
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclopentadienyl cations
Sameera
Ranasinghe
a,
Caleb D.
Martin
 *a and
Jason L.
Dutton
*b
*a and
Jason L.
Dutton
*b
aBaylor University, Department of Chemistry and Biochemistry, One Bear Place #97348, Waco, TX 76798, USA. E-mail: Caleb_D_Martin@baylor.edu
bLa Trobe University, Department of Biochemistry and Chemistry, La Trobe Institute for Molecular Science, Melbourne, Victoria 3086, Australia. E-mail: j.dutton@latrobe.edu.au
First published on 26th December 2024
Abstract
This perspective covers the chemistry of cyclopentadienyl cations from the first synthetic attempts generating transient variants to their successful isolation earlier this year. They are highly reactive species that researchers struggled to isolate and characterize that stifled efforts to explore their reactivity. The recent isolation of a cyclopentadienyl cation enabled characterization and reactivity studies that make this an exciting time in the area that will undoubtedly inspire research in cyclic four π–electron systems.
Introduction
Unsaturated carbon ring systems are a main pillar of organic chemistry. In five membered rings, cyclopentadienide (Cp−) has been widely studied and used as an effective six π–electron ligand for transition metals. The neutral unsaturated five membered ring is a five π–electron radical (Cp˙) and cation is the cyclopentadienyl cation (Cp+) with four π–electrons. The first documented study on a cationic cyclopentadienyl species is a 1925 report by Ziegler1 with the successful isolation coming 99 years later in 2024 by Schulz and coworkers.2 Despite this lengthy history, there has not been a review focused on this subject. This perspective serves as a resource for the key developments over nearly 100 years leading up to this year's isolation of a bonafide cyclopentadienyl cation.The electronic structure of Cp+ compounds is influenced by the substitution on the carbon atoms. The π-orbitals of the cyclopentadienyl cation are depicted in Fig. 1. The state with two degenerate SOMOs is a triplet with the electrons delocalized throughout the ring. There are two singlet states, both in which the degeneracy has been removed that results in a dienylic or allylic form, differing in the energetic ordering of the orbitals.3–5 The dienylic form is characteristic of their isoelectronic relatives, boroles, where the less electronegative boron atom reduces symmetry and breaks the degeneracy of the Hückel orbitals.6–9 In most cases the triplet state and the two isomeric singlet states are close to degenerate (within 3 kcal mol−1). In this perspective, the schemes depict the ground state electronic structures of each species as described in the respective reports.
 | ||
| Fig. 1 Structures of the triplet and singlet states of Cp+ with their π-orbital diagrams. | ||
The parent cyclopentadienyl cation
The first report of a synthetic attempt at the parent cyclopentadienyl cation (Cp+) was in 1972 by Breslow in a reaction of 5-iodocyclopentadiene with silver perchlorate at −15 °C, ultimately resulting in the silver ion catalyzing Diels–Alder dimerization (Fig. 2a).10 The following year, using 5-chlorocyclopentadiene or 5-bromocyclopentadiene, halide abstraction by SbF5 at −195 °C enabled observation of Cp+ by EPR spectroscopy revealing that the ground state is a triplet (Fig. 2b). Theoretical studies corroborated the experimental observations with triplet Cp+ calculated as 7.3 kcal mol−1 more stable than the singlet state.11 | ||
| Fig. 2 (a) Synthetic attempt at Cp+. (b) Successful low temperature generation of Cp+. | ||
Since the original synthetic reports, theoretical methods and computing power have become more advanced. Recent theoretical studies confirm that the triplet form is the ground state in the parent Cp+ cation, as well as other derivatives.12–14 The two isomers of the singlet excited state are computed to be nearly degenerate and are predicted to rapidly interconvert via a bond pseudorotation process.3
Chlorocyclopentadienyl cations
In 1964, Breslow reported the room temperature reaction of hexachlorocyclopentadiene with SbF5 to generate the pentachlorocyclopentadienyl cation (C5Cl5+, Fig. 3).15 The EPR spectrum of a frozen glass at −196 °C and variable temperature experiments indicate the ground state of C5Cl5+ is a C5 symmetric triplet, although with the singlet state lying slightly higher in energy that has also been supported by modern theoretical studies.3 Quenching with methanol implied that C5Cl5+ is only generated in ∼1% yield which prevented magnetic susceptibility analyses. Conducting the reaction of SbF5 and C5Cl6 in SO2, or with other Lewis acids to abstract the halide (AlCl3, BF3, SbCl5), did not result in observation of the triplet species by EPR spectroscopy.15 Malischewski and co-workers in 2020 reinvestigated this system and found that low temperature recrystalization from the reaction of perchloro or perbromocyclopentadiene with antimony or arsenic pentafluoride returned products arising from 2 + 2 cycloaddition processes.16 | ||
| Fig. 3 (a) Generation and observation of trace C5Cl5+. (b) Bulk outcome of 2 + 2 cycloaddition reaction. | ||
In 2017, Sander and co-workers photolyzed diazo tetrachlorocyclopentadiene in noble gas matrices to generate a triplet carbene that reacts with BF3 to produce a zwitterionic adduct (Fig. 4).17 The zwitterion features a borate and a positively charged C5 ring. From the IR and UV-vis data, as well as absence of EPR signals, the ground state was assigned as the singlet. Calculations indicate the allylic singlet state is 2.3 kcal mol−1 lower than the triplet state. Reaction with a second equivalent of BF3 resulted in binding to the fluoroborate while irradiation with low energy light (650 nm or the source of the IR spectrometer) induced fluoride migration from the boron to the adjacent carbon center to furnish the corresponding cyclopentadiene.
 | ||
| Fig. 4 Generation of the antiaromatic singlet cyclopentadienyl zwitterion and its reactivity. | ||
Pentaalkylcyclopentadienyl cations
In 2002, Lambert and co-workers claimed the reaction of pentamethylcyclopentadiene (HCp*) with [CPh3][B(C6F5)4] resulted in hydride abstraction to generate Cp*+ as a [B(C6F5)4]− salt (Fig. 5).18 Although they reported a single crystal X-ray diffraction structure, their claims of a stable antiaromatic compound were suspicious as their structure had two of the ring carbon atoms pyramidalized, contrary to the expected behaviour for an all sp2 hybridized ring. Furthermore, the compound was stable to ambient conditions and no EPR signal was detected, inconsistent with Schleyer's calculations from a few years earlier predicting a triplet ground state favoured over the singlet by 4.35 kcal mol−1.13 The compound is also colourless, inconsistent with a compound with a small HOMO–LUMO gap that would be expected for either singlet isomer. The work was scrutinized by the groups of Bertrand,19 Cowley,20 and Müller.21 Upon close examination of the solid-state structure, the material obtained by Lambert was identified as the dihydro variant of Cp*+, the pentamethylcyclopentenyl cation. Upon publication of the three papers, including one that Lambert was a coauthor,19 Lambert published a statement acknowledging their error and retracted the conclusion of the original claim.22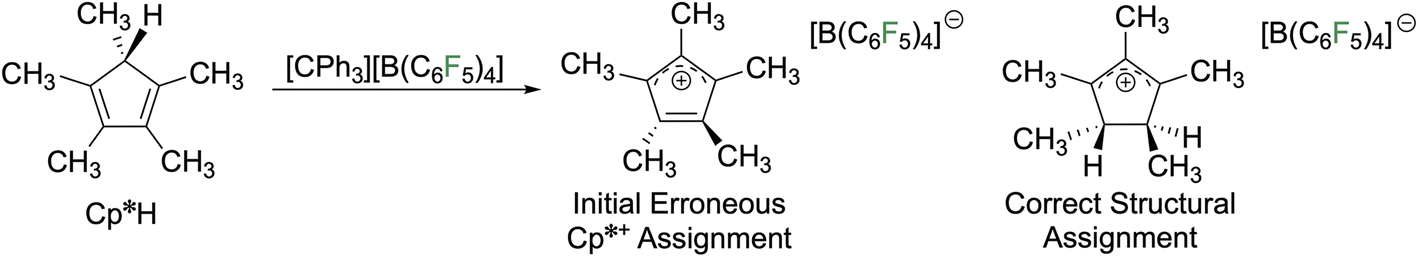 | ||
| Fig. 5 Claimed synthesis of Cp*+ and correction of the product assignment. | ||
Pentaarylcyclopentadienyl cations
In 1925, Ziegler reported the reaction of concentrated sulfuric acid with pentaphenylcyclopentadienol to give a purple compound that they claimed to be the pentaphenylcyclopentadienyl cation (C5Ph5+).1 In 1959, Krapcho conducted spectroscopic studies that agreed with the assignment as C5Ph5+ (Fig. 6a).23 Breslow reinvestigated this reaction in 1961 and concluded that C5Ph5+ is transient and rearranges to a mixture composed of triphenylcyclopentadienophenanthrene (48%), HCpPh5 (25%), a 3-cyclopentenone (5%), and an unidentified oxygen-containing compound (21%).24 A follow up study on C5Ph5+ generated by treating pentaphenylcyclopentadienol with BF3 gas in CH2Cl2 at −60 °C enabled observation and reactivity studies (Fig. 6b).25 The UV/vis spectrum features an intense λmax at 650 nm (ε = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) that begins changing above −40 °C where decomposition ensues. Quenching the reaction with methanol furnished the methyl ether with the yield being above 90%. Adding excess cycloheptatriene (C7H8) to the C5Ph5+ solution generated HC5Ph5. In the BF3 route, the decomposition products differ from the H2SO4 route and were described as “unidentified fluorinated materials.” The BF3 synthetic route was applied to generate a series of cations with various organic groups on the alcoholic carbon of pentaphenylcyclopentadienol (p-CH3O–C6H4, p-Cl–C6H4, p-CH3–C6H4, 2-naphthyl, and methyl). The compounds were quenched with cycloheptatriene that generated a mixture of tautomers of cyclopentadienes isolated in good yields for aryl species (52–77%) but low for the methyl variant (20%) rationalized by the acidity of methyl protons adjacent to the cationic π-system. They state the tautomers are in equilibrium but were unable to determine the major tautomer.25,26 The EPR spectra in frozen solutions at 77 K reveal that the phenyl, p-Cl–C6H4, p-CH3–C6H4, and CH3 variants exhibit spectra consistent with a triplet state.26 However, for the p-CH3O–C6H4 and 2-naphthyl species the triplet is not observed at any temperature. Generation of C5Ph5+ from AlCl3 instead of BF3 or using SO2 or CH2Cl2 as a solvent did not change the spectrum observed. From temperature dependent studies, it was determined that triplet C5Ph5+ is an excited state but only slightly higher in energy than the singlet. The singlet–triplet energy gap is temperature dependent (0.35 kcal mol−1 at 77 K and 1.15 kcal mol−1 at 170 K). The p-Cl–C6H4 species has similar singlet–triplet energy gaps of 0.30 kcal mol−1 at 82 K and 1.40 kcal mol−1 at 174 K while the p-CH3–C6H4 species has similar temperature variance but with a 1.3 kcal mol−1 higher energy gap and the methyl species was not sufficiently stable for variable temperature studies. Solution magnetic susceptibility measurements by Evans' method indicate less than 2.2% of C5Ph5+ exists in the triplet state and 1.1% of the p-Cl–C6H4 variant with no triplet detected for any of the other species. The relative populations were rationalized with the EPR studies on frozen solutions as EPR spectroscopy is more sensitive than Evans' method allowing for detection of the triplet of the p-CH3–C6H4 species. The lowering of energy of the singlet state for the pentaaryl derivatives with respect to the parent system and pentachloro derivative was rationalized by the larger size of the pentaaryl systems. There are more atoms for electron delocalization that reduces the electron pairing energy penalty for the singlet state.25,26
000 M−1 cm−1) that begins changing above −40 °C where decomposition ensues. Quenching the reaction with methanol furnished the methyl ether with the yield being above 90%. Adding excess cycloheptatriene (C7H8) to the C5Ph5+ solution generated HC5Ph5. In the BF3 route, the decomposition products differ from the H2SO4 route and were described as “unidentified fluorinated materials.” The BF3 synthetic route was applied to generate a series of cations with various organic groups on the alcoholic carbon of pentaphenylcyclopentadienol (p-CH3O–C6H4, p-Cl–C6H4, p-CH3–C6H4, 2-naphthyl, and methyl). The compounds were quenched with cycloheptatriene that generated a mixture of tautomers of cyclopentadienes isolated in good yields for aryl species (52–77%) but low for the methyl variant (20%) rationalized by the acidity of methyl protons adjacent to the cationic π-system. They state the tautomers are in equilibrium but were unable to determine the major tautomer.25,26 The EPR spectra in frozen solutions at 77 K reveal that the phenyl, p-Cl–C6H4, p-CH3–C6H4, and CH3 variants exhibit spectra consistent with a triplet state.26 However, for the p-CH3O–C6H4 and 2-naphthyl species the triplet is not observed at any temperature. Generation of C5Ph5+ from AlCl3 instead of BF3 or using SO2 or CH2Cl2 as a solvent did not change the spectrum observed. From temperature dependent studies, it was determined that triplet C5Ph5+ is an excited state but only slightly higher in energy than the singlet. The singlet–triplet energy gap is temperature dependent (0.35 kcal mol−1 at 77 K and 1.15 kcal mol−1 at 170 K). The p-Cl–C6H4 species has similar singlet–triplet energy gaps of 0.30 kcal mol−1 at 82 K and 1.40 kcal mol−1 at 174 K while the p-CH3–C6H4 species has similar temperature variance but with a 1.3 kcal mol−1 higher energy gap and the methyl species was not sufficiently stable for variable temperature studies. Solution magnetic susceptibility measurements by Evans' method indicate less than 2.2% of C5Ph5+ exists in the triplet state and 1.1% of the p-Cl–C6H4 variant with no triplet detected for any of the other species. The relative populations were rationalized with the EPR studies on frozen solutions as EPR spectroscopy is more sensitive than Evans' method allowing for detection of the triplet of the p-CH3–C6H4 species. The lowering of energy of the singlet state for the pentaaryl derivatives with respect to the parent system and pentachloro derivative was rationalized by the larger size of the pentaaryl systems. There are more atoms for electron delocalization that reduces the electron pairing energy penalty for the singlet state.25,26
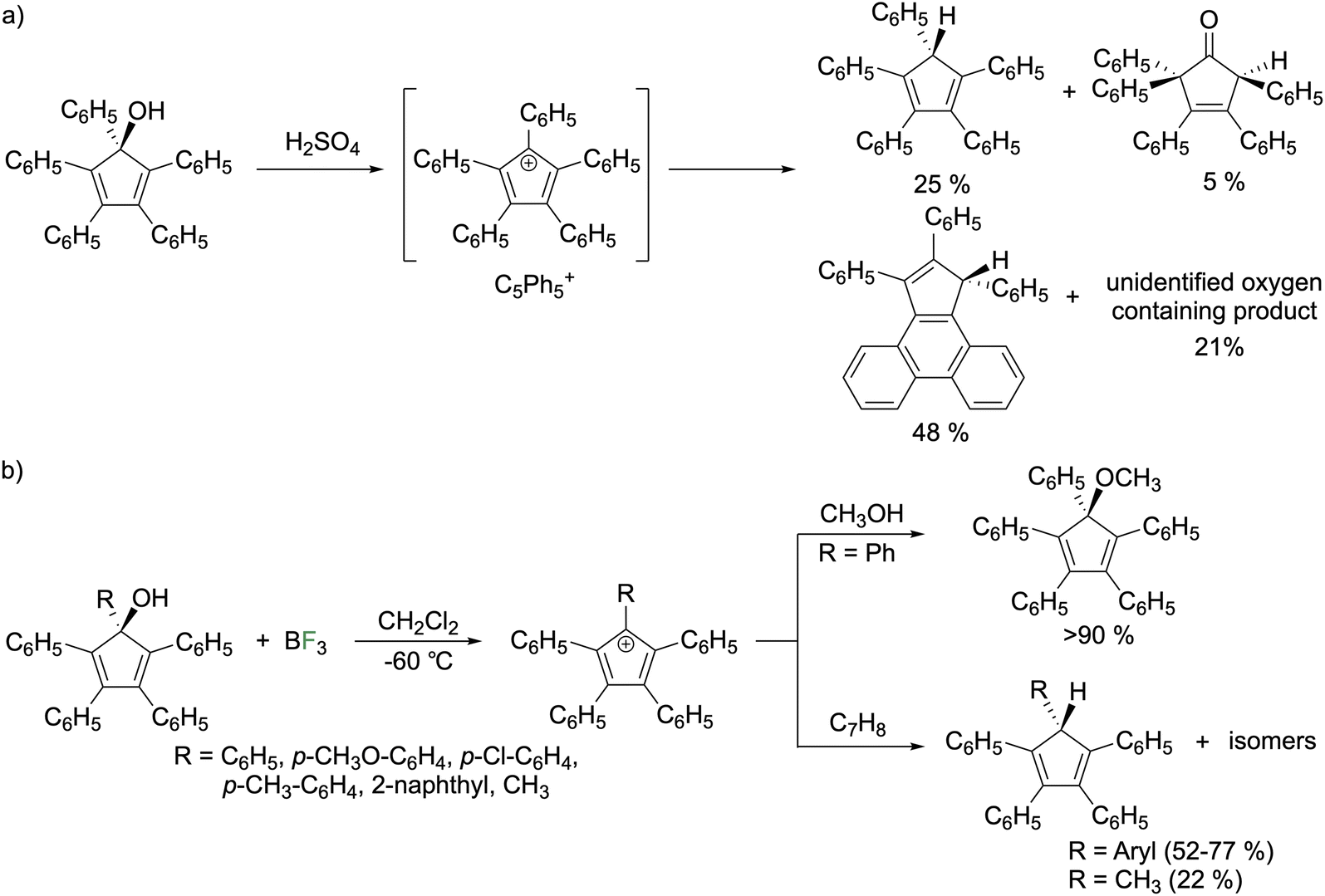 | ||
| Fig. 6 (a) Generation of C5Ph5+ from the protonation of cyclopentadienol and (b) from alcohol abstraction by BF3. | ||
In 2014, variants of C5Ph5+ with –C6F5 groups in place of phenyl groups on four of the carbon atoms and a methyl or phenyl on the other carbon were computationally investigated with the goal of increasing the singlet–triplet gap to identify an isolable target (Fig. 7).5 In the phenyl variant, the singlet state is predicted to be stable by 0.26 kcal mol−1 and by 2.61 kcal mol−1 in the methyl variant. Installing trifluoromethyl groups on the carbons adjacent to the formal carbocationic center revealed the stronger electron withdrawing groups drastically stabilized the singlet state with singlet–triplet gaps of nearly 10 kcal mol−1. Substitution of electron withdrawing groups at four carbons effectively renders these carbon atoms more electron deficient than the fifth to mimic the electronic structure of a borole ring (computed singlet–triplet gap of pentaphenylborole = 15 kcal mol−1 in favor of the singlet). Counterintuitively, the study showed that use of electron withdrawing groups to stabilize an electron poor Cp+ cation was a potentially viable strategy.
 | ||
| Fig. 7 Theoretically studied Cp+ species bearing electron withdrawing groups and their calculated singlet–triplet energy gaps, with the singlet being lower in energy in all cases. | ||
Earlier this year, Schulz and Haberhauer used five pentafluorophenyl groups as substituents to isolate a stable cyclopentadienyl cation, C5(C6F5)5+.2 Their synthetic approach was inspired by Ziegler and Breslow's early work by using a pentaaryl substituted alcohol,1,26 in this case pentakis(pentafluorophenyl)cyclopentadienol (Fig. 8a). Attempts to convert the alcohol to C5(C6F5)5+ with triflic anhydride in the presence or absence of bases, HBr in acetic acid, or PCl5 did not result in any reaction. Using super acidic conditions, similar to those used by Olah to generate the tert-butyl carbocation,27 reaction of the alcohol with SbF5·SO2 generated the targeted C5(C6F5)5+ paired with an [Sb3F16]− anion that was isolable as a dark blue material, the same color as pentaphenylborole.28 The triplet state is calculated to be favored in the gas phase by 2–6 kcal mol−1, depending on the computational method, however the molecule is EPR silent and the NMR spectrum is consistent with a diamagnetic singlet species. Stabilization of the observed singlet state was rationalized from interactions with the C6F6 solvent or the anion, which was supported by calculations, albeit only by a few kcal mol−1. A key factor in the relative stability of C5(C6F5)5+ as compared to C5Ph5+ is proposed to arise from the absence of C–H bonds. While it seems counterintuitive to stabilize an electron poor species by adding electron withdrawing groups, the –C6F5 groups shut down decomposition pathways that C–H bonds are known to be vulnerable to.
 | ||
Fig. 8 (a) Synthesis of C5(C6F5)5+ and (b) of  , (c) reduction of C5(C6F5)5+ to , (c) reduction of C5(C6F5)5+ to  . . | ||
Reaction of the pentakis(pentafluorophenyl)cyclopentadienol precursor with AlBr3 and bromoethane in benzene formed the neutral radical, 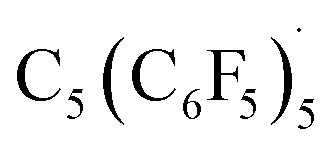 (Fig. 8b). The radical could be oxidized by SbF5·SO2 and XeF2 to furnish C5(C6F5)5+. The electron affinity of C5(C6F5)5+ is tremendously high, evidenced by the reverse reaction occurring with very poor reducing agents including alkanes and ortho-difluorobenzene (Fig. 8c).
(Fig. 8b). The radical could be oxidized by SbF5·SO2 and XeF2 to furnish C5(C6F5)5+. The electron affinity of C5(C6F5)5+ is tremendously high, evidenced by the reverse reaction occurring with very poor reducing agents including alkanes and ortho-difluorobenzene (Fig. 8c).
Single crystal X-ray diffraction studies on crystals of [C5(C6F5)5][Sb3F16] grown from C6F6 resulted in two structures that differ by their solid state interactions.2 In one, there are contacts between the anion and C5(C6F5)5+ whereas for the other, a C6F6 solvate interacts with the cation. Both structures feature a planar central C5 ring and with two localized π bonds [C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C range 1.359(8) to 1.378(5) Å] despite the symmetrical substitution pattern indicating that C5(C6F5)5+ exists as an antiaromatic singlet cation. Solid state magnetic susceptibility measurements also suggest singlet behavior. Reactivity studies showed high Lewis acidity of C5(C6F5)5+ with coordination of the weak Lewis base CO, analogous to borole chemistry.29,30 This was corroborated by theoretical calculations of fluoride and hydride affinities for which higher values were obtained than those for the perfluorinated tritylium cation C(C6F5)3+,28 indicating that C5(C6F5)5+ should be a potent Lewis acid.
C range 1.359(8) to 1.378(5) Å] despite the symmetrical substitution pattern indicating that C5(C6F5)5+ exists as an antiaromatic singlet cation. Solid state magnetic susceptibility measurements also suggest singlet behavior. Reactivity studies showed high Lewis acidity of C5(C6F5)5+ with coordination of the weak Lewis base CO, analogous to borole chemistry.29,30 This was corroborated by theoretical calculations of fluoride and hydride affinities for which higher values were obtained than those for the perfluorinated tritylium cation C(C6F5)3+,28 indicating that C5(C6F5)5+ should be a potent Lewis acid.
Conclusions
The 99 year journey from Ziegler generating a cyclopentadienyl cation to the isolation of C5(C6F5)5+ is a reminder to synthetic chemists to pursue what the literature describes as fleeting compounds or cryogenic matrix observed species. Clearly the substitution pattern on the organic groups appended to the 5-membered ring is the critical factor in achieving an isolable molecule. This was the case for the isoelectronic borole chemistry in which the substitution on the BC4 ring greatly impacts the stability, optical properties, and reactivity. The future of cyclopentadienyl chemistry is exciting, and we look forward to advancements to come.Data availability
There is no original data associated with this perspective article.Author contributions
S. Ranasinghe, C. D. Martin, and J. L. Dutton all contributed to literature searches and the composition of this perspective.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
J. L. D. thanks the Australian Research Council for support (FT16010007). C. D. M. is grateful to the Welch Foundation (Grant No. AA-2203-20240404) and the National Science Foundation (Award No. 2349851) for their generous support of this work.References
- K. Ziegler and B. Schnell, Justus Liebigs Ann. Chem., 1925, 445, 266–282 CrossRef CAS.
- Y. Schulte, C. Wölper, S. M. Rupf, M. Malischewski, D. J. SantaLucia, F. Neese, G. Haberhauer and S. Schulz, Nat. Chem., 2024, 16, 651–657 CrossRef CAS PubMed.
- W. Zou, M. Filatov and D. Cremer, Int. J. Quantum Chem., 2012, 112, 3277–3288 CrossRef CAS.
- H. J. Wörner and F. Merkt, Angew. Chem., Int. Ed., 2009, 48, 6404–6424 CrossRef.
- K. J. Iversen, D. J. D. Wilson and J. L. Dutton, Chem.–Eur. J., 2014, 20, 14132–14138 CrossRef CAS PubMed.
- H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903–10914 RSC.
- H. Braunschweig, I. Krummenacher and J. Wahler, in Advances in Organometallic Chemistry, Academic Press, 2013, vol. 61 Search PubMed.
- J. H. Barnard, S. Yruegas, K. Huang and C. D. Martin, Chem. Commun., 2016, 52, 9985–9991 RSC.
- J. He, F. Rauch, M. Finze and T. B. Marder, Chem. Sci., 2020, 12, 128–147 RSC.
- R. Breslow and J. M. Hoffman Jr, J. Am. Chem. Soc., 1972, 94, 2110–2111 CrossRef CAS.
- M. Saunders, R. Berger, A. Jaffe, J. M. McBride, J. O'Neill, R. Breslow, J. M. Hoffmann, C. Perchonock and E. Wasserman, et al. , J. Am. Chem. Soc., 1973, 95, 3017–3018 CrossRef CAS.
- P.-K. Lo and K.-C. Lau, J. Phys. Chem. A, 2014, 118, 2498–2507 CrossRef CAS PubMed.
- B. Reindl and P. V. R. Schleyer, J. Comput. Chem., 1998, 19, 1402–1420 CrossRef CAS.
- M. Stojanović, J. Aleksić and M. Baranac-Stojanović, Chemistry, 2021, 3, 765–782 CrossRef.
- R. Breslow, R. Hill and E. Wasserman, J. Am. Chem. Soc., 1964, 86, 5349–5350 CrossRef CAS.
- S. M. Rupf, P. Prohm and M. Malischewski, Chem. Commun., 2020, 56, 9834–9837 RSC.
- P. Costa, I. Trosien, J. Mieres-Perez and W. Sander, J. Am. Chem. Soc., 2017, 139, 13024–13030 CrossRef CAS PubMed.
- J. B. Lambert, L. Lin and V. Rassolov, Angew. Chem., Int. Ed., 2002, 41, 1429–1431 CrossRef CAS.
- M. Otto, D. Scheschkewitz, T. Kato, M. M. Midland, J. B. Lambert and G. Bertrand, Angew. Chem., Int. Ed., 2002, 41, 2275–2276 CrossRef CAS.
- J. N. Jones, A. H. Cowley and C. L. B. Macdonald, Chem. Commun., 2002, 1520–1521 RSC.
- T. Müller, Angew. Chem., Int. Ed., 2002, 41, 2276–2278 CrossRef.
- J. B. Lambert, Angew. Chem., Int. Ed., 2002, 41, 2278 CrossRef CAS.
- S. M. Bloom and A. P. Krapcho, Chem. Ind., 1959, 882 CAS.
- R. Breslow and H. W. Chang, J. Am. Chem. Soc., 1961, 83, 3727–3728 CrossRef.
- W. A. Yager, R. Breslow and H. W. Chang, J. Am. Chem. Soc., 1963, 85, 2033–2034 Search PubMed.
- R. Breslow, H. W. Chang, R. Hill and E. Wasserman, J. Am. Chem. Soc., 1967, 89, 1112–1119 Search PubMed.
- G. A. Olah, E. B. Baker, J. C. Evans, W. S. Tolgyesi, J. S. McIntyre and I. J. Bastien, J. Am. Chem. Soc., 1964, 86, 1360–1373 CrossRef.
- J. J. Eisch, N. K. Hota and S. Kozima, J. Am. Chem. Soc., 1969, 91, 4575–4577 CrossRef.
- A. Fukazawa, J. L. Dutton, C. Fan, L. G. Mercier, A. Y. Houghton, Q. Wu, W. E. Piers and M. Parvez, Chem. Sci., 2012, 3, 1814–1818 Search PubMed.
- Z. Wang, Y. Zhou, T. B. Marder and Z. Lin, Org. Biomol. Chem., 2017, 15, 7019–7027 RSC.
| This journal is © The Royal Society of Chemistry 2025 |