
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing electrocatalytic hydrogen evolution via engineering unsaturated electronic structures in MoS2
Qingqing
Zhou†
a,
Hao
Hu†
b,
Zhijie
Chen
 c,
Xiao
Ren
*a and
Ding
Ma
*a
c,
Xiao
Ren
*a and
Ding
Ma
*a
aBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: dma@pku.edu.cn
bCollege of Environment, Zhejiang University of Technology, Hangzhou 310012, PR China
cSchool of Civil and Environmental Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 18th December 2024
Abstract
The search for efficient, earth-abundant electrocatalysts for the hydrogen evolution reaction (HER) has identified unsaturated molybdenum disulfide (MoS2) as a leading candidate. This review synthesises recent advancements in the engineering of MoS2 to enhance its electrocatalytic properties. It focuses on strategies for designing an unsaturated electronic structure on metal catalytic centers and their role in boosting the efficiency of the hydrogen evolution reaction (HER). It also considers how to optimize the electronic structures of unsaturated MoS2 for enhanced catalytic performance. This review commences with an examination of the fundamental crystal structure of MoS2; it elucidates the classical unsaturated electron configurations and the intrinsic factors that contribute to such electronic structures. Furthermore, it introduces popular strategies for constructing unsaturated electronic structures at the atomic level, such as nanostructure engineering, surface chemical modification and interlayer coupling engineering. It also discusses the challenges and future research directions in the study of MoS2 electronic structures, with the aim of broadening their application in sustainable hydrogen production.
1 Introduction
As global demand for clean energy continues to rise, the development of efficient hydrogen production has emerged as a critical focus. Water electrolysis is a particularly promising approach due to its efficiency and environmentally friendly nature.1–3 However, its implementation depends heavily on the development of high-performance and durable catalysts for the hydrogen evolution reaction (HER).4,5 Traditionally, precious metals like platinum (Pt) have been the catalysts of choice due to their exceptional HER activity, which is attributed to their moderate binding interaction with key adsorbates, such as intermediate hydrogen species (H*).6,7 However, the scarcity and high cost of Pt and other noble metals have driven increasing interest in the search for earth-abundant alternatives.8–10 This shift is crucial to making hydrogen production more economically viable and scalable for widespread clean energy applications.Using density functional theory (DFT) calculations, Hinnemann and colleagues identified earth-abundant materials with catalytic properties similar to platinum (i.e., near-zero free energy for H* binding) for HER.11 Among these candidates, molybdenum disulfide (MoS2) was predicted as a promising HER catalyst, despite practical bulk MoS2 being regarded as a poor electrocatalyst.12,13 This apparent contradiction was later clarified by Jaramillo and Ib Chorkendorff, who demonstrated that hydrogen evolution predominantly occurs at the edge sites of layered MoS2, where unsaturated electronic structures are present.14 In contrast, the basal plane of MoS2, with its fully saturated electronic configuration, contributes negligibly to HER due to a lack of active sites.15,16 These findings underscore the significance of engineering unsaturated electronic structure of MoS2, supporting Norskov's perspective that the electrocatalytic performance of a material is fundamentally determined by its electronic structure.
Beyond exploring the atomic-level structure–activity relationship, numerous studies have focused on maximizing the intrinsic catalytical performance of MoS2 while developing additional electric properties to contribute HER progress. For example, Jin and Cui pioneered the use of Li+ intercalation to successfully tune the electronic states of MoS2, transforming semiconducting 2H-MoS2 into metallic 1T-MoS2. Their research demonstrated that Li+ intercalation introduces single electrons into the Mo d-orbitals, achieving a new stable state through d-orbital re-splitting. This transformation significantly enhances hydrogen species activation and improves charge transfer kinetics.12,17 In a complementary study, Tsai and colleagues introduced sulfur defects in the basal plane of MoS2, generating high-energy dangling bonds that increased the mass activity of the catalyst. Their findings suggest that thermodynamically unstable, coordinatively unsaturated sites are more prone to adsorbing H* intermediates, playing a key role in the HER process.18 Beyond structural modifications to the 2H phase of MoS2, researchers have explored the introduction of various metal and non-metal dopants to fine-tune the local coordination environment, electron density, and energy level structure of the catalytic centers. For instance, DFT calculations by Cui predicted how different transition metal dopants (e.g., Co, Fe, Ni, and Cu) impact the HER performance of MoS2.19 These strategies demonstrate that engineering unsaturated electron configurations in MoS2 can significantly enhance its electrochemical properties, paving the way for more efficient HER catalysts.
While it has vaguely perceived that unsaturated sites in MoS2 are somehow linked to its electrocatalytic performance, the precise nature of this relationship remains unclear. To address this, researchers have conducted extensive studies to establish design principles for MoS2 catalysts with high HER activity, particularly focusing on the interaction between intermediate hydrogen species and catalytic centers.20–23 Previous reviews have categorized modification methods for MoS2 catalysts, aiming for advanced technologies that offer high catalytic activity, stability, preparation efficiency, and etc. In contrast, our review provides a systematic summary of the unsaturated electronic structures in modified MoS2 catalysts from the perspective of band structure and electronic orbital arrangement, which are decisive factors in determining electrocatalytic performance. Additionally, we provide practical guidance for how to construct active catalytic centers with idea unsaturated electronic structures: (1) through nanostructure engineering, single-layer or multi-exposed-edge MoS2 materials are synthesized without altering their chemical composition. Additionally, stress regulation is employed to introduce or relieve lattice strain, facilitating the formation of active defects in the distorted structure. (2) Surface chemical modification strategies-such as creating vacancy defects, heteroatom substitution, heterogeneous doping, and incorporating heterogeneous crystal domains-are used to modify the local chemical environment of the catalytic centers. These methods regulate the chemical properties of MoS2 at the micro-regional level, thereby enhancing its HER performance. (3) Semiconductor/metal contact effects have emerged as a promising approach for constructing robust and highly efficient catalytic electrodes. By modulating the van der Waals gap between MoS2 catalytic layers and metal supports, the orbital overlap and hybridization at the interface can regulate the in-plane electronic structure of MoS2, thereby promoting the exposure of active centers and enhancing HER efficiency. Finally, we discuss the challenges and future research directions in optimizing the electronic structures of MoS2 catalysts, with the aim of promoting their broader application in sustainable hydrogen production.
2 Fundamentals of MoS2 electrocatalyst for HER
2.1 HER mechanism
HER is a two-electron/proton coupling process that occurs on the cathode surface during water electrolysis, with the reaction mechanism dependent on the pH of the solution.24,25 As outlined in Table 1, the HER involves two main steps: adsorption (Volmer) and desorption (Heyrovsky/Tafel). First, the Volmer step initiates the reaction, where hydrogen ions (in acidic environments) or water molecules (in alkaline environments) are electrochemically adsorbed onto the catalyst surface, forming an intermediate hydrogen species (Hads). The Heyrovsky and Tafel steps describe two desorption pathways for Hads. The Heyrovsky step, predominant at low Hads surface coverage, involves Hads reacting with another hydrogen ion or water molecule to form H2. The Tafel step, occurring at high Hads surface coverage, involves the combination of two Hads to produce H2.26 It is clear that an effective HER catalyst is essential for optimizing these adsorption and activation pathways.| Acid solution | Alkaline solution | |
|---|---|---|
| Volmer step | M + H+ + e− → Hads | M + H2O + e− → Hads + 2OH− |
| Heyrovsky step | M–Hads + H+ + e− → H2 | M–Hads + H2O + e− → H2 + OH− |
| Tafel step | 2M–Hads → H2 + 2M | 2M–Hads → H2 + 2M |
| Overall reaction | 2H+ + 2e− → H2 | 2H2O + 2e− → H2 + 2OH− |
2.2 Basics of MoS2 crystal
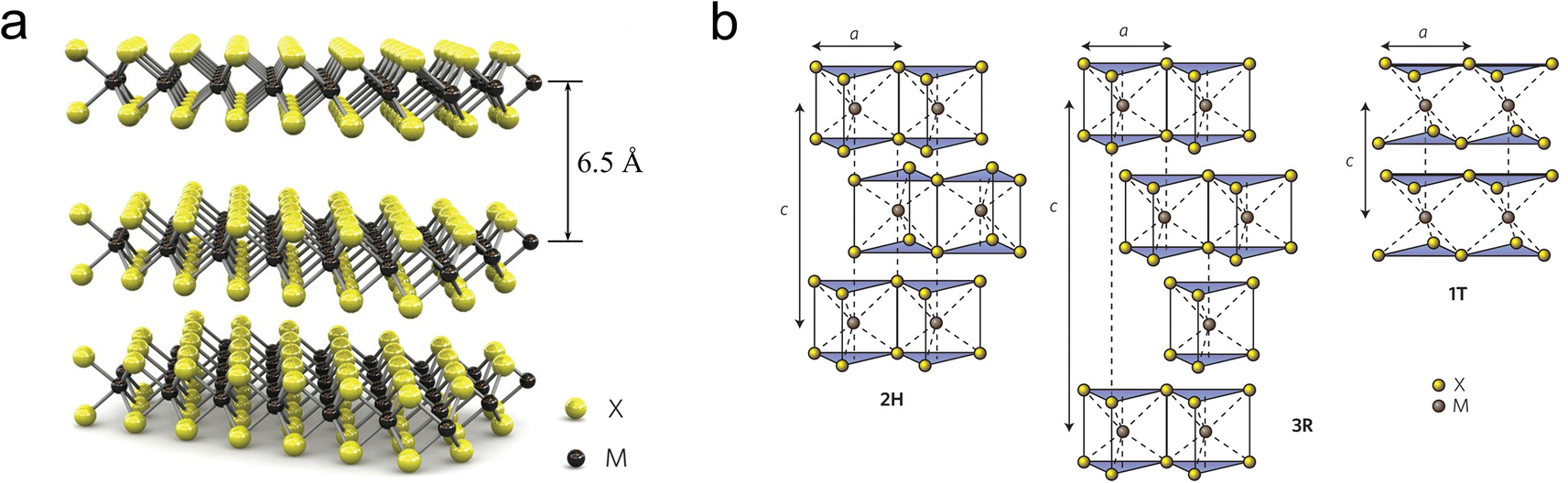 | ||
| Fig. 1 (a) Schematic diagram of the MoS2 crystal structure, (b) schematic diagram of the structures of 2H phase MoS2 (hexagonal symmetry, two layers per repeating unit, trigonal prism coordination), 3R phase MoS2 (rhombohedral symmetry, three layers per repeating unit, trigonal prism coordination) and 1T phase MoS2 (tetragonal symmetry, one layer per repeating unit, octahedral coordination), the stacking index c represents the number of layers in each stacking sequence, and the interlayer spacing is about 6.5 Å. | ||
As depicted in Fig. 1b, depending on the specific coordination of Mo atoms, MoS2 cell can exist in either a trigonal prismatic or octahedral configuration.30 Based on the phase structure and stacking sequence, MoS2 has three standard crystal structures: 2H phase (H stands for hexagonal) with two S–Mo–S molecular units stacking in an AB manner along the vertical (c-axis) direction; 3R (R stands for rhombohedral) with three S–Mo–S units stacked in an ABC manner along the c-axis; 1T phase (T stands for trigonal) with a monolayer S–Mo–S molecular per unit cell.31,32 The energy level splitting, electron filling, and spin states of the Mo d-orbitals vary with the coordination structure, leading to differences in electrical properties, magnetic properties, and thermodynamic stability.33,34 Among these, 2H-MoS2 is the most stable, while the 1T and 3R phases exhibit metastable behavior.35
Due to the varying orientations and shapes of the d orbitals in molybdenum ions, the d orbitals of the central metal atom experience electrostatic interactions with ligands that differ in both direction and energy depending on the crystal field environment. To clarify, 1T-phase MoS2 adopts an octahedral molecular configuration with an Oh point group, where six S ligands are positioned at the vertices of a symmetric octahedron surrounding the Mo atom. The electron clouds of the dz2 and dx2−y2 orbitals are located on the connecting line of Mo–S and directly face the negatively charged ligands, resulting in stronger repulsive interactions and higher energy levels, eg band.36,37 On the contrary, the electron clouds of the dxy, dyz, and dxz orbitals with spatial deviation from the Mo–S axis and avoid the ligands, leading to weaker repulsive interactions and lower energy t2g band (Fig. 2b). These degenerate orbitals remain non-bonding, allowing for quasi-continuous occupation by fermions, thereby giving MoS2 metallic properties. However, for nonlinear molecules, if their ground electronic state is orbitally degenerate, the molecule becomes unstable. In such cases, the Jahn–Teller distortion effect occurs to relieve the degeneracy.
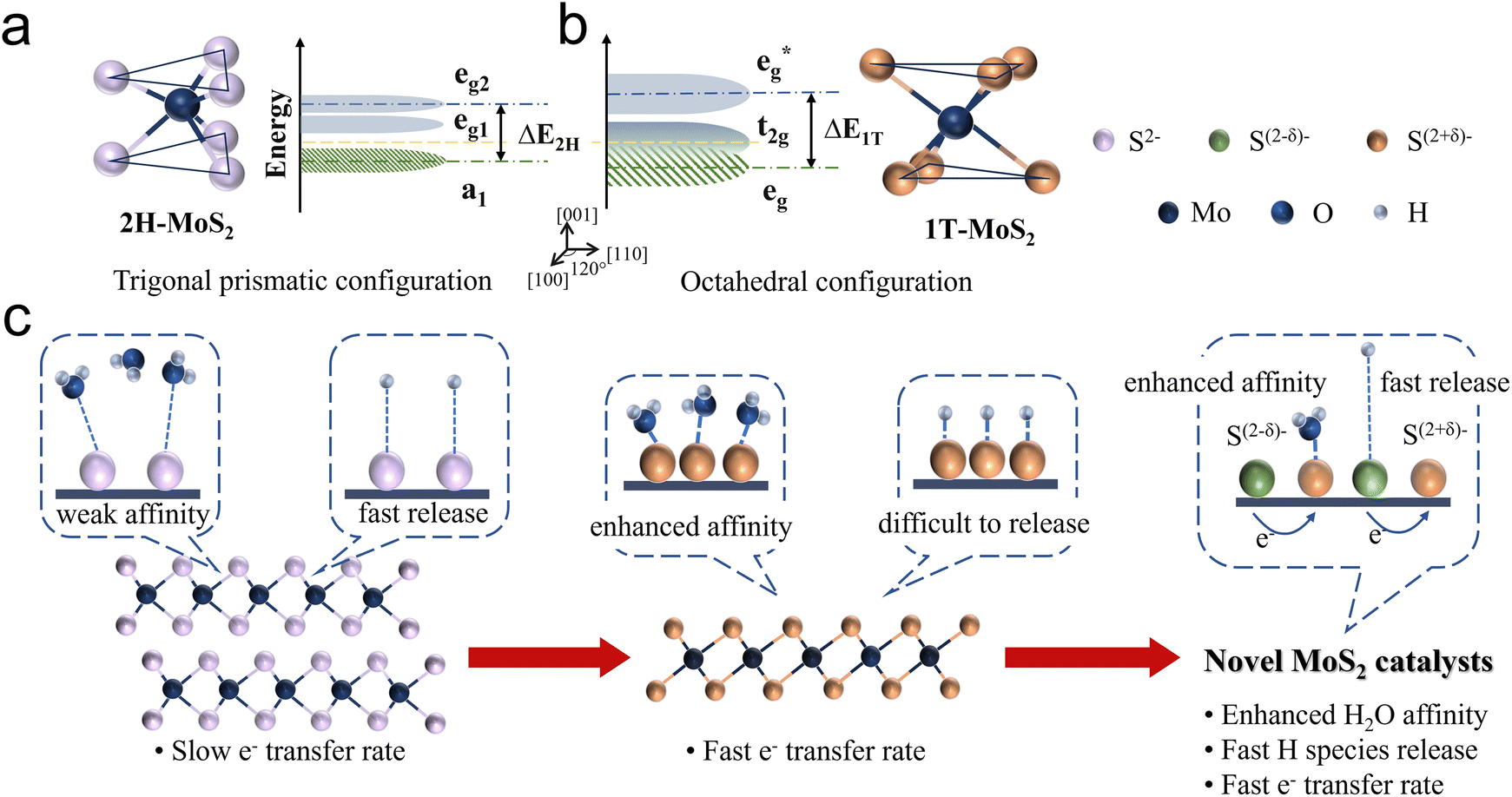 | ||
| Fig. 2 The unit cell structure, band structure and Fermi level position of (a) 2H phase MoS2 and (b) 1T phase MoS2. (c) HER catalytic behavior on 2H phase MoS2, 1T phase MoS2, and the principle to modify MoS2. | ||
The molecular configuration of 2H-MoS2 is such a distorted octahedron. By rotating one triangular face by 60° relative to the opposite face, a trigonal prismatic molecular configuration, point group D3h, is obtained. In this trigonal prismatic configuration, due to the lack of symmetrical alignment between the S ligands and the Mo 4d orbitals, the d orbitals tend to overlap “sideways” resulting in repulsion from all ligands. This causes the molecular orbitals to split into bonding and antibonding bands, creating an empty band gap. In this case, the d orbitals split into three bands: eg2, eg1, and a1, corresponding to the dxz, dyz; dxy, dx2−y2; and dz2, respectively (Fig. 2a).
As discussed above, the 2H and 1T crystalline phases of MoS2, the two most widely applied phases, exhibit fundamentally distinct solid-state physical and electronic properties, resulting in notable differences in HER catalytic performance. Regarding the HER process (Fig. 2c), the semiconducting 2H phase is characterised by a significant band gap and limited electron mobility. Additionally, its strong bonding within the basal plane provides excellent thermodynamic stability but renders it catalytically inert. Consequently, the HER activity of 2H-MoS2 primarily originates from its exposed edge sites, where unsaturated electronic states facilitate the adsorption and conversion of intermediates. This highlights edge engineering as a critical strategy for enhancing the catalytic performance of the 2H phase. In contrast, the metallic 1T phase offers excellent electronic conductivity, enabling rapid electron transfer during the HER process. Furthermore, the high density of states near the Fermi level strengthens interactions between 1T-MoS2 and water molecules, promoting efficient adsorption and activation of intermediates. However, the 1T phase suffers from structural instability, often requiring stabilization strategies such as intercalation or heteroatom doping to maintain its catalytic performance under operational conditions.
2.3 Relationship between electron configuration of MoS2 and its electrocatalytic activity
Generally, the basal plane of MoS2 exhibits limited catalytic activity due to its saturated electronic configuration.38,39 However, modifying the coordination environment of the Mo centers and tuning the energy levels of their d orbitals offer significant opportunities for creating high-activity, unsaturated MoS2 configurations, thereby enhancing its catalytic potential for HER.40,41 Numerous efforts have been made in this direction, as presented in as presented in Fig. 3 and Table 2. To clarify, in the ground-state 2H-MoS2 with a trigonal prismatic structure, the crystal field splitting energy is greater than the electron pairing energy. Following the Aufbau principle, electrons first occupy the low-energy dz2 orbital with two electrons of opposite spins, resulting in diamagnetism. Interestingly, when the host Mo atom is substituted by noble metals such as Pt and Rh, electrons are lost from the dz2 orbital due to the higher electronegativity of these metals.42,43 Consequently, this results in a low-spin unsaturated state that exhibits metallic-like properties. | ||
| Fig. 3 Orbital electron unsaturation strategy for MoS2 electrocatalysts based on crystal field theory. | ||
| Catalysts | Phase | Modification approach | Description of band structure and electrons state | Conductivity | Ref. |
|---|---|---|---|---|---|
| Pristine MoS2 | 2H | — | Three bands: a1 (dz2), eg1 (dxy, dx2−y2), and eg2 (dyz, dxz) | Semi-conductor | 34 |
| Electron state: paired electrons fully occupy the lowest a1 band | |||||
| Pt-MoS2 | 2H | Mo loses electrons through substitution with highly electronegative noble metals | Three bands: a1 (dz2), eg1 (dxy, dx2−y2), and eg2 (dyz, dxz) | Metallic-like | 42 |
| Electron state: single-spin electron half-fill the lowest a1 band | |||||
| FD-MoS2 | 2H | Mo losing electrons by making Frenkel disorder | Three bands: a1 (dz2), eg1 (dxy, dx2−y2), and eg2 (dyz, dxz) | Metallic-like | 42 |
| Electron state: single-spin electron half-fill the lowest a1 band | |||||
| Vs-MoS2 | 2H | Adding electrons by making S vacancy | Three bands: a1 (dz2), eg1 (dxy, dx2−y2), and eg2 (dyz, dxz) | Metallic-like | 43 |
| Electron state: paired electrons fully occupy the lowest a1 band, and single-spin electron incompletely occupy the higher eg1 band | |||||
| 1T-MoS2 | 1T | — | Two bands: t2g (dxy, dyz, dxz) and eg (dx2−y2, dz2) | Metallic | 34 |
| Electron state: two parallel-spin electrons incompletely occupy the degenerate t2g band | |||||
| CoO@1T-MoS2 | 1T | Adding electrons by co-substituting Mo and S with Co and O atoms | Two bands: t2g (dxy, dyz, dxz) and eg (dx2−y2, dz2) | Metallic | 44 |
| Electron state: three parallel-spin electrons half-fill the degenerate t2g band | |||||
| 1T′-MoS2 | Stretch 1T phase | Re-split band structure by intercalating alkine ions | Two major bands: t2g (dxy, dyz, dxz) and eg (dx2−y2, dz2) | Metallic-like | 45 |
| Electron state: two parallel-spin electrons half-fill the lower t2g band | |||||
| 1T′′-MoS2 | Stretch 1T phase | Re-split band structure by chemical exfoliation | Two major bands: t2g (dxy, dyz, dxz) and eg (dx2−y2, dz2) | Metallic-like | 46 |
| Electron state: two parallel-spin electrons half-fill the lower t2g band | |||||
| 1T′′′-MoS2 | Compress 1T phase | Re-split band structure by de-intercalating of alkine ions | Two major bands: t2g (dxy, dyz, dxz) and eg (dx2−y2, dz2) | Semi-conductor | 45 |
| Electron state: paired electrons fully occupy the lower t2g band |
Conversely, donating additional electrons to the eg band is more challenging due to the presence of a band gap. Creating vacancies can be a viable approach, as it destabilizes the lattice. This destabilization may trigger a transition from the 2H to the 1T phase upon electron absorption. However, the behavior of electrons differs significantly in the octahedral case. First, the crystal field splitting energy is lower than the electron pairing energy, allowing electrons to preferentially fill all orbitals of similar energy while avoiding pairing. As a result, the incomplete occupation of the t2g orbitals in 1T-MoS2 leads to an intermediate spin state, which demonstrates moderate activity but low stability.
Doping 1T-MoS2 with electron-donating atoms (e.g., Co, Mn, Fe) can achieve a half-filled t2g band, promoting a higher spin state and enhanced stability.44,47 Furthermore, significant distortion occurs when 1T-MoS2 undergoes external strain, such as compression or stretching. This strain can induce the spontaneous conversion of the 1T phase into 1T′ and 1T′′ phases, thereby lowering its energy through Jahn–Teller deformation effects.40,46 Typically, these distorted phases feature zigzag Mo–Mo chains and the formation of Mo–Mo bonds, such as dimerized Mo (1T′′) or trimerized Mo (1T′′′) atoms. These structures may act as key catalytic sites during the HER process, although they are sometimes overlooked in catalytic evaluation.
3 Nanostructure engineering
The differences in atomic arrangement and geometry contribute to the diverse physical and chemical properties of MoS2 catalysts. This includes variations in polymorphic structures (e.g., 2H and 1T phases), as well as band structure and crystal plane exposure, which are influenced by the number of layers and the structure of MoS2 catalysts.48,49 In recent years, geometry and strain engineering of nanomaterials has attracted great attention by adjusting the properties and electrocatalytic performance of MoS2 by regulating the crystal atomic arrangement and deformation.50,51 This section outlines recent advances in the development of monolayer materials, edge dangling bond engineering, and stress regulation strategies for the design of advanced MoS2 catalysts.3.1 Atomically thin structure
Bulk MoS2, held together by van der Waals (vdW) interactions, exhibits the properties of an indirect bandgap semiconductor.52,53 Differently, the monolayer MoS2 obtained by Novoselov et al. through mechanical exfoliation in 2005 is considered to have a direct band gap with metallic properties.54 Therefore, it was found that the electrical properties of MoS2 are highly related to the number of layers, and few-layer or monolayer MoS2 shows great potential in electrocatalysis due to its unique electrical properties.55,56 As depicted in Fig. 4a–g, to achieve controllable synthesis of monolayer MoS2, Zhang's group has focused on exploring the fabrication of atomically thin monolayer MoS2 films. These efforts include the epitaxial growth of monolayer WS2 on sapphire and the growth of monolayer MoS2 of various sizes on Au foil via low-pressure chemical vapor deposition (CVD), both of which can serve as outstanding electrocatalysts for HER.57–59 Based on this, Zhang and coauthors successfully prepared a four-layer 2H-TaS2 film on an Au foil, exposing edge active sites by controlling the number of layers. This approach achieved Pt-like hydrogen evolution efficiency, with a Tafel slope of 33–42 mV dec−1 and an exchange current density of ∼179.47 μA cm−2 (Fig. 4h and i).60 Additionally, Ye et al. achieved high-yield monolayer MoS2 films by hydrolyzing a lithium salt-containing MoS2 precursor, resulting in a monolayer MoS2 catalyst with a catalytic activity of 1563.6 μmol h−1.61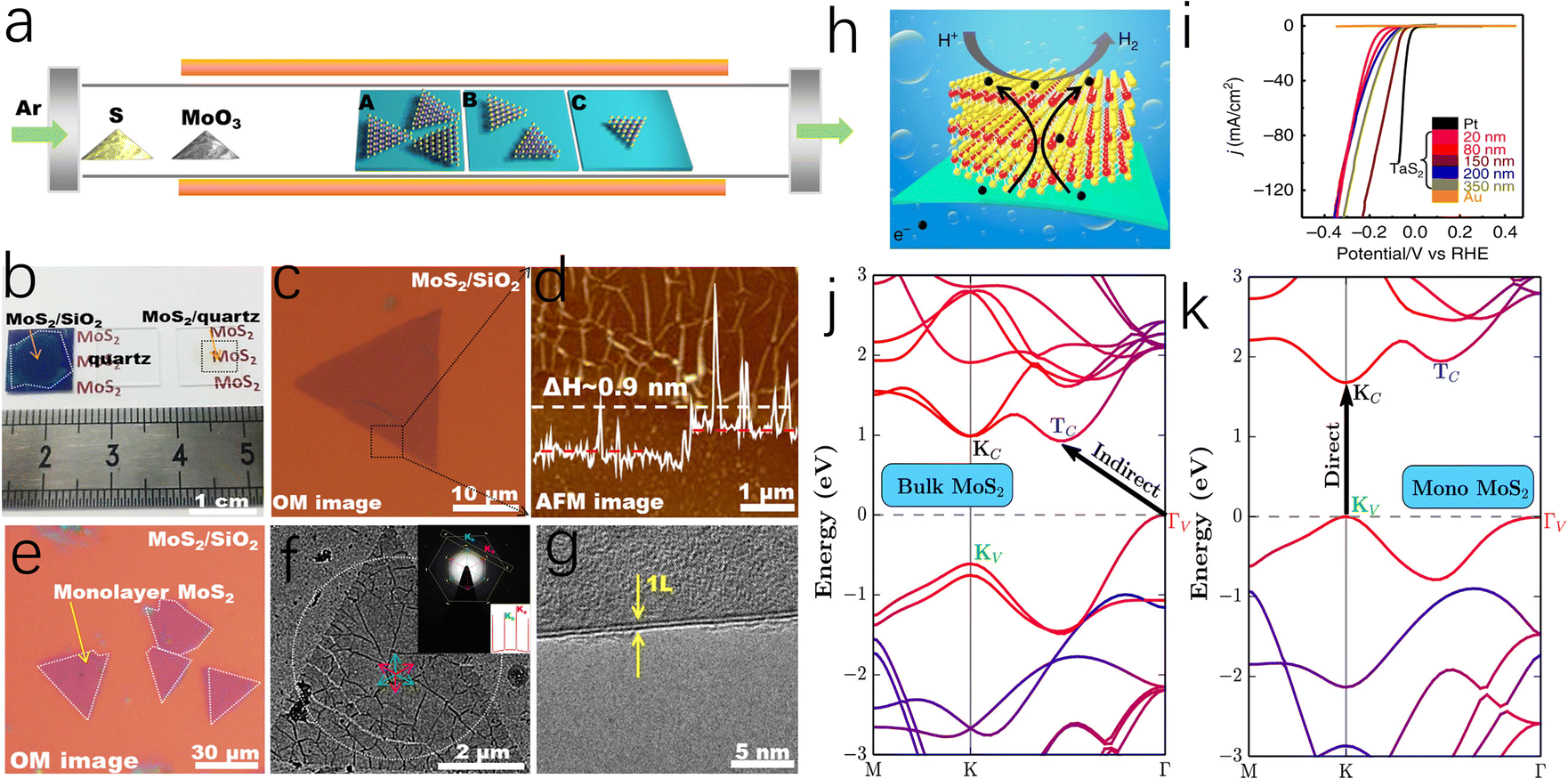 | ||
| Fig. 4 (a) Schematic representation of the dependence of coverage or flake size on different precursor-substrate distances. (b) Photographs of monolayer MoS2 films on SiO2/Si, a bare quartz substrate, and MoS2 on quartz. (c) A triangular MoS2 flake transferred onto SiO2/Si substrate. (d) Atomic force microscopy (AFM) image captured from the dashed box in (c) and corresponding section-view along the dashed arrow showing the monolayer feature of the flake (∼0.9 nm in apparent height). (e) Optical images of submonolayer MoS2 flakes after transference onto SiO2/Si and quartz substrates. (f) TEM image of a MoS2 triangle transferred on a carbon film supported on copper grids. (g) TEM image on a folded edge indicating the monolayer nature of the MoS2 flake. (h) Schematic illustration of the HER process of 2H-TaS2/Au foils. (i) Polarization curves (iR-corrected) of as-grown 2H-TaS2 with different thicknesses, Au foil, and commercial Pt. Band structure for MoS2 of the bulk (j) and the monolayer (k). The band structure has been projected onto constituting atomic species with the blue color representing S and red representing Mo. The band-edge states relevant to optical transition (KV, KC, ΓV, and TC) are labeled and the band gap transitions are indicated. | ||
To elucidate the mechanism by which the electronic properties of MoS2 are regulated by the number of layers, Zunger et al. employed first-principles calculations and revealed that in bulk MoS2, the valence band maximum and conduction band minimum are located at the Tv and Tc points in momentum space, respectively.62 However, in monolayer MoS2, these points shift to the K point in the Brillouin zone, inducing a bandgap transition, as illustrated in Fig. 4j and k. Secondly, the monolayer structure of MoS2 stabilizes the edge regions by eliminating constraints from adjacent layers, which facilitates the exposure of edge sites and the introduction of structural defects, thereby accelerating the HER process. Although monolayer or few layer MoS2 catalysts exhibit excellent electrical properties and a high ratio of exposed active edges, the exfoliation and preparation of such MoS2 catalysts remain long-standing challenges, making it unsuitable for the large-scale production of cost-effective HER catalysts.
3.2 Morphology control
The highly reactive edges of MoS2 are typically difficult to stabilize, leading bulk MoS2 to terminate with smooth, inert surfaces.63 To overcome this limitation, researchers have exploited the anisotropy and morphological diversity of MoS2 nanosheets to expose and stabilize active edge sites.64 For instance, as depicted in Fig. 5a, Cui et al. grew vertically MoS2 nanoarray on a Mo substrate via a CVD method, effectively increasing the reactive area and promoting the contact between the active edges and the electrolyte.65 This approach enables MoS2 to achieve an intrinsic turnover frequency (TOF) of 0.013 s−1 without additional energy input, which is close to the experimental fit value (0.016 s−1) for pure edge sites reported by Chorkendorff et al.14 In addition, Yang and co-workers also prepared a similar Mo-based MoS2 electrocatalyst, reaching remarkable stability over more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in 0.5 M H2SO4.66,67
000 cycles in 0.5 M H2SO4.66,67
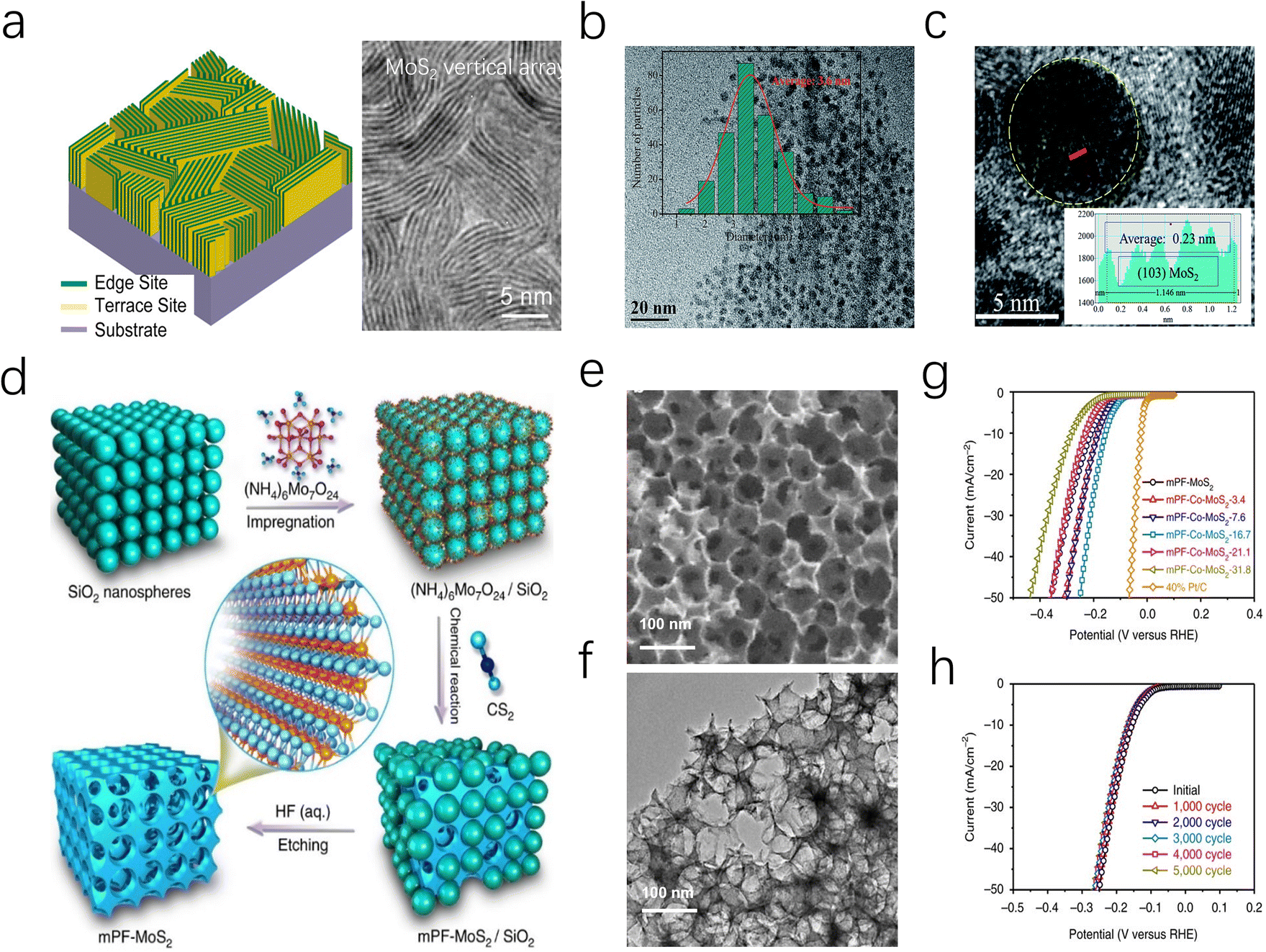 | ||
| Fig. 5 (a) (left) TEM images of MoS2 and MoSe2 films with edge-terminated structures. (right) Idealized structure of edge-terminated molybdenum chalcogenide films with the layers aligned perpendicular to the substrate. (b) TEM image of the MoS2 QDs, the inset displays the HRTEM image of the same. (c) The size distribution of the MoS2 QDs in (b). The red solid line is the simulated Gaussian curve. (d) Schematic illustration of the fabrication of mesoporous MoS2 foam (mPF-MoS2). (e and f) SEM and TEM images of mPF-MoS2. (g) HER polarization curves for mPF-Co-MoS2 with different Co doping contents in comparison with mPF-MoS2 and 40% Pt/C. (h) Durability measurement of mPF-Co-MoS2. | ||
In addition to utilizing metal foils, MoS2 nanosheets can also be fabricated into mesoporous foams (mPF-MoS2-SiO2) through SiO2 template etching. The resulting catalyst features an average pore size of ∼30 nm, which effectively enhances the transport of H3O+ and H+ ions. Moreover, the directional growth of MoS2 nanosheets around the mesopores contributes to edge stabilization, resulting in a catalyst that demonstrates cycle durability exceeding 5000 cycles (Fig. 5d–h). Preparing MoS2 as highly dispersed nanoparticles is a feasible approach to obtaining a large number of accessible edge sites.68 Jiang and coauthors successfully synthesized highly dispersed MoS2 nanoparticles on reduced graphene oxide-modified carbon nanotube/polyimide films (PI/CNT-RGO-MoS2) through an electrochemical method. The study indicates that PI/CNT-RGO facilitates the intimate growth of MoS2 particles on its surface, while the coupling between RGO and MoS2 enhances electron transfer between the edge sites and the substrate, enabling the catalyst to exhibit a low overpotential of 90 mV at a current density of 10 mA cm−2 and a Tafel slope of 61 mV dec−1.69
Reducing the dimension of 2D MoS2 to 0D quantum dots (MoS2 QDs) is another advanced strategy to achieve an ultra-high specific surface area and a unique edge-defect-rich structure (Fig. 5b and c). Moreover, monolayer MoS2 QDs can eliminate the interlayer barrier in the vertical direction, allowing more free electrons to transfer along the MoS2 surfaces. Under acidic conditions, the MoS2 QDs catalyst developed by Ren exhibits an overpotential of 39 mV at a current density of 10 mA cm−2, which is 14 times lower than that of bulk MoS2.70 More interestingly, Jaramillo et al. reported a unique double-helical MoS2 structure (DG) MoS2 film, which exposed a greater fraction of catalytically active edge sites through high surface curvature and promoted the diffusion process of H2 bubbles.63 Various morphology designs not only increase the active specific surface area of MoS2 catalysts but also expand their application potential. On one hand, morphology control allows for the manipulation of the micro-interface environment of electrodes, improving mass transfer behavior of the reactants. Additionally, in Amper-scale HER applications, nanoarray morphologies are expected to facilitate efficient H2 bubble desorption and diffusion, addressing kinetic challenges posed by massive hydrogen accumulation.
3.3 Strain engineering
Beyond controlling the number of layers and stacking patterns, applying local lattice strain or pressure engineering is a powerful approach for fine-tuning the band structure and electrical properties of TMDs. As depicted in Fig. 6a and b, Nayak et al. investigated the electronic structure and lattice vibrational dynamics of pristine 2H-MoS2 and distorted monolayer 1T-MoS2 using diamond anvil cell experiments and DFT calculations.71,72 Their study revealed that inevitable lattice distortion occurs, leading to an electronic transition from semiconducting to metallic at a pressure of approximately 19 GPa (Fig. 6c and d). The eventual metallization occurs due to the overlap of the valence and conduction bands, driven by S–S interactions as the interlayer gap decreases. Later, Zhao et al. demonstrated the z-axis in the MoS2 crystal cell is much more compressible than a-axis during the compression process, due to the weak vdW interaction between layers.73 Therefore, the eventual metallization arises from the overlap of the valence and conduction bands owing to the reduction of interlayer gap, which makes the MoS2 improved conductivity and faster HER kinetic process.74 | ||
| Fig. 6 (a) A 3D illustration of multilayered MoS2 in a DAC pressure medium for compression experiments. (b) Theoretical charge density isosurfaces of multilayered MoS2, charge accumulation (green) and depletion (orange) at the in-plane. (c) Theoretical calculation of the pressure-dependent band gap of multilayered MoS2. Inset: the unit cell of MoS2. (d) Pressure-dependent electrical resistivity of MoS2. Three characteristic regions have been identified: semiconducting (SC), intermediate state (IS) and metallic regions. (Inset) Theoretically calculated pressure-dependent electrical resistivity. (e) Schematic models of single-layered MoS2 with 2H and 1T phases in basal plane and cross-section views. Mo, blue; top S, orange; bottom S′, purple. (f) Transformation of 2H to 1T phase caused by S-plane sliding; the sliding of the Mo plane leads to the transformation of the 2H to the 2H’ phase. (g and h) Atomic movements during 2H to 1T phase transformation in single-layered MoS2 at T = 600 °C. | ||
In addition to applying crystal pressure, researchers have found that electron beam irradiation can also relieve lattice stress in MoS2 by disrupting lattice symmetry. This process causes atoms to migrate to lower energy sites, leading to structural reconstruction. As depicted in Fig. 6e and f, Lin and co-workers employed electron beam irradiation to disrupt the local stress equilibrium within the 2H-MoS2 lattice.75 This process induced the migration of Mo and S atoms, leading to the formation of a distorted 1T phase at the reconstructed domains. The resulting phase exhibited a triangular morphology with well-defined 2H–1T phase boundaries (Fig. 6g and h). Moreover, Katagiri and colleagues successfully demonstrated phase transformation patterning of MoS2 at room temperature using a 160 MeV nm−2 electron beam. Due to the interaction between the excited electron beam and the lattice, the 1T/2H in-plane Schottky junctions produced by this technique exhibited a lower barrier (0.13–0.18 eV) than the theoretical value (0.47 eV). Furthermore, the MoS2 crystal domains minimized electron scattering and barriers, thereby enhancing electron mobility while improving the kinetics of electrocatalytic reactions.76 Fine-tuning the micro-regional configuration and electronic properties of MoS2 through intracrystalline stress can promote the formation of unsaturated active sites and induce specific phase structures. However, due to the complexity of the required technology and high equipment costs, this approach is often coupled with theoretical calculations to gain deeper insights into the structural evolution and catalytic activity mechanisms.
4 Surface chemical modification
The redistributed electronic landscape of MoS2 alters the interactions between the electrocatalytic centers and intermediate hydrogen species when the host catalyst comes into contact with additives (such as atoms, ions, or molecules) or when vacancies are introduced.77,78 Surface chemical modification is an effective approach for the regulation of the crystal field and ligand field of MoS2, which can greatly affect the physical–chemical properties and significantly improve its catalytic activity. To date, the ligand structure of MoS2 can be modulated through vacancy defect, atomic doping, phase transition engineering and heterostructure engineering to achieve significant changes in their electrical nature, magnetic properties, and electrocatalysis performance.15,40,44,79 This section provides a detailed discussion of these approaches and highlights several representative examples.4.1 Vacancy defect
Introducing vacancy defects is a straightforward and effective strategy for creating additional active sites and fine-tuning the coordination environment of catalytic centers, making it a valuable method for catalyst modification. Typically, these defects are generated by disrupting the crystal periodic structure without the need for external additives.79 As depicted in Fig. 7a–c, Wang et al. precisely controlled the concentration of S-vacancy on the MoS2 surface by H2O2 etching, revealing the fact that the residual carriers left in S-vacancy prefer transferring into adjacent Mo atoms, which leading the Mo sites stronger H* attraction as a result of de delocalized electrons.46 Frenkel-defect (FD) is one of the typical vacancy defects among many related works. For example, Liu Song's group used a simple argon gas annealing treatment to exploit this defect in MoS2 and boost hydrogen evolution. It is revealed by DFT calculations that the FD can raise a unique charge landscape and appropriate sites for H* species. By comparison to the single atomic Pt doped MoS2 (Pt-MoS2), the overpotential of FD-MoS2 decreased from 211 mV to 164 mV at a current density of 10 mA cm−2.43 Subsequently, they prepared antisite defect structures with a concentration of 4% (where two sulfur atoms or a single sulfur atom occupy the position of a molybdenum atom, denoted as S2Mo-MoS2 or SMo-MoS2) within the monolayer MoS2 basal plane through calcination in an H2/Ar atmosphere, and investigated the role of these antisite defects in promoting the HER process (Fig. 7d). According to DFT analysis, the formation of antisite defects disrupts the original coordination of catalytically S atoms, resulting in a distinctly different and asymmetric charge density distribution in MoS2 (Fig. 7e). This change induces a transition from a semiconductor to a metallic state by introducing electronic states at the Fermi level, thereby enhancing H* adsorption.80 | ||
| Fig. 7 (a) Schematic of the chemical etching process to introduce single S-vacancies. (b) STEM images of CVD-grown single-layer MoS2 thin films after etching. The yellow dashed circles represent S vacancies. (c) Free energy versus the reaction coordinates of MoS2 with diverse S-vacancy states. Inset: corresponding MoS2 models in which Mo atoms and the upper S-vacancies are symbolized by the numbers 1–16 and the letters A–P, respectively. (d) Schematic diagram of the formation process of the antisite defect. (e) Total and partial density of states (DOS) of pristine MoS2, VMo-MoS2, and S2Mo-MoS2. The Fermi level is set to zero and denoted by the black dotted line. | ||
It is worth noting that specific types of vacancies can be generated by doping with designated heteroatoms, for example, metal dopants are more likely to expose S vacancies. Therefore, fully leveraging the synergistic effects of metal heteroatoms and vacancy defects can lead to remarkable improvements in HER performance. More examples are reflected in alkali metals that have strong interactions with S atoms, such as Zn, Fe, Co, Pd, and Cu.78,81–83 The concentration of missing S atoms can be easily controlled by controlling the amount of alkali metals.82,84 In addition, as low-loaded noble metals are expected to boost HER performance, the synergistic effect of noble metals and vacancy defects is highlighted in reducing the H* adsorption/desorption energy barrier and improving HER performance.
4.2 In-plane substitution
In-plane substitutional doping of MoS2 can be achieved through two strategies: replacing host Mo and S atoms, which is the main way to achieve in-plane charge rearrangement.85 Non-metal elements such as C, N, O, and P. are used as the main in-plane substituents due to their easy operation and low cost.86–89 Ge and co-authors replaced the S atoms in MoS2 with O atoms through a simple wet chemical method. The high electronegativity of O induced electron outflow from the adjacent S atoms, thereby constructing an efficient HER 1T-MoS2 catalyst. Additionally, O atoms tend to form hydrogen bonds with H2O in the electrolyte, enhancing the adsorption and dissociation of H2O on the catalyst surface. While the high electronegativity of non-metal elements is an effective strategy for driving charge transfer from host S atoms, an excessive electronegativity gap can lead to localized charge accumulation and slow H* desorption process.88 Jiang et al. addressed this issue by Co-doping MoS2 with both Se and O atoms, whose electronegativities are 2.55 and 3.5, respectively. Studies have shown that Se–O co-doped MoS2 exhibits moderate energy level splitting due to the synergistic effect of Se–O on the electronic structure. Furthermore, the smaller atomic diameter of Se can induce lattice distortion by replacing partial S atoms, resulting in Se-MoS2 electrocatalyst exhibiting a low Tafel slope of ∼47 mV dec−1 under acidic conditions.90To avoid the violent host–guest electron interaction, it is feasible to further awaken the inertness of metal sites. The synergistic effect of metal-nonmetal diatomic is developed to stimulate the utilization of MoS2. Transition metals (TMs), with their abundant d-orbital electron states and spin magnetism, are well-suited for adjusting the Fermi level of MoS2 by forming s–p hybridized orbitals with S atoms.40 As depicted in Fig. 8a, Deng and coworkers synthesized a Co and Se co-doped MoS2 nanostructured foam electrocatalyst (Co/Se-MoS2-NF) by confining Co atoms within the sublayer and Se atoms within the surface lattice of MoS2 using a one-step hydrothermal method. In this configuration, Co atoms serve as substitutional dopants for sublayer Mo, activating the inert S sites, while Se atoms are confined in the surface to stabilize the basal plane. The synergistic effect of Co and Se is evident in the optimized thermodynamics and structural stability of the HER sites (Fig. 8b and c). The near-zero free energy significantly accelerates the formation of H2 formation, while the balanced electronic structure ensures the long-term stability of the active sites. Tests revealed that Co/Se-MoS2-NF exhibited a low Tafel slope of 67 mV dec−1 and a long-term performance over 360 h at a high current density of 1000 mA cm−2 in 0.5 M H2SO4.91
 | ||
| Fig. 8 (a) Atomic resolution HAADF-STEM image of Co/Se-MoS2-NF. Inset: magnification of the white dashed area in (a). (b) H* adsorption structures at the (b) Mo edge, (c) plane, and (d) S edge positions of Co/Se-MoS2. (c) H* adsorption free energies (ΔGH*) at the plane, Mo edge, and S edge positions of pure MoS2, Co-doped, Se-doped, and Co/Se co-doped MoS2, denoted as MoS2, Co-MoS2, Se-MoS2, and Co/Se-MoS2, respectively. The hydrogen coverage at the Mo edge is defined based on the terminated S or Se monomer positions, while the hydrogen coverage at the S edge is defined based on the terminated S or Se dimer positions, which are the active sites for HER. (d) Schematic diagram of Pd and Ru dual-doped MoS2−xOHy phase. The blue, yellow, purple, green, and red spheres represent Pd, S, Mo, Ru, and O atoms, respectively. (e) (Left) HER mechanism of Pd, Ru-MoS2−xOHy in acidic media; (Right) HER mechanism of Pd, Ru-MoS2−xOHy in alkaline media. | ||
More recently, bimetallic atoms pair has also been widely used for the tandem regulation of the HER process. Li et al. developed a Ni, Co bimetallic atoms dual modified MoS2 catalyst (Ni, Co-MoS2) to enhance HER activity in alkaline solutions. The study showed that Ni atoms serve as initial activation sites for the H2O, while Co atoms facilitate the adsorption and desorption kinetics of H intermediates by influencing the electronic structure of the neighboring S. The Ni, Co-MoS2 catalyst exhibited a lower Tafel slope of 78 mV dec−1.92 Similarly, Han and coauthors developed a Cu and Pd dual-doped MoS2 catalyst (Cu–Pd-MoS2) using a two-step approach. The study showed that Cu enhanced the conductivity, while Pd facilitated the phase transition from 2H to 1T phase, thereby improving both the electron transfer and the number of active sites.93 Additionally, Luo et al. engineered a di-anionic surface (Pd, Ru-MoS2−xOHy) on MoS2 to modulate the HER mechanism (Fig. 8d). The study demonstrates that by replacing S sites with –OH anions, a strong non-covalent hydrogen interaction was established, effectively attracting hydronium ions and water molecules. Furthermore, the hydroxyl network works synergistically with adjacent metal atoms (M–OH), overcoming the kinetic barriers associated with water dissociation in alkaline media (Fig. 8e). As a result, the Pd, Ru-MoS2−xOHy catalyst exhibits superior kinetic performance in both acidic and alkaline conditions, with Tafel slopes of 85 mV dec−1 and 121 mV dec−1, respectively.94
4.3 Embedding doping
Driven by the high chemical potential of the reactants, the interstitial spaces within the MoS2 lattice become occupied by free atoms.95–97 These structural additives can subsequently modify the catalytic behavior by tuning the Fermi level and the density of states of neighboring atoms. Interestingly, these additives may directly alter the reaction pathway or indirectly influence the chemical process by facilitating the proper adsorption of H* by adjacent atoms.98,99 However, the instability of these structural additives and their strong dependence on the coordination environment pose challenges to heterogeneous catalysis. Noble metal single atoms are widely considered to be excellent regulators of HER, and the potential candidates for heterotopic doping as well. As depicted in Fig. 9a, Shi and coauthors utilized a photoreduction method to load dispersed Pt single atoms onto a 1T′-MoS2 catalyst. Structural characterization revealed that the loaded Pt atoms predominantly exist in three forms: substitutional Pt (Ptsub), Pt adsorbed on top of S (Ptads-S), and Pt adsorbed on top of Mo (Ptads-Mo). Among these, the Ptads-Mo sites exhibited nearly zero free energy for hydrogen evolution (Fig. 9b), resulting in a mass activity of 85 ± 23 A mgPt−1 for the s-Pt/1T′-MoS2 catalyst at an overpotential of −50 mV.100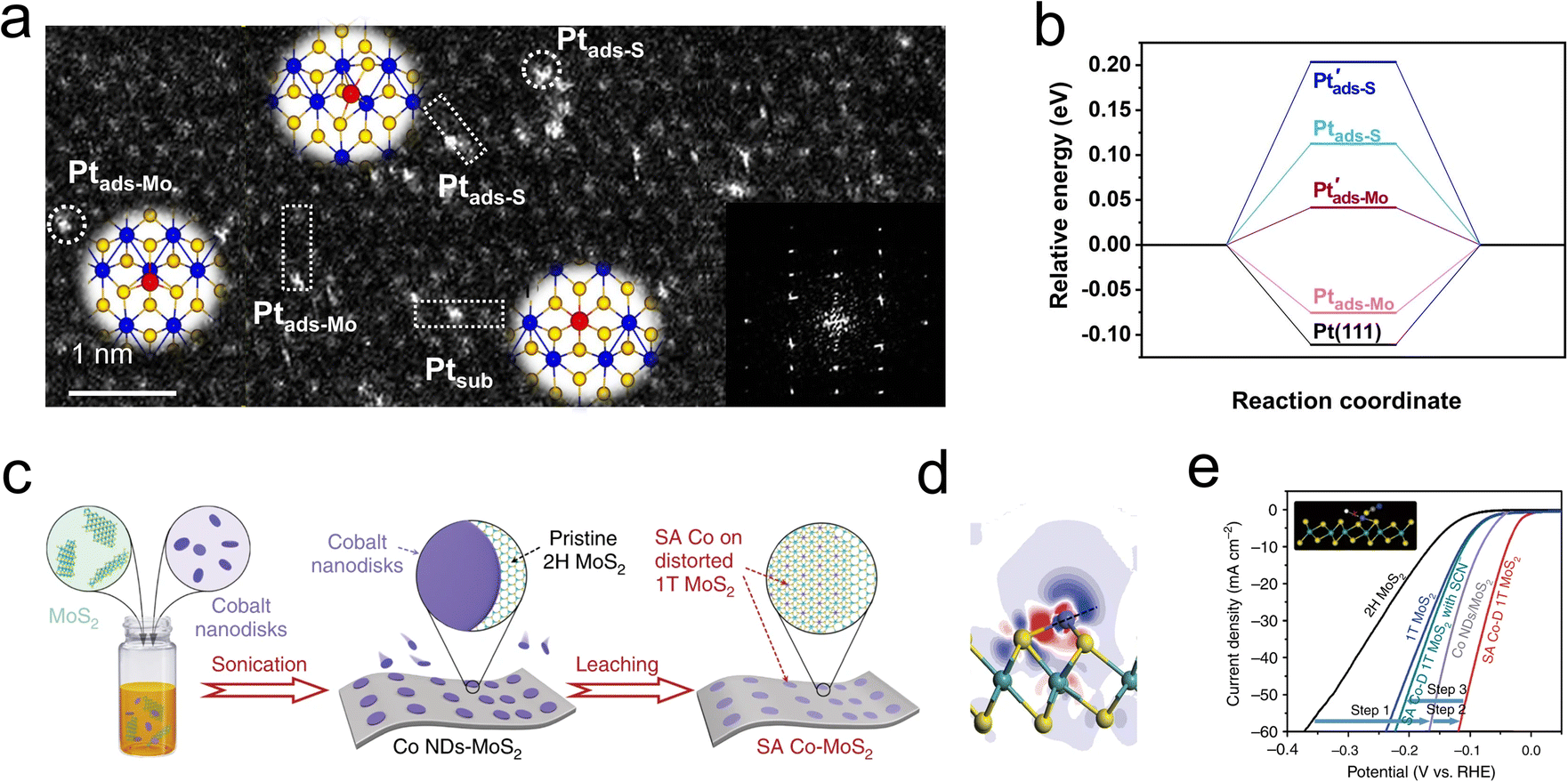 | ||
| Fig. 9 (a) Atomic-resolution HAADF-STEM image of s-Pt/1T′-MoS2 showing the isolated Pt atomically dispersed on 1T′-MoS2 NS. Three kinds of single-atomically dispersed Pt, denoted as Ptsub, Ptads-S and Ptads-Mo, respectively, are enclosed by the white dotted rectangles. (Inset) Simulated atomic structure diagram and corresponding FFT pattern. (b) Calculated ΔGH* diagrams for HER. (c) Schematic diagram of the fabrication process for SA Co-D 1T MoS2. (d) The electron density difference of 3 × 3 case. (e) HER polarization plots of MoS2, 1T MoS2 prepared by lithiation, Co NDs/MoS2 and SA Co-D 1T MoS2 without and with SCN-ions. The inset shows that the cobalt HER active centers are blocked by SCN-ions. | ||
Utilizing the d-orbital effects of transition metal Co, Qi et al. employed cyclic voltammetry (CV) to deposit Co nanodisks (NDs) onto the MoS2 surface (Fig. 9c). The incorporated Co atoms were found to occupy positions atop Mo atoms rather than at MoS2 defects or edge sites, leading to the formation of a distorted 1T phase (Fig. 9d). Research indicates that these single Co atoms act as active centers, directly participating in the binding of H intermediates on the MoS2. The resulting Co-D 1T MoS2 demonstrated outstanding HER performance as illustrated in Fig. 9e, with a low Tafel slope of 32 mV dec−1 and long-term stability over 2400 h.101 To develop a highly stable interstitial doping system, Zhou et al. incorporated carbon atoms with an ultra-small atomic radius and high chemical potential into the sublayer interstices of MoS2. The study focused on the structure–performance relationship of the HER in the interstitial carbon-doped MoS2 electrocatalyst (Cia-MoS2). Through hydrothermal synthesis, the atomic C was strategically embedded into the fcc sites of the MoS2 cell, resulting in a favorable local charge landscape and a 1T phase that promoted HER activity. Unlike previous studies, the Cia did not directly accelerate the HER process but instead served as a structural stabilizer for the emerging metallic phase. The synthesized Cia-MoS2 catalyst showed an 81% reduction in overpotential at 10 mA cm−2 compared to pristine MoS2.99 The introduction of heterotopic structural additives is increasingly being developed to enhance the stability of active phases, or provide additional adsorption sites and conductivity. These atypical doping sites can effectively alter reaction pathways and are generally suitable for additives with high dispersion, small size, and large chemical potential differences from the host catalyst.
4.4 Heterostructure construction
When 2D materials are combined through regions with different atomic arrangement orientations, rich physical and chemical properties have emerged, and the transition zone is called domain wall.102,103 Fansy phenomenon can be seen near the dislocation region, e.g., effective charge transfer, impurity band, intermediate structure and inhomogeneous electronic structure.104,105 Hong et al. developed a chemical vapor deposition technique to synthesize a single-layer MoS2 with a 1T–2H heterostructure, in which the 1T phase was as high as 61.5%. DFT calculations revealed that the strong electronic coupling was observed at the heterogeneous interface, and the electrons spontaneously transfer from the 2H phase to the 1T phase. This phenomenon was lead an increased electronic density around 1T phase while a depletion of electrons around the 2H phase, causes the transition region a low HER barrier (−0.156 eV) and fast kinetic process (Tafel slope is 78 mV dec−1).106 Along this line, more heterogeneous structure can be organized by MoS2 and other materials. Such as 1T0.81-MoS2@Ni2P HER catalyst studied by Liu et al. Firstly, the high electronegativity of P drives pristine MoS2 partial shift to 1T phase; then the electrons were transfer from the MoS2 into Ni2S or Ni2P regions, leading a rich electronic density environment around the domain (Fig. 10a and b). According to the rich heterogeneous interfaces, the as synthesized 1T0.81-MoS2@Ni2P catalyst accelerates the H2O activation, as well as the generation of intermediate H species (Fig. 10c and d), which possesses an extremely low overpotential in acidic (38.9 mV) and alkaline (95 mV) solutions at 10 mA cm−2, corresponding to a considerable Tafel slope of 41 and 42 mV dec−1, respectively.107 | ||
| Fig. 10 (a) The deformation of the electronic density of 2Hphase-MoS2@NiS2 and 1Tphase-MoS2@NiS2 interface, in which yellow/green isosurfaces correspond to positive/negative spin densities (0.00295308 e Å−3). (b) Band structure and density of states (DOS) for 1Tphase-MoS2@NiS2. (c) Free-energy diagrams for HER on the 2Hphase-MoS2, 1Tphase-MoS2, pure Ni2P, pure NiS2, 1T/2Hmix-MoS2, 2Hphase-MoS2@Ni2P, 2Hphase-MoS2@NiS2, 1Tphase-MoS2@NiS2 and 1Tphase-MoS2@Ni2P interface edge. (d) Schematics showing water activation, *H intermediate formation and hydrogen generation on multi-heterojunction electrocatalysts. (e) The HR-TEM images of the Mo5N6-MoS2 heterojunction by in-plane chemical bonding of MoS2 (100) basal planes and Mo5N6 (110) plane. (f) Schematic diagrams of the energy band of Schottky-type Mo5N6-MoS2 heterojunction (Evac = vacuum energy, Ec = conduction band, Ev = valence band, Ef = Fermi level, Eg = energy gap, W = work function, and Φ = depletion region). (g) Energy profiles for water dissociation and free energy diagrams for HER on 2H-MoS2 basal plane, 2H-MoS2 edge plane, Mo5N6 (110), and Mo5N6-MoS2 heterointerface. | ||
Constructing in-plane contact between metal and semiconductor is another effective approach to generate a built-in electric field using Schottky barriers. Pi et al. prepared the Mo5N6-MoS2 heterojunction nanosheets (Mo5N6-MoS2/HCNRs) on conductive hollow carbon nanoribbons by a simple hydrothermal method (Fig. 10e). The results showed that there was a Fermi level difference in the metal/semi-contact, which led to the electron transfer from 2H-MoS2 to Mo5N6 (Fig. 10f). In addition, the lattice dislocations led to abundant defective sites, which increased the number of active sites and charge transfer process, giving the catalyst a low overpotential of 53 mV at 10 mA cm−2 and a Tafel slope of 37.9 mV dec−1 (Fig. 10g).108 Heterogeneous structures are typically characterized by abundant crystal domains, and fully utilizing these interfaces can significantly enhance the electrical properties of catalysts. In addition, domain walls are often available for H* species adsorption and activation, which play a role in increasing active sites.
5 Interlayer coupling engineering
Layered intercalation is an effective method to modulate interlayer coupling and crystal field in MoS2.109 By widening the vdW gap, the crystal field of MoS2 switches from tetrahedral to octahedral coordination, leading to a re-splitting of energy levels.23,77 In addition, direct growth of MoS2 on metal support has been emerging as a novel approach to synthesize catalytic complexes. Taking advantage of the Fermi level difference between metals and semi, a large number of electrons can be transferred to MoS2 through metal supports, thus promoting the reconstruction of active centers.22,110–112 This section focuses on two typical interlayer couplings and their applications for enhancing the HER performance of MoS2 catalysts.5.1 Modulation of vdW gap
2D layered MoS2 are attracting each layer by weak physical vdW interaction (shown as Fig. 11c), and the properties of MoS2 catalysts directly endued by the height of vdW gap.113 Existing strategies for expanding the interlayer gap are mainly based on electrochemical intercalation techniques, whereby additive ions, molecules, and nanoparticles are driven into the vdW gaps of MoS2.34,114 | ||
| Fig. 11 (a) Zero-valent intercalation of metal nanoparticles by an in situ reduction strategy. (b) LSV curves of metal intercalated MoS2 catalysts. (c) Intrinsic van der Waals distance and van der Waals gap of MoS2 (van der Waals distance = 6.2 Å, van der Waals gap = 1.8 Å). (d) Schematic illustrations of the vdW gap engineering achieved by adsorption of water molecules, and the cross-sectional STEM image of the vdW interface formed by vdW gap engineering, showing a clear vdW gap while maintaining atomically sharp and clean interfaces. Scale bars, 5 nm (STEM image). | ||
Alkali metal ions are popular in modifying the vdW gap and chemical properties of MoS2, as they have extremely small diameter and low ionization potential.115 For example, Cui et al. reported a vertically aligned MoS2 electrocatalyst intercalated by Li+ for rapid HER. First of all, the Li+ acts as a gap extender, which directly changes the vdW interaction and alters the electronic properties. Secondly, Li+ transfers extra electrons to MoS2 due to its high reducibility, thus changing the spin–orbit coupling of Mo 3d orbital. Last, it was proved that the 2H phase MoS2 shifts into 1T phase when the concentration of Li+ is over 0.28.19 In addition, the capillary action of MoS2 powder and the molten K+ was used for the modulation of the vdW gap. It was reported the added K atoms can be driven into the vdW gaps to prepare complex artificial superlattices, due to the difference between the electron affinity of MoS2 (4.45 eV) and the ionization potential of K+ (4.34 eV).26 Based on this, as depicted in Fig. 11a and b, Chen et al. achieved the intercalation of several metal nanoparticles into a Li+-intercalated 1T′ LixMoS2 catalyst by in situ reduction of noble metal ion precursors. In this process, the wider vdW gaps provided channels for the diffusion of target noble species, endowing the novel catalyst with a higher redox potential. Unlike the in-plane doping, the intercalation of alkali metal ions species reacts with surface S atoms and forms M–S bonds (M = K, Na, Pt, Au), which suppresses electron scattering and maintains a high electron concentration around the d orbitals of Mo.
However, the introduction of alkali metal ions was realized through a hysteresis diffusion-limited process, leading to a fuzzy crystalline phase and reduced structural stability.116 To overcome these drawbacks, a self-intercalation approach was developed by Loh and his colleagues, where the host metal atoms move into the vdW gaps by exploiting their high chemical potential. It was found that the self-driven intercalant introduces additional spin-split bands, which can modulate the electronic properties of the carrier transport.117 An advanced solution was proposed by Zou et al., who exploited the vdW gaps to confine small pre-adsorbed water molecules. By controlling the saturation vapour pressure of water, the ‘vdW water gap’ was monotonic increasing up to 53.6 Å (Fig. 11d). This gap-engineering approach highlights the potential of vdW water gaps to modulate electronic properties and more utility for diode and electrochemical devices.113
5.2 Semiconductor/metal contacting
Generally, 2D films are grown on support like metal (e.g., Au, Pd, Cu, and Mo) or oxides (e.g., mica, sapphire, and silica) for robust action.20 Much effort has been put into exploring various supports, and a strong interaction between Au foil and MoS2 films has been discovered in 2014. It is revealed that the electronic coupling effects between Au and MoS2 and eliminate the charge transfer resistance, thus promote the catalysts reaching a high exchange current density of 38.1 μA cm−2.59 Obviously, semi/metal contacting effects play a vital role in design Schottky barrier and regulate the conductivity. Voiry et al. pioneered to concern the relationship of semi/metal contact effects on HER activity,22 and Charlie Tsai and coauthors studied how different supports influence the Gibbs free energy of Mo edge sites. Their results show that the HER reactivity of edged-Mo sites can be influenced by tuning the contact effects between semi-MoS2 films and different metal supports, where the electronic interaction induced Fermi level shift and reduced interface resistance.118 Along this line, as depicted in Fig. 12a, several metals such as In, Ti, Au, Pd, and Mo were used as simulated support to contact with the surface and edges part of monolayer MoS2. The orbital overlap, Schottky barrier, and tunnel barriers of TMDs and metal contact effects were disclosed by DFT calculations. Interestingly, the separate distance (d) of the metal-edge is much smaller than the distance of metal-surface in all combinations, thus strong covalent bonds can form between the metal and edge site. In addition, an atomic orbital overlap of sulfides and metal can be found in each case, which may lead to a reduction of the Schottky barrier and inner resistance of the monomer catalyst.119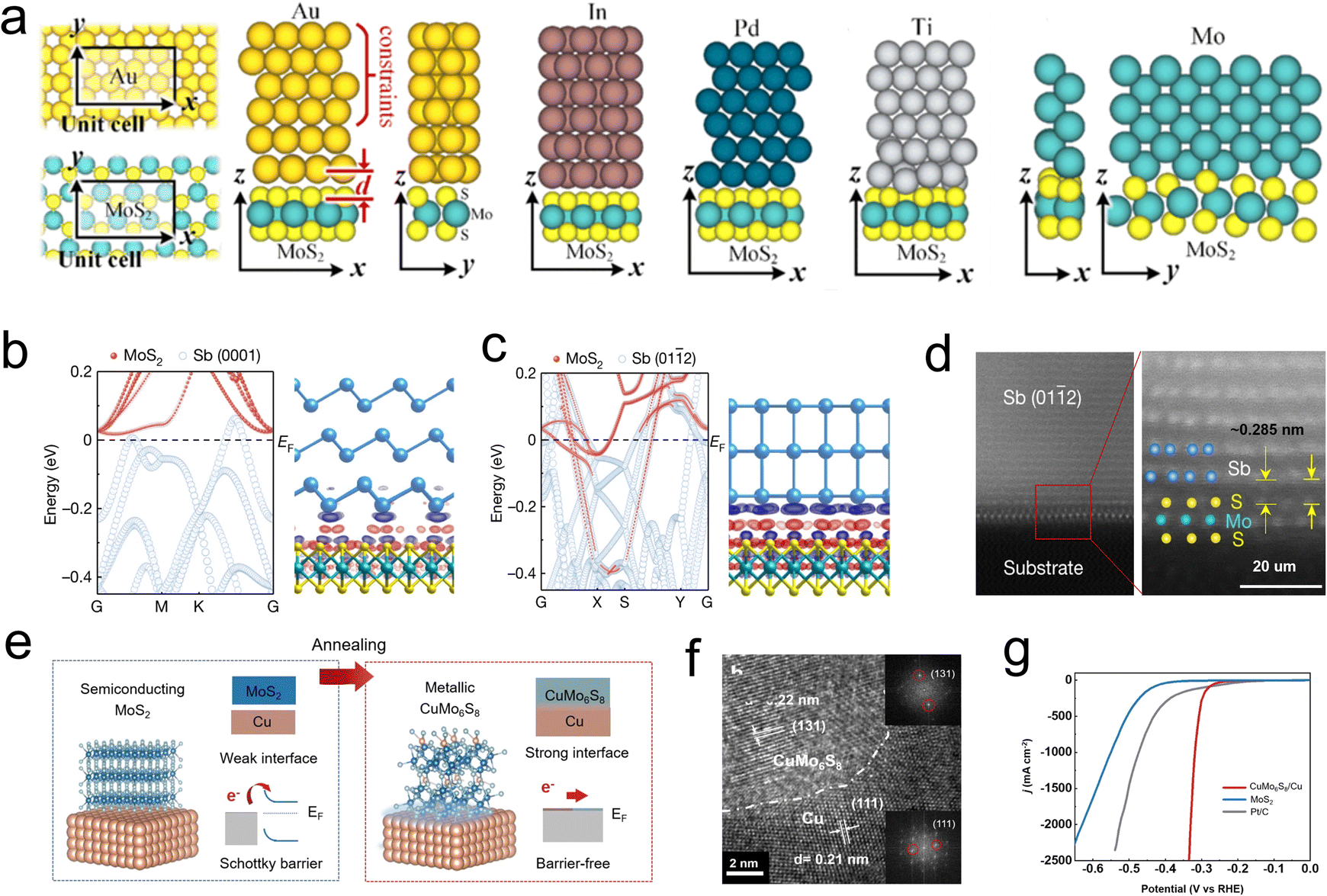 | ||
| Fig. 12 (a) Optimized geometries of top contacts to MoS2: Au-MoS2 (in different views), In-MoS2, Pd-MoS2, Ti-MoS2, Mo-MoS2 (in different views). d is defined as the physical separation (the z component of the nearest core-to-core distance between the metal atoms and the chalcogenide atoms). (b) Atomic projected electronic band structure of Sb (0001)–MoS2 contact and charge density near junction EF. (c) Atomic projected electronic band structure of Sb (0112)–MoS2 contact and charge density near junction EF (red, positive; blue, negative). (d) (Left) Cross-section HAADF-STEM image of the Sb (0112)–MoS2 contact. Scale bar, 2 nm. (Right) Zoom-in atomic-resolution image from the red box in (c). The Mo, S and Sb atoms are overlayed on the image. The vdW gap of 0.285 nm is marked. Scale bar, 1 nm. (e) A schematic showing the preparation process of CuMo6S8/Cu electrode. (f) Cross-sectional high-resolution transmission electron microscopy (HRTEM) zoom-in view of the CuMo6S8/Cu electrode. The inset of h is the corresponding fast Fourier transform patterns of CuMo6S8 (top) and Cu (bottom). (g) Polarization curves of three electrodes including CuMo6S8/Cu, MoS2, and Pt/C. All tests are done in 1 M KOH at a scan rate of 1 mV s−1 with 85% iR correction. | ||
More recently, Li et al. studied the contact effect between Sb with different plane orientations Sb (0001) and Sb (0112) prepared by electron beam evaporation and monolayer MoS2 (Fig. 12b–d). It is observed that the Mo d orbital has a large real-space overlap with the Sb (0112) pz orbital in the vertical direction, resulting in strong Sb (0112)–MoS2 hybridization and forcing the interface resistance to approach the quantum limit. In contrast, no obvious electron localization was observed between Mo d orbital and Sb (0001) p orbital.120 This work emphasizes the significance of crystal plane orientation in the preparation of zero contact resistance composite catalyst. Directly construct carrier pathway between metal support and MoS2 active component also considered an effective method to tune semi/metal contact interface. For example, Shin and Liu developed a highly mechanically stable novel electrocatalyst by growing MoS2 nanosheets on a self-support Cu foil, where the MoS2 are vertically embedded in the Cu (Fig. 12e). Differently, there is a composite region between the MoS2 and Cu, CuxMo6S8 (CMS), which is known as the Chevrel phase (Fig. 12f). It was shown that the CMS region serves as an effective charge transfer channel to promote the carriers transfer into the MoS2 active component, thus exhibiting a low overpotential of ∼334 mV and sustained stability of over 100 h at a current density of 2500 mA cm−2 (Fig. 12g).121,122 Similar studies were reported by Liu's group, who achieved in situ growth of a vertically oriented TaS2 active layer on a metallic Ta by an oriented solid-phase synthesis method. It was confirmed that electrons can directly inject into TaS2 layers without passing the vdW gap. Additionally, this novel integrated structure demonstrates excellent mechanical stability, with the Ta/TaS2 catalyst operating for over 200 hours while maintaining a low overpotential of 398 mV at a high current density of 2000 mA cm−2. These advances pave the way to apply this monomer catalyst to boost HER process.123
6 Challenges and outlooking
Although the unsaturated electronic structure is regarded as the key feature of MoS2 for achieving efficient HER, the long-term electrocatalytic stability is often compromised by the emergence of active structural configurations. Fig. 13a and b illustrate some common cases associated with HER performance. Based on current electrocatalytic test data, it is evident that TMDs based on Mo element tend to perform better under acidic conditions,99,101,107,122,124–128 whereas catalysts containing Ni element exhibit greater durability in alkaline environments.92,107,108,129–132 These findings provide valuable guidance for selecting appropriate catalysts for large-scale HER processes. As the study of constructing and regulating the structure–activity relationship of MoS2-like catalysts at the atomic level is still in its early stages, we propose several research topics to assist researchers to navigate through the scientific opportunities in this area (Fig. 14). | ||
| Fig. 13 HER performance of various catalysts under (a) acidic conditions and (b) alkaline conditions. | ||
 | ||
| Fig. 14 The potential development researches from modern theoretical analysis, advanced synthesis and characterization, and possible applications in electrocatalysis for MoS2. | ||
6.1 Electronic structure evolution of central metal during electrocatalysis and decay
The evolution of the electron states of d-orbital of the central metal site in transition metal sulfide catalysts during electrocatalysis remains a significant challenge.133 Some studies have reported that utilize low-temperature or in situ characterization approaches can allow for the semi-quantitative analysis of the local electronic structure and unsaturated states of transition metals. In a reaction process involving electrons transfer, electron transitions within the central metal, electron donation from dopants to the central metal, and the transfer of delocalized electrons between the central metal and ligands can alter the redox properties of the metal sites, thus, the formation, adsorption, or desorption of intermediates and products. Unlike other catalytic mechanisms, the presence of additional energy from magnetocaloric effects associated with intermediate or high-spin electrons could enhance reaction kinetics or intermediate mobility. This could lead to the appearance of “mirages” in electrocatalytic reactions that thermodynamic theory would deem impossible. Hence, researches on the influence of spin effects in transition metal catalysts on reaction activity and selectivity may become a critical focus in future catalysis research.6.2 Role of unsaturated electronic structure in the interaction between reactants/products and catalyst centers
Unsaturated electronic structures are typically accompanied by unpaired electrons, which significantly impact the magnetic properties and polarity of local catalytic centers. As a result, investigating the role of electron deficiency in spin states, magnetism, polarity, and conductivity in MoS2 could offer valuable insights into reaction mechanisms and Gibbs free energy pathways, especially in the context of MoS2 catalysts applied to more complex reactions. For instance, when products are present in a non-polar bubble state, adjusting the polarity of catalytic centers may be crucial for enhancing the desorption and diffusion of bubble-phase products. Despite bubble trouble becomes a growing focus on this topic in large-scale HER processes, research from this perspective remains limited.6.3 Advantages of metal/semiconductor contacts in constructing highly active electronic structures
In heterogeneous electrocatalysis, harnessing the interfacial contact effects between supported metals and semiconductor catalysts to modulate band positions, charge transfer, and structural stability of electrocatalysts remain a significant challenge. Recent studies suggest that as the van der Waals gap between 2D layered catalysts narrows, it can promote the formation of new chemical bonds or delocalized electrons, thereby creating internal pathways for electron transfer. Thus, exploring and harnessing the interfacial contact effects between supported metals and semiconductor catalysts may offers a promising “two birds with one stone” strategy for monomer catalysts in electrocatalysis-simultaneously improving catalyst stability and optimizing the active electronic structure.6.4 Advanced characterization methods to study unsaturated electronic structures
In the exploration of unsaturated electronic structures in 2D materials (such as MoS2), advanced characterization methods are crucial. Commonly used techniques include Raman spectroscopy, X-ray absorption spectroscopy (XAS), X-ray photoelectron spectroscopy (XPS), and electron paramagnetic resonance (EPR). These approaches provide valuable information about electronic structure, chemical environment, and unpaired electrons, but they face challenges when applied to light-sensitive 2D materials. For instance, while Raman spectroscopy is sensitive to unsaturated electronic structures, the signals are often weak at low-energy excitation source and susceptible to interference from fluorescence. XAS and XPS can offer semi-quantitative data on electronic states, but they mainly capture average structures, making it difficult to precisely characterize local defects or active sites. Future advancements may involve combining high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) with electron energy loss spectroscopy (EELS) to enhance spatial resolution, along with computational simulations (such as DFT calculations) to predict electron deficiency impact on the electrocatalytic performance.6.5 Utilizing unsaturated sites to couple hydrogen reduction with other important reactions
By employing various strategies, both electrophilic and nucleophilic active sites on MoS2 can be designed to meet the adsorption and activation requirements of different chemically active species. Introducing sulfur vacancies or other defects in the 2H phase MoS2 creates metallic-like sites with unpaired electrons, which effectively adsorb and activate electrophilic species such as CO2 and CO. Additionally, by doping high-electronegativity noble metals (such as Pt and Au), positively charged nucleophilic Mo sites (Moδ+) can be engineered on the MoS2 surface to interact with electron-rich reactants. Under appropriate stoichiometric conditions, these active sites facilitate in situ hydrogenation reactions, yielding high-value products such as methanol, formic acid, or other hydrocarbons. This approach demonstrates that MoS2 not only exhibits excellent catalytic performance in HER but also holds promise for applications in hydrogenation, CO2 reduction reactions, and other significant processes.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Author contributions
All of the authors contributed to the manuscript preparation. Q. Z., X. R., and D. M. conceived the outline of the manuscript. Q. Z., and H. H. wrote the original draft of the manuscript. Z. C., X. R., and D. M. discussed and helped revise the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (52202202 and 22472005) and the Provincial Key Research and Development Project of Sichuan (2024YFHZ0040).References
- M. Wagner, B. Meyer, M. Setvin, M. Schmid and U. Diebold, Nature, 2021, 592, 722–725 CrossRef PubMed.
- W. Huang, Q. He, Y. P. Hu and Y. G. Li, Angew. Chem., Int. Ed., 2019, 131, 8768–8772 CrossRef.
- T. Binninger and M. L. Doublet, Energy Environ. Sci., 2022, 15, 2519–2528 RSC.
- K. L. Hu, T. Ohto, Y. Nagata, M. Wakisaka, Y. Aoki, J. Fujita and Y. Ito, Nat. Commun., 2021, 12, 203 CrossRef PubMed.
- P. Garrido-Barros, J. Derosa, M. J. Chalkley and J. C. Peters, Nature, 2022, 609, 71–76 CrossRef PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- H. Y. Kim, J. M. Kim, Y. Ha, J. Woo, A. Byun, T. J. Shin, K. H. Park, H. Y. Jeong, H. Kim, J. Y. Kim and S. H. Joo, ACS Catal., 2019, 9, 11242–11254 CrossRef.
- A. S. Dudnik, V. L. Weidner, A. Motta, M. Delferro and T. J. Marks, Nat. Chem., 2014, 6, 1100–1107 CrossRef PubMed.
- Q. Q. Zhou, X. L. Zhou, R. H. Zheng, Z. F. Liu and J. D. Wang, Sci. Total Environ., 2022, 806, 150088 CrossRef.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef.
- M. Fang, Y. Q. Peng, P. W. Wu, H. Wang, L. X. Xing, N. Wang, C. M. Tang, L. Meng, Y. K. Zhou, L. Du and S. Y. Ye, Front. Energy, 2024, 18, 308–329 CrossRef.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef PubMed.
- Y. H. Jia, Y. C. Zhang, H. Q. Xu, J. Li, M. Gao and X. T. Yang, ACS Catal., 2024, 14, 4601–4637 CrossRef.
- H. S. Meng, Z. J. Chen, J. L. Zhu, B. You, T. Y. Ma, W. Wei, S. Vernuccio, J. Xu and B. J. Ni, Adv. Funct. Mater., 2024, 34, 2405270 CrossRef.
- H. T. Wang, Z. Y. Lu, S. C. Xu, D. S. Kong, J. J. Cha, G. Y. Zheng, P. C. Hsu, K. Yan, D. Bradshaw, F. B. Prinz and Y. Cui, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 19701–19706 CrossRef.
- C. Tsai, H. Li, S. Park, J. Park, H. S. Han, J. K. Nørskov, X. L. Zheng and F. Abild-Pedersen, Nat. Commun., 2017, 8, 15113 CrossRef.
- H. T. Wang, C. Tsai, D. S. Kong, K. Chan, F. Abild-Pedersen, J. K. Nørskov and Y. Cui, Nano Res., 2015, 8, 566–575 CrossRef.
- J. Zhang, Q. Y. Zhang and X. L. Feng, Adv. Mater., 2019, 31, 1808167 CrossRef PubMed.
- K. Fujisawa, B. R. Carvalho, T. Zhang, N. Perea-Lopez, Z. Lin, V. Carozo, S. L. L. M. Ramos, E. Kahn, A. Bolotsky, H. Liu, A. L. Elias and M. Terrones, ACS Nano, 2021, 15, 9658–9669 CrossRef PubMed.
- D. Voiry, R. Fullon, J. U. Yang, C. C. C. Silva, R. Kappera, I. Bozkurt, D. Kaplan, M. J. Lagos, P. E. Batson, G. Gupta, A. D. Mohite, L. Dong, D. Er, V. B. Shenoy, T. Asefa and M. Chhowalla, Nat. Mater., 2016, 15, 1003–1009 CrossRef.
- S. Park, C. Kim, S. O. Park, N. K. Oh, U. Kim, J. Lee, J. Seo, Y. Yang, H. Y. Lim, S. K. Kwak, G. Kim and H. Park, Adv. Mater., 2020, 32, 2001889 CrossRef.
- Z. J. Chen, S. N. Yun, L. Wu, J. Q. Zhang, X. D. Shi, W. Wei, Y. W. Liu, R. J. Zheng, N. Han and B. J. Ni, Nano-Micro Lett., 2023, 15, 4 CrossRef.
- Z. J. Chen, X. G. Duan, W. Wei, S. B. Wang and B. J. Ni, Nano Energy, 2020, 78, 105270 CrossRef.
- W. Yang and S. W. Chen, Chem. Eng. J., 2020, 393, 124726 CrossRef.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef PubMed.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef.
- Y. Li, K. A. N. Duerloo, K. Wauson and E. J. Reed, Nat. Commun., 2016, 7, 10671 CrossRef.
- H. M. Wang, C. H. Li, P. F. Fang, Z. L. Zhang and J. Z. Zhang, Chem. Soc. Rev., 2018, 47, 6101 RSC.
- Y. R. Lim, J. K. Han, Y. Yoon, J. B. Lee, C. Jeon, M. Choi, H. Chang, N. Park, J. H. Kim, Z. Lee, W. Song, S. Myung, S. S. Lee, K. S. An, J. H. Ahn and J. S. Lim, Adv. Mater., 2019, 31, 1901405 CrossRef PubMed.
- D. Zhu, J. Liu, Y. Zhao, Y. Zheng and S. Z. Qiao, Small, 2019, 15, 1805511 CrossRef.
- H. Yang, S. Kim, W. M. Chhowalla and Y. H. Lee, Nat. Phys., 2017, 13, 931–937 Search PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef.
- R. M. A. Khalil, F. Hussain, A. M. Rana, M. Imran and G. Murtaza, Phys. E Low-dimens. Syst. Nanostruct., 2019, 106, 338–345 CrossRef.
- Q. Yue, J. Kang, Z. Z. Shao, X. A. Zhang, S. L. Chang, G. Wang, S. Q. Qin and J. B. Li, Phys. Lett. A, 2012, 376, 1166–1170 CrossRef.
- Q. Z. Huang, J. L. Shen, Y. Lu, R. D. Ye and S. Gong, J. Phys. Chem. C, 2023, 127, 17406–17414 CrossRef.
- L. A. Zavala, K. Kumar, V. Martin, F. Maillard, F. Maugé, X. Portier, L. Oliviero and L. Dubau, ACS Catal., 2023, 13, 1221–1229 CrossRef.
- F. Yang, P. Hu, F. F. Yang, B. Chen, F. Yin, K. Hao, R. Sun, L. Gao, Z. Sun, K. Wang and Z. Yin, Small, 2023, 19, 2301468 CrossRef.
- Y. Cao, ACS Nano, 2021, 15, 11014–11039 CrossRef.
- Y. W. Zhang, Q. Wu, J. Z. Y. Seow, Y. J. Jia, X. Ren and Z. C. J. Xu, Chem. Soc. Rev., 2024, 53, 8123–8136 RSC.
- Y. Y. Linghu, N. Li, Y. P. Du and C. Wu, Phys. Chem. Chem. Phys., 2019, 21, 9391 RSC.
- J. Xu, G. L. Shao, X. Tang, F. Lv, H. Y. Xiang, C. F. Jing, S. Liu, S. Dai, Y. G. Li, J. Luo and Z. Zhou, Nat. Commun., 2022, 13, 2193 CrossRef CAS PubMed.
- Y. C. Huang, Y. H. Sun, X. L. Zheng, T. Aoki, B. Pattengale, J. Huang, X. He, W. Bian, S. Younan, N. Williams, J. Hu, J. X. Ge, N. Pu, X. X. Yan, X. Q. Pan, L. J. Zhang, Y. G. Wei and J. Gu, Nat. Commun., 2019, 10, 982 CrossRef CAS PubMed.
- L. Y. Zhang, M. Li, A. Q. Zou, S. H. Yu, T. Xiong, L. Wang, J. J. He, Q. Fu, K. Sun, D. H. C. Chua and J. M. Xue, ACS Appl. Energy Mater., 2019, 2, 493–502 CrossRef CAS.
- X. Wang, Y. W. Zhang, H. N. Si, Q. H. Zhang, J. Wu, L. Gao, X. F. Wei, Y. Sun, Q. L. Liao, Z. Zhang, K. Ammarah, L. Gu, Z. Kang and Y. Zhang, J. Am. Chem. Soc., 2020, 142, 4298–4308 CrossRef CAS PubMed.
- Y. Q. Fang, X. Z. Hu, W. Zhao, J. Pan, D. Wang, K. J. Bu, Y. L. Mao, S. F. Chu, P. Liu, T. Y. Zhai and F. Q. Huang, J. Am. Chem. Soc., 2019, 141, 790–793 Search PubMed.
- T. Zhang, Y. P. Liu, J. Yu, Q. T. Ye, L. Yang, Y. Li and H. J. Fan, Adv. Mater., 2022, 34, 2202195 CrossRef.
- G. L. Shao, M. Q. Yang, H. Y. Xiang, S. Luo, X. X. Xue, H. M. Li, X. Zhang, S. Liu and Z. Zhou, Nano Res., 2023, 16, 1670–1678 CrossRef.
- T. Zhang, Q. T. Ye, Z. Y. Han, Q. Y. Liu, Y. P. Liu, D. S. Wu and H. J. Fan, Nat. Commun., 2024, 15, 6508 CrossRef PubMed.
- J. W. Wang, L. Q. He, Y. H. Zhang, H. Y. Nong, S. N. Li, Q. K. Wu, J. Y. Tan and B. L. Liu, Adv. Mater., 2024, 36, 2314145 CrossRef PubMed.
- V. I. Klimov, A. A. Mikhailovsky, D. W. McBranch, C. A. Leatherdale and M. G. Bawendi, Phys. Rev. B:Condens. Matter Mater. Phys., 2000, 61, R13349 CrossRef.
- F. Wang, T. A. Shifa, X. Zhan, Y. Huang, K. Liu, Z. Cheng, C. Jiang and J. He, Nanoscale, 2015, 7, 19764–19788 RSC.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U.S.A., 2005, 102, 10451–10453 CrossRef PubMed.
- Q. Q. Ji, C. Li, J. L. Wang, J. J. Niu, Y. Gong, Z. P. Zhang, Q. Y. Fang, Y. Zhang, J. P. Shi, L. Liao, X. S. Wu, L. Gu, Z. F. Liu and Y. F. Zhang, Nano Lett., 2017, 17, 4908 CrossRef PubMed.
- Z. P. Zhang, J. J. Niu, P. F. Yang, Y. Gong, Q. Q. Ji, J. P. Shi, Q. Y. Fang, S. L. Jiang, H. Li, X. B. Zhou, L. Gu, X. S. Wu and Y. F. Zhang, Adv. Mater., 2017, 29, 1702359 CrossRef PubMed.
- Y. Zhang, Y. F. Zhang, Q. Q. Ji, J. Ju, H. T. Yuan, J. P. Shi, T. Gao, D. L. Ma, M. X. Liu, Y. B. Chen, X. J. Song, H. Y. Hwang, Y. Cui and Z. F. Liu, ACS Nano, 2013, 7, 8963–8971 CrossRef PubMed.
- J. G. Song, J. S. Park, W. Lee, T. Choi, H. Jung, C. W. Lee, S. H. Hwang, J. M. Myoung, J. H. Jung, S. H. Kim, C. L. Matras and H. Kim, ACS Nano, 2014, 8, 8617 CrossRef.
- J. P. Shi, D. L. Ma, G. F. Han, Y. Zhang, Q. Q. Ji, T. Gao, J. Y. Sun, X. J. Song, C. Li, Y. S. Zhang, X. Y. Lang, Y. F. Zhang and Z. F. Li, ACS Nano, 2014, 8, 10196–10204 CrossRef CAS.
- J. P. Shi, X. N. Wang, S. Zhang, L. F. Xiao, Y. H. Huan, Y. Gong, Z. P. Zhang, Y. C. Li, X. B. Zhou, M. Hong, Q. Y. Fang, Q. Zhang, X. F. Liu, L. Gu, Z. F. Liu and Y. F. Zhang, Nat. Commun., 2017, 8, 958 CrossRef.
- K. Chang, X. Hai, H. Pang, H. B. Zhang, L. Shi, G. G. Liu, H. M. Liu, G. X. Zhao, M. Li and J. H. Ye, Adv. Mater., 2016, 28, 10033–10041 CrossRef CAS.
- L. J. Zhang and A. Zunger, Nano Lett., 2015, 15, 949–957 CrossRef CAS.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- Z. Y. Huang, X. Qi, H. Yang, C. Y. He, X. L. Wei, X. Y. Peng and J. X. Zhong, J. Phys. D, 2015, 48, 205302 CrossRef.
- D. S. Kong, H. T. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341–1347 CrossRef CAS PubMed.
- X. Geng, W. Wu, N. Li, W. Sun, J. Armstrong, A. Al-hilo, M. Brozak, J. Cui and T. P. Chen, Adv. Funct. Mater., 2014, 24, 6123 CrossRef CAS.
- Y. Yang, H. Fei, G. Ruan, C. Xiang and J. M. Tour, Adv. Mater., 2014, 26, 8163 CrossRef CAS PubMed.
- J. Deng, H. B. Li, S. H. Wang, D. Ding, M. S. Chen, C. Liu, Z. Q. Tian, K. S. Novoselov, C. Ma, D. H. Deng and X. H. Bao, Nat. Commun., 2017, 8, 14430 CrossRef CAS.
- Y. M. Jiang, X. Li, S. J. Yu, L. P. Jia, X. J. Zhao and C. M. Wang, Adv. Funct. Mater., 2015, 25, 2693–2700 CrossRef CAS.
- X. P. Ren, L. Q. Pang, Y. X. Zhang, X. D. Ren, H. B. Fan and S. Z. Liu, J. Mater. Chem. A, 2015, 20, 10693–10697 RSC.
- A. P. Nayak, T. Pandey, D. Voiry, J. Liu, S. T. Moran, A. Sharma, C. Tan, C. H. Chen, L. J. Li, M. Chhowalla, J. F. Lin, A. K. Singh and D. Akinwande, Nano Lett., 2015, 15, 346–353 CrossRef CAS.
- A. P. Nayak, S. Bhattacharyya, J. Zhu, J. Liu, X. Wu, T. Pandey, C. Q. Jin, A. K. Singh, D. Akinwande and J. F. Lin, Nat. Commun., 2014, 5, 3731 CrossRef CAS PubMed.
- Z. Zhao, H. J. Zhang, H. T. Yuan, S. B. Wang, Y. Lin, Q. S. Zeng, G. Xu, Z. X. Liu, G. K. Solanki, K. D. Patel, Y. Cui, H. Y. Hwang and W. L. Mao, Nat. Commun., 2015, 6, 7312 CrossRef CAS.
- H. Y. Yu, G. B. Liu, J. J. Tang, X. D. Xu and W. Yao, Sci. Adv., 2017, 3, e1701696 CrossRef.
- Y. C. Lin, D. O. Dumcenco, Y. S. Huang and K. Suenaga, Nat. Nanotechnol., 2014, 9, 391–396 CrossRef CAS PubMed.
- Y. Katagiri, T. Nakamura, A. Ishii, C. Ohata, M. Hasegawa, S. Katsumoto, T. Cusati, A. Fortunelli, G. Iannaccone and G. Fiori, Nano Lett., 2016, 16, 3788–3794 CrossRef CAS.
- X. W. Guo, E. H. Song, W. Zhao, S. M. Xu, W. L. Zhao, Y. J. Lei, Y. Q. Fang, J. J. Liu and F. Q. Huang, Nat. Commun., 2022, 13, 5954 CrossRef.
- W. Z. Wu, C. Y. Niu, C. Wei, P. Y. Jia, C. Li and P. Q. Xu, Angew. Chem., Int. Ed., 2019, 131, 2051–2055 CrossRef.
- C. Y. Feng, Z. P. Wu, K. W. Huang, J. H. Ye and H. B. Zhang, Adv. Mater., 2022, 34, 2200180 CrossRef PubMed.
- J. Xu, X. X. Xue, G. L. Shao, C. F. Jing, S. Dai, K. He, P. P. Jia, S. Wang, Y. F. Yuan, J. Luo and J. Lu, Nat. Commun., 2023, 14, 7849 CrossRef.
- Y. Zhou, J. Zhang, E. H. Song, J. H. Lin, J. D. Zhou, K. Suenaga, W. Zhou, Z. Liu, J. J. Liu, J. Lou and H. J. Fan, Nat. Commun., 2020, 11, 2253 CrossRef.
- Z. Y. Luo, Y. X. Ouyang, H. Zhang, M. L. Xiao, J. J. Ge, Z. Jiang, J. L. Wang, D. M. Tang, X. Z. Cao, C. P. Liu and W. Xing, Nat. Commun., 2018, 9, 2120 CrossRef PubMed.
- Y. G. Liu, S. J. Guan, X. S. Du, Y. R. Chen, Y. Yang, K. L. Chen, Z. W. Zheng, X. Wang, X. Q. Shen, C. L. Hu and X. B. Li, Energy Fuels, 2023, 37, 5370–5377 CrossRef.
- J. M. Zhang, X. P. Xu, L. Yang, D. J. Cheng and D. P. Cao, Small Methods, 2019, 3, 1900653 CrossRef.
- B. Liu, Y. Cheng, B. Cao, M. Hu, P. Jing, R. Gao, Y. Du, J. Zhang and J. Liu, Appl. Catal., B, 2021, 298, 120630 CrossRef.
- Y. P. Zang, S. W. Niu, Y. S. Wu, X. S. Zheng, J. Y. Cai, J. Ye, Y. F. Xie, Y. Liu, J. B. Zhou, J. F. Zhu, X. J. Liu, G. M. Wang and Y. Qian, Nat. Commun., 2019, 10, 1217 CrossRef PubMed.
- Q. Yang, Z. Wang, L. Dong, W. Zhao, Y. Jin, L. Fang, B. Hu and M. Dong, J. Phys. Chem. C, 2019, 123, 10917–10925 CrossRef.
- J. Ge, D. Zhang, J. Jin, X. Han, Y. Wang, F. Zhang and X. Lei, Mater. Today Energy, 2021, 22, 100854 CrossRef.
- Y. Shi, D. Zhang, H. Miao, X. Wu, Z. Wang, T. Zhan, J. Lai and L. Wang, Sci. China: Chem., 2022, 65, 1829–1837 CrossRef.
- L. Jiang, Y. J. Zhang, X. H. Luo, L. Yu, H. X. Li and Y. J. Li, Chem. Eng. J., 2021, 425, 130611 CrossRef CAS.
- Z. L. Zheng, L. Yu, M. Gao, X. Y. Chen, W. Zhou, C. Ma, L. H. Wu, J. F. Zhu, X. Y. Meng, J. T. Hu, Y. C. Tu, S. S. Wu, J. Mao, Z. Q. Tian and D. H. Deng, Nat. Commun., 2020, 11, 3315 CrossRef CAS.
- H. Li, F. Yu, X. Ling, H. Wan, M. Zhang, Y. Zhou, J. Wei, F. Lu, X. Zhang, X. Zeng and M. Zhou, Nanotechnology, 2021, 32, 445703 CrossRef CAS PubMed.
- D. Han, Z. Luo, Y. Li, N. Gao, J. Ge, C. Liu and W. Xing, Appl. Surf. Sci., 2020, 529, 147117 CrossRef CAS.
- Z. Y. Luo, H. Zhang, Y. Q. Yang, X. Wang, Y. Li, Z. Jin, Z. Jiang, C. P. Liu, W. Xing and J. J. Ge, Nat. Commun., 2020, 11, 1116 CrossRef CAS.
- Y. T. Luo, S. Q. Zhang, H. Y. Pan, S. J. Xiao, Z. L. Guo, L. Tang, U. Khan, B. F. Ding, M. Li, Z. Y. Cai, Y. Zhao, W. Lv, Q. L. Feng, X. L. Zou, J. H. Lin, H. M. Cheng and B. L. Liu, ACS Nano, 2020, 14, 767–776 CrossRef PubMed.
- J. Wang, C. Cheng, Q. Yuan, H. Yang, F. Q. Meng, Q. H. Zhang, L. Gu, J. L. Cao, L. G. Li, S. C. Haw, Q. Shao, L. Zhang, T. Cheng, F. Jiao and X. Q. Huang, Chem, 2022, 8, 1673–1687 Search PubMed.
- J. Yang, Y. F. Cao, S. Y. Zhang, Q. W. Shi, S. Y. Chen, S. C. Zhu, Y. S. Li and J. F. Huang, Small, 2023, 19, 2207295 CrossRef.
- V. P. Pham and G. Y. Yeom, Adv. Mater., 2016, 28, 9024–9059 CrossRef PubMed.
- Q. Q. Zhou, Z. Y. Wang, H. D. Yuan, J. D. Wang and H. Hu, Appl. Catal., B, 2023, 332, 122750 CrossRef.
- Z. Y. Shi, X. Zhang, X. Q. Lin, G. G. Liu, C. Y. Ling, S. B. Xi, B. Chen, Y. Y. Ge, C. L. Tan, Z. C. Lai, Z. Q. Huang, X. Y. Ruan, L. Zhai, L. J. Li, Z. J. Li, X. X. Wang, G. H. Nam, J. W. Liu, Q. Y. He, Z. Q. Guan, J. L. Wang, C. S. Lee, A. R. J. Kucernak and H. Zhang, Nature, 2023, 621, 300–305 CrossRef.
- K. Qi, X. Q. Cui, L. Gu, S. S. Yu, X. F. Fan, M. C. Luo, S. Xu, N. B. Li, L. R. Zheng, Q. H. Zhang, J. Y. Ma, Y. Gong, F. Lv, K. Wang, H. H. Huang, W. Zhang, S. J. Guo, W. T. Zheng and P. Liu, Nat. Commun., 2019, 10, 5231 CrossRef.
- H. Hu, Q. Q. Zhou, Z. Y. Wang, J. D. Wang, Y. M. Chen and Y. X. Han, Chin. Sci. Bull., 2024, 69, 4299–4310 CrossRef.
- H. Xiong, C. F. Du, Z. L. Ma, R. C. Zhi, S. S. Hao, X. Y. Zhao, Z. Liu, F. Xu and H. Q. Wang, Adv. Funct. Mater., 2024, 34, 2402298 CrossRef.
- W. S. Chen, J. J. Gu, Y. P. Du, F. Song, F. X. Bu, J. H. Li, Y. Yuan, R. C. Luo, Q. L. Liu and D. Zhang, Adv. Funct. Mater., 2020, 30, 2000551 CrossRef.
- Z. M. Sun, L. Lin, M. W. Yuan, H. Y. Yao, Y. J. Deng, B. H. Huang, H. F. Li, G. B. Sun and J. Zhu, Nano Energy, 2022, 101, 107563 CrossRef.
- Z. A. Hong, W. T. Hong, B. C. Wang, Q. Cai, X. He and W. Liu, Chem. Eng. J., 2023, 460, 141858 CrossRef.
- M. Q. Liu, J. A. Wang, W. Klysubun, G. G. Wang, S. Sattayaporn, F. Li, Y. W. Cai, F. C. Zhang, J. Yu and Y. Yang, Nat. Commun., 2021, 12, 5260 CrossRef.
- C. R. Pi, X. X. Li, X. M. Zhang, H. Song, Y. Zheng, B. Gao, A. Kızılaslan, P. K. Chu and K. F. Huo, Small, 2022, 18, 2201137 CrossRef PubMed.
- Y. H. Xue, Q. Zhang, W. J. Wang, H. Cao, Q. H. Yang and L. Fu, Adv. Energy Mater., 2017, 7, 1602684 CrossRef.
- Y. C. Wu, D. f. Li, C. L. Wu, H. Y. Hwang and Y. Cui, Nat. Rev. Mater., 2023, 8, 41–53 CrossRef.
- D. Xu, S. N. Zhang, J. S. Chen and X. H. Li, Chem. Rev., 2023, 123, 1–30 CrossRef.
- Z. J. Chen, T. Y. Ma, W. Wei, W. Y. Wong, C. Zhao and B. J. Ni, Adv. Mater., 2024, 36, 2401568 CrossRef.
- C. Liu, X. M. Zou, Y. W. Lv, X. Q. Liu, C. Ma, K. L. Li, Y. Liu, Y. Chai, L. Liao and J. He, Nat. Nanotechnol., 2024, 19, 448–454 CrossRef.
- H. T. Yuan, H. T. Wang and Y. Cui, Acc. Chem. Res., 2015, 48(1), 81–90 CrossRef PubMed.
- X. Huang, Z. Y. Zeng and H. Zhang, Chem. Soc. Rev., 2013, 42, 1934–1946 RSC.
- Z. X. Chen, K. Leng, X. X. Zhao, S. Malkhandi, W. Tang, B. B. Tian, L. Dong, L. R. Zheng, M. Lin, B. S. Yeo and K. P. Loh, Nat. Commun., 2017, 8, 14548 CrossRef.
- X. X. Zhao, P. Song, C. C. Wang, A. C. Riis-Jensen, W. Fu, Y. Deng, D. Y. Wan, L. X. Kang, S. C. Ning, J. D. Dan, T. Venkatesan, Z. Liu, W. Zhou, K. S. Thygesen, X. Luo, S. J. Pennycook and K. P. Loh, Nature, 2020, 581, 171–177 CrossRef PubMed.
- C. Tsai, F. A. Pedersen and J. K. Nørskov, Nano Lett., 2014, 14, 1381–1387 CrossRef PubMed.
- J. H. Kang, W. Liu, D. Sarkar, D. Jena and K. Banerjee, Phys. Rev. X, 2014, 4, 031005 Search PubMed.
- W. S. Li, X. S. Gong, Z. H. Yu, L. Ma, W. J. Sun, S. Gao, Ç. Köroğlu, W. F. Wang, L. Liu, T. T. Li, H. K. Ning, D. X. Fan, Y. F. Xu, X. C. Tu, T. Xu, L. T. Sun, W. H. Wang, J. P. Lu, Z. H. Ni, J. Li, X. D. Duan, P. Wang, Y. F. Nie, H. Qiu and X. R. Wang, Nature, 2023, 613, 274–279 CrossRef PubMed.
- C. Bae, T. A. Ho, H. Kim, S. Lee, S. Lim, M. Kim, H. Yoo, J. M. Montero-Moreno, J. H. Park and H. Shin, Sci. Adv., 2017, 3, e1602215 CrossRef PubMed.
- H. M. Liu, R. K. Xie, Y. T. Luo, Z. C. Cui, Q. M. Yu, Z. Q. Gao, Z. Y. Zhang, F. N. Yang, X. Kang, S. Y. Ge, S. H. Li, X. F. Gao, G. L. Chai, L. Liu and B. L. Liu, Nat. Commun., 2022, 13, 6382 CrossRef CAS PubMed.
- Q. M. Yu, Z. Y. Zhang, S. Y. Qiu, Y. T. Luo, Z. B. Liu, F. N. Yang, H. M. Liu, S. Y. Ge, X. L. Zou, B. F. Ding, W. C. Ren, H. M. Cheng, C. H. Sun and B. L. Liu, Nat. Commun., 2021, 12, 6051 CrossRef CAS PubMed.
- M. Cheng, N. Cao, Z. Wang, K. Wang, T. C. Pu, Y. K. Li, T. L. Sun, X. Y. Yue, W. K. Ni, W. X. Dai, Y. He, Y. Shi, P. Zhang, Y. H. Zhu and P. F. Xie, ACS Nano, 2024, 18, 10582–10595 CrossRef CAS PubMed.
- T. Ma, H. Cao, S. Li, S. J. Cao, Z. Y. Zhao, Z. H. Wu, R. Yan, C. D. Yang, Y. Wang, P. A. van Aken, L. Qiu, Y. G. Wang and C. Cheng, Adv. Mater., 2022, 34, 2206368 CrossRef CAS.
- L. N. Liu, J. X. Wu, L. Y. Wu, M. Ye, X. Z. Liu, Q. Wang, S. Y. Hou, P. F. Lu, L. F. Sun, J. Y. Zheng, L. Xing, L. Gu, X. W. Jiang, L. M. Xie and L. Y. Jiao, Nat. Mater., 2018, 17, 1108–1114 CrossRef CAS.
- E. E. Benson, H. Y. Zhang, S. A. Schuman, S. U. Nanayakkara, N. D. Bronstein, S. Ferrere, J. L. Blackburn and E. M. Miller, J. Am. Chem. Soc., 2018, 140, 441–450 CrossRef CAS PubMed.
- I. S. Kwon, I. H. Kwak, T. T. Debela, H. G. Abbas, Y. C. Park, J. P. Ahn, J. Park and H. S. Kang, ACS Nano, 2020, 14, 6295–6304 CrossRef CAS.
- H. B. Wu, B. Y. Xia, L. Yu, X. Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed.
- H. J. Liu, Q. He, H. L. Jiang, Y. X. Lin, Y. K. Zhang, M. Habi, S. G. Chen and L. Song, ACS Nano, 2017, 11, 11574–11583 CrossRef CAS PubMed.
- G. F. Chen, T. Y. Ma, Z. Q. Liu, N. Li, Y. Z. Su, K. Davey and S. Z. Qiao, Adv. Funct. Mater., 2016, 26, 3314 CrossRef CAS.
- Y. Q. Xue, X. J. Bai, Y. Y. Xu, Q. Yan, M. Zhu, K. Zhu, K. Ye, J. Yan, D. X. Cao and G. L. Wang, Composites, Part B, 2021, 224, 109229 CrossRef CAS.
- C. H. An, W. Kang, Q. B. Deng and N. Hu, Rare Met., 2022, 41, 378–384 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |