
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into facile methane activation by a spin forbidden reaction with Ta+ ions in the gas phase†
Yang
Liu
 a,
Milan
Ončák
*b,
Tucker W. R.
Lewis
c,
Marcel
Meta
d,
Shaun G.
Ard
c,
Nicholas S.
Shuman
*c,
Jennifer
Meyer
d,
Albert A.
Viggiano
c and
Hua
Guo
*a
a,
Milan
Ončák
*b,
Tucker W. R.
Lewis
c,
Marcel
Meta
d,
Shaun G.
Ard
c,
Nicholas S.
Shuman
*c,
Jennifer
Meyer
d,
Albert A.
Viggiano
c and
Hua
Guo
*a
aDepartment of Chemistry and Chemical Biology, Center for Computational Chemistry, University of New Mexico, Albuquerque, New Mexico 87131, USA. E-mail: hguo@unm.edu
bUniversität Innsbruck, Institut für Ionenphysik und Angewandte Physik, Technikerstraße 25, 6020 Innsbruck, Austria
cAir Force Research Laboratory, Space Vehicles Directorate, Kirtland Air Force Base, New Mexico 87117, USA
dRPTU Kaiserslautern-Landau, Fachbereich Chemie and Forschungszentrum OPTIMAS, Erwin-Schrödinger Str. 52, 67663 Kaiserslautern, Germany
First published on 10th February 2025
Abstract
The activation of methane (CH4) by transition-metal cations in the gas phase provides a model for understanding the impact of electronic spin on reactivity, with implications in single atom catalysis. In this work, we present a mixed quantum-classical trajectory surface hopping study on the nominally spin-forbidden reaction Ta+ + CH4 → TaCH2+ + H2. To facilitate the dynamics calculations, full twelve-dimensional PESs for three low-lying spin (quintet, triplet, and singlet) states are constructed using a machine learning method from density functional theory data. Furthermore, we report the temperature dependence of the rate coefficients for the Ta+ + CH4 → TaCH2+ + H2 reaction measured using the selected ion flow tube (SIFT) technique. The measured rate coefficient has a near zero temperature dependence and is approximately 50% of the capture limit at room temperature. Our theoretical results with a Gaussian-binning treatment of the product zero-point energy reproduced the experimental rate coefficient and the temperature dependence. Satisfactory agreement is also obtained between theory and differential cross sections measured recently using molecular beams combined with velocity map imaging. Specifically, our multi-state calculations confirm the indirect mechanism of this reaction with long-lived reaction intermediate after passing through the initial barrier and reveal that the kinetic bottleneck in this reaction is intersystem crossing between the quintet and triplet states. Furthermore, the energy disposal in the TaCH2+ (both singlet and triplet) and H2 products is found to be largely statistical due to the long lifetime of the exit-channel complex.
Introduction
Methane (CH4), the primary component of natural gas and methane clathrates, is a crucial hydrocarbon feedstock for fuel and chemical production. However, its low volumetric energy density results in high transportation and storage costs, limiting its use as a chemical energy source. The conversion of methane into value-added chemicals and fuels is critical not only for economic reasons but also for addressing environmental concerns related to greenhouse gas emissions. Consequently, significant research efforts have focused on converting methane into more energy-dense fuels or higher-value chemicals.1,2 All these conversions begin with the activation of methane, which involves the weakening or breaking one of its four carbon–hydrogen bonds.3 However, CH4 is quite inert due to its strong C–H bonds, with a bond dissociation energy (BDE) of 4.5 eV.4 In addition, methane has zero dipole moment and quadrupole moment, low polarizability (2.593 Å3), and a high ionization energy of 12.6 eV.5 These factors contribute to its low reactivity in chemical reactions, making catalysts essential for efficient chemical conversions. Common catalysts for methane activation are transition metals dispersed on oxide supports.6–8 A major challenge in catalyst design is that nearly all newly formed bonds are weaker than the original C–H bond in methane. Therefore, highly active catalysts often lack selectivity. Recently, an increasing number of reports have suggested efficient methane activation with atomically dispersed transition metals.9–11 These studies indicated that such single atom catalysis (SAC) sensitively depends on the immediate environment of the metal ion, which controls its partial charge and presumably spin.A unique approach to understand the SAC is to investigate the activation of small molecules by transition metal ions in the gas phase.12–14 For methane activation, it is known that many third-row transition metal cations (M+) can readily dehydrogenate methane at room temperature to produce MCH2+,15–31 which can then, e.g., serve as an intermediate in the conversion of CH4 to CH2O. From a thermochemical perspective, the exothermic formation of M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2+ is viable only if the metal–methylidene bond strength D0(MCH2) is greater than the heat of dehydrogenation of methane, which is 4.743 ± 0.001 eV determined by the Active Thermochemical Tables (ATcT) approach.22,32,33 Irikura and Beauchamp observed reactions between methane and Hf+, Ta+, W+, Re+, Os+, Ir+, Pt+, and Au+ and found that Ta+, W+, Os+, Ir+, and Pt+ reacted at room temperature.16 Later, Shayesteh et al. investigated the reactions of methane with 59 atomic metal cations at room temperature and found that the H2 elimination channel occurred with As+, Nb+, Ta+, W+, Os+, Ir+, and Pt+ ions.29 In this work, we focus on the reaction of CH4 activation by the open-shell transition-metal Ta+ ions:
CH2+ is viable only if the metal–methylidene bond strength D0(MCH2) is greater than the heat of dehydrogenation of methane, which is 4.743 ± 0.001 eV determined by the Active Thermochemical Tables (ATcT) approach.22,32,33 Irikura and Beauchamp observed reactions between methane and Hf+, Ta+, W+, Re+, Os+, Ir+, Pt+, and Au+ and found that Ta+, W+, Os+, Ir+, and Pt+ reacted at room temperature.16 Later, Shayesteh et al. investigated the reactions of methane with 59 atomic metal cations at room temperature and found that the H2 elimination channel occurred with As+, Nb+, Ta+, W+, Os+, Ir+, and Pt+ ions.29 In this work, we focus on the reaction of CH4 activation by the open-shell transition-metal Ta+ ions:
| Ta+ + CH4 → TaCH2+ + H2 | (R1) |
This reaction is highly efficient, with thermal rates comparable to gas kinetic.29,30 However, as shown in Fig. 1, the reaction has a significant barrier and high endoergicity on the lowest quintet state (5F). The facile reactivity can only be explained by efficient spin–orbit-mediated transitions from the high-spin quintet state to the low-spin triplet (and possibly singlet) excited state of the entrance channel, both of which have a submerged barrier. This so-called multi-state or two-state reactivity (TSR)34–36 is a characteristic feature of many transition-metal-mediated processes where low-lying excited states play a crucial role.
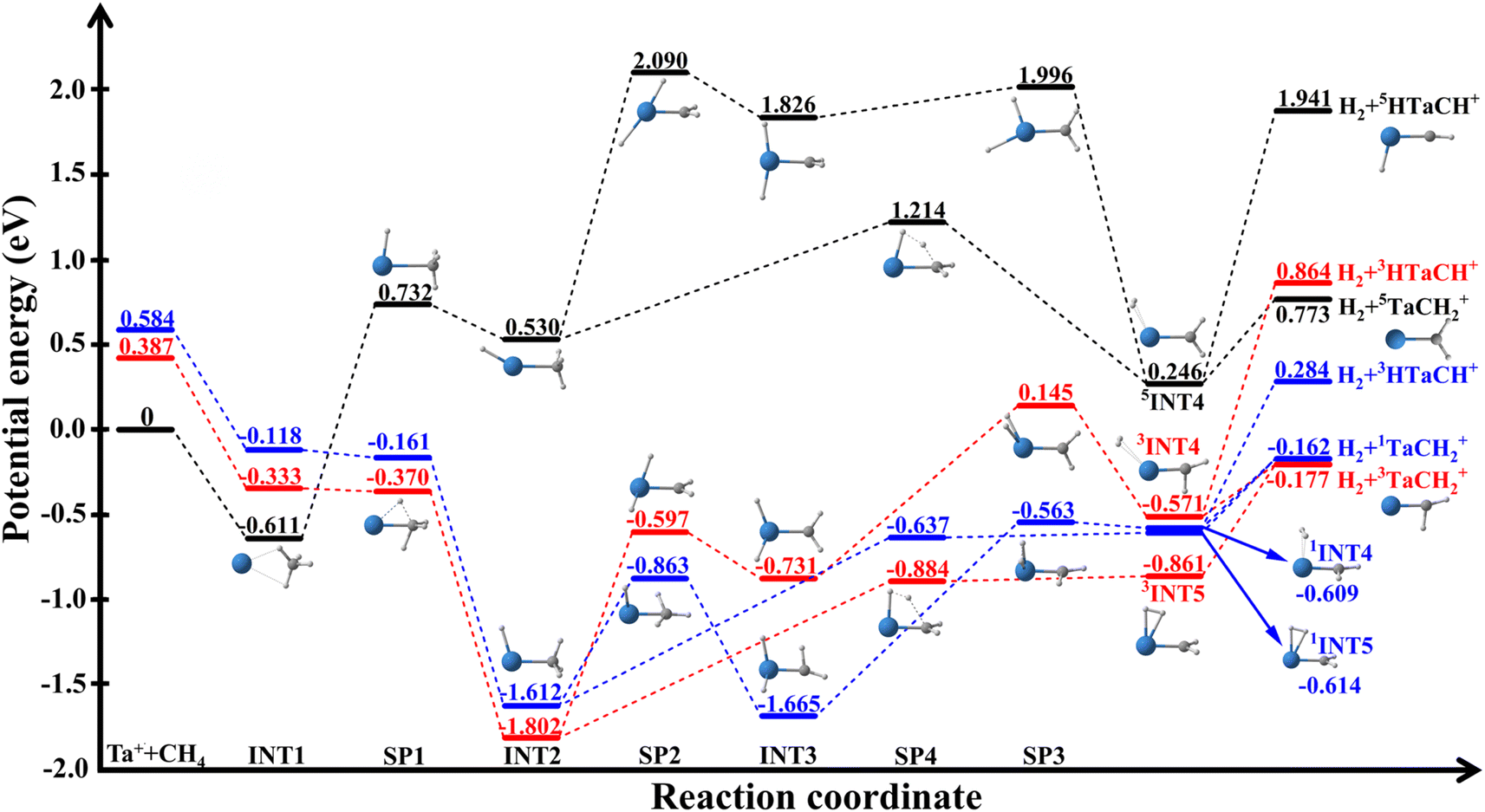 | ||
| Fig. 1 Schematic of the Ta+ + CH4 → TaCH2+ + H2 reaction on the quintet, triplet, and singlet state surfaces (black, red, and blue, respectively). The energies with ZPE correction are calculated at the B3LYP/DZ level. The ZPE correction induces the energy of some saddle points to fall below the local minima. | ||
Parke et al.28 investigated (R1) using guided ion beam techniques. The observed decrease in the reaction cross section with increasing kinetic energy suggests that the reaction proceeds through an intermediate, offering valuable dynamic insights. Furthermore, supported by theoretical calculations, they concluded that the relatively low reaction efficiency (18–44%) is attributed to the spin-forbidden nature of the reaction. Very recently, Meta et al. investigated the dynamics of (R1) using crossed-beam velocity map imaging, providing detailed chemical dynamic information about (R1) for the first time.37 The measured differential cross sections (DCSs) show that the product ions are dominated by isotropic scattering around the center-of-mass, indicating that the reaction is dominated by a complex-forming mechanism even at collision energies up to 1.3 eV. However, information about the identity of the long-lived complex and/or intermediate that governs this indirect mechanism is lacking. Little is known about the origin of the associated bottleneck that leads to this long lifetime. As shown in Fig. 1, the reaction begins on the quintet surface, forming a pre-reaction well (5INT1) before the quintet saddle point (5SP1). The formation of tantalum carbene (TaCH2+) in both the singlet and triplet product channels involves breaking two C–H bonds, forming two new bonds, and releasing molecular hydrogen. After crossing the triplet saddle point (3SP1), the system undergoes a series of complex transformations to reach the product asymptotes. This mechanism is akin to a classic two-step process. First, oxidative addition occurs as the tantalum ion inserts into a C–H bond, followed by the migration of a second hydrogen atom to the metal center to form INT3. Then, reductive elimination of molecular hydrogen occurs from the metal center. Alternatively, the system can bypass INT3 via a four-centered transition state, SP4, to form INT5 and subsequently reach the products. The post-transition state well (INT2) following the first H-atom transfer may have a long lifetime because it must overcome a relatively large barrier (SP2 or SP4) associated with significant molecular rearrangement to other wells (INT3, INT4 and INT5), before forming the products. Since the collision energy in the crossed beam experiment allow the access to these wells, energy partition in the products sheds light on the molecular rearrangements in these reaction intermediates. To understand the microscopic mechanism and dynamics of such a complex reactive system, dynamical simulations are essential to determine whether the indirect mechanism is associated with the reactant complex [Ta(CH4)]+ (INT1), one of the intermediate complexes [HTaCH3]+ (INT2) and [H2TaCH2]+ (INT3), or the product complex [(H2)TaCH2]+ (INT4 and INT5).
The molecular beam experiment also measured the product kinetic energy distributions, revealing low kinetic energy for the products, which indicates that the products' internal degrees of freedom (DOFs) are highly excited. However, it remains unclear which product(s) (TaCH2+ and/or H2) and which DOF(s) (vibration and/or rotation) are excited. It is also uncertain whether the reactivity of the reaction is governed by intersystem crossing (ISC) between the quintet and triplet states or by 3SP1. Furthermore, the contribution of the singlet state to the reaction is unknown and cannot be easily determined experimentally due to instrumental limitations.
To address these mechanistic questions, we present here a comprehensive kinetic and dynamic study of this reaction, which is made possible by full-dimensional potential energy surfaces (PESs) of the quintet, triplet, and singlet states, based on approximately 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 density functional theory (DFT) points. The particular density functional used in these calculations has been benchmarked with high-level ab initio methods. The dynamic calculations were performed using the mixed quantum-classical trajectory surface hopping method, along with ab initio spin–orbit couplings determined from multi-reference configuration interaction (MRCI) calculations. This strategy has been used by us previously to study Ta+ + CO2 and Nb+ + CO2 reactions with much success.38,39 A similar dynamical study has also been reported for the FeO+ + H2 reaction, which also proceeds through a TSR mechanism.40 In addition, we remeasured thermal rate coefficients using a selected ion flow tube (SIFT) across a much wider temperature range than reported to date.
300 density functional theory (DFT) points. The particular density functional used in these calculations has been benchmarked with high-level ab initio methods. The dynamic calculations were performed using the mixed quantum-classical trajectory surface hopping method, along with ab initio spin–orbit couplings determined from multi-reference configuration interaction (MRCI) calculations. This strategy has been used by us previously to study Ta+ + CO2 and Nb+ + CO2 reactions with much success.38,39 A similar dynamical study has also been reported for the FeO+ + H2 reaction, which also proceeds through a TSR mechanism.40 In addition, we remeasured thermal rate coefficients using a selected ion flow tube (SIFT) across a much wider temperature range than reported to date.
Results and discussion
Electronic structure calculations
The reaction pathways for the quintet, triplet, and singlet states are shown in Fig. 1, and the corresponding zero-point energy (ZPE)-corrected energies and frequencies of all stationary points calculated at the B3LYP/DZ level listed in Table S1.† The recent higher-level calculations at the CCSD(T)//B3LYP level37 and available experimental values5 are also included for comparison. The optimized geometries for the quintet, triplet, and singlet states are presented in Fig. S1–S3,† respectively. The B3LYP/DZ method provides very similar predictions to the CCSD(T)//B3LYP calculations, showing reasonable agreement within about 0.3 eV, except for the energy of the singlet Ta+ + CH4 asymptote. At the B3LYP/DZ level, the energy (0.584 eV) is significantly lower than the value calculated at the CCSD(T)//B3LYP level (1.328 eV)37 and the experimental measurement (1.216 eV).5 The underestimate for the singlet Ta+ + CH4 asymptote might be from the mis-convergence in the DFT calculations due to the multireference character. However, we believe this discrepancy has very limited impacts on our dynamic results due to the high energy of this channel, similar to the Ta+/Nb+ + CO2 systems.38,39As depicted in Fig. 1, the reaction between quintet Ta+ ion (5Ta+) and CH4 proceeds through a pre-reaction well (5INT1, −0.611 eV) and must circumvent a very high barrier (5SP1, 0.732 eV). INT1 is a complex formed due largely to electrostatic interaction, as evidenced by the comparable well depth in different spin states. The barrier features the insertion of the metal into a C–H bond. The product asymptotes are also very high, making the reaction an endoergic process. Hence, this adiabatic pathway is expected to be viable only at very high temperatures.
Alternatively, the reaction can also advance nonadiabatically via efficient ISC from the quintet to the triplet state, where 3SP1 is submerged. After crossing 3SP1, the reaction enters a very deep well (3INT2, −1.802 eV), which is formed by an oxidative addition reaction in which the tantalum ion inserts into a C–H bond, causing the hydrogen atom to migrate from the carbon to the tantalum atom. Starting from 3INT2, there are two ways to reach the products: one is 3INT2 → 3SP2 → 3INT3→ 3SP3 → 3INT4 → products, and the other is 3INT2 → 3SP4 → 3INT5 → products. In both reaction paths, the formation of the products is up-hill. Similar reaction paths can also be found on the singlet state surface.
The adiabatic reaction channel in the quintet state is endothermic, with a reaction energy of 0.773 eV at the B3LYP/DZ level. In contrast, the nonadiabatic reaction channel is slightly exothermic in the triplet and singlet states, with reaction energies of −0.177 eV and −0.162 eV, respectively, at the B3LYP/DZ level, they are close to the values at the CCSD(T)//B3LYP level (−0.297 and −0.302 eV).37 The reaction energy of the triplet state at the B3LYP/DZ level also agrees reasonably well with the experimental measurement of −0.10 ± 0.02 eV by Armentrout and coworkers.28 Independently, the TaCH2+ photodissociation experiment of Metz and coworkers18 sets a lower bound on the exothermicity of the reaction at −0.23 eV, which is consistent with our values.
We further note that all PESs leads also to the HTaCH+ + H2 products, as shown in Fig. 1. Due to the high energies, however, the H2 + HTaCH+ asymptote represents only a minor channel, and will not be discussed further.
Potential energy surfaces
About 64300 quintet, triplet, and singlet energies were calculated at the level of B3LYP/DZ level and represented using the permutation-invariant-polynomial neural-network (PIP-NN) method to generate the corresponding spin-specific twelve-dimensional PESs. For the quintet, triplet, and singlet PESs, the final root mean square errors (RMSEs) are 33.8, 41.5, and 40.4 meV, respectively, and the maximum errors are 573.0, 737.9, and 501.1 meV. A graphical representation of the errors throughout the entire energy range up to 7 eV is shown in Fig. S4a and S4b.† The majority of points have a fitting error under 20 meV. The energies, geometries, and harmonic frequencies of the stationary points obtained on the PESs can be found in Table S1 and Fig. S1–S3.† These static properties were well reproduced on the newly constructed PESs, except for the energy of 5INT2, which differs by 0.213 eV between B3LYP/DZ and the PES. However, due to the high barrier associated with the quintet state, this channel is negligible and thus it will not affect the dynamic results. Fig. S5–S7† depict the potential energies along the minimum energy paths (MEPs) from SP1 to SP4 on the quintet, triplet, and singlet PESs, respectively.
Kinetics and mechanism
Fig. 2 illustrates the comparison of calculated rate coefficients with previous experimental data at room temperature29,30 and the new ones measured using SIFT. The newly measured rate coefficient at room temperature (300 K) is in good agreement with previous results,29,30 as shown in Fig. 2. The temperature dependence over this range is very mild, and the rate coefficients are at about 40% of the Langevin–Gioumousis–Stevenson capture limit. This weak temperature dependence is consistent with the energy dependence reported from a prior guided ion beam measurement28 showing cross sections at low collision energies (Ec) at a constant fraction of the Langevin–Gioumousis–Stevenson value. Importantly, the facile reactivity is consistent with an exothermic reaction lacking an activation energy. However, the features of the PES responsible for reducing reactivity below the capture limit are not clear from experiment alone. The theoretical results (FSSH in the figure) are larger than the experiment, which is due to the fact that there are many trajectories with vibrational energy below the ZPE level of the H2 product. Given the fact that the nonadiabatic reaction pathway is nearly thermoneutral and H2 has a large vibrational frequency, it is not surprising that ZPE violation is quite acute in this system. To mitigate this problem, the one-dimensional Gaussian binning (1GB) method, an approach to address the ZPE leakage issue (see Methods for detail), was used. As shown in Fig. 2, the 1GB corrected rate coefficients (FSSH + 1GB) are in much better agreement with the experiment.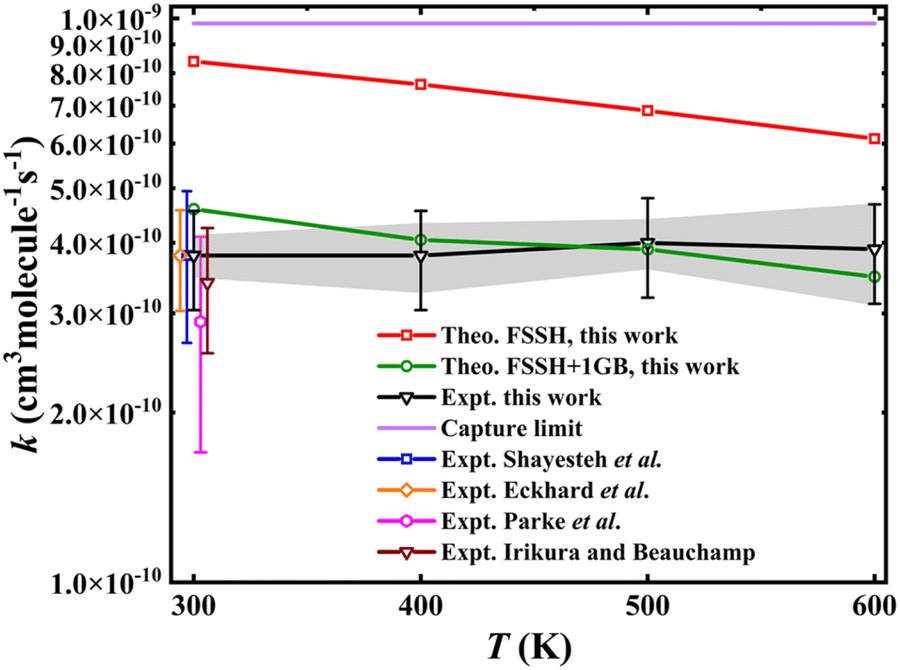 | ||
| Fig. 2 Comparison of the calculated and experimentally measured rate coefficients16,28–30 for the Ta+ + CH4 → TaCH2+ + H2 reaction. The error bars on the newly measured experimental rates indicate systematic uncertainties, while the shaded gray region denotes uncertainties for comparisons across different temperatures. The experimental rates from the literature, originally measured at 300 K, are slightly shifted to enhance clarity in the comparisons. | ||
While the thermal rate coefficient of the Ta+ + CH4 → TaCH2+ + H2 reaction constitutes a large percentage of the capture limit, the differences indicate a kinetic bottleneck for the reaction, in agreement with the results from recent dynamics experiments. To determine whether the ISC from quintet to triplet states or the saddle points on the triplet state (3SP1) is the controlling factor for this reaction, a critical point in any TSR system, we analysed the trajectories calculated at Ec = 0.7, 1.0, and 1.3 eV. The energies were chosen to allow for direct comparison with experiment. Fig. 3 show the distributions of the number of the transitions from quintet to triplet and from triplet to singlet for the three pathways: nonreactive (black), 5Ta+ + CH4 → 3TaCH2+ + H2 (red), and 5Ta+ + CH4 → 1TaCH2+ + H2 (blue). For nonreactive trajectories, only a tiny minority of trajectories, 1.7, 1.6, and 1.5% at Ec = 0.7, 1.0, and 1.3 eV, respectively, underwent a quintet → triplet → quintet transition. For most reactive trajectories, only a single transition from quintet to triplet is needed for the reaction to happen. In a small subset of trajectories, 2 to 10 transitions are required. The distribution of these transitions declined rapidly as the number of transitions increased. Consequently, from the above analysis, we can safely conclude that the ISC is the rate-limiting step in this reaction. This reinforces the conclusion of Parke et al.28 that the relatively low efficiency of the reaction is a result of its spin-forbidden nature, as discussed above. Fig. S8† shows a comparison of the geometries of 3SP1 and the minimum energy crossing point (MECP) between the quintet and triplet states. As seen, the MECP is located before 3SP1 but they have very similar structures. Furthermore, the energy of the MECP is −0.274 eV at the B3LYP/DZ level, which is slightly lower than that of 3SP1 (−0.191 eV, without ZPE correction). This indicates that overcoming 3SP1 is relatively easy for this reaction once the crossing occurs, suggesting that the energy barrier on the triplet state is not the kinetic bottleneck. In contrast, in the prototypical FeO+ + H2 reaction,40 the energy of MECP is 0.351 eV lower than that of the saddle point, resulting in that the saddle point is the rate-limiting step rather than the spin-state change at low reaction energy.
 | ||
| Fig. 3 Distributions of the number of ISC transitions from the quintet to triplet (solid line) and from the triplet to singlet (dotted line) for the nonreactive (black), 5Ta+ + CH4 → 3TaCH2+ + H2 (blue), and 5Ta+ + CH4 → 1TaCH2+ + H2 (red) reactions at three collision energies, respectively. Note that the reverse transitions from triplet to quintet (3 → 5) and from singlet to triplet (1 → 3) are necessary for the nonreactive channel (black lines). | ||
Cross sections and product energy disposal
The velocity distributions of the product ion TaCH2+ are displayed in Fig. 4(a)–(c) for experimental data37 and Fig. 4(d)–(f) for calculated data, at three collision energies. The superimposed circles represent the maximum possible energy partitioned into the product kinetic energy, starting from the quintet (green) and triplet (orange) reactants.37 It is apparent that some of the experimental events fall outside the velocity space defined by the kinematic cut-off, indicating an overestimation of the kinetic energy of the product ion TaCH2+.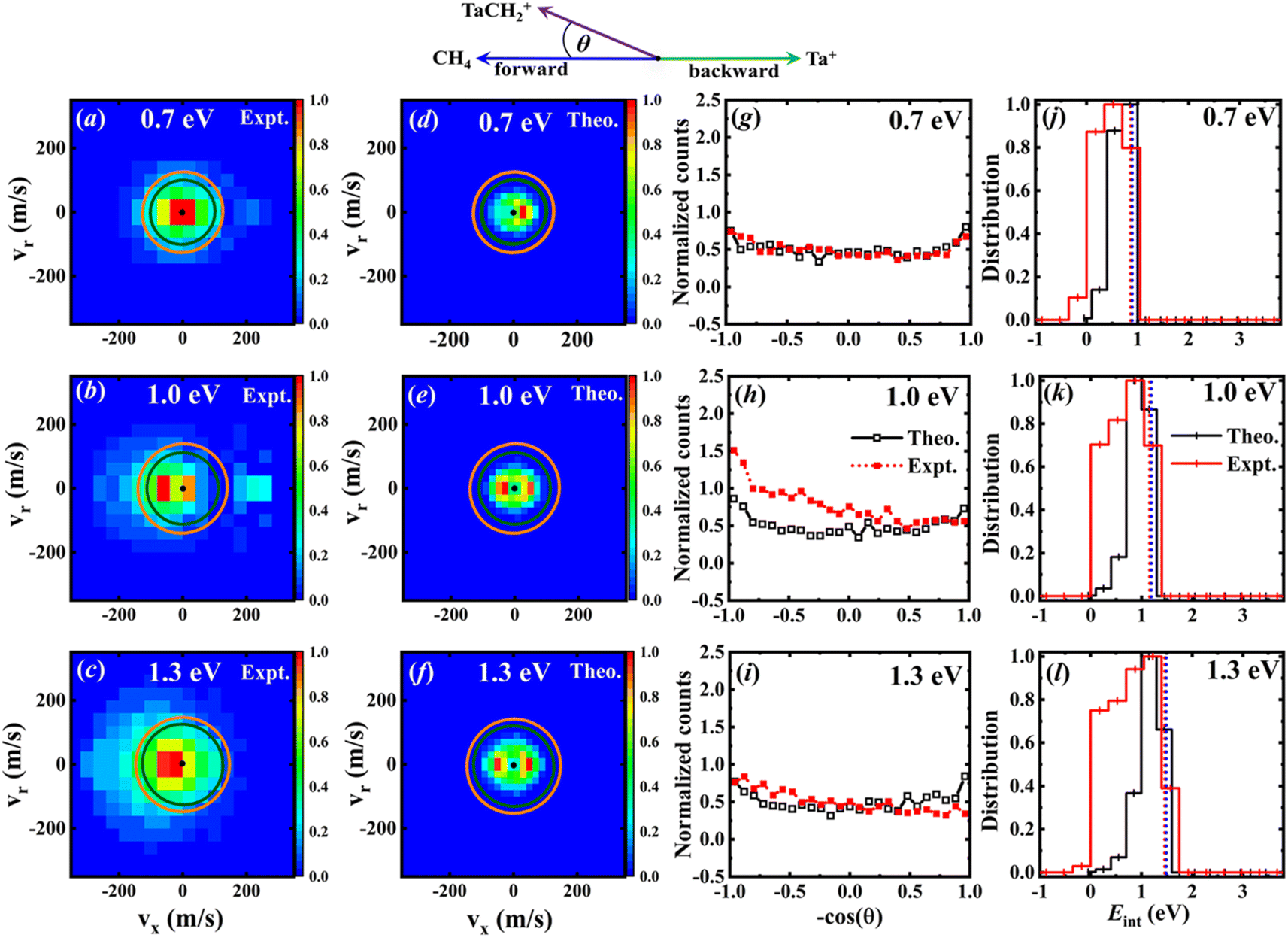 | ||
| Fig. 4 (a)–(c) The experimental37 velocity distributions of the TaCH2+ product ion at Ec = 0.7, 1.0, and 1.3 eV, respectively. (d–f) Similar to (a)–(c) but for the theoretical data. Histograms are normalized with the bin of highest intensity set to one. (g–i) The corresponding integrated angular distributions from simulations with the experimental data37 included for comparison. The definition of the scattering angle is also illustrated by the simplified Newton diagram. All distributions normalized to an area of one. The dotted lines in j–l are the calculated available energies for the triplet (red) and singlet (blue) product channels (since these values are nearly identical, the lines overlap). Experimental distributions are truncated at the kinematic cut-off for better comparison. | ||
The calculated energy distributions of the internal energy align well with experimental measurements at the position of the peak and the right side of the peak. Nonetheless, the experimental distributions are broader than the theoretical ones. On the left side of the peak, and surprisingly, the experiments show some negative internal energy distributions which is due to Eint being calculated with respect to the kinematic cut-off. The experimental and theoretical internal energies of the products are obtained using the formula:
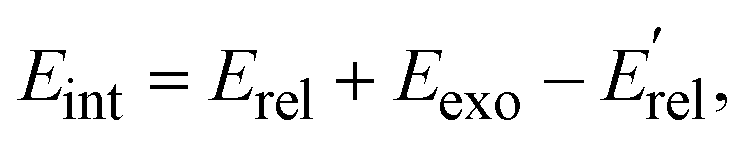
 represent the relative collision energy, reaction exothermicity, and the product relative translational energy, respectively. The negative contribution to Eint may result from missing contributions to the energy balance, such as electronically excited states of Ta+ or the internal energy of CH4, as discussed in ref. 37, which includes a detailed analysis on the experimental energy spread. The experimental and theoretical velocity distributions of the product ion TaCH2+ show predominantly isotropic scattering around the center of mass, but discrepancies exist between the two. The calculated and measured DCSs at Ec = 0.7, 1.0, and 1.3 eV are given in Fig. 4(g)–(i) as a function of cos(θ). The calculated velocity distributions are forward-backward symmetric at all three collision energies and agree well with the experimental at Ec = 0.7 eV. However, at Ec = 1.0 and 1.3 eV, the experimental DCSs have a forward bias. The differences in the DCS at high collision energies are also present in the Ta+/Nb+ + CO2 systems,38,39 possible explanations include the presence of additional electronic states at high collision energies and the underestimation of DFT calculations for the singlet state reactant asymptote. The calculated energy distributions of the internal energy, shown in Fig. 4(j)–(l), agree well with the experimental measurements at the position of the peak, but the experimental distributions are broader than the theoretical ones on the left side of the peak.
represent the relative collision energy, reaction exothermicity, and the product relative translational energy, respectively. The negative contribution to Eint may result from missing contributions to the energy balance, such as electronically excited states of Ta+ or the internal energy of CH4, as discussed in ref. 37, which includes a detailed analysis on the experimental energy spread. The experimental and theoretical velocity distributions of the product ion TaCH2+ show predominantly isotropic scattering around the center of mass, but discrepancies exist between the two. The calculated and measured DCSs at Ec = 0.7, 1.0, and 1.3 eV are given in Fig. 4(g)–(i) as a function of cos(θ). The calculated velocity distributions are forward-backward symmetric at all three collision energies and agree well with the experimental at Ec = 0.7 eV. However, at Ec = 1.0 and 1.3 eV, the experimental DCSs have a forward bias. The differences in the DCS at high collision energies are also present in the Ta+/Nb+ + CO2 systems,38,39 possible explanations include the presence of additional electronic states at high collision energies and the underestimation of DFT calculations for the singlet state reactant asymptote. The calculated energy distributions of the internal energy, shown in Fig. 4(j)–(l), agree well with the experimental measurements at the position of the peak, but the experimental distributions are broader than the theoretical ones on the left side of the peak.
The nearly isotropically scattered product ion TaCH2+ indicates that the reaction is dominated by a complex-forming mechanism, suggesting a relatively long lifetime of a reaction complex. To identify the long-lived complex/intermediate of this reaction, we calculated the lifetimes of the pre-reaction well and post-reaction well, based on the same criteria as in the Ta+ + CO2 system.38 For INT1, the time is measured between when the Ta–C distance becomes less than 4.5 Å and when the system crosses SP1, defined as the Ta–C distance less than 2.4 Å. This indicates that the system enters the post-transition state well. The lifetime of the post-transition state wells ends when the center of mass (COM) distance between the products exceeds 4.5 Å. We define the system as entering INT2 when only one of the C–H distances becomes longer than 1.4 Å. Similarly, the system is considered to enter INT3 when two of the C–H distances exceed 1.4 Å, provided that the distance between two hydrogen atoms farthest from the carbon atom exceeds 1.1 Å. Finally, if the distance between the two hydrogen atoms decreases to less than 1.1 Å, the system is assumed to reach INT4 or INT5 (the two structures are indistinguishable in this work). The average lifetimes of pre-reaction well are 0.63, 0.27, and 0.13 ps at Ec = 0.7, 1.0 and 1.3 eV, respectively, while for post-reaction well, we found INT2 and INT3 both have much longer lifetimes. They are 8.21, 7.47, and 9.41 ps for INT2 and 6.37, 4.98, and 5.46 ps for INT3 at the three collision energies, respectively. The lifetime of INT4 (or/and INT5) is 0.32, 0.25, 0.23 ps at the three collision energies, respectively. This is quite different from those observed in the Ta+/Nb+ + CO2 systems in which the pre-reaction well has the longer lifetimes.38,39 The complex-forming mechanism can also be demonstrated by analysing the relationship between the scattering angle and the impact parameter, as shown in Fig. S9,† which reveals no correlation between these variables, indicating the presence of a long-lived intermediate. This intermediate contributes to the forward-backward symmetric scattering patterns observed in our theoretical results.
The calculated average energy partitioning of the two products as a function of collision energy is shown in Fig. 5(a). The highly excited product internal energy is consistent with the experimentally observed low kinetic energy. The results indicate that both the vibrational and rotational modes of TaCH2+ are highly excited, while that of the molecular hydrogen H2 product is quite moderate. However, this picture is somewhat deceiving as the former has six vibrational modes and three rotational modes, rather than the single vibrational and two rotational modes for H2. As illustrated in Fig. 5(b), the average amount of energy partitioned into the different types of motion divided by the number of associated degrees-of-freedom (DOFs) are quite comparable, suggesting equipartitioning of the available energy in product energy disposal. This is apparently a consequence of the long lifetime of the post-reaction well, where energy randomization must have taken place. The energy disposal in the title reaction is very different from that in the Ta+ + CO2 and Nb+ + CO2 reactions, where the energy disposal into the internal DOFs of MO+ is dominant.38,39 The long lifetime also results in a larger distribution of singlet state products for the reaction compared to the Ta+/Nb+ + CO2 reactions because it increases the chance of crossing from triplet to singlet. Unlike the transitions from quintet to triplet, the distributions of transition numbers from the triplet to singlet state do not show a sharp peak but instead display a uniform distribution across the transition numbers, see Fig. 3. This is different from what we observed in the Ta+/Nb+ + CO2 systems. The product branching is approximately 0.7/0.3 for 3TaCH2+/1TaCH2+ at the three collision energies, whereas in the Ta+/Nb+ + CO2 reactions, it is only about 0.9/0.1 for 3TaO+/1TaO+ and 3NbO+/1NbO+.38,39
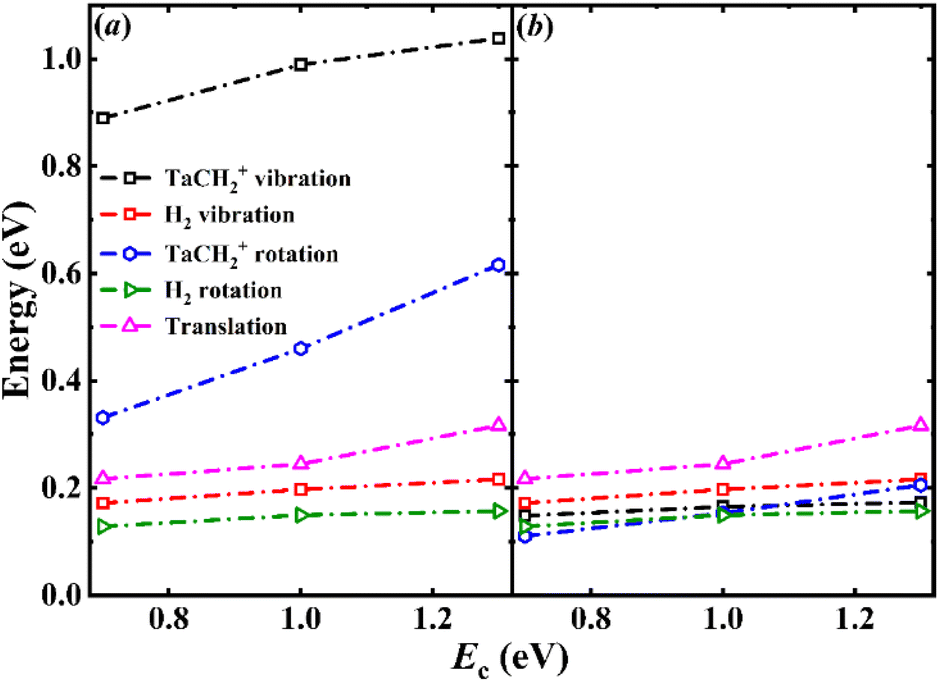 | ||
| Fig. 5 (a) Product (3,1TaCH2+ + H2) energy partitioning to different product motion as a function of the collision energy. (b) The average energy partitioning per degree-of-freedom, namely, the average energy in each mode for the two products. | ||
Fig. S10(a) and (b)† show the experimental37 and theoretical velocity distribution of TaCD2+, respectively, at the Ec = 1.2 eV and the corresponding integrated angular distributions are given in Fig. S10(c) and (d).† As seen, the similar scattering signatures to TaCH2+ are observed for TaCD2+, suggesting that the Ta+ + CD4 reaction is also controlled by the indirect mechanism. The distribution of the internal energy, as shown in Fig. S10(e) and (f),† in the Ta+ + CD4 reaction is also very similar to the one in the Ta+ + CH4 reaction.
Conclusions
To summarize, the first set of full-dimensional PESs for the quintet, triplet, and singlet states of the Ta+ + CH4 → TaCH2+ + H2 reaction were developed at the B3LYP/DZ level based on PIP-NN fits of 64300 points. Multi-state nonadiabatic dynamical calculations were performed on these PESs using the FSSH method, with spin–orbital couplings (SOCs) determined from MRCI calculations. The calculated thermal rate coefficients are found to be in good agreement with the newly acquired experimental data in the temperature range between 300 and 600 K. However, this good agreement was only achieved after ZPE correction based on the 1GB scheme. The significant ZPE violation can be attributed to the near thermoneutrality of the reaction, and the large vibrational frequency of the H2 product. Additionally, the intersystem crossing between the quintet and triplet states, rather than the triplet saddle point, is the kinetic bottleneck of this reaction because of the very close proximity of the crossing point to the triplet saddle point.
The dynamical calculations predict product angular distributions consistent with the molecular beam measurements, although minor differences exist at relatively high collision energies. Our calculations support the experimental finding that the title reaction is predominantly governed by an indirect mechanism, even at higher collision energies. Additional evidence supporting this conclusion is that we found that there is no correlation between the scattering angle and the impact parameter as discussed above. More importantly, our analysis attributes the indirect mechanism to two long-lived reaction intermediates in the exit channel. Also consistent with the slow relative recoil velocity observed in the experiment, the internal DOFs of the TaCH2+ product are found to be excited. However, the energy disposal into the products was found to be equally partitioned into each individual degree-of-freedom, representing a statistical distribution, thanks to the long lifetime of the reaction intermediate, which allows efficient and complete energy randomization. The long lifetime also leads to the nonnegligible contribution of the singlet state to the reaction.
We emphasize that the DFT treatment of the electronic structure is an approximation, which ignores many important details such as spin–orbit coupling and multi-reference characteristics. Nevertheless, the reasonably good agreement with available experimental data supports the validity of the theoretical model used to describe the kinetics and dynamics of this prototypical model reaction for the activation of methane. It is clearly shown that the reaction is facilitated by a “spin-forbidden” mechanism, in which a barrierless pathway is in-turn facilitated by ISC from the quintet state to the triplet state, followed by mixing between the triplet and singlet states. This two-state reactivity mechanism is expected to be instructive in understanding the catalytic capacity of transition metals in more complex environments such as single atom catalysis.
Methods
Electronic structure calculations
The geometries, energies, and harmonic frequencies of all stationary points are calculated at the B3LYP/DZ level,41,42 using the aug-cc-pVDZ basis set for C and H and the ECP60MDF_AVDZ basis set for Ta.43 This level of theory is also used to construct the quintet, triplet, and singlet PESs for the Ta+ + CH4 system. To prevent the B3LYP/DZ calculations from converging to electronically excited states, which can occur due to the numerous low-lying excited states in the Ta+ + CH4 system, the wave function stability analysis was performed before each calculation. This ensured that the ground electronic state for the relevant spin multiplicity was correctly retrieved. All DFT calculations were performed with the Gaussian quantum chemical program.44The SOC calculations were performed at the multi-reference configurational interaction with the same basis set as in DFT (MRCI/DZ).45,46 The complete active space self-consistent field (CASSCF) method47,48 was used to provide the wave functions and reference states for subsequent MRCI calculations. The CASSCF calculations employed an active space of 6 electrons in 7 orbitals (6e, 7o), considering two singlet, two triplet, and two quintet molecular terms (18 states in total). We selected seven geometries near the quintet/triplet MECP from the minimum energy path (MEP) on the triplet state and performed the SOC calculations. For each point, the root-mean-square of the sum of the MRCI SOC matrix elements was used to provide an averaged value.49 As shown in Fig. S11,† the SOC values of the seven points show no significant variation. The average value of these points was used for our dynamical calculations. MRCI calculations were performed using Molpro software,50 and the MECP search was conducted with the EasyMECP program,51 modified to allow for wave function stabilization prior to each DFT calculation.
Potential energy surfaces
To cover all relevant configuration spaces for the Ta+ + CH4 → TaCH2+ + H2 reaction, about 64300 points were sampled and calculated the quintet, triplet, and singlet state energies at the B3LYP/DZ level. The PIP-NN method52,53 was employed to fit these energy points in the full twelve dimensions. The NN input layer employs permutation-invariant polynomials (PIPs) to enforce the symmetry of the four identical hydrogen atoms. For the Ta+ + CH4 system, 1052 PIPs up to fifth order were employed in the NN input layer. The Morse-like variables 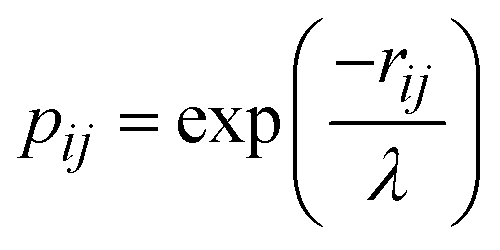 were used to construct the PIPs,54 where rij represents the internuclear distance and λ is an adjustable constant set to 1.8 Å in this work. After several tests, a neural network structure of 1052-5-100 was chosen, which results in 5966 fitting parameters for each spin-specific PES. To prevent overfitting, the “early stopping” method was employed, which involved randomly splitting the dataset into 90% for training and 5% each for validation and testing. This strategy ensures that the model remains generalizable and effective across various data subsets. The fittings are accepted only when the RMSEs for the training, validation, and test sets were comparable, effectively preventing false extrapolation. The Levenberg–Marquardt algorithm was utilized for NN training.55
were used to construct the PIPs,54 where rij represents the internuclear distance and λ is an adjustable constant set to 1.8 Å in this work. After several tests, a neural network structure of 1052-5-100 was chosen, which results in 5966 fitting parameters for each spin-specific PES. To prevent overfitting, the “early stopping” method was employed, which involved randomly splitting the dataset into 90% for training and 5% each for validation and testing. This strategy ensures that the model remains generalizable and effective across various data subsets. The fittings are accepted only when the RMSEs for the training, validation, and test sets were comparable, effectively preventing false extrapolation. The Levenberg–Marquardt algorithm was utilized for NN training.55
Since both H2 and CH4 have no dipole, very small quadrupoles, and small polarizability. No special treatment of the electrostatic interaction was applied in the long range. However, we have carefully checked the PIP-NN PESs near the asymptote and found them to be smooth and possess no artificial features as depicted in Fig. S12.†
Trajectory surface hopping
The dynamical calculations were performed using Tully's fewest switches surface hopping (FSSH) method.56 This semiclassical treatment of nonadiabatic dynamics involves using classical mechanics to describe the nuclear motion on each electronic state PES, with the electronic motion governed by quantum mechanics. The FSSH algorithm determines the transition probability from one electronic state to another at each timestep, utilizing the electronic wavefunctions and nonadiabatic coupling elements. Then, a random number, drawn uniformly between 0 and 1, is used to compare against this probability to decide if a hop should take place. When a hop happens, the nuclear momenta are altered to ensure total energy conservation. In the FSSH calculations, the SOC values between the quintet and triplet, the quintet and singlet, and the triplet and singlet are 280.7, 0.0, and 192.6 cm−1, respectively. The 2023 version of the ANT program57 was employed to perform the FSSH calculations.A trajectory is initiated with the reactants at an initial separation of 12 Å and terminated when the reactants or products reached the same separation distance. All trajectories start on the quintet reactant asymptote. The maximum impact parameter (bmax) is set to match the initial separation between the collision partners, ensuring it is large enough for convergence of the results. The impact parameter (b) is calculated using the equation b = bmaxξ1/2, with ξ being a random number uniformly distributed between 0 and 1. For trajectory integration, the Bulirsch–Stoer method with adaptive step size was employed.
The thermal rate coefficients are calculated at the temperatures of 300, 400, 500, and 600 K. The translational, vibrational, and rotational degrees of freedom of the reactants were sampled using the Boltzmann distribution at the specified temperature. The dynamic calculations were performed at the experimental collision energies (Ec) of 0.7, 1.0, and 1.3 eV, with CH4 in its ground ro-vibrational state. To keep the statistical error under 5.0%, 1.0 × 104 trajectories for thermal rate calculations and 2.0 × 105 trajectories were initiated for dynamical calculations. Almost all trajectories maintain energy conservation within a 10−3 eV criterion.
The following equation was used to calculate the reactive integral cross section (ICS) for the product channel TaCH2+ + H2:
| σr(Ec) = πbmax2Pr(Ec). | (1) |
 .
.
The differential cross section (DCS) is calculated using the following formula:
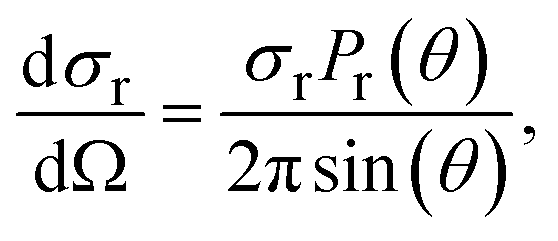 | (2) |
The following experimental definition of the scattering angle37 was used:
 | (3) |
The following formula was used to calculate the thermal rate coefficient:
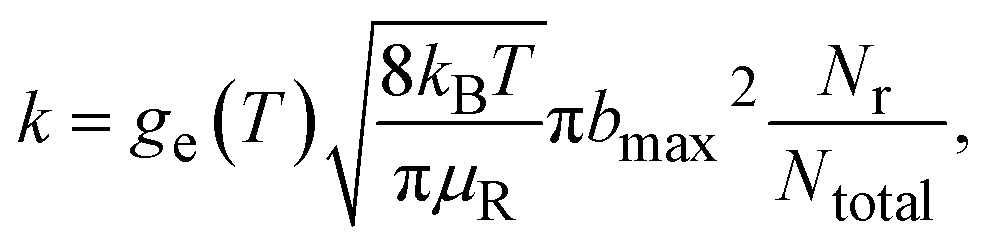 | (4) |
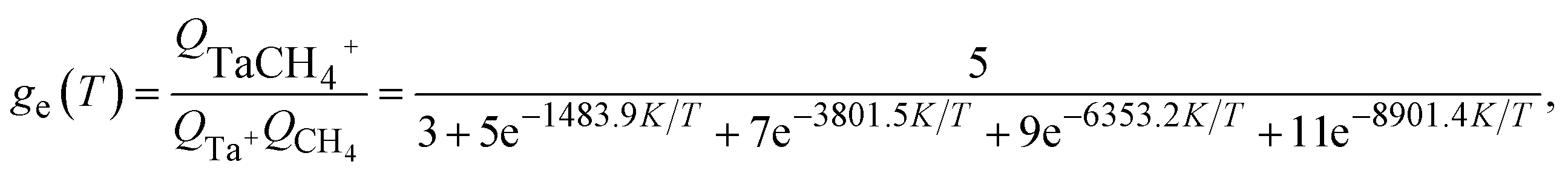 | (5) |
In quasi-classical trajectory simulations, molecular vibrations are not quantized, and the vibrational energy is allowed to fall below ZPE. To address the ZPE leakage issue, trajectories that violate ZPE can be removed, a technique known as hard-ZPE correction. A more reasonable approach is the Gaussian binning (GB) method,58 which incorporates quantum effects into the quasi-classical results by assigning a weight to each reactive trajectory. However, as the number of vibrational modes increases, the computational cost of GB grows exponentially, making it unsuitable for polyatomic products. To address this problem, Czakó and Bowman proposed the 1GB method,59 in which the following Gaussian weight is attached to each trajectory:
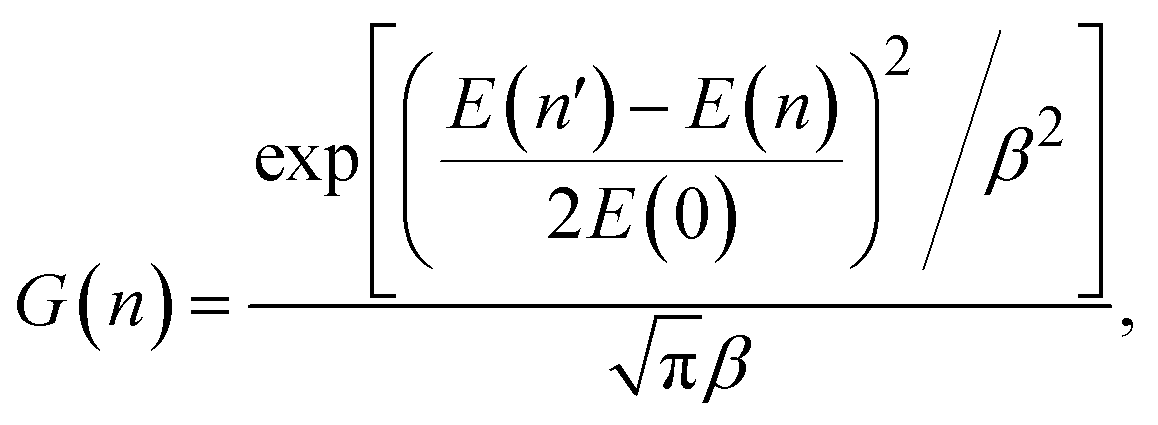 | (6) |
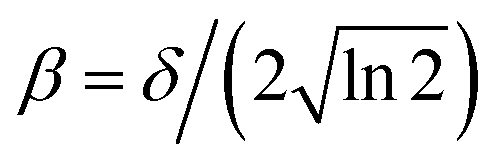 , where δ is the full width at half maximum, set to 0.5 in this work after the testing. The weight for the corresponding vibrational states of the two products (TaCH2+ and H2) is calculated as the product of two Gaussians, G(n) = G(n1,n2,n3,n4,n5,n6) × G(n7).
, where δ is the full width at half maximum, set to 0.5 in this work after the testing. The weight for the corresponding vibrational states of the two products (TaCH2+ and H2) is calculated as the product of two Gaussians, G(n) = G(n1,n2,n3,n4,n5,n6) × G(n7).
The reaction probability is calculated using the following formula, as suggested by Bonnet and Espinosa-García:60
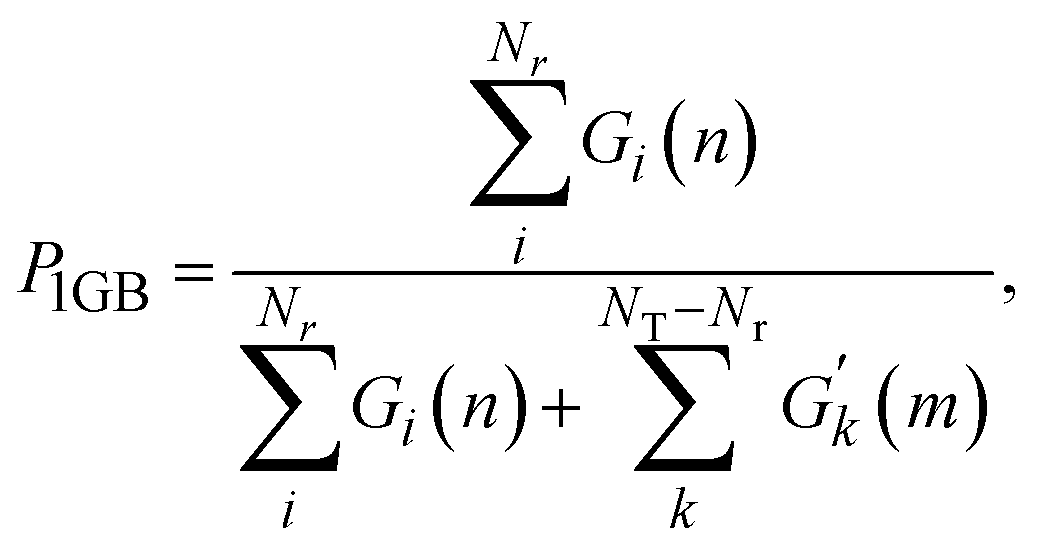 | (7) |
SIFT
Thermal rate constants were measured using a selected-ion flow tube apparatus, similar to described elsewhere.61 Briefly, Ta+ ions were formed using a laser vaporization source via impinging the second harmonic of an Nd:YAG laser operating at 100 Hz onto a rotating, translating tantalum rod (ESPI metals, 99.9%). Ions were entrained into an expansion of argon (99.999%, Matheson) through a pulsed valve (Iota One) with an 0.2 mm aperture also operating at 100 Hz. Ions were mass-selected using a quadrupole mass filter and transported via a series of quadrupole ion guides to the entrance of a 7 cm diameter, 1 m long, stainless steel flow tube and injected against a pressure gradient via a Venturi inlet into a 102 m s−1 flow of helium (99.999%, Matheson). 59 cm prior to the terminus of the flow tube, reactant gas (methane, AirGas 99.99%) was added using a mass flow controller (MKS) via a 1/8′′ diameter finger inlet. At the terminus of the flow tube, the core of the flow was sampled via a 3 mm aperture in a rounded, carbon-coated nosecone. Ions entered a higher vacuum region and were transported using a quadrupole ion guide to the entrance of an orthogonally-accelerated reflectron time-of-flight mass spectrometer. Relative reactant and product ion concentrations were monitored as a function of methane concentration in the flow tube. Rate constants were derived in the standard manner assuming pseudo-first order kinetics.Only the rate constants for decay of the primary Ta+ ion are reported here, with discussion of the significant sequential chemistry reserved for a later publication.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written by Y. L. and H. G. with contributions of all authors. All authors have given approval to the final version of the manuscript. M. O., S. G. A., N. S. S., J. M. and H. G designed the project. Y. L. and M. O performed the calculations. T. W. R. L., S. G. A. and N. S. S. performed the SIFT experiments. All authors participated in the data analysis. H. G. directed the project.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work at UNM was supported by Air Force Office of Scientific Research (FA9550-22-1-0350 to H. G.). Support for AFRL authors was provided by the Air Force Office of Scientific Research under AFOSR-22RVCOR009. J. M. acknowledges support by the Deutsche Forschungsgemeinschaft DFG (Project number 500279291). The computation was performed at CARC (Center for Advanced Research Computing) at UNM and at the HPC infrastructure LEO of the University of Innsbruck. The views expressed are those of the authors and do not reflect the official guidance or position of the Department of the Air Force, the Department of Defense (DoD), or the U.S. government.References
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- R. Horn and R. Schlögl, Catal. Lett., 2015, 145, 23–39 CrossRef CAS.
- A. A. Latimer, H. Aljama, A. Kakekhani, J. S. Yoo, A. Kulkarni, C. Tsai, M. Garcia-Melchor, F. Abild-Pedersen and J. K. Nørskov, Phys. Chem. Chem. Phys., 2017, 19, 3575–3581 RSC.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 2005 Search PubMed.
- A. Kramida, Y. Ralchenko, J. Reader and NIST ASD Team, NIST Atomic Spectra Database (version 5.12), 2024, https://physics.nist.gov/asd Search PubMed.
- J. Wei and E. Iglesia, J. Catal., 2004, 224, 370–383 CrossRef CAS.
- Z. Liu, D. C. Grinter, P. G. Lustemberg, T.-D. Nguyen-Phan, Y. Zhou, S. Luo, I. Waluyo, E. J. Crumlin, D. J. Stacchiola, J. Zhou, J. Carrasco, H. F. Busnengo, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, Angew. Chem., Int. Ed., 2016, 55, 7455–7459 Search PubMed.
- P. G. Lustemberg, R. M. Palomino, R. A. Gutiérrez, D. C. Grinter, M. Vorokhta, Z. Liu, P. J. Ramírez, V. Matolín, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, J. Am. Chem. Soc., 2018, 140, 7681–7687 Search PubMed.
- Z. Zuo, S. Liu, Z. Wang, C. Liu, W. Huang, J. Huang and P. Liu, ACS Catal., 2018, 8, 9821–9835 CrossRef CAS.
- M. Akri, S. Zhao, X. Li, K. Zang, A. F. Lee, M. A. Isaacs, W. Xi, Y. Gangarajula, J. Luo, Y. Ren, Y.-T. Cui, L. Li, Y. Su, X. Pan, W. Wen, Y. Pan, K. Wilson, L. Li, B. Qiao, H. Ishii, Y.-F. Liao, A. Wang, X. Wang and T. Zhang, Nat. Commun., 2019, 10, 5181 CrossRef PubMed.
- Y. Tang, Y. Wei, Z. Wang, S. Zhang, Y. Li, L. Nguyen, Y. Li, Y. Zhou, W. Shen, F. F. Tao and P. Hu, J. Am. Chem. Soc., 2019, 141, 7283–7293 CrossRef CAS PubMed.
- D. K. Böhme and H. Schwarz, Angew. Chem., Int. Ed., 2005, 44, 2336–2354 CrossRef PubMed.
- S. M. Lang and T. M. Bernhardt, Phys. Chem. Chem. Phys., 2012, 14, 9255–9269 RSC.
- H. Schwarz, Catal. Sci. Techno., 2017, 7, 4302–4314 RSC.
- S. W. Buckner, T. J. MacMahon, G. D. Byrd and B. S. Freiser, Inorg. Chem., 1989, 28, 3511–3518 CrossRef CAS.
- K. K. Irikura and J. Beauchamp, J. Phys. Chem., 1991, 95, 8344–8351 CrossRef CAS.
- J. C. Weisshaar, Acc. Chem. Res., 1993, 26, 213–219 CrossRef CAS.
- R. B. Metz, Int. Rev. Phys. Chem., 2004, 23, 79–108 Search PubMed.
- J. Roithová and D. Schröder, Chem. Rev., 2010, 110, 1170–1211 CrossRef PubMed.
- H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 10096–10115 CrossRef CAS PubMed.
- H. Schwarz, J. Chem., 2014, 54, 1413–1431 CAS.
- K. K. Irikura and J. L. Beauchamp, J. Am. Chem. Soc., 1991, 113, 2769–2770 CrossRef CAS.
- M. Pavlov, M. R. A. Blomberg, P. E. M. Siegbahn, R. Wesendrup, C. Heinemann and H. Schwarz, J. Phys. Chem. A, 1997, 101, 1567–1579 CrossRef CAS.
- X.-G. Zhang, R. Liyanage and P. B. Armentrout, J. Am. Chem. Soc., 2001, 123, 5563–5575 CrossRef CAS PubMed.
- M. M. Armentrout, F.-X. Li and P. B. Armentrout, J. Phys. Chem. A, 2004, 108, 9660–9672 CrossRef CAS.
- P. B. Armentrout, S. Shin and R. Liyanage, J. Phys. Chem. A, 2006, 110, 1242–1260 CrossRef CAS PubMed.
- F.-X. Li, X.-G. Zhang and P. B. Armentrout, Int. J. Mass Spectrom., 2006, 255–256, 279–300 CrossRef CAS.
- L. G. Parke, C. S. Hinton and P. B. Armentrout, J. Phys. Chem. C, 2007, 111, 17773–17787 CrossRef CAS.
- A. Shayesteh, V. V. Lavrov, G. K. Koyanagi and D. K. Bohme, J. Phys. Chem. A, 2009, 113, 5602–5611 CrossRef CAS PubMed.
- J. F. Eckhard, T. Masubuchi, M. Tschurl, R. N. Barnett, U. Landman and U. Heiz, J. Phys. Chem. A, 2021, 125, 5289–5302 CrossRef CAS PubMed.
- P. B. Armentrout, L. Parke, C. Hinton and M. Citir, ChemPlusChem, 2013, 78, 1157–1173 CrossRef CAS PubMed.
- B. Ruscic, R. E. Pinzon, M. L. Morton, G. von Laszevski, S. J. Bittner, S. G. Nijsure, K. A. Amin, M. Minkoff and A. F. Wagner, J. Phys. Chem. A, 2004, 108, 9979–9997 CrossRef CAS.
- B. Ruscic, R. E. Pinzon, G. von Laszewski, D. Kodeboyina, A. Burcat, D. Leahy, K. A. Amin, M. Minkoff and A. F. Wagner, J. Phys.: Conf. Ser., 2005, 16, 561 CrossRef CAS.
- P. B. Armentrout, Science, 1991, 251, 175–179 CrossRef CAS PubMed.
- D. Schröder, S. Shaik and H. Schwarz, Acc. Chem. Res., 2000, 33, 139–145 CrossRef PubMed.
- H. Schwarz, Int. J. Mass Spectrom., 2004, 237, 75–105 CrossRef CAS.
- M. Meta, M. E. Huber, M. Birk, M. Wedele, M. Ončák and J. Meyer, Faraday Discuss., 2024, 251, 587–603 RSC.
- Y. Liu, M. Ončák, J. Meyer, S. G. Ard, N. S. Shuman, A. A. Viggiano and H. Guo, J. Am. Chem. Soc., 2024, 146, 14182–14193 CrossRef CAS PubMed.
- Y. Liu, M. Ončák, J. Meyer, S. G. Ard, N. S. Shuman, A. A. Viggiano and H. Guo, J. Phys. Chem. A, 2024, 128, 6943–6953 CrossRef CAS PubMed.
- S. Essafi, D. P. Tew and J. N. Harvey, Angew. Chem., Int. Ed., 2017, 56, 5790–5794 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- H.-J. Werner and P. J. Knowles, J. Chem. Phys., 1988, 89, 5803–5814 CrossRef CAS.
- P. J. Knowles and H.-J. Werner, Chem. Phys. Lett., 1988, 145, 514–522 CrossRef CAS.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- P. E. M. Siegbahn, J. Almlöf, A. Heiberg and B. O. Roos, J. Chem. Phys., 1981, 74, 2384–2396 CrossRef.
- D. Danovich and S. Shaik, J. Am. Chem. Soc., 1997, 119, 1773–1786 CrossRef CAS.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS.
- J. Rodríguez-Guerra, I. Funes-Ardoiz and F. Maseras, EasyMECP. Zenodo, version v0.3.2, 2018 Search PubMed.
- B. Jiang and H. Guo, J. Chem. Phys., 2013, 139, 054112 CrossRef PubMed.
- J. Li, B. Jiang and H. Guo, J. Chem. Phys., 2013, 139, 204103 CrossRef PubMed.
- B. J. Braams and J. M. Bowman, Int. Rev. Phys. Chem., 2009, 28, 577–606 Search PubMed.
- M. T. Hagan and M. B. Menhaj, IEEE Trans. Neural Network., 1994, 5, 989–993 Search PubMed.
- J. C. Tully, J. Chem. Phys., 1990, 93, 1061–1071 CrossRef CAS.
- Y. Shu, L. Zhang, J. Zheng, Z. H. Li, A. W. Jasper, D. A. Bonhommeau, R. Valero, R. Meana-Pañeda, S. L. Mielke, Z. Varga and D. G. Truhlar, ANT 2023.B: A Program for Adiabatic and Nonadiabatic Trajectories, version v2; Zenodo, 2023 Search PubMed.
- L. Bonnet and J.-C. Rayez, Chem. Phys. Lett., 2004, 397, 106–109 CrossRef CAS.
- G. Czakó and J. M. Bowman, J. Chem. Phys., 2009, 131, 244302 CrossRef PubMed.
- L. Bonnet and J. Espinosa-Garcia, J. Chem. Phys., 2010, 133, 164108 CrossRef CAS PubMed.
- M. E. Huber, T. W. R. Lewis, M. Meta, S. G. Ard, Y. Liu, B. C. Sweeny, H. Guo, M. Ončák, N. S. Shuman and J. Meyer, Phys. Chem. Chem. Phys., 2024, 26, 8670–8680 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08457h |
| This journal is © The Royal Society of Chemistry 2025 |