
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient and selective hydroboration of alkenes catalyzed by an air-stable (PPCF3P)CoI2 precatalyst†
Matthew C. Fitzsimmons,
Maria C. Seith,
Curtis E. Moore and
Christine M. Thomas*
and
Christine M. Thomas*
Department of Chemistry and Biochemistry, The Ohio State University, 100 W. 18th Ave., Columbus, OH 43210, USA. E-mail: thomasc@chemistry.ohio-state.edu
First published on 26th February 2025
Abstract
Alkene hydroboration provides a convenient route to generate organoborane synthons and recent efforts to develop catalysts for this and many other organic transformations have involved a shift to Earth-abundant first row transition metals. Herein, we report the synthesis of a new bench-stable CoII precatalyst, (PPCF3P)CoI2 (1), which was found to function as a highly active alkene hydroboration catalyst in the presence of an activator. The substrate scope was probed through exploring a collection of electronically and sterically distinct alkenes with a wide range of substitution patterns and functional groups. A single species is spectroscopically observed during catalysis, and activation of the CoII precatalyst with KBEt3H in the presence of styrene and in the absence of HBpin affords this species, (PPCF3P)Co(η2-styrene)H (2), which has been isolated, characterized, and demonstrated to function as an active catalyst for alkene hydroboration in the absence of additional activators. A plausible mechanism involving a CoI-hydride active species is proposed based on catalytic and stoichiometric experiments.
Introduction
Organoboranes have been established as privileged synthons due to the wide range of transformations that they can undergo, including the Brown hydroboration–oxidation sequence and carbon–carbon bond formation reactions such as Suzuki–Miyaura coupling.1–3 The versatility of organoborane reagents in the synthesis of complex molecules necessitates a diverse chemical “toolkit” capable of accessing large libraries of organoboranes. The hydroboration of alkenes catalyzed by transition metal complexes is known to afford high atom economy and selectivity.4 While most of these reports take advantage of noble metal catalysts, recent decades have seen significant attention paid to the more abundant first-row transition metals like cobalt or iron, in part due to concerns regarding the long-term usage of noble metals, which are relatively scarce and often more toxic than first-row transition metals.5–9In the case of first-row transition metals, pincer ligands have become ubiquitous as a means to tune the reactivity of the metal center towards the substrate of interest, while imparting high thermal stability and steric bulk to discourage undesirable side reactions.10–12 A selection of recent literature examples of pincer-ligated cobalt complexes and their catalytic alkene hydroboration activity is provided in Fig. 1. In 2013, Chirik and co-workers reported a pyridine diimine based CoI alkyl precatalyst, (MesPDI)CoCH3, that gave full conversion of terminal alkenes to boronate ester products in 15 minutes without activators. Furthermore, sterically hindered internal alkenes, both endocyclic and linear, are functionalized under mild conditions with Chirik's system (1 mol%, 50 °C, 16 h) via an isomerization-hydroboration sequence.13 Lu et al. demonstrated enantioselective anti-Markovnikov hydroboration of α-methylstyrene derivatives using a mixed donor oxazoline/iminopyridine cobalt precatalyst (5 mol%) when activated by 3 equiv. NaBH(sec-Bu)3 (25 °C, 5 h).14 In 2017, Thomas and co-workers reported a bipyridyl-oxazoline supported cobalt dichloride complex that, when activated by sodium tert-butoxide, afforded excellent Markovnikov selectivity for the hydroboration of terminal alkenes under mild conditions.15 A CoII complex ligated by a redox-active pyrimidinediimine ligand (PPymDipp) reported by the Roşca group in 2023 enabled the conversion of terminal alkenes to the anti-Markovnikov hydroboration product under mild conditions.16 Among the most active Co pincer catalysts reported to date is the (POCOP)CoIICl compound reported by Huang et al. in 2023, which proved to be capable of converting terminal alkenes to the linear hydroboration product with ppm catalyst loadings. Much like the Chirik catalyst, the conversion of internal alkenes required more forcing conditions (1–5 mol%, 80–120 °C, 24 h) and was found to proceed through chain-walking to a terminal alkene that is released in a final hydroboration step.17 A triazine-derived (PNP)CoCl2 complex synthesized by Lewandowski and Hreczycho in 2023 demonstrated the selective anti-Markovnikov hydroboration of the vinyl alkene in allyldimethyl(vinyl)silane without activators (5 mol%, 60 °C, 18 h); in the presence of NaBEt3H (5 mol%) and (PNP)CoCl2 (2.5 mol%), both vinyl and allyl double bonds were functionalized with gentle warming (40 °C).18
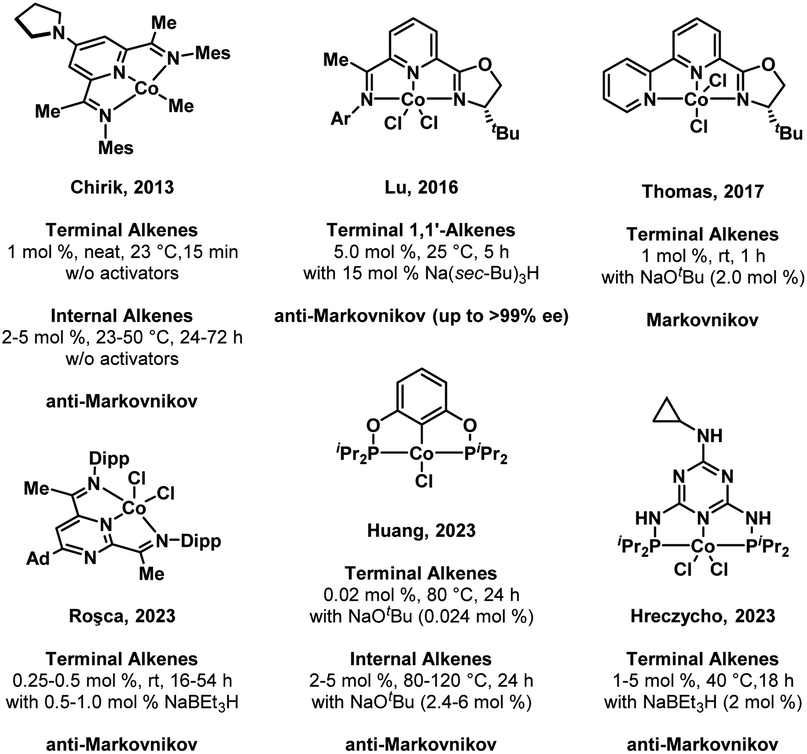 | ||
| Fig. 1 A selection of the most active cobalt complexes reported for the catalytic hydroboration of alkenes. | ||
A commonality among the catalysts/precatalysts in Fig. 1 is the use of pincer ligands that are either redox-active or possess strongly donating ligands intended to enforce a strong field ligand environment and low-spin electronic configuration. In this study, we incorporate a π-accepting diamidophosphine fragment into a pincer ligand. Prior work by our group has demonstrated that the N-heterocyclic phosphine-containing cobalt complexes (PPHP)CoI2 and (PPClP)CoCl2 are effective precatalysts for the hydrogenation and hydroboration of terminal alkenes, respectively, where the (PPH/ClP) ligand is a bis(phosphine) pincer ligand with an N-heterocyclic chloro- or hydrophosphine in the central position.19,20 These transformations were found to proceed under mild conditions upon activation of the cobalt complex with two equiv. KBEt3H. The catalytic activity and substrate scope of these prior systems were primarily found to be limited by a rapid dimerization process upon substrate depletion that yields the catalytically inactive species [(PPP)CoH]2, where the central phosphine has been converted to a phosphide via loss of the H or Cl substituent under catalytic conditions. This dimer formation was found to be irreversible. Seeking to prevent dimerization of the purported cobalt hydride active species while increasing π-acceptor interactions, we posited that installation of a trifluoromethyl group on the central phosphorus atom would afford increased stability to the catalytically active species. Furthermore, we envisioned that the CF3 group would provide a convenient method to monitor reactions using 19F NMR spectroscopy, providing valuable mechanistic information. Herein, we describe an air-stable CoII compound ligated by a trifluoromethyl-derived (PPCF3P) ligand that is significantly more active than the (PPClP)CoCl2 system while greatly increasing the scope of the alkene hydroboration reaction to include substrates with a wider range of functional groups and alkene substitution patterns.
Results and discussion
The synthesis of the (PPCF3P) pincer ligand was achieved by nucleophilic trifluoromethylation of the (PPClP) ligand precursor using TMSCF3 in the presence of KF and 18-crown-6 according to protocols developed by Ruppert, Prakash, and others (Fig. 2).21–23 Although our previous (PPClP)CoCl2 hydroboration catalyst featured cobalt-bound chlorides,20 we chose to metallate the (PPCF3P) ligand with CoI2 as we have generally found cobalt iodide compounds to enjoy greater solubility in non-polar solvents than their chloride analogues. Addition of (PPCF3P) to a stirring solution of CoI2 (1.1 equiv.) in THF affords (PPCF3P)CoI2 (1) in 90% yield as a golden-brown solid with modest solubility in polar organic solvents (Fig. 2a). The 1H NMR spectrum of 1 contains 7 broad paramagnetically shifted resonances between 9.3 and 2.5 ppm in accordance with a paramagnetic Cs-symmetric CoII complex with a few resonances that are too broad to observe due to proximity to the paramagnetic center; 1 exhibits no discernible 19F or 31P resonances. The magnetic moment of 1 was determined by solution-state Evans' method, revealing a μeff value (1.78 μB) consistent with the spin-only value (1.73 μB) for an S = ½ spin state and a low spin CoII center. Single crystals of 1 were grown by slow evaporation of a concentrated CH2Cl2 solution at room temperature. The solid-state structure of 1 (Fig. 2b) confirms that the ligand is bound to a CoI2 fragment with all three phosphines in a co-planar arrangement with a nearly ideal square pyramidal geometry at cobalt (τ5 = 0.04).24 The trifluoromethyl substituent of the central phosphorus atom adopts an anti-periplanar conformation with respect to the apical iodide ligand. The distance between cobalt and the central phosphorus atom was found to be 2.1034(6) Å, commensurate with a high degree of π-backbonding from cobalt to the diamido-substituted phosphorus center. This Co–P distance is >0.1 Å shorter than the Co–P distances associated with the phosphine sidearms and is consistent with the Co–P distances in related (PPHP)CoI2 (2.0855(9) Å) and (PPClP)CoCl2 (2.0719(9) Å) complexes.19,20 Complex 1 is remarkably stable to air and moisture: samples stored under atmospheric conditions for 6 months show no signs of degradation by 1H NMR spectroscopy.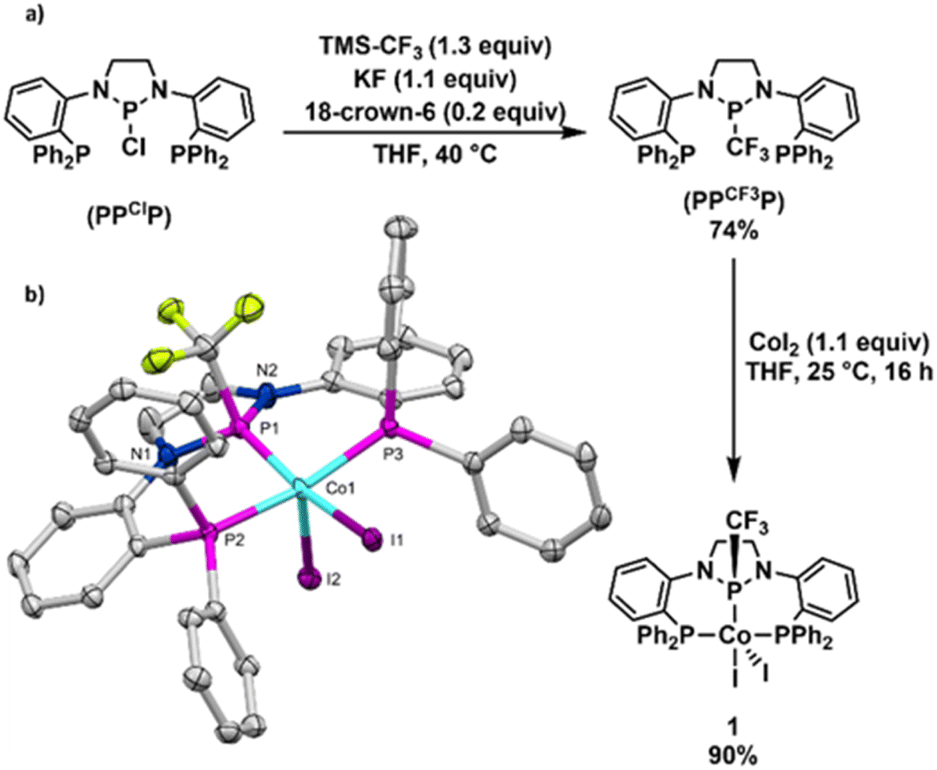 | ||
| Fig. 2 (a) Synthesis of trifluoromethyl phosphine ligand (PPCF3P) and CoII precatalyst (PPCF3P)CoI2 (1). (b) Solid-state structure of 1. All hydrogen atoms and three CH2Cl2 solvate molecules are omitted for clarity and only one position of the disordered CF3 group is shown for simplicity. Selected interatomic distances (Å): Co–P1, 2.1034(6); Co–P2, 2.2404(6); Co–P3, 2.2388(6); Co–I1, 2.5753(3); Co–I2, 2.7033(3). | ||
With complex 1 in hand, we began evaluating its catalytic activity for alkene hydroboration using styrene as a model substrate (Fig. S40–S50†). Initially, we found that combining styrene with HBpin (1.1 equiv.) in the presence of 1 mol% pre-catalyst 1 and 2 mol% KBEt3H (added as 100 mM solution in THF) resulted in rapid conversion to the linear, anti-Markovnikov boronate ester product in 94% yield (GC-FID) at room temperature in 45 min (Table 1, entry 1). Analysis of the crude reaction mixture using GC-MS revealed a small amount of the hydrogenation product, ethylbenzene, but no evidence for formation of the Markovnikov product. In a series of control experiments, no product formation was observed in the absence of 1 and/or KBEt3H (Table S2,† entries 1–3).20 As reported in 2020 by S. P. Thomas and co-workers, nucleophilic decomposition of HBpin can generate BH3 in situ, catalyzing hydroboration reactivity.25 The use of TMEDA (10 mol%) as an additive during catalysis has been shown to effectively chelate the resulting boron decomposition products. In the case of 1/KBEt3H, the addition of TMEDA (10 mol%) was found to have no discernible effect on catalytic outcomes for the hydroboration of styrene (Table S2,† entry 5). Finally, we screened the use of Cs2CO3 as a milder and more user-friendly activator. Despite the near complete lack of observed solubility of Cs2CO3 (0.2 mol%) under standard conditions (0.1 mol% 1, C6H6, 25 °C), near total conversion of styrene was observed with no loss in product selectivity, although a longer reaction time was required as a result of solubility limitations (Table S2,† entry 6). Given that activation requires both reduction from CoII to CoI and generation of a hydride-containing active species (vide infra), it is intriguing that a less reducing, non-nucleophilic, and hydride-free base could accomplish catalyst activation. We posit that interaction between HBpin and Cs2CO3 generates the requisite hydride source, but future work will be needed to further understand the method by which activation using Cs2CO3 is accomplished.
|
|||||
|---|---|---|---|---|---|
| Entry | n | Solventb | Temp. (°C) | Time | Conv.a (%) |
| a Conversion determined by GC-FID. Values in parentheses represent isolated yields.b THF is also introduced into these reactions through addition of a 100 mM solution of KBEt3H in THF.c NMR yield.d Compound 2 used directly as a catalyst in place of 1 and KBEt3H. | |||||
| 1 | 1 | C6H6 | 25 | 45 min | 94 |
| 2 | 1 | Toluene | 25 | 60 min | 96 |
| 3 | 1 | THF | 25 | 85 min | 94 |
| 4 | 0.5 | C6H6 | 25 | 80 min | 96 |
| 5 | 0.5 | C6H6 | 37 | 40 min | >99 |
| 6 | 0.5 | C6H6 | 50 | 12 min | >99 |
| 7 | 0.3 | C6H6 | 25 | 195 min | 99 |
| 8 | 0.1 | C6H6 | 25 | 6 h | (94) |
| 9 | 0.1 | C6H6 | 50 | <90 min | >99 |
| 10 | 0.03 | Neat | 50 | <5 h | 98 |
| 11 | 0.01 | Neat | 25 | 48 h | 83c |
| 12 | 0.01 | Neat | 50 | 12 h | 98 (84) |
| 13 | 0.5d | C6H6 | 25 | 60 min | 95 |
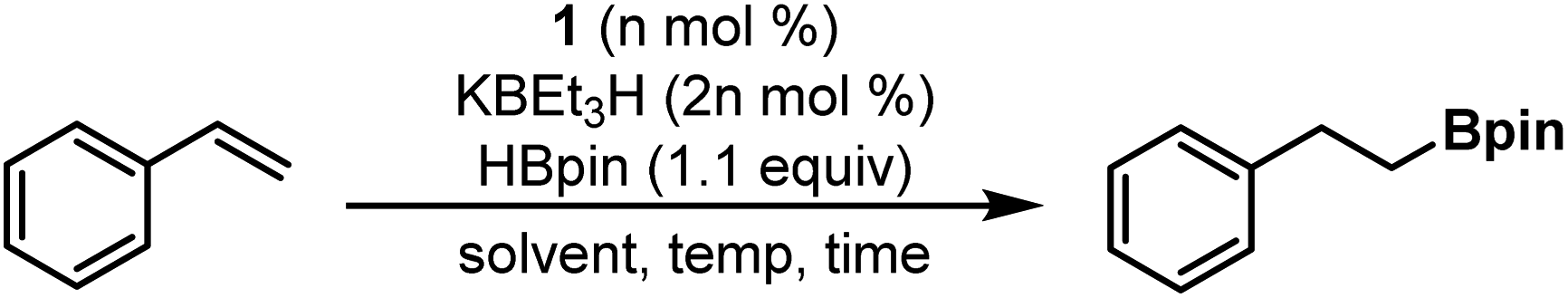
In efforts to optimize reaction conditions, it was found that using toluene as an alternative solvent (Table 1, entry 2) did not alter the reaction profile significantly, but employing THF as the reaction solvent (entry 3) resulted in much slower conversion of starting material. Reducing the catalyst loading to 0.5 mol% (entries 4–6) resulted in excellent conversion, with the reaction rate increasing at elevated temperatures: styrene hydroboration proceeded to >99% conversion in 12 minutes when the reaction was warmed to 50 °C (entry 6). The catalyst loading could be further reduced to 0.1 mol% (entries 7 and 8) without significant impact on catalytic outcomes when reactions were run at room temperature, although reaction times increased substantially at lower catalyst loading. In general, solvent-free conditions were well-tolerated, allowing reactions at even lower catalyst loading (0.03 mol%) to proceed to completion with gentle heating (entry 10). Reactions run with a precatalyst loading of 0.01 mol% were sluggish at room temperature (>24 h, entry 11), but high conversion to the linear hydroboration product was still observed when the reaction mixture was analyzed after approximately 48 hours. Moreover, heating a reaction with 0.01 mol% loading to 50 °C was found to provide significant enhancement to the reaction rate (entry 12), affording 98% conversion (GC-FID, 84% isolated) of the starting material after 12 hours (TON = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). This represents a 100-fold increase in catalytic activity when compared to the previously reported (PPClP)CoCl2 precatalyst.20 Hoping to establish mild conditions across a wide range of substrates, we ultimately chose to proceed with a general loading of 0.1 mol% 1 for the remainder of our catalytic evaluation.
000). This represents a 100-fold increase in catalytic activity when compared to the previously reported (PPClP)CoCl2 precatalyst.20 Hoping to establish mild conditions across a wide range of substrates, we ultimately chose to proceed with a general loading of 0.1 mol% 1 for the remainder of our catalytic evaluation.
It is notable that catalysis with 1/KBEt3H was not found to be impacted by the presence of the inhibitor (butylated hydroxytoluene) in commercial bottles of styrene and, in contrast to some of the previously reported cobalt-based hydroboration catalysts,13,17,20 all further alkene reagents could be used as-received without observable impact on catalytic activity. The robustness of the catalytic system towards adventitious moisture and air was established by the addition of air (1 mL, Table S1† entry 4) or water (1.5 mol%, Table S1† entry 5) after activation with KBEt3H; both were not found to inhibit catalysis and isolated yields greater than 85% could be obtained under these conditions. This suggests that the species responsible for catalysis is highly resistant to undesirable deactivation in the presence of adventitious impurities. Lastly, we note that the hydroboration of styrene reflected in Table 1 entry 1 was performed on a nearly 1 g scale (0.86 g styrene), demonstrating that the catalytic conditions were amenable to preparative reaction scales (see ESI†).
Having established optimal catalytic reaction conditions, we began to evaluate the substrate scope using electronically differentiated styrene derivatives (Table 2). In most cases during our substrate evaluations, time and temperature were optimized unless otherwise noted. Electron-deficient para-substituted styrene derivatives 4-bromostyrene and 4-chlorostyrene were hydroborated in 16 h under solvent-free conditions and afforded 88% and 85% isolated yields, respectively (entries 2 and 3). In comparison to styrene itself, 4-bromo/4-chlorostyrene required longer reaction times (16 h) to completely consume the starting alkene. Hydroboration of 4-fluorostyrene also proceeded smoothly but more slowly than styrene, affording 91% yield after 3 h but with an increased loading of 1 (0.5 mol%, entry 4). Hydroboration of 4-trifluoromethylstyrene was found to proceed slowly at room temperature, even when the catalyst loading was increased to 0.2 mol%, affording a 74% yield of the anti-Markovnikov product by NMR after 24 hours (entry 5). The more sluggish reactivity of 4-trifluoromethylstyrene is likely resultant from significant electron-withdrawing effects from the trifluoromethyl group, as CF3 is well-accepted as a stronger electron-withdrawing group than F.26,27 Much like previously reported catalysts, the nitro group of 4-nitrostyrene (entry 6) was not tolerated by this system, and attempted catalytic reactions afforded only mixtures of starting material and unidentified organic products. We hypothesize that the incompatibility is a function of undesired reactivity between the metal complex and the pendent nitro group, rather than the electronic character of the alkene alone. Styrene derivatives bearing electron-donating groups in the para position, such as 4-methylstyrene and 4-vinylanisole were hydroborated in excellent yields of 96% and 96%, respectively (Table 2, entries 7 and 8). Hydroboration of 4-methylstyrene, when performed under identical conditions to styrene (entry 9), revealed no clear effect of the Me group on the rate of reaction, reaching complete conversion of starting material in 6 hours as determined by GC-FID.
|
||||
|---|---|---|---|---|
| Entry | R | Solventa | Time (h) | Yieldb (%) |
| a THF is also introduced into these reactions through addition of a 100 mM solution of KBEt3H in THF.b Isolated yields unless otherwise noted.c Catalyst loading 0.5 mol%.d Yield determined by quantitative NMR spectroscopy.e Catalyst loading 0.2 mol%.f Conversion determined by GC-FID. | ||||
| 1 | H | Neat | 6 | 94 |
| 2 | Br | Neat | 16 | 88 |
| 3 | Cl | Neat | 16 | 85 |
| 4 | F | C6D6 | 3 | 91c,d |
| 5 | CF3 | C6D6 | 24 | 74d,e |
| 6 | NO2 | C6D6 | 24 | 0 |
| 7 | Me | C6H6 | 16 | 96 |
| 8 | OMe | C6H6 | 24 | 96d |
| 9 | Me | Neat | 6 | >99f |
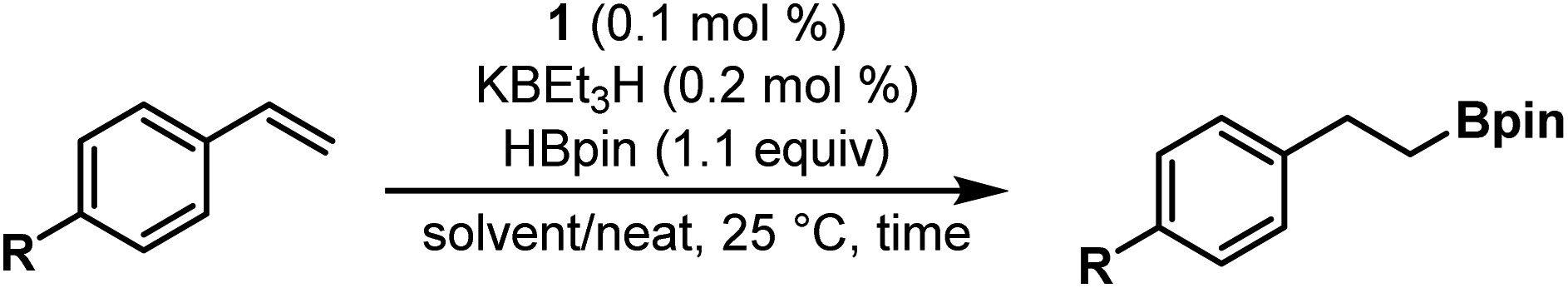
In addition to vinyl arenes, precatalyst 1 was also found to be active for terminal aliphatic alkene substrates (Table 3). Under standard solvent-free reaction conditions (0.1 mol% 1, 0.2 mol% KBEt3H, rt) 3-phenylpropene was found to convert to product A completely in <1 hour as evidenced by the complete absence of starting material when reaction aliquots were analyzed by GCMS-FID, resulting in a 93% isolated yield of the anti-Markovnikov product. Hydroboration of allyl trimethylsilane also proceeds smoothly under standard conditions in neat substrate, affording product B in >99% yield in 6 h. Catalytic hydroboration of dodecene using 1/KBEt3H was also explored and, in a similar manner to 3-phenylpropene, catalysis was noted to reach completion within 2 hours and pure C was obtained after a short silica filtration (>99% yield). Reducing the loading of 1 (0.01 mol%) with respect to dodecene was also successful, affording a 97% isolated yield of the hydroboration product in less than 14 hours at room temperature (TON = 10000). The recently reported (POCOP)CoCl system achieves similarly high isolated yields of the analogous terminal aliphatic alkene substrate, 1-octene, but required elevated temperature (80 °C), a longer reaction time (24 h), and a higher catalyst loading (0.02 mol%, TON = 5000).17 Indeed, to our knowledge, 1/KBEt3H represents the most efficient room temperature hydroboration catalyst for such terminal aliphatic alkenes.16,17
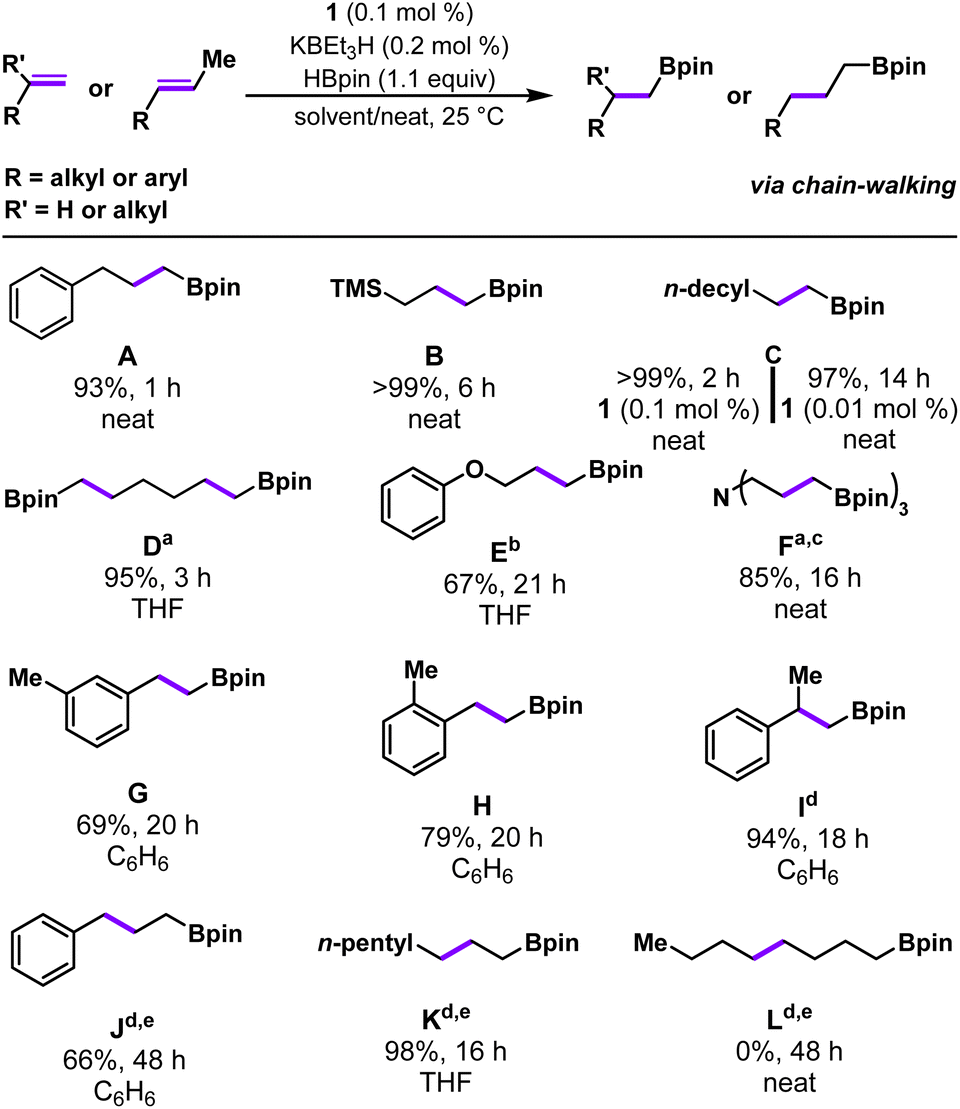
Next, we began to explore substrates with potentially coordinating functional groups (Table 3). 1,5-Hexadiene was chosen as a probe for ring-closing reactivity and to determine the compatibility of substrates that can potentially form chelates with the activated complex. Catalytic hydroboration reactions with 1,5-hexadiene were run with 0.2 mol% catalyst loading, reflecting 0.1 mol% per alkene and catalysis proceeded to completion in three hours, furnishing the doubly-hydroborated product D in 95% isolated yield. Allyl phenyl ether was observed to convert much more slowly than other substrates under standard catalytic conditions, ultimately requiring 21 hours using 0.2 mol% 1 to achieve a 67% yield of E, with a significant amount of starting material remaining. We observed even lower yields of E from allyl phenyl ether when benzene was used as a solvent (Fig. S76†), a similar finding to the previously reported (PPClP)CoI2/KBEt3H alkene hydroboration system.20 We suspect that this solvent dependence is attributed to coordination of the ether functionality of the alkene substrate to the cobalt active species in the absence of coordinating solvent. The presence of a tertiary amine within the substrate was well-tolerated, even under neat reaction conditions. Functionalization of triallylamine was performed with 0.3 mol% cobalt, reflecting a final loading of 0.1 mol% per alkene. Reactions with triallylamine reached completion within 16 hours and the final product F was isolated after acidification as the ammonium salt in 85% isolated yield. In comparison, Huang's (POCOP)CoCl system afforded product F in high yield (81%) with lower catalyst loading (0.02 mol%) but higher temperature (80 °C).17
We then sought to explore a more structurally diverse series of substrates that probe the effects of increased steric encumbrance around the alkene (Table 3). It was found that both 3-methylstyrene and 2-methylstyrene were amenable to this system with 69% and 79% isolated yield of G and H, respectively, after 20 hours. Increased catalyst loading and elevated temperature were not required; however, the reactions required longer reaction times to reach completion compared to styrene. Complex 1 was also found to be active for the catalytic hydroboration of a 1,1-di-substituted alkene, α-methylstyrene. Under standard conditions, this more sterically hindered substrate resulted in slower conversion times; however, increasing the catalyst loading to 0.5 mol% gave satisfactory results, affording the anti-Markovnikov hydroboration product I in 94% isolated yield in 18 h at room temperature. Precatalyst 1's activity towards 1,1′-disubstituted alkenes is superior to the (POCOP)CoCl catalyst, which required 2 mol% catalyst loading and 24 h at 120 °C to reach a similar yield.17 Moreover, a higher yield is achieved with 1 at lower loadings compared to Lu's asymmetric hydroboration catalysts, although the latter catalyst was evaluated for a much wider range of 1,1′-disubstituted alkenes and exhibited impressive enantioselectivity.14 On the other hand, 1/KBEt3H is less active than a chiral imidopyridine/oxazoline CoI–Me compound reported by Huang et al., which operates without an activator and affords product with high enantioselectivity.28
Catalytic hydroboration of internal alkene substrates is typically more challenging, but trans-alkene substrates with alkenes in the 2-position were successfully hydroborated using 1/KBEt3H with increased temperature (48 °C) and catalyst loading (0.5 mol%), exclusively generating linear products with the boronate ester in the terminal position. For example, trans-2-octene was successfully hydroborated to the linear, anti-Markovnikov product K with gentle heating in THF (48 °C, 0.5 mol%). The exclusive formation of the terminal boronate ester products can be explained by a chain-walking mechanism whereby reversible alkene insertion and β-hydride elimination generates the more reactive terminal Co-alkyl species, which irreversibly forms the terminal hydroboration product (Scheme 1). Trans-2-octene represents a substrate that was previously inaccessible for hydroboration/hydrogenation using (PPHP)Co or (PPClP) complexes prepared by our laboratory.19,20 Hydroboration of conjugated internal alkenes, owing to their increased steric bulk and decreased π-system reactivity, represents a gap in the available literature for first-row catalytic systems; selective generation of the linear, chain-walked product was completely unprecedented for substrates such as trans-β-methylstyrene until the recent report by Huang et al. (2 mol% (POCOP)CoCl, 120 °C, 24 h).17 Catalytic hydroboration of trans-β-methylstyrene with 1/KBEt3H was carried out under the standard conditions for internal alkenes (0.5 mol%, 48 °C), affording exclusively the linear anti-Markovnikov product J in 66% isolated yield. The position of the internal alkene appears to be crucial, however, as a hydroboration reaction under identical conditions (0.5 mol% 1, 48 °C) with trans-4-octene was found to afford a mixture containing primarily starting material with no boronate ester products L observed by GCMS. Attempts to obtain satisfactory yields through increasing catalyst loading (1 mol%) or increasing the reaction temperature (48 °C) were successful in the detection of a small quantity of hydroboration product after three days (Fig. S74†). We hypothesize that hydroboration activity with trans-4-octene is sluggish owing to the both the increased steric bulk of the alkene, disfavoring binding of the alkene to the Co center, and the slow nature of the multiple isomerization steps required to achieve catalytic turnover, as observed by Obligacion and Chirik in 2013.13
 | ||
| Scheme 1 Terminal selective hydroboration mechanism with trans-2-alkenes via a reversible alkene insertion/β-hydride elimination pathway. | ||
Having established the high catalytic activity, we next sought to probe the identity of the (PPCF3P)Co-containing species present during catalysis using the P–CF3 functional group as a spectroscopic handle. While monitoring a catalytic reaction (1.0 mol% 1, 1.1 equiv. HBpin) by 19F NMR spectroscopy, a single resonance (d, −62 ppm) was observed for the catalytically active species. To identify the (PPCF3P)-containing species observed during catalysis, a stoichiometric reaction was performed in the absence of HBpin. Treatment of 1 with 2 equiv. KBEt3H in the presence of styrene afforded a single, bright-yellow metal complex with a 19F NMR resonance identical to that observed during catalysis (d, −61.9 ppm). Spectroscopic and structural characterization revealed this species to be the styrene adduct of a CoI hydride, (PPCF3P)Co(η2-styrene)H (2) (Scheme 2). The 1H{31P} spectrum in combination with 2D NMR experiments (COSY) allowed the assignment of the benzylic and distal protons of the bound styrene molecule, which experience a significant upfield shift (Fig. S12†). The benzylic proton of the bound styrene molecule is located at 3.92 ppm, while the protons on the terminal carbon of styrene are observed at 3.10 and 2.72 ppm. A characteristic hydride signal is observed for complex 2 as a well resolved multiplet (ddd) at −14.4 ppm in the 1H NMR spectrum (Fig. S7†). The 31P{1H} NMR spectrum of 2 reveals inequivalent sidearm phosphines (47.1 and 44.8 ppm) and a central phosphorus signal at 124.2 ppm (Fig. S9†).
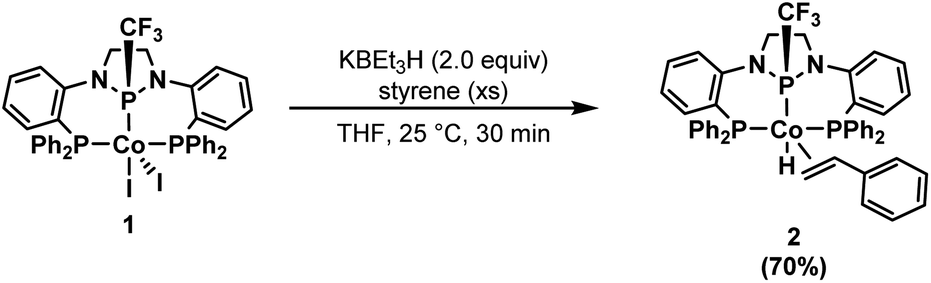 | ||
| Scheme 2 Synthesis of (PPCF3P)Co(η2-styrene)H (2), the primary metal-containing species observed spectroscopically during catalytic turnover. | ||
Complex 2 enjoys significantly increased solubility in nonpolar organic solvents compared to 1 and X-ray quality single crystals were grown from a concentrated n-pentane solution stored at −35 °C. The solid-state structure of 2 (Fig. 3) supports the connectivity proposed using NMR spectroscopy, namely that the styrene adopts an η2 binding mode rather than inserting into the cobalt–hydride bond to generate a cobalt-alkyl. The geometry about the Co center is trigonal bipyramidal, with the hydride ligand trans to the central phosphorus atom of the pincer ligand and the η2-styrene ligand in an equatorial position syn to the P–CF3 moiety. The bond distance between the vinylic carbons of the styrene fragment is indicative of a slight activation (1.399(8) Å) of the carbon–carbon double bond compared to free styrene (1.338 Å).29
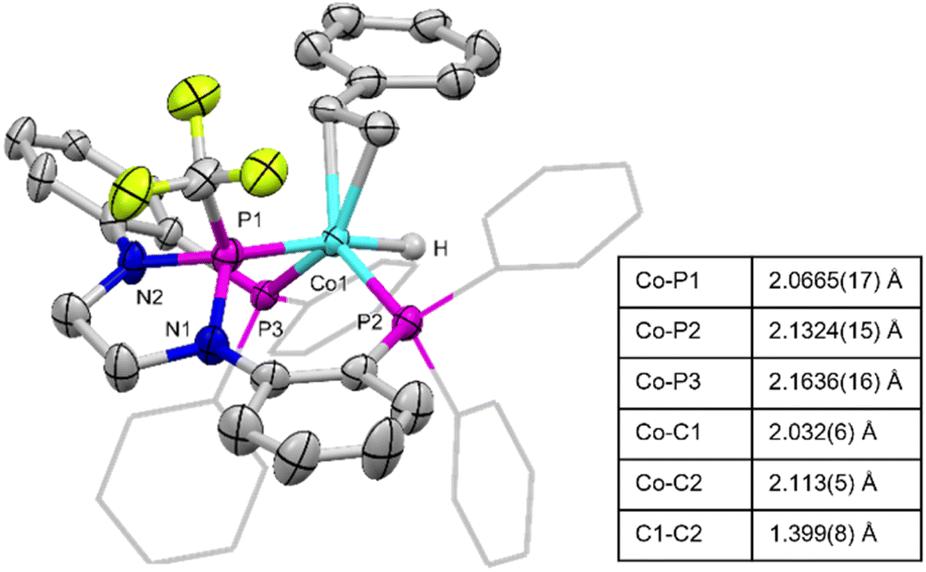 | ||
| Fig. 3 Solid-state structure of (PPCF3P)Co(η2-styrene)H (2). Hydrogen atoms, except for the Co-bound hydride, have been omitted for clarity and only one of two independent molecules in the asymmetric unit is shown for simplicity. Selected interatomic distances are for one of the two similar independent molecules in the asymmetric unit of 2. | ||
Gratifyingly, when spectroscopically pure 2 (0.5 mol%) was added to a mixture of styrene and HBpin (1.1 equiv.) 95% conversion of the starting material to hydroboration product was observed by GCMS after 60 min without additional activators (Table 1, entry 13, Fig. S68†), comparable to the catalytic activity observed with 1/KBEt3H. This prompted us to assess whether the catalyst remains in an active form upon substrate depletion: Hydroboration of styrene was carried out with 1 (0.1 mol%) at room temperature and allowed to stir for 24 h, resulting in complete substrate consumption. The catalytic reaction mixture was subsequently recharged with additional styrene (1 equiv.) and HBpin (1.1 equiv.) two times, each time allowing catalysis to proceed for 24 h. No loss in catalytic activity was observed over these 3 cycles in a 72 h period (Fig. S69†). The stability and ability to recycle the catalyst after the substrates are consumed in the 1/KBEt3H system represents an improvement compared to the previously reported (PPHP)CoI2 and (PPClP)Cl2 catalysts.19,20 In the latter cases, a catalytically inactive dimeric species, [(PPP)CoH]2, was formed upon alkene depletion at the end of catalysis, preventing recyclability and limiting catalytic activity towards alkenes that react more slowly with the active species. Thus, the more robust P–CF3 linkage provides a distinct advantage compared to previously reported (PPRP)Co precatalysts by preventing deactivation via P-substituent loss and dimerization through the resulting phosphido functionality.
Encouraged by this result, we explored the use of 2 as a catalyst for the transformation of substrates that were poorly tolerated due to incompatibility with the KBEt3H activator (Scheme 3). Hydroboration of 4-acetoxystyrene was initially met with difficulty due to undesired reactivity of the substrate with KBEt3H. The hydride source was found to cleave the phenoxy-ester moiety, and ultimately led to apparent inhibition of the catalyst as evidenced by the presence of significant alkene starting material (Fig. S73†). We attribute the side-product formation to the generation of more reactive borane or borohydride species from the reaction of HBpin with the KBEt3H activator itself.25,30 Catalytic hydroboration reactions of 4-acetoxystyrene using isolated 2 (0.5 mol%, rt, 18 h) proceeded much more smoothly, affording greatly reduced formation of phenolic organoboranes resulting from ester cleavage and an 83% yield of the alkene hydroboration product (Fig. S75†). While synthetically accessible, the activated complex 2 is more reactive to adventitious impurities than 1 and more difficult to handle owing to air sensitivity. It is for this reason that we sought a viable one-pot methodology, whereby 1 (0.1 mol%) was stirred with styrene (3–4 mol%) and KBEt3H (0.2 mol%) for 10 minutes prior to the addition of 4-acetoxystyrene and HBpin (1.1 equiv.). This telescoping strategy successfully enabled the conversion of approximately 71% the alkene starting material over a 48 h period with relatively high selectivity for the linear hydroboration product (45% isolated yield, Scheme 3). Notably, the one-pot telescoping procedure was not as effective as using complex 2 as a catalyst directly.
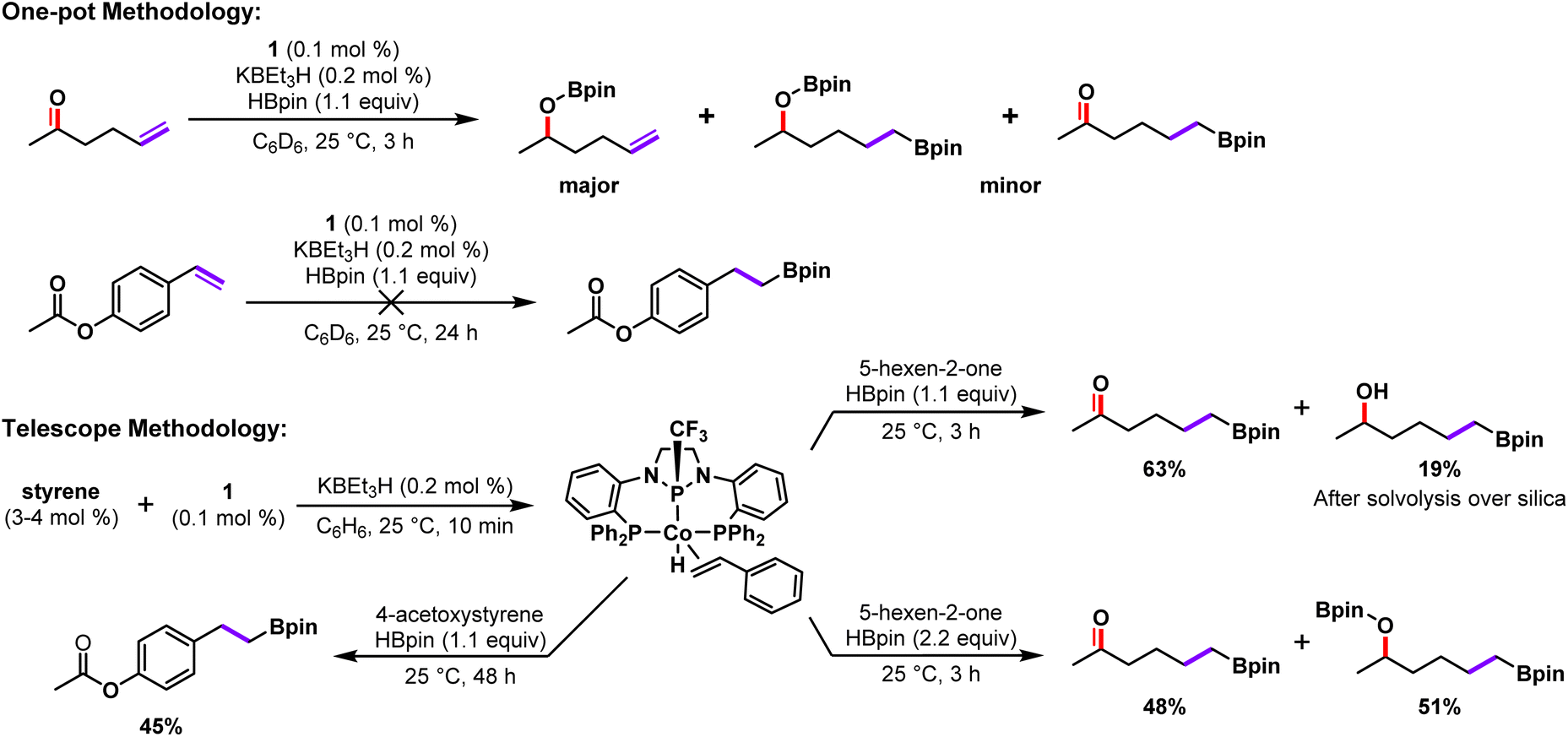 | ||
| Scheme 3 Divergent experimental outcomes for the hydroboration of carbonyl containing substrates using either 1/KBEt3H or pre-generation of 2 prior to the introduction of substrate. | ||
Similarly, the ketone functionality of 5-hexen-2-one is known to rapidly react with HBpin and catalytic amounts of KBEt3H (or nucleophiles) to afford the corresponding alcohol after work up.25,30,31 Thus, when employing the standard in situ catalyst activation protocol, hydroboration of the alkene functional group only occurred when 2 or more equiv. HBpin were used and was accompanied by reduction of the ketone as well (Scheme 3, Fig. S71 and S72†). Employing the same pre-activation strategy as previously described, telescoped samples of 2 improved selectivity for the desired alkene hydroboration product (63%), with greatly diminished reduction of the ketone (19%) when 1 equiv. HBpin is used (Scheme 3).
The isolation of 2, observation of this species during catalysis, and its competence as a catalyst without activators strongly suggests its identity as the catalytic resting state. It appears that the addition of KBEt3H results in both a one-electron reduction of the CoII precatalyst 1 and a transmetallation to form a CoI-hydride active species that rapidly binds an L-type ligand, such as styrene, to form a stabilized adduct (e.g. Scheme 2). Attempts to isolate a cobalt hydride complex in the absence of a coordinating alkene results in the observance of a mixture containing a transiently stable hydride complex, but the latter has not been isolable in our hands (Fig. S77 and S78†). To trap such a CoI–H species using a non-alkene L-type ligand, 1 was activated with two equiv. KBEt3H for 15 minutes, followed by the addition of 3 equiv. PMe3, resulting in the formation of (PPCF3P)Co(PMe3)H (3, Scheme 4). The 31P{1H} NMR spectrum of 3 reveals three broad resonances in a 1:2:1 ratio centered at 132.1, 41.1, and −2.5 ppm corresponding to the central phosphorus atom, the two phosphine sidearms, and a bound PMe3 ligand, respectively. The hydride resonance of 3 is well-resolved in the 1H NMR spectrum and observed at −11.67 ppm (ddt). Crystals of 3 suitable for X-ray diffraction were isolated by crystallization from a concentrated solution of n-pentane at −35 °C. The solid-state structure of 3 (Fig. S81†) confirms that the PMe3 ligand occupies the vacant coordination site on the same face of the complex as the trifluoromethyl group (dihedral angle = 34.2°). The distance between Co and the central phosphorus atom (2.0375(18) Å) is shorter than in 1 or 2, again, indicative of the strongly π-accepting nature of the central diamidophosphine ligand, with the Co–P distance decreasing as the electron density of the CoI center increases.
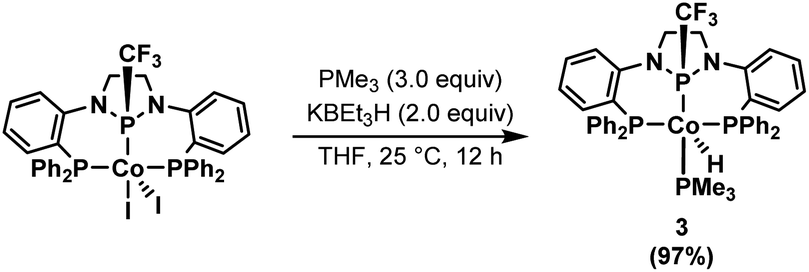 | ||
| Scheme 4 Synthesis of (PPCF3P)Co(PMe3)H (3) from 1. | ||
The PMe3 ligand in 3 precludes alkene binding to the purported (PPCF3P)CoH active species operative in catalysis. Complex 3 is not an active catalyst for the hydroboration of styrene; no product was detected after 1 hour at room temperature using 0.5 mol% 3 as a catalyst (Table S2,† entry 4). Moreover, catalysis was shown to be inhibited by the addition of PMe3 to an in-progress styrene hydroboration reaction (Fig. S67†).
The culmination of catalytic observations and stoichiometric reactions allows us to propose a straightforward mechanism for catalytic hydroboration starting with precatalyst 1 (Scheme 5). Precatalyst 1 is first activated with 2 equiv. KBEt3H to generate a (PPCF3P)CoIH species, which rapidly binds the alkene substrate. The η2-alkene may then undergo 1,2- or 2,1-insertion to furnish either a linear or branched cobalt alkyl complex. During our study, no branched products were detected, but the observed chain-walking that occurs with internal alkenes suggests that reversible insertion/β-hydride elimination is operative for isomerization.19 Moreover, previous studies performed by our laboratory using DBpin for the hydroboration of styrene with (PPClP)CoCl2/KBEt3H showed the incorporation of deuterium into both the benzylic and distal positions of styrene.19 To complete the catalytic cycle, 1,2-alkene insertion is followed by addition of HBpin via either σ-bond metathesis or oxidative addition and reductive elimination steps, resulting in the release of hydroboration product and regeneration of the postulated CoI–H active species. Since only anti-Markovnikov products are observed, the C–B bond-forming step must only be possible from the linear 1,2-insertion product, which we attribute to steric factors.
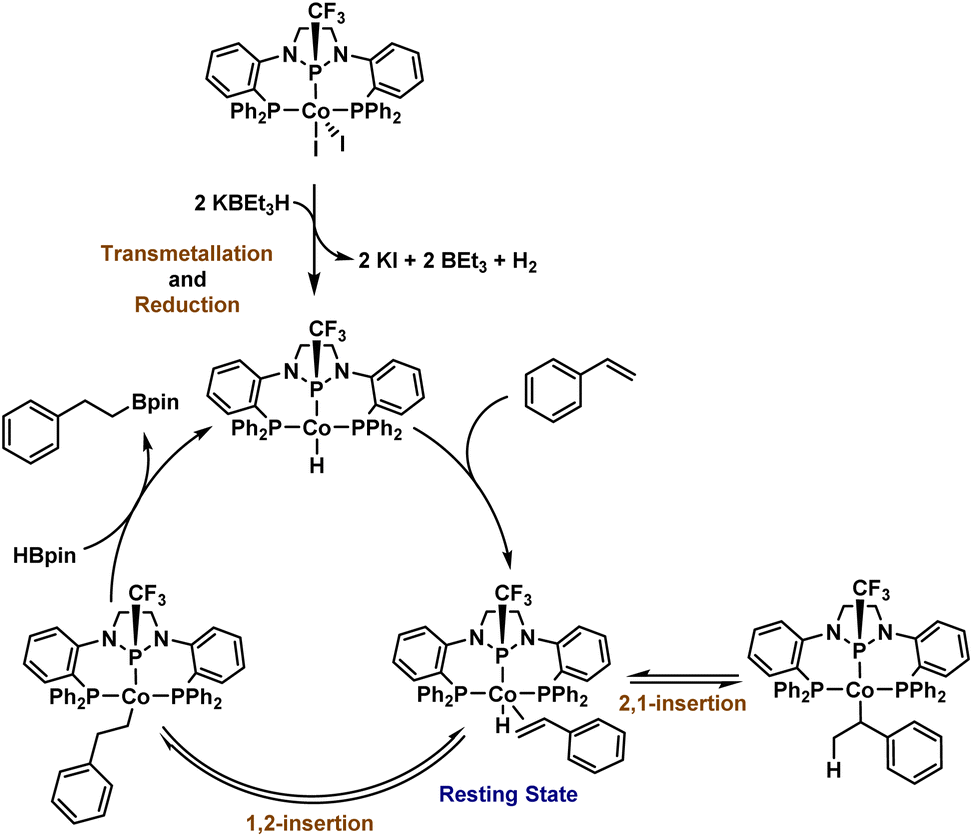 | ||
| Scheme 5 Proposed mechanism for the catalytic hydroboration of styrene. | ||
Conclusions
In summary, the synthesis and characterization of a new CoII complex, (PPCF3P)CoI2 (1), supported by a novel diphosphine pincer ligand with a central N-heterocyclic trifluoromethylphosphine moiety has been described. Through the scope of this study, we have demonstrated that complex 1 is an active and bench-stable precatalyst for the hydroboration of alkenes in the presence of two equivalents of KBEt3H. The addition of KBEt3H to 1 generates a CoI-hydride active species that rapidly binds styrene to generate (PPCF3P)Co(styrene)H (2), which was observed spectroscopically as the catalyst resting state.The catalytic activity, robustness, and substrate scope of 1 represent a significant improvement on the previous (PPH/ClP)CoX2 systems reported by our laboratory. The superiority of 1 can be attributed to the observation that the P–CF3 bond remains intact throughout and at the end of catalysis allowing the catalyst to remain active when the substrate binds slowly or is depleted at the end of catalysis. To our knowledge, 1 represents one of the most active first-row metal hydroboration catalysts, achieving high conversions with electronically differentiated vinylarene derivatives, demonstrating rapid room temperature turnover with aliphatic alkenes, and exhibiting excellent activity towards internal and 1,1-substituted alkenes at mild elevated temperatures. The 1/KBEt3H system is more active than Huang's (POCOP)CoCl catalyst, achieving full conversion in shorter times and with lower temperatures and catalyst loadings for vinylarenes, 1,1′-disubstituted alkenes, and aliphatic substrates.17 Chirik's (MesPDI)CoCH3 catalyst, on the other hand, poses significant advantages over 1/KBEt3H in terms of its activity towards remote C–H functionalization of substituted and internal alkenes;13 1/KBEt3H is limited to terminal or 2-alkenes. Substrates containing coordinating functional groups such as amines, ethers, or additional alkenes are well-tolerated by the 1/KBEt3H system. Catalytic hydroboration of substrates that are incompatible with the KBEt3H activator itself is readily accessible by first generating the styrene-stabilized CoI-hydride complex 2 in situ, then telescoping this reaction mixture into a hydroboration reaction by adding HBpin and the desired alkene substrate.
One of the most prominent advantages of 1 is its bench-stability as well as the tolerance of the 1/KBEt3H catalytic system to air/moisture exposure. The latter renders protocols involving 1/KBEt3H both scalable and user-friendly to those researchers without access to the specialized equipment or skillset for working with highly air- and moisture-sensitive compounds.
Although many examples of catalytic hydroboration of alkenes have been published using Co pincer catalysts, most propose mechanisms similar to that shown in Scheme 5 based on indirect evidence for cobalt-hydride species through deuterium labelling/scrambling studies. The isolation of 2 and the ability to observe it spectroscopically in situ, is therefore unique among cobalt hydroboration catalysts, and provides solid evidence for the intermediacy of a cobalt(I) hydride active species. The observation of 2 as the catalyst resting state might seem to imply that alkene insertion is rate-limiting, however, it is equally likely that HBpin addition is rate-determining and alkene insertion simply represents a reversible equilibrium process in which the alkene/hydride species is thermodynamically downhill. Investigations are currently underway to further elucidate the catalytic mechanism of hydroboration using the (PPCF3P)CoI2/KBEt3H system, to interrogate the role that incorporation of a diamidophosphine π-acceptor fragment into a pincer ligand plays in the remarkable catalytic activity observed, and to understand the donor/acceptor properties of diamidophosphine ligands in general.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1–3 have been deposited at the Cambridge Crystallographic Data Center (CCDC) under CCDC 2382421–2382423.†Author contributions
C. M. T. supervised and acquired funding for the project. M. C. F. and M. C. S. conducted the experiments and data analysis. C. E. M. collected and refined data for the reported single-crystal structures. M. C. F. and C. M. T. contributed to writing, reviewing, and editing the manuscript and all authors gave approval to the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the U.S. National Science Foundation under grants CHE-2101002 and CHE-2349302 and associated supplemental funding to M. C. F. under the MPS-GRSV program.References
- H. Brown and B. C. Rao, J. Org. Chem., 1957, 22, 1137–1138 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, 2009 Search PubMed.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem, 2018, 2, 15–34 CrossRef CAS PubMed.
- P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed.
- C. M. Macaulay, S. J. Gustafson, J. T. I. Fuller, D.-H. Kwon, T. Ogawa, M. J. Ferguson, R. McDonald, M. D. Lumsden, S. M. Bischof, O. L. Sydora, D. H. Ess, M. Stradiotto and L. Turculet, ACS Catal., 2018, 8, 9907–9925 CrossRef CAS.
- J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef CAS.
- A. D. Ibrahim, S. W. Entsminger and A. R. Fout, ACS Catal., 2017, 7, 3730–3734 CrossRef CAS.
- M. Vogt and R. Langer, Eur. J. Inorg. Chem., 2020, 2020, 3885–3898 CrossRef CAS.
- M. A. W. Lawrence, K.-A. Green, P. N. Nelson and S. C. Lorraine, Polyhedron, 2018, 143, 11–27 CrossRef CAS.
- B. Lee, T. P. Pabst, G. Hierlmeier and P. J. Chirik, Organometallics, 2023, 42, 708–718 CrossRef CAS PubMed.
- J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 19107–19110 CrossRef CAS PubMed.
- H. Zhang and Z. Lu, ACS Catal., 2016, 6, 6596–6600 CrossRef CAS.
- J. Peng, J. H. Docherty, A. P. Dominey and S. P. Thomas, Chem. Commun., 2017, 53, 4726–4729 RSC.
- J. S. Doll, M. L. Heldner, M. Scherr, J. Ballmann and D.-A. Roşca, ACS Catal., 2023, 13, 8770–8782 CrossRef CAS.
- J. Liu, J. Du, F. Yu, L. Gan, G. Liu and Z. Huang, ACS Catal., 2023, 13, 7995–8003 CrossRef CAS.
- D. Lewandowski and G. Hreczycho, Inorg. Chem. Front., 2023, 10, 3656–3663 RSC.
- M. C. Fitzsimmons, A. Yessengazin, G. P. Hatzis, J. E. Stevens, C. E. Moore and C. M. Thomas, Organometallics, 2023, 42, 1439–1443 CrossRef CAS.
- A. M. Poitras, L. K. Oliemuller, G. P. Hatzis and C. M. Thomas, Organometallics, 2021, 40, 1025–1031 CrossRef CAS.
- G. K. S. Prakash, R. Krishnamurti and G. A. Olah, J. Am. Chem. Soc., 1989, 111, 393–395 CrossRef CAS.
- W. Volbach and I. Ruppert, Tetrahedron Lett., 1983, 24, 5509–5512 CrossRef CAS.
- I. Tworowska, W. Dąbkowski and J. Michalski, Angew. Chem., Int. Ed., 2001, 40, 2898–2900 CrossRef CAS PubMed.
- A. G. Blackman, E. B. Schenk, R. E. Jelley, E. H. Krenske and L. R. Gahan, Dalton Trans., 2020, 49, 14798–14806 RSC.
- A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2020, 10, 13479–13486 CrossRef CAS.
- R. Su, K. Xie, Y. Liang, K. N. Houk and F. Liu, J. Org. Chem., 2023, 88, 893–900 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- L. Zhang, Z. Zuo, X. Wan and Z. Huang, J. Am. Chem. Soc., 2014, 136, 15501–15504 CrossRef CAS PubMed.
- D. W. Crandell, S. B. Muñoz, J. M. Smith and M.-H. Baik, Chem. Sci., 2018, 9, 8542–8552 RSC.
- I. P. Query, P. A. Squier, E. M. Larson, N. A. Isley and T. B. Clark, J. Org. Chem., 2011, 76, 6452–6456 CrossRef CAS PubMed.
- L. C. A. De Carvahlo, Y. Peres, M. Dartiguenave, Y. Dartiguenave and A. L. Beauchamp, Organometallics, 1985, 4, 2021–2028 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2382421, 2382422 and 2382423. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08818b |
| This journal is © The Royal Society of Chemistry 2025 |