
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnifying principles for the design and evaluation of natural product-inspired compound collections†
Frederik Simonsen
Bro
 and
Luca
Laraia
*
and
Luca
Laraia
*
Department of Chemistry, Technical University of Denmark, 2800, Kongens Lyngby, Denmark. E-mail: luclar@kemi.dtu.dk
First published on 24th January 2025
Abstract
Natural products play a major role in the discovery of novel bioactive compounds. In this regard, the synthesis of natural product-inspired and -derived analogues is an active field that is further developing. Several strategies and principles for the design of such compounds have been developed to streamline their access and synthesis. This perspective describes how individual strategies or their elements can be combined depending on the project goal. Illustrative examples are shown that demonstrate the blurred lines between approaches and how they can work in concert to discover new biologically active molecules. Lastly, a general set of guidelines for choosing an appropriate strategy combination for the specific purpose is presented.
 Frederik Simonsen Bro | Frederik Simonsen Bro achieved his BSc in chemistry at the Technical University of Denmark (DTU) in 2019 which included a bachelor's project with Prof. Robert Madsen working on iron-catalysed dehydrogenation of alcohols. In 2021 he completed his MSc in chemistry at DTU carrying out his master's project with Assoc. Prof. Luca Laraia in the synthesis of an alkaloid-inspired library. Subsequently, he carried out his PhD studies in the same lab focussing on the development of inhibitors of sterol transport proteins through synthesis of sterol-inspired libraries, which he completed in 2024, and continued as a postdoctoral researcher in the same group. |
 Luca Laraia | Luca Laraia received his MSci at Imperial College London (2009) before completing a PhD in chemical biology at the University of Cambridge (2014). He subsequently moved to the Max Planck Institute of Molecular Physiology as an Alexander von Humboldt fellow and subsequently project leader, before embarking on his independent career at the Technical University of Denmark as an assistant professor. Since 2021 he has been an associate professor for chemical biology and medicinal chemistry. His group's work lies at the interface of chemistry and biology, including the synthesis of natural product-inspired compounds and the study and modulation of sterol-mediated processes. |
Introduction
Natural products (NPs) are an important source of bioactive small molecules. They have co-evolved with their biosynthetic proteins, thus exploring biologically relevant chemical space and encoding inherent biological relevance, as a result of their ability to bind biomolecules and cross cell membranes. In many, though not all, cases they have also evolved to be stable, at least for the duration of their intended bioactivity. Consequently, NPs were the first examples of therapeutics. NPs, their derivatives, and compounds inspired by them are and have been the foundation of organic and medicinal chemistry and play a major role in drug discovery.1–8 Importantly, one third of approved drugs since 1981 fall into one of these categories, highlighting the historical and continuing impact of NPs in this area.9Despite the obvious benefits of NPs, there are limitations in terms of drug discovery. Accessing natural products by isolation or total synthesis (TS) can sometimes be laborious and involve inefficient processes, while often not delivering enough material for biological evaluation and structure–activity relationship (SAR) analysis. The reasons for this are low concentration and extraction from complex mixtures when isolating NPs from natural sources, and (often) multi-step and low yielding total syntheses of NPs. Lastly, the selection criteria for nature are different from the selection criteria in drug discovery. As such, NPs have evolved for their producing organisms, not human therapeutic applications.5,7 Though NPs cover biologically relevant chemical space, their restricted natural selection means that they only cover a limited fraction of NP-like chemical space.10 Thus, the vast majority of biologically relevant, NP-like chemical space remains to be explored. In fact, investigation of the surrounding chemical space of an NP can be more beneficial than investigating the NP alone in terms of drug discovery.2 Consequently, strategies to synthesise NP-derived and -inspired compounds in a practical and efficient way are in demand to navigate new NP-like chemical space and obtain highly bioactive compounds that can serve as drug candidates or tool compounds.
To meet the demand, several strategies to synthesise compounds derived from or inspired by NPs have emerged. Diversity-oriented synthesis (DOS)11,12 focusses on characteristics typical of NPs, including a high fraction of sp3-hybridised carbons (Fsp3) and several stereogenic centres, but is not necessarily based on an NP or NP scaffold. The similar privileged-substructure-based DOS (pDOS)13,14 is based on a privileged scaffold with proven biological relevance that is not necessarily derived from an NP. For DOS and pDOS, molecular scaffold diversity is a key point, which is also the case for activity-directed synthesis (ADS).15,16 The compounds resulting from the strategies including pseudo-natural product (PNP) synthesis,17,18 biology-oriented synthesis (BIOS),19,20 function-oriented synthesis (FOS),21 and pharmacophore-directed retrosynthesis (PDR)22 are all based on NP fragments, scaffolds, or pharmacophores. The total synthesis (TS) of NPs is guided by target molecules (TMs). However, the focus on a single TM limits the exploration of chemical space. This has been a driving force for the establishment of synthetic approaches that investigate the chemical space surrounding a guiding NP, which include complexity-to-diversity (CtD),23 dynamic retrosynthetic analysis (DRA),2,24 diverted total synthesis (DTS),25,26 two-phase synthesis (TPS),27 and analogue-oriented synthesis (AOS).28 The recently described diverse PNP (dPNP) strategy29 combines PNP and DOS/CtD, and thus originates from NP fragments (see Fig. S1–S13† for graphical illustrations, explanations and examples of the individual strategies).
To navigate the plethora of approaches outlined above, we have found it helpful to separate them based on the qualitative similarity of the core frameworks generated to those found in NPs,30 as highlighted by some representative examples (Fig. 1).22,31–46 It is important to note that several quantitative computational approaches for assessing NP-likeness have been developed. For example, the NP-score uses the prevalence of specific atom-centred fragments to compare NPs to fully synthetic compounds.47 The NP character of compound collections is a continuum between fully synthetic compounds (no guiding NP) and unmodified NPs. Guided by retrosynthetic analysis, fully synthetic drugs and libraries of drug candidates can be synthesised by conventional synthesis (CS) and focussed or targeted library synthesis (FLS), respectively, and NPs are synthesised by TS.48,49 These are examples of target-oriented synthesis (TOS) which by definition lacks diversity due to the single target approach. Nonetheless, diversity might arise for FLS and TS when several targets are synthesised in the same study. This is particularly evident for divergent approaches in TS where a common intermediate is used in the synthesis of several members of an NP compound class.50 When a single compound of interest is not known, combinatorial library synthesis (CLS) provides quick access to many compounds. However, although combinatorial libraries can afford complexity giving the compounds slightly higher NP-character, structural diversity is often still limited.51 Here it should be noted that complexity alone does not guarantee bioactivity,52 nor is it always a clear predictor of properties that would be of interest to medicinal chemists, including solubility and oral bioavailability.53 Increased complexity has been correlated with increased selectivity;54 however, increased molecular weight and lipophilicity both correlate with increased promiscuity.55,56 As such, a careful balance of parameters should be targeted when employing such metrics in library design strategies. Overall, many ways to calculate complexity have been reported and we refer the reader to recent publications for a more detailed discussion and their application.52,53,57,58
 | ||
| Fig. 1 Continuum of qualitative similarity to NP frameworks of compounds designed via the different strategies with representative examples: conventional synthesis (CS): paracetamol (1); focussed library synthesis (FLS): K00135 (2);31 combinatorial library synthesis (CLS): 3;32 diversity-oriented synthesis (DOS): (R)-dosabulin (4);33 privileged-substructure-based diversity-oriented synthesis (pDOS): 5;34 diverse pseudo-natural product (dPNP): (−)-asteroxin-1 (6);35 pseudo-natural product (PNP): (+)-glupin (7);36 biology-oriented synthesis (BIOS): 8;37,38 function-oriented synthesis (FOS): 9;39 pharmacophore-directed retrosynthesis (PDR): 10;22 complexity-to-diversity (CtD): ferroptocide (11);40 dynamic retrosynthetic analysis (DRA): (−)-O6C-20-nor-salA ((−)-12);45,46 diverted total synthesis (DTS): cycloproparadicicol (13);41,42 total synthesis (TS): strychnine (14).43,44 TOS = target-oriented synthesis. | ||
The increased focus on diversity and privileged scaffolds in the DOS and pDOS strategies brings the resulting compounds closer to NPs than combinatorial approaches. However, there is no strict requirement for the compounds to be NP-like, and design considerations are often governed by the synthetic accessibility of the resulting products. The privileged scaffolds incorporated can also be fully synthetic in origin, providing key differences to strategies delivering compounds with a higher qualitative similarity to NPs. The PNP strategy is based on the recombination of NP fragments, and as such the resulting scaffolds are not typically found in NPs, even though their constituent fragments are. This gives them a lower NP score, according to the definition of Ertl et al.47 BIOS is for the most part based on actual NP scaffolds, thus bringing the resulting analogues closer to NPs compared to DOS and PNP. Since dPNPs are the result of different combinations of PNP and diversification strategies, they are not necessarily NP scaffolds and may thus be less likely to have frameworks found in NPs than both PNP and BIOS-derived compounds. The FOS, PDR, CtD, DRA, DTS, TPS, and AOS strategies can give compounds that are very close to, or some distance from, actual NPs. For example, a key difference of CtD to other strategies is the frequent use of ring distortion reactions, which can sometimes steer compounds far away from NPs in chemical space. While being beneficial in terms of targeting unexplored chemical space, it carries an inherently greater degree of uncertainty as to the utility of the resulting compounds in biological screens. It is difficult to say which strategy provides compounds with greater NP character, as the resemblance to the parent/guiding NP varies from case to case, thus placing them somewhere in between BIOS and TS.
It should be noted that the definitions of most strategies are open to a degree of interpretation and often overlap, making the distinction between standalone strategies and umbrella terms difficult. For example, CtD could be considered a subset of DOS or its own strategy. Additionally, while TPS is rooted in TS, in principle it can be applied to diversify natural product skeletons/scaffolds. Lastly, DOS and FOS can be grouped together with TOS as general umbrella terms.59 In principle, any strategy aiming for structural diversity in an efficient manner could be described as DOS,11,12 while any strategy that delivers compounds which recapitulate or even enhance the activity of a natural product through a simplified scaffold can be described as FOS.21,60,61 Despite this, principles for the design of DOS and FOS libraries applied as standalone strategies have been described, and representative examples are included in Fig. 1. We view this flexibility as an advantage in enabling chemists to make bolder choices in their library syntheses.
Several excellent reviews and perspectives have been published on the design of NP-inspired compound collections using specific strategies and approaches.2,4,5,7,18,24,62–67 These showcase recent findings, examples, and thoughts in the field relating to individual approaches. In this perspective, our goal is to identify unifying principles across a range of library synthesis approaches. We will highlight such principles with appropriate case studies and make the argument that the existing strategies for NP-inspired compound collections are not necessarily mutually exclusive, but rather complementary, with significant benefits existing from a more open approach by combining strategies or elements from them according to the project goal compared to the use of individual strategies in isolation. The choice of strategies for prospective projects will vary based on whether one seeks to develop new chemistry to increase the chemical and biological diversity of a screening collection, identify entirely new chemical matter for a target/phenotype, or improve potency or other properties for a ligand of a known target or phenotype. Therefore, we will also develop guidelines for assessing which combination of design strategies is most beneficial based on the project goal.
Current approaches: different or complementary?
The different approaches and strategies outlined so far have developed as a consequence of different project goals and information available at the project outset. Key considerations include the availability of target or phenotype information, as well as the availability of known ligands, and particularly NPs, as starting points for design. However, the common denominator for all the approaches and strategies is the use of NPs themselves or their characteristics to develop and identify bioactive molecules, whether this is in a targeted or completely unbiased approach. The need for well-defined strategies and the differentiation between them provides theoretical frameworks that simplify and structure a project, ideally allowing fast access to desired compounds. Differentiation between strategies should thus make it easier to find the right approach for a specific project goal. However, we have found that in many cases the approach ultimately used by research groups implicitly combines components from several different strategies under a larger “umbrella” approach, even though the initial strategy was presented as a single defined approach. More recently, research groups including our own have explicitly targeted the combination of strategies for specific applications.To highlight how various approaches work well together and have more similarities than differences, we have chosen illustrative examples based on different combinations of strategies and the outcomes they present. We have chosen to structure the initial discussion based on chemical strategies, rather than biological outcome, with the latter being addressed in a subsequent section (vide infra). In this regard, the synthesis of six alkaloid-inspired libraries (Scheme 1)68 highlights the use of multiple strategies. These include the synthesis of nicotine (19) analogues (16 and 17) and spirocyclic analogues (24) containing a benzylic-substituted pyrrolidine as found in anisomycin (20) using a complexity-generating Pd-catalysed aminoarylation reaction starting from 15 or 23. Following tert-butyloxycarbonyl (Boc) deprotection of 16, compounds 17 could undergo different diversification reactions to access additional analogues including sulfonamide 18. In a similar fashion, 24 could also be diversified into additional analogues such as sulfonamide 25. In terms of synthetic design, this can be classified as substrate-based DOS. However, the simplified nicotine analogues could be considered as BIOS analogues and the pyrrolidine analogues as PNP (anisomycin fragment) or pDOS (pyrrolidine as a privileged scaffold).69 The analogues could be further diversified using traditional diversification methodologies. Similarly, the authors also diversified the horsfiline (21) scaffold 26 using Ullmann–Goldberg cross-coupling to give 27. Following benzyl deprotection, 28 could then undergo either reductive amination affording 29 or urea formation to give carbamide (30). This shares ideas from PNP and BIOS by using the horsfiline scaffold as a starting point for analogue synthesis. Furthermore, applying different reagents from 28 to generate different analogues could be classed as reagent-based DOS. The last example using the daphnezomine M (22)-like scaffold 31 also used the reagent-based DOS line of thought. Reductive amination afforded 32, amide coupling afforded 33, and the reaction with isocyanate afforded carbamide 34. In total, six different libraries with different scaffolds were synthesised. Overall, this elegantly demonstrates substrate- and reagent-based DOS with the incorporation of ideas from BIOS and PNP using alkaloid scaffolds to enhance biological relevance.
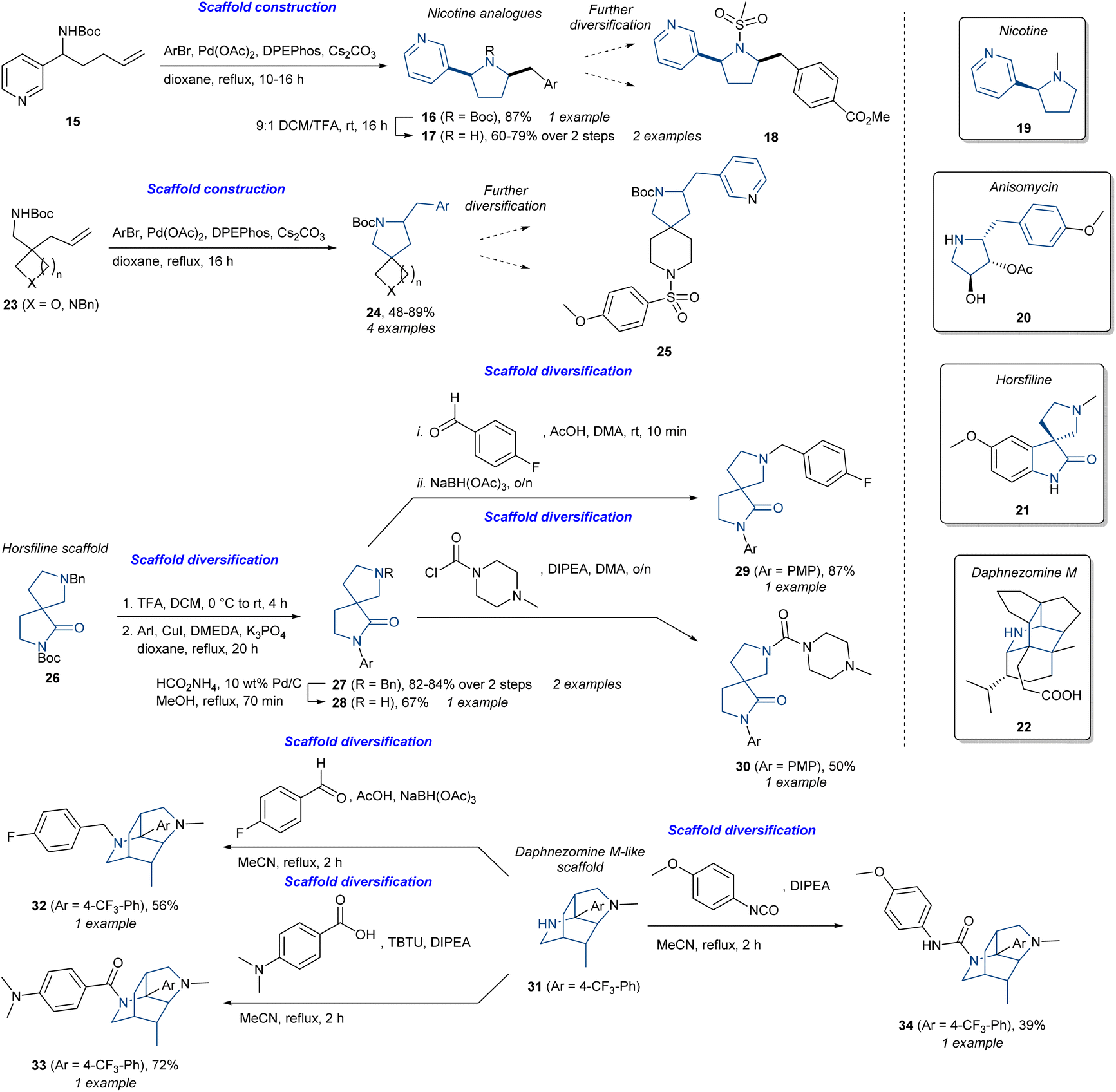 | ||
| Scheme 1 Synthesis of alkaloid-inspired compounds.68 DIPEA = N,N-diisopropylethylamine, DMEDA = N,N′-dimethylethylenediamine, DPEPhos = bis[(2-diphenylphosphino)phenyl]ether, PMP = p-methoxyphenyl, TBTU = N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate. | ||
Similar published work also shares the idea of a complexity-generating reaction followed by diversity focused reactions in a reagent-based DOS fashion.54,55 Focus on diversity is also a key point in activity-directed synthesis, where chemical space is explored by using reactions with multiple and diverse possible outcomes.15,16 The overall idea of generating diversity from a single substrate is embedded in CtD as well, where NPs with (preferably many) diversification vectors are used to create NP-derived analogues using ring distortion, ring formation, and diversification reactions. CtD limits itself to NPs that are not the end point, but rather are complex starting points that can be diversified.23 Thus, CtD could be seen as an example of reagent-based DOS on NPs, where simply by modifying the reagents one can access significant scaffold diversity. The idea of creating diversity from a complex starting material is thus found in both CtD and in work not related to an actual NP that could consequently be viewed as “CtD on non-NPs”. For example, pDOS has been combined with CtD by using ring distortion and ring formation to form diverse medium/macro- and bridged heterocyclic compounds containing the privileged scaffold pyrimidine.70
Employing an NP as a starting point for complexity works well conceptually with other strategies. For example, fragment-sized NPs like the cinchona alkaloids quinine and quinidine can work well in DOS, BIOS, or PNP campaigns.71 Significant effort has been made to make quinine-derived analogues (“quinalogs”72) using different strategies (Scheme 2). Diverse and complex macrocycles can be accessed directly from quinine using a combination of CtD and DOS strategies,73 which are both useful strategies in the synthesis of diverse macrocycles.74 Initially, quinine (35) is functionalised with terminal alkene-containing linkers via Steglich esterification to give esters 36 which were then cyclised to form the quinine macrocycles 37 using ring-closing metathesis (RCM). Four other macrocycles were also accessed from other quinine-derived building blocks. This approach has a strong resemblance to the build/couple/pair (B/C/P) strategy: “building” quinine building blocks, intermolecular “coupling” with other functionalised building blocks, and intramolecular “pairing” of the functional groups to form the macrocycles. Macrocycles remain a desired moiety since they have proven to be a privileged class of molecules for modulating challenging targets such as protein–protein interactions in drug discovery.75,76 In this context, a complex quinidine (38)-inspired 20-membered macrocycle, tantalosin-I (39), that induced autophagy by induction of microtubule-associated protein 1A/1B light chain 3 (LC3) lipidation through disruption of a particular part of the endosomal sorting complexes required for transport (ESCRT) called the IST1-CHMP1B complex was recently reported.77,78 Additional work showed how you could use the relatively small NPs quinine and quinidine in a PNP setting.79,80 The NPs were initially transformed to the corresponding ketones 40 in two steps by Rh-catalysed isomerisation of the terminal alkene to the internal alkene followed by a Malaprade–Lemieux–Johnson oxidation. Ketones are a strategic functional group in the synthesis of NP-inspired compounds since they serve as a suitable coupling partner for fusion with other scaffolds.81 They reported the synthesis of edge-fused indoles and azaindoles 41 from quinine and quinidine via a one-pot imine condensation and a Hegedus–Mori–Heck reaction. This led to the identification of an autophagy and lipid kinase VPS34 inhibitor, azaquindole-1 (41a). Moreover, the spiro-fused chromanones 42 were accessed through a Kabbe reaction. Thus, different PNPs could be accessed simply by changing the reagents, the principle of reagent-based DOS. In this work there was no specific biological target in mind and the compounds were screened phenotypically. Importantly, tantalosin-I (39) and azaquindole-1 (41a) show different bioactivity to each other and to the parent NP, highlighting the value of diversifying a relatively large building block, the cinchona alkaloid framework. All the analogues in Scheme 2 come from a synthetically easily accessible quinine- or quinidine-scaffold, thus having a resemblance to BIOS-derived compounds. This shows how the different strategies can overlap in a beneficial way. Another great example of this is the work on indotropanes.82 It can be seen as a BIOS library20,83 but also shares ideas from the PNP strategy.84 Whether you define it as one or the other, the final outcome was the identification of a novel class of hedgehog-signalling inhibitors and the myosin light chain kinase 1 (MLCK1) inhibitor, myokinasib.
 | ||
| Scheme 2 Synthesis of quinine- and quinidine-inspired compounds73,80 and identification of the autophagy inducer, tantalosin-1 (39),77,78 and autophagy inhibitor, azaquindole-1 (41a).79 DABCO = 1,4-diazabicyclo[2.2.2]octane. DCC = N,N′-dicyclohexylcarbodiimide, DMAP = 4-dimethylaminopyridine, Mes = mesityl. | ||
Similarly, in addition to the abovementioned cinchona alkaloids, stemona-inspired compounds have also been targeted using diverse strategies (Scheme 3).85–87 This work is particularly interesting as it can be considered an early example of the PNP strategy before it was formally defined, with elements from BIOS and (p)DOS. Here, the core scaffolds containing a strategic ketone are synthesised from scratch and not derived from an NP as above. Firstly, a Lewis acid-catalysed Diels–Alder reaction between diene 43 and dienophile 44 afforded the bicyclic azido diketones 45 which could undergo a Schmidt reaction by further treatment with a Lewis acid. Careful choice of Lewis acid and its equivalents allowed for the tandem Diels–Alder/Schmidt reaction to go all the way to the tricycle. Epimerisation with base of the alkyl group of the diastereoisomeric mixture of α-alkylated tricyclic ketones afforded the thermodynamically favoured β-alkyl stemona scaffold 46. This could then be subjected to a reductive Friedländer quinoline synthesis to afford edge-fused quinoline-analogues 47 in a PNP-fashion. By definition the PNP approach only allows the fusion of NP fragments. However, fusion with privileged scaffolds not found in NPs, as implied in the pDOS approach, to access a large number of scaffold combinations, can enable the identification of compounds that modulate diverse targets and processes in a selective way. Even though this work predates the PNP concept, this line of thought can be seen in the access of the spiro-fused 3,4-dihydroquinoxalines 48, a privileged scaffold, via a multicomponent reaction (MCR) with o-phenylenediamines, the ketone 46, and isocyanides.88 The 3,4-dihydroquinoxaline 48a was found to have a binding affinity (inhibitory constant (Ki)) of 431 nM to the 5-hydroxytryptamine (serotonin) 1A receptor (5-HT1A). Furthermore, subjecting PNPs to general synthetic diversification strategies as in DOS can give access to even more diverse and biologically relevant compounds. This idea is visible in the synthesis of the carbamates 49, which is an important structural motif in medicinal chemistry.89 The ketone 46 was diastereoselectively reduced with L-selectride® to give an alcohol which was then reacted with isocyanates to give the carbamates 49. Other analogues were accessed from another tricyclic ketone 50, synthesised from 46via an aza-Wittig reaction. The tricyclic ketone scaffold 50 is not found in NPs, but it is NP-like in terms of complexity. From this ketone, reductive amination yielded the tertiary amines 51 where analogue 51a showed a Ki of 2 nM to the sigma-1 receptor (σ1R) and 175 nM to the sigma-2 receptor (σ2R). Several other analogues with different functionalities were also accessed and in total the library consisted of 104 stemona analogues. Using the tricyclic core of stemona alkaloids in the synthesis shares a lot of ideas from BIOS. In addition, some similarity to reagent-based DOS is obvious since the analogues are derived from two core ketone scaffolds by changing reagents and conditions. One may even classify this example as an exhaustive DTS identifying 46 as the advanced intermediate. In fact, 46 has been used in a TS en route to the stemona alkaloid neostenine (52).90
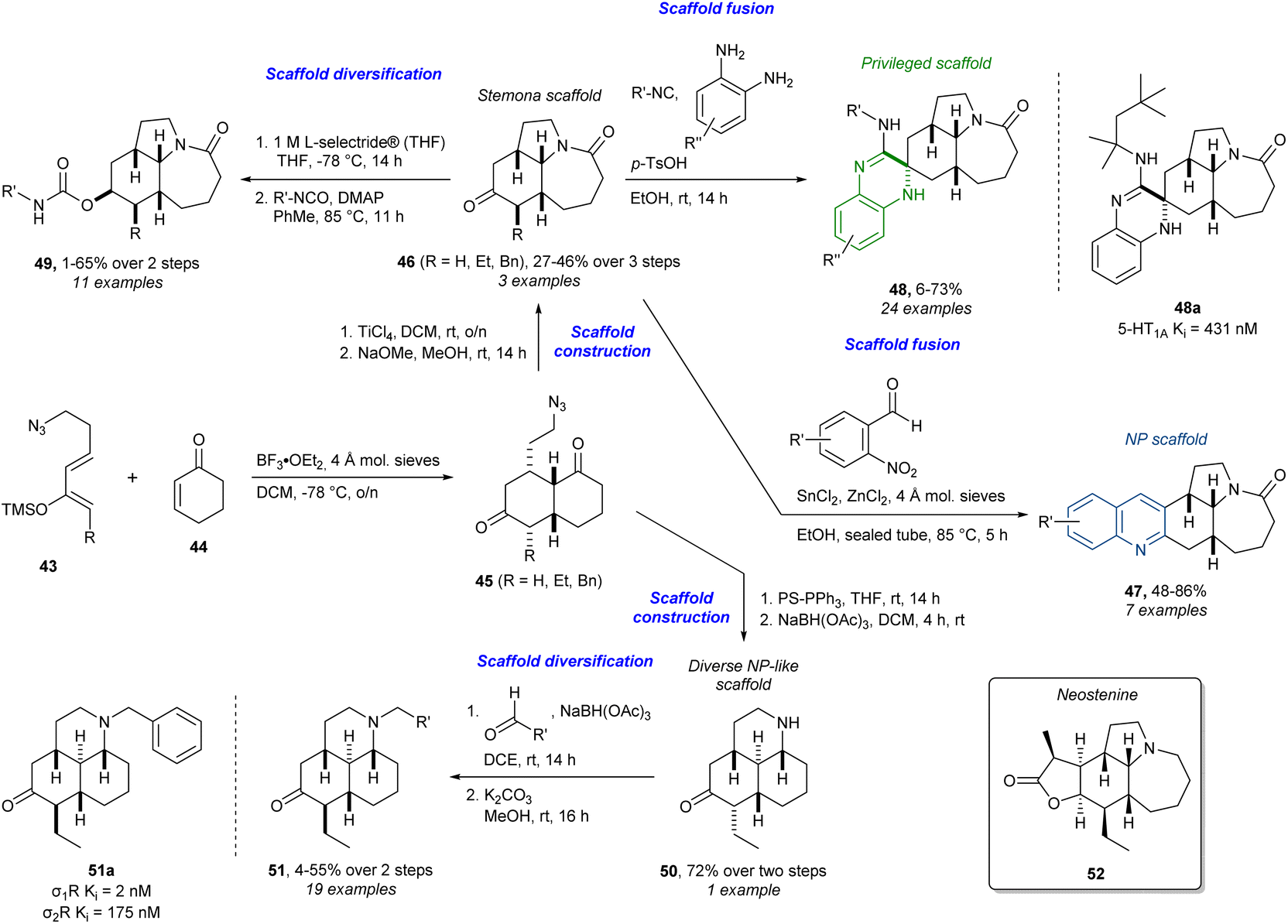 | ||
| Scheme 3 Synthesis of stemona-inspired compounds and identification of 2-HT1A ligand 48a and σ1R and σ2R ligand 51a.85–87 L-selectride® = lithium tri-sec-butylborohydride, PS-PPh3 = polystyrene bound triphenyl phosphine. | ||
Our own work on a tropane- and quinuclidine alkaloid-inspired compound collection91 is conceptually similar to the above. Using the commercially available quinuclidine scaffold 53 and tropane scaffold 57 several scaffold fusions could be carried out, again taking advantage of a reactive ketone (Scheme 4). Using the Kabbe reaction, the spirochromanones 54 and 58 could be accessed. The spirochromanone-quinuclidine analogue 54a was identified as a selective dual serotonin 2B (5-HT2B) and 2C receptor (5-HT2C) antagonist which was termed (S)-SCQ1. Chromanone-tropane 58 could be further diversified condensing with 2-aminobenzamide to give spirocyclic 2,3-dihydroquinazolinone 60. From the quinuclidine ketone 53, the Pictet–Spengler reaction afforded the spiro-fused tetrahydro-β-carboline (tryptoline) 55. Additionally, the spirophthalides 56 were accessed from 2-bromobenzoic acid in a three-step sequence going through a lithium–halogen exchange, nucleophilic attack, and intramolecular Fischer esterification. Lastly, the tropane 57 could be converted to the N-tosylhydrazone which could participate in a 1,3-dipolar (3 + 2) cycloaddition with chalcone to give the spiropyrazoline 61, a privileged scaffold. The total number of analogues was 58 including six additional other scaffolds not presented here. The overall strategy was presented as a mixture of PNP and DOS (reagent-based); however, the use of privileged scaffolds and core alkaloid skeletons makes the resemblance to pDOS and BIOS striking. Together with the aforementioned stemona alkaloid library, this is a representative example of the construction of compound libraries with specific target(s) in mind. This contrasts with the phenotypic screening approach as described in the synthesis of, for example, the “quinalogs”.
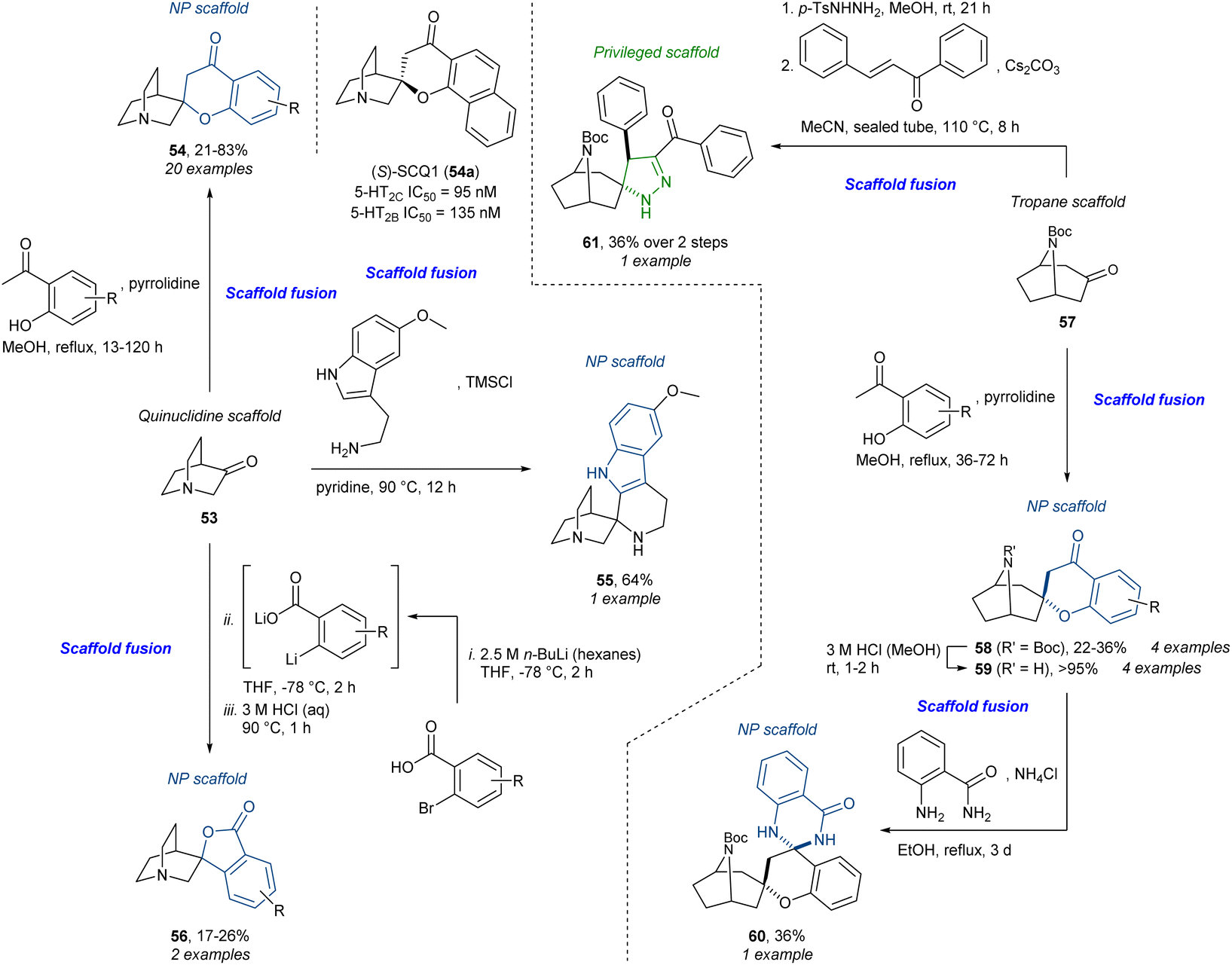 | ||
| Scheme 4 Synthesis of tropane- and quinuclidine alkaloid-inspired compounds and identification of dual 2-HT2B/C antagonist (S)-SCQ1 (54a).91 TMS = trimethylsilyl. | ||
As illustrated above, the incorporation of privileged scaffolds and diversity-generating strategies ((p)DOS) into a PNP approach can be very beneficial. Our recent work92 is an example of the PNP strategy where privileged scaffolds are introduced to access additional scaffolds. By the fusion of a trans-decalin sterol scaffold with several NP scaffolds and “unnatural” privileged scaffolds, a range of sterol-inspired analogues was synthesised (Scheme 5). The ketone 62 was synthesised as a key precursor containing the trans-sterol scaffold. The Fischer indole synthesis afforded the indoles 64. Furthermore, the quinoline-fused analogues 65 could be synthesised following a microwave irradiation (MWI) assisted Friedländer quinoline synthesis. Via the α-bromoketone 66, the imidazothiadiazoles 67 were isolated. The Pinner pyrimidone synthesis yielded the analogues 69 from the β-ketoester 68. Lastly, the β-ketoaldehyde 70 was used in a Knorr pyrazole synthesis affording 71. In addition, nine other scaffolds were accessed affording 65 sterol-inspired compounds in total. The pyrazole-fused analogues led to the identification of the potent and selective Aster-C inhibitor, (–)-astercin-1 (71a). Interestingly, the active enantiomer has the “unnatural” AB-ring stereochemistry. This indicated the importance of synthesising the library as racemic mixtures from the beginning. This is important for libraries where the goal is biological, as well as chemical, diversity, as this is more likely to be obtained by doubling the total number of compounds and probing enantiomeric differences. This is relevant for libraries where analogues are derived from scaffolds that are synthesised de novo in contrast to analogues derived from an NP source directly, since the majority of NPs are naturally produced as single enantiomers.
 | ||
| Scheme 5 Synthesis of sterol-inspired compounds and identification of Aster-C inhibitor (–)-astercin-1 (71a).92 DIPEA = N,N-diisopropylethylamine, MEM = 2-methoxyethoxymethyl. | ||
Following the trans-decalin sterol-inspired library, we decided to target a cis-decalin sterol-inspired compound collection,35 the reasoning being that compound libraries with diverse diastereochemical attributes can result in diverse biological profiles and different biological activity.93,94 In addition to similar edge-fused analogues targeted in the trans-decalin library, additional analogues were targeted using the CtD strategy. In this context, there is no reason why the CtD strategy should be limited to NPs.95–97 NP fragments or NP-inspired compounds that are accessible in sufficient quantities are also excellent substrates for the ring distortion reactions used in the CtD approach. Indoles have proven to be useful scaffolds for the CtD strategy.98–102 Thus, we employed a ring distortion strategy on the PNPs to access dPNPs (Scheme 6). The cis-fused decalone 72 was synthesised and used as the primary sterol scaffold. The indoles 73 were accessed by the Fischer indole synthesis in a similar manner to the trans-fused library. The indoles could be ring-expanded to afford the ketolactams 74 in a Witkop oxidation. The resulting ketolactams could be ring-contracted upon treating with base yielding 75 though the Camps quinolone synthesis. Oxidative ring dearomatisation of the indoles gave the 3-hydroxyindolenines 76. The ring contraction through an oxidative rearrangement of the indoles afforded spiropseudoindoxyl 77. The spirooxepinoindoles 78 were obtained by a different oxidative ring contraction in tandem with an intramolecular ring-forming condensation. The cis-fused sterol-inspired library consisted of 69 compounds in total. The morpholine-substituted spirooxepinoindole (–)-asteroxin-1 (6) was identified as a potent and selective Aster-A inhibitor. Again, the active enantiomer featured the unnatural stereochemistry at the AB-fusion, though one can argue that the resulting scaffold scarcely resembles a steroid in structure, while retaining its bioactivity features. In this case, modifying the oxidants afforded a range of diverse scaffolds from a PNP, where one could argue that the CtD component of the library is an example of reagent-based DOS. Importantly, the work on trans- and cis-fused sterol-inspired compounds is another example of a target-based screening campaign, where biological diversity was sought within a specific class of proteins, rather than across the whole proteome.
 | ||
| Scheme 6 Synthesis of sterol-inspired compounds and identification of Aster-A inhibitor (–)-asteroxin-1 (6).35 Oxone® = KHSO5·0.5KHSO4·0.5K2SO4, NBS = N-bromosuccinimide, NCS = N-chlorosuccinimide. | ||
In addition to our own work, several other research groups have also combined PNPs with diversity strategies (DOS) and ring distortion (CtD) in an explicit manner.29,103,104 In this context dPNPs were first defined as the combination of PNP and DOS/CtD giving compound collections that incorporate both biological relevance and scaffold diversity.29 In this work (Scheme 7), the indoles 79 underwent photocatalysed ring rearrangement to give pseudoindoxyls 80. These products could be fused to give 81 through an intramolecular Buchwald–Hartwig cross-coupling. The starting indoles 79 could also be subjected to a Pd-catalysed carbonylation/intramolecular indole dearomatisation cascade to give the ring-spiro-fused indolylindanones 82. The analogue 82a was identified as an inhibitor of Hedgehog (Hh) signalling. The analogues 82 could be further diversified by ring-edge-fusion to give indoline-indanone-isoquinolines 83via a combined Pd-catalysed arylation and amidation. Additionally, reduction of the indolenine functionality in 82 afforded the spiro-indoline-indanones 84 with high diastereoselectivity. Further diversification of this class was achieved by substitution at the nitrogen using different halides to give 85. A number of other compound classes were produced to afford a compound collection of 154 analogues in total. This work is a good example of using diversity strategies from DOS (here B/C/P in particular) to make diverse scaffolds which by design fall under the category of PNPs. Thus, the diversity is introduced in the design of the PNPs. This is slightly different from the work on (–)-asteroxin-1 where the diversity is introduced to the resulting PNPs via ring distortions afterwards and not directly into the design of the initial PNPs.
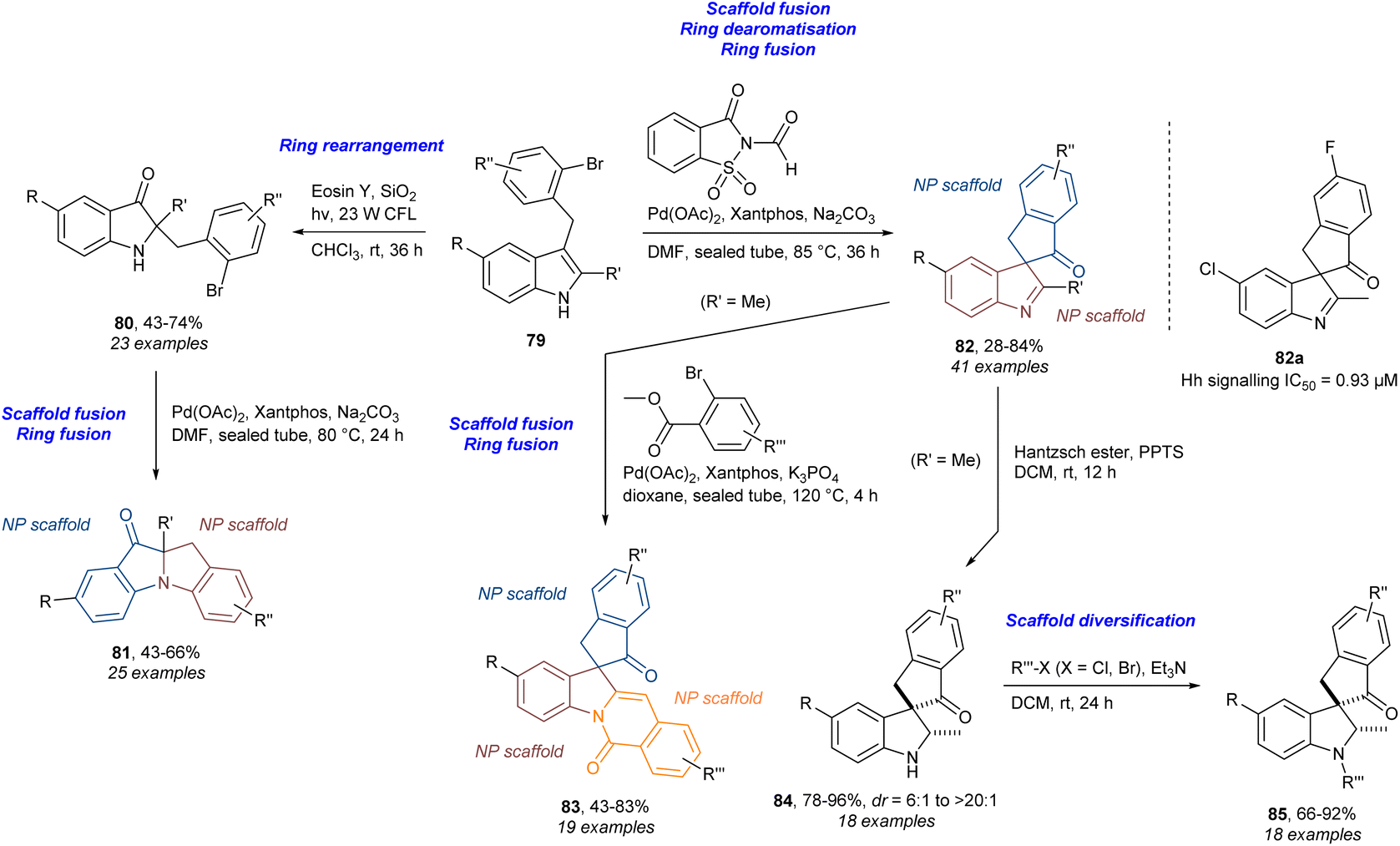 | ||
| Scheme 7 Synthesis of dPNPs and identification of Hh signalling inhibitor 82a.29 CFL = compact fluorescent lamp, Eosin Y = 2-(2,4,5,7-tetrabromo-6-oxido-3-oxo-3H-xanthen-9-yl)benzoate, Hantzsch ester = diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate, PPTS = pyridinium p-toluenesulfonate, Xantphos = (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane). | ||
As shown above, taking certain elements from NPs to generate NP-inspired compounds can certainly result in biologically relevant compounds. Nevertheless, the actual TS of NPs and their simplified derivatives has long been used to access bioactive compounds and generate SAR information about a certain pharmacologically active NP. However, while beneficial, important areas of chemical space may be missed due to synthetic limitations.64 Alongside the strategies focused on generating a wide range of structurally different analogues, modern pragmatic takes on TS such as DTS, FOS, DRA, and PDR have also developed in order to streamline the process of investigating the chemical space surrounding an NP. These share important similarities with other strategies, especially BIOS. Recent work presents the synthesis of salvinorin A (salA) analogues using DRA to explore the chemical space around this NP (Scheme 8).45,46,105 SalA is a potent κ-opioid receptor agonist. However, its TS has been troublesome due to its complexity and instability which in turn have also made exploration of the chemical space around it and SAR studies difficult. The NP was treated as a dynamic TM to reduce synthetic complexity while retaining molecular complexity. This led to the realisation that a rational removal of the C20 methyl would ease the synthesis and stabilise the resulting compounds. The goal of easing the synthesis of the NP is also a key point in FOS and PDR. Additionally, the authors used molecular docking (another key attribute in FOS) to evaluate 20-nor-salvinorin A, which suggested that it would have similar binding to salA. Thus, they synthesised the common intermediate 87 in eight steps from Hagemann's ester (86) which could be diversified in a DTS-fashion through a Heck reaction followed by lactonisation to afford the first generation of salA analogues 88 which were more stable than salA. More importantly, the analogue (±)-20-nor-salA (88a) showed similar potency and selectivity. They then synthesised (±)-O6C-20-nor-salA ((±)-12) in five steps from 87 and identified that replacement of the O6 with a carbon further stabilised the compound while retaining the potency and selectivity. They then developed an asymmetric synthesis of 12 starting from (+)-89 which could be synthesised with a 99% enantiomeric excess (ee). From (+)-89 the common intermediate 90 was accessed in five steps. This intermediate was diversified by a Hayashi conjugate addition to give the second generation of salA analogues 91 including the enantioenriched (–)-12. Further diversification of the second vector, the ketone, allowed for synthesis of oximes and alcohols (92) through condensation and nucleophilic addition/reduction, respectively. Consequently, a salA analogue with improved potency (92a) was identified. Lastly, ring expansion of 91 in a CtD-manner through the Beckmann rearrangement and Baeyer–Villiger oxidation afforded the corresponding lactam or lactone (93), respectively. This second generation of salA analogues allowed for further SAR study and exploration of the salA chemical space through a common and diversifiable intermediate similar to DTS and AOS. In addition, some of the added functionalities are NP fragments or privileged scaffolds showing some resemblance to PNP and BIOS in this diversification step. In total, five first generation and 29 second generation salA analogues were synthesised.
 | ||
| Scheme 8 Synthesis of salvinorin A analogues and identification of multiple KOR agonists.45,46,105 BenzP* = 1,2-bis(tert-butylmethylphosphino)benzene, BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl, COD = 1,5- cyclooctadiene, QuinoxP* = 2,3-bis(tert-butylmethylphosphino)quinoxaline, (S)-Trifer = 1,1′-bis{1-[(R)-ferrocenyl-2-(S)-ethyl-1-(diethylamino)phenyl]-(R)-phosphino}ferrocene, XPhos = dicyclohexyl[2′,4′,6′-tris(propan-2-yl)[1,1′- biphenyl]-2-yl]phosphane. | ||
In recent work methyl deletion is also used in a FOS of 26-nor-germanicol (94) and 26-nor-lupeol (95) (Scheme 9).106 Here the methyl is removed to remove the synthetically difficult vicinal quaternary stereogenic centre from the synthesis. The authors speculated that the biological function of lupeol and germanicol would be retained in the nor-derivatives. They desired to reduce synthetic complexity while retaining function which is very similar to ideas of DRA and PDR. Initially, the common intermediate 96 was synthesised which allowed access to the germanicol analogue, 26-nor-germanicol (94), in six steps and the lupeol analogue, 26-nor-lupeol (95), in 12 steps overall. The synthesis of 95 went through 97 (six steps from 96) and 98 (four steps from 97) which was converted into 95 in two steps. Unfortunately, biological screening of the 26-nor analogues 94 and 95 was not feasible due to dimethyl sulfoxide (DMSO) solubility issues. Interestingly, screening the intermediates en route to the TMs, similar to the PDR, led to the identification of two substituted unnatural ent-estranes as androgen receptor (AR) antagonists with similar (98) and enhanced (97) potency compared to lupeol. Thus, these less structurally complex intermediates retain or improve function. The late intermediate 96 allows for the synthesis of other 26-nor analogues similar to a DTS approach to further gain SAR information.
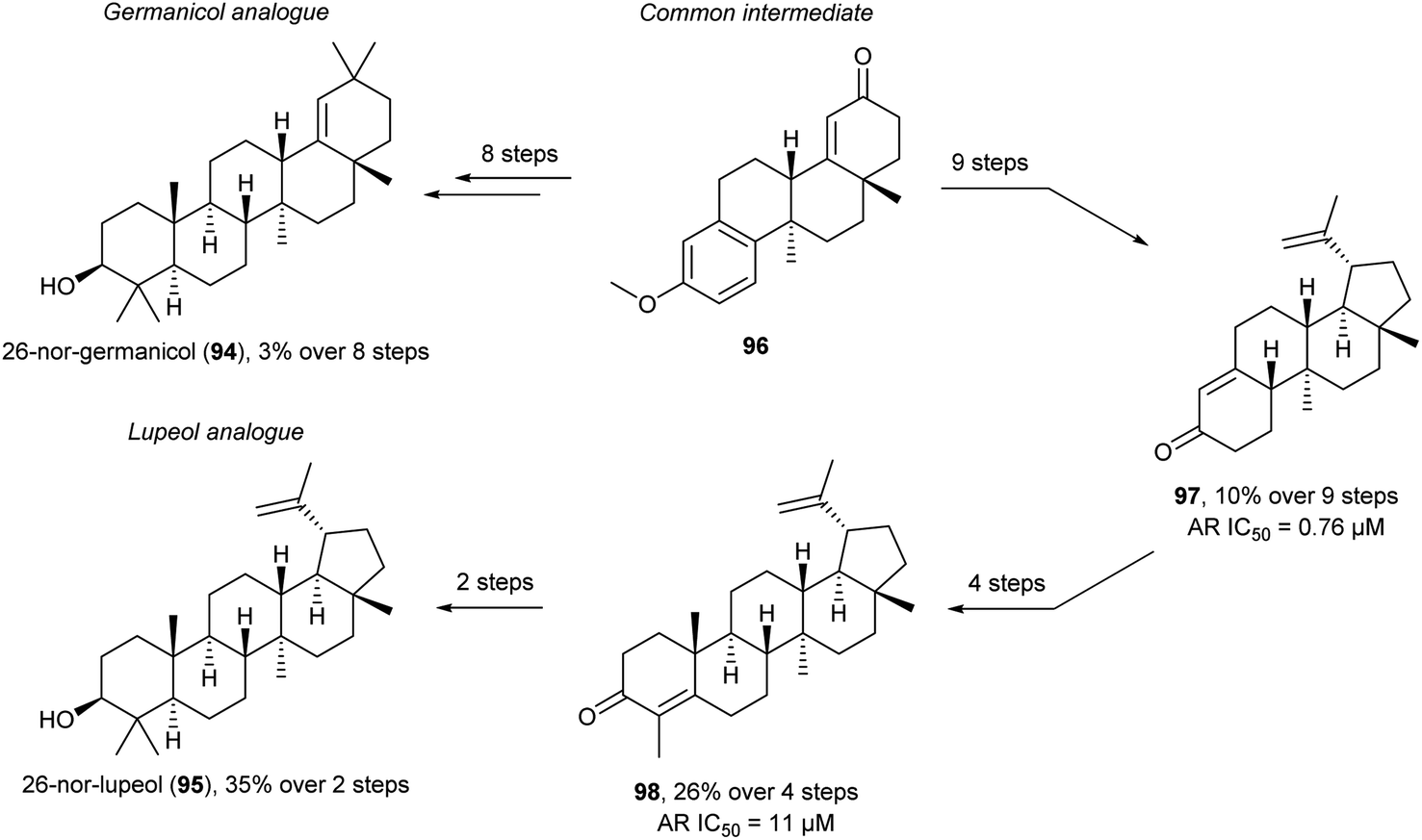 | ||
| Scheme 9 Synthesis of 26-nor-germanicol (94) and 26-nor-lupeol (95) and identification of AR antagonists 97 and 98.106 | ||
The work on latrunculin analogues107 should also be mentioned. In this work, the authors simplify and streamline the synthesis including yet another methyl deletion. They identify a simplified analogue that shares similar actin-biding properties to the most active member of the latrunculin family. The work is presented as a DTS but ideas from FOS, DRA, PDA, and even BIOS are easy to identify. The research on analogues of sinularia NPs108 led to the identification of new compounds with interesting cytotoxicities and selectivities against cancer cell lines. The compounds contain the tricyclic cores as found in sinularia NPs but in general less complex, and one of the compounds could serve as a useful intermediate towards additional analogues. The work is published as an example of PDR, but again it shares a lot of similarities to DTS, AOS, FOS, PDA, and BIOS.
The majority of examples outlined so far focus on terpenes and alkaloids as guiding NP targets for library synthesis. However, polyketides and more specifically polyether ionophores (PEIs) have received considerably less attention. This is most likely in large part due to the absence of a bioactivity-defining scaffold, requiring larger synthetic efforts to approach NP complexity. Recent work has addressed this challenge by deconstructing natural PEIs to smaller fragments and reconstructing them in new ways to create structural diversity and access so-called “hybrid polyethers”.109 With this structural optimisation they retain the antibacterial activity found in the natural PEIs but achieve improved antibacterial selectivity (Scheme 10). The lasalocid acid fragment 99 was obtained from commercially available lasalocid. Using slightly different conditions, the Z-triethylsilyl (TES) enol ether 100 and a mixture of E-(100′) and Z-TES enol ether (2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z) were obtained. Then Mukaiyama aldol addition to the synthesised nonthmicin and ecteinamycin fragment 101 yielded the anti (102) or syn (102′) product depending on the starting material. Then silyl deprotection and ester hydrolysis afforded 103. Through DCC-activation of the carboxylic acid, it was coupled to fragment 104 which is proposed to be the group responsible for cation binding in certain polyether ionophores. This yielded the final hybrid polyethers 105 and the new potent and selective antibiotic 105a. This is an excellent example of streamlining the synthesis for efficient diversification and investigation of the chemical space and biology related to the NPs, key arguments in FOS, DRA, and PDR. The use of coupling of NP fragments and fragments with a known biological function have strong resemblance to PNP and BIOS.
:1 E/Z) were obtained. Then Mukaiyama aldol addition to the synthesised nonthmicin and ecteinamycin fragment 101 yielded the anti (102) or syn (102′) product depending on the starting material. Then silyl deprotection and ester hydrolysis afforded 103. Through DCC-activation of the carboxylic acid, it was coupled to fragment 104 which is proposed to be the group responsible for cation binding in certain polyether ionophores. This yielded the final hybrid polyethers 105 and the new potent and selective antibiotic 105a. This is an excellent example of streamlining the synthesis for efficient diversification and investigation of the chemical space and biology related to the NPs, key arguments in FOS, DRA, and PDR. The use of coupling of NP fragments and fragments with a known biological function have strong resemblance to PNP and BIOS.
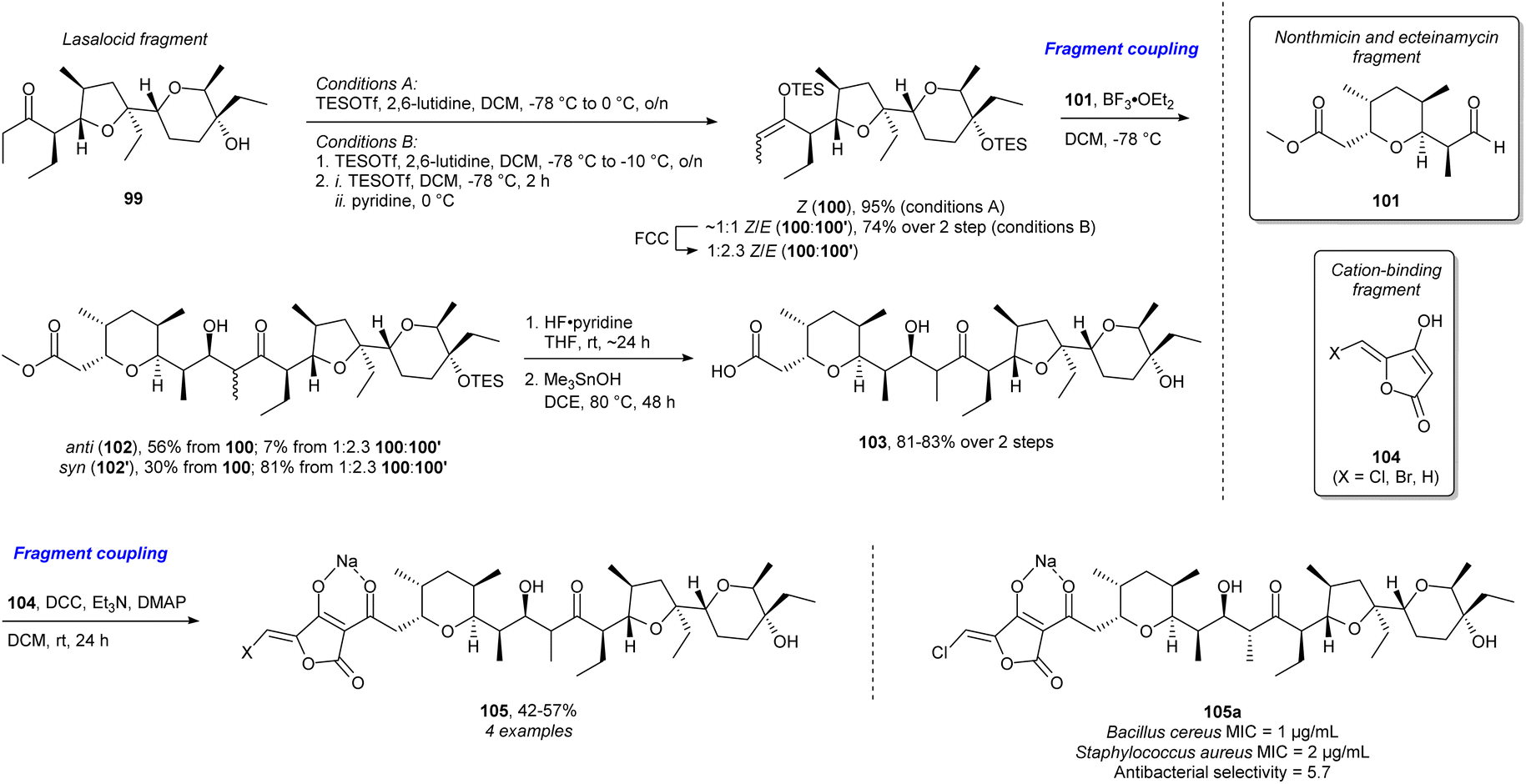 | ||
| Scheme 10 Synthesis of “hybrid polyethers” and identification of the potent and selective antibiotic 105a.109 FCC = flash column chromatography, DCC = N,N′-dicyclohexylcarbodiimide, DMAP = 4-dimethylaminopyridine, MIC = minimum inhibitory concentration. | ||
The described examples show the benefit of rational changes to the retrosynthetic analysis and/or synthesis to reduce synthetic complexity while maintaining structural complexity and biological function (FOS/DRA/PDR) coupled with diversity methodologies to make analogues of the NP (DTS/AOS/DOS) in the pursuit of “supernatural products”.110,111 The examples also illustrate how the boundaries between the different synthetic strategies can be blurred or even broken down, and that this is more likely to be a benefit than a problem. In terms of biological screening, the majority are target-based and influenced by the known bioactivity of the guiding/parent NP since they are screened against the targets known for the guiding NP. However, compound collections may also be made to target a biological phenotype with multiple associated targets or used across various target classes and phenotypes altogether. Therefore, the choice of chemical starting point(s) and synthetic strategies must be carefully considered.
Targeted combination of approaches for a specific project goal?
To conclude the perspective, we have chosen to provide a prospective view to aid researchers seeking to design a strategy for their specific goal, whether this be a target- or phenotype-based screen, or a synthetic chemistry campaign aimed at broad coverage of chemical and biological space. In this context we concur with Shenvi's assertion that the “purpose dictates strategy”.7 We have outlined a generalised approach based on a flow chart to ask researchers to consider the project goal and the information available to them at the start, when choosing individual strategies or a combination thereof (Fig. 2). | ||
| Fig. 2 Flow chart for choosing the optimal strategies based on the project goals. | ||
For example, if the desired outcome is a ligand for a specific target, one must determine whether the target has any known ligands. If that is the case, then the ligand(s) can be broken down into one or two primary scaffolds or fragments with functionalisable handles. Then, the chemical diversity localised around the primary scaffold/fragment should be targeted. This can be achieved by general diversity-generating approaches like (p)DOS and/or fusion with relevant scaffolds as in PNP, as illustrated by the previous work on stemona alkaloids85,86 and our own work on alkaloid- and sterol-inspired compounds.35,91,92 This strategy applies to all types of ligands. Additionally, if the ligand is an NP, the NP-driven approaches such as DTS, AOS, DRA, CtD, PDR, and FOS are also very good options. If very specific SAR questions need to be answered by densely populating an area of chemical space, then DTS and AOS are useful. If the NP is in high abundance, CtD is an option to quickly access diverse analogues of the NP. Furthermore, when the pharmacophore is known for the NP, it can be used as a primary fragment in a (p)DOS/BIOS/PNP approach but is also applicable in a FOS/PDR campaign. If the NP is in low abundance, unstable, or hard to access synthetically, breaking it down to useful fragment(s) as above or following a DRA approach may be beneficial as showcased in the work on salA analogues.45,46,105
On the other hand, if no ligands are known or an entirely new chemotype is sought, one can employ an X-ray crystal structure or AlphaFold112,113 to model a binding site and predict a pharmacophore model which can be targeted by synthesis. The predicted pharmacophore can then be used as a starting point in a (p)DOS/PNP campaign. Alternatively, a fragment-based screen can be a cost-efficient approach to generate new scaffolds and starting points while covering a larger proportion of chemical space, around which a (p)DOS/PNP approach can be centred. Notably, even the fragments themselves can be designed by DOS to increase chemical diversity and Fsp3 content in the fragment collection.114 This has already been used in some cases115 including the reported synthesis of fluorinated Fsp3-rich fragments.116
In addition to a target-based approach, a phenotype of interest can also be selected and screened against. If there are known modulators of the phenotype, then diverse fragments thereof could be picked. The fragments are fused with diverse secondary fragments similar to the case with known ligands. Similarly, the NP-based approaches are also applicable if the modulators are NPs. If no modulators of the phenotype of interest are known, a chemical diversity-driven approach could be applied, similarly to (p)DOS. Compounds with suitable physicochemical properties to reach the target site should be prioritised. In this regard, using NP-inspired starting points as in BIOS/(d)PNP can be beneficial to reach biologically relevant space. This approach is also useful if no specific phenotype is targeted, but an unbiased phenotypic evaluation such as cell painting is used, as shown with the work on quinalogs.79,80 Cell painting is a particularly powerful method to assess the bioactivity of a compound collection with no specific target or phenotype in mind, in an unbiased way. Here, cells are stained with multiple dyes covering different organelles, and changes to staining patterns after compound treatment can be measured. Importantly, with a large and diverse reference set of compounds at hand, it is possible to identify modes of action and even target(s) of new compounds, by comparison. Recent bioactivity clusters that can rapidly be discerned are lysosomotropism117 and cholesterol homeostasis,118 modulation of tubulin polymerisation,119 mitochondrial stress,120 modulation of potassium channels,121 and pyrimidine biosynthesis,122 amongst others. It should be noted that in approaches with no known ligands or modulators, high-throughput screening (HTS) is always an option to identify starting points if time and budget allows.
In all library synthesis strategies where bioactive scaffolds or fragments are designed from scratch, the goal is typically to synthesise 10–20 fragment combinations or scaffolds, with approximately 5–20 analogues each for a total of 50–400 compounds. Fragments, or the resulting analogues, should ideally contain diverse substituents including electron-withdrawing groups (EWGs), electron-donating groups (EDGs), hydrophobic and hydrophilic groups, and ideally hydrogen bond donors (HBDs) and hydrogen bond acceptors (HBAs), at different positions. Additionally, substituents with different steric bulk can be explored. This includes flexible or locked and linear or branched groups. Lastly, the compounds should be synthesised as racemic mixtures to reduce bias and obtain twice the number of compounds for biological screening in approaches where compounds are synthesised de novo. In strategies where compounds are directly derived from available NPs, this criterion is, in most cases, not possible to fulfil. In our experience the above criteria are both necessary and sufficient to find bioactive compounds for a given target or phenotype of interest, even from a relatively modestly sized library of <100 compounds.
Outlook and conclusions
In addition to the considerations outlined thus far, we see great benefits in adopting recent developments in the field, including C–H and late-stage functionalisation, as well as single-atom and skeletal editing strategies, to streamline analogue synthesis and further expand the diversity of a given collection, but also for the medicinal chemistry optimisation of the hit compounds into leads. Efficiently combining library synthesis with C–H/late-stage functionalisation has proven effective as shown from the work on oxazatwistane-derived analogues.123 It can provide access to new analogues from a vector/diversification point that is not obvious. Several strategies for C–H functionalisation are known including free-radical, metal-catalysed, photochemical, electrochemical, and chemoenzymatic reactions.124 Chemoenzymatic synthesis has proven to be a very useful strategy for C–H functionalisation. Very recent work combined chemoenzymatic synthesis with DOS and CtD, which was termed chemoenzymatic DOS (CeDOS).125 Chemoenzymatic C–H functionalisation of the NP parthenolide enabled the synthesis of a diverse parthenolide-based library using divergent chemical routes. With chemoenzymatic synthesis applied successfully to TS of NPs,126–133 this is a powerful example of the combination of chemoenzymatic synthesis with a diversifying strategy such as DOS. Two main strategies can be considered when utilising chemoenzymatic synthesis in library synthesis and medicinal chemistry.134 The first is the early-stage synthesis of novel building blocks combined with a general diversifying strategy to access new analogues. The second is the late-stage diversification of NP-inspired advanced key intermediates or analogues. Late-stage single-atom and skeletal editing is an attractive approach in medicinal chemistry to quickly elucidate SAR of hit and lead compounds.135 Removal, addition, or exchange of a single atom in a molecule is often achieved by modifying the synthesis of the compound from an early point in the route or by a totally different route. However, recent advantages in direct atom deletion, insertion, and exchange136–153 can in some cases remove the need for new retrosynthetic analysis and provide new diverse compounds more efficiently. Additionally, subtle changes to the overall molecular shape and not just single atoms can have a large impact on the function and properties of compounds. Efficient methods to access isomeric chemical space by “shapeshifting” have also been reported.154 This example bears some resemblance to CtD strategies, which may also be considered skeletal editing, in a broad sense. Even combining chemoenzymatic methods with skeletal editing can be a powerful tool as showcased by the recent example which allowed for ring expansion at aliphatic C–H sites.155 With the current and continuously growing number of methods in the area of late-stage C–H functionalisation and skeletal editing, we see these becoming more integrated into the synthesis of NP-inspired compounds and lead optimisation and in accessing new chemically and biologically relevant space.In summary, we have outlined how a large variety of strategies are available to access diverse NP-derived and -inspired compound collections. We emphasise how combining several strategies or elements thereof, depending on the specific need and purpose, can be beneficial in the pursuit of new bioactive molecules in an efficient manner. We hope that the ideas outlined here will serve to help chemists push the boundaries in the synthesis of natural-product inspired, biologically relevant compound collections.
Abbreviations
| 5-HT1A | 5-Hydroxytryptamine 1A |
| 5-HT2B | 5-Hydroxytryptamine 2B receptor |
| 5-HT2C | 5-Hydroxytryptamine 2C receptor |
| σ1R | Sigma-1 receptor |
| σ2R | Sigma-2 receptor |
| Ac | Acetyl |
| ADS | Activity-directed synthesis |
| AOS | Analogue-oriented synthesis |
| AR | Androgen receptor |
| Ar | Aryl |
| B/C/P | Build/couple/pair |
| BenzP* | 1,2-Bis(tert-butylmethylphosphino)benzene |
| BINAP | 2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl |
| BIOS | Biology-oriented synthesis |
| Bn | Benzyl |
| Boc | tert-Butyloxycarbonyl |
| CeDOS | Chemoenzymatic diversity-oriented synthesis |
| CFL | Compact fluorescent lamp |
| CLS | Combinatorial library synthesis |
| COD | 1,5-Cyclooctadiene |
| conc. | Concentrated |
| CS | Conventional synthesis |
| CtD | Complexity-to-diversity |
| Cy | Cyclohexyl |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DCE | Dichloroethane |
| DCM | Dichloromethane |
| DIPEA | N,N-Diisopropylethylamine |
| DMA | Dimethylacetamide |
| DMAP | 4-Dimethylaminopyridine |
| DMEDA | N,N′-Dimethylethylenediamine |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DOS | Diversity-oriented synthesis |
| DPEPhos | bis[(2-Diphenylphosphino)phenyl]ether |
| dPNP | Diverse pseudo-natural product |
| dr | Diastereoisomeric ratio |
| DRA | Dynamic retrosynthetic analysis |
| DTS | Diverted total synthesis |
| DTU | Technical University of Denmark |
| EC50 | Half maximal effective concentration |
| EDG | Electron-donating group |
| ee | Enantiomeric excess |
| ESCRT | Endosomal sorting complexes required for transport |
| Et | Ethyl |
| EWG | Electron-withdrawing group |
| FBDD | Fragment-based drug-discovery |
| FCC | Flash column chromatography |
| FLS | Focussed library synthesis |
| FOS | Function-oriented synthesis |
| Fsp3 | Fraction of sp3-hybridised carbons |
| HBA | Hydrogen bond acceptor |
| HBD | Hydrogen bond donor |
| HFIP | Hexafluoroisopropanol |
| Hh | Hedgehog |
| HTS | High-throughput screening |
| IC50 | Half maximal inhibitory concentration |
| K i | Inhibitory constant |
| LC3 | Microtubule-associated protein 1A/1B light chain 3 |
| MCR | Multicomponent reaction |
| Me | Methyl |
| MEM | 2-Methoxyethoxymethyl |
| Mes | Mesityl |
| MIC | Minimum inhibitory concentration |
| MLCK1 | Myosin light chain kinase 1 |
| mol. sieves | Molecular sieves |
| MWI | Microwave irradiation |
| NBS | N-Bromosuccinimide |
| n-Bu | n-Butyl |
| NCS | N-Chlorosuccinimide |
| NP | Natural product |
| o/n | Overnight |
| pDOS | Privileged-substructure-based diversity-oriented synthesis |
| PDR | Pharmacophore-directed retrosynthesis |
| PEI | Polyether ionophore |
| Ph | Phenyl |
| PMP | para-Methoxyphenyl |
| PNP | Pseudo-natural product |
| PPTS | Pyridinium para-toluenesulfonate |
| PS | Polystyrene |
| p-Ts | para-Tosyl |
| QuinoxP* | 2,3-bis(tert-Butylmethylphosphino)quinoxaline |
| RCM | Ring-closing metathesis |
| rt | Room temperature |
| salA | Salvinorin A |
| SAR | Structure–activity relationship |
| (S)-Trifer | 1,1′-bis{1-[(R)-Ferrocenyl-2- (S)-ethyl-1-(diethylamino)phenyl]–(R)-phosphino}ferrocene |
| TBTU | N,N,N′,N′-Tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate |
| t-Bu | tert-Butyl |
| TES | Triethylsilyl |
| Tf | Trifyl |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| TM | Target molecule |
| TMS | Trimethylsilyl |
| TOS | Target-oriented synthesis |
| TPS | Two-phase synthesis |
| TS | Total synthesis |
| Xantphos | (9,9-Dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) |
| XPhos | Dicyclohexyl[2′,4′,6′-tris(propan-2-yl)[1,1′– biphenyl]-2-yl]phosphane |
Data availability
The supplementary figures with graphical illustrations, explanations, and examples of the individual strategies are available in the ESI.†Author contributions
F. S. B. and L. L. conceived the perspective. F. S. B. produced all figures. F. S. B. and L. L. conducted the literature search and wrote the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Novo Nordisk Foundation (NNF21OC0067188), the European Research Council (ERC, ChemBioChol, 101041783) and DTU for funding. We thank Prof. Herbert Waldmann and Dr Daniel Foley for critical reading of the manuscript and helpful comments.References
- L. Laraia and H. Waldmann, Drug Discovery Today:Technol., 2017, 23, 75–82 CrossRef PubMed.
- B. J. Huffman and R. A. Shenvi, J. Am. Chem. Soc., 2019, 141, 3332–3346 CrossRef CAS PubMed.
- M. Grigalunas, A. Burhop, A. Christoforow and H. Waldmann, Curr. Opin. Chem. Biol., 2020, 56, 111–118 CrossRef CAS PubMed.
- M. Grigalunas, S. Brakmann and H. Waldmann, J. Am. Chem. Soc., 2022, 144, 3314–3329 CrossRef CAS PubMed.
- R. J. Young, S. L. Flitsch, M. Grigalunas, P. D. Leeson, R. J. Quinn, N. J. Turner and H. Waldmann, JACS Au, 2022, 2, 2400–2416 CrossRef CAS PubMed.
- J. Liu, M. Grigalunas and H. Waldmann, in Annual Reports in Medicinal Chemistry, Academic Press, 2023, vol. 61, pp. 1–53 Search PubMed.
- R. A. Shenvi, ACS Cent. Sci., 2024, 10, 519–528 CrossRef CAS PubMed.
- S. B. Bharate and C. W. Lindsley, J. Med. Chem., 2024, 67, 20723–20730 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- C. R. Pye, M. J. Bertin, R. S. Lokey, W. H. Gerwick and R. G. Linington, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5601–5606 CrossRef CAS PubMed.
- M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46–58 CrossRef.
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 CrossRef.
- S. Oh and S. B. Park, Chem. Commun., 2011, 47, 12754–12761 RSC.
- J. Kim, H. Kim and S. B. Park, J. Am. Chem. Soc., 2014, 136, 14629–14638 CrossRef CAS PubMed.
- G. Karageorgis, S. Warriner and A. Nelson, Nat. Chem., 2014, 6, 872–876 CrossRef CAS PubMed.
- G. Karageorgis, S. Liver and A. Nelson, ChemMedChem, 2020, 15, 1776–1782 CrossRef CAS PubMed.
- G. Karageorgis, D. J. Foley, L. Laraia and H. Waldmann, Nat. Chem., 2020, 12, 227–235 CrossRef CAS PubMed.
- G. Karageorgis, D. J. Foley, L. Laraia, S. Brakmann and H. Waldmann, Angew. Chem., Int. Ed., 2021, 60, 15705–15723 CrossRef CAS PubMed.
- S. Wetzel, R. S. Bon, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 10800–10826 CrossRef CAS PubMed.
- H. van Hattum and H. Waldmann, J. Am. Chem. Soc., 2014, 136, 11853–11859 CrossRef CAS PubMed.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS.
- M. E. Abbasov, R. Alvariño, C. M. Chaheine, E. Alonso, J. A. Sánchez, M. L. Conner, A. Alfonso, M. Jaspars, L. M. Botana and D. Romo, Nat. Chem., 2019, 11, 342–350 CrossRef CAS PubMed.
- R. W. Huigens III, K. C. Morrison, R. W. Hicklin, T. A. Flood Jr, M. F. Richter and P. J. Hergenrother, Nat. Chem., 2013, 5, 195–202 CrossRef.
- S. Woo and R. A. Shenvi, Acc. Chem. Res., 2021, 54, 1157–1167 CrossRef CAS PubMed.
- J. T. Njardarson, C. Gaul, D. Shan, X.-Y. Huang and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 1038–1040 CrossRef CAS.
- R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2006, 71, 8329–8351 CrossRef CAS.
- Y. Ishihara and P. S. Baran, Synlett, 2010, 12, 1733–1745 Search PubMed.
- Z. A. Könst, A. R. Szklarski, S. Pellegrino, S. E. Michalak, M. Meyer, C. Zanette, R. Cencic, S. Nam, V. K. Voora, D. A. Horne, J. Pelletier, D. L. Mobley, G. Yusupova, M. Yusupov and C. D. Vanderwal, Nat. Chem., 2017, 9, 1140–1149 CrossRef.
- S. Bag, J. Liu, S. Patil, J. Bonowski, S. Koska, B. Schölermann, R. Zhang, L. Wang, A. Pahl, S. Sievers, L. Brieger, C. Strohmann, S. Ziegler, M. Grigalunas and H. Waldmann, Nat. Chem., 2024, 16, 945–958 CrossRef CAS PubMed.
- A. Schuffenhauer, P. Ertl, S. Roggo, S. Wetzel, M. A. Koch and H. Waldmann, J. Chem. Inf. Model., 2007, 47, 47–58 CrossRef CAS PubMed.
- V. Pogacic, A. N. Bullock, O. Fedorov, P. Filippakopoulos, C. Gasser, A. Biondi, S. Meyer-Monard, S. Knapp and J. Schwaller, Cancer Res., 2007, 67, 6916–6924 CrossRef CAS.
- S. D. Larsen, C. F. Stachew, P. M. Clare, J. W. Cubbage and K. L. Leach, Bioorg. Med. Chem. Lett., 2003, 13, 3491–3495 CrossRef CAS PubMed.
- B. M. Ibbeson, L. Laraia, E. Alza, C. J. O' Connor, Y. S. Tan, H. M. L. Davies, G. McKenzie, A. R. Venkitaraman and D. R. Spring, Nat. Commun., 2014, 5, 3155 CrossRef PubMed.
- J. Kim, J. Jung, J. Koo, W. Cho, W. S. Lee, C. Kim, W. Park and S. B. Park, Nat. Commun., 2016, 7, 13196 CrossRef CAS PubMed.
- F. S. Bro, L. Depta, N. J. Dekker, H. P. Bryce-Rogers, M. L. Madsen, K. F. Præstegaard, T. Petersson, T. Whitmarsh-Everiss, M. Kubus and L. Laraia, ACS Cent. Sci., 2025, 11, 136–146 CrossRef CAS PubMed.
- J. Ceballos, M. Schwalfenberg, G. Karageorgis, E. S. Reckzeh, S. Sievers, C. Ostermann, A. Pahl, M. Sellstedt, J. Nowacki, M. A. Carnero Corrales, J. Wilke, L. Laraia, K. Tschapalda, M. Metz, D. A. Sehr, S. Brand, K. Winklhofer, P. Janning, S. Ziegler and H. Waldmann, Angew. Chem., Int. Ed., 2019, 58, 17016–17025 CrossRef CAS.
- A. Nören-Müller, I. Reis-Corrêa, H. Prinz, C. Rosenbaum, K. Saxena, H. J. Schwalbe, D. Vestweber, G. Cagna, S. Schunk, O. Schwarz, H. Schiewe and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10606–10611 CrossRef PubMed.
- I. R. Corrêa Jr, A. Nören-Müller, H.-D. Ambrosi, S. Jakupovic, K. Saxena, H. Schwalbe, M. Kaiser and H. Waldmann, Chem.–Asian J., 2007, 2, 1109–1126 CrossRef PubMed.
- P. A. Wender, J. L. Sloane, Q. H. Luu-Nguyen, Y. Ogawa, A. J. Shimizu, S. M. Ryckbosch, J. H. Tyler and C. Hardman, J. Org. Chem., 2020, 85, 15116–15128 CrossRef CAS PubMed.
- E. Llabani, R. W. Hicklin, H. Y. Lee, S. E. Motika, L. A. Crawford, E. Weerapana and P. J. Hergenrother, Nat. Chem., 2019, 11, 521–532 CrossRef CAS PubMed.
- K. Yamamoto, R. M. Garbaccio, S. J. Stachel, D. B. Solit, G. Chiosis, N. Rosen and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 1280–1284 CrossRef CAS.
- X. Lei and S. J. Danishefsky, Adv. Synth. Catal., 2008, 350, 1677–1681 CrossRef CAS PubMed.
- R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker and K. Schenker, J. Am. Chem. Soc., 1954, 76, 4749–4751 CrossRef CAS.
- R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker and K. Schenker, Tetrahedron, 1963, 19, 247–288 CrossRef CAS.
- S. Hirasawa, M. Cho, T. F. Brust, J. J. Roach, L. M. Bohn and R. A. Shenvi, Bioorg. Med. Chem. Lett., 2018, 28, 2770–2772 CrossRef CAS PubMed.
- S. J. Hill, N. Dao, V. Q. Dang, E. L. Stahl, L. M. Bohn and R. A. Shenvi, ACS Cent. Sci., 2023, 9, 1567–1574 CrossRef CAS PubMed.
- P. Ertl, S. Roggo and A. Schuffenhauer, J. Chem. Inf. Model., 2008, 48, 68–74 CrossRef CAS PubMed.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS.
- D. R. Spring, Org. Biomol. Chem., 2003, 1, 3867–3870 RSC.
- D. L. Boger and C. E. Brotherton, J. Org. Chem., 1984, 49, 4050–4055 CrossRef CAS.
- R. J. Spandl, A. Bender and D. R. Spring, Org. Biomol. Chem., 2008, 6, 1149–1158 RSC.
- A. Krzyzanowski, A. Pahl, M. Grigalunas and H. Waldmann, J. Med. Chem., 2023, 66, 12739–12750 CrossRef CAS PubMed.
- T. I. Oprea and C. Bologa, J. Med. Chem., 2023, 66, 12710–12714 CrossRef CAS PubMed.
- P. A. Clemons, N. E. Bodycombe, H. A. Carrinski, J. A. Wilson, A. F. Shamji, B. K. Wagner, A. N. Koehler and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18787–18792 CrossRef CAS.
- M. P. Gleeson, A. Hersey, D. Montanari and J. Overington, Nat. Rev. Drug Discovery, 2011, 10, 197–208 CrossRef CAS PubMed.
- A. R. Leach and M. M. Hann, Curr. Opin. Chem. Biol., 2011, 15, 489–496 CrossRef CAS PubMed.
- T. Böttcher, J. Chem. Inf. Model., 2016, 56, 462–470 CrossRef PubMed.
- R. M. Demoret, M. A. Baker, M. Ohtawa, S. Chen, C. C. Lam, S. Khom, M. Roberto, S. Forli, K. N. Houk and R. A. Shenvi, J. Am. Chem. Soc., 2020, 142, 18599–18618 CrossRef CAS.
- P. A. Wender, Nat. Prod. Rep., 2014, 31, 433–440 RSC.
- P. A. Wender, Tetrahedron, 2013, 69, 7529–7550 CrossRef CAS PubMed.
- S. Ichikawa, Chem. Rec., 2016, 16, 1106–1115 CrossRef CAS PubMed.
- I. Pavlinov, E. M. Gerlach and L. N. Aldrich, Org. Biomol. Chem., 2019, 17, 1608–1623 RSC.
- S. E. Motika and P. J. Hergenrother, Nat. Prod. Rep., 2020, 37, 1395–1403 RSC.
- N. J. Truax and D. Romo, Nat. Prod. Rep., 2020, 37, 1436–1453 RSC.
- A. Nelson and G. Karageorgis, RSC Med. Chem., 2021, 12, 353–362 RSC.
- B. O. Alkubaisi, A. I. Shahin, R. A. Zenati, A. Ravi, R. Alchami, M. Alkalla, R. Khaled, M. I. El-Gamal and T. H. Al-Tel, ChemMedChem, 2023, 18, e202300117 CrossRef CAS PubMed.
- X.-F. Cheng, M. Grigalunas and H. Waldmann, Arkivoc, 2024, 5, 202312153 Search PubMed.
- P. Craven, A. Aimon, M. Dow, N. Fleury-Bregeot, R. Guilleux, R. Morgentin, D. Roche, T. Kalliokoski, R. Foster, S. P. Marsden and A. Nelson, Bioorg. Med. Chem., 2015, 23, 2629–2635 CrossRef CAS PubMed.
- G. Li Petri, M. V. Raimondi, V. Spanò, R. Holl, P. Barraja and A. Montalbano, Top. Curr. Chem., 2021, 379, 34 CrossRef CAS.
- Y. Choi, S. Lee, H. Kim and S. B. Park, Front. Chem., 2022, 10, 841250 CrossRef CAS PubMed.
- F. P. Player and D. J. Foley, Asian J. Org. Chem., 2024, 13, e202400397 CrossRef CAS.
- A. Lawer, F. P. Player, V. M. Avery and D. J. Foley, Adv. Synth. Catal., 2024, 366, 2090–2100 CrossRef CAS.
- J. J. Ciardiello, H. L. Stewart, H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Bioorg. Med. Chem., 2017, 25, 2825–2843 CrossRef CAS PubMed.
- K. T. Mortensen, T. J. Osberger, T. A. King, H. F. Sore and D. R. Spring, Chem. Rev., 2019, 119, 10288–10317 CrossRef CAS.
- A. K. Yudin, Chem. Sci., 2014, 6, 30–49 RSC.
- D. Garcia Jimenez, V. Poongavanam and J. Kihlberg, J. Med. Chem., 2023, 66, 5377–5396 CrossRef CAS.
- G. Niggemeyer, A. Knyazeva, R. Gasper, D. Corkery, P. Bodenbinder, J. J. Holstein, S. Sievers, Y.-W. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2022, 61, e202114328 CrossRef CAS.
- A. Knyazeva, S. Li, D. P. Corkery, K. Shankar, L. K. Herzog, X. Zhang, B. Singh, G. Niggemeyer, D. Grill, J. D. Gilthorpe, M. Gaetani, L.-A. Carlson, H. Waldmann and Y.-W. Wu, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2317680121 CrossRef CAS PubMed.
- D. J. Foley, S. Zinken, D. Corkery, L. Laraia, A. Pahl, Y.-W. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2020, 59, 12470–12476 CrossRef CAS.
- M. Grigalunas, A. Burhop, S. Zinken, A. Pahl, J.-M. Gally, N. Wild, Y. Mantel, S. Sievers, D. J. Foley, R. Scheel, C. Strohmann, A. P. Antonchick and H. Waldmann, Nat. Commun., 2021, 12, 1883 CrossRef CAS PubMed.
- D. J. Foley and H. Waldmann, Chem. Soc. Rev., 2022, 51, 4094–4120 RSC.
- R. Narayan, J. O. Bauer, C. Strohmann, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 12892–12896 CrossRef CAS PubMed.
- R. Narayan, M. Potowski, Z.-J. Jia, A. P. Antonchick and H. Waldmann, Acc. Chem. Res., 2014, 47, 1296–1310 CrossRef CAS PubMed.
- T. Schneidewind, S. Kapoor, G. Garivet, G. Karageorgis, R. Narayan, G. Vendrell-Navarro, A. P. Antonchick, S. Ziegler and H. Waldmann, Cell Chem. Biol., 2019, 26, 512–523 CrossRef CAS PubMed.
- K. J. Frankowski, B. Neuenswander and J. Aubé, J. Comb. Chem., 2008, 10, 721–725 CrossRef CAS PubMed.
- K. J. Frankowski, V. Setola, J. M. Evans, B. Neuenswander, B. L. Roth and J. Aubé, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6727–6732 CrossRef CAS PubMed.
- K. J. Frankowski, V. Setola, J. M. Evans, B. Neuenswander, B. L. Roth and J. Aubé, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15526–15527 CrossRef.
- A. Shaabani, A. Maleki, H. Mofakham and H. R. Khavasi, J. Comb. Chem., 2008, 10, 323–326 CrossRef CAS PubMed.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- K. J. Frankowski, J. E. Golden, Y. Zeng, Y. Lei and J. Aubé, J. Am. Chem. Soc., 2008, 130, 6018–6024 CrossRef CAS PubMed.
- R. Yao, A. A. Jensen, H. P. Bryce-Rogers, K. Schultz-Knudsen, L. Zhou, N. P. Hovendal, H. Pedersen, M. Kubus, T. Ulven and L. Laraia, J. Med. Chem., 2023, 66, 11536–11554 CrossRef CAS PubMed.
- T. Whitmarsh-Everiss, A. H. Olsen and L. Laraia, Angew. Chem., Int. Ed., 2021, 60, 26755–26761 CrossRef CAS PubMed.
- B. Melillo, J. Zoller, B. K. Hua, O. Verho, J. C. Borghs, S. D. Jr. Nelson, M. Maetani, M. J. Wawer, P. A. Clemons and S. L. Schreiber, J. Am. Chem. Soc., 2018, 140, 11784–11790 CrossRef CAS.
- K. A. Scott, N. Ropek, B. Melillo, S. L. Schreiber, B. F. Cravatt and E. V. Vinogradova, Curr. Res. Chem. Biol., 2022, 2, 100028 CrossRef CAS.
- D. J. Foley, P. G. E. Craven, P. M. Collins, R. G. Doveston, A. Aimon, R. Talon, I. Churcher, F. von Delft, S. P. Marsden and A. Nelson, Chem.–Eur. J., 2017, 23, 15227–15232 CrossRef CAS PubMed.
- C. Townley, L. McMurray, S. P. Marsden and A. Nelson, Bioorg. Med. Chem. Lett., 2022, 62, 128631 CrossRef CAS PubMed.
- E. A. Okolo, A. Pahl, S. Sievers, C. M. Pask, A. Nelson and S. P. Marsden, Chem.–Eur. J., 2023, 29, e202203992 CrossRef CAS PubMed.
- S. Liu, J. S. Scotti and S. A. Kozmin, J. Org. Chem., 2013, 78, 8645–8654 CrossRef CAS PubMed.
- N. G. Paciaroni, R. Ratnayake, J. H. Matthews, V. M. Norwood IV, A. C. Arnold, L. H. Dang, H. Luesch and R. W. Huigens III, Chem.–Eur. J., 2017, 23, 4327–4335 CrossRef CAS PubMed.
- V. Srinivasulu, P. Schilf, S. Ibrahim, M. A. Khanfar, S. M. Sieburth, H. Omar, A. Sebastian, R. A. AlQawasmeh, M. J. O'Connor and T. H. Al-Tel, Nat. Commun., 2018, 9, 4989 CrossRef.
- V. Srinivasulu, S. M. Sieburth, M. A. Khanfar, I. A. Abu-Yousef, A. Majdalawieh, M. Ramanathan, A. Sebastian and T. H. Al-Tel, J. Org. Chem., 2021, 86, 12872–12885 CrossRef CAS PubMed.
- G. Srikanth, A. Ravi, A. Sebastian, J. Joseph, M. A. Khanfar, M. I. El-Gamal, R. A. Al-Qawasmeh, I. A. Shehadi, S. McN. Sieburth, I. A. Abu-Yousef, A. F. Majdalawieh and T. H. Al-Tel, Eur. J. Org Chem., 2023, 26, e202300080 CrossRef CAS.
- J. Liu, J. Flegel, F. Otte, A. Pahl, S. Sievers, C. Strohmann and H. Waldmann, Angew. Chem., Int. Ed., 2021, 60, 21384–21395 CrossRef CAS.
- V. Porte, B. C. van Veen, H. Zhang, P. Piacentini, S. A. Matheu, S. Woolford, K. R. Sokol, S. Shaaban, H. Weinstabl and N. Maulide, Org. Lett., 2024, 26, 4873–4876 CrossRef CAS.
- J. J. Roach, Y. Sasano, C. L. Schmid, S. Zaidi, V. Katritch, R. C. Stevens, L. M. Bohn and R. A. Shenvi, ACS Cent. Sci., 2017, 3, 1329–1336 CrossRef CAS.
- Z. D. Stempel, H. S. Radomska, C. C. Coss and G. C. Micalizio, Org. Lett., 2024, 26, 3054–3059 CrossRef CAS PubMed.
- A. Fürstner, D. Kirk, M. D. B. Fenster, C. Aïssa, D. De Souza and O. Müller, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8103–8108 CrossRef PubMed.
- N. J. Truax, S. Ayinde, K. Van, J. O. Liu and D. Romo, Org. Lett., 2019, 21, 7394–7399 CrossRef CAS PubMed.
- S. Lin, H. Liu, E. B. Svenningsen, M. Wollesen, K. M. Jacobsen, F. D. Andersen, J. Moyano-Villameriel, C. N. Pedersen, P. Nørby, T. Tørring and T. B. Poulsen, Nat. Chem., 2021, 13, 47–55 CrossRef CAS PubMed.
- K. K. Wan and R. A. Shenvi, Synlett, 2016, 27, 1145–1164 CrossRef CAS.
- Z.-C. Wu and D. L. Boger, Nat. Prod. Rep., 2020, 37, 1511–1531 RSC.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- H. F. Klein, D. J. Hamilton, I. J. P. de Esch, M. Wijtmans and P. O'Brien, Drug Discovery Today, 2022, 27, 2484–2496 CrossRef CAS PubMed.
- S. L. Kidd, T. J. Osberger, N. Mateu, H. F. Sore and D. R. Spring, Front. Chem., 2018, 6, 460 CrossRef.
- N. S. Troelsen, E. Shanina, D. Gonzalez-Romero, D. Danková, I. S. A. Jensen, K. J. Śniady, F. Nami, H. Zhang, C. Rademacher, A. Cuenda, C. H. Gotfredsen and M. H. Clausen, Angew. Chem., Int. Ed., 2020, 59, 2204–2210 CrossRef CAS PubMed.
- L. Laraia, G. Garivet, D. J. Foley, N. Kaiser, S. Müller, S. Zinken, T. Pinkert, J. Wilke, D. Corkery, A. Pahl, S. Sievers, P. Janning, C. Arenz, Y. Wu, R. Rodriguez and H. Waldmann, Angew. Chem., Int. Ed., 2020, 59, 5721–5729 CrossRef CAS PubMed.
- T. Schneidewind, A. Brause, B. Schölermann, S. Sievers, A. Pahl, M. G. Sankar, M. Winzker, P. Janning, K. Kumar, S. Ziegler and H. Waldmann, Cell Chem. Biol., 2021, 28, 1780–1794 CrossRef CAS.
- M. Akbarzadeh, I. Deipenwisch, B. Schoelermann, A. Pahl, S. Sievers, S. Ziegler and H. Waldmann, Cell Chem. Biol., 2022, 29, 1053–1064 CrossRef CAS.
- S. Rezaei Adariani, D. Agne, S. Koska, A. Burhop, C. Seitz, J. Warmers, P. Janning, M. Metz, A. Pahl, S. Sievers, H. Waldmann and S. Ziegler, J. Med. Chem., 2024, 67, 13252–13270 CrossRef CAS PubMed.
- T. Whitmarsh-Everiss, Z. Wang, C. Hauberg Hansen, L. Depta, E. Sassetti, O. Rafn Dan, A. Pahl, S. Sievers and L. Laraia, ChemBioChem, 2023, 24, e202200555 CrossRef CAS PubMed.
- B. Schölermann, J. Bonowski, M. Grigalunas, A. Burhop, Y. Xie, J. G. F. Hoock, J. Liu, M. Dow, A. Nelson, C. Nowak, A. Pahl, S. Sievers and S. Ziegler, ChemBioChem, 2022, 23, e202200475 CrossRef PubMed.
- L. Laraia, K. Ohsawa, G. Konstantinidis, L. Robke, Y.-W. Wu, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 2145–2150 CrossRef CAS PubMed.
- H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS PubMed.
- A. R. Bortz, J. M. Bennett and R. Fasan, Chem, 2024, 10, 3488–3502 CAS.
- X. Zhang, E. King-Smith and H. Renata, Angew. Chem., Int. Ed., 2018, 57, 5037–5041 CrossRef CAS PubMed.
- J. Li, F. Li, E. King-Smith and H. Renata, Nat. Chem., 2020, 12, 173–179 CrossRef CAS PubMed.
- X. Zhang, E. King-Smith, L.-B. Dong, L.-C. Yang, J. D. Rudolf, B. Shen and H. Renata, Science, 2020, 369, 799–806 CrossRef CAS PubMed.
- F. Li and H. Renata, J. Am. Chem. Soc., 2021, 143, 18280–18286 CrossRef CAS PubMed.
- F. Li, H. Deng and H. Renata, J. Am. Chem. Soc., 2022, 144, 7616–7621 CrossRef CAS PubMed.
- J. Li, F. Chen and H. Renata, J. Am. Chem. Soc., 2022, 144, 19238–19242 CrossRef CAS PubMed.
- Y. Jiang and H. Renata, Nat. Chem., 2024, 16, 1531–1538 CrossRef CAS PubMed.
- C. N. Stout and H. Renata, J. Am. Chem. Soc., 2024, 146, 21815–21823 CrossRef PubMed.
- F. Li, H. Deng and H. Renata, Nat. Synth., 2023, 2, 708–718 CrossRef CAS.
- C. Ma, C. W. Lindsley, J. Chang and B. Yu, J. Med. Chem., 2024, 67, 11459–11466 CrossRef CAS PubMed.
- B. D. Dherange, P. Q. Kelly, J. P. Liles, M. S. Sigman and M. D. Levin, J. Am. Chem. Soc., 2021, 143, 11337–11344 CrossRef CAS PubMed.
- S. Liu and X. Cheng, Nat. Commun., 2022, 13, 425 CrossRef CAS PubMed.
- J. C. Reisenbauer, O. Green, A. Franchino, P. Finkelstein and B. Morandi, Science, 2022, 377, 1104–1109 CrossRef CAS PubMed.
- S. C. Patel and N. Z. Burns, J. Am. Chem. Soc., 2022, 144, 17797–17802 CrossRef CAS PubMed.
- E. E. Hyland, P. Q. Kelly, A. M. McKillop, B. D. Dherange and M. D. Levin, J. Am. Chem. Soc., 2022, 144, 19258–19264 CrossRef CAS PubMed.
- G. L. Bartholomew, F. Carpaneto and R. Sarpong, J. Am. Chem. Soc., 2022, 144, 22309–22315 CrossRef CAS PubMed.
- J. Wang, H. Lu, Y. He, C. Jing and H. Wei, J. Am. Chem. Soc., 2022, 144, 22433–22439 CrossRef CAS.
- T. J. Pearson, R. Shimazumi, J. L. Driscoll, B. D. Dherange, D.-I. Park and M. D. Levin, Science, 2023, 381, 1474–1479 CrossRef CAS PubMed.
- J. Woo, C. Stein, A. H. Christian and M. D. Levin, Nature, 2023, 623, 77–82 CrossRef CAS PubMed.
- Q. Cheng, D. Bhattacharya, M. Haring, H. Cao, C. Mück-Lichtenfeld and A. Studer, Nat. Chem., 2024, 16, 741–748 CrossRef CAS PubMed.
- J. Luo, Q. Zhou, Z. Xu, K. N. Houk and K. Zheng, J. Am. Chem. Soc., 2024, 146, 21389–21400 CrossRef PubMed.
- F.-P. Wu, J. L. Tyler, C. G. Daniliuc and F. Glorius, ACS Catal., 2024, 14, 13343–13351 CrossRef CAS.
- F.-P. Wu, M. Lenz, A. Suresh, A. R. Gogoi, J. L. Tyler, C. G. Daniliuc, O. Gutierrez and F. Glorius, Chem. Sci., 2024, 15, 15205–15211 RSC.
- D. Kim, J. You, D. H. Lee, H. Hong, D. Kim and Y. Park, Science, 2024, 386, 99–105 CrossRef CAS PubMed.
- A. Conboy and M. F. Greaney, Chem, 2024, 10, 1940–1949 CAS.
- S. Liu, Y. Yang, Q. Song, Z. Liu, P. Sivaguru, Y. Zhang, G. de Ruiter, E. A. Anderson and X. Bi, Nat. Commun., 2024, 15, 9998 CrossRef CAS PubMed.
- A.-S. Paschke, Y. Brägger, B. Botlik, E. Staudinger, O. Green and B. Morandi, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-prwm8.
- B. Ghosh, P. Kafle, R. Mukherjee, R. Welles, D. Herndon, K. M. Nicholas, Y. Shao and I. Sharma, Science, 2025, 387, 102–107 CrossRef CAS PubMed.
- A. Sanchez, A. Gurajapu, W. Guo, W.-Y. Kong, C. J. Laconsay, N. S. Settineri, D. J. Tantillo and T. J. Maimone, J. Am. Chem. Soc., 2023, 145, 13452–13461 CrossRef CAS PubMed.
- R. Fasan, J. Bennett and A. Bortz, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-9shgl.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08017c |
| This journal is © The Royal Society of Chemistry 2025 |