and H. Sun, LlaSMol: Advancing Large Language Models for Chemistry with a Large-Scale, Comprehensive, High-Quality Instruction Tuning Dataset, arXiv, 2024, preprint, arXiv:2402.09391 [cs], DOI:10.48550/arXiv.2402.09391, http://arxiv.org/abs/2402.09391.
- Y. Labrak, A. Bazoge, E. Morin, P.-A. Gourraud, M. Rouvier and R. Dufour, BioMistral: A collection of open-source pretrained large language models for medical domains, arXiv, 2024, preprint, arXiv:2402.10373, DOI:10.48550/arXiv.2402.10373, http://arxiv.org/abs/2402.10373.
- S. Pieri, S. Shaji Mullappilly, F. S. Khan, R. Muhammad Anwer, S. Khan, T. Baldwin and H. Cholakkal, BiMediX: Bilingual medical mixture of experts LLM, arXiv, 2024, preprint, arXiv:2402.13253, DOI:10.48550/arXiv.2402.13253, http://arxiv.org/abs/2402.13253.
- X. Zhao, Q. Zhao and T. Tanaka, EpilepsyLLM: Domain-specific large language model fine-tuned with epilepsy medical knowledge, arXiv, 2024, preprint, arXiv:2401.05908, DOI:10.48550/arXiv.2401.05908, http://arxiv.org/abs/2401.05908.
- Z. Chen, M. Varma, J.-B. Delbrouck, M. Paschali, L. Blankemeier, D. Van Veen, J. M. J. Valanarasu, A. Youssef, J. Paul Cohen, E. P. Reis, E. B. Tsai, A. Johnston, C. Olsen, T. M. Abraham, S. Gatidis, A. S. Chaudhari and C. Langlotz, CheXagent: Towards a foundation model for chest X-ray interpretation, arXiv, 2024, preprint, arXiv:2401.12208, DOI:10.48550/arXiv.2401.12208, http://arxiv.org/abs/2401.12208.
- G. W. Kyro, A. Morgunov, R. I. Brent and V. S. Batista, ChemSpaceAL: An Efficient Active Learning Methodology Applied to Protein-Specific Molecular Generation, J. Chem. Inf. Model., 2024, 64(3), 653–665, DOI:10.1021/acs.jcim.3c01456.
- Y. Luo, J. Zhang, S. Fan, K. Yang, Y. Wu, M. Qiao and Z. Nie, BioMedGPT: Open multimodal generative pre-trained transformer for BioMedicine, arXiv, 2023, preprint, arXiv:2308.09442, DOI:10.48550/arXiv.2308.09442.
- T. Xie, Y. Wan, W. Huang, Z. Yin, Y. Liu, S. Wang, Q. Linghu, C. Kit, C. Grazian, W. Zhang, I. Razzak and B. Hoex, DARWIN series: Domain specific large language models for natural science, arXiv, 2023, preprint, arXiv:2308.13565, DOI:10.48550/arXiv.2308.13565, http://arxiv.org/abs/2308.13565.
- C. Wu, W. Lin, X. Zhang, Y. Zhang, Y. Wang and W. Xie, PMC-LLaMA: Towards building open-source language models for medicine, arXiv, 2023, preprint, arXiv:2304.14454, DOI:10.48550/arXiv.2304.14454, http://arxiv.org/abs/2304.14454.
- R. Luo, L. Sun, Y. Xia, T. Qin, S. Zhang, H. Poon and T.-Y. Liu, BioGPT: generative pre-trained transformer for biomedical text generation and mining, Briefings Bioinf., 2022, 23(6), 1477–4054, DOI:10.1093/bib/bbac409.
- N. C. Frey, R. Soklaski, S. Axelrod, S. Samsi, R. Gómez-Bombarelli, C. W. Coley and V. Gadepally, Neural scaling of deep chemical models, Nat. Mach. Intell., 2023, 5(11), 1297–1305, DOI:10.1038/s42256-023-00740-3.
- V. Bagal, R. Aggarwal, P. K. Vinod and U. D. Priyakumar, MolGPT: Molecular generation using a Transformer-Decoder model, J. Chem. Inf. Model., 2022, 62(9), 2064–2076, DOI:10.1021/acs.jcim.1c00600.
- S. Adilov, Generative Pre-Training from molecules, ChemRxiv, 2021, preprint, DOI:10.26434/chemrxiv-2021-5fwjd.
- J. W.-H. Li and J. C. Vederas, Drug Discovery and Natural Products: End of an Era or an Endless Frontier?, Science, 2009, 325(5937), 161–165, DOI:10.1126/science.1168243.
- D. J. Newman and G. M. Cragg, Natural Products As Sources of New Drugs over the 30 Years from 1981 to 2010, J. Nat. Prod., 2012, 75(3), 311–335, DOI:10.1021/np200906s.
- M. A. Farha and E. D. Brown, Strategies for target identification of antimicrobial natural products, Nat. Prod. Rep., 2016, 33(5), 668–680, 10.1039/C5NP00127G.
- A. K. Nigam, R. Pollice, M. Krenn, G. dos P. Gomes and A. Aspuru-Guzik, Beyond generative models: superfast traversal, optimization, novelty, exploration and discovery (STONED) algorithm for molecules using SELFIES, Chem. Sci., 2021, 12(20), 7079–7090, 10.1039/D1SC00231G.
- H. A. Gandhi and A. D. White, Explaining molecular properties with natural language, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-v5p6m-v3.
- Y. Du, A. R. Jamasb, J. Guo, T. Fu, C. Harris, Y. Wang, C. Duan, P. Liò, P. Schwaller and T. L. Blundell, Machine learning-aided generative molecular design, Nat. Mach. Intell., 2024, 1–16, DOI:10.1038/s42256-024-00843-5.
- N. Houlsby, A. Giurgiu, S. Jastrzebski, B. Morrone, Q. De Laroussilhe, A. Gesmundo, M. Attariyan and S. Gelly, Parameter-efficient transfer learning for nlp, in International conference on machine learning, PMLR, 2019, pp. 2790–2799 Search PubMed.
- A. S. Fuhr and B. G. Sumpter, Deep generative models for materials discovery and machine learning-accelerated innovation, Front. Mater., 2022, 9, 865270, DOI:10.3389/fmats.2022.865270.
- R. Han, H. Yoon, G. Kim, H. Lee and Y. Lee, Revolutionizing medicinal chemistry: The application of artificial intelligence (ai) in early drug discovery, Pharmaceuticals, 2023, 16(9), 1259, DOI:10.3390/ph16091259.
- N.-M. Koutroumpa, K. D. Papavasileiou, A. G. Papadiamantis, G. Melagraki and A. Afantitis, A systematic review of deep learning methodologies used in the drug discovery process with emphasis on in vivo validation, Int. J. Mol. Sci., 2023, 24(7), 6573, DOI:10.3390/ijms24076573.
- D. B. Kell, S. Samanta and N. Swainston, Deep learning and generative methods in cheminformatics and chemical biology: navigating small molecule space intelligently, Biochem. J., 2020, 477(23), 4559–4580, DOI:10.1042/BCJ20200781.
- C. Bilodeau, W. Jin, T. Jaakkola, R. Barzilay and K. F. Jensen, Generative models for molecular discovery: Recent advances and challenges, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12(5), e1608, DOI:10.1002/wcms.1608.
- A. Gangwal, A. Ansari, I. Ahmad, A. K. Azad, V. Kumarasamy, V. Subramaniyan and L. S. Wong, Generative artificial intelligence in drug discovery: basic framework, recent advances, challenges and opportunities, Front. Pharmacol., 2024, 15, 1331062, DOI:10.3389/fphar.2024.1331062.
- M. Vogt, Using deep neural networks to explore chemical space, Expert Opin. Drug Discovery, 2022, 17(3), 297–304, DOI:10.1080/17460441.2022.2019704.
- S. Talluri, M. A. Kamal and R. Rao Malla, Novel computational methods for cancer drug design, Curr. Med. Chem., 2024, 31(5), 554–572, DOI:10.2174/0929867330666230403100008.
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy, M. Veselov, A. Kadurin, S. Johansson, H. Chen, S. Nikolenko, A. Aspuru-Guzik and A. Zhavoronkov, Molecular sets (MOSES): A benchmarking platform for molecular generation models, Front. Pharmacol., 2020, 11, 565644, DOI:10.3389/fphar.2020.565644.
- N. Brown, M. Fiscato, M. H. S. Segler and A. C. Vaucher, GuacaMol: Benchmarking models for de novo molecular design, J. Chem. Inf. Model., 2019, 59(3), 1096–1108, DOI:10.1021/acs.jcim.8b00839.
- K. Preuer, P. Renz, T. Unterthiner, S. Hochreiter and G. Klambauer, Fréchet chemnet distance: a metric for generative models for molecules in drug discovery, J. Chem. Inf. Model., 2018, 58(9), 1736–1741 CrossRef PubMed.
- S. Haroon, C. A. Hafsath and A. S. Jereesh, Generative Pre-trained Transformer (GPT) based model with relative attention for de novo drug design, Comput. Biol. Chem., 2023, 106, 107911, DOI:10.1016/j.compbiolchem.2023.107911.
- J. Wang, J. Mao, M. Wang, X. Le and Y. Wang, Explore drug-like space with deep generative models, Methods, 2023, 210, 52–59, DOI:10.1016/j.ymeth.2023.01.004.
- J. Mao, J. Wang, A. Zeb, K.-H. Cho, H. Jin, J. Kim, O. Lee, Y. Wang and K. Tai No, Transformer-Based Molecular Generative Model for Antiviral Drug Design, J. Chem. Inf. Model., 2024, 64(7), 2733–2745, DOI:10.1021/acs.jcim.3c00536.
- W. Zhang, K. Zhang and J. Huang, A Simple Way to Incorporate Target Structural Information in Molecular Generative Models, J. Chem. Inf. Model., 2023, 63(12), 3719–3730, DOI:10.1021/acs.jcim.3c00293.
- X. Wang, C. Gao, P. Han, X. Li, W. Chen, A. R. Patón, S. Wang and P. Z. PETrans, De Novo Drug Design with Protein-Specific Encoding Based on Transfer Learning, Int. J. Mol. Sci., 2023, 24(2), 1146, DOI:10.3390/ijms24021146.
- Y. Yoshikai, T. Mizuno, S. Nemoto and H. Kusuhara, A novel molecule generative model of VAE combined with Transformer for unseen structure generation, arXiv, 2024, preprint, arXiv:2402.11950, DOI:10.48550/arXiv.2402.11950, http://arxiv.org/abs/2402.11950.
- X. Yan, C. Gu, Y. Feng and J. Han, Predicting Drug-drug Interaction with Graph Mutual Interaction Attention Mechanism, Methods, 2024, 223, 16–25, DOI:10.1016/j.ymeth.2024.01.009.
- T. Shen, J. Guo, Z. Han, G. Zhang, Q. Liu, X. Si, D. Wang, S. Wu and J. Xia, AutoMolDesigner for Antibiotic Discovery: An AI-Based Open-Source Software for Automated Design of Small-Molecule Antibiotics, J. Chem. Inf. Model., 2024, 64(3), 575–583, DOI:10.1021/acs.jcim.3c01562.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Quantifying the chemical beauty of drugs, Nat. Chem., 2012, 4(2), 90–98 CrossRef.
- G. Subramanian, B. Ramsundar, V. Pande and R. A. Denny, Computational modeling of β-secretase 1 (bace-1) inhibitors using ligand based approaches, J. Chem. Inf. Model., 2016, 56(10), 1936–1949 CrossRef PubMed.
- G. Xu, M. C. Abad, P. J. Connolly, M. P. Neeper, G. T. Struble, B. A. Springer, S. L. Emanuel, N. Pandey, R. H. Gruninger and M. Adams, et al., 4-amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent erbb-2/egfr dual kinase inhibitors, Bioorg. Med. Chem. Lett., 2008, 18(16), 4615–4619 CrossRef PubMed.
- L. Yu, L. He, B. Gan, R. Ti, Q. Xiao, X. Yang, H. Hu, L. Zhu, S. Wang and R. Ren, Structural insights into sphingosine-1-phosphate receptor activation, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(16), e2117716119 CrossRef.
- P. Xu, S. Huang, H. Zhang, C. Mao, X. E. Zhou, Xi Cheng, I. A. Simon, D.-D. Shen, H.-Y. Yen and C. V. Robinson, et al., Structural insights into the lipid and ligand regulation of serotonin receptors, Nature, 2021, 592(7854), 469–473 CrossRef.
- J. Sun, N. Jeliazkova, V. Chupakhin, J.-F. Golib-Dzib, O. Engkvist, L. Carlsson, J. Wegner, H. Ceulemans, I. Georgiev and V. Jeliazkov, et al., Excape-db: an integrated large scale dataset facilitating big data analysis in chemogenomics, J. Cheminf., 2017, 9, 1–9 Search PubMed.
- Z. Han, C. Gao, J. Liu, J. Zhang and S. Qian Zhang, Parameter-efficient fine-tuning for large models: A comprehensive survey, arXiv, 2024, preprint, arXiv:2403.14608, DOI:10.48550/arXiv.2403.14608, http://arxiv.org/abs/2403.14608.
- N. Ding, Y. Qin, G. Yang, F. Wei, Z. Yang, Y. Su, S. Hu, Y. Chen, C.-M. Chan, W. Chen, J. Yi, W. Zhao, X. Wang, Z. Liu, H.-T. Zheng, J. Chen, Y. Liu, J. Tang, J. Li and M. Sun, Parameter-efficient fine-tuning of large-scale pre-trained language models, Nat. Mach. Intell., 2023, 5(3), 220–235, DOI:10.1038/s42256-023-00626-4.
- E. J. Hu, Y. Shen, P. Wallis, Z. Allen-Zhu, Y. Li, S. Wang, L. Wang and W. Chen, LoRA: Low-rank adaptation of large language models, arXiv, 2021, preprint, arXiv:2106.09685, DOI:10.48550/arXiv.2106.09685, http://arxiv.org/abs/2106.09685.
- A. Guzman-Pando, G. Ramirez-Alonso, C. Arzate-Quintana and J. Camarillo-Cisneros, Deep learning algorithms applied to computational chemistry, Mol. Diversity, 2024, 28(4), 2375–2410, DOI:10.1007/s11030-023-10771-y.
- J. C. Fromer and C. W. Coley, Computer-aided multi-objective optimization in small molecule discovery, Patterns, 2023, 4(2), 100678, DOI:10.1016/j.patter.2023.100678.
- M. Vogt, Exploring chemical space — Generative models and their evaluation, Artif. Intell. Life Sci., 2023, 3, 100064, DOI:10.1016/j.ailsci.2023.100064.
- M. Goel, R. Aggarwal, B. Sridharan, P. K. Pal and U. Deva Priyakumar, Efficient and enhanced sampling of drug-like chemical space for virtual screening and molecular design using modern machine learning methods, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2023, 13(2), e1637, DOI:10.1002/wcms.1637.
- S. Honda, S. Shi and H. R. Ueda, SMILES transformer: Pre-trained molecular fingerprint for low data drug discovery, arXiv, 2019, preprint, arXiv:1911.04738, DOI:10.48550/arXiv.1911.04738, http://arxiv.org/abs/1911.04738.
- T. Sagawa and R. Kojima, ReactionT5: a large-scale pre-trained model towards application of limited reaction data, arXiv, 2023, preprint, arXiv:2311.06708, DOI:10.48550/arXiv.2311.06708, http://arxiv.org/abs/2311.06708.
- Y. Fang, N. Zhang, Z. Chen, L. Guo, X. Fan and H. Chen, Domain-agnostic molecular generation with chemical feedback, arXiv, 2023, preprint, arXiv:2301.11259, DOI:10.48550/arXiv.2301.11259, http://arxiv.org/abs/2301.11259.
- D. Christofidellis, G. Giannone, J. Born, O. Winther, T. Laino and M. Manica, Unifying molecular and textual representations via multi-task language modelling, arXiv, 2023, preprint, arXiv:2301.12586, DOI:10.48550/arXiv.2301.12586, http://arxiv.org/abs/2301.12586.
- A. C. Vaucher, F. Zipoli, J. Geluykens, V. H. Nair, P. Schwaller and T. Laino, Automated extraction of chemical synthesis actions from experimental procedures, Nat. Commun., 2020, 11(1), 3601, DOI:10.1038/s41467-020-17266-6.
- C. Edwards, T. Lai, K. Ros, H. Garrett, K. Cho and H. Ji, Translation between molecules and natural language, arXiv, 2022, preprint, arXiv:2204.11817, DOI:10.48550/arXiv.2204.11817, http://arxiv.org/abs/2204.11817.
- C. Edwards, C. X. Zhai and H. Ji, Text2Mol: Cross-modal molecule retrieval with natural language queries, in Proceedings of the 2021 Conference on Empirical Methods in Natural Language Processing, ed. M.-F. Moens, X. Huang, L. Specia and S. Wen-tau Yih, Association for Computational Linguistics, Punta Cana, Dominican Republic, 2021, pp. 595–607, DOI:10.18653/v1/2021.emnlp-main.47.
- R. A. Shenvi, Natural product synthesis in the 21st century: Beyond the mountain top, ACS Cent. Sci., 2024, 10(3), 519–528, DOI:10.1021/acscentsci.3c01518.
- Q. Ai, F. Meng, J. Shi, B. Pelkie and C. W. Coley. Extracting Structured Data from Organic Synthesis Procedures Using a Fine-Tuned Large Language Model, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-979fz.
- E. J. Corey, Robert Robinson Lecture. Retrosynthetic thinking—essentials and examples, Chem. Soc. Rev., 1988, 17, 111–133, 10.1039/CS9881700111.
- J. B. Nerenberg, D. T. Hung, P. K. Somers and S. L. Schreiber, Total synthesis of the immunosuppressive agent (-)-discodermolide, J. Am. Chem. Soc., 1993, 115(26), 12621–12622, DOI:10.1021/ja00079a066.
- J. Nam and J. Kim, Linking the Neural Machine Translation and the Prediction of Organic Chemistry Reactions, arXiv, 2016, preprint, arXiv:1612.09529 [cs], DOI:10.48550/arXiv.1612.09529, http://arxiv.org/abs/1612.09529.
- B. Liu, B. Ramsundar, K. Prasad, J. Shi, J. Gomes, Q. L. Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models, ACS Cent. Sci., 2017, 3(10), 1103–1113, DOI:10.1021/acscentsci.7b00303.
- N. Schneider, N. Stiefl and G. A. Landrum, What's what: The (nearly) definitive guide to reaction role assignment, J. Chem. Inf. Model., 2016, 56(12), 2336–2346, DOI:10.1021/acs.jcim.6b00564.
- M. Gunther, Software could revolutionise chemistry, 2016, https://www.chemistryworld.com/news/software-could-revolutionise-chemistry/1017236.article.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice and B. A. Grzybowski, Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory, Chem, 2018, 4(3), 522–532, DOI:10.1016/j.chempr.2018.02.002.
- M. H. S. Segler and M. P. Waller, Neural-symbolic machine learning for retrosynthesis and reaction prediction, Chemistry, 2017, 23(25), 5966–5971, DOI:10.1002/chem.201605499.
- V. R. Somnath, C. Bunne, C. W. Coley, A. Krause and R. Barzilay, Learning Graph Models for Retrosynthesis Prediction, arXiv, 2021, preprint, arXiv:2006.07038 [cs, stat], DOI:10.48550/arXiv.2006.07038, http://arxiv.org/abs/2006.07038.
- G. P. Wellawatte and P. Schwaller, Extracting human interpretable structure-property relationships in chemistry using XAI and large language models, arXiv, 2023, preprint, arXiv:2311.04047, DOI:10.48550/arXiv.2311.04047, http://arxiv.org/abs/2311.04047.
- A. Cadeddu, E. K. Wylie, J. Jurczak, M. Wampler-Doty and B. A. Grzybowski, Organic Chemistry as a Language and the Implications of Chemical Linguistics for Structural and Retrosynthetic Analyses, Angew. Chem., Int. Ed., 2014, 53(31), 8108–8112, DOI:10.1002/anie.201403708.
- P. Schwaller, T. Gaudin, D. Lányi, C. Bekas and T. Laino, “Found in Translation”: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models, Chem. Sci., 2018, 9(28), 6091–6098, 10.1039/C8SC02339E.
- W. Jin, W. C. Connor, R. Barzilay and T. Jaakkola, Predicting Organic Reaction Outcomes with Weisfeiler-Lehman Network, arXiv, 2017, preprint, arXiv:1709.04555 [cs, stat], DOI:10.48550/arXiv.1709.04555, http://arxiv.org/abs/1709.04555.
- J. Bradshaw, M. J. Kusner, B. Paige, M. H. S. Segler and J. M. Hernández-Lobato, A Generative Model For Electron Paths, arXiv, 2019, preprint, arXiv:1805.10970 [physics, stat], DOI:10.48550/arXiv.1805.10970, http://arxiv.org/abs/1805.10970.
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy, Chem. Sci., 2020, 11(12), 3316–3325, 10.1039/C9SC05704H.
- S. Chen and Y. Jung, Deep Retrosynthetic Reaction Prediction using Local Reactivity and Global Attention, JACS Au, 2021, 1(10), 1612–1620, DOI:10.1021/jacsau.1c00246.
- A. M. Westerlund, S. Manohar Koki, S. Kancharla, A. Tibo, L. Saigiridharan, M. Kabeshov, R. Mercado and S. Genheden, Do Chemformers Dream of Organic Matter? Evaluating a Transformer Model for Multistep Retrosynthesis, J. Chem. Inf. Model., 2024, 64(8), 3021–3033, DOI:10.1021/acs.jcim.3c01685.
- S. Zheng, J. Rao, Z. Zhang, J. Xu and Y. Yang, Predicting Retrosynthetic Reactions Using Self-Corrected Transformer Neural Networks, J. Chem. Inf. Model., 2020, 60(1), 47–55, DOI:10.1021/acs.jcim.9b00949.
- D. Mark Lowe, Extraction of chemical structures and reactions from the literature, 2012, http://www.dspace.cam.ac.uk/handle/1810/244727.
- J. Li, L. Fang and J.-G. Lou, Retro-BLEU: quantifying chemical plausibility of retrosynthesis routes through reaction template sequence analysis, Digital Discovery, 2024, 3(3), 482–490, 10.1039/D3DD00219E.
- K. Papineni, S. Roukos, W. Todd and W.-J. Zhu, BLEU: a method for automatic evaluation of machine translation, in Proceedings of the 40th Annual Meeting on Association for Computational Linguistics – ACL ’02, ed. P. Isabelle, E. Charniak and D. Lin, Association for Computational Linguistics, Philadelphia, Pennsylvania, USA, 2002, pp. 311–318, DOI:10.3115/1073083.1073135.
- C.-Y. Lin, ROUGE: A Package for Automatic Evaluation of Summaries, in Text Summarization Branches Out, Association for Computational Linguistics, Barcelona, Spain, 2004, pp. 74–81, https://aclanthology.org/W04-1013 Search PubMed.
- L. David, A. Thakkar, R. Mercado and O. Engkvist, Molecular representations in AI-driven drug discovery: a review and practical guide, J. Cheminform., 2020, 12(1), 56, DOI:10.1186/s13321-020-00460-5.
- G. L. W. Hart, T. Mueller, C. Toher and S. Curtarolo, Machine learning for alloys, Nat. Rev. Mater., 2021, 6(8), 730–755, DOI:10.1038/s41578-021-00340-w.
- O. Eraso, D. Bolaños, N. Echeverri, C. O. Donneys, T. Ameri and J. D. Perea, A present scenario of the computational approaches for ternary organic solar cells, J. Renewable Sustainable Energy, 2023, 15(6), 062702, DOI:10.1063/5.0172426.
- Yu-C. Lo, S. E. Rensi, T. Wen and R. B. Altman, Machine learning in chemoinformatics and drug discovery, Drug Discovery Today, 2018, 23(8), 1538–1546, DOI:10.1016/j.drudis.2018.05.010.
- D. Flam-Shepherd and A. Aspuru-Guzik, Language models can generate molecules, materials and protein binding sites directly in three dimensions as XYZ, CIF and PDB files, arXiv, 2023, preprint, arXiv:2305.05708, DOI:10.48550/arXiv.2305.05708, http://arxiv.org/abs/2305.05708.
- K. Rajan, A. Zielesny and C. Steinbeck, DECIMER 1.0: deep learning for chemical image recognition using transformers, J. Cheminform., 2021, 13(1), 61, DOI:10.1186/s13321-021-00538-8.
- J. T. Carstensen and F. Attarchi, Decomposition of Aspirin in the Solid State in the Presence of Limited Amounts of Moisture III: Effect of Temperature and a Possible Mechanism, J. Pharm. Sci., 1988, 77(4), 318–321, DOI:10.1002/jps.2600770407.
- P. Seidl, A. Vall, S. Hochreiter and G. Klambauer, Enhancing Activity Prediction Models in Drug Discovery with the Ability to Understand Human Language, arXiv, 2023, preprint, arXiv:2303.03363, DOI:10.48550/arXiv.2303.03363.
- H. Xu, A. Woicik, H. Poon, R. B. Altman and S. Wang, Multilingual translation for zero-shot biomedical classification using BioTranslator, Nat. Commun., 2023, 14(1), 738, DOI:10.1038/s41467-023-36476-2.
- S. Liu, J. Wang, Y. Yang, C. Wang, L. Liu, H. Guo and C. Xiao, ChatGPT-powered Conversational Drug Editing Using Retrieval and Domain Feedback, arXiv, 2023, preprint, arXiv:2305.18090, DOI:10.48550/arXiv.2305.18090.
- Z. Liu, W. Zhang, Y. Xia, L. Wu, S. Xie, T. Qin, M. Zhang and T.-Y. Liu, MolXPT: Wrapping Molecules with Text for Generative Pre-training, arXiv, 2023, preprint, arXiv:2305.10688, DOI:10.48550/arXiv.2305.10688.
- P. Liu, Y. Ren, J. Tao and Z. Ren, Git-mol: A multi-modal large language model for molecular science with graph, image and text, Comput. Biol. Med., 2024, 171, 108073, DOI:10.1016/j.compbiomed.2024.108073.
- J. Fang, S. Zhang, C. Wu, Z. Liu, S. Li, K. Wang, W. Du and X. Wang, MolTC: Towards Molecular Relational Modeling In Language Models, arXiv, 2024, preprint, arXiv:2402.03781, DOI:10.48550/arXiv.2402.03781.
- H. Zhang, J. Wu, S. Liu and S. Han, A pre-trained multi-representation fusion network for molecular property prediction, Inf. Fusion, 2024, 103, 102092, DOI:10.1016/j.inffus.2023.102092.
- H. Zhu, T. Xiao and V. G. Honavar, 3M-Diffusion: Latent Multi-Modal Diffusion for Text-Guided Generation of Molecular Graphs, arXiv, 2024, preprint, arXiv:2403.07179, DOI:10.48550/arXiv.2403.07179.
- C. Gao, W. Bao, S. Wang, J. Zheng, L. Wang, Y. Ren, L. Jiao, J. Wang and X. Wang, DockingGA: enhancing targeted molecule generation using transformer neural network and genetic algorithm with docking simulation, Brief. Funct. Genom., 2024, 23(5), 595–606, DOI:10.1093/bfgp/elae011.
- P. Zhou, J. Wang, C. Li, Z. Wang, Y. Liu, S. Sun, J. Lin, L. Wang and X. Zeng, Instruction Multi-Constraint Molecular Generation Using a Teacher-Student Large Language Model, arXiv, 2024, preprint, arXiv:2403.13244, DOI:10.48550/arXiv.2403.13244.
- H. Gong, Q. Liu, S. Wu and L. Wang, Text-Guided Molecule Generation with Diffusion Language Model, arXiv, 2024, preprint, arXiv:2402.13040, DOI:10.48550/arXiv.2402.13040.
- E. Soares, E. Vital Brazil, K. F. A. Gutierrez, R. Cerqueira, D. Sanders, K. Schmidt and D. Zubarev, Beyond Chemical Language: A Multimodal Approach to Enhance Molecular Property Prediction, arXiv, 2023, preprint, arXiv:2306.14919 [physics, q-bio], DOI:10.48550/arXiv.2306.14919, http://arxiv.org/abs/2306.14919.
- M. Riedl, S. Mukherjee and M. Gauthier, Descriptor-free deep learning QSAR model for the fraction unbound in human plasma, Mol. Pharm., 2023, 20(10), 4984–4993, DOI:10.1021/acs.molpharmaceut.3c00129.
- MIMIC-III documentation, 2021, https://mimic.mit.edu/docs/iii/, accessed: 2024-3-25.
- Y. Liu, M. Ott, N. Goyal, J. Du, M. Joshi, D. Chen, O. Levy, M. Lewis, L. Zettlemoyer and V. Stoyanov, RoBERTa: A robustly optimized BERT pretraining approach, arXiv, 2019, preprint, arXiv:1907.11692, DOI:10.48550/arXiv.1907.11692, http://arxiv.org/abs/1907.11692.
- H. Smith, Z. Zhang, J. Culnan and P. Jansen, ScienceExamCER: A high-density fine-grained science-domain corpus for common entity recognition, arXiv, 2019, preprint, arXiv:1911.10436, DOI:10.48550/arXiv.1911.10436, http://arxiv.org/abs/1911.10436.
- M. C. Swain and J. M. Cole, ChemDataExtractor: A toolkit for automated extraction of chemical information from the scientific literature, J. Chem. Inf. Model., 2016, 56(10), 1894–1904, DOI:10.1021/acs.jcim.6b00207.
- S. Ibanez, chemie-turk: Mechanical turk on your own machine for chemical literature annotation, https://github.com/asibanez/chemie-turk.
- W. Zhang, Q. Wang, X. Kong, J. Xiong, S. Ni, D. Cao, B. Niu, M. Chen, R. Zhang, Y. Wang, L. Zhang, X. Li, Z. Xiong, Q. Shi, Z. Huang, Z. Fu and M. Zheng, Fine-tuning large language models for chemical text mining, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2023-k7ct5-v2.
- K. Chen, H. Cao, J. Li, Y. Du, M. Guo, X. Zeng, L. Li, J. Qiu, P. Ann Heng and G. Chen, An autonomous large language model agent for chemical literature data mining, arXiv, 2024, preprint, arXiv:2402.12993, DOI:10.48550/arXiv.2402.12993, http://arxiv.org/abs/2402.12993.
- X. Wang, L. Huang, S. Xu and K. Lu, How does a generative large language model perform on domain-specific information extraction? – a comparison between GPT-4 and a rule-based method on band gap extraction, J. Chem. Inf. Model., 2024, 64(20), 7895–7904, DOI:10.1021/acs.jcim.4c00882.
- M. Schilling-Wilhelmi, M. Ríos-García, S. Shabih, M. V. Gil, S. Miret, C. T. Koch, J. A. Márquez and K. M. Jablonka, From text to insight: Large language models for materials science data extraction, arXiv, 2024, preprint, arXiv:2407.16867, DOI:10.48550/arXiv.2407.16867, http://arxiv.org/abs/2407.16867.
- P. Shetty, A. C. Rajan, C. Kuenneth, S. Gupta, L. Prerana Panchumarti, L. Holm, C. Zhang and R. Ramprasad, A general-purpose material property data extraction pipeline from large polymer corpora using natural language processing, npj Comput. Mater., 2023, 9(1), 52, DOI:10.1038/s41524-023-01003-w.
- M. Shoeybi, M. Patwary, R. Puri, P. LeGresley, J. Casper and B. Catanzaro, Megatron-LM: Training multi-billion parameter language models using model parallelism, arXiv, 2019, preprint, arXiv:1909.08053, DOI:10.48550/arXiv.1909.08053, http://arxiv.org/abs/1909.08053.
- P. Rajpurkar, J. Zhang, K. Lopyrev and P. Liang, SQuAD: 100,000+ questions for machine comprehension of text, arXiv, 2016, preprint, arXiv:1606.05250, DOI:10.48550/arXiv.1606.05250, http://arxiv.org/abs/1606.05250.
- Y. Peng, Q. Chen and Z. Lu, An empirical study of multi-task learning on BERT for biomedical text mining, arXiv, 2020, preprint, arXiv:2005.02799, DOI:10.48550/arXiv.2005.02799, http://arxiv.org/abs/2005.02799.
- J. Welbl, N. F. Liu and M. Gardner, Crowdsourcing multiple choice science questions, arXiv, 2017, preprint, arXiv:1707.06209, DOI:10.48550/arXiv.1707.06209, http://arxiv.org/abs/1707.06209.
- Y. Song, S. Miret, H. Zhang and B. Liu, HoneyBee: Progressive instruction finetuning of large language models for materials science, arXiv, 2023, preprint, arXiv:2310.08511, DOI:10.48550/arXiv.2310.08511, http://arxiv.org/abs/2310.08511.
- H. Zhang, Y. Song, Z. Hou, S. Miret and B. Liu, HoneyComb: A flexible LLM-based agent system for materials science, arXiv, 2024, preprint, arXiv:2409.00135, DOI:10.48550/arXiv.2409.00135, http://arxiv.org/abs/2409.00135.
- S. Auer, D. A. C. Barone, C. Bartz, E. G. Cortes, M. Y. Jaradeh, O. Karras, M. Koubarakis, D. Mouromtsev, D. Pliukhin, D. Radyush, I. Shilin, M. Stocker and E. Tsalapati, The SciQA scientific question answering benchmark for scholarly knowledge, Sci. Rep., 2023, 13(1), 7240, DOI:10.1038/s41598-023-33607-z.
- D. Jin, E. Pan, N. Oufattole, W.-H. Weng, H. Fang and P. Szolovits, What disease does this patient have? a large-scale open domain question answering dataset from medical exams, arXiv, 2021, preprint, arXiv:2009.13081, DOI:10.48550/arXiv.2009.13081, http://arxiv.org/abs/2009.13081.
- A. Pal, L. Kumar Umapathi and M. Sankarasubbu, MedMCQA : A large-scale multi-subject multi-choice dataset for medical domain question answering, arXiv, 2022, preprint, arXiv:2203.14371, DOI:10.48550/arXiv.2203.14371, http://arxiv.org/abs/2203.14371.
- C. M. C. Nascimento and A. S. Pimentel, Do Large Language Models Understand Chemistry? A Conversation with ChatGPT, J. Chem. Inf. Model., 2023, 63(6), 1649–1655, DOI:10.1021/acs.jcim.3c00285.
- T. Humphry and A. L. Fuller, Potential ChatGPT Use in Undergraduate Chemistry Laboratories, J. Chem. Educ., 2023, 100(4), 1434–1436, DOI:10.1021/acs.jchemed.3c00006.
- M. E. Emenike and B. U. Emenike, Was This Title Generated by ChatGPT? Considerations for Artificial Intelligence Text-Generation Software Programs for Chemists and Chemistry Educators, J. Chem. Educ., 2023, 100(4), 1413–1418, DOI:10.1021/acs.jchemed.3c00063.
- S. Fergus, M. Botha and M. Ostovar, Evaluating Academic Answers Generated Using ChatGPT, J. Chem. Educ., 2023, 100(4), 1672–1675, DOI:10.1021/acs.jchemed.3c00087.
- Z. Zheng, A. H. Alawadhi, S. Chheda, S. Ephraim Neumann, N. Rampal, S. Liu, H. L. Nguyen, Y.-H. Lin, Z. Rong, J. Ilja Siepmann, L. Gagliardi, A. Anandkumar, C. Borgs, J. T. Chayes and O. M. Yaghi, Shaping the Water-Harvesting Behavior of Metal-Organic Frameworks Aided by Fine-Tuned GPT Models, J. Am. Chem. Soc., 2023, 145(51), 28284–28295, DOI:10.1021/jacs.3c12086.
- Z. Zheng, O. Zhang, C. Borgs, J. T. Chayes and O. M. Yaghi, ChatGPT Chemistry Assistant for Text Mining and the Prediction of MOF Synthesis, J. Am. Chem. Soc., 2023, 145, 18048–18062, DOI:10.1021/jacs.3c05819.
- Z. Xie, X. Evangelopoulos, Ö. H. Omar, A. Troisi, A. I. Cooper and L. Chen, Fine-tuning GPT-3 for machine learning electronic and functional properties of organic molecules, Chem. Sci., 2024, 15(2), 500–510, 10.1039/D3SC04610A.
- Z. Zheng, Z. Rong, N. Rampal, C. Borgs, J. T. Chayes and O. M. Yaghi, A GPT-4 Reticular Chemist for Guiding MOF Discovery, Angew. Chem., Int. Ed., 2023, 62(46), e202311983, DOI:10.1002/anie.202311983.
- J. Deb, L. Saikia, K. D. Dihingia and G. Narahari Sastry, Chatgpt in the material design: Selected case studies to assess the potential of chatgpt, J. Chem. Inf. Model., 2024, 64(3), 799–811, DOI:10.1021/acs.jcim.3c01702.
- B. S. Bloom, Taxonomy of Educational Objectives: The Classification of Educational Goals; Handbook. Cognitive Domain, McKay, 1968 Search PubMed.
- B. S. Bloom, A taxonomy for learning, teaching and assessing: A revision of Bloom's taxonomy of educational objectives, Longman, 2010 Search PubMed.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. John Hart, T. F. Jamison and K. F. Jensen, A robotic platform for flow synthesis of organic compounds informed by AI planning, Science, 2019, 365(6453), eaax1566, DOI:10.1126/science.aax1566.
- P. S. Gromski, J. M. Granda and L. Cronin, Universal Chemical Synthesis and Discovery with ‘The Chemputer’, Trends Chem., 2020, 2(1), 4–12, DOI:10.1016/j.trechm.2019.07.004.
- F. Grisoni, B. J. H. Huisman, A. L. Button, M. Moret, K. Atz, D. Merk and G. Schneider, Combining generative artificial intelligence and on-chip synthesis for de novo drug design, Sci. Adv., 2021, 7(24), eabg3338, DOI:10.1126/sciadv.abg3338.
- B. Goldman, S. Kearnes, T. Kramer, P. Riley and W. Patrick Walters, Defining Levels of Automated Chemical Design, J. Med. Chem., 2022, 65(10), 7073–7087, DOI:10.1021/acs.jmedchem.2c00334.
- G. Schneider, Automating drug discovery, Nat. Rev. Drug Discovery, 2018, 17(2), 97–113, DOI:10.1038/nrd.2017.232.
- J. Paul Janet, L. Mervin and O. Engkvist, Artificial intelligence in molecular de novo design: Integration with experiment, Curr. Opin. Struct. Biol., 2023, 80, 102575, DOI:10.1016/j.sbi.2023.102575.
- C. W. Coley, N. S. Eyke and K. F. Jensen, Autonomous Discovery in the Chemical Sciences Part I: Progress, Angew. Chem., Int. Ed., 2020, 59(51), 22858–22893, DOI:10.1002/anie.201909987.
- C. W. Coley, N. S. Eyke and K. F. Jensen, Autonomous Discovery in the Chemical Sciences Part II: Outlook, Angew. Chem., Int. Ed., 2020, 59(52), 23414–23436, DOI:10.1002/anie.201909989.
- A. Thakkar, S. Johansson, K. Jorner, D. Buttar, J.-L. Reymond and O. Engkvist, Artificial intelligence and automation in computer aided synthesis planning, React. Chem. Eng., 2021, 6(1), 27–51, 10.1039/D0RE00340A.
- Y. Shen, J. E. Borowski, M. A. Hardy, R. Sarpong, A. G. Doyle and T. Cernak, Automation and computer-assisted planning for chemical synthesis, Nat. Rev. Methods Primers, 2021, 1(1), 23, DOI:10.1038/s43586-021-00022-5.
- Y. Liu, L. Sun, H. Zhang, L. Shang and Y. Zhao, Microfluidics for Drug Development: From Synthesis to Evaluation, Chem. Rev., 2021, 121(13), 7468–7529, DOI:10.1021/acs.chemrev.0c01289.
- K. Darvish, M. Skreta, Y. Zhao, N. Yoshikawa, S. Som, M. Bogdanovic, Y. Cao, H. Han, H. Xu, A. Aspuru-Guzik, A. Garg and F. Shkurti, ORGANA: A robotic assistant for automated chemistry experimentation and characterization, arXiv, 2024, preprint, arXiv:2401.06949, DOI:10.48550/arXiv.2401.06949, http://arxiv.org/abs/2401.06949.
- T. Šalamon, Design of Agent-based Models: Developing Computer Simulations for a Better Understanding of Social Processes, ed. Tomáš Bruckner, Repin, CZE, 2011 Search PubMed.
- Z. Xi, W. Chen, X. Guo, W. He, Y. Ding, B. Hong, M. Zhang, J. Wang, S. Jin, E. Zhou, R. Zheng, X. Fan, X. Wang, L. Xiong, Y. Zhou, W. Wang, C. Jiang, Y. Zou, X. Liu, Z. Yin, S. Dou, R. Weng, W. Cheng, Z. Qi, W. Qin, Y. Zheng, X. Qiu, X. Huang and T. Gui, The rise and potential of large language model based agents: A survey, arXiv, 2023, preprint, arXiv:2309.07864, DOI:10.48550/arXiv.2309.07864, http://arxiv.org/abs/2309.07864.
- S. Gao, A. Fang, Y. Huang, V. Giunchiglia, A. Noori, J. R. Schwarz, Y. Ektefaie, J. Kondic and M. Zitnik, Empowering biomedical discovery with AI agents, arXiv, 2024, preprint, arXiv:2404.02831, DOI:10.48550/arXiv.2404.02831, http://arxiv.org/abs/2404.02831.
- L. Wang, C. Ma, X. Feng, Z. Zhang, H. Yang, J. Zhang, Z. Chen, J. Tang, X. Chen, Y. Lin, W. X. Zhao, Z. Wei and J. Wen, A survey on large language model based autonomous agents, Front. Comput. Sci., 2024, 18(6), 2095–2236, DOI:10.1007/s11704-024-40231-1.
- L. Weng, LLM powered autonomous agents, 2023, https://lilianweng.github.io/posts/2023-06-23-agent/, accessed: 2024-1-22.
- T. R. Sumers, S. Yao, K. Narasimhan and T. L. Griffiths, Cognitive architectures for language agents, arXiv, 2023, preprint, arXiv:2309.02427, DOI:10.48550/arXiv.2309.02427, http://arxiv.org/abs/2309.02427.
- B. Wang, X. Liang, J. Yang, H. Huang, S. Wu, P. Wu, L. Lu, Z. Ma and Z. Li, Enhancing large language model with self-controlled memory framework, arXiv, 2023, preprint, arXiv:2304.13343, DOI:10.48550/arXiv.2304.13343, http://arxiv.org/abs/2304.13343.
- Y. Zhang, Z. Yu, W. Jiang, Y. Shen and J. Li, Long-term memory for large language models through topic-based vector database, in 2023 International Conference on Asian Language Processing (IALP), IEEE, 2023, DOI:10.1109/ialp61005.2023.10337079.
- X. Zhu, Y. Chen, H. Tian, C. Tao, W. Su, C. Yang, G. Huang, B. Li, L. Lu, X. Wang, Y. Qiao, Z. Zhang and J. Dai, Ghost in the minecraft: Generally capable agents for open-world environments via large language models with text-based knowledge and memory, arXiv, 2023, preprint, arXiv:2305.17144, DOI:10.48550/arXiv.2305.17144, http://arxiv.org/abs/2305.17144.
- W. Zhong, L. Guo, Q. Gao, H. Ye and Y. Wang, MemoryBank: Enhancing large language models with long-term memory, arXiv, 2023, preprint, arXiv:2305.10250, DOI:10.48550/arXiv.2305.10250, http://arxiv.org/abs/2305.10250.
- Y. Han, C. Liu and P. Wang, A comprehensive survey on vector database: Storage and retrieval technique, challenge, arXiv, 2023, preprint, arXiv:2310.11703, DOI:10.48550/arXiv.2310.11703, http://arxiv.org/abs/2310.11703.
- A. Zhao, D. Huang, Q. Xu, M. Lin, Y.-J. Liu and G. Huang, ExpeL: LLM agents are experiential learners, arXiv, 2023, preprint, arXiv:2308.10144, DOI:10.48550/arXiv.2308.10144, http://arxiv.org/abs/2308.10144.
- ANN-benchmarks, https://ann-benchmarks.com/, accessed: 2024-2-1.
- K. Hatalis, D. Christou, J. Myers, S. Jones, K. Lambert, A. Amos-Binks, Z. Dannenhauer and D. Dannenhauer, Memory matters: The need to improve Long-Term memory in LLM-Agents, Proceedings of the AAAI Symposium Series, 2023, 2(1), 277–280, DOI:10.1609/aaaiss.v2i1.27688 , https://ojs.aaai.org/index.php/AAAI-SS/article/view/27688.
- J. Sung Park, J. C. O'Brien, C. J. Cai, M. R. Morris, P. Liang and M. S. Bernstein, Generative agents: Interactive simulacra of human behavior, arXiv, 2023, preprint, arXiv:2304.03442, DOI:10.48550/arXiv.2304.03442, http://arxiv.org/abs/2304.03442.
- S. S. Raman, V. Cohen, E. Rosen and I. Idrees, Planning with large language models via corrective re-prompting, Foundation Models, 2022, https://openreview.net/pdf?id=cMDMRBe1TKs Search PubMed.
- S. Dhuliawala, M. Komeili, J. Xu, R. Raileanu, X. Li, A. Celikyilmaz and J. Weston, Chain-of-verification reduces hallucination in large language models, arXiv, 2023, preprint, arXiv:2309.11495, DOI:10.48550/arXiv.2309.11495, http://arxiv.org/abs/2309.11495.
- W. Huang, F. Xia, T. Xiao, H. Chan, J. Liang, P. Florence, A. Zeng, J. Tompson, I. Mordatch, Y. Chebotar, P. Sermanet, N. Brown, T. Jackson, L. Luu, S. Levine, K. Hausman and B. Ichter, Inner monologue: Embodied reasoning through planning with language models, arXiv, 2022, preprint, arXiv:2207.05608, DOI:10.48550/arXiv.2207.05608, http://arxiv.org/abs/2207.05608.
- T. Kojima, S. S. Gu, M. Reid, Y. Matsuo and Y. Iwasawa, Large language models are zero-shot reasoners, 2022, https://proceedings.neurips.cc/paper_files/paper/2022/file/8bb0d291acd4acf06ef112099c16f326-Paper-Conference.pdf Search PubMed.
- J. Wei, X. Wang, D. Schuurmans, M. Bosma, E. Chi, F. Xia, Q. Le and D. Zhou, Chain of thought prompting elicits reasoning in large language models, arXiv, 2022, preprint, arXiv:2201.11903, DOI:10.48550/arXiv.2201.11903, http://arxiv.org/abs/2201.11903.
- X. Wang, J. Wei, D. Schuurmans, Q. Le, E. Chi, S. Narang, A. Chowdhery and D. Zhou, Self-consistency improves chain of thought reasoning in language models, arXiv, 2022, preprint, arXiv:2203.11171, DOI:10.48550/arXiv.2203.11171, http://arxiv.org/abs/2203.11171.
- S. Yao, J. Zhao, D. Yu, N. Du, I. Shafran, K. Narasimhan and Y. Cao, ReAct: Synergizing reasoning and acting in language models, arXiv, 2022, preprint, arXiv:2210.03629, DOI:10.48550/arXiv.2210.03629, http://arxiv.org/abs/2210.03629.
- H. Shibo, Y. Gu, H. Ma, J. J. Hong, Z. Wang, D. Z. Wang and Z. Hu, Reasoning with language model is planning with world model, arXiv, 2023, preprint, arXiv:2305.14992, DOI:10.48550/arXiv.2305.14992, http://arxiv.org/abs/2305.14992.
- H. Liu, C. Sferrazza and P. Abbeel, Chain of hindsight aligns language models with feedback, arXiv, 2023, preprint, arXiv:2302.02676, DOI:10.48550/arXiv.2302.02676, http://arxiv.org/abs/2302.02676.
- S. Yao, D. Yu, J. Zhao, I. Shafran, T. L. Griffiths, Y. Cao and K. Narasimhan, Tree of thoughts: Deliberate problem solving with large language models, arXiv, May 2023, preprint, arXiv:2305.10601, DOI:10.48550/arXiv.2305.10601, http://arxiv.org/abs/2305.10601.
- J. Kang, R. Laroche, X. Yuan, A. Trischler, X. Liu and J. Fu, Think before you act: Decision transformers with working memory, arXiv, 2023, preprint, arXiv:2305.16338, DOI:10.48550/arXiv.2305.16338, http://arxiv.org/abs/2305.16338.
- C. Qian, S. Liang, Y. Qin, Y. Ye, X. Cong, Y. Lin, Y. Wu, Z. Liu and M. Sun, Investigate-consolidate-exploit: A general strategy for inter-task agent self-evolution, arXiv, 2024, preprint, arXiv:2401.13996, DOI:10.48550/arXiv.2401.13996, http://arxiv.org/abs/2401.13996.
- R. Anil, A. M. Dai, O. Firat, M. Johnson, D. Lepikhin, A. Passos, S. Shakeri, E. Taropa, P. Bailey, Z. Chen, E. Chu, J. H. Clark, L. El Shafey, Y. Huang, K. Meier-Hellstern, G. Mishra, E. Moreira, M. Omernick, K. Robinson, S. Ruder, Y. Tay, K. Xiao, Y. Xu, Y. Zhang, G. H. Abrego, J. Ahn, A. Jacob, P. Barham, J. Botha, J. Bradbury, S. Brahma, K. Brooks, M. Catasta, Y. Cheng, C. Cherry, C. A. Choquette-Choo, A. Chowdhery, C. Crepy, S. Dave, M. Dehghani, S. Dev, J. Devlin, M. Díaz, N. Du, E. Dyer, V. Feinberg, F. Feng, V. Fienber, M. Freitag, X. Garcia, S. Gehrmann, L. Gonzalez, G. Gur-Ari, S. Hand, H. Hashemi, L. Hou, J. Howland, A. Hu, J. Hui, J. Hurwitz, M. Isard, A. Ittycheriah, M. Jagielski, W. Jia, K. Kenealy, M. Krikun, S. Kudugunta, C. Lan, K. Lee, B. Lee, E. Li, M. Li, W. Li, Y. Li, J. Li, H. Lim, H. Lin, Z. Liu, F. Liu, M. Maggioni, A. Mahendru, J. Maynez, V. Misra, M. Moussalem, Z. Nado, J. Nham, E. Ni, A. Nystrom, A. Parrish, M. Pellat, M. Polacek, A. Polozov, R. Pope, S. Qiao, E. Reif, B. Richter, P. Riley, A. C. Ros, A. Roy, B. Saeta, R. Samuel, R. Shelby, A. Slone, D. Smilkov, D. R. So, D. Sohn, S. Tokumine, D. Valter, V. Vasudevan, K. Vodrahalli, X. Wang, P. Wang, Z. Wang, T. Wang, J. Wieting, Y. Wu, K. Xu, Y. Xu, L. Xue, P. Yin, J. Yu, Q. Zhang, S. Zheng, C. Zheng, W. Zhou, D. Zhou, S. Petrov and Y. Wu, PaLM 2 technical report, arXiv, 2023, preprint, arXiv:2305.10403, DOI:10.48550/arXiv.2305.10403, http://arxiv.org/abs/2305.10403.
- K. Cobbe, V. Kosaraju, M. Bavarian, M. Chen, H. Jun, L. Kaiser, M. Plappert, J. Tworek, H. Jacob, R. Nakano, C. Hesse and J. Schulman, Training verifiers to solve math word problems, arXiv, 2021, preprint, arXiv:2110.14168, DOI:10.48550/arXiv.2110.14168, http://arxiv.org/abs/2110.14168.
- Z. Jie, J. Li and W. Lu, Learning to reason deductively: Math word problem solving as complex relation extraction, arXiv, 2022, preprint, arXiv:2203.10316, DOI:10.48550/arXiv.2203.10316, http://arxiv.org/abs/2203.10316.
- Y. Lan, L. Wang, Q. Zhang, Y. Lan, B. Tian Dai, Y. Wang, D. Zhang and Ee-P. Lim, MWPToolkit: An open-source framework for deep learning-based math word problem solvers, arXiv, 2021, preprint, arXiv:2109.00799, DOI:10.48550/arXiv.2109.00799, http://arxiv.org/abs/2109.00799.
- M. Besta, N. Blach, A. Kubicek, R. Gerstenberger, M. Podstawski, L. Gianinazzi, J. Gajda, T. Lehmann, H. Niewiadomski, P. Nyczyk and T. Hoefler, Graph of thoughts: Solving elaborate problems with large language models, arXiv, 2023, preprint, arXiv:2308.09687, DOI:10.48550/arXiv.2308.09687, http://arxiv.org/abs/2308.09687.
- B. Sel, A. Al-Tawaha, V. Khattar, R. Jia and M. Jin, Algorithm of thoughts: Enhancing exploration of ideas in large language models, arXiv, 2023, preprint, arXiv:2308.10379, DOI:10.48550/arXiv.2308.10379, http://arxiv.org/abs/2308.10379.
- T. Liang, Z. He, W. Jiao, X. Wang, Y. Wang, R. Wang, Y. Yang, Z. Tu and S. Shi, Encouraging divergent thinking in large language models through multi-agent debate, arXiv, 2023, preprint, arXiv:2305.19118, DOI:10.48550/arXiv.2305.19118, http://arxiv.org/abs/2305.19118.
- Y. Du, S. Li, A. Torralba, J. B. Tenenbaum and I. Mordatch, Improving factuality and reasoning in language models through multiagent debate, arXiv, 2023, preprint, arXiv:2305.14325, DOI:10.48550/arXiv.2305.14325, http://arxiv.org/abs/2305.14325.
- C.-M. Chan, W. Chen, Y. Su, J. Yu, W. Xue, S. Zhang, J. Fu and Z. Liu, ChatEval: Towards better LLM-based evaluators through multi-agent debate, arXiv, 2023, preprint, arXiv:2308.07201, DOI:10.48550/arXiv.2308.07201.
- C. H. Song, J. Wu, C. Washington, B. M. Sadler, W.-L. Chao and Y. Su, LLM-planner: Few-shot grounded planning for embodied agents with large language models, arXiv, 2022, preprint, arXiv:2212.04088, DOI:10.48550/arXiv.2212.04088, http://arxiv.org/abs/2212.04088.
- B. Liu, Y. Jiang, X. Zhang, Q. Liu, S. Zhang, J. Biswas and P. Stone, LLM+P: Empowering large language models with optimal planning proficiency, arXiv, 2023, preprint, arXiv:2304.11477, DOI:10.48550/arXiv.2304.11477, http://arxiv.org/abs/2304.11477.
- A. Madaan, N. Tandon, P. Gupta, S. Hallinan, L. Gao, S. Wiegreffe, U. Alon, N. Dziri, S. Prabhumoye, Y. Yang, S. Gupta, B. Prasad Majumder, K. Hermann, S. Welleck, A. Yazdanbakhsh and P. Clark, Self-refine: Iterative refinement with self-feedback, arXiv, 2023, preprint, arXiv:2303.17651, DOI:10.48550/arXiv.2303.17651, http://arxiv.org/abs/2303.17651.
- Z. Xi, S. Jin, Y. Zhou, R. Zheng, S. Gao, T. Gui, Q. Zhang and X. Huang, Self-polish: Enhance reasoning in large language models via problem refinement, arXiv, 2023, preprint, arXiv:2305.14497, DOI:10.48550/arXiv.2305.14497, http://arxiv.org/abs/2305.14497.
- Z. Wang, S. Cai, G. Chen, A. Liu, X. Ma and Y. Liang, Describe, explain, plan and select: Interactive planning with large language models enables open-world multi-task agents, arXiv, 2023, preprint, arXiv:2302.01560, DOI:10.48550/arXiv.2302.01560, http://arxiv.org/abs/2302.01560.
- A. Deshpande, V. Murahari, T. Rajpurohit, A. Kalyan and K. Narasimhan, Toxicity in ChatGPT: Analyzing persona-assigned language models, arXiv, 2023, preprint, arXiv:2304.05335, DOI:10.48550/arXiv.2304.05335, http://arxiv.org/abs/2304.05335.
- S. Hong, M. Zhuge, J. Chen, X. Zheng, Y. Cheng, C. Zhang, J. Wang, Z. Wang, S. Ka S. Yau, Z. Lin, L. Zhou, C. Ran, L. Xiao, C. Wu and J. Schmidhuber, MetaGPT: Meta programming for a multi-agent collaborative framework, arXiv, 2023, preprint, arXiv:2308.00352, DOI:10.48550/arXiv.2308.00352, http://arxiv.org/abs/2308.00352.
- G. Li, H. A. Al Kader Hammoud, H. Itani, D. Khizbullin and B. Ghanem, CAMEL: Communicative agents for “mind” exploration of large language model society, arXiv, 2023, preprint, arXiv:2303.17760, DOI:10.48550/arXiv.2303.17760, http://arxiv.org/abs/2303.17760.
- S. Jinxin, Z. Jiabao, W. Yilei, X. Wu, L. Jiawen and H. Liang, CGMI: Configurable general multi-agent interaction framework, arXiv, 2023, preprint, arXiv:2308.12503, DOI:10.48550/arXiv.2308.12503, http://arxiv.org/abs/2308.12503.
- C. Qian, X. Cong, W. Liu, C. Yang, W. Chen, Y. Su, Y. Dang, J. Li, J. Xu, D. Li, Z. Liu and M. Sun, Communicative agents for software development, arXiv, 2023, preprint, arXiv:2307.07924, DOI:10.48550/arXiv.2307.07924, http://arxiv.org/abs/2307.07924.
- Y. Shao, L. Li, J. Dai and X. Qiu, Character-LLM: A trainable agent for role-playing, arXiv, 2023, preprint, arXiv:2310.10158, DOI:10.48550/arXiv.2310.10158, http://arxiv.org/abs/2310.10158.
- K. Pei, B. Wen, Z. Feng, X. Liu, X. Lei, J. Cheng, S. Wang, A. Zeng, Y. Dong, H. Wang, J. Tang and M. Huang, CritiqueLLM: Scaling LLM-as-critic for effective and explainable evaluation of large language model generation, arXiv, 2023, preprint, arXiv:2311.18702, DOI:10.48550/arXiv.2311.18702, http://arxiv.org/abs/2311.18702.
- L. Wang, J. Zhang, H. Yang, Z. Chen, J. Tang, Z. Zhang, X. Chen, Y. Lin, R. Song, W. Xin Zhao, J. Xu, Z. Dou, J. Wang and J.-R. Wen, User behavior simulation with large language model based agents, arXiv, 2023, preprint, arXiv:2306.02552, DOI:10.48550/arXiv.2306.02552, http://arxiv.org/abs/2306.02552.
- P. A. Lisa, E. C. Busby, N. Fulda, J. R. Gubler, C. Rytting and D. Wingate, Out of one, many: Using language models to simulate human samples, Polit. Anal., 2023, 31(3), 337–351, DOI:10.1017/pan.2023.2.
- A. Kirillov, E. Mintun, N. Ravi, H. Mao, C. Rolland, L. Gustafson, T. Xiao, W. Spencer, A. C. Berg, W.-Y. Lo, P. Dollár and R. Girshick, Segment anything, arXiv, 2023, preprint, arXiv:2304.02643, DOI:10.48550/arXiv.2304.02643, http://arxiv.org/abs/2304.02643.
- AI Open, GPT-4V(ision) system card, 2023, https://cdn.openai.com/papers/GPTV_System_Card.pdf.
- H. Liu, C. Li, Q. Wu and Y. J. Lee, Visual instruction tuning, arXiv, 2023, preprint, arXiv:2304.08485, DOI:10.48550/arXiv.2304.08485, http://arxiv.org/abs/2304.08485.
- Y. Zhao, Z. Lin, D. Zhou, Z. Huang, J. Feng and B. Kang, BuboGPT: Enabling visual grounding in multi-modal LLMs, arXiv, 2023, preprint, arXiv:2307.08581, DOI:10.48550/arXiv.2307.08581, http://arxiv.org/abs/2307.08581.
- C. Lyu, M. Wu, L. Wang, X. Huang, B. Liu, Z. Du, S. Shi and Z. Tu, Macaw-LLM: Multi-modal language modeling with image, audio, video and text integration, arXiv, 2023, preprint, arXiv:2306.09093, DOI:10.48550/arXiv.2306.09093, http://arxiv.org/abs/2306.09093.
- C. Wang, W. Luo, Q. Chen, H. Mai, J. Guo, S. Dong, X. Xiaohua, Z. Li, M. Lin and S. Gao, Tool-LMM: A large multi-modal model for tool agent learning, arXiv, 2024, preprint, arXiv:2401.10727, DOI:10.48550/arXiv.2401.10727, http://arxiv.org/abs/2401.10727.
- D. Gao, L. Ji, L. Zhou, K. Q. Lin, J. Chen, Z. Fan and M. Zheng Shou, AssistGPT: A general multi-modal assistant that can plan, execute, inspect and learn, arXiv, 2023, preprint, arXiv:2306.08640, DOI:10.48550/arXiv.2306.08640, http://arxiv.org/abs/2306.08640.
- G. Wang, Y. Xie, Y. Jiang, A. Mandlekar, C. Xiao, Y. Zhu, L. Fan and A. Anandkumar, Voyager: An open-ended embodied agent with large language models, arXiv, 2023, preprint, arXiv:2305.16291, DOI:10.48550/arXiv.2305.16291, http://arxiv.org/abs/2305.16291.
- M. Ahn, A. Brohan, N. Brown, Y. Chebotar, C. Omar, B. David, C. Finn, C. Fu, K. Gopalakrishnan, K. Hausman, A. Herzog, D. Ho, J. Hsu, J. Ibarz, B. Ichter, A. Irpan, E. Jang, R. J. Ruano, K. Jeffrey, S. Jesmonth, N. J. Joshi, J. Ryan, D. Kalashnikov, Y. Kuang, K.-H. Lee, S. Levine, Y. Lu, L. Luu, C. Parada, P. Pastor, J. Quiambao, K. Rao, J. Rettinghouse, D. Reyes, P. Sermanet, N. Sievers, C. Tan, A. Toshev, V. Vincent, F. Xia, T. Xiao, P. Xu, S. Xu, M. Yan and A. Zeng, Do as I can, not as I say: Grounding language in robotic affordances, arXiv, 2022, preprint, arXiv:2204.01691, DOI:10.48550/arXiv.2204.01691, http://arxiv.org/abs/2204.01691.
- M. Chen, J. Tworek, H. Jun, Q. Yuan, H. P. de Oliveira Pinto, J. Kaplan, H. Edwards, Y. Burda, N. Joseph, G. Brockman, A. Ray, R. Puri, G. Krueger, M. Petrov, H. Khlaaf, G. Sastry, P. Mishkin, B. Chan, S. Gray, N. Ryder, M. Pavlov, A. Power, L. Kaiser, M. Bavarian, C. Winter, P. Tillet, F. P. Such, D. Cummings, M. Plappert, F. Chantzis, E. Barnes, A. Herbert-Voss, W. H. Guss, A. Nichol, A. Paino, N. Tezak, J. Tang, I. Babuschkin, S. Balaji, S. Jain, W. Saunders, C. Hesse, A. N. Carr, J. Leike, J. Achiam, V. Misra, E. Morikawa, A. Radford, M. Knight, M. Brundage, M. Murati, K. Mayer, P. Welinder, B. McGrew, D. Amodei, S. McCandlish, I. Sutskever and W. Zaremba, Evaluating large language models trained on code, arXiv, 2021, preprint, arXiv:2107.03374, DOI:10.48550/arXiv.2107.03374, http://arxiv.org/abs/2107.03374.
- Y. Qin, S. Hu, Y. Lin, W. Chen, N. Ding, G. Cui, Z. Zeng, Y. Huang, C. Xiao, C. Han, Yi R. Fung, Y. Su, H. Wang, C. Qian, R. Tian, K. Zhu, S. Liang, X. Shen, B. Xu, Z. Zhang, Y. Ye, B. Li, Z. Tang, J. Yi, Y. Zhu, Z. Dai, L. Yan, X. Cong, Y. Lu, W. Zhao, Y. Huang, J. Yan, X. Han, X. Sun, D. Li, J. Phang, C. Yang, T. Wu, H. Ji, Z. Liu and M. Sun, Tool learning with foundation models, arXiv, 2023, preprint, arXiv:2304.08354, DOI:10.48550/arXiv.2304.08354.
- R. Nakano, H. Jacob, S. Balaji, J. Wu, L. Ouyang, C. Kim, C. Hesse, S. Jain, V. Kosaraju, W. Saunders, X. Jiang, K. Cobbe, T. Eloundou, G. Krueger, K. Button, M. Knight, B. Chess and J. Schulman, WebGPT: Browser-assisted question-answering with human feedback, arXiv, 2021, preprint, arXiv:2112.09332, DOI:10.48550/arXiv.2112.09332, http://arxiv.org/abs/2112.09332.
- T. Schick, J. Dwivedi-Yu, R. Dessì, R. Raileanu, M. Lomeli, L. Zettlemoyer, N. Cancedda and T. Scialom, Toolformer: Language models can teach themselves to use tools, arXiv, 2023, preprint, arXiv:2302.04761, DOI:10.48550/arXiv.2302.04761, http://arxiv.org/abs/2302.04761.
- A. Parisi, Z. Yao and N. Fiedel, TALM: Tool augmented language models, arXiv, 2022, preprint, arXiv:2205.12255, DOI:10.48550/arXiv.2205.12255, http://arxiv.org/abs/2205.12255.
- C. Qian, C. Xiong, Z. Liu and Z. Liu, Toolink: Linking toolkit creation and using through chain-of-solving on open-source model, arXiv, 2023, preprint, arXiv:2310.05155, DOI:10.48550/arXiv.2310.05155, http://arxiv.org/abs/2310.05155.
- E. Karpas, O. Abend, Y. Belinkov, B. Lenz, O. Lieber, N. Ratner, Y. Shoham, H. Bata, Y. Levine, K. Leyton-Brown, D. Muhlgay, N. Rozen, E. Schwartz, G. Shachaf, S. Shalev-Shwartz, A. Shashua and M. Tenenholtz, MRKL systems: A modular, neuro-symbolic architecture that combines large language models, external knowledge sources and discrete reasoning, arXiv, 2022, preprint, arXiv:2205.00445, DOI:10.48550/arXiv.2205.00445, http://arxiv.org/abs/2205.00445.
- Z. Chen, K. Zhou, B. Zhang, Z. Gong, W. X. Zhao and Ji-R. Wen, ChatCoT: Tool-augmented chain-of-thought reasoning on chat-based large language models, arXiv, 2023, preprint, arXiv:2305.14323, DOI:10.48550/arXiv.2305.14323, http://arxiv.org/abs/2305.14323.
- T. Cai, X. Wang, T. Ma, X. Chen and D. Zhou, Large language models as tool makers, arXiv, 2023, preprint, arXiv:2305.17126, DOI:10.48550/arXiv.2305.17126, http://arxiv.org/abs/2305.17126.
- C. Qian, C. Han, Yi R. Fung, Y. Qin, Z. Liu and H. Ji, CREATOR: Disentangling abstract and concrete reasonings of large language models through tool creation, arXiv, 2023, preprint, arXiv:2305.14318, DOI:10.48550/arXiv.2305.14318, http://arxiv.org/abs/2305.14318.
- L. Yuan, Y. Chen, X. Wang, Yi R. Fung, H. Peng and H. Ji, CRAFT: Customizing LLMs by creating and retrieving from specialized toolsets, arXiv, 2023, preprint, arXiv:2309.17428, DOI:10.48550/arXiv.2309.17428, http://arxiv.org/abs/2309.17428.
- A. Hilmy Abiyyu, Flaticon, https://www.flaticon.com/authors/hilmy-abiyyu-a, accessed: 2024-5-1.
- L. Islam Ani, Flaticon, https://www.flaticon.com/authors/laisa-islam-ani, accessed: 2024-5-1.
- Freepik, Flaticon, https://www.flaticon.com/authors/freepik, accessed: 2024-5-1.
- Kiranshastry, Flaticon, https://www.flaticon.com/authors/kiranshastry, accessed: 2024-5-1.
- M. D. Skarlinski, S. Cox, J. M. Laurent, J. D. Braza, M. Hinks, M. J. Hammerling, M. Ponnapati, S. G. Rodriques and A. D. White, Language agents achieve superhuman synthesis of scientific knowledge, arXiv, 2024, preprint, arXiv:2409.13740, DOI:10.48550/arXiv.2409.13740, http://arxiv.org/abs/2409.13740.
- Y. Chiang, E. Hsieh, C.-H. Chou and J. Riebesell, LLaMP: Large language model made powerful for high-fidelity materials knowledge retrieval and distillation, arXiv, 2024, preprint, arXiv:2401.17244, DOI:10.48550/arXiv.2401.17244, http://arxiv.org/abs/2401.17244.
- P. Ma, T.-H. Wang, M. Guo, Z. Sun, J. B. Tenenbaum, D. Rus, C. Gan and W. Matusik, LLM and simulation as bilevel optimizers: A new paradigm to advance physical scientific discovery, arXiv, 2024, preprint, arXiv:2405.09783, DOI:10.48550/arXiv.2405.09783, http://arxiv.org/abs/2405.09783.
- Y. Qu, K. Huang, H. Cousins, W. A. Johnson, D. Yin, M. Shah, D. Zhou, R. Altman, M. Wang and L. Cong, CRISPR-GPT: An LLM agent for automated design of gene-editing experiments, bioRxiv, 2024, preprint, DOI:10.1101/2024.04.25.591003.
- H. Liu, Y. Li, J. Jian, Y. Cheng, J. Lu, S. Guo, J. Zhu, M. Zhang, M. Zhang and H. Wang, Toward a team of AI-made scientists for scientific discovery from gene expression data, arXiv, 2024, preprint, arXiv:2402.12391, DOI:10.48550/arXiv.2402.12391, http://arxiv.org/abs/2402.12391.
- H. W. Sprueill, C. Edwards, K. Agarwal, M. V. Olarte, U. Sanyal, C. Johnston, H. Liu, H. Ji and S. Choudhury, ChemReasoner: Heuristic search over a large language model's knowledge space using quantum-chemical feedback, arXiv, 2024, preprint, arXiv:2402.10980, DOI:10.48550/arXiv.2402.10980, http://arxiv.org/abs/2402.10980.
- Y. Ma, Z. Gou, H. Junheng, R. Xu, S. Wang, L. Pan, Y. Yang, Y. Cao and A. Sun, SciAgent: Tool-augmented language models for scientific reasoning, arXiv, 2024, preprint, arXiv:2402.11451, DOI:10.48550/arXiv.2402.11451, http://arxiv.org/abs/2402.11451.
- Y. Shao, Y. Jiang, T. A. Kanell, P. Xu, K. Omar and M. S. Lam, Assisting in writing wikipedia-like articles from scratch with large language models, arXiv, 2024, preprint, arXiv:2402.14207, DOI:10.48550/arXiv.2402.14207, http://arxiv.org/abs/2402.14207.
- C. Völker, T. Rug, K. Maik Jablonka and S. Kruschwitz, LLMs can design sustainable concrete – a systematic benchmark. 2024, https://www.researchsquare.com/article/rs-3913272/v1.
- A. Ghafarollahi and M. J. Buehler, ProtAgents: Protein discovery via large language model multi-agent collaborations combining physics and machine learning, arXiv, 2024, preprint, arXiv:2402.04268, DOI:10.48550/arXiv.2402.04268, http://arxiv.org/abs/2402.04268.
- J. Lála, O. O'Donoghue, A. Shtedritski, S. Cox, S. G. Rodriques and A. D. White, PaperQA: Retrieval-augmented generative agent for scientific research, arXiv, 2023, preprint, arXiv:2312.07559, DOI:10.48550/arXiv.2312.07559, http://arxiv.org/abs/2312.07559.
- S. Cox, M. Hammerling, J. Lála, J. Laurent, S. Rodriques, M. Rubashkin and A. White, WikiCrow: Automating synthesis of human scientific knowledge, 2023, https://www.futurehouse.org/wikicrow, accessed: 2024-2-15.
- M. Ansari and S. M. Moosavi, Agent-based learning of materials datasets from scientific literature, arXiv, 2023, preprint, arXiv:2312.11690, DOI:10.48550/arXiv.2312.11690, http://arxiv.org/abs/2312.11690.
- M. H. Prince, H. Chan, A. Vriza, T. Zhou, V. K. Sastry, M. T. Dearing, R. J. Harder, R. K. Vasudevan and M. J. Cherukara, Opportunities for retrieval and tool augmented large language models in scientific facilities, arXiv, 2023, preprint, arXiv:2312.01291, DOI:10.48550/arXiv.2312.01291, http://arxiv.org/abs/2312.01291.
- Y. Liu, S. Chen, H. Cheng, M. Yu, R. Xiao, A. Mo, Y. Tang and Y. Huang, CoQuest: Exploring research question co-creation with an LLM-based agent, arXiv, 2023, preprint, arXiv:2310.06155, DOI:10.48550/arXiv.2310.06155, http://arxiv.org/abs/2310.06155.
- O. O'Donoghue, A. Shtedritski, J. Ginger, R. Abboud, A. E. Ghareeb, J. Booth and S. G. Rodriques, BioPlanner: Automatic evaluation of LLMs on protocol planning in biology, arXiv, 2023, preprint, arXiv:2310.10632, DOI:10.48550/arXiv.2310.10632, http://arxiv.org/abs/2310.10632.
- N. Janakarajan, T. Erdmann, S. Swaminathan, T. Laino and J. Born, Language models in molecular discovery, arXiv, 2023, preprint, arXiv:2309.16235, DOI:10.48550/arXiv.2309.16235, http://arxiv.org/abs/2309.16235.
- Y. Kang and J. Kim, ChatMOF: An autonomous AI system for predicting and generating metal-organic frameworks, arXiv, 2023, preprint, arXiv:2308.01423, DOI:10.48550/arXiv.2308.01423, http://arxiv.org/abs/2308.01423.
- B. Mouriño, E. Moubarak, J. Van Herck, S. Majumdar and X. Zhang, i-digest: v1.0, 2023, https://zenodo.org/record/8080962.
- B. Rankovic, A. M. Bran and P. Schwaller, BOLLaMa: BOLLaMA interface working with CHAOS, 2023, https://zenodo.org/record/8096827.
- S. Kruschwitz, C. Völker and G. A. Zia, Text2Concrete, 2023, https://zenodo.org/record/8091195.
- M. C. Ramos, S. Cox and A. White, MAPI_LLM: MAPI_LLM first release, 2023, https://zenodo.org/record/8097336.
- M. C. Ramos, S. S. Michtavy, M. D. Porosoff and A. D. White. Bayesian optimization of catalysts with in-context learning, arXiv, 2023, preprint, arXiv:2304.05341, DOI:10.48550/arXiv.2304.05341, http://arxiv.org/abs/2304.05341.
- G. M. Hocky and A. D. White, Natural language processing models that automate programming will transform chemistry research and teaching, Digital discovery, 2022, 1(2), 79–83, 10.1039/d1dd00009h.
- A. D. White, G. M. Hocky, H. A. Gandhi, M. Ansari, S. Cox, G. P. Wellawatte, S. Sasmal, Z. Yang, K. Liu, Y. Singh and W. J. P. Ccoa, Assessment of chemistry knowledge in large language models that generate code, Digital Discovery, 2023, 2(2), 368–376, 10.1039/D2DD00087C.
- A. Kristiadi, F. Strieth-Kalthoff, M. Skreta, P. Poupart, A. Aspuru-Guzik and G. Pleiss, A sober look at LLMs for material discovery: Are they actually good for bayesian optimization over molecules?, arXiv, 2024, preprint, arXiv:2402.05015, DOI:10.48550/arXiv.2402.05015, http://arxiv.org/abs/2402.05015.
- B. Ranković and P. Schwaller, BoChemian: Large language model embeddings for Bayesian optimization of chemical reactions, 2023, https://openreview.net/pdf?id=A1RVn1m3J3.
- K. Jablonka, Q. Ai, A. H. Al-Feghali, S. Badhwar, J. D. Bocarsly Andres Bran, S. Bringuier, L. Brinson, K. Choudhary, D. Circi, S. Cox, W. D. Jong, M. L. Evans, N. Gastellu, J. Genzling, M. Gil, A. Gupta, Z. Hong, A. Imran, S. Kruschwitz, A. Labarre, J. L'ala, T. Liu, S. Ma, S. Majumdar, G. Merz, N. Moitessier, E. Moubarak, B. Mouriño, B. G. Pelkie, M. Pieler, M. C. Ramos, B. Rankovi’c, S. G. Rodriques, J. N. Sanders, P. Schwaller, M. Schwarting, J.-X. Shi, B. Smit, B. Smith, J. V. Heck, C. Volker, L. T. Ward, S. Warren, B. Weiser, S. Zhang, X. Zhang, G. A. J. Zia, A. Scourtas, K. Schmidt, I. T. Foster, A. D. White and B. Blaiszik, 14 examples of how LLMs can transform materials science and chemistry: a reflection on a large language model hackathon, Digital discovery, 2023, 2(5), 1233–1250, 10.1039/D3DD00113J.
- B. Ranković, R.-R. Griffiths, H. B. Moss and P. Schwaller, Bayesian optimisation for additive screening and yield improvements in chemical reactions – beyond one-hot encoding, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2022-nll2j-v3.
- B. Weiser, J. Genzling, N. Gastellu, S. Zhang, T. Liu, A. Al-Feghali, N. Moitessier, A. Labarre and S. Ma, LLM-Guided-GA: LLM-Guided-GA digital discovery release, 2023, https://zenodo.org/record/8125541.
- D. Circi and S. Badhwar, InsightGraph: InsightGraph, 2023, https://zenodo.org/record/8092575.
- M. Zaabi, W. Hariri and N. Smaoui, A review study of ChatGPT applications in education, in 2023 International Conference on Innovations in Intelligent Systems and Applications (INISTA), IEEE, 2023, pp. 1–5, DOI:10.1109/inista59065.2023.10310439.
- E. Kasneci, K. Sessler, S. Küchemann, M. Bannert, D. Dementieva, F. Fischer, U. Gasser, G. Groh, S. Günnemann, E. Hüllermeier, S. Krusche, G. Kutyniok, T. Michaeli, C. Nerdel, J. Pfeffer, O. Poquet, M. Sailer, A. Schmidt, T. Seidel, M. Stadler, J. Weller, J. Kuhn and G. Kasneci, ChatGPT for good? on opportunities and challenges of large language models for education, Learn. Individ. Differ., 2023, 103(102274), 102274, DOI:10.1016/j.lindif.2023.102274.
- A. Hellas, J. Leinonen, S. Sarsa, C. Koutcheme, L. Kujanpaa and J. Sorva, Exploring the responses of large language models to beginner programmers' help requests, arXiv, 2023, preprint, arXiv:2306.05715, DOI:10.48550/arXiv.2306.05715, http://arxiv.org/abs/2306.05715.
- Y. Dan, Z. Lei, Y. Gu, Y. Li, J. Yin, J. Lin, L. Ye, Z. Tie, Y. Zhou, Y. Wang, A. Zhou, Z. Zhou, Q. Chen, J. Zhou, L. He and X. Qiu, EduChat: A large-scale language model-based chatbot system for intelligent education, arXiv, 2023, preprint, arXiv:2308.02773, DOI:10.48550/arXiv.2308.02773, http://arxiv.org/abs/2308.02773.
- T. Jie, J. Hou, Z. Wu, S. Peng, Z. Liu, Y. Xiang, B. Gu, N. Filla, Y. Li, N. Liu, X. Chen, K. Tang, T. Liu and X. Wang, Assessing large language models in mechanical engineering education: A study on mechanics-focused conceptual understanding, arXiv, 2024, preprint, arXiv:2401.12983, DOI:10.48550/arXiv.2401.12983, http://arxiv.org/abs/2401.12983.
- P. Lewis, E. Perez, A. Piktus, F. Petroni, V. Karpukhin, N. Goyal, H. Kuttler, M. Lewis, W.-T. Yih, T. Rocktaschel, S. Riedel and D. Kiela, Retrieval-augmented generation for knowledge-intensive NLP tasks, arXiv, 2020, preprint, arXiv:2005.11401, DOI:10.48550/arXiv.2005.11401, http://arxiv.org/abs/2005.11401.
- K. Chen, J. Li, K. Wang, Y. Du, J. Yu, J. Lu, L. Li, J. Qiu, J. Pan, Y. Huang, Q. Fang, P. Ann Heng and G. Chen, Chemist-X: Large language model-empowered agent for reaction condition recommendation in chemical synthesis, arXiv, 2023, preprint, arXiv:2311.10776, DOI:10.48550/arXiv.2311.10776, http://arxiv.org/abs/2311.10776.
- J. B. Ingraham, M. Baranov, Z. Costello, K. W. Barber, W. Wang, I. Ahmed, F. Vincent, D. M. Lord, C. Ng-Thow-Hing, E. R. Van Vlack, S. Tie, X. Vincent, S. C. Cowles, A. Leung, J. V. Rodrigues, C. L. Morales-Perez, A. M. Ayoub, R. Green, K. Puentes, O. Frank, N. V. Panwar, O. Fritz, R. R. Adam, A. L. Beam, F. J. Poelwijk and G. Grigoryan, Illuminating protein space with a programmable generative model, Nature, 2023, 623(7989), 1070–1078, DOI:10.1038/s41586-023-06728-8.
- K. E. Wu, K. K. Yang, R. van den Berg, S. Alamdari, J. Y. Zou, A. X. Lu and A. P. Amini, Protein structure generation via folding diffusion, Nat. Commun., 2024, 15(1), 1059, DOI:10.1038/s41467-024-45051-2.
- X. Tang, Q. Jin, K. Zhu, T. Yuan, Y. Zhang, W. Zhou, M. Qu, Y. Zhao, J. Tang, Z. Zhang, A. Cohan, Z. Lu and M. Gerstein, Prioritizing safeguarding over autonomy: Risks of LLM agents for science, arXiv, 2024, preprint, arXiv:2402.04247, DOI:10.48550/arXiv.2402.04247, http://arxiv.org/abs/2402.04247.
- Y. Ruan, C. Lu, N. Xu, J. Zhang, J. Xuan, J. Pan, Q. Fang, H. Gao, X. Shen, N. Ye, Q. Zhang and Y. Mo, Accelerated end-to-end chemical synthesis development with large language models, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-6wmg4.
- Z. Zheng, O. Zhang, C. Borgs, J. T. Chayes and O. M. Yaghi, ChatGPT Chemistry Assistant for Text Mining and the Prediction of MOF Synthesis, J. Am. Chem. Soc., 2023, 145, 18048–18062, DOI:10.1021/jacs.3c05819.
- Z. Zheng, Z. He, O. Khattab, N. Rampal, M. A. Zaharia, C. Borgs, J. T. Chayes and O. M. Yaghi, Image and data mining in reticular chemistry powered by GPT-4V, Digital discovery, 2024, 3(3), 491–501, 10.1039/d3dd00239j.
- Z. Zheng, O. Zhang, Ha L. Nguyen, N. Rampal, A. H. Alawadhi, Z. Rong, T. Head-Gordon, C. Borgs, J. T. Chayes and O. M. Yaghi, ChatGPT research group for optimizing the crystallinity of MOFs and COFs, ACS Cent. Sci., 2023, 9(11), 2161–2170, DOI:10.1021/acscentsci.3c01087.
- N. Nascimento, P. Alencar and D. Cowan, Self-adaptive large language model (LLM)-based multiagent systems, in 2023 IEEE International Conference on Autonomic Computing and Self-Organizing Systems Companion (ACSOS-C), IEEE, pp. 104–109, 2023, DOI:10.1109/ACSOS-C58168.2023.00048.
- S. K. Niazi and Z. Mariam, Recent advances in Machine-Learning-Based chemoinformatics: A comprehensive review, Int. J. Mol. Sci., 2023, 24(14), 11488, DOI:10.3390/ijms241411488.
- A. D. McNaughton, G. Ramalaxmi, A. Kruel, C. R. Knutson, R. A. Varikoti and N. Kumar, CACTUS: Chemistry agent connecting tool-usage to science, arXiv, 2024, preprint, arXiv:2405.00972, DOI:10.48550/arXiv.2405.00972, http://arxiv.org/abs/2405.00972.
- Y. Kang, H. Park, B. Smit and J. Kim, A multi-modal pre-training transformer for universal transfer learning in metal–organic frameworks, Nat. Mach. Intell., 2023, 5(3), 309–318, DOI:10.1038/s42256-023-00628-2.
- M. Manica, J. Born, J. Cadow, D. Christofidellis, A. Dave, D. Clarke, Y. G. N. Teukam, G. Giannone, S. C. Hoffman, M. Buchan, V. Chenthamarakshan, T. Donovan, H. H. Hsu, F. Zipoli, S. Oliver, A. Kishimoto, L. Hamada, I. Padhi, K. Wehden, L. McHugh, A. Khrabrov, P. Das, S. Takeda and J. R. Smith, Accelerating material design with the generative toolkit for scientific discovery, npj Comput. Mater., 2023, 9(1), 1–6, DOI:10.1038/s41524-023-01028-1.
- rxn4chemistry: Python wrapper for the IBM RXN for chemistry API, https://github.com/rxn4chemistry/rxn4chemistry.
- P. Gaiński, L. Maziarka, T. Danel and S. Jastrzebski, HuggingMolecules: An Open-Source library for Transformer-Based molecular property prediction (student abstract), Proceedings of the AAAI Conference on Artificial Intelligence, 2022, 36(11), 12949–12950, DOI:10.1609/aaai.v36i11.21611.
- G. Landrum, Rdkit documentation, Release, 1(1-79):4, 2013, pp. 1533–3752, https://media.readthedocs.org/pdf/rdkit/latest/rdkit.pdf.
- Y. Zheng, H. Y. Koh, J. Ju, A. T. N. Nguyen, L. T. May, G. I. Webb and S. Pan, Large language models for scientific synthesis, inference and explanation, arXiv, 2023, preprint, arXiv:2310.07984, DOI:10.48550/arXiv.2310.07984, http://arxiv.org/abs/2310.07984.
- Q. Wang, D. Downey, H. Ji and T. Hope, SciMON: Scientific inspiration machines optimized for novelty, arXiv, 2023, preprint, arXiv:2305.14259, DOI:10.48550/arXiv.2305.14259, http://arxiv.org/abs/2305.14259.
- X. Gu and M. Krenn, Generation and human-expert evaluation of interesting research ideas using knowledge graphs and large language models, arXiv, 2024, preprint, arXiv:2405.17044, DOI:10.48550/arXiv.2405.17044, http://arxiv.org/abs/2405.17044.
- H. W. Sprueill, C. Edwards, M. V. Olarte, U. Sanyal, H. Ji and S. Choudhury, Monte carlo thought search: Large language model querying for complex scientific reasoning in catalyst design, arXiv, 2023, preprint, arXiv:2310.14420, DOI:10.48550/arXiv.2310.14420, http://arxiv.org/abs/2310.14420.
- G. Zhou, Z. Gao, Q. Ding, H. Zheng, H. Xu, Z. Wei, L. Zhang and G. Ke, Uni-Mol: A universal 3D molecular representation learning framework, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2022-jjm0j-v4.
- S. Miret and N. M. Anoop Krishnan, Are LLMs ready for real-world materials discovery?, arXiv, 2024, preprint, arXiv:2402.05200, DOI:10.48550/arXiv.2402.05200, http://arxiv.org/abs/2402.05200.
- Y. Du, C. Duan, A. Bran, A. Sotnikova, Y. Qu, H. Kulik, A. Bosselut, J. Xu and P. Schwaller, Large language models are catalyzing chemistry education, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-h722v.
- J. Zhang, Y. Fang, X. Shao, H. Chen, N. Zhang and X. Fan, The future of molecular studies through the lens of large language models, J. Chem. Inf. Model., 2024, 64(3), 563–566, DOI:10.1021/acs.jcim.3c01977.
- J. A. OpenAI, S. Adler, S. Agarwal, L. Ahmad, I. Akkaya, F. L. Aleman, D. Almeida, J. Altenschmidt, S. Altman, S. Anadkat, R. Avila, I. Babuschkin, S. Balaji, V. Balcom, B. Paul, H. Bao, M. Bavarian, J. Belgum, I. Bello, J. Berdine, G. Bernadett-Shapiro, C. Berner, L. Bogdonoff, O. Boiko, M. Boyd, A.-L. Brakman, G. Brockman, T. Brooks, B. Miles, K. Button, T. Cai, R. Campbell, A. Cann, B. Carey, C. Carlson, R. Carmichael, B. Chan, C. Chang, F. Chantzis, D. Chen, S. Chen, R. Chen, J. Chen, M. Chen, B. Chess, C. Cho, C. Chu, H. W. Chung, D. Cummings, J. Currier, Y. Dai, C. Decareaux, T. Degry, N. Deutsch, D. Deville, A. Dhar, D. Dohan, S. Dowling, S. Dunning, A. Ecoffet, A. Eleti, T. Eloundou, D. Farhi, L. Fedus, N. Felix, S. P. Fishman, J. Forte, I. Fulford, L. Gao, E. Georges, C. Gibson, V. Goel, T. Gogineni, G. Goh, R. Gontijo-Lopes, J. Gordon, G. Morgan, S. Gray, R. Greene, J. Gross, S. S. Gu, Y. Guo, C. Hallacy, J. Han, J. Harris, Y. He, M. Heaton, J. Heidecke, C. Hesse, A. Hickey, W. Hickey, P. Hoeschele, B. Houghton, K. Hsu, S. Hu, X. Hu, J. Huizinga, S. Jain, S. Jain, J. Jang, A. Jiang, R. Jiang, H. Jin, D. Jin, S. Jomoto, B. Jonn, H. Jun, T. Kaftan, L. Kaiser, K. Ali, I. Kanitscheider, N. S. Keskar, T. Khan, K. Logan, J. W. Kim, C. Kim, Y. Kim, J. H. Kirchner, J. Kiros, M. Knight, D. Kokotajlo, L. Kondraciuk, A. Kondrich, A. Konstantinidis, K. Kosic, G. Krueger, V. Kuo, M. Lampe, I. Lan, T. Lee, J. Leike, J. Leung, D. Levy, C. M. Li, R. Lim, M. Lin, S. Lin, M. Litwin, T. Lopez, R. Lowe, P. Lue, A. Makanju, K. Malfacini, S. Manning, T. Markov, Y. Markovski, B. Martin, K. Mayer, A. Mayne, B. McGrew, S. M. McKinney, C. McLeavey, McM. Paul, J. McNeil, D. Medina, A. Mehta, M. Jacob, L. Metz, A. Mishchenko, P. Mishkin, V. Monaco, E. Morikawa, D. Mossing, M. Tong, M. Murati, O. Murk, D. Mély, A. Nair, R. Nakano, R. Nayak, A. Neelakantan, R. Ngo, H. Noh, L. Ouyang, C. O'Keefe, J. Pachocki, A. Paino, J. Palermo, A. Pantuliano, G. Parascandolo, J. Parish, E. Parparita, A. Passos, M. Pavlov, A. Peng, A. Perelman, F. d. A. B. Peres, M. Petrov, H. P. de Oliveira Pinto, M. Pokorny, M. Pokrass, V. H. Pong, T. Powell, A. Power, B. Power, E. Proehl, R. Puri, A. Radford, J. Rae, A. Ramesh, C. Raymond, F. Real, K. Rimbach, C. Ross, B. Rotsted, H. Roussez, N. Ryder, M. Saltarelli, T. Sanders, S. Santurkar, G. Sastry, H. Schmidt, D. Schnurr, J. Schulman, D. Selsam, K. Sheppard, T. Sherbakov, J. Shieh, S. Shoker, P. Shyam, S. Sidor, E. Sigler, M. Simens, J. Sitkin, K. Slama, I. Sohl, B. Sokolowsky, Y. Song, N. Staudacher, F. P. Such, N. Summers, I. Sutskever, J. Tang, N. Tezak, M. B. Thompson, P. Tillet, T. Amin, E. Tseng, P. Tuggle, N. Turley, J. Tworek, J. F. C. Uribe, A. Vallone, A. Vijayvergiya, C. Voss, C. Wainwright, J. J. Wang, A. Wang, B. Wang, J. Ward, J. Wei, C. J. Weinmann, A. Welihinda, P. Welinder, J. Weng, L. Weng, M. Wiethoff, D. Willner, C. Winter, S. Wolrich, H. Wong, L. Workman, S. Wu, J. Wu, M. Wu, K. Xiao, T. Xu, S. Yoo, K. Yu, Q. Yuan, W. Zaremba, R. Zellers, C. Zhang, M. Zhang, S. Zhao, T. Zheng, J. Zhuang, W. Zhuk and B. Zoph, GPT-4 technical report, arXiv, 2023, preprint, arXiv:2303.08774, DOI:10.48550/arXiv.2303.08774, http://arxiv.org/abs/2303.08774.
- W. Gao and C. W. Coley, The synthesizability of molecules proposed by generative models, J. Chem. Inf. Model., 2020, 12, 5714–5723, DOI:10.1021/acs.jcim.0c00174.
- P. Liu, J. Tao and Z. Ren, Scientific language modeling: A quantitative review of large language models in molecular science, arXiv, 2024, preprint, arXiv:2402.04119, DOI:10.48550/arXiv.2402.04119, http://arxiv.org/abs/2402.04119.
- P. Schwaller, B. Hoover, J.-L. Reymond, H. Strobelt and T. Laino, Extraction of organic chemistry grammar from unsupervised learning of chemical reactions, Sci. Adv., 2021, 7(15), eabe4166, DOI:10.1126/sciadv.abe4166.
- O. Schilter, M. Alberts, F. Zipoli, A. C. Vaucher, P. Schwaller and T. Laino, Unveiling the secrets of 1H-NMR spectroscopy: A novel approach utilizing attention mechanisms, 2023, https://openreview.net/pdf?id=4ilKwquW51.
- S. Aryal, T. Do, B. Heyojoo, S. Chataut, B. Dip Shrestha Gurung, V. Gadhamshetty and E. Gnimpieba, Leveraging multi-AI agents for cross-domain knowledge discovery, arXiv, 2024, preprint, arXiv:2404.08511, DOI:10.48550/arXiv.2404.08511, http://arxiv.org/abs/2404.08511.
- F. Bohm, Y. Gao, C. M. Meyer, O. Shapira, I. Dagan and I. Gurevych, Better rewards yield better summaries: Learning to summarise without references, arXiv, 2019, preprint, arXiv:1909.01214, DOI:10.48550/arXiv.1909.01214, http://arxiv.org/abs/1909.01214.
- S. Pan, V. Lialin, S. Muckatira and A. Rumshisky, Let's reinforce step by step, arXiv, 2023, preprint, arXiv:2311.05821, DOI:10.48550/arXiv.2311.05821, http://arxiv.org/abs/2311.05821.
- B. Hu, C. Zhao, P. Zhang, Z. Zhou, Y. Yang, Z. Xu and B. Liu, Enabling intelligent interactions between an agent and an LLM: A reinforcement learning approach, arXiv, 2023, preprint, arXiv:2306.03604, DOI:10.48550/arXiv.2306.03604, http://arxiv.org/abs/2306.03604.
- Z. Xu, C. Yu, F. Fang, Y. Wang and Y. Wu, Language agents with reinforcement learning for strategic play in the werewolf game, arXiv, 2023, preprint, arXiv:2310.18940, DOI:10.48550/arXiv.2310.18940, http://arxiv.org/abs/2310.18940.
- M. R. Morris, J. Sohl-dickstein, N. Fiedel, T. Warkentin, A. Dafoe, A. Faust, C. Farabet and S. Legg, Levels of AGI for operationalizing progress on the path to AGI, arXiv, 2023, preprint, arXiv:2311.02462, DOI:10.48550/arXiv.2311.02462, http://arxiv.org/abs/2311.02462.
- Types of artificial intelligence, https://www.ibm.com/think/topics/artificial-intelligence-types, 2024, accessed: 2024-10-9.
- P. Langley, J. E. Laird and S. Rogers, Cognitive architectures: Research issues and challenges, Cogn. Syst. Res., 2009, 10(2), 141–160, DOI:10.1016/j.cogsys.2006.07.004.
- B. Goertzel, Artificial general intelligence: Concept, state of the art and future prospects, J. Artif. Gen. Intell., 2014, 5(1), 1–48, DOI:10.2478/jagi-2014-0001.
- Y. Ruan, H. Dong, A. Wang, S. Pitis, Y. Zhou, J. Ba, Y. Dubois, C. J. Maddison and T. Hashimoto, Identifying the risks of LM agents with an LM-emulated sandbox, arXiv, 2023, preprint, arXiv:2309.15817, DOI:10.48550/arXiv.2309.15817, http://arxiv.org/abs/2309.15817.
- Z. Ji, N. Lee, R. Frieske, T. Yu, D. Su, Y. Xu, E. Ishii, Y. Jin Bang, A. Madotto and P. Fung, Survey of hallucination in natural language generation, ACM Comput. Surv., 2023, 55(12), 1–38, DOI:10.1145/3571730.
- Z. Cai, B. Chang and W. Han, Human-in-the-loop through chain-of-thought, arXiv, 2023, preprint, arXiv:2306.07932, DOI:10.48550/arXiv.2306.07932, http://arxiv.org/abs/2306.07932.
- H. Xiao and P. Wang, LLM a*: Human in the loop large language models enabled a* search for robotics, arXiv, 2023, preprint, arXiv:2312.01797, DOI:10.48550/arXiv.2312.01797, http://arxiv.org/abs/2312.01797.
- I. Drori and D. Te’eni, Human-in-the-Loop AI reviewing: Feasibility, opportunities and risks, J. Assoc. Inf. Syst., 2024, 25(1), 98–109, DOI:10.17705/1jais.00867.
- N. J. Szymanski, B. Rendy, Y. Fei, R. E. Kumar, T. He, D. Milsted, M. J. McDermott, M. Gallant, E. D. Cubuk, A. Merchant, H. Kim, A. Jain, C. J. Bartel, K. Persson, Y. Zeng and G. Ceder, An autonomous laboratory for the accelerated synthesis of novel materials, Nature, 2023, 624(7990), 86–91, DOI:10.1038/s41586-023-06734-w.
- M. Peplow, Robot chemist sparks row with claim it created new materials, Nature, 2023 DOI:10.1038/d41586-023-03956-w , https://www.nature.com/articles/d41586-023-03956-w.
- S. Hong, Y. Lin, B. Liu, B. Liu, B. Wu, D. Li, J. Chen, J. Zhang, J. Wang, L. Zhang, L. Zhang, M. Yang, M. Zhuge, T. Guo, T. Zhou, W. Tao, W. Wang, X. Tang, X. Lu, X. Zheng, X. Liang, Y. Fei, Y. Cheng, Z. Xu and C. Wu, Data interpreter: An LLM agent for data science, arXiv, 2024, preprint, arXiv:2402.18679, DOI:10.48550/arXiv.2402.18679, http://arxiv.org/abs/2402.18679.
- D. Qi and J. Wang, CleanAgent: Automating data standardization with LLM-based agents, arXiv, 2024, preprint, arXiv:2403.08291, DOI:10.48550/arXiv.2403.08291, http://arxiv.org/abs/2403.08291.
- X. Yee Tai, H. Zhang, Z. Niu, S. D. R. Christie and J. Xuan, The future of sustainable chemistry and process: Convergence of artificial intelligence, data and hardware, Energy AI, 2020, 2(100036), 100036, DOI:10.1016/j.egyai.2020.100036.
- C. Zheng, I. Jalan, P. Cost, K. Oliver, A. Gupta, S. Misture, J. A. Cody and C. J. Collison, Impact of Alkyl Chain Length on Small Molecule Crystallization and Nanomorphology in Squaraine-Based Solution Processed Solar Cells, J. Phys. Chem. C, 2017, 121(14), 7750–7760, DOI:10.1021/acs.jpcc.7b01339.
| This journal is © The Royal Society of Chemistry 2025 |