
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular-locking modification enables efficient asymmetric donor–acceptor–donor′ type ultraviolet emitters for high-performance OLEDs with reduced efficiency roll-off and high color purity†
Shengnan Wanga,
Rui Zhanga,
Runjie Dinga,
Hao Huanga,
Haoyuan Qia,
Yuchao Liu a,
Shian Ying*a,
Dongge Ma*b and
Shouke Yan*ac
a,
Shian Ying*a,
Dongge Ma*b and
Shouke Yan*ac
aKey Laboratory of Rubber-Plastics, Ministry of Education, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: shian0610@126.com
bState Key Laboratory of Luminescent Materials and Devices, Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates, Center for Aggregation-Induced Emission, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: msdgma@scut.edu.cn
cState Key Laboratory of Chemical Resource Engineering, College of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: skyan@mail.buct.edu.cn
First published on 26th February 2025
Abstract
Developing high-performance ultraviolet organic light-emitting diodes with low efficiency roll-off and high color purity remains challenging due to their inherent wide-bandgap characteristics. In this work, we present an intramolecular noncovalent bond locking strategy to modulate donor–acceptor–donor′ (D–A–D′) type ultraviolet fluorophores (mPImCZ2F, mPIoCZ2F and mPImCP2F) with a hot-exciton mechanism. Notably, these asymmetric emitters exhibit significantly enhanced bipolar transport capacity and fluorescence efficiency compared to their counterparts. Among them, mPIoCZ2F exhibits a more remarkable intramolecular locking effect due to multiple C–H⋯F interactions and ortho-substitution-induced steric hindrance, which endows it with a higher radiation rate, narrower emission spectrum, and more balanced charge transport. Consequently, the mPIoCZ2F-based non-doped device achieves an electroluminescence (EL) peak at 393 nm with a maximum external quantum efficiency (EQE) of 6.62%. Moreover, in the doped device, mPIoCZ2F emits stable ultraviolet light with an EL peak at 391 nm and a full width at half maximum (FWHM) of 40 nm, corresponding to color coordinates of (0.167, 0.025). It also exhibits an exceptionally high EQE of 8.71% and minimal efficiency roll-off (7.95% at 1000 cd m−2), ranking among the best EL efficiencies reported for UV-OLEDs at high brightness levels.
1 Introduction
Organic light-emitting diodes (OLEDs) possess numerous remarkable advantages such as ultra-wide viewing angles, high contrast, rapid response time, lightweight design, superior energy efficiency, and unparalleled flexibility, which have endowed them with broad application prospects in the fields of high-end televisions, smartphones, tablets, as well as wearable devices.1–8 Since the invention of OLEDs in 1987,1 significant research has been conducted to develop red, green, and blue (RGB) devices that are highly efficient, exhibit low efficiency roll-off, and possess good color purity.9–14 However, ultraviolet (UV) OLEDs with short-wavelength emissions below 400 nm, which have potential applications in lithography, medicine, and high-density information storage, and as photoexcitation sources, lag behind RGB devices in the above aspects due to their intrinsic wide bandgap characteristics (exceeding 3.1 eV).15,16One of the critical factors in achieving ideal UV pure organic emitters lies in the effective utilization of triplet excitons. Although first-generation traditional fluorophores can achieve low efficiency roll-off in OLEDs due to the rapid radiation decay of singlet excitons, their electroluminescence (EL) efficiency remains limited. Thermally activated delayed fluorescence (TADF) and hot-exciton emitters can efficiently convert “non-radiative” triplet excitons into emissive singlet excitons through efficient reverse intersystem crossing (RISC) and high-lying RISC (hRISC) processes, respectively, enabling the harvesting of 100% of the electrically generated excitons.5,17 Distinct from TADF-OLEDs which generally suffer from severe bimolecular annihilation processes caused by the relatively long-lived triplet excitons,18 hot-exciton emitters possess pronounced advantages in the construction of efficient UV materials, attributed to their exceptionally rapid hRISC rates coupled with weakened intramolecular charge transfer (ICT) effects, which is conducive to not only mitigating the accumulation and quenching of triplet excitons but also improving good color purity in OLEDs.19 For example, She et al. introduced two boron–oxygen-embedded hot-exciton emitters,20 in which BO-bPh-based OLEDs achieved a high maximum external quantum efficiency (EQEmax) of 11.3% with an EL peak at 394 nm and a small full-width at half maximum (FWHM) of 48 nm, corresponding to high color purity with Commission Internationale de l'Eclairage (CIE) coordinates of (0.166, 0.021). Tang and Lu have independently reported UV OLEDs doped with POPCN-2CP and CTPPI,21,22 achieving EQEmax values of 7.9% and EL peaks at 396 nm, respectively, aligned with CIE coordinates of (0.166, 0.033) and (0.16, 0.04).
Effective carrier injection and transport constitute another crucial factor for achieving high-performance UV OLEDs, despite the inherent challenge posed by the wide-bandgap nature of UV emitters. A typical molecular design concept for alleviating this problem is to construct donor–acceptor (D–A) type bipolar UV emitters.23–25 Tang et al. developed a donor–acceptor–donor (D–A–D) type hot-exciton emitter 2BuCz–CNCz, achieving a EQEmax of 10.79% with an EL peak at 396 nm and a small FWHM of 33 nm.26 Recently, Liu and co-workers reported a highly efficient D–A–D type hot-exciton emitter CDFDB,27 where the doped device achieved a record EQEmax of 12% with an EL peak at 398 nm and a FWHM of 55 nm. Our group reported a series of UV and near-UV emitters based on space-crowded D–A–D type,25,28,29 and donor′–donor–acceptor (D′–D–A) type molecular structures, realizing a considerable EQEmax of 7.85% with a FWHM value of 41 nm in a doped device.30 However, although some advanced UV-OLEDs have achieved an EQEmax of around 12%, they degrade to below 5% at a brightness of 1000 cd m−2, due to the increase in exciton quenching processes at high current densities. Furthermore, it is challenging to achieve high color purity with a small CIEy coordinate while maintaining high efficiency and low roll-off in UV-OLEDs.
Herein, we present an intramolecular noncovalent bond locking strategy to assist asymmetric donor–acceptor–donor′ (D–A–D′) type emitters, embedded with a 1,4-difluorobenzene acceptor bridge, for the development of efficient UV hot-exciton materials and OLEDs. As depicted in Fig. 1, on one hand, the wide bandgap group phenyl-phenanthro[9,10-d]imidazole (PPI), an effective electron-donating unit for constructing hot-exciton emitters,31 with excellent thermal stability, high fluorescence efficiency, and large singlet–triplet energy splitting (ΔEST) was selected as the donor (D) group. Meanwhile, meta-positioned PPI derivatives can retain excellent bipolar transport properties while maintaining wide bandgap emission.19 Phenylcarbazole and its derivative (1,3-bis(N-carbazolyl)benzene) possessing weaker electron-donating ability served as the second auxiliary donor (D′). On the other hand, the target materials exhibit multiple intramolecular C–H⋯F interactions and pronounced steric hindrances induced by meta- and ortho-linkages, which not only restrict the molecular conjugation length, enabling effective short-wavelength emission, but also minimize intermolecular interactions and spectral broadening. Consequently, the asymmetric D–A–D′ type emitters, namely mPImCZ2F, mPIoCZ2F, and mPImCP2F, demonstrate superior bipolar transport capabilities and exhibit higher EL efficiencies in non-doped devices compared to the symmetric 1,4-difluorobenzene-bridged D–A–D type molecules, which utilize phenylcarbazole and its derivative as donors. The mPIoCZ2F emitter featuring an ortho-linkage in the A–D′ component emits stable UV light with an EL peak at 393 nm, achieving an impressive EQE of 6.62% together with CIE coordinates of (0.160, 0.034). Notably, in the doped device, the mPImCZ2F-based device achieves a high EQEmax of 8.13% and a value of 7.43% at 1000 cd m−2, accompanied by an EL peak at 389 nm, a FWHM of 42 nm and CIE coordinates of (0.167, 0.028). The mPIoCZ2F-based device, featuring a small FWHM of 40 nm and CIE coordinates of (0.167, 0.025), exhibits a more remarkable EQE of 8.71% and maintains a high value of 7.95% at 1000 cd m−2. To the best of our knowledge, this slight efficiency roll off represents one of the best results reported for UV-OLEDs with a CIEy value smaller than 0.04, demonstrating the effectiveness of this molecular design strategy.
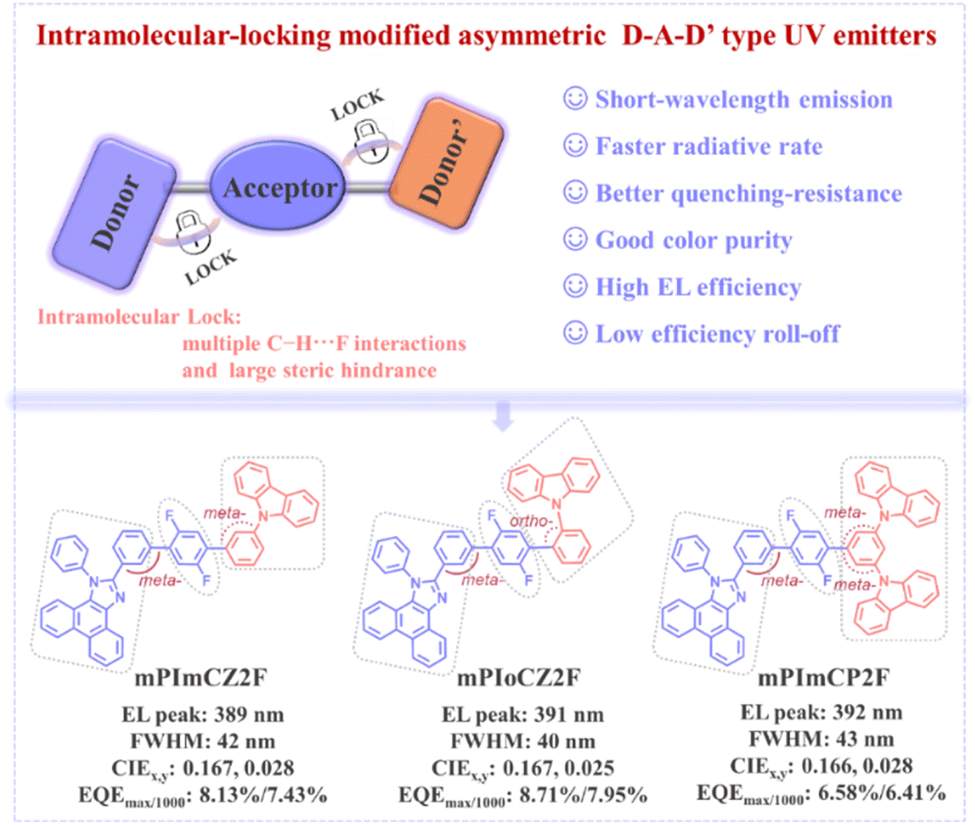 | ||
| Fig. 1 proposed molecular design strategy based on steric hindrance engineering of asymmetric D–A–D′ type hot-exciton emitters and chemical structures of mPImCZ2F, mPIoCZ2F and mPImCP2F. | ||
2 Results and discussion
2.1. Molecular synthesis and characterization
The synthesis routes for mPImCZ2F, mPIoCZ2F, and mPImCP2F are illustrated in Scheme S1.† All the compounds were obtained through the tetrakis(triphenylphosphine)palladium(0) catalyzed Suzuki cross-coupling reaction. Meanwhile, symmetric D–A–D type molecules, specifically DmCZ2F and DmCP2F, were synthesized for comparative purposes, utilizing 1,4-difluorobenzene as the acceptor and phenylcarbazole and 1,3-bis(N-carbazolyl)benzene as donors. After purification by column chromatography and temperature-gradient vacuum sublimation, the target materials were characterized by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry. The detailed procedures and structural characterization data are given in the ESI.†2.2. Theoretical calculations
To gain insight into the molecular geometries and electronic properties, density functional density (DFT) and time-dependent DFT (TD-DFT) calculations in a vacuum were performed using Gaussian 09 software at the B3LYP/6-31G(d, p) level. As depicted in Fig. 2, mPImCZ2F, mPIoCZ2F, and mPImCP2F show moderately twisted geometric configurations in the ground (S0) state, with the torsion angles ranging from 29.21–38.85° and 36.97–50.87° in the D–A and A–D′ segments, respectively. The dihedral angles between benzene and connected units in the D and D′ of target materials show a small change compared to the optimized S0 geometries of PPI and phenylcarbazole (Fig. S1†), endowing the emitters with an appropriate degree of π-conjugation. As shown by the reduced density gradient (RDG) analysis (Fig. S2†), all the emitters exhibit a considerable steric hindrance effect (brown regions) and multiple intramolecular noncovalent interactions (green regions), which can effectively reduce the structural relaxation in the excited states to enhance the fluorescence efficiency and spectral narrowing. Owing to the ortho-substitution of phenylcarbazole, mPIoCZ2F exhibits more pronounced intramolecular interactions, resulting in a significant distortion within the A–D′ segment. Meanwhile, there exists a shorter C–H⋯F distance of 2.180 Å in the D–A segment of mPIoCZ2F (Fig. S3†), which decreases the dihedral angle between PPI and difluorobenzene, consequently showing a slight reduction in the bandgap for mPIoCZ2F (3.69 eV). The calculated C–H⋯F distances in mPImCZ2F and mPImCP2F are 2.333 to 2.397 Å, shorter than their van der Waals radii (2.55 Å).27 The synergistic effect of C–H⋯F interactions and steric hindrance achieves intramolecular locking, which endows the emitters with appropriate molecular rigidity and π-conjugation length, ensuring effective luminous efficiency, while simultaneously alleviating significant issues of redshift in the emission wavelength and spectral broadening. Interestingly, mPImCZ2F, mPIoCZ2F, and mPImCP2F exhibit similar frontier molecular orbital (FMO) distributions. The highest occupied molecular orbitals (HOMOs), unlike those in symmetrical molecules (Fig. S4†), are predominantly localized on the PPI unit without extending towards the N-linked benzene ring, while the lowest unoccupied molecular orbitals (LUMOs) are primarily distributed on the 1,4-difluorobenzene acceptor and connected benzene rings, resulting in an effective orbital overlap in the C-linked benzene of the PPI unit. Additionally, the HOMO−1 distributions of all the emitters are mainly concentrated on the D′ component, whereas their LUMO+1 distributions are mostly delocalized across the entire PPI segment with only minor residuals on the central terphenyl unit, due to the electron-withdrawing ability of PPI induced by the sp2 hybridized nitrogen atom. | ||
| Fig. 2 Molecular geometries in the ground state, FMO distributions and calculated energy levels of mPImCZ2F, mPIoCZ2F and mPImCP2F. | ||
Natural transition orbitals (NTOs) analysis for mPImCZ2F, mPIoCZ2F, and mPImCP2F were carried out to investigate their excited states properties using the Multiwfn program.32 As shown in Fig. S5–S7,† and summarized in Tables S1–S3,† mPImCZ2F, mPIoCZ2F, and mPImCP2F exhibit extremely similar hole and particle distributions in the S0 → S1 transition; their distributions overlap well in the C-linked benzene of the PPI unit (overlap integrals of 0.2740, 0.2714, and 0.2841 for mPImCZ2F, mPIoCZ2F, and mPImCP2F) while showing some separation in the D–A section. This result suggests the hybridization between LE and CT states in their S1 states, showing the typical HLCT characteristic.33–38 Meanwhile, their S2 states exhibit HLCT characteristics with notable orbital overlap on the phenylene within the A–D′ segment, accompanied by some spatial separations between the difluorobenzene and carbazole units. Nevertheless, the hole and particle distributions of their T1 states demonstrate substantial overlap primarily on the PPI unit, indicating that these T1 states are LE-dominated excited states. The calculated energy splits (ΔES1T1) between T1 and S1 are as large as 0.68, 0.60, and 0.64 eV for mPImCZ2F, mPIoCZ2F, and mPImCP2F, respectively, making it difficult to achieve an effective RISC channel from T1 to S1. Interestingly, the high-lying T5 and T6 states for mPImCZ2F, T4, T5, T6, and T7 states for mPIoCZ2F, and the T7 state for mPImCP2F exhibit HLCT characteristics where the CT components in these high-lying triplet states could facilitate triplet-to-singlet conversion. As depicted in Fig. S8,† mPImCZ2F exhibits smaller energy gaps between S1/S2 and T5/T6 states than ΔES1T1, while the spin–orbit coupling (SOC) matrix elements are larger than those between T1 and S1. The S1 state of mPIoCZ2F shows smaller energy gaps (−0.041–0.126 eV) and considerable SOC values (0.095–0.207 cm−1) with T4–7 states, whereas mPImCP2F exhibits the energy gaps of −0.144–0.055 eV and SOC constants of 0.251–0.476 cm−1. In accordance with Fermi's golden and Kasha's rules,39 these observations can facilitate the hRISC processes occurring at higher excited states, potentially boosting the utilization of triplet excitons in the EL process. The larger SOC values of mPIoCZ2F and mPImCP2F, compared to those of mPImCZ2F, suggest that the hRISC processes in mPIoCZ2F and mPImCP2F are significantly more efficient, resulting in a higher exciton utilization efficiency in the OLEDs.
2.3. Photophysical properties
To investigate the photophysical properties, ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectra of mPImCZ2F, mPIoCZ2F, and mPImCP2F, were measured in diluted toluene solution (10 μM) at room temperature (Fig. 3a), and the detailed data are summarized in Table 1. All the three molecules exhibit very similar absorption profiles, where the absorption peaks observed at 290 and 363 nm correspond to the n–π* transition of the carbazole group and π–π* transition of the PPI unit, and the absorption bands at around 320 nm can be ascribed to the π–π* transition of the phenylcarbazole group.40,41 The optical band gaps (Egs) are estimated to be 3.35, 3.32 and 3.32 eV for mPImCZ2F, mPIoCZ2F, and mPImCP2F, respectively, based on the onset of their absorption spectra. Unlike the reference molecules DmCZ2F and DmCP2F exhibiting broad, structureless PL profiles (Fig. S9†), mPImCZ2F, mPIoCZ2F, and mPImCP2F exhibit similar PL spectra with obvious vibronic fine structures and UV emissions at 369 and 388 nm for mPImCZ2F, 371 and 390 nm for mPIoCZ2F, 371 and 390 nm for mPImCP2F, indicating LE state dominated emission. It is noteworthy that the fine structure of their PL spectra closely resembles that of the PPI unit (with emission peaks at 369 and 387 nm, Fig. S9b†), suggesting that the PL spectra of mPImCZ2F, mPIoCZ2F, and mPImCP2F could be attributed to the radiative transition of the PPI unit, resulting in exceptionally high photoluminescence quantum yields (PLQYs) exceeding 70% in solution. Moreover, all of them exhibit good color purity with narrow FWHMs of 33, 31, and 40 nm for mPImCZ2F, mPIoCZ2F, and mPImCP2F. Based on the emission peaks observed in the low-temperature fluorescence and phosphorescence spectra measured in frozen toluene solution at 77 K (Fig. 3b), the energy levels of the S1 and T1 states are estimated to range from 3.20 to 3.35 eV and 2.40 to 2.59 eV, respectively, yielding the following sequence of ΔES1T1 values: mPImCZ2F (0.61 eV) < mPImCP2F (0.72 eV) < mPIoCZ2F (0.95 eV). This implies that the intersystem crossing (ISC) process from S1 to T1 is potentially the least efficient in mPIoCZ2F, as a larger ΔES1T1 generally renders ISC less likely. Meanwhile, the large ΔES1T1 values exceeding 0.6 eV, coupled with the nanosecond-scale lifetimes observed in solution (Fig. S10†), conclusively rule out the TADF mechanism.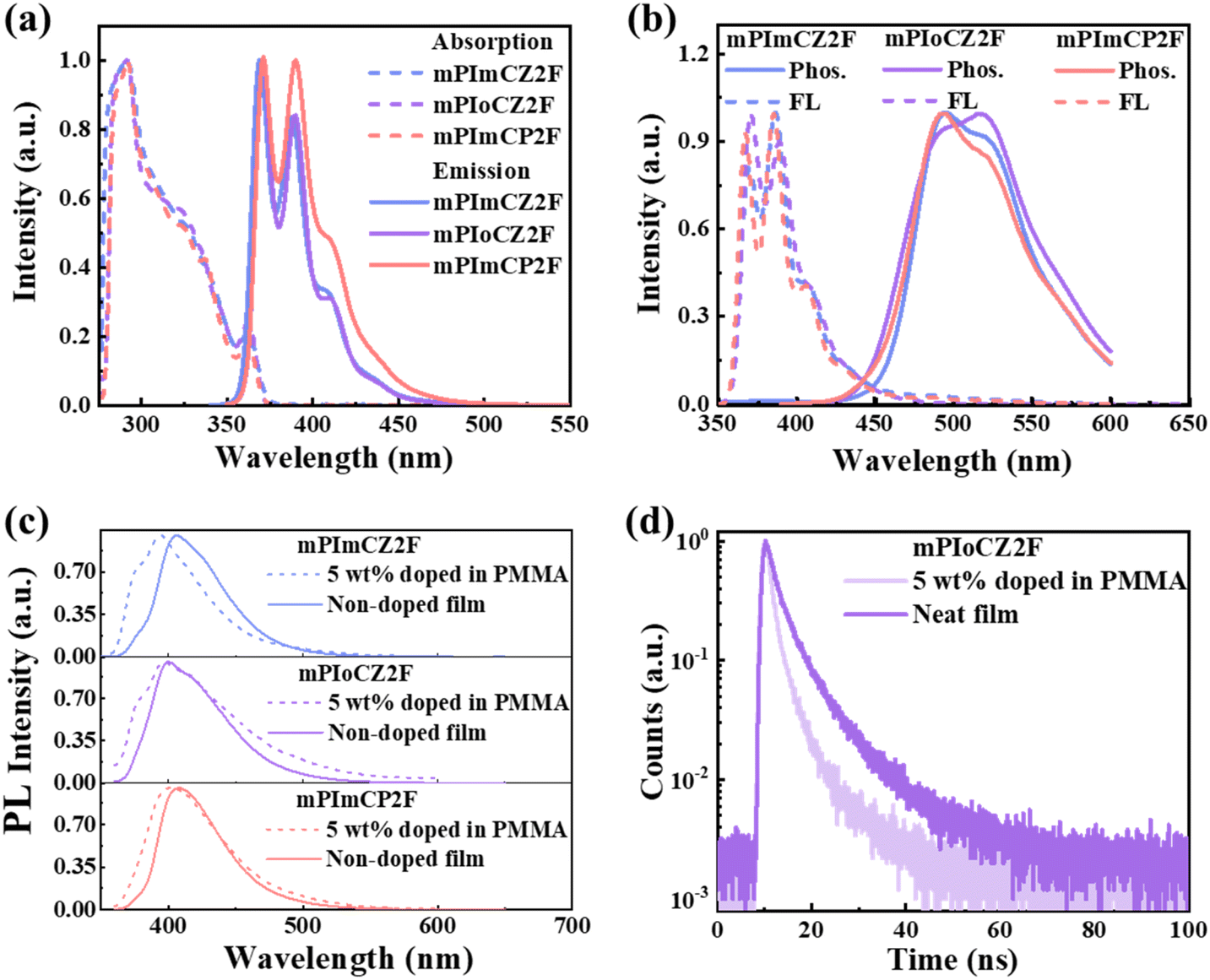 | ||
| Fig. 3 (a) Absorption and PL spectra of mPImCZ2F, mPIoCZ2F, and mPImCP2F in dilute toluene solution (10 μM) at room temperature. (b) Low-temperature fluorescence (FL) and phosphorescence (Phos.) spectra in ultra-cooled dilute toluene at 77 K. (c) PL spectra of mPImCZ2F, mPIoCZ2F, and mPImCP2F in neat films and doped films where they were dispersed in a polymethyl methacrylate (PMMA) matrix at a doping ratio of 5 wt%. (d) Transient PL decay spectra of neat and doped films for mPIoCZ2F. | ||
| Compound | λPLa [nm] | FWHMa [nm] | ΦPLa [%] | λfb [nm] | Φfb [%] | Egc [eV] | HOMO/LUMOd [eV] | ES1/ET1/ΔES1T1e [eV] | Tdf [°C] |
|---|---|---|---|---|---|---|---|---|---|
| a λPL, FWHM, ΦPL: emission peak, full width at half maximum, and PLQY measured in THF solution at 300 K.b λf and Φf: Emission peak and PLQY measured in non-doped and doped films at 300 K.c Eg: optical bandgap calculated from the absorption onset.d HOMO energy level: obtained from CV measurement. LUMO energy level: calculated using ELUMO = EHOMO + Eg.e ES1 and ET1: the energy levels of S1 and T1 estimated from the peaks of low-temperature fluorescence and phosphorescence spectra at 77 K, ΔES1T1 = ES1 − ET1.f Td: decomposition temperature at 5% weight loss measured by TGA. | |||||||||
| mPImCZ2F | 369, 388 | 33 | 83 | 407/394 | 34/70 | 3.35 | −5.31/−1.96 | 3.20/2.59/0.61 | 457 |
| mPIoCZ2F | 371, 390 | 31 | 72 | 400/396 | 53/48 | 3.32 | −5.32/−2.00 | 3.35/2.40/0.95 | 437 |
| mPImCP2F | 371, 390 | 40 | 76 | 408/402 | 35/54 | 3.32 | −5.37/−2.05 | 3.22/2.50/0.72 | 463 |
Furthermore, the photophysical properties of their neat and doped films were investigated, where they were dispersed in a polymethyl methacrylate matrix at a doping ratio of 5 wt%. Owing to the meta-connection mode in the D–A segment, the fluorescence emission of all the doped and neat films occurs in the near-UV and UV regions with the emission peaks located in the 394 to 408 nm range. As the substitution of the D′ unit is at the ortho-position, the intramolecular conjugation and planar rigidity of mPIoCZ2F are effectively restricted, leading to a slight emission blueshift. The PLQY values of asymmetrical molecules in both neat and doped films are higher than those of symmetrical molecules (Table 1 and S4†), with the PLQYs of mPImCZ2F, mPIoCZ2F, and mPImCP2F determined to be 34%, 53%, and 35% in neat films, and 70%, 48% and 54% in doped films, respectively. Meanwhile, the transient PL decay curves of their doped and neat films reveal that all the three emitters exhibit double-exponential compositions containing a fast-decay component with lifetimes of 1.30–3.88 ns and a long-lived component with lifetimes of 6.01–8.35 ns (Fig. 3d and S11†). The former can be attributed to prompt fluorescence emanating from singlet excitons, whereas the latter can be identified as delayed fluorescence resulting from hRISC processes, which convert triplet excitons into singlet excitons. This finding aligns well with theoretical calculations. Combining the PLQYs, the detailed kinetic parameters of their neat and doped films are subsequently calculated and summarized in Table S5.† It is noteworthy that the calculated rates of the hRISC process for mPImCZ2F, mPIoCZ2F, and mPImCP2F exceed 2 × 107 s−1, surpassing those of high efficiency TADF materials.42,43 Compared with mPImCZ2F and mPImCP2F in neat films, mPIoCZ2F not only exhibits a higher radiative transition rate (kr) exceeding 1.1 × 108 s−1 but also achieves a lower intersystem crossing rate, which can boost the triplet exciton utilization in non-doped OLEDs. Moreover, all the emitters exhibit higher krs in doped films, which is attributed to the suppression of the aggregation effect and enhanced molecular rigidity induced by intramolecular locking interaction.
To further reveal their excited state properties, the PL spectra in different-polarity solvents were measured (Fig. S12†). As the solvent polarity increases from n-hexane to ethyl ether, the fine structure of PL spectra of mPImCZ2F, mPIoCZ2F, and mPImCP2F gradually becomes blurred, while the emission peak exhibits minimal shifts. When the solvent polarity further increases from ethyl ether to acetonitrile, although the vibration fine structure still exists in the short-wavelength region (≤400 nm), the PL spectra of these three materials exhibit distinct characteristics in the long-wavelength region (410–500 nm). In highly polar solvents like N,N-dimethylacetamide (DMF), acetone and acetonitrile, the emission of mPImCZ2F is stronger than that of mPIoCZ2F in the long-wavelength region, while mPImCP2F exhibits a broad and structureless emission profile with peaks at 454–464 nm. This indicates that the S1 excited states of all three emitters are dominated by a LE state, accompanied by a certain proportion of the CT component. Furthermore, the proportion of the CT component gradually increases in the sequence of mPIoCZ2F, mPImCZ2F, and mPImCP2F. However, the S1 states of the symmetrical molecules DmCZ2F and DmCP2F exhibit obvious CT sate characteristics, as evidenced by significant emission redshifts of 86 nm and 88 nm, respectively, when the solvent polarity increases from n-hexane to acetonitrile. These results demonstrate that the asymmetrical D–A–D′ structure, assisted by steric hindrance, can effectively modulate the LE/CT components, achieving substantial fluorescence efficiency and high color purity while maintaining UV emission.
2.4. Thermal, electrochemical and charge transport properties
The thermal characteristics of mPImCZ2F, mPIoCZ2F, and mPImCP2F were assessed through thermogravimetric analysis (TGA). As depicted in Fig. S13,† their decomposition temperatures (Tds) at 5% weight loss are notably high, reaching 457, 437 and 463 °C, respectively, which are advantageous for their application in OLEDs utilizing the vacuum thermal deposition process. According to the onsets of oxidation potentials measured by cyclic voltammetry (CV) using ferrocene as the reference (Fig. S14†), the HOMO energy levels of mPImCZ2F, mPIoCZ2F, and mPImCP2F are calculated to be −5.31, −5.32 and −5.37 eV. The LUMO energy levels are determined to be −1.96, −2.00, and −2.05 eV, respectively, using the equation of LUMO = HOMO + Eg.To deeply understand the bipolar transport capacity of these asymmetric D–A–D′ type emitters, electron-only and hole-only devices (EODs and HODs) for mPImCZ2F, mPIoCZ2F, and mPImCP2F were fabricated. It can be observed from current density–voltage curves (Fig. S15†) that the hole or electron currents of EODs and HODs increase significantly as the voltage increases, indicting that all three emitters exhibit a notable bipolar transport characteristic. The charge mobility that is dependent on the electric field can be calculated through the application of the space-charge-limited current (SCLC) method, utilizing the Mott–Gurney and Poole–Frenkel formulae.23,44–46 As presented in Fig. 4, S16 and Table S6,† compared to the symmetrical molecules DmCZ2F and DmCP2F, mPImCZ2F, mPIoCZ2F, and mPImCP2F demonstrate electron and hole mobilities that are two orders of magnitude higher. Under an electric field of 5 × 105 V cm−1, the electron mobilities reach 2.31 × 10−5, 1.49 × 10−5, and 2.21 × 10−5 cm2 V−1 s−1 for mPImCZ2F, mPIoCZ2F, and mPImCP2F, respectively. Meanwhile, mPIoCZ2F exhibits a higher hole mobility of 4.29 × 10−6 cm2 V−1 s−1 compared to mPImCZ2F (hole mobility: 1.95 × 10−7 cm2 V−1 s−1) and mPImCP2F (hole mobility: 1.32 × 10−6 cm2 V−1 s−1), demonstrating balanced charge carriers and a wide recombination zone during the EL process. Consequently, it can effectively mitigate exciton–exciton quenching, singlet-polaron annihilation (SPA), and triplet-polaron annihilation (TPA), thereby achieving high-brightness performance in OLEDs. Notably, compared to some reported emitters based on the para-position of the PPI unit,31,44 the meta-linkage of the PPI unit in the D–A segment not only limits the molecular conjugation length but also preserves the high electron and hole mobilities, which is favourable for constructing highly bipolar UV materials and non-doped devices with low efficiency roll-off.
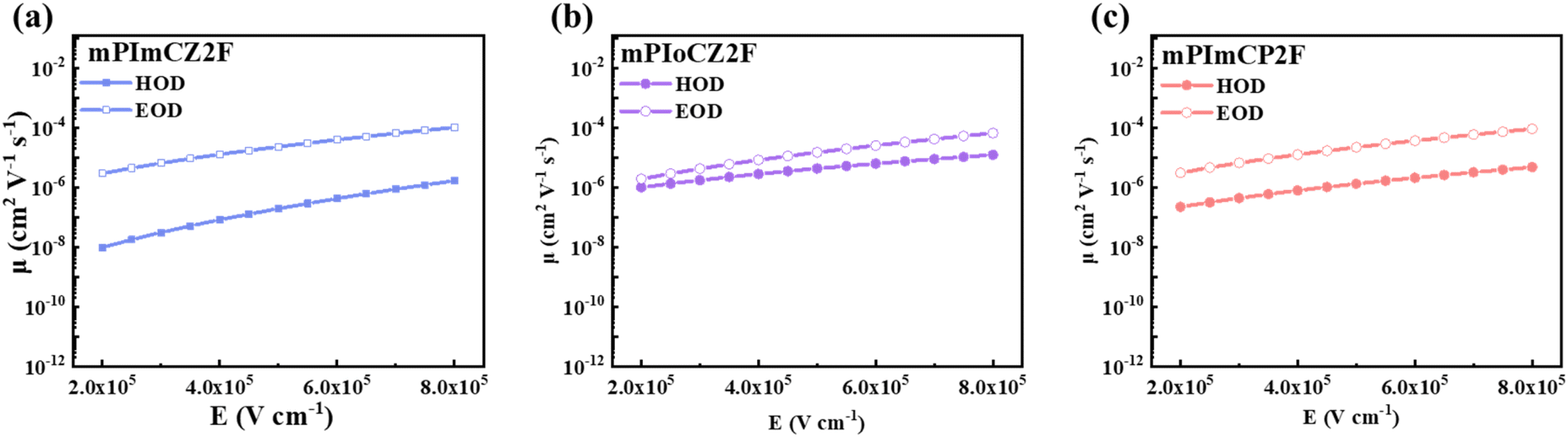 | ||
| Fig. 4 Charge mobilities versus electric field curves for mPImCZ2F (a), mPIoCZ2F (b), and mPImCP2F (c), respectively. | ||
2.5. EL performance
Enlightened by their excellent performance in potential hRISC channel and exceptional bipolar transport, we further evaluated the EL performance of mPImCZ2F, mPIoCZ2F, mPImCP2F in non-doped OLEDs with the device architecture of ITO/HATCN (20 nm)/TPAC (45 nm)/TCTA (10 nm)/emissive layer/TPBi (30 nm)/LiF (1 nm)/Al (100 nm), in which indium tin oxide (ITO) and aluminum (Al) serve as the anode and cathode; hexaazatriphenylenehexacarbonitrile (HATCN) and lithium fluoride (LiF) function as the hole- and electron-injection layers; tris(4-carbazoyl-9-ylphenyl)amine (TCTA) serves as an exciton-blocking layer; di-(4-(N,N-ditolyl-amino)-phenyl)cyclohexane (TAPC) and 1,3,5-tris (1-phenyl-1H-benzimidazol-2-yl) benzene (TPBi) function as the hole- and electron-transport layers, respectively. The optimized emissive layers for N1, N2, and N3 devices are respectively composed of 20 nm thick mPImCZ2F, 20 nm thick mPIoCZ2F, and 15 nm thick mPImCP2F. Meanwhile, the reference devices (N4 and N5) were fabricated using 20 nm thick DmCZ2F and DmCP2F as emissive layers. The energy level alignment and chemical structure of the materials used in the OLEDs are shown Fig. S17.† The EL performances of non-doped devices are shown in Fig. 5, S18, and S19† and summarized in Table 2.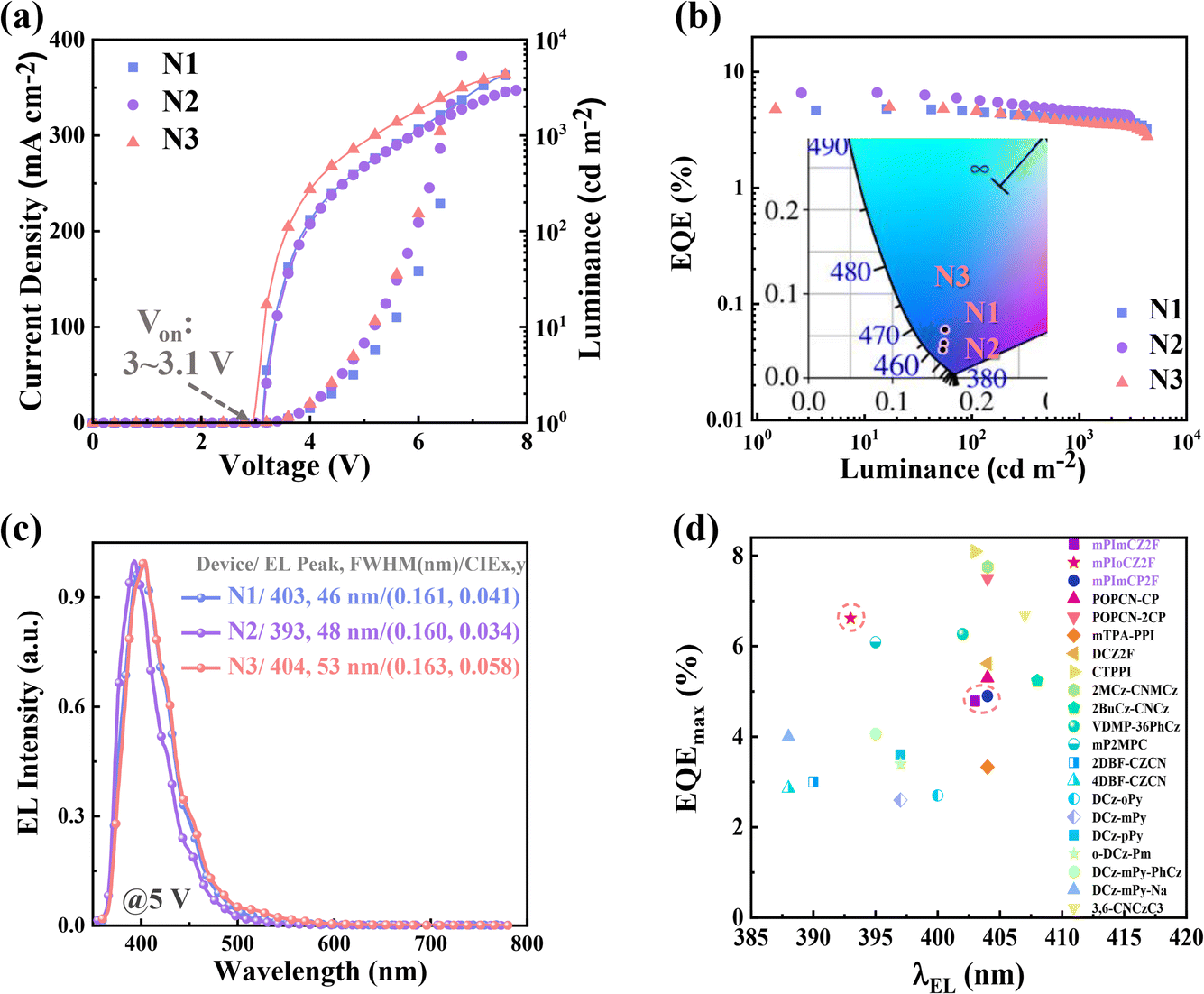 | ||
| Fig. 5 (a) Current density–voltage–luminance characteristics of the devices N1, N2, and N3. (b) EQE versus luminance curves of the devices N1, N2, and N3 (Inset: CIE coordinates at 5 V). (c) EL spectra of the devices N1, N2, and N3 at 5 V. (d) Summary of the EQEmax values of representative non-doped fluorescent OLEDs with an EL peak wavelength ≤ 410 nm, and the details are listed in Table S7.† | ||
| Devices | EML | Von/1000a [V] | CEmax/1000b [cd A−1] | PEmax/1000c [lm W−1] | EQEmax/1000d [%] | J90e [mA cm−2] | λELf [nm] | FWHMg [nm] | CIE (x, y)h |
|---|---|---|---|---|---|---|---|---|---|
| a Von/1000: turn-on voltage at the luminescence of 1 and 1000 cd m−2.b CEmax/1000: maximum current efficiency and the value taken at 1000 cd m−2.c PEmax/1000: maximum power efficiency and the value taken at 1000 cd m−2.d EQEmax/1000: maximum EQE and the value taken at 1000 cd m−2.e J90: the current density at which the external quantum efficiency (EQE) drops to 90%.f λEL: emission peak in the EL spectrum.g FWHM: full width at half maximum of the EL spectrum at 5 V.h CIE: Commission International de l'Éclairage (CIE) coordinates at 5 V. | |||||||||
| N1 | mPImCZ2F | 3.1/5.8 | 0.97/0.74 | 0.95/0.40 | 4.80/3.90 | 27 | 403 | 46 | (0.161, 0.041) |
| N2 | mPIoCZ2F | 3.1/6.0 | 0.83/0.52 | 0.81/0.28 | 6.62/4.55 | 11 | 393 | 48 | (0.160, 0.034) |
| N3 | mPImCP2F | 3.0/5.2 | 5.50/0.95 | 5.75/0.57 | 4.99/3.67 | 8 | 404 | 53 | (0.163, 0.058) |
| N4 | DmCZ2F | 4.0/7.9 | 0.58/0.57 | 0.23/0.23 | 2.96/2.80 | 5.4 | 403 | 52 | (0.168, 0.040) |
| N5 | DmCP2F | 3.4/— | 0.86/— | 0.80/— | 1.35/— | — | 412 | 61 | (0.167, 0.081) |
| D1 | CBP:mPImCZ2F | 3.5/6.9 | 0.50/0.50 | 0.23/0.22 | 8.13/7.43 | 238 | 389 | 42 | (0.167, 0.028) |
| D2 | CBP:mPIoCZ2F | 3.5/6.6 | 0.47/0.47 | 0.23/0.22 | 8.71/7.95 | 232 | 391 | 40 | (0.167, 0.025) |
| D3 | CBP:mPImCP2F | 3.4/6.6 | 0.48/0.48 | 0.24/0.23 | 6.58/6.41 | 299 | 392 | 43 | (0.166, 0.028) |
All the non-doped devices (N1–3) with asymmetrical D–A–D′ type molecules as emitters exhibit turn-on voltages of 3.0–3.1 V (Fig. 5a), lower than the Eg values, indicating effective charge carrier injection, transport, and recombination in these devices. This can be attributed to the stepped progression of the HOMO energy level, the low injection energy barrier, and the bipolar transport capacities of the emitters. The maximum luminance values of 4302, 2968, and 4253 cd m−2 are achieved in the devices N1, N2, and N3, respectively. It is worth noting that the EL performances of devices N1–3 are far superior to those of devices N4–5 based on DmCZ2F and DmCP2F. The device N2 based on mPIoCZ2F achieves the most outstanding EL performance with an EQEmax value of 6.62%, while the EQEmax values of the devices N1 and N3 based on mPImCZ2F and mPImCP2F are 4.80% and 4.99%, respectively (Fig. 5b). At the luminance levels of 100 and 1000 cd m−2, the EQE values remain 5.75% and 4.55% in the N2 device, which are superior to the EQE values of N1 and N3 devices. This can be attributed to the high and balanced hole and electron mobilities of mPIoCZ2F. The non-doped devices N1 and N3 exhibit stable deep-blue emission with the CIE coordinates of (0.161, 0.041) and (0.163, 0.058) (Fig. 5b), which are close to the broadcast service television 2020 and 709 blue standards CIE coordinates of (0.131, 0.046) and (0.15, 0.06) for ultra-high-definition and high-definition displays. More amazingly, the mPIoCZ2F-based device N2 emits stable UV light with an EL peak at 393 nm and a FWHM of 48 nm, corresponding to the CIE coordinates of (0.160, 0.034) (Fig. 5b and c). To our knowledge, the EL performance of non-doped device N2 is one of the best results for non-doped UV-OLEDs with an EL peak smaller than 400 nm (Fig. 5d, and Table S7†).
As is well known, the host–guest doping technique is beneficial for suppressing the concentration quenching effect, thereby reducing efficiency roll-off,47–49 while promoting the blue shift of emission spectra. Compared to the neat films, the doped films exhibited exceptional values of kr and PLQY, prompting us to delve into the investigation of their EL performance in doped OLEDs. With the device structure of ITO/HATCN (20 nm)/TPAC (45 nm)/TCTA (10 nm)/emissive layer (20 nm)/TPBi (30 nm)/LiF (1 nm)/Al (100 nm), the doped OLEDs (D1, D2, and D3) were optimized and fabricated by doping 5 wt% mPImCZ2F, 10 wt% mPIoCZ2F, 10 wt% mPImCP2F respectively into the 4,4′-bis(N-carbazolyl)−1,1′-biphenyl (CBP) host matrix. EL performances of doped devices are shown in Fig. 6, S20, and S21† and summarized in Table 2.
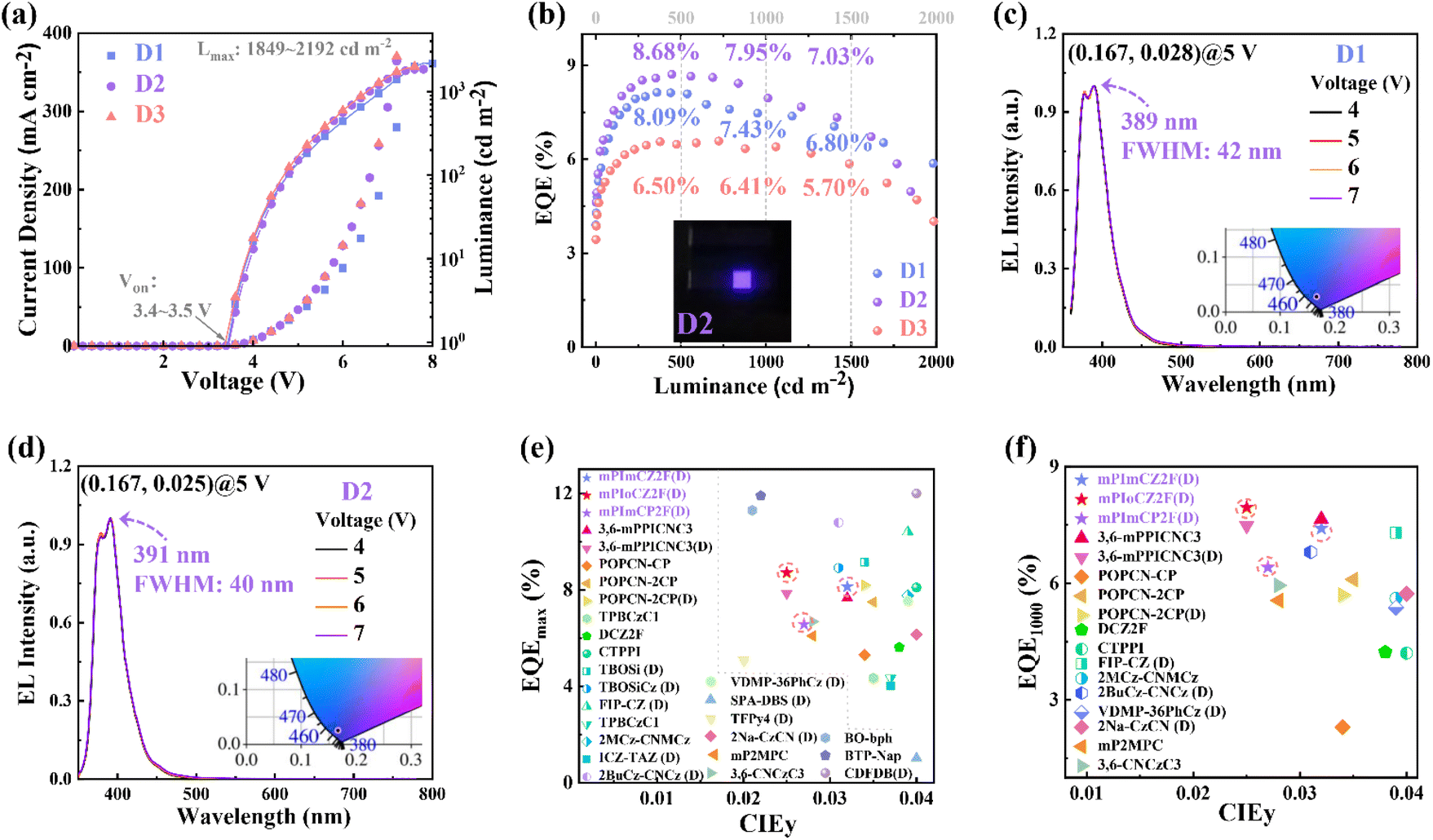 | ||
| Fig. 6 (a) Current density–voltage–luminance characteristics of the devices D1, D2, and D3. (b) EQE versus luminance curves of the devices D1, D2, and D3 (Inset Photograph of the device D2). (c and d) EL spectra of the devices D1 and D2 at different voltages (Inset CIE coordinates at 5 V). (e and f) Summary of EQEmax and EQE at 1000 cd m−2 for the representative reported non-doped and doped (D) fluorescent OLEDs with a CIEy ≤ 0.04, and the details are listed in Table S8.† | ||
As depicted in Fig. 6 and S20,† all the three doped devices not only exhibit exceptional spectral stability at voltages ranging from 4 to 7 V, but also emit UV light with the emission maxima/FWHM of 389/42, 391/40, and 392/43 nm for D1, D2, and D3, respectively, corresponding to ascendant color purity with the CIE coordinates of (0.167, 0.028), (0.167, 0.025) and (0.166, 0.028). The narrow emission could be attributed to the cooperative effect between large steric hindrance and intramolecular C–H⋯F interactions, which restricts the structural relaxation. Excitedly, compared to non-doped devices, the EL efficiencies have achieved significant enhancement, and the magnitude of efficiency roll-off at high brightness levels has also been notably improved. The turn-on voltages of 3.4–3.5 V and the maximum luminance of 1849–2192 cd m−2 are achieved. The devices D1, D2, and D3 exhibit EQEmax values of 8.13%, 8.71%, and 6.58% at 360, 446, and 722 cd m−2, respectively, maintaining high EQEmax values of 7.43%, 7.95%, and 6.41% at a brightness of 1000 cd m−2. Even at a high brightness of 1500 cd m−2, the devices D1–3 still maintain 6.80%, 7.03%, and 5.70%, respectively. Here, J90 is defined as the current density at which the EQEmax falls to 90%,50 which is used to evaluate the efficiency roll-off of the OLEDs. The J90 values of devices D1, D2, and D3 are calculated to be 238, 232, and 299 mA cm−2, respectively, which are superior to those of the vast majority of fluorescent and phosphorescent OLEDs.50 Such excellent EL efficiencies at high brightness levels can be ascribed to the reasonable device structure, high hRISC rates, and enhanced krs. To our knowledge, although the EQEmax attained by the mPIoCZ2F-based doped device D2 does not surpass the highest value reported for UV-OLEDs with a CIEy of ≤0.04, the EQE of 7.95% achieved at 1000 cd m−2 remains one of the highest values recorded in such UV-OLEDs (Fig. 6e and f).
Assuming a light outcoupling efficiency of 20–30% and balanced injection of electrons and holes in the devices, the exciton utilization efficiencies (EUEs) of D1, D2, and D3 are calculated to be 39–58%, 60–91%, and 41–61%, exceeding the upper limit of exciton utilization of 25% for traditional fluorescent OLEDs.51 The single-exponential decay lifetimes in the nanosecond range of three emitters can exclude the TADF mechanism, while the triplet–triplet annihilation (TTA) mechanism can be ruled out by the large ΔES1T1 values and a good linear relationship between the luminance and current density (Fig. S22†). Based on the analysis of theoretical calculations and photophysical properties, the primary mechanism underlying their exceptional EL efficiencies should be the “hot-exciton” mechanism, which involves the hRISC process from high-energy triplet to singlet states. The high EUE achieved in device D2 can be attributed to the combined effects of a faster radiative transition rate (kr) exceeding 2 × 108 s−1, multi-channel hRISC, and an inefficient ISC process from S1 to T1 in the mPIoCZ2F molecule.
3 Conclusions
In summary, by incorporating an intramolecular noncovalent bond locking strategy into an asymmetric D–A–D′ molecular structure, three exceptional hot-exciton UV materials, specifically mPImCZ2F, mPIoCZ2F, and mPImCP2F, were successfully designed and synthesized. All the three emitters exhibited excellent thermal stability, enhanced photoluminescence efficiency, and good bipolar transport capability, while maintaining effective UV emission and high hRISC rates. Compared to mPImCZ2F and mPImCP2F, mPIoCZ2F with its ortho-linkage in the A–D′ segment exhibits fast kr and an inefficient ISC process, achieving a commendable EQE of 6.62% with CIE coordinates of (0.160, 0.034) in non-doped OLEDs. Furthermore, the doped device utilizing mPIoCZ2F attains an exceptional EQE of 8.71% with CIE coordinates of (0.167, 0.025), maintaining a robust value of 7.95% at 1000 cd m−2 with a low efficiency roll-off. This represents a record-breaking achievement among UV-OLEDs with a CIEy ≤ 0.04 at a high brightness level. This work underscores the significant potential of the steric hindrance-assisted asymmetric D–A–D′ molecular design approach for the development of efficient UV emitters and high-performance OLEDs with high efficiency, low roll-off, and excellent color purity.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Wang, S., Zhang, R., Ding, R., Huang, H., Qi, H., and Ying, S. – conceptualization, investigation, OLED devices, writing – original draft; Wang, S., Huang, H., and Qi, H. – theoretical calculations, investigation; Wang, S., Huang, H., Qi, H., and Ying, S. – formal analysis, OLED devices; Wang, S., Zhang, R., Ding, R., Huang, H., and Qi, H. – investigation (thermal properties); Wang, S., Huang, H., and Qi, H. – investigation (transient PL properties); Liu, Y., Ying, S., Ma, D., and Yan, S. – project administration, supervision; Ying, S., Ma, D., and Yan, S. – writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Natural Science Foundation of Shandong Provincial (no. ZR2023QE078, ZR2022ZD37, and ZR2019ZD50), the Natural Science Foundation of Qingdao (no. 23-2-1-75-zyyd-jch), the National Natural Science Foundation of China (no. 52002804, 52103220, 52103017, and 22022501), and the Science and Technology Support Plan for Youth Innovation of Colleges and Universities in Shandong Province (2023KJ097).Notes and references
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- V. Jankus, C. J. Chiang, F. Dias and A. P. Monkman, Adv. Mater., 2013, 25, 1455–1459 CrossRef CAS PubMed.
- C. K. Moon, K. Suzuki, K. Shizu, C. Adachi, H. Kaji and J. J. Kim, Adv. Mater., 2017, 29, 1606448 CrossRef PubMed.
- M. Y. Lai, C. H. Chen, W. S. Huang, J. T. Lin, T. H. Ke, L. Y. Chen, M. H. Tsai and C. C. Wu, Angew. Chem., Int. Ed., 2008, 47, 581–585 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- X. Qiu, S. Ying, C. Wang, M. Hanif, Y. W. Xu, Y. Li, R. Y. Zhao, D. H. Hu, D. G. Ma and Y. G. Ma, J. Mater. Chem. C, 2019, 7, 592–600 RSC.
- S. Ying, Y. B. Wu, Q. Sun, Y. F. Dai, D. Z. Yang, X. F. Qiao, J. S. Chen and D. G. Ma, Appl. Phys. Lett., 2019, 114, 033501 CrossRef.
- S. Ying, Q. Sun, Y. F. Dai, D. Z. Yang, X. F. Qiao and D. G. Ma, Mater. Chem. Front., 2019, 3, 640–649 RSC.
- D. Wan, J. Zhou, Y. Yang, G. Meng, D. Zhang, L. Duan and J. Ding, Adv. Mater., 2024, 36, 2409706 CrossRef CAS PubMed.
- Y. T. Lee, C. Y. Chan, N. Matsuno, S. Uemura, S. Oda, M. Kondo, R. W. Weerasinghe, Y. Hu, G. N. I. Lestanto, Y. Tsuchiya, Y. Li, T. Hatakeyama and C. Adachi, Nat. Commun., 2024, 15, 3174 CrossRef CAS PubMed.
- M. Hayakawa, X. Tang, Y. Ueda, H. Eguchi, M. Kondo, S. Oda, X. C. Fan, G. N. Iswara Lestanto, C. Adachi and T. Hatakeyama, J. Am. Chem. Soc., 2024, 146, 18331–18340 CrossRef CAS PubMed.
- H. Wang, J. X. Chen, Y. Z. Shi, X. Zhang, L. Zhou, X. Y. Hao, J. Yu, K. Wang and X. H. Zhang, Adv. Mater., 2023, 36, 2307725 CrossRef PubMed.
- L. Liang, C. Qu, X. Fan, K. Ye, Y. Zhang, Z. Zhang, L. Duan and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316710 CrossRef CAS PubMed.
- W. Yuan, Q. Jin, M. Du, L. Duan and Y. Zhang, Adv. Mater., 2024, 36, 2410096 CrossRef CAS PubMed.
- S. Chen and H. Xu, Chem. Soc. Rev., 2021, 50, 8639 RSC.
- M. Shimizu and T. Sakurai, Aggregate, 2021, 3, e144 CrossRef.
- Y. Xu, X. Liang, X. Zhou, P. Yuan, J. Zhou, C. Wang, B. Li, D. Hu, X. Qiao, X. Jiang, L. Liu, S.-J. Su, D. Ma and Y. Ma, Adv. Mater., 2019, 31, 1807388 CrossRef PubMed.
- Y. Luo, S. Li, Y. Zhao, C. Li, Z. Pang, Y. Huang, M. Yang, L. Zhou, X. Zheng, X. Pu and Z. Lu, Adv. Mater., 2020, 32, 2001248 CrossRef CAS PubMed.
- L. Peng, J. C. Lv, S. Xiao, Y. M. Huo, Y. C. Liu, D. G. Ma, S. Ying and S. K. Yan, Chem. Eng. J., 2022, 450, 138339 CrossRef CAS.
- G. Li, K. Xu, J. Zheng, X. Fang, W. Lou, F. Zhan, C. Deng, Y.-F. Yang, Q. Zhang and Y. She, J. Am. Chem. Soc., 2024, 146, 1667–1680 CrossRef CAS PubMed.
- J. Chen, H. Liu, J. Guo, J. Wang, N. Qiu, S. Xiao, J. Chi, D. Yang, D. Ma, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202116810 CrossRef CAS PubMed.
- Z. Zhong, X. Zhu, X. Wang, Y. Zheng, S. Geng, Z. Zhou, X. J. Feng, Z. Zhao and H. Lu, Adv. Funct. Mater., 2022, 32, 2112969 CrossRef CAS.
- J. C. Lv, S. P. Song, J. Li, L. Peng, Y. Z. Li, Y. C. Liu, D. G. Ma, S. Ying and S. K. Yan, Adv. Opt. Mater., 2024, 12, 2301413 CrossRef CAS.
- H. Y. Qi, S. N. Wang, Z. X. Gao, D. Y. Xie, J. Li, Y. C. Liu, S. F. Xue, S. Ying, D. G. Ma and S. K. Yan, ACS Mater. Lett., 2024, 6, 3844 CrossRef CAS.
- Y. M. Huo, L. Peng, S. Y. He, R. Chen, H. Huang, F. Y. He, Y. W. Ming, Y. C. Liu, S. Ying and S. K. Yan, Dyes Pigm., 2024, 226, 112128 CrossRef CAS.
- H. Zhang, G. Li, X. Guo, K. Zhang, B. Zhang, X. Guo, Y. Li, J. Fan, Z. Wang, D. Ma and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 22241–22247 CrossRef CAS PubMed.
- K. Zhang, Z. Zhou, D. Liu, Y. Chen, S. Zhang, J. Pan, X. Qiao, D. Ma, S. Su, W. Zhu and Y. Liu, Angew. Chem., Int. Ed., 2024, 63, e202407502 CrossRef CAS PubMed.
- Y. Huo, H. Qi, S. He, J. Li, S. Song, J. Lv, Y. Liu, L. Peng, S. Ying and S. Yan, Aggregate, 2023, 226, 112128 Search PubMed.
- Y. Huo, J. Lv, Y. Xie, L. Hua, Y. Liu, Z. Ren, T. Li, S. Ying and S. Yan, ACS Appl. Mater. Interfaces, 2022, 14, 57092–57101 CrossRef CAS PubMed.
- H. Qi, D. Xie, Z. Gao, S. Wang, L. Peng, Y. Liu, S. Ying, D. Ma and S. Yan, Chem. Sci., 2024, 15, 11053 RSC.
- S. Wang, L. Peng, F. He, Y. Ming, H. Qi, Y. Liu, D. Ma, S. Ying and S. Yan, Adv. Opt. Mater., 2024, 12, 202400503 Search PubMed.
- T. Lu and F. Chen, Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- H. Zhou, R. Wang, M. Sun, Y. Zhou, L. Zhang, J. Song, Q. Sun, S. T. Zhang, W. Yang and S. Xue, Chem. Sci., 2024, 15, 18601 RSC.
- C. Liao, S. Wang, B. Chen, Q. Xie, J. Feng, J. Bai, X. Li and H. Liu, Angew. Chem., Int. Ed., 2024, 64, e202414905 CrossRef PubMed.
- G. Wu, X. Ge, Z. Yang, Y. Liu, Z. Chen, Y. Wang, G. Li, D. Guo, H. Deng, J. Zhao and Z. Chi, Chem. Eng. J., 2024, 497, 154659 CrossRef CAS.
- B. Ma, B. Zhang, H. Zhang, Y. Huang, L. Liu, B. Wang, D. Yang, D. Ma, B. Z. Tang and Z. Wang, Adv. Sci., 2024, 11, e2407254 CrossRef PubMed.
- L. Peng, Y. M. Huo, L. Hua, J. C. Lv, Y. C. Liu, S. A. Ying and S. K. Yan, J. Mater. Chem. C, 2022, 10, 9621–9627 RSC.
- C. Du, H. Liu, Z. Cheng, S. Zhang, Z. Qu, D. Yang, X. Qiao, Z. Zhao and P. Lu, Adv. Funct. Mater., 2023, 33, 202304854 Search PubMed.
- M. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li and P. Lu, Chem. Sci., 2015, 6, 3797–3840 RSC.
- Z. Gao, G. Cheng, F. Shen, S. Zhang, Y. Zhang, P. Lu and Y. Ma, Laser Photonics Rev., 2014, 8, L6–L10 CrossRef CAS.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photonics, 2020, 14, 643–649 CrossRef CAS.
- Y. Zou, M. Yu, Y. Xu, Z. Xiao, X. Song, Y. Hu, Z. Xu, C. Zhong, J. He, X. Cao, K. Li, J. Miao and C. Yang, Chem, 2024, 10, 1485–1501 CAS.
- L. Peng, Y. M. Huo, S. Y. He, Y. C. Liu, Z. J. Ren, S. A. Ying and S. K. Yan, J. Mater. Chem. C, 2022, 10, 11642–11652 RSC.
- P. N. Murgatroyd, J. Phys. D: Appl. Phys., 1970, 3, 151 CrossRef.
- S. Wang, H. Qi, H. Huang, J. Li, Y. Liu, S. Xue, S. Ying, C. Shi and S. Yan, Mater. Chem. Front., 2025, 9, 55–64 RSC.
- T. Fan, Q. Liu, H. Zhang, X. Wang, D. Zhang and L. Duan, Adv. Mater., 2024, 36, 202408816 Search PubMed.
- C. Liao, B. Chen, Q. Xie, X. Li, H. Liu and S. Wang, Adv. Mater., 2023, 35, 202305310 Search PubMed.
- W. Zheng, F. Zhu and D. Yan, Adv. Opt. Mater., 2024, 2402335, DOI:10.1002/adom.202402335.
- S. Diesing, L. Zhang, E. Zysman-Colman and I. D. W. Samuel, Nature, 2024, 627, 747–753 Search PubMed.
- C. Du, H. Liu, Z. Cheng, S. Zhang, Z. Qu, D. Yang, X. Qiao, Z. Zhao and P. Lu, Adv. Funct. Mater., 2023, 33, 2304854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08473j |
| This journal is © The Royal Society of Chemistry 2025 |