
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of fully synthetic FKBP12-mTOR molecular glues†
Robin C. E.
Deutscher‡
a,
Christian
Meyners‡
a,
Maximilian L.
Repity
a,
Wisely Oki
Sugiarto
a,
Jürgen M.
Kolos
a,
Edvaldo V. S.
Maciel
a,
Tim
Heymann
a,
Thomas M.
Geiger
a,
Stefan
Knapp
 bcd,
Frederik
Lermyte
a and
Felix
Hausch
*ae
bcd,
Frederik
Lermyte
a and
Felix
Hausch
*ae
aInstitute for Organic Chemistry and Biochemistry, Technical University Darmstadt, Peter-Grünberg-Straße 4, 64287 Darmstadt, Germany. E-mail: felix.hausch@tu-darmstadt.de
bInstitut für Pharmazeutische Chemie, Goethe-University Frankfurt, Biozentrum, Max-von-Laue-Str. 9, 60438 Frankfurt am Main, Germany
cStructural Genomics Consortium, Goethe-University Frankfurt, Buchmann Institute for Life Sciences, Max-von-Laue-Str. 15, 60438 Frankfurt am Main, Germany
dGerman Cancer Consortium (DKTK)/German Cancer Research Center (DKFZ), DKTK Site Frankfurt-Mainz, 69120 Heidelberg, Germany
eCentre for Synthetic Biology, Technical University of Darmstadt, 64287 Darmstadt, Germany
First published on 13th January 2025
Abstract
Molecular glues are a new drug modality with the potential to engage otherwise undruggable targets. However, the rational discovery of molecular glues for desired targets is a major challenge and most known molecular glues have been discovered by serendipity. Here we present the first fully synthetic FKBP12-mTOR molecular glues, which were discovered from a FKBP-focused, target-unbiased ligand library. Our biochemical screening of >1000 in-house FKBP ligands yielded one hit that induced dimerization of FKBP12 and the FRB domain of mTOR. The crystal structure of the ternary complex revealed that the hit targeted a similar surface on the FRB domain compared to natural product rapamycin but with a radically different interaction pattern. Structure-guided optimization improved potency 500-fold, and led to compounds which initiate FKBP12-FRB complex formation in cells. Our results show that molecular glues targeting flat surfaces can be discovered by focused screening and support the use of FKBP12 as a versatile presenter protein for molecular glues.
Introduction
For a long time, intracellular proteins without suitable ligand binding pockets have been considered undruggable. The discovery of molecular glues as a drug modality challenged that notion.1 Through the help of an additional protein – a presenter protein – the available binding surface of the molecular glue–protein-complex can become large enough to bind even flat, featureless target protein surfaces with high affinity.2–4 If the presenter protein is an E3 ligase, the degradation of the target protein through the proteasome machinery can be enabled, providing molecular glue degraders.5,6 Unfortunately, both molecular glues and molecular glue degraders are still largely discovered by serendipity as approaches to identify them by a more rational strategy are rare.7 The first and most prominent examples for the serendipitous discovery of molecular glues are the clinically used immunosuppressants rapamycin 1, FK506 2 and cyclosporin A 3 (Fig. 1).8–10 Being among the first of their kind, their cellular function was discovered first, followed by the identification of the presenter protein, and finally, the target protein itself. | ||
| Fig. 1 Natural product molecular glues rapamycin 1, FK506 2, cyclosporine A 3 and WDB002 4. FKBP12 or Cyp18 binding moieties are highlighted in slate blue. | ||
FK506 2 and cyclosporin A 3 are now known to bind to FKBP12 (FK506 binding protein 12) and cyclophilin 18 (Cyp18), respectively, and their binary complexes bind to calcineurin, blocking access to its substrate binding site.11–13 Rapamycin 1 binds to FKBP12 and then their complex binds to the FRB (FKBP-rapamycin 1 binding) domain of mTOR (mechanistic target of rapamycin 1), thereby inhibiting functions of the mTORC1 complex.14
FKBP12 and Cyp18 might be preferred presenter proteins as nature used them repeatedly for molecular glues, with additional examples being WDB002 4 (Fig. 1), inducing FKBP12-CEP250 complexes,15 and sanglifehrin A, which was shown to induce Cyp18-IMPDH2 complexes.16,17 For the natural product Antascomicin B,18 we recently showed that it can stabilize the interaction between the larger FKBP51 and the kinase Akt.19 Furthermore, there are several other FKBP12-binding natural products (e.g. Meridamycin)20 that can be considered orphan molecular glues, as their postulated ternary target proteins have not yet been identified.21 Recently, rapamycin 1 analog libraries (rapafucins) have been developed by Liu and coworkers22 as potential synthetic FKBP-based molecular glues, which led to inhibitors for hENT1,22 GLUT1,23 and PAANIB-1.24 Based on early work by WarpDriveBio, the company Revolution Medicines developed the Cyp18-based covalent-reactive KRASG12C inhibitors RMC-4998 and RMC-6291,25 with the latter currently being investigated in a phase I clinical trial (NCT05462717).26 Based on the scaffold of RMC-6291, the pan-RAS inhibitors RMC-7977 and RMC-6236 were developed,27,28 the latter of which is also investigated in a phase I clinical trial (NCT05379985).29 Besides molecular glues based on immunophilins, significant progress in nondegradative molecular glues was also made recently with 14-3-3 protein stabilizers30 and molecular glues to form a complex of MEK and RAF.31 As of today there is no universally applicable strategy to systematically identify molecular glue hits32 and little is known about the prospects for subsequent optimization.
Results and discussion
To explore the likelihood to discover novel molecular glues from scratch, we used FKBP12 and the FRB domain of mTOR as a well-established model system. We opted for a HTRF (homogeneous time-resolved fluorescence) screening assay using a His-eGFP-FKBP12 and GST-tagged FRB constructs. To enable the detection of weak initial hits, we optimized the assay conditions to allow for high compound concentrations (Fig. S2†). Using this assay, we screened our internal compound library containing >1000 FKBP focused ligands (Fig. 2A), originally developed for human FKBP51 or bacterial FKBPs (Fig. S3†).33,34,36–47 Three hits, compounds 5,336![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 and 735 (Fig. 2B), were identified to induce the HTRF signal in a dose-dependent manner (Fig. 2C). However, only compound 7 dose-dependently induced higher fluorescence polarization, indicative of ternary complex formation, in an orthogonal fluorescence polarization (FP) assay with fluorescein-labelled FKBP12 in the presence of high concentrations of FRB (Fig. 2D). For compound 6, we were able to attribute the strong activity in the HTRF-assay to compound-induced binding of His-eGFP-FKBP12 directly to the anti-GST antibody (Fig. S4†). The desired activity of compound 7 was further validated by in vitro photocrosslinking experiments using FKBP12 site-specifically labelled with a photocrosslinking moiety (Fig. 2E). The band in the western blot at around 55 kDa in the presence of FKBP12, GST-FRB and compound 7 indicates the formed ternary complex. The observation was made with photoreactive, diazirine labelled FKBP12Q53C as well as two additional diazirine labelled FKBP12 mutants (Fig. S5†). Recent studies have demonstrated the value of native MS in studying artificially-induced ternary complex formation and stoichiometry.48–52 Our native MS analysis of the FKBP12-7-FRB ternary complex (Fig. 2F) and additional compounds (Fig. S6†) provided a direct identification of the intact protein complexes and corresponding subunits. With these three experiments we firmly validated the weak molecular glue activity of compound 7.
34 and 735 (Fig. 2B), were identified to induce the HTRF signal in a dose-dependent manner (Fig. 2C). However, only compound 7 dose-dependently induced higher fluorescence polarization, indicative of ternary complex formation, in an orthogonal fluorescence polarization (FP) assay with fluorescein-labelled FKBP12 in the presence of high concentrations of FRB (Fig. 2D). For compound 6, we were able to attribute the strong activity in the HTRF-assay to compound-induced binding of His-eGFP-FKBP12 directly to the anti-GST antibody (Fig. S4†). The desired activity of compound 7 was further validated by in vitro photocrosslinking experiments using FKBP12 site-specifically labelled with a photocrosslinking moiety (Fig. 2E). The band in the western blot at around 55 kDa in the presence of FKBP12, GST-FRB and compound 7 indicates the formed ternary complex. The observation was made with photoreactive, diazirine labelled FKBP12Q53C as well as two additional diazirine labelled FKBP12 mutants (Fig. S5†). Recent studies have demonstrated the value of native MS in studying artificially-induced ternary complex formation and stoichiometry.48–52 Our native MS analysis of the FKBP12-7-FRB ternary complex (Fig. 2F) and additional compounds (Fig. S6†) provided a direct identification of the intact protein complexes and corresponding subunits. With these three experiments we firmly validated the weak molecular glue activity of compound 7.
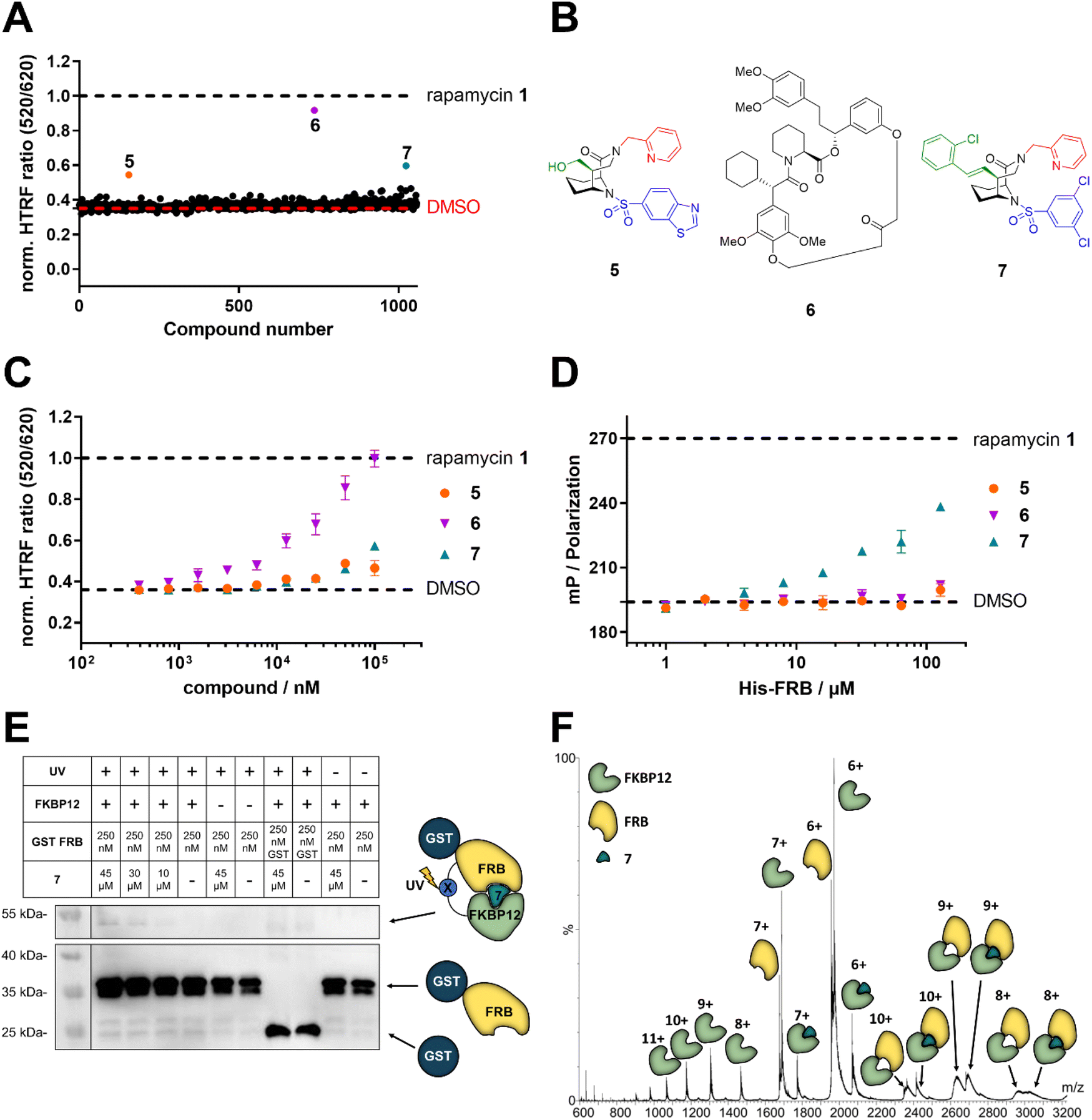 | ||
| Fig. 2 Identification of compound 7 as a FKBP12-FRB molecular glue. (A) Initial HTRF screening for the compound-induced formation of the ternary FKBP12-FRB complex using 100 μM His-eGFP-FKBP12, 20 nM GST-FRB and 1 nM terbium-labelled anti-GST antibody, data are represented as mean. (B) Structure of the three initial screening hits 5,33634 and 7.35 (C) Compounds 5, 6, and 7 dose-dependently increase the HTRF signal indicative of induced proximity between His-eGFP-FKBP12 and the terbium-labelled antibody/GST-FRB complex, data are represented as mean ± SEM. (D) Compound 7, but not 5 or 6, increases polarization in a FRB dose-dependent fluorescence polarization assay using 20 nM fluorescein-labelled FKBP12E140C and 5 μM compound, data are represented as mean ± SEM. (E) Anti-GST western blot of photoreactive, diazirine labelled FKBP12Q53C mutants photo-crosslinked with GST-FRB. UV light-induced GST-reactive bands at a size of approx. 55 kDa are indicative of the ternary complex of compound 7, FKBP12 and FRB being formed in vitro. (F) Native mass spectrum of the FKBP12-7-FRB complex acquired under soft conditions to maintain the non-covalent interactions at a concentration of 17 μM of each sample component (1:1:1), in 200 mM NH4OAc (pH 6.8). The ternary complex is present in three charge states (+8, +9, and +10). The presence of additional peaks of intermediary species (e.g., protein subunits, etc.) is typical as no isolation of a specific peak was performed. MS main parameters included: capillary voltage, 1.5 kV; source temperature, 30 °C; desolvation temperature, 200 °C; trap collision energy, 5 V; and transfer collision energy, 2 V. (A, C, and D) Rapamycin 1 and DMSO were used as positive and negative controls, respectively. | ||
To clarify the molecular binding mode, we determined the cocrystal structure of the FKBP12-7-FRB ternary complex (Fig. 3A). The binding of compound 7 to FKBP12 was similar as observed with related ligands from the [4.3.1]-bicyclic sulfonamide class53 and all key interactions were conserved (e.g. hydrogen bonds to the backbone NH of Ile56 or to the phenol of Tyr82, Fig. 3B).
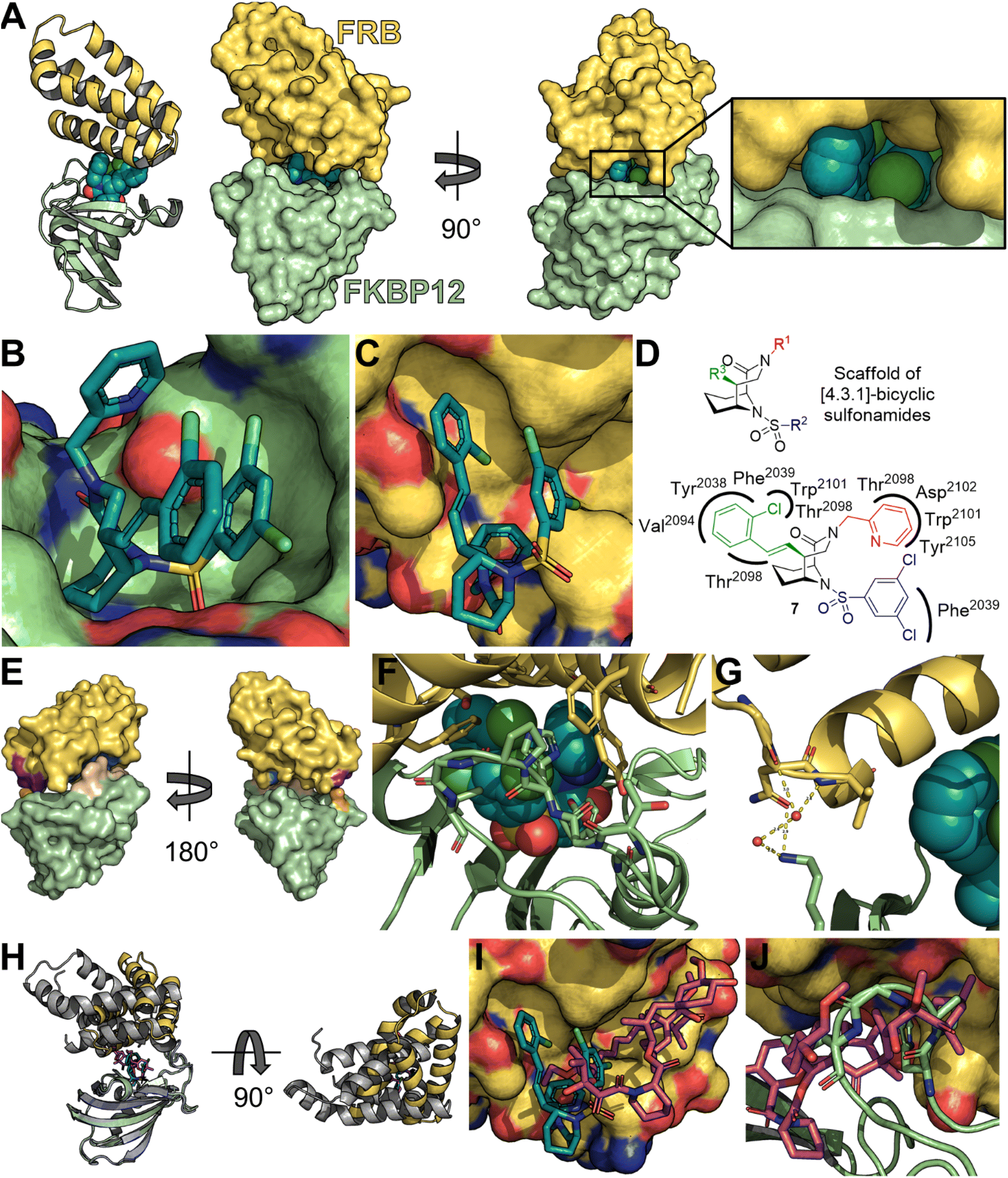 | ||
| Fig. 3 Cocrystal structure of the FKBP12-7-FRB ternary complex (PDB:8PPZ). (A) Structure of overall complex of compound 7 (spheres in dark cyan), FKBP12 (surface or cartoon in light green) and FRB (surface or cartoon in yellow). (B) Binding mode of compound 7 (dark cyan sticks) towards FKBP12 (light green surface) in the ternary complex. FRB omitted for clarity. (C) Binding mode of compound 7 (dark cyan sticks) towards FRB (yellow surface) in the ternary complex. FKBP12 omitted for clarity. (D) Scaffold of [4.3.1]-bicyclic sulfonamides with R1-position substituents in red, R2-position substituents in blue and R3-position substituents in green and two-dimensional interaction map of compound 7 with the FRB domain of mTOR. (E) FKBP12 (shown as green surface) and FRB (shown as yellow surface) with direct amino acid contacts colored in wheat and orange for FKBP12 and marine and purple for FRB (primary and secondary interaction sites, respectively). (F) Complex of FKBP12, FRB and compound 7 (dark cyan spheres) with amino acids participating in primary and secondary direct contacts shown as sticks. (G) Complex of FKBP12, FRB and compound 7 (dark cyan spheres) with amino acids participating in secondary direct contacts between FKBP12 and FRB shown as sticks. Water and water-mediated hydrogen bonds are shown as red spheres and yellow dashes. (H) Overlay of FKBP12 of the ternary complexes of compound 7, FKBP12 and FRB (PDB:8PPZ) with the ternary complex of rapamycin 1, FKBP12 and FRB (PDB:1NSG). FKBP12 molecules were partially omitted for clarity. Rapamycin 1 (magenta sticks) and compound 7 (dark cyan sticks) lead to different orientations of FRB (yellow for complex with compound 7, grey in complex with rapamycin 1). (I) Overlay of FRB of the cocrystal structures of compound 7 (dark cyan sticks, FKBP12 omitted for clarity) with the cocrystal structure of rapamycin 1 (magenta sticks, FRB in gray), highlighting the different binding mode of both complexes. (J) Overlay of FRB of the cocrystal structure of compound 7 (FKBP12 shown in green cartoon and sticks, FRB as yellow surface, 7 not shown) with the cocrystal structure of rapamycin 1 (magenta sticks, PDB:1NSG). | ||
The interactions between compound 7 and FRB were largely of hydrophobic nature (Fig. 3C and D). All three substituents (R1, R2 and R3) of the [4.3.1]-bicyclic core engaged in contacts with the FRB domain (Fig. 3C). The R1-pyridine formed van-der-Waals contacts with Thr2098, Trp2101, Asp2102and Tyr2105. One chlorine and the para-position of the R2-phenyl ring formed van-der-Waals contacts with Phe2039. The R3-substituent of compound 7 formed most interactions with the FRB domain, incl. van-der-Waals contacts to Tyr2038, Phe2039, Val2094, Thr2098 and Trp2101.
Several direct contacts between FKBP12 and FRB were observed, located in two regions (Fig. 3E). The major contacts were formed between the 80s loop of FKBP12 (Tyr82 and Thr85-Ile90) and the side chains of Ser2035, Phe2039, Trp2101, Tyr2105, and Phe2108 of FRB (Fig. 3F). This included a direct hydrogen bond from the phenol group of Tyr2105 (FRB) to the backbone carbonyl of Gly86 (FKBP12). In the second region, the amine group of Lys44 of FKBP12 formed a hydrogen bond to the primary amide carbonyl bond of Asn2093 (FRB), as well as a hydrogen bond to Gly2092, which was mediated by two water molecules (Fig. 3G). The side chain of Lys44 of FKBP12 also formed van-der-Waals contacts with Val2094 of the FRB domain.
The comparison with the known FKBP12-rapamycin 1-FRB ternary complex (PDB:1NSG)54 revealed that the FKBP12-7 and FKBP12-rapamycin 1 binary complexes target a similar surface region on FRB. However, the specific interactions radically differed since the orientation of the FRB was rotated by 90° between the two ternary complexes (Fig. 3H). While the binding surface on the FRB domain partially matched for compound 7 and rapamycin 1, both also formed unique interactions with parts of the FRB-domain (Fig. 3I). Interestingly, in the FKBP12-7-FRB complex the 80s loop of FKBP12 mimicked some of the interactions formed by the conjugated triene moiety of rapamycin 1 in the FKBP12-1-FRB complex (Fig. 3J).
The total binding interface, calculated with PISA,55 between the FKBP12-7 complex and the FRB-domain was 632 Å2, which was similar to the interaction surface between the FKBP12-rapamycin 1 complex and FRB (698 Å2). However, the contributions of the compounds vs. FKBP12 differed substantially. While in the FKBP12-7-FRB complex, 194 Å2 of the contact surface were contributed by compound 7 and 428 Å2 by ‘direct’ contacts of FKBP12, in the FKBP12-rapamycin 1-FRB complex 395 Å2 were contributed by rapamycin 1 and 303 Å2 by FKBP12.
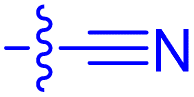
To increase the weak potency of the initial hit 7 utilizing the structure of the ternary complex, we studied the role of the chlorine pointing into a small cavity between FKBP12 and the FRB domain (Fig. 3A insert, chlorine shown as green sphere). To explore this position, we substituted one of the meta chlorines with small substituents such as bromine, nitrile, and acetylene (Scheme 1). This led to compounds 9a/b and 10a/b with slightly improved potencies for ternary complex induction (Table 1). Gratifyingly, the extension of the acetylene by an additional substituent like allyl, phenyl rings and heterocycles substantially enhanced the ternary complex formation 12- to 500-fold. Although addition of an allyl group (10c) already brought potency down below 10 μM, a full phenyl ring (10l) enhanced the potency 175-fold. Hydrophobic substituents on the phenyl ring, for example methyl (10o), were better tolerated, while more hydrophilic substituents, e.g. hydroxy (10i) and nitrile (10h) reduced ternary complex formation. Thiophenes (10j, 10k), thiazoles (10f, 10g) and methylthiophenes (10e, 10m, 10n) all induced formation of the ternary complex with <2 μM potency in the FP-assay. Similarly to the phenyl rings, more hydrophilic substituents perform worse regarding the ternary complex potency. All analogs retained high affinity to purifies FKBP12 alone (Ki < 12 nM) and occupied FKBP12 inside human cells with an ICnanoBRET50 between 40 and 7500 nM (Table 1). The affinity gains of extending from the chlorine likely originate from displacing unfavorable water atoms and hydrophobic interactions, as the small pocket seems to be of hydrophobic nature, which is supported by the higher ternary complex affinity of the hydrophobic substituents in comparison to more hydrophilic ones.
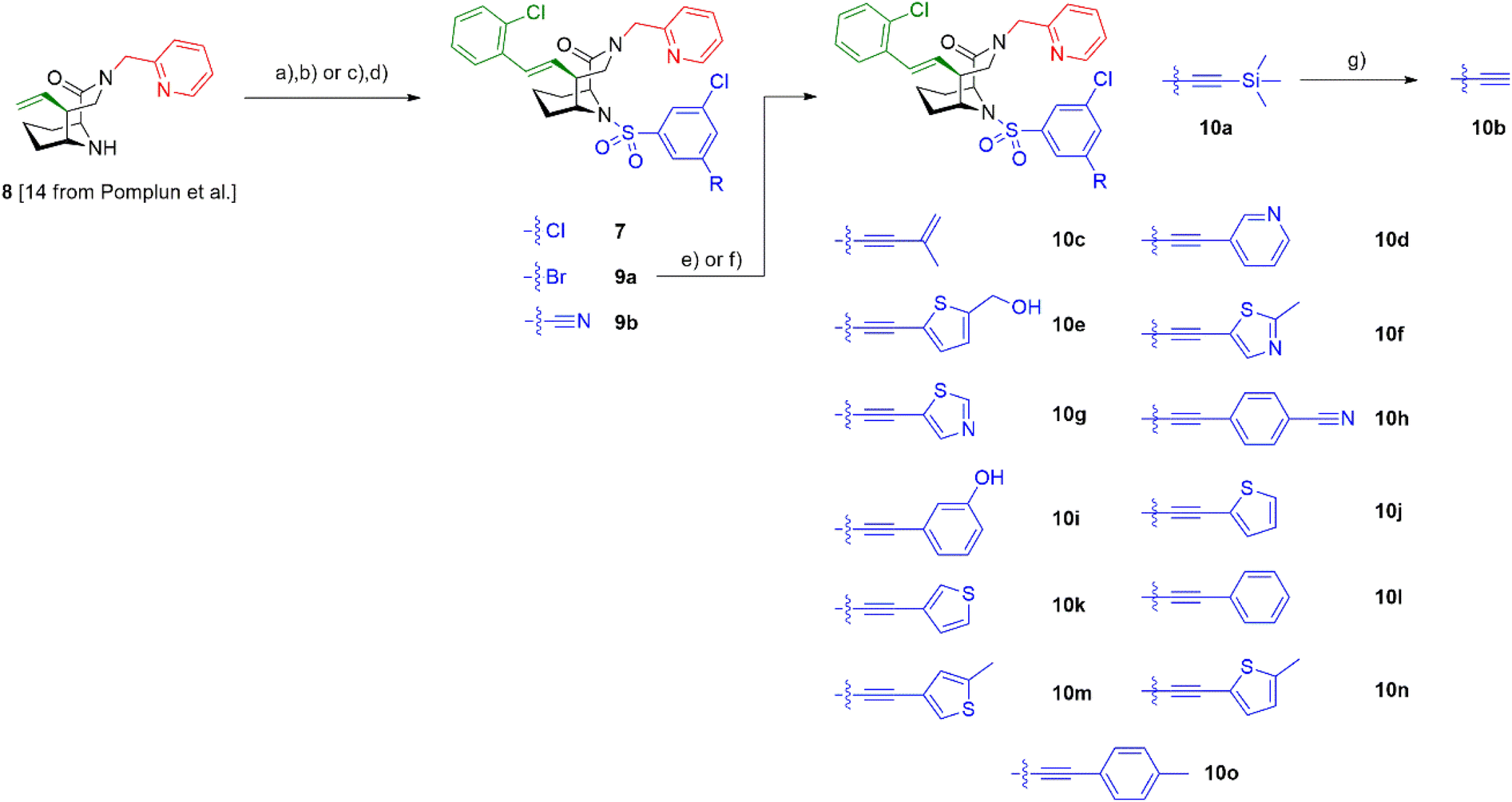 | ||
| Scheme 1 Synthesis of [4.3.1]-bicyclic sulfonamide analogs. Reagents and conditions: (a) sulfonyl chloride, DIPEA, MeCN, rt, compound 7:18, 48% yield, 9b: 16 h, 49% yield; (b) 1-bromo-2-chlorobenzene, K2CO3, Pd(dppf)Cl2·CH2Cl2, 100 °C, compound 7: dioxane, 40 h, 57% yield, 9b: DMF, 18 h, 25% yield; (c) 1-bromo-2-chlorobenzene, K2CO3, Pd(dppf)Cl2·CH2Cl2, dioxane:H2O = 20:1, 100 °C, 19 h, 78% yield; (d) 3-bromo-5-chlorosulfonyl chloride, DIPEA, MeCN, rt, 46 h, 55% yield; (e) alkyne, Pd(PPh3)4 or Pd(dppf)Cl2·CH2Cl2, CuI, TMEDA, 80–90 °C, 2.5–38 h, 34–92% yield; (f) TMS-alkyne, Pd(dppf)Cl2·CH2Cl2, CuCl, TMEDA:DMF = 1:1, 80 °C, 14.5–22 h, 53–78% yield; (g) K2CO3, MeOH, rt, 2.5 h, 94% yield. | ||
| No. | Human FKBP12, KFPi/nM56 | ECternary FP50/μM | FKBP12 ICNanoBRET50/nM57 | ECternary nanoBRET50/μM |
|
|---|---|---|---|---|---|
| Rapamycin 1 | 0.6 | 0.039 ± 0.006 | 30.3 ± 1.5 | 1.8 ± 0.16 | — |
| 7 | 6.3 | 93 ± 21 | 81.2 ± 16.3 | n.b. |

|
| 9a | 5.8 | 56 ± 10 | 40.6 ± 5.3 | n.m. |

|
| 9b | 3.6 | 54 ± 6 | 47.8 ± 10.7 | n.m. |
|
| 10a | 11 | 50 ± 5 | 405 ± 219 | n.b. |
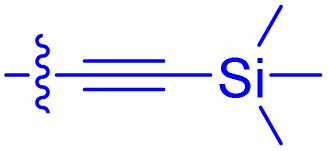
|
| 10b | 13 | 63 ± 6 | 101 ± 19 | n.m. |
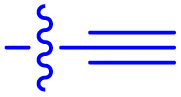
|
| 10c | 5.1 | 7.8 ± 2.6 | 146 ± 28.5 | n.b. |
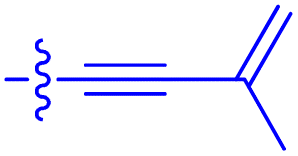
|
| 10d | 4.5 | 4.1 ± 0.4 | 25.5 ± 3.0 | 50.5 ± 9.8 |

|
| 10e | 6.9 | 2.0 ± 0.2 | 57.3 ± 16.8 | 38.8 ± 1.7 |

|
| 10f | 1.8 | 1.9 ± 0.2 | 259 ± 37 | 28.2 ± 1.3 |
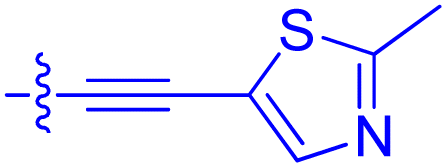
|
| 10g | 4.7 ± 1.8 | 1.8 ± 0.1 | 49.2 ± 4.0 | 26.0 ± 1.6 |

|
| 10h | 0.8 | 1.5 ± 0.2 | 66.9 ± 22.5 | 31.7 ± 2.5 |

|
| 10i | 0.4 | 1.3 ± 0.2 | 264 ± 36.8 | 172 ± 36 |
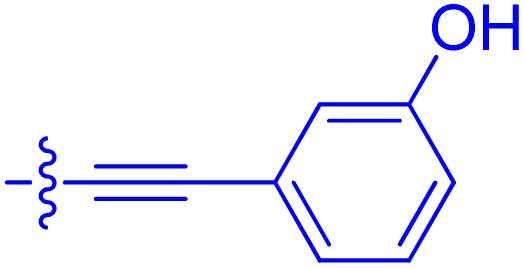
|
| 10j | 7.2 ± 1.7 | 0.63 ± 0.06 | 799 ± 183 | 57.5 ± 3.6 |
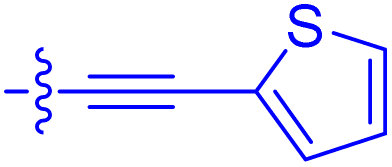
|
| 10k | 4.1 ± 0.6 | 0.56 ± 0.03 | 314 ± 21 | 26.3 ± 1.3 |
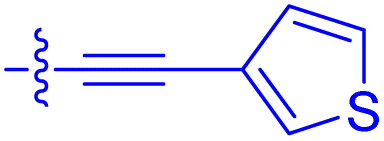
|
| 10l | 4.5 | 0.53 ± 0.07 | 527 ± 77 | 32.9 ± 2.5 |

|
| 10m | 6.0 | 0.23 ± 0.03 | 952 ± 147 | 31.7 ± 2.1 |
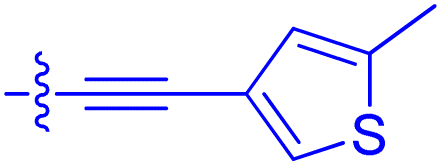
|
| 10n | 4.8 ± 0.8 | 0.18 ± 0.02 | 1330 ± 195 | 42.1 ± 2.8 |
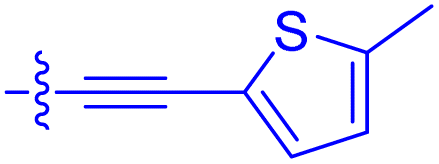
|
| 10o | 2.6 | 0.17 ± 0.02 | 7460 ± 2100 | 109 ± 6.3 |

|

Unfortunately, we were unable to solve cocrystal structures of more advanced molecular glues as the binding site for the larger substituents is occupied by a neighboring protein of the next unit cell in the ternary complex structure of 7. Notably, the optimized compounds 10d and 10g strictly relied on FKBP12 to engage FRB, as we were unable to detect binding to the rapamycin binding site up to a concentration of 10 μM (Fig. S7†).
To test if the synthetic FKBP12-FRB molecular glues were active in cells, we performed a NanoBRET assay using nanoLuc-tagged FKBP12 and HaloTag-tagged FRB (Fig. 4A and Table 1). Compounds 10d–o all dose-dependently induced the FKBP12-FRB complex in HEK293T cells, although more weakly than rapamycin 1. This was FKBP12 binding-dependent since pretreatment with a high affinity FKBP12 ligand abolished the NanoBRET signal (Fig. 4B). For all active analogs, the induction of the ternary complex in cells consistently occurred at similar concentrations (25–170 nM), which were substantially lower than the ternary complex formation potencies determined biochemically. We attribute this to a combination of intracellular FKBP12 occupancy (reflected by the competitive NanoBRET assay) and the potency of the FKBP12-compound pre-complex to bind to FRB (reflected by the biochemically determined ternary complex formation, ECternary FP50). The higher apparent potency for intracellular ternary complex formation can be explained by an excess of FKBP12-nLuc over FRB-Halo. Thereby, only a small fraction of FKBP12 occupancy is sufficient to produce a maximal NanoBRET signal. Interestingly, we observed a clear threshold for intracellular ternary complex formation, which was determined by biochemical ternary complex formation potency. Compounds with an ECternary FP50 > 4.1 μM did not induced ternary intracellular NanoBRET signals while all compounds with an ECternary FP50 < 4.1 μM did.
 | ||
| Fig. 4 Cellular characterization of FKBP12-FRB molecular glues. (A) Compounds 10d and 10g-induced FKBP12-FRB ternary complex formation in HEK293T cells determined by NanoBRET assay using C-terminal NanoLuc-tagged FKBP12 and C-terminal HaloTag-tagged FRB, while compound 10c did not. (B) Compound 10g-induced FKBP12-FRB ternary complex formation in HEK293T cells is abolished by pre-treatment with a potent FKBP12 inhibitor (18S-Me from Kolos et al.33). | ||
Conclusions
In conclusion, our screening approach enabled us to identify a novel molecular glue targeting the flat surface of the FRB-domain of mTOR. Screening at high target protein concentrations was crucial to identify an initially very weak hit, which would have been difficult to detect by other approaches. Although our approach was unbiased regarding the binding site on FRB, the identified molecular glues target a similar region on FRB as rapamycin 1. The surface on FRB around Tyr2028/Phe2039, Val2094–Thr2098, and Trp2101–Phe2105, while not a priori apparent, thus appeared to represent a preferred region for protein–protein contacts. Indeed, this site has been suggested to assist in the binding of mTOR targets such as S6K and PRAS40, as well as phosphatidic acid (PA).59 The preference for this region was not due to specific contacts with FKBP12, since in the context with compound 7, FKBP12 engaged FRB in a completely different manner than in context with rapamycin 1. However, as for rapamycin 1, direct FKBP12-FRB contacts were crucial to dramatically enhance the affinity of the FKBP12-compound 7 complex to FRB compared to FRB-binding of the compound alone.60,61 The substantially higher affinity of the FKBP12-rapamycin 1 complex over the FKBP12-compound 7 complex for FRB is likely due to energetically more favorable interactions, where the rapamycin-FRB interactions appear to be more extensive and elaborate compared to the compound 7-FRB contacts. Aided by the crystal structure we were able to improve the affinity of our initial hit by rational design up to 500-fold, leading to compounds which bind the FRB domain of mTOR at nanomolar concentrations in cells.Our findings have several implications for the discovery of molecular glues
(i) Molecular glues might be less rare than initially thought as we found one hit within a relatively small, focused library.
(ii) Screening approaches with high compound and presenter protein concentrations were necessary to find such weak molecular glue hits. Biochemical approaches seemed to be most adequate as weak activity is easier to detect compared e.g. to cellular assays.62,63
(iii) The use of a focused library targeted to the presenter protein (FKBP12 in our case) likely facilitated the identification of molecular glues substantially since part of the recognition problem was already pre-engineered.
(iv) Weak initial molecular glue hits can be used as a starting point for rational design to get more potent molecular glues. Even for weak molecular glue hits, the ternary complex structure can be obtained, which facilitates optimization substantially.25
(v) Shallow hydrophobic surfaces seem to be a preferred interaction site for molecular glues, in line with the binding modes of rapamycin 1, FK506 2 and WBD002 4.11–13,64
(vi) At large excess of the presenter protein over the target protein, only a small fractional occupancy of the presenter protein might be sufficient to evoke the effect.27
(vii) The expression levels of the presenter protein represent a threshold beyond which weak molecular glues cannot work in cells.27
(viii) The choice of the presenter protein is likely a key factor. FKBP12 (like Cyp18) might be a privileged presenter protein featuring high abundance in many tissues,65 absence of negative effects by binding of FKBP12 alone, availability of potent ligands as docking scaffolds, and numerous exit vectors on the latter. These features likely contributed to the prevalence of FKBPs (and cyclophilins) as presenter proteins in nature and support their use to target otherwise undruggable proteins in drug discovery.
Data availability
All associated experimental details are provided in the ESI.† Crystallographic data for compound number 7 have been deposited at the PBD under accession number 8PPZ.Author contributions
R. C. E. D. data curation, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review and editing; C. M. conceptualization, data curation, formal analysis, investigation, methodology, writing – review and editing; M. L. R. data curation, formal analysis, investigation, methodology; W. O. S. data curation, formal analysis, investigation, methodology; J. M. K. data curation, investigation; E. V. S. M. data curation, investigation, methodology, visualization, writing – review and editing; T. H. data curation, investigation; T. M. G. investigation, formal analysis, writing – review and editing; S. K. resources; F. L. funding acquisition, supervision, writing – review and editing F. H. conceptualization, funding acquisition, project management, supervision, visualization, writing – original draft, writing – review and editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding by the iGLUE project of the PROXIDRUGS consortium (03ZU1109EB) and by the DFG (433472263 to F. H., 524226614 to F. L.). The synchrotron data were collected at beamline operated by EMBL Hamburg at the PETRA III storage ring (DESY, Hamburg, Germany).66 We would like to thank David von Stetten for the assistance in using the beamline. Edvaldo V. S. Maciel was supported by Humboldt fellowship. Synapt instrument was funded by DFG grant 461372424.Notes and references
- S. L. Schreiber, Cell, 2021, 184, 3 CrossRef CAS PubMed.
- H. Rui, et al. , RSC Chem. Biol., 2023, 4, 192 RSC.
- T. M. Geiger, et al. , Curr. Res. Chem. Biol., 2022, 2, 100018 CrossRef CAS.
- S. L. Schreiber, Cell Chem. Biol., 2024, 31, 1050 CrossRef CAS PubMed.
- Z. Kozicka and N. H. Thomä, Cell Chem. Biol., 2021, 28, 1032 CrossRef CAS PubMed.
- J. M. Sasso, et al. , Biochemistry, 2023, 62, 601 CrossRef CAS PubMed.
- S. Liu, et al. , J. Am. Chem. Soc., 2023, 145, 23281 CrossRef CAS PubMed.
- K. Mahalati and B. D. Kahan, Clin. Pharmacokinet., 2001, 40, 573 CrossRef CAS PubMed.
- M. Suthanthiran, et al. , Am. J. Kidney Dis., 1996, 28, 159 CrossRef CAS PubMed.
- S. L. Schreiber, Science, 1991, 251, 283 CrossRef CAS PubMed.
- J. Liu, et al. , Cell, 1991, 66, 807 CrossRef CAS PubMed.
- S. Gaali, et al. , Curr. Med. Chem., 2011, 18, 5355 CrossRef CAS PubMed.
- S. L. Schreiber, Cell, 1992, 70, 365 CrossRef CAS PubMed.
- E. J. Brown, et al. , Nature, 1994, 369, 756 CrossRef CAS PubMed.
- U. K. Shigdel, et al. , Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 17195 CrossRef CAS PubMed.
- C.-F. Chang, et al. , Angew Chem. Int. Ed. Engl., 2021, 60, 17045 CrossRef CAS PubMed.
- K. H. Pua, et al. , Cell Rep., 2017, 18, 432 CrossRef CAS PubMed.
- T. Fehr, et al. , J. Antibiot., 1996, 49, 230 CrossRef CAS PubMed.
- S. C. Schäfer, et al. , Bioorg. Med. Chem. Lett., 2024, 104, 129728 CrossRef PubMed.
- G. M. Salituro, et al. , Tetrahedron Lett., 1995, 36, 997 CrossRef CAS.
- M. Y. Summers, et al. , J. Antibiot., 2006, 59, 184 CrossRef CAS PubMed.
- Z. Guo, et al. , Nat. Chem., 2019, 11, 254 CrossRef CAS PubMed.
- Z. Guo, et al. , Angew Chem. Int. Ed. Engl., 2019, 58, 17158 CrossRef CAS PubMed.
- H. Park, et al. , Cell, 2022, 185, 1943–1959 CrossRef CAS PubMed.
- C. J. Schulze, et al. , Science, 2023, 381, 794 CrossRef CAS PubMed.
- P. A. Jänne, et al. , Mol. Cancer Ther., 2023, 22, 1 CrossRef.
- M. Holderfield, et al. , Nature, 2024, 629, 919 CrossRef CAS PubMed.
- U. N. Wasko, et al. , Nature, 2024, 629, 927 CrossRef CAS PubMed.
- J. Jiang, et al. , Cancer Discovery, 2024, 14, 994 CrossRef PubMed.
- P. Pitasse-Santos, et al. , Curr. Opin. Struct. Biol., 2024, 86, 102822 CrossRef CAS PubMed.
- M. B. Ryan, et al. , Cancer Discovery, 2024, 14, 1190 CrossRef PubMed.
- G. A. Holdgate, et al. , SLAS Discovery, 2024, 29, 100136 CrossRef CAS PubMed.
- J. M. Kolos, et al. , Chem. Sci., 2021, 12, 14758 RSC.
- M. Bauder, et al. , J. Med. Chem., 2021, 64, 3320 CrossRef CAS PubMed.
- J. Kolos, PhD thesis, Technical University of Darmstadt, 2023.
- R. Gopalakrishnan, et al. , J. Med. Chem., 2012, 55, 4123 CrossRef CAS PubMed.
- R. Gopalakrishnan, et al. , J. Med. Chem., 2012, 55, 4114 CrossRef CAS PubMed.
- S. Gaali, et al. , Nat. Chem. Biol., 2015, 11, 33 CrossRef CAS PubMed.
- S. Gaali, et al. , J. Med. Chem., 2016, 59, 2410 CrossRef CAS PubMed.
- X. Feng, et al. , J. Med. Chem., 2020, 63, 231 CrossRef CAS PubMed.
- X. Feng, et al. , J. Med. Chem., 2015, 58, 7796 CrossRef CAS PubMed.
- M. Bischoff, et al. , Org. Lett., 2014, 16, 5254 CrossRef CAS PubMed.
- M. Bischoff, et al. , Chemistry, 2020, 26, 4677 CrossRef CAS PubMed.
- S. Pomplun, et al. , J. Med. Chem., 2018, 61, 3660 CrossRef CAS PubMed.
- S. Pomplun, et al. , Angew Chem. Int. Ed. Engl., 2015, 54, 345 CrossRef CAS PubMed.
- A. M. Voll, et al. , Angew Chem. Int. Ed. Engl., 2021, 60, 13257 CrossRef CAS PubMed.
- Y. Wang, et al. , J. Med. Chem., 2013, 56, 3922 CrossRef CAS PubMed.
- C. J. A. Verhoef, et al. , Chem. Sci., 2023, 14, 6756 RSC.
- L. M. Sternicki and S.-A. Poulsen, Anal. Chem., 2023, 95, 18655 CrossRef CAS PubMed.
- C. Jackson and R. Beveridge, Analyst, 2024, 149, 3178 RSC.
- X. Huang, et al. , J. Am. Chem. Soc., 2023, 145, 14716 CrossRef CAS PubMed.
- I. M. M. Ahmed and R. Beveridge, Rapid Commun. Mass Spectrom., 2023, 37, e9604 CrossRef CAS PubMed.
- P. L. Purder, et al. , JACS Au, 2023, 3, 2478 CrossRef CAS PubMed.
- J. Liang, et al. , Acta Crystallogr., Sect. D:Biol. Crystallogr., 1999, 55, 736 CrossRef CAS PubMed.
- E. Krissinel and K. Henrick, J. Mol. Biol., 2007, 372, 774 CrossRef CAS PubMed.
- C. Kozany, et al. , Chembiochem, 2009, 10, 1402 CrossRef CAS PubMed.
- M. T. Gnatzy, et al. , Chembiochem, 2021, 22, 2257 CrossRef CAS PubMed.
- T. Machleidt, et al. , ACS Chem. Biol., 2015, 10, 1797 CrossRef CAS PubMed.
- H. Yang, et al. , Nature, 2017, 552, 368 CrossRef CAS PubMed.
- A. M. März, et al. , Mol. Cell. Biol., 2013, 33, 1357 CrossRef PubMed.
- L. A. Banaszynski, et al. , J. Am. Chem. Soc., 2005, 127, 4715 CrossRef CAS PubMed.
- E. S. Wang, et al. , Nat. Chem. Biol., 2021, 17, 711 CrossRef CAS PubMed.
- E. Sijbesma, et al. , J. Am. Chem. Soc., 2019, 141, 3524 CrossRef CAS PubMed.
- Comment: The only reported rapamycin mimic with a non-FKBP-targeted scaffold is WRX606, identified by in silico screening, R. Shams, A. Matsukawa, Y. Ochi, Y. Ito and H. Miyatake, J. Med. Chem., 2022, 65, 1329 CrossRef CAS PubMed , However, we did not observe FKBP12 binding or FKBP12-FRB dimerization in our biochemical assays for this compound (see ESI Fig. S8†)..
- P. S. Marinec, et al. , Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1336 CrossRef CAS PubMed.
- M. Cianci, et al. , J. Synchrotron Radiat., 2017, 24, 323 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06917j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |