
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArylation of gold nanoclusters and insights into structure-related CO2 reduction reaction performances†
Chen Zhu‡
a,
Bo Li‡b,
Chen Lia,
Luyao Lua,
Hao Li a,
Xinhua Yuana,
Xi Kang*a,
De-en Jiang*b and
Manzhou Zhu*a
a,
Xinhua Yuana,
Xi Kang*a,
De-en Jiang*b and
Manzhou Zhu*a
aDepartment of Chemistry and Centre for Atomic Engineering of Advanced Materials, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei, Anhui 230601, P. R. China. E-mail: kangxi_chem@ahu.edu.cn; zmz@ahu.edu.cn
bDepartment of Chemical and Biomolecular Engineering, Vanderbilt University, 2301 Vanderbilt Place, Nashville, Tennessee, USA. E-mail: de-en.jiang@vanderbilt.edu
First published on 12th May 2025
Abstract
Research on arylgold complexes and ligand-protected gold nanoclusters has proceeded independently thus far due to the difficulty in controllably introducing aryl groups to synthesize arylgold nanoclusters. Herein we synthesized an arylgold Au15 nanocluster, Au15(DPPOE)3(S-PhpMe)4(Ph)2, thereby bridging the two independent research fields. Tetraarylborates were exploited as arylating agents to transfer aryl groups onto the nanocluster kernel, triggering the arylation of the Au15 cluster while maintaining the molecular framework. Furthermore, two other arylgold Au15 nanoclusters with halogenated surfaces were controllably synthesized by substituting the arylating agent NaBPh4 with its benzene ring-halide derivatives. In addition, the change in the electronic structure from Au-SR to Au-aryl and the energetics of the arylation process from Au15-SR to Au15-Ph were elucidated computationally. Furthermore, the catalytic capability of the two Au15 nanoclusters with nuanced ligand differences was investigated in the electrochemical reduction of CO2, and the comparable reactivity of the two cluster-based nanocatalysts was theoretically rationalized. Our findings have cross-fertilized the fields of arylgold complexes and gold nanoclusters, pointing toward a new avenue of exploration for novel arylgold nanoclusters.
1 Introduction
Gold nanoclusters have received extensive attention due to their consistently monodisperse sizes and accurately characterized structures.1–6 These ultrasmall gold nanoparticles exhibit strong quantum-confinement effects and discrete electronic energy levels, which are manifested in their structure-dependent physicochemical properties.7–16 Structurally, gold nanoclusters consist of metallic kernels containing “free electrons” that are protected by motif surfaces that include “peripheral ligands”.17–20 Among them, the peripheral ligands play a significant role in directing the surface chemistry of metal nanoclusters, which further determines their intracluster structures, intercluster interactions, and physicochemical properties.21–25 To date, several types of peripheral ligands have been employed to stabilize gold nanoclusters, including thiolate,26–28 phosphine,29–31 halogens,32–34 alkynyl,35–37 carbene,38–40 and pyridine.41,42 Due to the diverse binding capacities and coordination modes between different peripheral ligands and gold atoms, ligand engineering, as well as ligand exchange, has served as a versatile approach for regulating the electronic/geometric structures of gold nanoclusters.43,44The construction of gold nanoclusters is always associated with the reduction of their AuI complexes; for example, the relationships between AuI-PPh3 complexes and phosphine-stabilized clusters,45,46 AuxI(SR)y complexes and thiolate-stabilized clusters,47,48 AuxI(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) R)y complexes and alkynyl-stabilized clusters,49 etc. The chemistry of arylgold has been developed for more than half a century to anchor arene groups onto gold atoms, giving rise to arylgold complexes with widespread applications in catalysis and emitting devices.50–54 However, little has been achieved in constructing arylgold complex-related arylgold nanoclusters that contain Au0 kernels and free electrons. To our knowledge, there is only one case of arylgold nanoclusters being achieved through ligand exchange with a maintained template—the neutral Au10(C6F5)4(PPh3)5, prepared by reacting Au9(PPh3)8 with arylgold Au(C6F5)2 complexes, and the phenyl-stabilized Au44 was synthesized directly via a one-pot reducing procedure.55,56 The attainment of arylated gold nanoclusters with atomic precision is highly desirable to expand their research scope and allow the fabrication of new cluster members with novel structures, compositions and performances. One idea is to leverage the knowledge and chemistry of arylgold complexes. Indeed, tetraarylborates or their derivatives have been extensively exploited for the construction of arylgold complexes (or other arylmetal complexes) via C–B bond cleavage followed by aryl-gold bond formation;57–59 however, whether such an arylation strategy works for arylgold nanoclusters remains unexplored.
R)y complexes and alkynyl-stabilized clusters,49 etc. The chemistry of arylgold has been developed for more than half a century to anchor arene groups onto gold atoms, giving rise to arylgold complexes with widespread applications in catalysis and emitting devices.50–54 However, little has been achieved in constructing arylgold complex-related arylgold nanoclusters that contain Au0 kernels and free electrons. To our knowledge, there is only one case of arylgold nanoclusters being achieved through ligand exchange with a maintained template—the neutral Au10(C6F5)4(PPh3)5, prepared by reacting Au9(PPh3)8 with arylgold Au(C6F5)2 complexes, and the phenyl-stabilized Au44 was synthesized directly via a one-pot reducing procedure.55,56 The attainment of arylated gold nanoclusters with atomic precision is highly desirable to expand their research scope and allow the fabrication of new cluster members with novel structures, compositions and performances. One idea is to leverage the knowledge and chemistry of arylgold complexes. Indeed, tetraarylborates or their derivatives have been extensively exploited for the construction of arylgold complexes (or other arylmetal complexes) via C–B bond cleavage followed by aryl-gold bond formation;57–59 however, whether such an arylation strategy works for arylgold nanoclusters remains unexplored.
In this work, we report the arylation of gold nanoclusters (Scheme 1). The starting [Au15(DPPOE)3(S-PhpMe)6]+ nanocluster [Au15-SR for short; DPPOE = bis(2-diphenylphosphinophenyl)ether] has two weak surface Au-SPhpMe bonds. The introduction of NaBPh4 into the solution of Au15-SR results in the cleavage of C–B bonds of [BPh4]−, triggered by cluster catalysis. The dissociated phenyl groups attack the two weak Au–S bonds on Au15-SR and displace the corresponding two S-PhpMe groups from the cluster surface, leading to the generation of an arylgold [Au15(DPPOE)3(S-PhpMe)4(Ph)2]+ nanocluster (Au15-Ph for short). We further show that this approach can also be used to synthesize arylgold cluster derivatives, [Au15(DPPOE)3(S-PhpMe)4(Ph-F)2]+ and [Au15(DPPOE)3(S-PhpMe)4(Ph-Cl)2]+ (Au15-Ph-F and Au15-Ph-Cl for shorts, respectively). In addition, we employ computational chemistry to understand the driving force behind the ligand displacement reaction on the cluster surface from Au-SR to Au-aryl. Besides, the catalytic performances of the two Au15 cluster-based nanocatalysts in the CO2 reduction reaction were investigated experimentally and theoretically. Overall, the findings of this work provide impetus for future experimental and theoretical developments in arylgold nanoclusters.
 | ||
| Scheme 1 Illustration of the arylation of gold nanoclusters. The Au15(DPPOE)3(SR)6 cluster precursor undergoes arylation to produce the arylgold Au15(DPPOE)3(SR)4(Ph)2 nanocluster in the presence of [BPh4]−. | ||
2 Results and discussion
The Au15-SR nanocluster was prepared by a one-pot synthetic procedure via reducing the Au-SR-DPPOE complexes with NaBH4 (see the ESI for more details; Fig. S1†). Despite several attempts, the crystallization and structural determination of the Au15-SR nanocluster were unsuccessful due to its instability, which was evaluated by monitoring its optical absorption as a function of time at room temperature. As shown in Fig. S2,† the characteristic UV-vis absorptions of Au15-SR gradually decreased in intensity after three hours and completely disappeared within 24 hours, indicating degradation. With the help of electrospray ionization-mass spectrometry (ESI-MS) and thermogravimetric analysis (TGA), we determined the chemical formula of the nanocluster to be [Au15(DPPOE)3(S-PhpMe)6]+ (Fig. S3 and S4†). To further confirm the valence state of Au15-SR, we performed ESI-MS with the addition of Cs+ cations. The mass results were consistent with those obtained without Cs+, further confirming the chemical formula of the Au15-SR nanocluster as [Au15(DPPOE)3(S-PhpMe)6]+ (Fig. S5†).Inspired by the arylation of tetraarylborates in constructing arylgold complexes, we attempted to anchor the aryl groups onto the Au15-SR surface to substitute them for the two detachable thiolate ligands. As shown in Fig. 1A, NaBPh4 was used as the arylating agent to stabilize the Au15 cluster framework (see more synthetic details in the ESI†). The ESI-MS results suggested the transformation of the nanocluster from Au15-SR to Au15-Ph as well as the substitution of the two detachable thiolate ligands with phenyl groups on the nanocluster surface (Fig. 1B and S6†), demonstrating the successful arylation of the Au15 nanocluster. Of note, in the only one reported case of the arylgold nanocluster, Au10(C6F5)4(PPh3)5, the arylation was achieved by introducing arylgold complexes into the nanocluster precursor, which altered the cluster framework. By comparison, we herein accomplished the template-maintained arylation of gold nanoclusters through a simple ligand-exchange-triggered arylation approach.
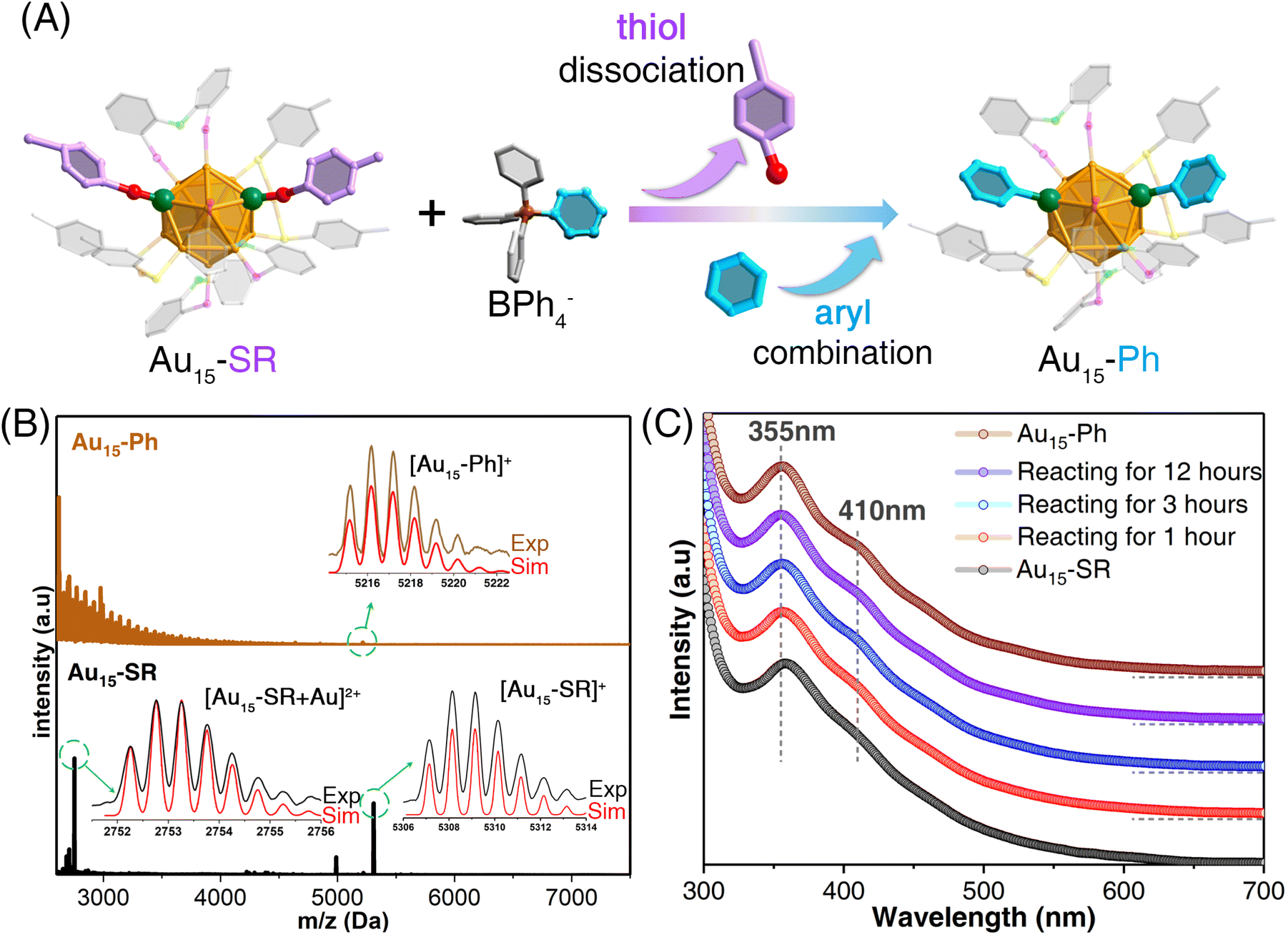 | ||
| Fig. 1 Arylation of the gold nanocluster. (A) Scheme illustrating the arylation of the Au15-SR precursor to produce the arylgold Au15-Ph nanocluster in the presence of [BPh4]−. Accompanying the cluster arylation, two thiolate ligands of Au15-SR were detached, and two aryl ligands were anchored onto the cluster surface. Color labels: orange/green = Au; yellow/red = S; magenta = P; green = O; grey = C; pink = C of the detachable thiolate ligands; cerulean = C of the aryl ligands. Some C and H atoms are omitted for clarity. (B) ESI-MS results of the Au15-SR precursor and the arylgold Au15-Ph nanocluster in the presence of NaBPh4. The stray mass signals at ∼3000 Da stemmed from the introduction of [BPh4]−, which were absent in the mass spectrum of the Au15-Ph cluster (Fig. S6†). (C) Time-dependent optical absorption spectra of the transformation from Au15-SR to Au15-Ph. | ||
The optical absorptions of the Au15-SR and Au15-Ph nanoclusters were compared. An intense optical absorption peak for Au15-SR was detected at 355 nm (Fig. 1C). Following the cluster arylation, the 355 nm signal remained unchanged, and a characteristic band appeared at 410 nm. Optical absorption was also observed for the crystal of Au15-Ph with a similar peak intensity, demonstrating the high conversion efficiency of the cluster arylation. More importantly, the arylgold Au15-Ph displayed a robust cluster template that enabled crystallization (Fig. S7†), and the atomically precise structure of this arylgold nanocluster could be determined. Additionally, the stability of Au15-Ph in O2, at various temperatures, and in different solvents was tested. The nanocluster exhibited good stability in the presence of oxygen and in different solvents. However, the nanocluster decomposed gradually when the temperature exceeded 70 °C (Fig. S8†). Besides, the cyclic voltammetry tests of Au15-SR and Au15-Ph nanoclusters demonstrated that the energy gap between the first oxidation peak and the first reduction peak increased upon arylation of the gold nanocluster (Fig. S9†), which was consistent with the trend observed for the calculated HOMO–LUMO gap of the two nanoclusters (discussed below).
Structurally, the Au15-Ph nanocluster contained an icosahedral Au13 kernel (Fig. 2A), which was enwrapped by three bidentate DPPOE ligands and two dimeric Au1(SR)2 motif structures via Au–P and Au–S interactions, respectively, giving rise to an Au15(DPPOE)3(SR)4 structure (Fig. 2B–D). In this context, of the 12 surface Au atoms on this Au13 kernel, six Au atoms and four Au atoms were capped by phosphine and thiol ligands, respectively, while two Au atoms remained bare on Au15(DPPOE)3(SR)4 (Fig. 2D). Two aryl ligands, originating from the arylating agent NaBPh4, further stabilized the remaining two bare Au atoms, completing the overall structure of the arylgold Au15-Ph nanocluster (Fig. 2E–G). Considering their similar optical absorptions, we propose that the Au15-SR and Au15-Ph nanoclusters exhibited the same cluster framework; accordingly, the two detachable thiolate ligands of Au15-SR should also anchor onto its Au13 kernel independently, and their dissociation would expose the cluster kernel directly to the external environment, leading to the instability of the nanocluster. In contrast, the aryl ligands adhered onto the Au15-Ph cluster surface firmly with the assistance of several intracluster C–H⋯π interactions (Fig. 2H). Specifically, three types of C–H⋯π interactions were detected, including (i) the type I interaction between C–H from aryl ligands and π from phosphine ligands (Fig. 2I), (ii) the type II interaction between C–H from thiol ligands and π from aryl ligands, and (iii) the type III interaction between C–H from aryl ligands and π from thiol ligands (Fig. 2J). Collectively, each aryl ligand was locked by a combination of adjacent DPPOE and Au1(SR)2 ligands via multiple C–H⋯π interactions, realizing the high robustness of the Au15-Ph framework. In addition, we compared the bond lengths of the standard Au13 nanocluster with those of Au15-Ph and found that the Au13 kernel was more tightly arranged than that of Au15-Ph (Table S4†).
 | ||
| Fig. 2 Structural anatomy of the arylgold Au15-Ph nanocluster. (A) The innermost icosahedral Au13 kernel. (B) The bidentate DPPOE cap. (C) The dimeric Au1(SR)2 motif. (D) The Au15(DPPOE)3(SR)4 structure. (E) The aryl ligand. (F and G) The overall structure of the Au15-Ph nanocluster. (H–J) The intracluster C–H⋯π interactions stabilizing the aryl ligands on the nanocluster surface. The interactions are highlighted in colors corresponding to the π-donating groups. Color labels: orange/green = Au; yellow = S; magenta = P; green = O; grey = C; cerulean = C of the aryl ligands; pale magenta = C of the phosphine ligands; pale yellow = C of the thiol ligands; white = H. Some C and H atoms are omitted for clarity. | ||
We further expanded the cluster system of arylgold Au15 by substituting the arylating agent [BPh4]− with its benzene ring-halide derivatives—[B(Ph-F)4]− and [B(Ph-Cl)4]−. As shown in Fig. S10 and S11,† the ESI-MS and UV-vis results demonstrated the successful arylation of Au15-SR and the generation of [Au15(DPPOE)3(S-PhpMe)4(Ph-F)2]+ and [Au15(DPPOE)3(S-PhpMe)4(Ph-Cl)2]+ (Au15-Ph-F and Au15-Ph-Cl for short, respectively). All three arylgold Au15 nanoclusters exhibited the same molecular composition, [Au15(DPPOE)3(S-PhpMe)4(Ph-X)2]+ (X = H, F, or Cl), suggesting their closed-shell electronic structures with a nominal electron count of eight (i.e., 15 (Au) − 4 (SR) − 2 (Ph-X) − 1 (charge) = 8), tallying with the icosahedral unit in the cluster framework. Besides, the X-ray photoelectron spectroscopy (XPS) results showed that the binding energies of Au4f in the three arylgold nanoclusters showed remarkable blue-shifts relative to Au15-SR. (Fig. S12†). The stability of these Au15 nanoclusters bearing different aryl groups was analyzed, and both Au15-Ph-F and Au15-Ph-Cl exhibited excellent stability over 24 hours (Fig. S13†). Structurally, the Au15-Ph-F and Au15-Ph-Cl clusters displayed analogous geometric structures and intramolecular interactions to their initial arylgold Au15-Ph nanocluster (Fig. S14 and S15†); however, due to the substituent effects on the aryl ligands, the three clusters expressed slight differences in terms of their atomic packing and bond lengths. Specifically, the icosahedral Au13 kernels of the three nanoclusters were similar, considering their analogous average lengths of Au(icosahedral core)–Au(icosahedral core) and Au(icosahedral core)–Au(icosahedral surface) bonds (Fig. S16†). However, for the kernel–shell interactions of the cluster framework, a closer interaction was detected for both Au15-Ph-F and Au15-Ph-Cl relative to the Au15-Ph nanocluster due to the shortened bond lengths between Au atoms on the icosahedral surface and S/P/C atoms from peripheral ligands (Fig. S17 and S18†). In this context, the substituent group (i.e., F or Cl) increased the electron-withdrawing capability of the aryl ligand and strengthened the interactions between gold kernels of nanoclusters and peripheral ligands. Besides, the substituent effects also influenced the crystalline packing of these arylgold nanoclusters (Fig. S19†)—although all three cluster entities were crystallized in a triclinic crystal system with a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, they exhibited different crystalline parameters, including cell lengths, cell angles, crystal densities, etc.
space group, they exhibited different crystalline parameters, including cell lengths, cell angles, crystal densities, etc.
For the preparation of arylgold complexes (or other arylmetal complexes), tetraarylborates were extensively exploited as arylating agents to deliver aromatic groups via C–B bond cleavage. For the arylation process from Au15-SR to Au15-Ph, the initiation reaction should be the C–B bond cleavage of [BPh4]− catalyzed by the nanocluster, the same as the reaction between gold complexes and borate species (Fig. S20†).60,61 Indeed, the nuclear magnetic resonance (NMR) and gas chromatography mass spectrometry (GC-MS) results identified BPh3 and biphenyl as by-products of the nanocluster arylation, which were considered as fragment products from the C–B bond cleavage of [BPh4]− (Fig. S21–S24†). Besides, the GC results also identified the presence of benzene in the by-product (Fig. S25†). Interestingly, the Au15-SR nanocluster could catalyze the C–B bond cleavage of tetraarylborates, which further contributed to the nanocluster arylation. However, for the reported nanoclusters with similar thiol and DPPOE ligands, no C–B bond cleavage of the introduced [BPh4]− was observed (Fig. S26†).62 Indeed, the introduction of [BPh4]− in previous cluster research aimed at substituting small-sized counterions (e.g., Cl−) in positively charged nanoclusters for facilitating their crystallization. In this work, the two detachable thiolate ligands on Au15-SR led to its instability, while, the partially exposed gold kernel might endow the cluster molecule with high catalytic capacity towards the C–B bond cleavage of tetraarylborates. In addition, the dissociated aryl group from [BPh4]− further activated the Au15-SR molecule, triggering the complete dissociation of the two detachable thiolate ligands on the cluster surface by anchoring onto these two positions via Au–aryl interactions, giving rise to the final arylgold Au15-Ph nanocluster (Fig. S20†). In this context, the arylation of this Au15 cluster template could be accomplished. Of note, the Au–aryl interactions in Au15-Ph were much more robust than the Au–S interactions at the two ligand-exchanging sites, and the anti-arylation from Au15-Ph to Au15-SR was not successful (Fig. S27†).
To understand the thermodynamic driving force of the arylation process from Au15-SR to Au15-Ph, we employed density functional theory (DFT) calculations to calculate the reaction energy of the arylation process (see the ESI† for more details). Modelling the complete arylation process required determining the speciation of the side products involving SR, which was challenging due to the high complexity of the reaction system. To simplify the process, we used the phenyl group and the thiolate group in their radical states; in other words, the reaction was a measure of the relative binding energy of Au15-Ph versus Au15-SR. The structure of the Au15-Ph was taken from its crystal structure and the Au15-SR cluster was built by substituting -SR for the two -Ph groups. Geometry optimizations were performed before calculating the reaction energy. For Au15-Ph, the DFT optimized structure shows a small deviation (RMSD = 0.4 Å) compared to the crystal structure. The calculated reaction energy for the arylation process from Au15-SR to Au15-Ph was determined to be −33.85 kcal mol−1, suggesting that the reaction [Au15(DPPOE)3(SR)6]+ + 2Ph* → [Au15(DPPOE)3(SR)4Ph2]+ + 2SR* is very favorable; in other words, each Au-Ph bond was about 17 kcal mol−1 more stable than the corresponding Au-SR bond. This finding corroborated our experimental strategy of using phenyl groups to displace the weekly bonded -SR groups on Au15-SR. To understand the origin of the higher stability of Au15-Ph vs. Au15-SR, we analyzed the frontier orbitals of the two clusters. As can be seen from Fig. 3, the Au15-Ph cluster has a slighter larger band gap and a lower HOMO level, indicating its higher stability. More importantly, although both clusters are eight-electron systems according to the superatomic complex model, their HOMO orbitals exhibit very different characteristics: the HOMO of Au15-SR is dominated by the two weakly bound SR groups, while that of Au15-Ph features a combination of the P-type superatomic orbital of the metal core and the -Ph ligands. In other words, the more covalent or conjugated interactions between Au15 and the two -Ph ligands via the Au–C bonds stabilized the cluster, while the two -SR groups had weaker interactions with the valence electrons of the cluster core. The charge density difference plot of the Au15-Ph cluster confirmed the strong charge accumulation between the Au and C atom centers due to the formation of the covalent Au–C bond (Fig. S28†).
 | ||
| Fig. 3 Frontier orbital analyses of nanoclusters. Orbital diagram and HOMO/LUMO orbitals of [Au15(DPPOE)3(SR)6]+ (left) and [Au15(DPPOE)3(SR)4(Ph)2]+ nanoclusters (right). | ||
Considering the nuanced differences in ligands between Au15-SR and Au15-Ph nanoclusters, we investigated their capability in electrochemically reducing CO2 to CO (CO2RR). The electrocatalytic CO2RR was selected as the model reaction due to the outstanding efficiency of gold in this process and the promising prospects it offers for both fundamental and practical investigations.63 The linear sweep voltammetry (LSV) results revealed that Au15-SR displayed a higher current density and a lower onset potential than Au15-Ph in CO2-saturated aqueous 0.5 M KHCO3 solution, indicating that the Au15-Ph cluster-based catalyst showed lower CO2RR efficiency relative to Au15-SR (Fig. 4A). Then, we gathered the gas products produced by the CO2RR process and analyzed them by gas chromatography (GC). Gas chromatography detected only H2 and CO in gas products, demonstrating a total faradaic efficiency (FE) of approximately 100% for CO and H2. As depicted in Fig. 4B, the potential-dependent FE for CO formation was analyzed for two Au15 nanoclusters. Au15-SR revealed a slightly higher selectivity for CO production, achieving the highest remarkable FECO over 96.2% at −0.7 V, compared to 92.9% for Au15-Ph. In other words, Au15-SR and Au15-Ph nanoclusters generated CO across all potentials, but Au15-SR exhibited slightly higher CO production than Au15-Ph (Fig. 4B). In addition, the CO partial current density activity for both Au15-SR and Au15-Ph nanoclusters increased with growing overpotentials (Fig. 4C). Notably, Au15-SR exhibited approximately a two-fold enhancement over Au15-Ph at −0.8 or 0.7 V. The CO production efficiency of Au15-SR was much higher than that of Au15-Ph, showing a comparable turnover frequency (TOF) of the two nanoclusters (Fig. S29†). Additionally, electrochemical impedance spectroscopy (EIS) was performed on Au15-SR and Au15-Ph to access charge-transfer resistance, and the Au15-SR nanocluster exhibited a remarkably smaller semi-circular arc, suggesting its faster electrode kinetics (Fig. 4D). We proposed that the homogeneity of the Au15-SR cluster molecule resulted in smaller charge transfer resistance of the electrolyte and the catalyst, thereby making it easier for electrons to penetrate the catalyst. Furthermore, the electrocatalytic stability of Au15-SR and Au15-Ph was tested at −0.7 V, both showing high robustness over 8 hours (Fig. S30†), further evidenced by their maintained UV-vis, XPS, and transmission electron microscopy results (Fig. S31–S33†).
 | ||
| Fig. 4 Electrochemical performance of Au15-SR and Au15-Ph nanoclusters. (A) The LSV of Au15-SR and Au15-Ph nanoclusters. (B) The CO faradaic efficiency within the range of −1 to −0.6 V vs. the reversible hydrogen electrode (RHE). (C) The CO partial current density (jco). (D) The EIS of Au15-SR and Au15-Ph nanoclusters. | ||
DFT calculations were then carried out to understand the CO2RR mechanism of Au15-SR and Au15-Ph nanoclusters. Following previous studies on the electrocatalysis of gold nanoclusters, the active Au site was created by dethiolating one -SR ligand from nanoclusters.64,65 For Au15-SR, dethiolating the independently anchored ligand was energetically more favorable by 0.76 eV compared to the staple motif ligands (Fig. S34†), leaving the top Au atom from the Au13 kernel as the active site (Fig. 5A). By comparison, for Au15-Ph, the active site was located on the undercoordinated Au atom from the staple motif after dethiolation (Fig. 5B). The Gibbs free energy profile in Fig. 5C shows that CO2 reduction with Au15-SR was thermodynamically more favorable due to the weaker adsorption of intermediates and the lower limiting potential of 0.43 eV, while the stronger adsorption of *CO with Au15-Ph led to a higher limiting potential of 0.77 eV for CO desorption. The calculation results suggested that it is more favorable to form CO in Au15-SR than in Au15-Ph, in agreement with the experimental observations.
 | ||
| Fig. 5 DFT-optimized structures and the corresponding energetics of CO2RR. (A) Optimized structures of *COOH and *CO intermediates for singly dethiolated [Au15(DPPOE)3(SR)3(Ph)2]+ and (B) singly dethiolated [Au15(DPPOE)3(SR)5]+ nanoclusters. (C) Free energy profile of the CO2RR on dethiolated [Au15(DPPOE)3(SR)3(Ph)2]+ (blue color) and [Au15(DPPOE)3(SR)5]+ (red color) nanoclusters at 0 V vs. RHE. | ||
3 Conclusions
In summary, the arylation of gold nanoclusters has been accomplished. The Au15-SR nanocluster carried two detachable thiolate ligands on its surface, accounting for the instability of the cluster framework. On the positive side, the active surface environment of Au15-SR endowed this nanocluster with high catalytic capacity towards the C–B bond cleavage of tetraarylborates. The subsequently generated dissociative aryl groups would activate the surface structure of Au15-SR by anchoring aryl ligands onto the cluster kernel via Au-aryl interactions, giving rise to an arylgold Au15-Ph nanocluster. Besides, two cluster derivatives of arylgold Au15-Ph were developed by functionalizing the aryl ligands with halide substituents. In addition, DFT calculations confirmed the thermodynamic driving force underlying the arylation process from Au15-SR to Au15-Ph and rationalized the higher stability of the Au15-Ph cluster by comparing its frontier orbitals with those of Au15-SR. Furthermore, both Au15 cluster-based nanocatalysts displayed comparable faradaic efficiency in CO2RR, while Au15-SR exhibited superior CO generation efficiency over Au15-Ph, as derived from experimental results and rationalized by theoretical calculations. Overall, the findings in this work have significant implications for research in both metal nanoclusters and arylgold complexes, and we hope these observations will provide valuable insights for further studies of arylgold nanoclusters.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
C. Zhu conceived and carried out the experiments. B. Li performed the theoretical simulations. C. Li, L. Lu, H. Li, and X. Yuan assisted in the synthesis and CO2RR measurements. X. Kang, D.-e. Jiang, and M. Zhu supervised the project. All authors commented on and agreed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the NSFC (22371003, 22101001, and 22471001), the Ministry of Education, Natural Science Foundation of Anhui Province (2408085Y006), the University Synergy Innovation Program of Anhui Province (GXXT-2020-053), and the Scientific Research Program of Universities in Anhui Province (2022AH030009). D.-e. Jiang acknowledges Vanderbilt University for support.Notes and references
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 Search PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 Search PubMed.
- N. A. Sakthivel and A. Dass, Acc. Chem. Res., 2018, 51, 1774–1783 Search PubMed.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320, 238–250 Search PubMed.
- S. Kenzler and A. Schnepf, Chem. Sci., 2021, 12, 3116–3129 Search PubMed.
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 Search PubMed.
- Z. Liu, H. Tan, B. Li, Z. Hu, D.-e. Jiang, Q. Yao, L. Wang and J. Xie, Nat. Commun., 2023, 14, 3374 Search PubMed.
- W.-D. Si, C. Zhang, M. Zhou, W.-D. Tian, Z. Wang, Q. Hu, K.-P. Song, L. Feng, X.-Q. Huang, Z.-Y. Gao, C.-H. Tung and D. Sun, Sci. Adv., 2023, 9, eadg3587 Search PubMed.
- P. Luo, X.-J. Zhai, S. Bai, Y.-B. Si, X.-Y. Dong, Y.-F. Han and S.-Q. Zang, Angew. Chem., Int. Ed., 2023, 62, e202219017 Search PubMed.
- J. Zhao, A. Ziarati, A. Rosspeintner and T. Bürgi, Angew. Chem., Int. Ed., 2023, 63, e202316649 Search PubMed.
- J. Tang, S. Zhang, B.-W. Zhou, W. Wang and L. Zhao, J. Am. Chem. Soc., 2023, 145, 23442–23451 Search PubMed.
- X.-L. Pei, P. Zhao, H. Ube, Z. Lei, K. Nagata, M. Ehara and M. Shionoya, J. Am. Chem. Soc., 2022, 144, 2156–2163 Search PubMed.
- W. Fei, S. Antonello, T. Dainese, A. Dolmella, M. Lahtinen, K. Rissanen, A. Venzo and F. Maran, J. Am. Chem. Soc., 2019, 141, 16033–16045 Search PubMed.
- G. Yang, Z. Wang, F. Du, F. Jiang, X. Yuan and J. Y. Ying, J. Am. Chem. Soc., 2023, 145, 11879–11898 Search PubMed.
- C. Zhou, M. Wang, Q. Yao, Y. Zhou, C. Hou, J. Xia, Z. Wang, J. Chen and J. Xie, Aggregate, 2024, 5, e401 Search PubMed.
- Z. Liu, J. Chen, B. Li, D.-e. Jiang, L. Wang, Q. Yao and J. Xie, J. Am. Chem. Soc., 2024, 146, 11773–11781 Search PubMed.
- H. Häkkinen and M. Walter, J. Phys. Chem. B, 2006, 110, 9927–9931 Search PubMed.
- Y. Horita, S. Hossain, M. Ishimi, P. Zhao, M. Sera, T. Kawawaki, S. Takano, Y. Niihori, T. Nakamura, T. Tsukuda, M. Ehara and Y. Negishi, J. Am. Chem. Soc., 2023, 145, 23533–23540 Search PubMed.
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 Search PubMed.
- Y. Li and R. Jin, J. Am. Chem. Soc., 2020, 142, 13627–13644 Search PubMed.
- Z. Xu, H. Dong, W. Gu, Z. He, F. Jin, C. Wang, Q. You, J. Li, H. Deng, L. Liao, D. Chen, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2023, 62, e202308441 Search PubMed.
- G. Soldan, M. A. Aljuhani, M. S. Bootharaju, L. G. AbdulHalim, M. R. Parida, A.-H. Emwas, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 5749 Search PubMed.
- Y. Zhang, S.-R. He, Y. Yang, T.-S. Zhang, Z.-M. Zhu, W. Fei and M.-B. Li, J. Am. Chem. Soc., 2023, 145, 12164–12172 Search PubMed.
- T.-H. Chiu, J.-H. Liao, Y.-Y. Wu, J.-Y. Chen, Y. Chen, X. Wang, S. Kahlal, J.-Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2023, 145, 16739–16747 Search PubMed.
- V. Rück, N. K. Mishra, K. K. Sørensen, M. B. Liisberg, A. B. Sloth, C. Cerretani, C. B. Mollerup, A. Kjaer, C. Lou, K. J. Jensen and T. Vosch, J. Am. Chem. Soc., 2023, 145, 16771–16777 Search PubMed.
- M. Suyama, S. Takano and T. Tsukuda, J. Am. Chem. Soc., 2023, 145, 3361–3368 Search PubMed.
- N. A. Sakthivel, M. Shabaninezhad, L. Sementa, B. Yoon, M. Stener, R. L. Whetten, G. Ramakrishna, A. Fortunelli, U. Landman and A. Dass, J. Am. Chem. Soc., 2020, 142, 15799–15814 Search PubMed.
- L.-J. Liu, F. Alkan, S. Zhuang, D. Liu, T. Nawaz, J. Guo, X. Luo and J. He, Nat. Commun., 2023, 14, 2397 Search PubMed.
- X. Kang, X. Wei, S. Wang and M. Zhu, Inorg. Chem., 2020, 59, 8736–8743 Search PubMed.
- J. Chen, Q.-F. Zhang, T. A. Bonaccorso, P. G. Williard and L.-S. Wang, J. Am. Chem. Soc., 2014, 136, 92–95 Search PubMed.
- D. Arima and M. Mitsui, J. Am. Chem. Soc., 2023, 145, 6994–7004 Search PubMed.
- Y. Shichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 Search PubMed.
- Z.-H. Gao, J. Dong, Q.-F. Zhang and L.-S. Wang, Nanoscale Adv., 2020, 2, 4902–4907 Search PubMed.
- J.-J. Li, Z.-J. Guan, S.-F. Yuan, F. Hu and Q.-M. Wang, Angew. Chem., Int. Ed., 2021, 60, 6699 Search PubMed.
- N. Kobayashi, Y. Kamei, Y. Shichibu and K. Konishi, J. Am. Chem. Soc., 2013, 135, 16078–16081 Search PubMed.
- J.-J. Li, Z. Liu, Z.-J. Guan, X.-S. Han, W.-Q. Shi and Q.-M. Wang, J. Am. Chem. Soc., 2022, 144, 690–694 Search PubMed.
- L. Qin, F. Sun, X. Ma, G. Ma, Y. Tang, L. Wang, Q. Tang, R. Jin and Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 26136–26141 Search PubMed.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C.-T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Häkkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 Search PubMed.
- H. Shen, G. Deng, S. Kaappa, T. Tan, Y.-Z. Han, S. Malola, S.-C. Lin, B. K. Teo, H. Häkkinen and N. Zheng, Angew. Chem., Int. Ed., 2019, 58, 17731–17735 Search PubMed.
- V. K. Kulkarni, B. N. Khiarak, S. Takano, S. Malola, E. L. Albright, T. I. Levchenko, M. D. Aloisio, C.-T. Dinh, T. Tsukuda, H. Häkkinen and C. M. Crudden, J. Am. Chem. Soc., 2022, 144, 9000–9006 Search PubMed.
- S.-F. Yuan, Z. Lei, Z.-J. Guan and Q.-M. Wang, Angew. Chem., Int. Ed., 2021, 60, 5225–5229 Search PubMed.
- S.-F. Yuan, C.-Q. Xu, J. Li and Q.-M. Wang, Angew. Chem., Int. Ed., 2019, 58, 5906–5909 Search PubMed.
- X. Kang and M. Zhu, Chem. Mater., 2019, 31, 9939–9969 Search PubMed.
- C. A. Hosier and C. J. Ackerson, J. Am. Chem. Soc., 2019, 141, 309–314 Search PubMed.
- S. Kenzler, F. Fetzer, C. Schrenk, N. Pollard, A. R. Frojd, A. Z. Clayborne and A. Schnepf, Angew. Chem., Int. Ed., 2019, 58, 5902–5905 Search PubMed.
- Q.-F. Zhang, X. Chen and L.-S. Wang, Acc. Chem. Res., 2018, 51, 2159–2168 Search PubMed.
- H. Yang, Y. Wang, J. Yan, X. Chen, X. Zhang, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2014, 136, 7197–7200 Search PubMed.
- P. N. Gunawardene, J. F. Corrigan and M. S. Workentin, J. Am. Chem. Soc., 2019, 141, 11781–11785 Search PubMed.
- X.-K. Wan, Z.-J. Guan and Q.-M. Wang, Angew. Chem., Int. Ed., 2017, 56, 11494–11497 Search PubMed.
- A. Genoux, M. Biedrzycki, E. Merino, E. Rivera-Chao, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2021, 60, 4164–4168 Search PubMed.
- M.-C. Tang, C.-H. Lee, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 9341–9349 Search PubMed.
- M.-C. Tang, M.-Y. Leung, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 13115–13124 Search PubMed.
- L. Laurentius, S. R. Stoyanov, S. Gusarov, A. Kovalenko, R. Du, G. P. Lopinski and M. T. McDermott, ACS Nano, 2011, 5, 4219–4227 Search PubMed.
- F. Mirkhalaf, J. Paprotny and D. J. Schiffrin, J. Am. Chem. Soc., 2018, 128, 7400–7401 Search PubMed.
- A. Laguna, M. Laguna, M. C. Gimeno and P. G. Jones, Organometallics, 1992, 11, 2759–2760 Search PubMed.
- W.-D. Si, C. Zhang, M. Zhou, Z. Wang, L. Feng, C.-H. Tung and D. Sun, Sci. Adv., 2024, 10, eadm6928 Search PubMed.
- A. Sladek, S. Hofreiter, M. Paul and H. Schmidbaur, J. Organomet. Chem., 1995, 501, 47–51 Search PubMed.
- H. Salem, L. J. W. Shimon, G. Leitus, L. Weiner and D. Milstein, Organometallics, 2008, 27, 2293–2299 Search PubMed.
- W. V. Konze, B. L. Scott and G. J. Kubas, Chem. Commun., 1999, 1807–1808 Search PubMed.
- M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022–15045 Search PubMed.
- F. Yue, H. Ma, P. Ding, H. Song, Y. Liu and Q. Wang, ACS Cent. Sci., 2023, 9, 2268–2276 Search PubMed.
- C. Zhu, J. Xin, J. Li, H. Li, X. Kang, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202205947 Search PubMed.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Naira and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 Search PubMed.
- D. R. Alfonso, D. Kauffman and C. Matranga, J. Chem. Phys., 2016, 144, 184705 Search PubMed.
- F. Sun, L. Qin, Z. Tang, G. Deng, M. S. Bootharaju, Z. Wei, Q. Tang and T. Hyeon, Chem. Sci., 2023, 14, 10532–10546 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S33 and Tables S1–S5. CCDC 2307548–2307550. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01200g |
| ‡ C. Z. and B. L. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |