
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvatomorphic diversity dictates the stability and solubility of metal–organic polyhedra†
Ankit K.
Yadav
a,
Andrzej
Gładysiak
 *a,
Emma H.
Wolpert
b,
Alex M.
Ganose
b,
Bronson
Samel-Garloff
c,
Dipankar
Koley
c,
Kim E.
Jelfs
b and
Kyriakos C.
Stylianou
*a
*a,
Emma H.
Wolpert
b,
Alex M.
Ganose
b,
Bronson
Samel-Garloff
c,
Dipankar
Koley
c,
Kim E.
Jelfs
b and
Kyriakos C.
Stylianou
*a
aMaterials Discovery Laboratory (MaD Lab), Department of Chemistry, Oregon State University, Corvallis, OR 97331, USA. E-mail: andrzej.gladysiak@oregonstate.edu; kyriakos.stylianou@oregonstate.edu
bDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, White City Campus, 82 Wood Lane, W12 0BZ, UK
cDepartment of Chemistry, Oregon State University, Corvallis, OR 97331, USA
First published on 11th December 2024
Abstract
The reaction between molybdenum(II) acetate and 5-aminoisophthalic acid (H2Iso-NH2) afforded [Mo12O12(μ2-O)12(Iso-NH2)12]12−, a novel molybdenum(V) metal–organic polyhedron (MOP) with a triangular antiprismatic shape stabilized by intramolecular N–H⋯O hydrogen bonds. The synthesis conditions, particularly the choice of solvent and reaction time, led to the precipitation of the Mo(V)-MOP in five distinct crystalline forms. These forms vary in their packing arrangements, co-crystallized solvent molecules, and counter-cations, with three phases containing dimethylammonium (dma+) and the other two containing diethylammonium (dea+). Each solvatomorph exhibits unique physical properties, including differences in porosity, and stability. These properties were discerned through empirical observations and supported by density functional theory calculations. Remarkably, the solubility of these MOP solvatomorphs in water was determined for the first time, with values of 4.30(2) g L−1 for a (dma)12[Mo(V)-MOP] phase, and 10.25(7) g L−1 and 14.41(10) g L−1 for two (dea)12[Mo(V)-MOP] phases. Additionally, aqueous solutions of the Mo(V)-MOP were found to conduct electricity as weak electrolytes, showcasing their potential for applications in fields requiring partially ionized species.
Introduction
Polymorphism is the ability of a chemical substance to exist in multiple crystalline solid-state forms.1 In molecular crystals, the phenomenon often originates from the irregular shapes of molecules, allowing for various arrangements in three-dimensional (3D) space. For instance, while naphthalene molecules, with their regular shapes, tend to assemble in a singular manner in 3D space,2–4 effectively making this polyaromatic compound monomorphic, acridine, a structural analog of anthracene featuring a hollow structure due to the substitution of a central C–H group with an N atom, can exist in up to nine distinct crystalline forms.5–9 Stabilization of polymorphic forms in molecular crystals is frequently facilitated by arrays of intermolecular short contacts and hydrogen bonds, as evidenced by studies on glycine and urea.10–14 Such forms can be obtained through precise control of crystallization conditions from melt or solution, or by inducing transformations of less stable polymorphs through external stimuli.15,16 Although polymorphs of a given substance share the same chemical composition, they may exhibit significant variations in physicochemical properties such as melting point, density, hygroscopicity, mechanical properties, stability, and solubility. Additionally, when a compound is crystallized from solution or through vapor diffusion, solvent molecules may be incorporated into its crystal lattice. For example, acridine forms a hydrate by co-crystallizing with water in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.75 ratio,5,17 and a solvate by co-crystallizing with 1,2-diiodotetrafluorobenzene in a 7:4 ratio.18 While technically not polymorphs, these structures, originating from the same parent substance, are categorized as solvatomorphs or pseudopolymorphs due to their resemblance.19 The term “pseudopolymorphism” remains controversial, sparking a debate in the scientific community, with some authors opposing its use and others actively advocating for it.20
:0.75 ratio,5,17 and a solvate by co-crystallizing with 1,2-diiodotetrafluorobenzene in a 7:4 ratio.18 While technically not polymorphs, these structures, originating from the same parent substance, are categorized as solvatomorphs or pseudopolymorphs due to their resemblance.19 The term “pseudopolymorphism” remains controversial, sparking a debate in the scientific community, with some authors opposing its use and others actively advocating for it.20
The practical importance of investigating polymorphism is prominent in the context of pharmaceuticals.21 Differences in mechanical properties between polymorphs may turn out to be key during the manufacturing process. Additionally, a low solubility may negatively affect the time needed for the drug to act, potentially leading to suboptimal bioavailability and different metabolic pathways, as well as varying side effects. Both mechanical properties and solubility play a crucial role in the manufacture and use of paracetamol, a common analgesic, which can occur in three polymorphic forms. Crystals of paracetamol form I22,23 lack slip planes necessary for plastic deformation upon compaction, therefore, the current manufacturing practice is to mix these crystals with gelatin, polyvinylpyrrolidone, or starch prior to tableting.24 Conversely, crystals of the metastable paracetamol form II25 have well-developed slip planes that enable their direct compression not necessitating the use of such binding additives.26 Coincidentally, paracetamol-II dissolves in water at a rate greater than that of paracetamol-I.27 Furthermore, differences in stability between polymorphs can impact the shelf life of a drug. For example, hygroscopicity of form α of imatinib mesylate, a chemotherapeutic, negatively impacts its storage and processability, and for this reason, its non-hygroscopic form β is solely used for pharmaceutical formulation.28 The importance of polymorphism in the pharmaceutical industry is exacerbated by the possibility of extending intellectual property protection to polymorphic forms of drugs through patenting.28,29
As much as it is important for pharmaceuticals, polymorphism may also influence the properties of functional crystalline materials. Among them, metal–organic polyhedra (MOPs), also referred to as metal–organic cages, coordination nanocages, or nanoballs, constitute a remarkable class of molecular coordination compounds formed by metal ions or clusters and organic ligands.30–33 The arrangement of organic ligands around the metal ions leads to the generation of a void in the center of a coordination polyhedron, which is an emblematic feature of MOPs and a potential gas adsorption site.34–36 This intrinsic porosity, inherent to individual molecules, is further complemented by the extrinsic porosity of their crystals, stemming from MOP molecules not occupying the entire 3D space. Thanks to their permanent porosity, MOPs are considered functional porous materials and can be applied for gas capture and storage,34–37 gas separation,38,39 and heterogeneous catalysis.40 However, unlike other porous materials, MOPs may be soluble in water and organic solvents,41 facilitating their processability and purification with chromatography and recrystallization. Additionally, anionic and cationic MOPs42,43 can act as electrolytes, potentially applicable in electrochemical cells. Solid-state materials comprising of ionic MOPs have been shown to conduct electricity;44–46 however, the electrical conductivity of aqueous MOP solutions has not been studied to date.
In spite of their irregular shapes, research into polymorphism in MOPs has been limited.47–53 True polymorphs of MOPs are unlikely to exist, as such forms would necessitate identical structures and relative numbers of (i) MOP molecules, (ii) counterions balancing the charge of MOP molecules, (iii) solvent molecules coordinated to the metal atoms, and (iv) solvent molecules occluded in the intrinsic and extrinsic pores. Due to this structural complexity, the examples described herein include MOP pseudopolymorphs, which are phases featuring the same metal–organic cage skeletons, but different crystal symmetries and packing. In a pioneering work, Yaghi and coworkers obtained Cu24(m-BDC)24(DMF)14(H2O)10·(H2O)50(DMF)6(C2H5OH)6 (a-MOP-1, m-BDC2− = 1,3-benzenedicarboxylate, DMF = dimethylformamide) in form of anorthic (triclinic) crystals, which, when left in the mother liquor for three months, transformed into cubic crystals of Cu24(m-BDC)24(H2O)24·(H2O)42 (c-MOP-1). During this transformation, the coordinated and occluded solvent molecules underwent exchange, and the 3D arrangement of polyhedra in a-MOP-1 and c-MOP-1 was altered; however, the core structure of the Cu24(m-BDC)24 cage retained its integrity.47 Later, Cohen and coworkers demonstrated the ability to obtain mixtures of a-MOP-1 and c-MOP-1 at desired ratios using a polymeric modulator.48 Additionally, Bloch and coworkers employed another strategy to control MOP crystallization patterns.49 They first used NMR spectroscopy to confirm the identity of Cu4(3,3′-((5-formyl-1,3-phenylene)bis(ethyne-2,1-diyl))dibenzoate)4 in solution and then isolated the MOP in 5 different crystalline forms depending on the solvent used for crystallization and the history of the sample.49 Those mutually transformable solvatomorphs exhibited diverse flexibilities and porosities in their activated states.49 In another work, polymorphs of Cr24(TEI)24 (TEI2− = 5-triisopropylsilylethynylisophthalate) were obtained by tuning the reactant concentration in the starting solution, and were characterized by distinct sorption properties.50 Similarly, (Cu2)2(3,3′-[1,3-benzenediyldi-(ethynyl)]dibenzoate)4(DMF)4 can form either anorthic or monoclinic crystals, which exhibit remarkably different CO2 uptakes,51 while two activated forms of a MOP based on zinc(II) and [3 + 3] calixarene exhibit identical anorthic symmetry but different crystal packings; the two polymorphs show distinct structural flexibilities resulting in their differing potential for hydrogen isotope separation.52 Conversely, activated MUV-27-py-NH2 and its DMF and dichloromethane solvates are all isostructural.53 Although these examples showcase the ability of MOPs to occur in multiple polymorphic forms, it is still unclear, due to the limited amount of experimental data, how these structural differences translate into another important macroscopic property of MOP crystals, namely, their solubilities.
Solubility indicates the number of grams of a chemical substance that is capable of forming a homogeneous mixture (a solution) in 1 liter of a specific solvent. This fundamental macroscopic property has multiple practical implications such as the applicability for separation and purification mentioned earlier, and is also essential while considering the manufacturing, use, and environmental impact of a chemical. Metal–organic polyhedra are often reported without mentioning their solubility; in other cases, general information is provided on whether a given MOP is soluble or insoluble in a given solvent.41 For example, MOP-18 is highly soluble in chloroform, toluene, tetrahydrofuran, ethyl acetate, dimethylacetamide (DMA), and hot DMF, but insoluble in dimethyl sulfoxide, acetonitrile, methanol (MeOH), 1-butanol, and isoamyl alcohol.54 In another example, yes/no solubilities for as many as 15 calixarene-capped MOPs in 8 solvents have been reported.55 Techniques for how to chemically tune the solubility of MOPs have been identified.56 These include the pre-synthetic choice of the ligand,57,58 post-synthetic covalent,59 and coordination functionalization,60 as well as post-synthetic light-induced isomerization.61 As much as it is an essential physical property, there have only been a handful of attempts to determine the numerical values of MOP solubilities.62–65
Herein, we present a novel anionic molybdenum(V)-based MOP occurring in five crystalline forms (BVR-105-109; BVR MOPs discovered at Oregon State University) that differ in (i) crystal packings, (ii) identities of counter-cations, and/or (iii) identities of non-coordinated solvent molecules. These structural variations result in distinct macroscopic properties among the MOP solvatomorphs, including differences in their porosities, and stabilities. Additionally, we report, for the first time, the influence of solvatomorphism on the solubility of the MOP in water, and we identify MOP aqueous solutions as weak electrolytes.
Results and discussion
Molecular structure
The Mo(V)-MOP is an anionic molecule with the chemical formula of [Mo12O12(μ2-O)12(Iso-NH2)12]12− (Iso-NH22− = 5-aminoisophthalate). Structurally, it adopts a triangular antiprism shape with D3d symmetry, capable of accommodating a sphere with a diameter of up to 12 Å (Fig. 1a). The base of the antiprism forms an equilateral triangle with an edge length of 12.5 Å (depicted as orange sticks in Fig. 1b), while the sides of the antiprism consist of isosceles triangles with shorter edges of 10.7 Å (illustrated as red sticks in Fig. 1b). Chemically, the vertices of the antiprism are composed of Mo2-dimers connected by a single bond and two bridging oxo ligands. Each Mo atom is bonded to another oxo ligand through a short double bond (Fig. 1c). The average lengths of the Mo–Mo, Mo—(μ2-O), and Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds are 2.55, 1.93, and 1.68 Å, respectively. The base edges of the antiprism are formed by Iso-NH22− ligands, which coordinate to Mo2-dimers through one O atom from each carboxylate group, while the side faces of the antiprism are completed by intramolecular N–H⋯O hydrogen bonds (Fig. 1d). In contrast, the side edges of the antiprism consist of η2 chelating Iso-NH22− ligands on both sides (Fig. 1e). The MOP molecule interacts with surrounding small counter-cations via ionic and hydrogen bonds (Fig. 1f). Importantly, arrays of small cations block the aperture through which the internal MOP cavity can be accessed (highlighted by three cations on the yellow background in Fig. 1f). This arrangement suggests that the intrinsic porosity of the Mo(V)-MOP may not be readily available for gas adsorption.
O bonds are 2.55, 1.93, and 1.68 Å, respectively. The base edges of the antiprism are formed by Iso-NH22− ligands, which coordinate to Mo2-dimers through one O atom from each carboxylate group, while the side faces of the antiprism are completed by intramolecular N–H⋯O hydrogen bonds (Fig. 1d). In contrast, the side edges of the antiprism consist of η2 chelating Iso-NH22− ligands on both sides (Fig. 1e). The MOP molecule interacts with surrounding small counter-cations via ionic and hydrogen bonds (Fig. 1f). Importantly, arrays of small cations block the aperture through which the internal MOP cavity can be accessed (highlighted by three cations on the yellow background in Fig. 1f). This arrangement suggests that the intrinsic porosity of the Mo(V)-MOP may not be readily available for gas adsorption.
 | ||
| Fig. 1 Structure of the Mo(V)-MOP molecule. (a) Overall view with the central cavity rendered as a yellow sphere. (b) Simplification of the structure into a triangular antiprism. (c) Coordination scheme of the Mo2-dimer. (d) Ligands forming the base of the antiprism (orange rods in panel b) with intramolecular H-bonds shown as fragmented rods. (e) Ligands forming the sides of the antiprism (red rods in panel b). (f) Small counter-cations surrounding the Mo(V)-MOP molecule. Color code: C, light gray; H, dark gray, Mo, green; N, blue; O, red. | ||
The structural characteristics of the Mo(V)-MOP molecule, which remain consistent across all crystalline forms examined in this study, are evidenced by spectra obtained from its stable solid phases. The coordination environment of the Mo2-dimer (Fig. 1c) is typical for Mo in the +V oxidation state, equally identified in a recent report.53 X-ray photoelectron spectra (XPS, Fig. 2a and S1†) further supported this finding, with peaks observed at binding energies of 231.65 eV (Mo 3d5/2) and 234.78 eV (Mo 3d3/2), consistent with Mo(V) compounds reported in the literature.66–68 Fourier-transform infrared (FT-IR) spectra of our Mo(V)-MOP (Fig. 2b and S2†) reveal absorption bands corresponding to the MoO stretch (964 cm−1), Mo—(μ2-O)—Mo anti-symmetric stretch (747 cm−1), and Mo—(μ2-O)—Mo symmetric stretch (460 cm−1).69–71 Importantly, molybdenum(II) acetate [Mo2(OAc)4], the starting material, does not exhibit absorption at these wavenumbers, affirming the specificity of the observed peaks for the Mo(V)-MOP. In the ultraviolet-visible (UV-vis) spectra (Fig. 2c), the Mo(V)-MOP displays strong absorption peaks corresponding to a ligand-to-metal (bonding π of O2− to (dxz, dyz) of Mo) charge–transfer transition (315 nm), and a d–d transition (376 nm), in agreement with previous literature.72 These maxima are notably distinct from those observed for Mo2(OAc)4. Interestingly, upon dissolution of the Mo(V)-MOP in water, the UV-vis peaks undergo a hypsochromic shift to 290 and 350 nm, respectively (Fig. S3†).
 | ||
| Fig. 2 Spectroscopic features of the Mo(V)-MOP molecule. (a) X-ray photoelectron spectra of BVR-107. (b) Fourier-transform infrared and (c) ultraviolet-visible spectra of Mo2(OAc)4, BVR-107, BVR-108, and BVR-109. All sets of spectra were recorded on the respective solids. | ||
Solvatomorphic diversity
The Mo(V)-MOP molecule occurs in five crystalline phases, all synthesized from Mo2(OAc)4 and 5-aminoisophthalic acid (H2Iso-NH2) in various solvent mixtures such as DMF/MeOH, DMA/MeOH, or DEF/MeOH (DEF = diethylformamide) mixtures. Four Mo(V)-based MOP solvatomorphs, named as BVR-105, BVR-107, BVR-108, and BVR-109, were obtained by heating the precursors at 120 °C in sealed vials for 48 h followed by gradual cooling to room temperature. BVR-105 and BVR-107 crystallized as pure phases, whereas BVR-108 and BVR-109 crystallized together in the same vessel. Based on microscopic images, the contribution of BVR-109 to the total number of crystals in the mixture with BVR-108 was estimated to be 30% (Fig. S4†). Additionally, allowing the crystals of BVR-105 to remain in their mother liquor at room temperature for at least one week facilitated their transformation into a fifth Mo(V)-MOP solvatomorph, denoted as BVR-106. Only a few single crystals of BVR-106 could be isolated from each reaction mixture.In BVR-105, BVR-106, and BVR-107, the negative charge of the Mo(V)-MOP molecule is balanced by dimethylammonium (dma+) cations, while BVR-108 and BVR-109 incorporate diethylammonium (dea+) cations instead. Consistent with previous literature reports,43,73,74 we consider these small solvent-derived cations as guests in the lattice of large Mo(V)-MOP anions, rather than integral parts of binary salts. This interpretation allows us to treat the (dma)12[Mo(V)-MOP] and (dea)12[Mo(V)-MOP] phases as solvatomorphs of the Mo(V)-MOP molecule, which dominates the crystal lattice. Additionally, solution 1H NMR spectra (Fig. S5†) confirmed the co-crystallization of Mo(V)-MOP solvatomorphs with non-coordinated amide solvent molecules, contributing to the chemical compositions of the phases, reported in Table 1.
| BVR-105 | BVR-106 | BVR-107 | BVR-108 | BVR-109 | |
|---|---|---|---|---|---|
| Reaction medium | DMF, MeOH | DMF, MeOH | DMA, MeOH | DEF, MeOH | DEF, MeOH |
| Photomicrograph |

|

|

|

|

|
| Molecular formula | (dma)12[Mo12O12(μ2-O)12(Iso-NH2)12]·13.5 DMF | (dma)12[Mo12O12(μ2-O)12(Iso-NH2)12]·8.3 DMF | (dma)12[Mo12O12(μ2-O)12(Iso-NH2)12]·11 DMA | (dea)12[Mo12O12(μ2-O)12(Iso-NH2)12]·9.5 DEF | (dea)12[Mo12O12(μ2-O)12(Iso-NH2)12]·15.2 DEF |
| Formula weight (Da) | 5224.96 | 4844.89 | 5196.56 | 5535.82 | 6112.37 |
| Crystal system | Monoclinic | Hexagonal | Orthorhombic | Monoclinic | Triclinic |
| Space group | P21/n | P6/m | Cmce | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 23.4316(3) | 20.6212(3) | 37.4589(18) | 23.4926(3) | 18.9295(7) |
| b (Å) | 20.2535(3) | 20.6212(3) | 29.3487(9) | 21.0361(2) | 20.8274(3) |
| c (Å) | 24.6419(3) | 15.9529(3) | 20.1896(6) | 24.7119(3) | 31.7770(8) |
| α (°) | 90 | 90 | 90 | 90 | 84.010(2) |
| β (°) | 104.0860(10) | 90 | 90 | 103.6430(10) | 86.075(3) |
| γ (°) | 90 | 120 | 90 | 90 | 80.923(2) |
| V (Å3) | 11342.7(3) |
5874.9(2) | 22195.8(14) |
11867.9(2) |
12286.5(6) |
| Z, Z′ | 2, 1/2 | 1, 1/12 | 4, 1/4 | 2, 1/2 | 2, 1 |
| Symmetry of molecule in crystal (molecular symmetry = D3d) | C i | C 3i | C 2h | C i | C i |
| Stability in air | Hygroscopic | Hygroscopic | Bench-stable | Bench-stable | Bench-stable |
| BET surface area (m2 g−1) | N/A | N/A | 12 | 33 | 9 |
The (dma)12[Mo(V)-MOP] phases exhibit either monoclinic, hexagonal, or orthorhombic symmetry, while the (dea)12[Mo(V)-MOP] phases display monoclinic or triclinic symmetry (Table 1). Schematic representations of their crystal packings are shown in Fig. 3, illustrating unit-cell edges and the centroids of the Mo(V)-MOP molecules. In this figure, it is evident that the packings of BVR-105, BVR-107, or BVR-108 are characterized by the same number of nearest neighbors of each molecule (eight), while the number of nearest neighbors in BVR-106 and BVR-109 is six. Furthermore, the packings of BVR-105 ((dma)12[Mo(V)-MOP]) and BVR-108 ((dea)12[Mo(V)-MOP]) display a striking geometric similarity. Next, using the scheme shown in Fig. 3, we distinguish crystallographic planes where Mo(V)-MOP molecules align parallel to each other, facilitating 2D projections of the 3D structures. In BVR-106, this alignment occurs along the (001) plane (depicted in orange), while in BVR-109, it is along the (101) plane (also depicted in orange). In BVR-105 and BVR-108, these alignments occur along mutually orthogonal (101) and (01) planes, depicted respectively in green and red. In BVR-107, the alignments occur along mutually orthogonal (010) and (100) planes, also depicted respectively in green and red. These 2D projections are further visualized in Fig. 4, which presents explicit representations of the Mo(V)-MOP rather than symbolic centroids. The 2D projections of BVR-105 and BVR-108 reveal interactions between neighboring Mo(V)-MOP molecules dominated by edge-to-face N–H⋯O(C–O) and edge-to-edge N–H⋯O(Mo) hydrogen bonds. Conversely, Mo(V)-MOP molecules stacked along [010] do not form short contacts due to the presence of void space between them. The packing in BVR-107 exhibits remarkably similar characteristics. The molecules within this lattice adopt a C2h symmetry, which is higher than the Ci symmetry displayed by BVR-105 and BVR-108, yet lower than the D3d symmetry of the ideal molecule. The angle between the two orthogonal planes is strictly 90° (compared to 87° for BVR-105 and BVR-108). Apart from these minor differences, the packing scheme and the dominant interactions in BVR-107 mirror those observed in BVR-105 and BVR-108. On the contrary, the packings of BVR-106 and BVR-109 are strikingly different. The Mo(V)-MOP molecules populate the planes of densest packing [(001) in BVR-106 and (101) in BVR-109], where each molecule has 6 neighbors (compared to 4 neighbors in each plane for the other solvatomorphs described earlier), interacting through N–H⋯(μ2-O) hydrogen bonds. However, in directions perpendicular to these planes, the molecules are spaced apart, creating voids filled with heavily disordered solvent molecules that could not be localized from the diffraction data. The differences between the structures of BVR-106 and BVR-109 include different symmetries of the Mo(V)-MOP molecule in the crystal lattice (C3i and Ci, respectively), along with a shift between the consecutive planes of densest packing observed only in BVR-109.
 | ||
| Fig. 3 Scheme of the crystal packing of the five Mo(V)-MOP solvatomorphs. Positions of centroids of the Mo(V)-MOP molecules, represented as gray spheres, are shown with respect to the unit cells of the five solvatomorphs. Symmetrically independent distances between the centroids of neighboring Mo(V)-MOP molecules are given in angstroms. | ||
 | ||
| Fig. 4 Crystal packing of the five Mo(V)-MOP solvatomorphs viewed in two orthogonal planes. The planes correspond to those shown in Fig. 3 in 3D. Arrays of intermolecular MOP⋯MOP hydrogen bonds between neighboring Mo(V)-MOP molecules are shown to the right. Color code: C, light gray; H, dark gray, Mo, green; N, blue; O, red. The molecules and the H-bonds of interest are emphasized in orange. | ||
The purity of BVR-105 and BVR-107 allowed for the investigation of these phases in bulk, while a slight difference in the densities of BVR-108 and BVR-109 enabled the mechanical separation of their crystals. Pawley refinements of the PXRD patterns recorded on crystals ground in the presence of amide solvents (Fig. S6 and Table S1†) confirmed the bulk phase purity. The powders exhibited lower symmetries and different unit-cell angles, yet comparable unit-cell lengths and volumes to those derived from the respective single crystals (Table 1). It is likely that the Mo(V)-MOP crystals underwent plastic deformation upon grinding; however, their periodicity, determined by the size of individual molecules and their arrangement, remained intact. When activated under vacuum at 120 °C for 12 h, the crystallinity of the BVR-107, BVR-108, and BVR-109 powders decreased significantly (Fig. S7†), while BVR-105 and BVR-106 could not be isolated as dry powders since they dissolved in water absorbed from the air.
Origin of solvatomorphism
The formation of five different Mo(V)-MOP solvatomorphs originating from the same metal-and-ligand system involving Mo2(OAc)4 and H2Iso-NH2 stems from the varying synthetic conditions employed in our study. Notably, the pivotal role of time and temperature as synthetic parameters is evident, particularly illustrated by the formation of BVR-106, which occurs only when crystals of BVR-105 are left in their mother liquor at room temperature for an extended period. Additionally, the choice of solvent medium is another important synthetic parameter influencing the crystalline form of the product (Table 1). The role of DMF, DMA, or DEF in the synthesis of the Mo(V)-MOP solvatomorphs is two-fold: they facilitate the dissolution of reactants for effective combination and act as a source of dma+ and dea+ cations that balance the charge of the anionic Mo(V)-MOPs. A vast body of previous literature suggests that DMF hydrolyzes faster than DMA (Table S2†). At the same time, computational predictions suggest that complex processes involving constituent steps proceeding at different rates may lead to different resultant MOPs.75 Therefore, in our study, the varying hydrolysis rates of DMF, DMA, and DEF, which supply the system with dma+ or dea+, likely contribute to the diverse crystalline packings observed in the resultant Mo(V)-MOP variations. Unlike the amides, MeOH acts as an anti-solvent during the synthesis of Mo(V)-MOP, triggering its precipitation. We produced spectroscopic evidence that the Mo(V)-MOP molecules are formed in MeOH-free amide solutions without precipitation immediately after the reactants have been mixed. This observation is in line with previous studies on MOP formation kinetics: Cu12(Iso-OH)12 (Iso-OH2− = 5-hydroxyisophthalate) is formed in solution in tens of seconds,76 while the formation of Pd6(1,3,5-tris(4-pyridylmethyl)benzene)8 concludes within 5 min.77 Conversely, MOP crystallization processes typically require longer time frames; for instance, Mo24(Iso-OH)24 requires 12 days of heating at 85 °C to crystallize.78 In our study, freshly prepared reaction mixtures exhibit an orange color similar to that of the Mo(V)-MOP crystals (Fig. S8†). Furthermore, FT-IR (Fig. S9†) and UV-vis (Fig. S10†) spectra of these mixtures reveal characteristic peaks at 965, 760, 480 cm−1 (FT-IR), and 280–290, 350 nm (UV-vis), which are consistent with those observed for the Mo(V)-MOP (Fig. 2b, S2 and S3†). Heating the reaction mixtures at 120 °C does not result in any subsequent changes in the spectra.Stability
The solvatomorphs of Mo(V)-MOP exhibit strikingly diverse stabilities. BVR-105 and BVR-106 display hygroscopic behavior and, when separated from their mother liquors, uptake moisture from the air reaching a complete dissolution within minutes (thus limiting our solid-state characterization capabilities for these phases). Interestingly, when crystals of BVR-105 were left in the mother liquor for one week and then subjected to microscopic observation, a few crystals of BVR-106 were detected, and then, their number relative to BVR-105 increased over time. This observation suggests that BVR-105 spontaneously transforms into BVR-106, which is potentially driven by the greater stability of the latter. Density functional theory (DFT) calculations corroborated this observation, indicating that the BVR-106 phase possesses a lower total energy (Fig. 5). In contrast to the deliquescent nature of BVR-105 and BVR-106, all other solvatomorphs (BVR-107, BVR-108, and BVR-109) exhibit non-hygroscopic behavior, remaining bench-stable at room temperature for at least 5 months. Among these, BVR-107, the only bench-stable phase in the (dma)12[Mo(V)-MOP] family, was found to have the highest DFT-calculated relative total energy (Fig. 5), indicating that it is metastable under these conditions. In the (dea)12[Mo(V)-MOP] family, the PXRD patterns of the as-made and activated BVR-108 align, whereas BVR-109 nearly amorphized upon activation (Fig. S7†). Consistent with these findings, DFT calculations showed that BVR-108 is lower in energy than BVR-109, indicating that the former is thermodynamically favored (Fig. 5). | ||
| Fig. 5 Relative total energies of the (dma)12[Mo(V)-MOP] (left) and (dea)12[Mo(V)-MOP] (right) solvatomorphs calculated with DFT. Values expressed in kJ per mol of formula units (see Table 1). | ||
Two factors influencing the stability of the solvatomorphs can be inferred. First, the nature of the solvent molecules occupying the extrinsic pores is crucial; phases crystallized from DMF/MeOH exhibit hygroscopic behavior, whereas those derived from DMA/MeOH or DEF/MeOH are bench-stable. Second, the formal substitution of dma+ for dea+ within the lattice of the anionic Mo(V)-MOP transforms the hygroscopic phase (BVR-105) into a stable form (BVR-108), despite maintaining identical symmetry and packing. It is noteworthy that the stability observed in BVR-107, BVR-108, and BVR-109 can be disrupted by outgassing at 120 °C for 12 h carried out with the aim of pore activation. Despite their high extrinsic porosities anticipated from crystal structures, porosity measurements revealed relatively low but discernible gas uptakes (Fig. S11†) and BET surface areas (Table 1). BVR-107, BVR-108, and BVR-109 exhibit slight variations in thermal stability (Fig. S12†), and exposure to an extreme temperature of 600 °C results in the loss of Mo(V)-MOP chemical identity, as evidenced by the disappearance of characteristic FT-IR peaks (Fig. S13†).
Solubility and electrical conductivity in solution
The outstanding property of the Mo(V)-MOP is its solubility in water, ethanol, and DMF. Each of the bench-stable phases of the Mo(V)-MOP (Table 1) displays a distinct solubility in water (Fig. 6a). Comparative analysis with previously reported monomorphic MOP solubilities in water indicates that the solubility values for BVR-107, BVR-108, and BVR-109 are 100-1000 times greater than those of Zr-based MOPs, yet lower than those of certain Fe- and Cu-MOPs (Table S3†).62–64 Among the tested bench-stable phases, BVR-109 showed the highest value of 14.41(10) g L−1 followed by that of BVR-108, 10.25(7) g L−1, while 4.30(2) g L−1, the value for BVR-107, was the lowest one. This variance in solubility underscores the impact of solvatomorphism on MOP properties, presenting opportunities for tailored applications in aqueous environments. Spectroscopic data strongly suggest the integrity of Mo(V)-MOP cages is preserved in aqueous solutions. The 1H NMR spectra of BVR-107 and BVR-108 (Fig. S5†) show resonances in the aromatic region with chemical shifts markedly different from those of free H2Iso-NH2 (Fig. S14 and S15†). Furthermore, diffusion-ordered NMR spectra (DOSY, Fig. S16†) show that dma+ cations diffuse uniformly with the anionic Mo(V)-MOP cage in BVR-107, and the same holds true for dea+ and the Mo(V)-MOP in BVR-108.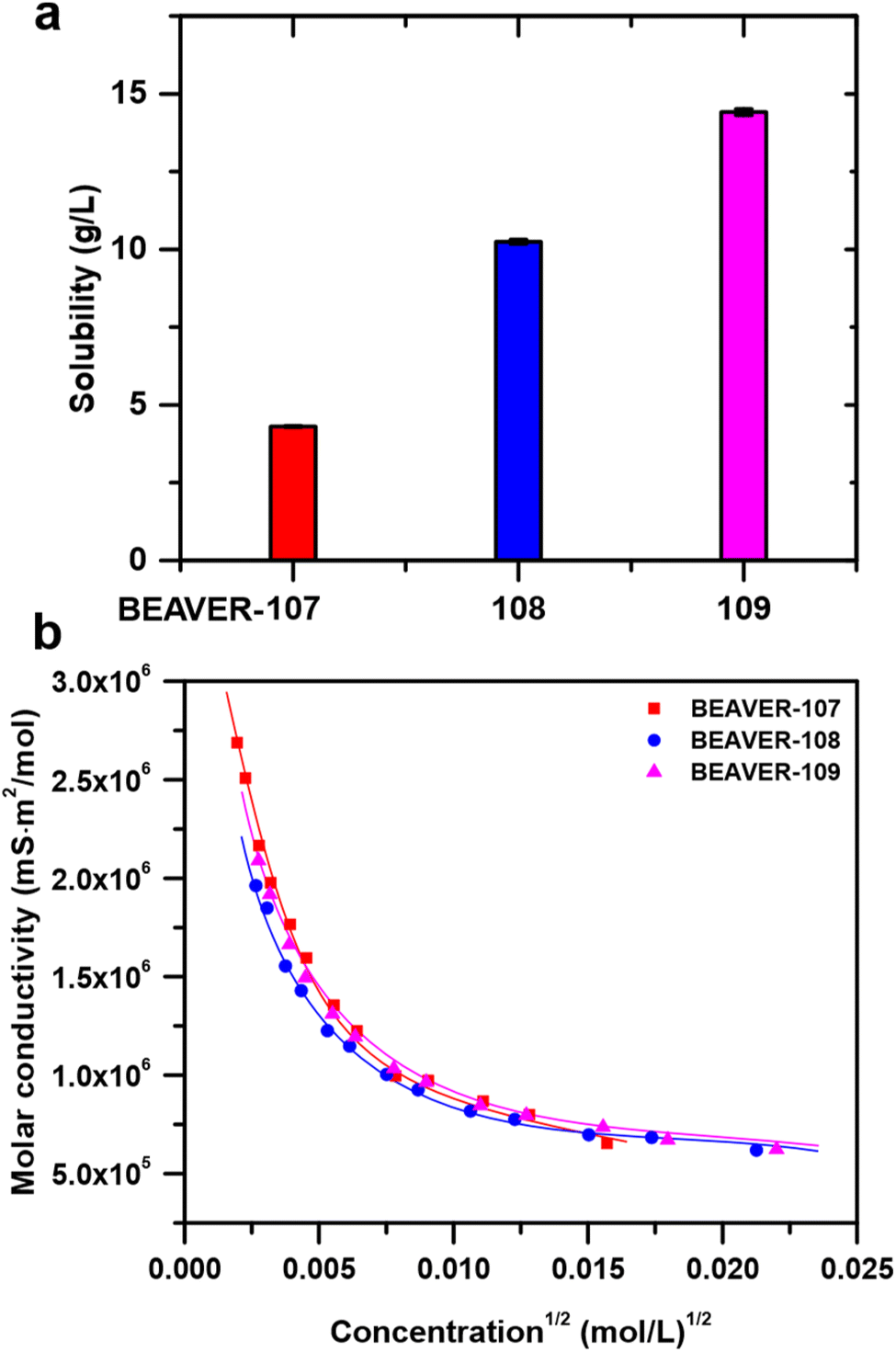 | ||
| Fig. 6 (a). Solubilities of Mo(V)-MOP solvatomorphs in water. (b) Molar conductivities of Mo(V)-MOP solvatomorphs dissolved in water plotted as a function of square root of molar concentration. | ||
Upon dissolution of Mo(V)-MOP phases, the molar conductivities sharply increase with dilution (Fig. 6b), deviating from Kohlrausch's law and classifying the Mo(V)-MOP as a weak electrolyte. Notably, the molar conductivities of BVR-107, BVR-108, and BVR-109 nearly overlap at each concentration (Fig. 6b), suggesting that while the physical properties of the Mo(V)-MOP solvatomorphs differ in the solid state, they are remarkably similar in solution. This observation leads us to hypothesize that the spatial arrangement of [Mo12O12(μ2-O)12(Iso-NH2)12]12− and dma+/dea+ ions in solution is random, resulting in nearly identical behavior for every dissolved Mo(V)-MOP phase. Fitting the values of molar conductivity to Ostwald's dilution law (Fig. S17†) reveals extremely high values of limiting molar conductivities and pKa values relevant to weak acids (Table 2).
| Limiting molar conductivity (mS m2 mol−1) | pKa | |
|---|---|---|
| BVR-107 | 3.76 × 106 | 5.19 |
| BVR-108 | 3.38 × 106 | 5.24 |
| BVR-109 | 3.21 × 106 | 5.06 |
Conclusions
The discovery of the novel Mo(V)-based metal–organic polyhedron [Mo12O12(μ2-O)12(Iso-NH2)12]12−, which co-crystallizes with either dma+ or dea+ ions, and DMF, DMA, or DEF solvent molecules yielding five solvatomorphic forms, contributes to the gradually growing group of MOPs exhibiting (pseudo)polymorphism.47–53 This phenomenon, as previously observed in other (pseudo)polymorphic varieties of MOPs,49–52 translates into significant differences in key physical properties such as porosities, and, most importantly, stabilities among the Mo(V)-MOP solvatomorphs. Among the (dma)12[Mo(V)-MOP] phases, orthorhombic BVR-107 synthesized in DMA/MeOH was found to be the only isolable phase, while the other two solvatomorphs (hexagonal BVR-106 and monoclinic BVR-105 that slowly transforms to it, both synthesized in DMF/MeOH) were hygroscopic and deliquescent. In turn, among the (dea)12[Mo(V)-MOP] phases synthesized in DEF/MeOH, monoclinic BVR-108 and triclinic BVR-109 were both found to be bench-stable. DFT calculations and observation of phase transformations were instrumental in assessing their thermodynamic stability. Our study also uniquely demonstrates, for the first time, the influence of solvatomorphism on the solubility of MOP crystals in water, which ranges from 4 to nearly 15 g L−1. Moreover, we have shown, also for the first time, that aqueous solutions of MOPs act as weak electrolytes, which, in turn, can potentially form buffer solutions relevant to chemical synthesis, electrochemical cells, and biological systems. Our current contribution represents an initial step toward understanding how to tune the MOPs solubility taking advantage of their (pseudo)polymorphism and how to make use of this knowledge in practical applications.Data availability
All data are available within the article (and its ESI†) and from the corresponding authors, A.G. and K.C.S., upon reasonable request.Author contributions
A. K. Y. performed all the experiments and analyzed the experimental data. A. G. refined the crystal structures, drew the figures, and wrote the manuscript with input from all the authors. E. H. W, A. M. G., and K. E. J. ran the DFT calculations. B. S.-G. and D. K. assisted in the interpretation of the electrical conductivity data. K. C. S. secured funding and coordinated the project.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
K. C. S. thanks the Department of Chemistry at Oregon State University (OSU) for support through start-up funding and the College of Science Teacher Scholar Award. A. K. Y., A. G., and K. C. S. thank Dr Lev Zakharov for their help with X-ray crystallography. K. E. J. thanks the EPSRC for funding (EP/V026887/1). We acknowledge computational resources and support provided by the Imperial College Research Computing Service. The authors acknowledge the Murdock Charitable Trust (Grant SR-2017297) for funding the acquisition of the single-crystal X-ray diffractometer and to the NMR facility at OSU for their invaluable support in experimental design.References
- J. Bernstein, Polymorphism in Molecular Crystals, Clarendon Press, Oxford, 2002 Search PubMed.
- S. C. Abrahams, J. M. Robertson and J. G. White, Acta Crystallogr., 1949, 2, 233–238 CrossRef CAS.
- F. P. A. Fabbiani, D. R. Allan, S. Parsons and C. R. Pulham, Acta Crystallogr., B, 2006, 62, 826–842 CrossRef PubMed.
- S. C. Abrahams, J. M. Robertson and J. G. White, Acta Crystallogr., 1949, 2, 238–244 CrossRef CAS.
- R. D. Lowde, D. C. Phillips and R. G. Wood, Acta Crystallogr., 1953, 6, 553–556 CrossRef CAS.
- D. Phillips, Acta Crystallogr., 1956, 9, 237–250 CrossRef CAS.
- D. C. Phillips, F. R. Ahmed and W. H. Barnes, Acta Crystallogr., 1960, 13, 365–377 CrossRef CAS.
- D. Musumeci, C. A. Hunter and J. F. McCabe, Cryst. Growth Des., 2010, 10, 1661–1664 CrossRef CAS.
- P. W. Stephens, E. Schur, S. H. Lapidus and J. Bernstein, Acta Crystallogr., E, 2019, 75, 489–491 CAS.
- E. V. Boldyreva, V. A. Drebushchak, T. N. Drebushchak, I. E. Paukov, Y. A. Kovalevskaya and E. S. Shutova, J. Therm. Anal. Calorim., 2003, 73, 409–418 CrossRef CAS.
- S. B. Hendricks, J. Am. Chem. Soc., 1928, 50, 2455–2464 CrossRef CAS.
- A. Olejniczak, K. Ostrowska and A. Katrusiak, J. Phys. Chem. C, 2009, 113, 15761–15767 CrossRef CAS.
- K. Roszak and A. Katrusiak, J. Phys. Chem. C, 2017, 121, 778–784 CrossRef CAS.
- K. Dziubek, M. Citroni, S. Fanetti, A. B. Cairns and R. Bini, J. Phys. Chem. C, 2017, 121, 2380–2387 CrossRef CAS.
- D. Paliwoda, K. F. Dziubek and A. Katrusiak, Cryst. Growth Des., 2012, 12, 4302–4305 CrossRef CAS.
- E. Patyk, J. Skumiel, M. Podsiadło and A. Katrusiak, Angew. Chem., Int. Ed., 2012, 51, 2146–2150 CrossRef CAS.
- E. Schur, J. Bernstein, A. Lemmerer and R. Vainer, Acta Crystallogr., E, 2011, 67, o2761 CrossRef CAS PubMed.
- D. Cinčić, T. Friščić and W. Jones, CrystEngComm, 2011, 13, 3224–3231 RSC.
- A. Nangia and G. R. Desiraju, Chem. Commun., 1999, 605–606 RSC.
- A. Nangia, Cryst. Growth Des., 2006, 6, 2–4 CrossRef CAS.
- N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Deliv. Rev., 2007, 59, 617–630 CrossRef CAS PubMed.
- P. Groth, in Chemische Krystallographie, Engelmann, Leipzig, 1919, vol. 4, pp. 240–241 Search PubMed.
- M. Haisa, S. Kashino, R. Kawai and H. Maeda, Acta Crystallogr., B, 1976, 32, 1283–1285 CrossRef.
- G. Nichols and C. S. Frampton, J. Pharm. Sci., 1998, 87, 684–693 CrossRef CAS.
- M. Haisa, S. Kashino and H. Maeda, Acta Crystallogr., B, 1974, 30, 2510–2512 CrossRef.
- P. Di Martino, A. M. Guyot-Hermann, P. Conflant, M. Drache and J. C. Guyot, Int. J. Pharm., 1996, 128, 1–8 CrossRef.
- Y.-T. Sohn, J. Kor. Pharm. Sci., 1990, 20, 97–103 CAS.
- Novartis AG, US Pat., 6894051B1, 2005 Search PubMed.
- BASF SE, US Pat., 8063092B2, 2011 Search PubMed.
- D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS PubMed.
- H. Vardhan, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 306, 171–194 CrossRef CAS.
- A. Bavykina, A. Cadiau and J. Gascon, Coord. Chem. Rev., 2019, 386, 85–95 CrossRef CAS.
- S. Lee, H. Jeong, D. Nam, M. S. Lah and W. Choe, Chem. Soc. Rev., 2021, 50, 528–555 RSC.
- A. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987–9014 CrossRef CAS PubMed.
- A. Carné-Sánchez, J. Martínez-Esaín, T. Rookard, C. J. Flood, J. Faraudo, K. C. Stylianou and D. Maspoch, ACS Appl. Mater. Interfaces, 2023, 15, 6747–6754 CrossRef PubMed.
- K. Kiaei, K. Brunson, A. Gładysiak, K. Smith, K. Hunter, A. Thomas, D. Radke, T. Zuehlsdorff and K. C. Stylianou, J. Mater. Chem. A, 2023, 11, 14265–14271 RSC.
- G. R. Lorzing, A. J. Gosselin, B. A. Trump, A. H. P. York, A. Sturluson, C. A. Rowland, G. P. A. Yap, C. M. Brown, C. M. Simon and E. D. Bloch, J. Am. Chem. Soc., 2019, 141, 12128–12138 CrossRef CAS PubMed.
- M. A. Andrés, A. Carné-Sánchez, J. Sánchez-Laínez, O. Roubeau, J. Coronas, D. Maspoch and I. Gascón, Chem.–Eur. J., 2020, 26, 143–147 CrossRef PubMed.
- I. Tejedor, M. A. Andrés, A. Carné-Sánchez, M. Arjona, M. Pérez-Miana, J. Sánchez-Laínez, J. Coronas, P. Fontaine, M. Goldmann, O. Roubeau, D. Maspoch and I. Gascón, ACS Appl. Mater. Interfaces, 2022, 14, 27495–27506 CrossRef CAS PubMed.
- H. Vardhan and F. Verpoort, Adv. Synth. Catal., 2015, 357, 1351–1368 CrossRef CAS.
- E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480–13544 CrossRef PubMed.
- M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469–471 CrossRef CAS.
- D. L. Caulder, R. E. Powers, T. N. Parac and K. N. Raymond, Angew. Chem., Int. Ed., 1998, 37, 1840–1843 CrossRef CAS.
- J. Lee, D.-W. Lim, S. Dekura, H. Kitagawa and W. Choe, ACS Appl. Mater. Interfaces, 2019, 11, 12639–12646 CrossRef CAS PubMed.
- L. Li, L. Shan, A. M. Sheveleva, M. He, Y. Ma, Y. Zhou, M. Nikiel, L. Lopez-Odriozola, L. S. Natrajan, E. J. L. McInnes, M. Schröder, S. Yang and F. Tuna, Mater. Adv., 2023, 4, 1941–1948 RSC.
- Y. J. Kim, S. Y. Ko, S. Kim, K. M. Choi and W.-H. Ryu, Small, 2023, 19, 2206561 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, J. B. Wachter, H. K. Chae, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4368–4369 CrossRef CAS PubMed.
- T.-H. Chen, L. Wang, J. V. Trueblood, V. H. Grassian and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 9646–9654 CrossRef CAS PubMed.
- W. M. Bloch, R. Babarao and M. L. Schneider, Chem. Sci., 2020, 11, 3664–3671 RSC.
- J. Park, Z. Perry, Y.-P. Chen, J. Bae and H.-C. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 28064–28068 CrossRef CAS PubMed.
- M. Jaya Prakash, M. Oh, X. Liu, K. N. Han, G. H. Seong and M. S. Lah, Chem. Commun., 2010, 46, 2049–2051 RSC.
- D. He, L. Zhang, T. Liu, R. Clowes, M. A. Little, M. Liu, M. Hirscher and A. I. Cooper, Angew. Chem., Int. Ed., 2022, 61, e202202450 CrossRef CAS PubMed.
- S. Delaporte, I. Abánades Lázaro, J. López-Cabrelles, E. C. Mazarakioti, S. Chebourou, I. J. Vitórica-Yrezábal, M. Giménez-Marqués and G. Mínguez Espallargas, Dalton Trans., 2023, 52, 15682–15687 RSC.
- H. Furukawa, J. Kim, K. E. Plass and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 8398–8399 CrossRef CAS PubMed.
- M. R. Dworzak, M. M. Deegan, G. P. A. Yap and E. D. Bloch, Inorg. Chem., 2021, 60, 5607–5616 CrossRef CAS PubMed.
- M. M. Deegan, A. M. Antonio, G. A. Taggart and E. D. Bloch, Coord. Chem. Rev., 2021, 430, 213679 CrossRef CAS.
- G. Liu, Z. Ju, D. Yuan and M. Hong, Inorg. Chem., 2013, 52, 13815–13817 CrossRef CAS PubMed.
- S. Mollick, S. Mukherjee, D. Kim, Z. Qiao, A. V. Desai, R. Saha, Y. D. More, J. Jiang, M. S. Lah and S. K. Ghosh, Angew. Chem., Int. Ed., 2019, 58, 1041–1045 CrossRef CAS PubMed.
- G. A. Taggart, A. M. Antonio, G. R. Lorzing, G. P. A. Yap and E. D. Bloch, ACS Appl. Mater. Interfaces, 2020, 12, 24913–24919 CrossRef CAS PubMed.
- A. Carné-Sánchez, J. Albalad, T. Grancha, I. Imaz, J. Juanhuix, P. Larpent, S. Furukawa and D. Maspoch, J. Am. Chem. Soc., 2019, 141, 4094–4102 CrossRef PubMed.
- J. Park, L.-B. Sun, Y.-P. Chen, Z. Perry and H.-C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 5842–5846 CrossRef CAS PubMed.
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS PubMed.
- J. Liu, C. R. P. Fulong, L. Hu, L. Huang, G. Zhang, T. R. Cook and H. Lin, J. Membr. Sci., 2020, 606, 118122 CrossRef CAS.
- M. G. Sullivan, G. E. Sokolow, E. T. Jensen, M. R. Crawley, S. N. MacMillan and T. R. Cook, Dalton Trans., 2023, 52, 338–346 RSC.
- C. R. P. Fulong, J. Liu, V. J. Pastore, H. Lin and T. R. Cook, Dalton Trans., 2018, 47, 7905–7915 RSC.
- A. Khademi, R. Azimirad, A. A. Zavarian and A. Z. Moshfegh, J. Phys. Chem. C, 2009, 113, 19298–19304 CrossRef CAS.
- C. Wu and J. Li, ACS Appl. Mater. Interfaces, 2017, 9, 41314–41322 CrossRef CAS PubMed.
- J. Wang, P. Cai, L. Jia, D. Yao, X. Xue, D. Li, L. Wang, Z. Lu, F. Hu and X. Zhang, J. Electron. Mater., 2022, 51, 1850–1856 CrossRef CAS.
- M. L. Larson and F. W. Moore, Inorg. Chem., 1963, 2, 881–882 CrossRef CAS.
- J. I. Dulebohn, T. C. Stamatakos, D. L. Ward and D. G. Nocera, Polyhedron, 1991, 10, 2813–2820 CrossRef CAS.
- J. Luo and L. Xu, Inorg. Chem., 2006, 45, 11030–11034 CrossRef CAS PubMed.
- P. C. H. Mitchell, J. Inorg. Nucl. Chem., 1963, 25, 963–970 CrossRef CAS.
- R. W. Saalfrank, R. Burak, A. Breit, D. Stalke, R. Herbst-Irmer, J. Daub, M. Porsch, E. Bill, M. Müther and A. X. Trautwein, Angew Chem. Int. Ed. Engl., 1994, 33, 1621–1623 CrossRef.
- S. Mann, G. Huttner, L. Zsolnai and K. Heinze, Angew Chem. Int. Ed. Engl., 1996, 35, 2808–2809 CrossRef CAS.
- X. Zhang, S. Takahashi, K. Aratsu, I. Kikuchi, H. Sato and S. Hiraoka, Phys. Chem. Chem. Phys., 2022, 24, 2997–3006 RSC.
- R. W. Larsen, J. Am. Chem. Soc., 2008, 130, 11246–11247 CrossRef CAS PubMed.
- D. K. Chand, K. Biradha, M. Fujita, S. Sakamoto and K. Yamaguchi, Chem. Commun., 2002, 2486–2487, 10.1039/B206625B.
- J.-R. Li, A. A. Yakovenko, W. Lu, D. J. Timmons, W. Zhuang, D. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2010, 132, 17599–17610 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2368993–2368997. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05037a |
| This journal is © The Royal Society of Chemistry 2025 |