
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Valorisation of citrus waste for sustainable synthesis of carbon-supported copper nanoparticles active in CO2 electroreduction†
Federica
De Luca
a,
Palmarita
Demoro
a,
Izuchica
Nduka
b,
Cristina
Italiano
c,
Salvatore
Abate.
 *a and
Rosa
Arrigo
*b
*a and
Rosa
Arrigo
*b
aERIC aisbl, CASPE/INSTM, Dpt. ChiBioFarAM University of Messina, Viale F. Stagno D'Alcontres 31, Messina, 98166, Italy. E-mail: abates@unime.it
bSchool of Science, Engineering and Environment, University of Salford, M5 4WT, Manchester, UK. E-mail: r.arrigo@salford.ac.uk
cNational Research Council of Italy (CNR-ITAE), Salita Santa Lucia Sopra Contesse 5, 98126, Messina, Italy
First published on 27th December 2024
Abstract
This study describes a microwave-assisted hydrothermal method to synthesise carbon-supported Cu-based electrocatalysts for CO2 conversion using citrus peels as both the carbon precursor and the reducing agent for Cu cations. XPS, TEM, and XRD analyses reveal the structural heterogeneity of the samples, resulting from a complex chemistry influenced by both the type of citrus peel used and the Cu salt precursor. As a result, mixed Cu/Cu2O nanoparticles form, which are immobilized on the surface or embedded within the carbon matrix. Orange peel-derived systems exhibit an optimal graphitic-to-defective carbon ratio, resulting in an optimal porosity, electron conduction, and Cu stabilisation, leading to superior CO2 reduction performance. A Cu sulphate-derived catalyst supported on orange peel-derived carbon yields the best performance for CO and methane production, shedding light on specific structural characteristics of the catalysts precursor state able to generate in situ an active phase with improved performance. This work demonstrates the potential of orange peel waste as a sustainable feedstock for the production of CO2 reduction electrocatalysts, offering a green strategy for waste valorisation and clean energy technologies.
Sustainability spotlightCarbon materials are increasingly important for their use in energy storage and conversion technologies. This is due to a tuneable surface chemistry and a high electrical conductivity, which make them ideal materials for hosting metal electroactive species. However, with the prospect of increased use, carbon must be sourced sustainably. Waste biomass offers a sustainable alternative to fossil carbon and is abundantly available. Their reuse as raw materials for carbon manufacturing would reduce the impact of their disposal in the environment as well as associated costs. However, the structure of the C materials and the immobilised metal active species needs to be tailored to the specific technological application. At the same time, waste biomass, such as citrus waste is highly heterogeneous in nature and rich in active chemical constituents, with specific chemical properties. Thus the conversion of waste biomass into carbon-based materials requires an optimised synthetic protocol. In this study, the application of citrus waste is explored for the synthesis of electrocatalytic carbon-supported Cu nanostructured materials for CO2 electrolysis to value added compounds. The CO2 electroreduction reaction is of pivotal importance to produce C-based fuels sustainably. Herein we advance the knowledge on how the biomass constituents as well as the Cu precursors interact during synthesis to yield electrocatalysts active in CO2 reduction. We identify specific elements that lead to improved performances establishing direction in materials design starting from waste biomass. This work represents the first necessary step for the effective and sustainable reuse of waste biomass and aligns with the UN SDG 12, responsible consumption and production. |
Introduction
The remarkable versatility of carbon for various applications is due to its diverse dimensionality, structure and texture.1 Properties such as a high surface area, tuneable surface chemistry, and a high electrical conductivity make carbon materials ideal candidates for hosting metal active species for various applications, including energy conversion and storage.2 In CO2 electroreduction by carbon-supported Cu-based nanoparticles (NPs), the choice of the carbon support significantly influences the product distribution.3–5 The presence of in-plane vacancies as well as heteroatom doping6,7 in the graphitic domains of the carbon support alter the electronic structure of the support itself and can induce specific interactions with immobilised metal active species. This, in turn, influences their behaviour towards the adsorption and activation of CO2 molecules, as well as the electron and proton transfer.8,9 Copper nanoparticles supported on graphene showed interesting activity and selectivity for CH4 production, attributed to the participation of the support via hydrogen spill-over to the metal sites.10 The selectivity towards ethylene was reported to increase when moving from single wall carbon nanotubes to reduced graphene oxide and onion-like carbon.3–5 The enhanced performance of onion-like carbon in C–C coupling was attributed to the unique catalyst design, where the shell of the onion-like carbon surrounding the Cu-based NPs was suggested to electro-reduce CO2 to CO, thereby increasing the surface concentration of CO on adjacent NPs.3 Waste biomass-derived carbon offers a sustainable alternative to fossil carbon (e.g. coal and charcoal) due to its inherent renewability and low cost.11,12 Global agricultural waste production is estimated to reach 500 million tons per annum.13 This vast quantity of underutilized resources represents a substantial and sustainable source of raw material. Additionally, the reuse of waste streams reduces operational costs associated with waste disposal.14 In a previous study,15 we demonstrated that the choice of the biomass as a feedstock for electrode material manufacturing is application-dependent and requires a rational approach for effective utilisation. This is due to the fact that biomass is rich in soluble chemical compounds that can be leveraged for the in situ generation of metallic electroactive species. Compared to impregnation and sol-immobilisation methods, this synthetic approach enables a more extensive interaction of solution-based Cu species with the C-precursor during the simultaneous formation of both the graphitic C matrix and the metal NPs, resulting in a composite material. This contrasts with impregnation methods, where the interactions between the C and metal species are confined to the external surface or accessible porosity of the C support particles. By exploring the use of citrus waste as a feedstock, we produced C-supported Cu-based-nanomaterials16,17 by a one pot synthesis and we have demonstrated that citrus waste is two-fold advantageous: (i) it serves as the carbon support for copper NPs, and (ii) it provides the reducing compounds (e.g. ascorbic acid) required for the reduction of solution-based cationic copper species. Noteworthily, orange peel contains a large quantity of ascorbic acid as well as other epicatechin compounds,15 which have yielded Cu-based electrocatalysts active in CO2 electroreduction to value-added compounds such as formic acid and oxalate. In this study, we further explore the one-pot microwave-assisted synthesis of carbon (C)-supported copper-based NPs with the aim to further enhance the performances of these electrocatalysts for the CO2 reduction reaction (CO2RR). To this end, we use commercially available citrus peels (lemon and orange) and evaluate the influence of various copper precursors (sulphate, nitrate, and chloride) on the structural characteristics of the supported NPs. We use microwave-assisted hydrothermal synthesis to reduce the synthesis time and achieve a more homogeneous temperature profile within the reactive mixture, compared to conventional heating.18 We expect that the molecular level interactions occurring between the peel carbonaceous matrix and the cationic and anionic species from the salt precursor solutions during synthesis will influence the topological and chemical characteristics of the synthetised materials, offering an opportunity for fine-tuning of the performance.19,20 This work aims to unveil these aspects with a comprehensive characterisation of the synthesised materials using scanning electron microscopy (SEM) for morphological analysis, X-ray photoelectron spectroscopy (XPS) for electronic structural analysis, and X-ray diffraction (XRD) and transmission electron microscopy (TEM) for structural characterisation. Moreover, we use liquid chromatography-mass spectrometry (LC-MS) to characterise the nature of the chemical species extracted from the peels during the hydrothermal synthesis, offering valuable insights to help explain our results and optimise the synthesis process for better control and scalability. This in-depth structural analysis, in conjunction with the evaluation of the electrocatalyst performances, aims to identify promising avenues for the sustainable valorisation of biomass waste for the development of functional materials for energy applications.Results and discussion
The bulk and surface chemical compositions of the samples are summarised in Table 1. The surface abundance of copper exhibits minor variations depending on the copper precursor employed and remains relatively invariant irrespective of the citrus peel utilized. The T-CuCl2-O sample presents the highest copper surface abundance of 5.30 wt%. The total copper content, as determined by energy-dispersive X-ray spectroscopy (EDS) is markedly higher than the surface abundance. For the samples with the highest copper loading, specifically T-CuSO4-L, the overall content reaches 47.2%. The discrepancies between the overall bulk content and the surface content are due to the varying detection sensitivities of the two methods. Specifically, XPS probes only the topmost layer, up to 10 nm in thickness, which means that the largest part of the particles is not detected, especially those within the pores of the carbon support. In contrast, these particles are the primary contributors to the EDS signal.| Sample | C | O | Cu | Cl | N | S |
|---|---|---|---|---|---|---|
| (a) Surface elemental analysis | ||||||
| T-CuCl2-L | 73.52 (±0.12%) | 20.72 (±0.15%) | 5.23 (±0.20%) | 0.52 (±0.10%) | — | — |
| T-CuCl2-O | 77.52 (±0.13%) | 16.18 (±0.16%) | 5.30 (±0.20%) | 1.01 (±0.10%) | — | — |
| T-Cu(NO3)2-L | 80.53 (±0.1%) | 13.81 (±0.1%) | 3.34 (±0.20%) | — | 2.31 (±0.19%) | — |
| T-Cu(NO3)2–O | 77.75 (±0.14%) | 17.07 (±0.18%) | 3.45 (±0.21%) | — | 1.73 (±0.21%) | — |
| T-CuSO4-L | 84.56 (±0.11%) | 13.73 (±0.19%) | 1.64 (±0.23%) | — | — | 0.07 (±0.03%) |
| T-CuSO4-O | 79.95 (±0.14%) | 15.67 (±0.15%) | 3.45 (±0.24%) | — | — | 0.93 (±0.15%) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| (b) Bulk elemental analysis | ||||||
| T-CuCl2-L | 31.2 (±0.1%) | 29.4 (±0.1%) | 36.4 (±0.1%) | 2.9 (±0.4%) | — | — |
| T-CuCl2-O | 35.5 (±0.1%) | 31.3 (±0.1%) | 33.2 (±0.1%) | — | — | — |
| T-Cu(NO3)2-L | 31.4 (±0.1%) | 31.2 (±0.1%) | 30.3 (±0.1%) | — | 6.7 (±0.2%) | 0.4 (±0.5%) |
| T-Cu(NO3)2–O | 28.9 (±0.1%) | 31.3 (±0.1%) | 34.5 (±0.1%) | — | 4.7 (±0.2%) | 0.5 (±0.5%) |
| T-CuSO4-L | 32.6 (±0.1%) | 19.3 (±0.1%) | 47.2 (±0.1%) | — | — | 0.8 (±0.5) |
| T-CuSO4-O | 33.3 (±0.1%) | 24.1 (±0.1%) | 41.4 (0.1%) | — | — | 1.1 (±0.5%) |
Fig. 1(a, c and e) displays the back-scattered electron (BSE)-SEM images of the electrocatalysts supported on orange peel-derived carbon prepared using chloride, nitrate, and sulphate precursors, respectively. Fig. 1(b, d and f) shows the SEM images of the electrocatalysts prepared with the same precursors but supported on lemon peel-derived carbon. Accordingly, the primary particles of the carbon supports are characterised by a flake-like morphology, consistent with a turbostratic structure typical of graphitic materials when there is disorder in the stacking of the layers. This stacking generates a hierarchical porous structure, highly heterogeneous in pore size and shape. Moreover, the primary carbon particles are highly heterogeneous in size, with the nitrate-derived samples appearing to contain the smallest primary carbon particles. The contrast variations observed in the SEM images, where the brighter regions correspond to Cu-based NPs, indicate that these are located on the side edges of the C flakes and are larger for the chloride-derived samples, more so when supported on the lemon peel-derived system. The SEM image of the nitrate precursor sample shows Cu-based NPS with the smallest particle size amongst the lemon peel-derived samples. The differences in particle dispersion from the SEM images are more subtle for the orange peel-derived electrocatalysts.
 | ||
| Fig. 1 SEM images recorded with a Phenom ProX Desktop instrument at 10 kV acceleration voltage and at ×2000 magnification in back-scattered electron (BSE) mode: (a) T-CuSO4-O (b) T-CuSO4-L (c) T-CuCl2-O, (d) T-CuCl2-L, (e) T-Cu(NO3)2–O, and (f) T-Cu(NO3)2-L. | ||
X-ray diffraction (XRD) patterns of the synthesized materials are presented in Fig. 2. Noteworthily, the samples exhibit characteristic diffraction peaks corresponding to crystalline Cu and Cu2O, along with a broad peak attributed to the carbonaceous graphitic-like support. The diffraction peaks at 2θ values of about 43.3°, 50.34°, and 74.08° correspond to the crystallographic planes (111), (200), and (220) of metallic Cu,21 respectively (JCPDS no. 04-0836). The peaks at 36.43° and 61.71° can be attributed to the (111) and (220) planes of Cu2O (JCPDS no. 05-0667).17 No additional crystalline phases were identified in the XRD patterns. Table 2 summarises the sizes of the planes in the different phases.
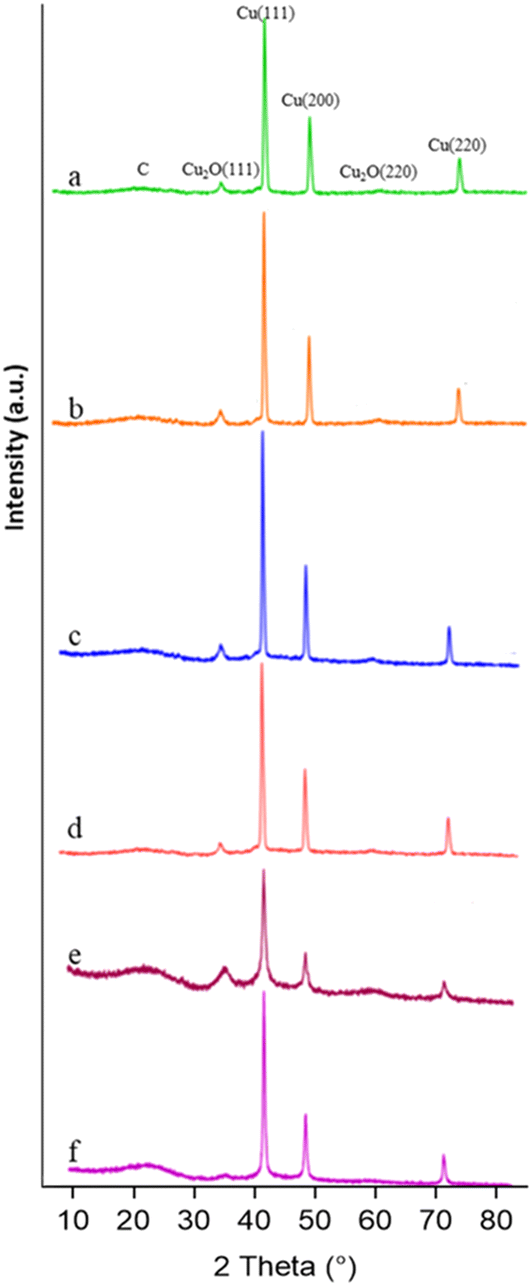 | ||
| Fig. 2 X-ray diffraction patterns of the as-prepared samples in the 2θ range of 10° to 85°. From top to bottom: (a) T-CuSO4-L, (b) T-CuSO4-O, (c) T-CuCl2-L, (d) T-CuCl2-O (e) T-Cu(NO3)2-O, and (f) T-Cu(NO3)2-L. | ||
| Sample | Cu2O (111) | Cu0 (111) | Cu0 (200) | Cu2O (220) | Cu0 (220) |
|---|---|---|---|---|---|
| T-CuCl2-L | 13.2 | 23.1 | 21.6 | 11.3 | 20.9 |
| T-CuCl2-O | 12.1 | 25.2 | 24.9 | 9.7 | 23.4 |
| T-(CuNO3)2-L | 9.0 | 19.2 | 20.7 | 15.6 | 9.4 |
| T-(CuNO3)2-O | 4.8 | 17.5 | 18.4 | 4.7 | 16.5 |
| T-CuSO4-L | 7.5 | 19.6 | 20.3 | 7.7 | 23.1 |
| T-CuSO4-O | 11.3 | 22.5 | 20.4 | 4.3 | 25.2 |
The analysis of the sizes of the different planes indicates larger metallic domains and smaller cuprous oxide domains when the chloride or sulphate precursors are used, more so for the orange peel-derived sample. The size variation of the different planes within the same phase further signifies morphological differences among the samples. When using the nitrate precursor, both Cu0 and Cu2O phases comparatively present the smallest crystallite size, consistent with the SEM analysis shown in Fig. 1.
The TEM analysis provides further insights into the structural characteristics of the samples.
Fig. 3 shows the TEM micrographs of the as-prepared T-CuSO4-O at different magnifications. Carbon is found throughout the sample, as indicated by the arrows in b and e. The Cu phase is composed of primary particles, which aggregate in a highly heterogeneous manner, forming rounded core–shell superstructures measuring approximately 100–200 nm in diameter (Fig. 3a–d). These structures feature a less dense or voided core, while the Cu-based primary particles are concentrated in the shell. The size of the primary particles qualitatively aligns with the length of the crystalline planes determined by XRD. The core–shell particles exhibit a lattice fringe spacing of 0.245 nm (Fig. 3d), indexed to the mainly exposed (111) crystal planes of Cu2O (JCPDS card no. 74-1230). In addition to the lattice fringes of Cu2O, spherical agglomerates show a lattice fringe spacing of 0.208 nm (Fig. 3f), corresponding to the (111) plane of metallic Cu (JCPDS card no. 01-1241). Fig. 3e shows that in some areas the primary particles are agglomerated with no specific order and decorate a carbon particle. Overall, these findings are consistent with the XRD analysis (Fig. 2), providing a comprehensive understanding of the morphological characteristics of the sample. The TEM analysis of the samples derived from the orange peel using Cu chloride and Cu nitrate as precursors is presented in Fig. S1.†
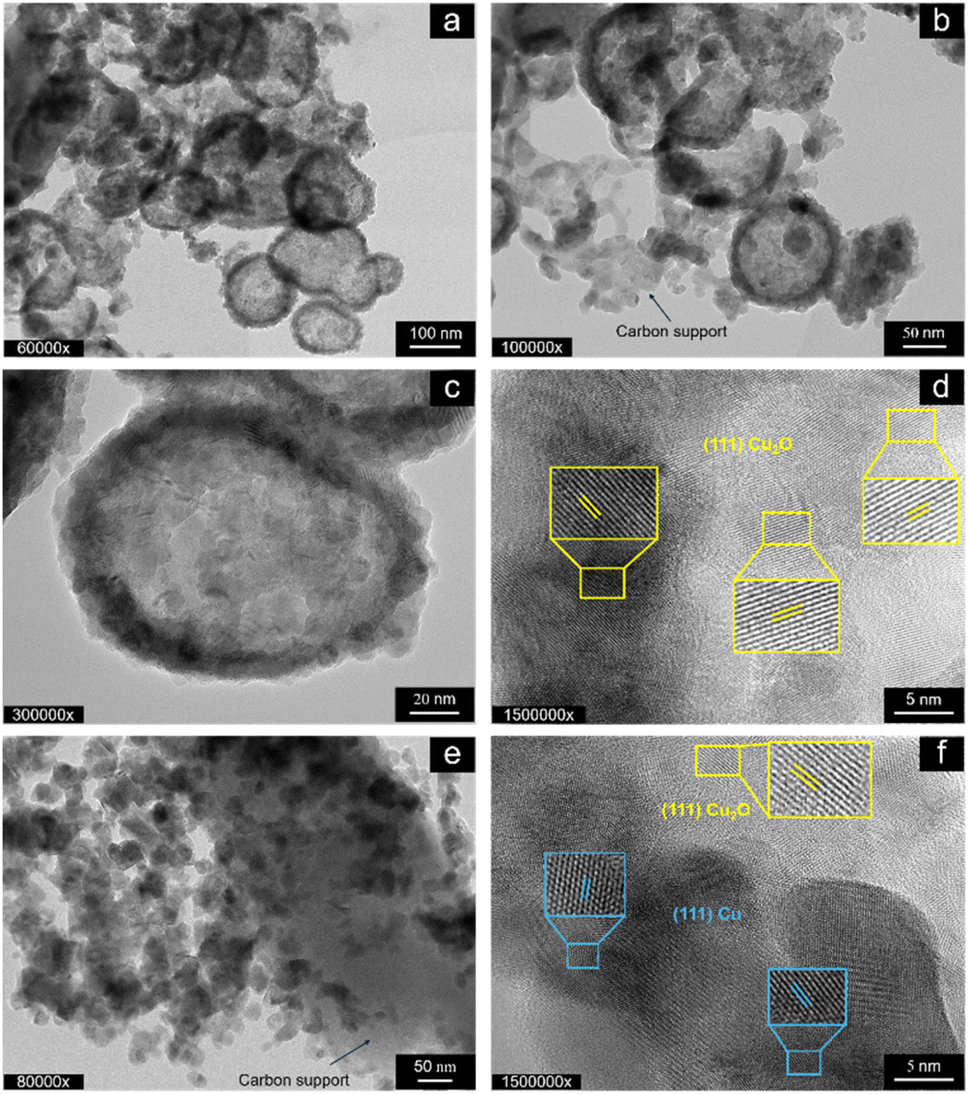 | ||
| Fig. 3 HR-TEM micrographs of as-prepared T-CuSO4-O. The sample is characterised by voided Cu2O/Cu NPs (a) with C particulate attached to the external surface of the NPs (b) exhibiting a filamentous morphology (as indicated by the arrow). The HRTEM image in (c) shows the size and arrangement of the primary particles forming the voided secondary particles, whereas (d) shows the lattice fringes characteristic of Cu2O. Additionally, the sample contains solid-core primary particles agglomerated around a larger C particles. The HRTEM image in (f) shows the lattice fringes characteristic of Cu2O and Cu within the primary particles agglomerates. | ||
Cu chloride yields rounded particles of varying sizes, spanning one order of magnitude in length, most of which are voided core-shell particles (Fig. S1a†). The particles with a solid core are found mainly at the surface of the carbon support particles (Fig. S1b†), suggesting that the voided particles are embedded within the pores of the carbon support, with voids forming due to the Kirkendall effect.22 This contrasts with T-CuSO4-O, which generally presents larger particles external to the carbon support. Fig. S1b† shows an example of Cu-based NPs with an ill-defined shape covering the carbon support, which exhibits graphite-like layers. The sample derived from Cu nitrate shows particles decorating the flake-like carbon particle (Fig. S1c†), which are generally solid in the core, although smaller voided particles are also observed. These nanoparticles are much smaller for the nitrate-derived sample than in the previous cases, consistent with the SEM images in Fig. 1. TEM analysis of lemon-peel derived samples is presented in Fig. S2.† The sulphate-derived sample (Fig. S2a†) shows the same characteristic particles, homogeneously distributed on the carbon support, with some voided particles and others featuring a metallic core with an oxide shell (Fig. S2b†). This sample differs significantly from T-CuSO4-O, which shows unique morphological and structural characteristics in Cu speciation among the investigated samples, indicating a specific interplay between the nature of the peel and the Cu precursor during synthesis. In contrast, the chloride- and nitrate-derived samples exhibit Cu-based nanoparticles with nanostructural and morphological characteristics similar to those of their orange peel-derived counterparts, indicating that the peel plays a less significant role than the anion in determining the properties in these cases.
The electronic structure of the fresh samples was investigated by XPS at the Cu 2p, C 1s, and O 1s core levels (Fig. 4).
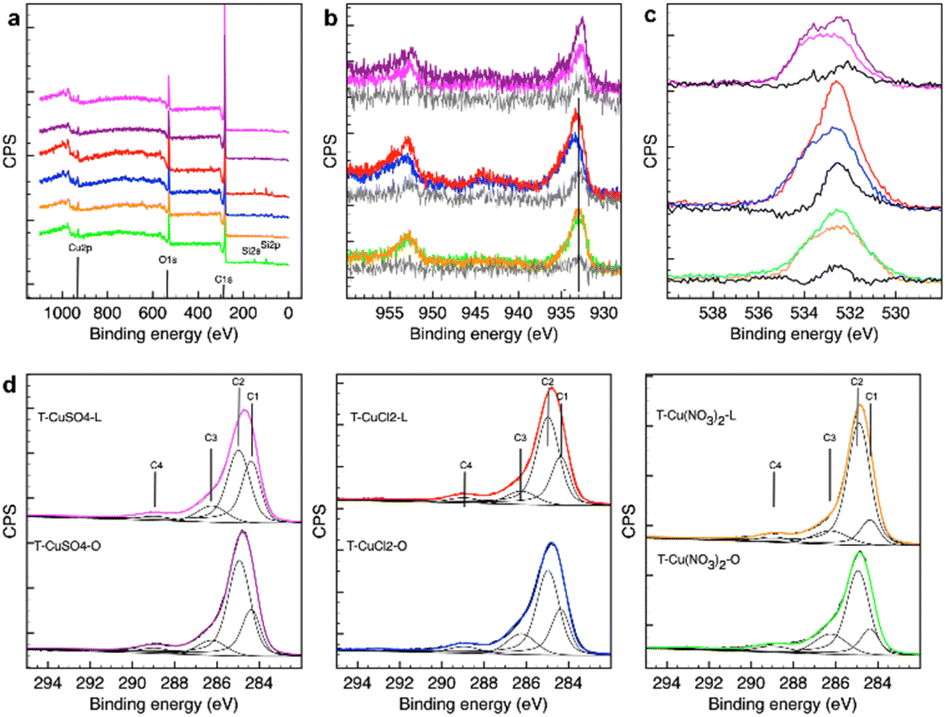 | ||
| Fig. 4 XPS analysis of the samples investigated using a PHI VersaProbe II (Physical Electronics), equipped with an Al Kα (1486.6 eV) X-ray source. (a) Survey spectra; (b) Cu 2p3/2; (c) O 1s, and (d) C 1s. The colour code is consistent in this figure as follows: T-CuSO4-L (magenta), T-CuSO4-O (purple), T-CuCl2-L (red), T-CuCl2-O (blue), T-Cu(NO3)2-L (orange), and T-Cu(NO3)2-O (green). | ||
The survey spectra in Fig. 4a indicate a surface composition consisting of mostly C, O and Cu, with Si impurities observed in some cases.
The Cu 2p XPS spectra (Fig. 4b) show a subtle difference in Cu chemical speciation for the samples derived from the nitrate precursor. The binding energy (BE) of the main 2p3/2 peak is found to be at 933 eV and no satellites features are present, indicating a reduced state of the catalysts. However, Cu0 and Cu2O are difficult to distinguish by Cu 2p XPS, with the Cu 2p3/2 peak for these species at a very similar BE of 932.6 eV and 932.4 eV, respectively. The value found in this work falls well within the range reported for Cu/Cu2O core–shell nanoparticles on thin C films (932.3 eV to 933.8 eV),23 where the larger the metallic core and the higher the Cu/C ratio, the lower the BE. A thin C overlayer on large Cu/Cu2O core-shell NPs would also provide a similar BE shift.15 Due to the surface sensitivity of the method, the XPS signal for these systems will be dominated by the C overlayer/Cu-based NP interphase region with a shift to lower values the thinner the C overlayer and the larger the metallic core. The samples prepared from a chloride precursor differ from the nitrate-derived samples in that they present a broader Cu2p3/2 peak toward the higher BE side as well as the satellite peak, whose shape is consistent with Cu(II) hydroxide species. Moreover, the difference spectrum for the chloride samples shows a peak at approximately 932.4–932.6 eV, consistent with a larger contribution of metallic and Cu2O species on the lemon peel-derived support. The results of the XRD analysis for these chloride-derived samples can be reconciled with the XPS findings by accounting for the different probing depths of the two techniques, where XRD provides bulk-sensitive information, reflecting the Cu phases throughout the sample volume including within the pores of the carbon support, whereas XPS provides the composition of the outermost layer. It is therefore possible to infer that T-CuCl2-L presents regions with a thinner carbon (C) overlayer or more exposed NPs supported on the external surface, thereby allowing us to probe a larger portion of the metal–metal oxide NPs by XPS. The SEM image in Fig. 1 shows that, compared to T-CuCl2-O, a higher abundance of the Cu NPs are located on the external surface of T-CuCl2-L, possibly due to the apparent lower porosity of this system.
The Cu 2p spectra of the sulphate-derived samples are more similar to those of the nitrate samples in terms of Cu speciation, although some intensity at around 935 eV suggests the existence of Cu(II) species in the former. We find that the orange peel-derived support contains a higher abundance of a component centred at 932.5 eV, consistent with a larger portion of Cu for this sample probed by XPS, and thus a thinner C overlayer and/or more exposed Cu phases, as confirmed by TEM analysis (Fig. 3). The XPS findings are also consistent with the XRD results and TEM analysis, indicating that the nitrate-derived samples exhibit the smallest particle sizes amongst the samples.
The O 1s XPS spectra for all the samples present a broad peak ranging from 530 eV to 536 eV. From the difference spectral analysis of all the samples, one could identify at least three peaks centred at approximately 531 eV, 532.6 eV and 533.5 eV (Fig. 4c). The O species due to Cu2O are generally found at a lower BE24 and therefore it is possible to infer that the O 1s spectrum is dominated by the O species on the C support. Noteworthily, whilst the O 1s profile for the orange-peel derived samples is very similar, regardless of the metal precursor used, a large variation is observed for the lemon peel-derived materials. A closer inspection indicates that the nitrate-derived sample on the orange peel support contains a larger abundance of a component centred at 532.6 eV BE than that on the lemon peel support, which is assigned to C–O species.25 T-CuCl2-O is also characterised by a higher abundance of more oxidised C–O species as evidenced by the components at higher BE. For the sulphate systems, T-CuSO4-O presents a higher abundance of a component at approximately 532 eV, which is either more reduced C–O species, or Cu(II)-bound oxygen species. Amongst the lemon-peel systems, the sample produced from the chloride precursor presents a higher abundance of a component at approximately 532 eV together with a species at 531 eV which can be attributed to the Cu(II)-bound OH species, consistent with more exposed Cu-phases.
The analysis of the C 1s XPS spectra provides more insights into the influence of the metal salt precursor on the graphitization process of the support during the thermal treatment. The spectra were fitted using a model developed earlier,26,27 which includes the following components: C1, 284.35 ± 0.01; C2, 284.90 ± 0.01; C3, 286.20 ± 0.01; C4, 288.80 ± 0.01. C1 is assigned to sp2 graphitic C; C2 is attributed to highly disordered graphite due to heteroatoms, with a large contribution from C in a sp3 bonding configuration; C3 is attributed to C–O species in cellulose28 and is expected to convert first into C2 and then into C1 as the structure reorders upon thermal annealing; C4 is due to carbonates.27 T-CuSO4-L presents the highest graphitic character among the samples. This is in contrast with a previous study,15 in which the metal loading was significantly lower as a result of a different reactive condition realised in a stirred reactor, suggesting an important role of the metal species in the deoxygenation reaction of the peel taking place during the hydrothermal synthesis as well as the subsequent thermal treatment. T-CuSO4-O contains a higher abundance of disordered graphite (C2) than T-CuSO4-L, but also a slightly higher amount of the C–O component (C3) from the starting peel precursor. A striking difference is observed for the samples from the nitrate precursor, which present the lowest graphitic character, regardless of the starting peel. This can be explained by the oxidative action of the nitrate species during the hydrothermal synthesis, which, by introducing oxygenated functionalities on the C backbone of the insoluble peel constituents, contrasts the condensation/dehydrogenation reactions which are a necessary step to form a condensed aromatic ring. It is interesting to note that these results qualitatively correlate with the thickness and lengths of the flake stacking in the C particles seen in the SEM images in Fig. 1 and in the TEM images in Fig. S1 and S2.† Accordingly, the nitrate-derived samples exhibit smaller carbon particles, and consequently smaller graphitic domains.
Moreover, a higher degree of graphitization is found together with larger Cu0 and Cu2O coherent domains (Table 2), which result from thermally induced particle growth, alongside some oxidised Cu(II) species, suggesting the presence of exposed and unprotected particles. In contrast, smaller particles are found for a more disordered C structure, indicating a stronger interaction between the Cu species and the carbon support matrix during the synthesis. Therefore, it is possible to infer that not only the chemical composition of the peel as earlier discovered,15 but also the nature of the anion influences the reducibility of the Cu species by the peel constituents, as well as the transformation of the peel itself during thermal treatment. From the TEM analysis, it is possible to deduce that during the hydrothermal synthesis, some of the particles are directly formed within the pores of the peel solid component, which undergoes restructuring influenced by the metal species as discussed earlier. This phenomenon is particularly evident in the lemon-peel-derived samples. The similarity of particle nanostructures derived from the same Cu precursor, regardless of the peel type, indicates that the migration of soluble, solvated anion-bound Cu species into the pores of the insoluble peel component is influenced by the anion size. Smaller anions, such as NO3− (179 pm), can access smaller pores, leading to the formation of smaller particles. In contrast, larger anions like Cl− (184 pm) and SO42− (258 pm) preferentially migrate into larger pores, resulting in higher Cu concentration in these regions. In smaller pores, a lower Cu concentration lead to the formation of voided particles through the Kinkerdall effect.22 However, it is also important to consider that particle formation can occur in solution due to the action of molecular or oligomeric constituents derived from the original peel. This phenomenon explains the presence of agglomerated solid-core particles deposited on the solid peel components without intimate interactions with them. These are the predominant form of Cu nanostructures in T-CuSO4-O, consistent with the larger anion size and increased extraction process of the constituents during the hydrothermal synthesis. To understand the nature of the species released into the solution, we used liquid chromatography coupled with mass spectrometry. We analysed the liquid phase of the suspension after the simulated hydrothermal treatment of the peel at the same pH of the synthesis but without the Cu precursor (Fig. S3†). Accordingly, the solutions from both peels contain a large variety of chemicals of similar nature. The most notable difference between the peels is that orange peel releases certain chemicals not detected in the case of the lemon peel. Peaks corresponding to masses of 580 m/z and 610 m/z are assigned to narirutin and hesperidin, respectively. Additional peaks with masses of 372, 402, and 432 m/z are consistent with sinensetin or tangeretin, hexamethoxyflavones (e.g., nobiletin), and heptamethoxyflavones (e.g., methoxynobiletin), respectively.29 It is their antioxidant ability that facilitates the reduction of ligated-Cu(II) species in the bulk of the solution, leading to its deposition on the insoluble constituents. To explain the formation of large spherical supra-structures, it seems logical to consider oligomeric carbon species in the suspension acting as a template for the Cu supra-structures. To summarise, we can distinguish between insoluble peel-mediated nucleation and growth of Cu-based particles and solution-based particle nucleation and growth of Cu-based particles. The former leads to particles within the pores, whereas the latter one leads to C-surface immobilised NPs.
The electrochemical behaviour of the samples was investigated using cyclic voltammetry (CV) (Fig. 5) to elucidate their redox properties within the potential range of 0 ÷ −2 V vs. Ag/AgCl.
 | ||
| Fig. 5 Cyclic voltammetry of lemon supported (a), and orange supported (b) electrodes in 0.1 M KHCO3 CO2-saturated solution. (c) Correlative analysis XPS data vs. ECSA. White symbols are related to the abundance of the elements as indicated and obtained by normalising the relevant peak area by using the sum of the peak areas of the C, O and Cu core levels. Coloured symbols are related to peak area ratios of the core levels (or in the case of the C core level, also individual components derived from the fitting, namely C1 and C2) according to the formulae indicated in the legend. | ||
The CV profiles revealed a reduction peak at approximately −0.86 V vs. the Ag/AgCl reference electrode (eqn (1)). This peak can be attributed to the one-electron reduction of Cu+ to Cu0, as described by using the following equation:
| Cu+ + H+ + e− → Cu0 | (1) |
| Sample | ECSA (cm2) |
|---|---|
| T-CuCl2-L | 14.2 |
| T-CuCl2-O | 28.7 |
| T-(CuNO3)2-L | 25.1 |
| T-(CuNO3)2-O | 26.0 |
| T-CuSO4-L | 17.3 |
| T-CuSO4-O | 30.0 |
Another important aspect to consider is that the higher graphitic disorder in these samples leads to an increased porosity (SEM images in Fig. 1). Fig. 5c presents an attempt to correlate the chemical composition of the electrocatalysts as determined by XPS with the corresponding ESCA values. We focus on the relationships between the ESCA value and the abundance of the C1 component (ordered sp2 C), the C2 component (disordered sp2–sp3 C), the total surface Cu abundance and the total surface O abundance as determined by XPS. It is evident that there is no clear trend between the ESCA values and the abundance of the different core levels, indicating a more complex interdependence. Moreover, the assumption that the porosity, represented by the C2 component (disordered sp2–sp3 C) over the C1 component (ordered sp2 C), with the additive contribution of O species and Cu species determine the ESCA, is not consistent with the experimental values (blue triangle data in Fig. 5c). However, the data suggest that high ESCA values are obtained with a high C2 value, where the highest ESCA is obtained with an intermediate abundance of the C1 graphitic peak. It follows that the C1 component plays a dual role, having an opposing effect on the ESCA value, which requires an optimal composition of the carbon matrix. On the one hand, a higher abundance of C1 may indicate reduced porosity; on the other hand it ensures good electron conduction which is paramount for electrocatalysis. Additionally, we analysed the abundance of Cu and O species as additive, direct, or inverse contributions to the ESCA value and found that they have only a minor effect on these correlations (as seen by the similar trends of the data points indicated by red circles and the black square in Fig. 5c). This suggests that not only the textural properties of the C matrix but also its electronic structural characteristics influence the ECSA value. This is consistent with a recent study on structural disorder and how this determines capacitance in nanoporous carbons.32
Next, we discuss the electrocatalytic performances of these systems in the CO2RR. In this work, we focus on KHCO3 as the electrolyte, rather than KOH,15 because the lower pH, attained with a CO2-saturated solution, favours multi-electron transfer in the CO2RR.33 Moreover, in a previous study, the use of KOH as the electrolyte led to the formation of carbonates near the catalyst surface causing catalyst passivation.34 The electrocatalytic tests in a CO2-saturated 0.1 M KHCO3 electrolyte under potentiostatic conditions were performed using a previously described custom-made three-electrode electrochemical cell.35 The CO2RR performance was evaluated over a 1 hour timeframe at applied potentials of −1.8 V and −2.0 V vs. the Ag/AgCl reference electrode. The data are summarised in Fig. 6 and Table 4.
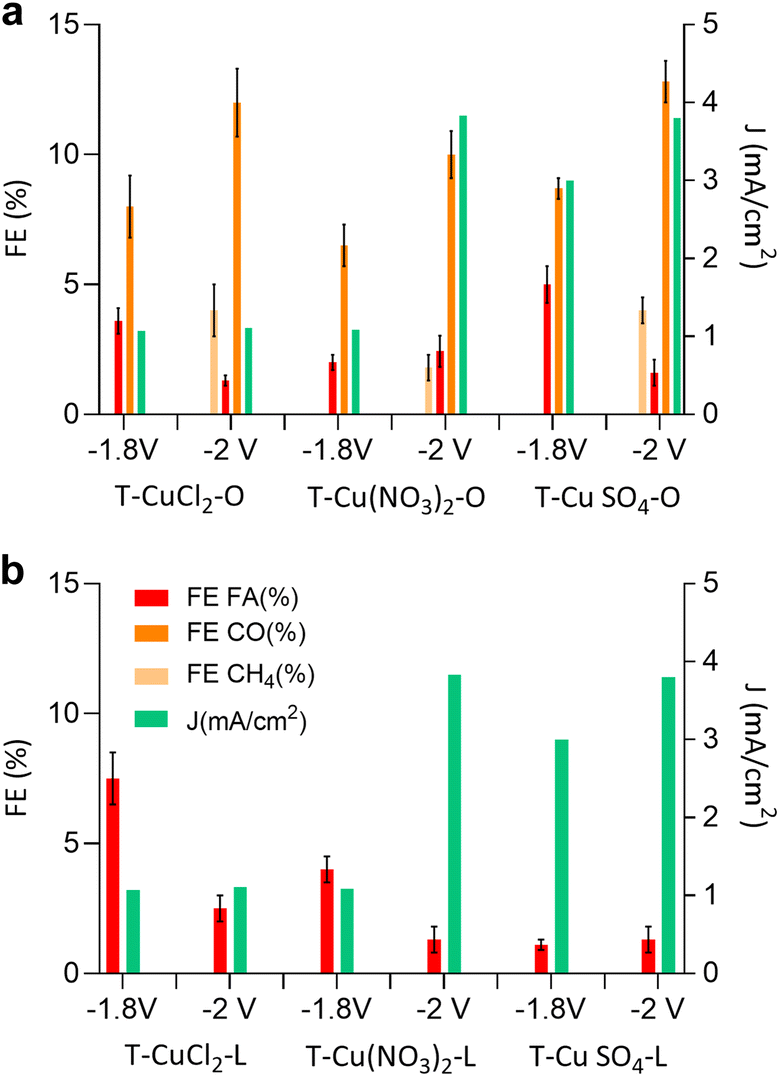 | ||
| Fig. 6 Current density (J), and faradaic efficiency of formic acid (FA), carbon monoxide (CO) and methane (CH4) of (a) orange peel-derived samples and (b) the lemon peel derived sample after 1 hour of testing at an applied potential of −1.8 and −2 V vs. Ag/AgCl. | ||
| Sample | FEFA (%) | FECO (%) | FECH4 (%) | J (mA cm−2) |
|---|---|---|---|---|
| −1.8 V vs. Ag/AgCl | ||||
| T-CuCl2-L | 7.5 (±1.0) | — | — | −1.07 |
| T-CuCl2-O | 3.6 (±0.5) | 8.0 (±1.2) | — | −3.08 |
| T-(CuNO3)2-L | 4.0 (±0.5) | — | — | −1.90 |
| T-(CuNO3)2–O | 2.0 (±0.3) | 6.5 (±0.8) | — | −3.10 |
| T-CuSO4-L | 1.1 (±0.2) | — | — | −3.00 |
| T-CuSO4-O | 5.0 (±0.7) | 8.7 (±0.4) | — | −2.90 |
|
||||
| −2 V vs. Ag/AgCl | ||||
| T-CuCl2-L | 2.5 (±0.5) | — | — | −1.11 |
| T-CuCl2-O | 1.3 (±0.2) | 12 (±1.3) | 4 (±1) | −4.35 |
| T-(CuNO3)2-L | 1.3 (±0.5) | — | — | −3.83 |
| T-(CuNO3)2–O | 2.4 (±0.6) | 10 (±0.9) | 1.8 (±0.5) | −4.10 |
| T-CuSO4-L | 1.3 (±0.5) | — | — | −3.80 |
| T-CuSO4-O | 1.6 (±0.5) | 12.8 (±0.8) | 4 (±0.5) | −4.00 |
The selectivity of the electrocatalyst, that is its ability to favor the CO2RR over the HER, is a crucial parameter for its performance. The observed product distribution appears to be strongly influenced by the support material. As shown in Fig. 6 and Table 4, electrocatalysts derived from lemon peel primarily produced formic acid (FA) at both applied potentials with the highest faradaic efficiency (FE) towards FA, reaching approximately 7.5% for T-CuCl2-L at −1.8 V vs. Ag/AgCl, whereas the performance deteriorated at more negative potentials. Conversely, the orange peel-derived catalysts exhibited a more diverse product distribution, including FA and CO, and at more negative potentials also CH4. T-CuSO4-O achieved the greatest FE of CO and CH4 at around 12.8% and 4%, respectively. The trend that emerges from Fig. 6 and Table 4 is as follows: the sum of FEs increases in the order T-Cu(NO3)2-O < T-CuCl2-O < T-CuSO4-O, consistent with the trend observed for ECSA. The observed differences in product selectivity might be attributed to different factors, all accounting for the chemical properties of the original peel and how this influences the structure of the resulting electrocatalyst. The importance of the nanostructure of the C support was already evidenced earlier,15 where the further annealing of the Cu electrocatalysts up to 800 °C resulted in a deterioration of the electrocatalytic performances, suggesting that the optimal balance between ordered sp2 C, needed for electron conduction, and defective sp3 carbon was an important aspect. In the present work, we are able to clarify the role of these defective C, which leads to an increase in the ECSA by introducing not only porosity but also optimal electron conduction, thereby resulting in improved performances. The ECSA value is proportionally relatable to the current density; however it is insufficient to ensure high selectivity to the CO2 reduction product, particularly to multi-electron transfer products such as methane. One such example is T-Cu(NO3)2-L having a relatively high ECSA, comparable to the orange-peel derived systems, but yielding FA at −2 V vs. Ag/AgCl with a relatively low FE of approximately 1.7%, despite the high current density observed. Therefore, other aspects besides the ECSA value need to be considered to explain the selectivity issue. A role played by the peel constituents concerns the influence on the Cu phase at the nanoscale, through the interactions of the insoluble cellulosic part with the Cu species in solution as well as the capping properties of the molecular constituents of the peel as the NPS are formed under hydrothermal conditions. These phenomena will influence the size of crystallographic planes (Fig. 2) and the electronic structure (Fig. 4b) of the Cu-based nanostructures as well as their location within the C matrix. This in turn will determine the accessibily of the Cu phases by the molecule of reactants (Fig. 1 and 3).
As shown in Fig. 7, we attempt to establish a correlation between the product selectivity and the structural features related to the Cu phases. It is important to note that these correlations, which are based on the initial state of the electrocatalyst will not provide direct information about the nature of the active state, but rather of a precursor state, which will be converted into the active state under CO2RR conditions. The nature of the active Cu state has been extensively studied using advanced experimental and computational methods, with contrasting observations regarding its influence on selectivity, making it an ongoing topic of debate.36,37 However, a recurring observation has led to the hypothesis that a mixed Cu/Cu2O phase is crucial for selective CO2 electrocatalysis.38 The transformation of Cu electrocatalysts into an ordered Cu(111) phase at cathodic potentials, driven by thermodynamics, is indeed detrimental for CO2 electrocatalysis.39 Therefore, the first step in materials design is to explore how to kinetically stabilize this mixed phase. In this work, the strategy involves entrapping the Cu phase within the C matrix. Thus, the analysis presented here can be considered an assessment of the readiness of the precursor state for selective electrocatalysis.
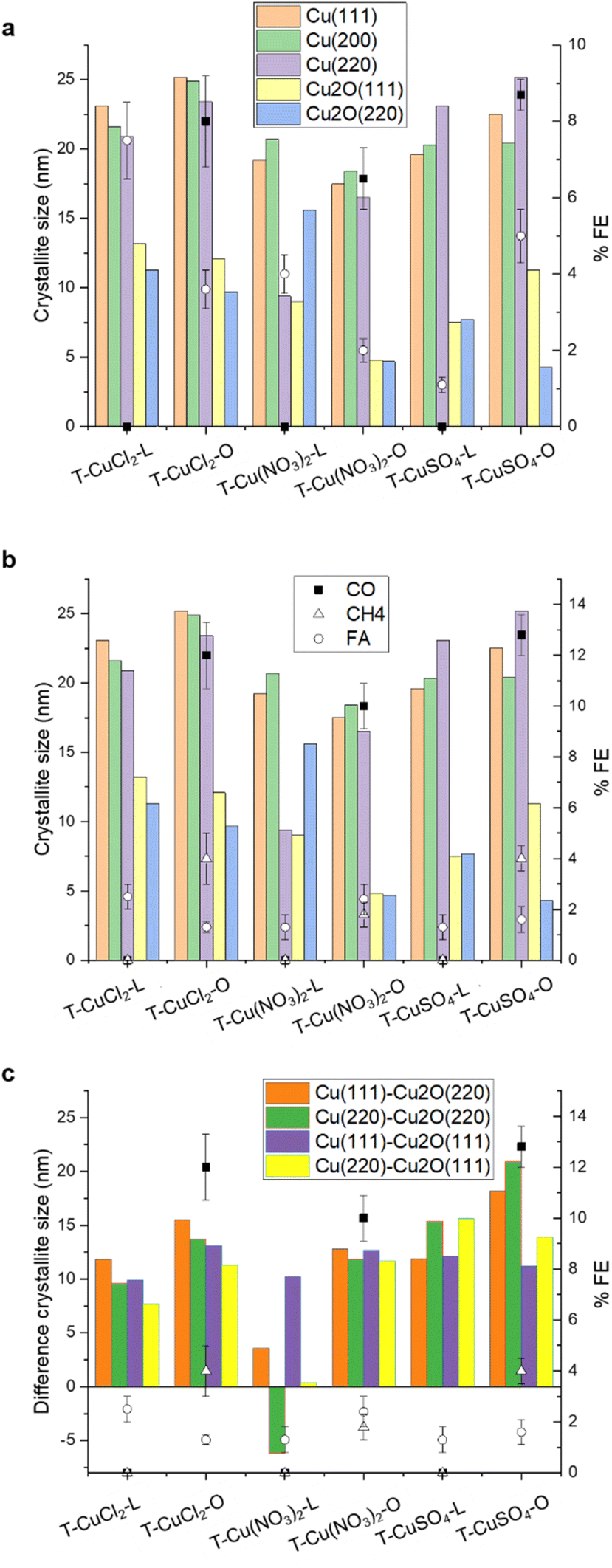 | ||
| Fig. 7 (a) Correlation between the FE obtained at −1.8 V vs. Ag/AgCl and the size of the planes as indicated, determined by XRD analysis; (b) correlation between the FE obtained at −2 V vs. Ag/AgCl and the size of the planes as indicated, determined by XRD analysis; (c) correlation between the FE obtained at −2 V vs. Ag/AgCl and the difference between the size of the planes as indicated. | ||
From Fig. 7a and b, it is evident that there is no linear correlation of the FE with any of the individual crystallographic components in the initial state of the electrocatalysts. We suggest that this is due to the exposure of the Cu phases contained in each electrocatalyst and how these transform in situ differently during the CO2RR due to a specific location and stabilisation within the C matrix. This is not surprising as it is well known from mechanistic studies using advanced theoretical and experimental approaches that many factors influence the catalytic performances in a complex interdependency, including the surface nanostructure and composition,40 mesoscale structural and textural characteristics,41 reaction conditions (pH, buffer strength, and ion effects),42 mass transport related effects,43 and local electric or magnetic fields.44 All these aspects would influence the availabilities of reagents and active site dynamics at the polarized interface. A limitation in assessing a structure activity/performance is the fact that the exposed metal active phase remains unknown for these partially encapsulated particles, but the selectivity issue can be discussed. Based on these results, we suggest that the apparent condition for achieving higher CO2RR Faraday efficiency for the formation of multi-electron reduction products is a higher Cu0/Cu2O crystallite sizes ratio, regardless of the absolute value of the crystallite size, whereas larger Cu2O crystallites favor FA formation. From Fig. 7c, it seems that in the case of the orange peel-derived samples, the higher the difference in crystallite size between the metallic and cuprous oxide planes, namely Cu(111)–Cu2O(220) and Cu(220)–Cu2O(220), the higher the total CO2RR Faraday efficiency as well as the FE towards methane at 2 V vs. Ag/AgCl. This correlation could be explained in terms of an optimal co-location of Cu2O/Cu planes, where the Cu(I)–Cu(0) sites at the interface are involved in the activation of CO2 and the metallic planes provide the H+ and e− needed for the hydrogenation reaction.45
The pores generated by the defects within the graphitic layers offer an avenue for particle stabilization. We observed that the particles are interacting at the edge sides of the bent graphite flakes (Fig. 1), where the bending causes charge localization leading to an increased stabilization of nanoparticles at the edge sites.46 The presence of a carbon overlayer on the surface of the Cu-based nanoparticles could be also a crucial aspect in the stabilization of the CO2RR active and selective phases, aligned with the well-established role of the carbon overlayer in regulating selectivity for metal nanoparticles in heterogeneous catalysis.47 However, the nature of the carbon component—whether as the support interacting with Cu phases or as the carbon overlayer on Cu phases—can also be expected to influence electrocatalysis.
For the lemon peel-derived systems, no clear correlation has been observed between the initial Cu phases and CO2 reduction reaction (CO2RR) performance at −2 V vs. Ag/AgCl. Similarly, there is no evident correlation with the electrochemical surface area (ESCA), and thus with the textural and electronic properties of the support. The results on the lemon peel samples are consistent with the high FA efficiency observed for reduced graphene oxide (rGO)-supported Cu nanoparticles, and attributed to oxygenated groups in rGO, limiting the electron transfer process.48 We postulate that the performances are dominated by the electrochemical properties of the support, together with an exposed Cu phase predominantly as Cu2O. Additionally, the exposed Cu2O phase may lack the stabilizing effect of the pores, leading to its rapid transformation under CO2RR reaction conditions into a metallic Cu system selective for the HER. TEM images reveal that most particles are embedded within the carbon matrix, rendering them inaccessible for catalysis. This finding implies a crucial role for the structural attributes of the carbon support in influencing product formation and selectivity, which depend on the choice of the biomass as well as the metal precursor.
Conclusions
This study explored the role of the copper precursors, specifically chloride, nitrate and sulphate, in the synthesis of C-supported Cu-based electrocatalysts active in the CO2RR, using citrus peel as a sustainable source of the C support. The inherent compositional complexity of citrus peels presents significant challenges, leading to highly heterogeneous materials and making synthesis difficult to control. Herein we aim to shed light on this complex chemistry as a critical aspect for the utilisation of citrus peel-derived biomass as a source for the manufacturing of energy materials. During the hydrothermal synthesis, the combined redox properties of the peel constituents, along with the redox properties of the counter anions (Cl−, NO3−, and SO42−) control the thermally induced restructuring and graphitization of the insoluble peel constituents, generating intrinsic porosity. The cumulative effect of the redox properties of the peel constituents and the nature of the anion will also determine the surface oxygenated moieties on the insoluble part, which act as anchoring sites for the metal nanoparticles, thereby influencing their location within the structure. At the same time, the molecular constituents of the peel facilitate the formation of Cu2O/Cu particles through their reducing and capping properties.The particle dispersion within the C matrix indicates that counter anion affects the final particle location, with the sulphate anion favouring the formation of particles on the external surface of the support. Consequently, the sulphate precursor produces an electrocatalyst with a higher FE for CO2 reduction products, including multi-electron reduction products like methane. However, the nature of the starting peel plays a major role in determining performances. Electrocatalysts derived from the orange peel exhibited superior characteristics compared to those synthesised using lemon peel. Orange peel-derived samples resulted in the formation of larger metallic domains in the Cu2O/Cu nanoparticles and an optimised structural composition of the C support, in terms of sp2 and sp3 character, which ensured electron conduction and porosity, respectively.
This optimization enhanced the electrochemically active surface area (ECSA) and promoted a diverse CO2 reduction product distribution, including CO, CH4, and formic acid.
Notably, the T-CuSO4-O catalyst achieved the highest faradaic efficiency for both CO (12.8%) and CH4 (4%) production at −2 V vs. Ag/AgCl. In contrast, lemon peel-supported catalysts primarily produced formic acid regardless of the ECSA value, indicating limited interfacial electron transfer, characteristic of a performance dominated by the carbon support. Additionally, excessive copper exposure due to a lack of protective C overlayer from the molecular constituents of the peel might induce phase transformations favouring the competing hydrogen evolution reaction.
Overall, this work highlights the potential of valorising citrus waste, particularly orange peels, to produce efficient CO2 reduction electrocatalysts and encourages further exploration of waste biomass sources for developing advanced electrocatalysts. While this study focused on CO2 electrocatalytic conversion, the chemistry unveiled is also relevant to other clean energy applications.
Experimental section
Synthesis of Cu catalyst samples: in our synthetic protocol, 18 mmol of the Cu salt (CuSO4 anhydrous powder, 99.99% purity from Sigma-Aldrich; Cu(NO3)2, hydrate powder, 99.999% Sigma-Aldrich; CuCl2 anhydrous powder 99%, Sigma-Aldrich) was dissolved in 30 mL of distilled water at 50 °C for 10 minutes. Two commercial citrus peels from Nutripowder were used, namely the orange peel and the lemon peel. 0.65 g of finely ground citrus peel powder, either from lemon or orange, was suspended in 4 mL of the Cu precursor solution and diluted to 8 mL with distilled water in a 10 mL microwave tube, and then irradiated at 68 °C for 11 minutes under continuous stirring using a CEM Discover SP Microwave Reactor. The suspension was allowed to cool for 2 minutes and centrifuged at 1500 rpm for 8 minutes. The solid material was washed with distilled water and ethanol, vacuum filtered and used for further analyses and testing. The sample notations are listed in Table 5.| ID sample | Description |
|---|---|
| T-CuCl-L | 500 °C – precursor-lemon |
| T-CuCl-O | 500 °C – precursor-orange |
| T-Cu(NO3)2-L | 500 °C – precursor-lemon |
| T-Cu(NO3)2-O | 500 °C – precursor-orange |
| T-CuSO4-L | 500 °C – precursor-lemon |
| T-CuSO4-O | 500 °C – precursor-orange |
Electrode preparation
The synthesized catalysts underwent thermal treatment in an argon atmosphere. This involved heating them to 500 °C for 2 hours with a controlled temperature ramp of 5 °C per minute. Following thermal activation, an ink was prepared for fabricating the working electrode. This was achieved by dispersing 8 mg of the catalyst (corresponding to a loading of 0.5 mg cm−2) within a mixture containing 20 μL of a 10 wt% Nafion solution (Aldrich) and 1.1 mL of anhydrous absolute ethanol (Carlo Erba). The mixture was then sonicated for 2 hours to ensure homogeneous distribution of the catalyst particles throughout the ink. The prepared catalyst ink was then deposited onto a SIGRACET GDL 28 BC gas diffusion layer with a geometric surface area of approximately 16 cm2 using a spray coating technique.Electrocatalytic test
This study investigated the electrochemical conversion of CO2 into liquid products using a two-compartment cell separated by a Nafion® 115 cation exchange membrane. The working electrode (WE) was composed of thermally treated samples with a geometric surface area of approximately 6 cm2. A 0.1 M KHCO3 solution saturated with CO2 was employed in the cathodic compartment, while the anodic compartment contained a 0.1 M KHCO3 electrolyte solution. CO2 gas (20 mL min−1) was flowed through the cathode tank for 20 minutes to ensure saturation of the electrolytic solution. A peristaltic pump circulated the electrolyte solutions between the compartments. The total solution volume (cathode + external tank + tubes) was 35 mL, with each compartment holding 7 mL. Amperometric detection experiments were conducted at constant applied voltages ranging from −1.8 V to −2 V vs. Ag/AgCl, with current density being monitored. In the anodic compartment, water co-electrolysis produced protons (H+) in situ. These protons migrated through the Nafion membrane towards the cathode where the CO2 reduction reaction occurred. The tests were carried out for two hours at each investigated potential. Liquid products were analysed using ion chromatography with a mobile phase of 0.05 mM H2SO4, a flow rate of 0.05 mL min−1, and a pressure of 5 MPa. The stationary phase consisted of a 25 cm organic acid column with a 7.8 mm internal diameter preceded by a 50 mm × 4 mm Metrosep Organic Acid Guard pre-column. A 944 professional UV/vis detector with a wavelength of 215 nm was used for product detection. Gaseous products accumulating in the headspace were analysed using an online micro gas chromatograph (Micro GC, Agilent 490) equipped with two columns: (i) PoraPLOT Q for volatile organic compounds and (ii) Molsieve 5A for separating inorganic compounds (H2, N2, O2, CO, and CH4). A thermal conductivity detector (TCD) was employed for analyte determination. Faraday efficiency is calculated the following eqn (2).| FE = (α × n × F)/Q | (2) |
485 C mol−1); Q is the average charge passed.
Cyclic voltammetry (CV)
Cyclic voltammetry in the potential range of 0 ÷ −2 V vs. Ag/AgCl at a scan rate of 50 mV s−1 was carried out in 0.1 M KHCO3 solution saturated with CO2.Electrochemical active surface area (ECSA)
Cyclic voltammetry is employed to estimate the electrochemically active surface area of the samples. The initial step involves determining the double-layer capacitance (CDL). To achieve this via CV, measurements were recorded at various scan rates within a non-faradaic potential window, as reported by Connor et al.31 The samples in Table 1 were used as working electrodes for recording CVs in a N2-saturated 0.1 M NaClO4 solution. The chosen non-faradaic potential range extends from 0.05 V to −0.35 V vs. Ag/AgCl, employing scan rates of 10, 20, 50, 80, and 120 mV s−1. The formula presented in eqn (3) was used to calculate the CDL.| I = CDL × v | (3) |
The linear relationship between the two parameters makes it easy to assess the CDL value. It is calculated by taking the absolute value of the slope of the regressed straight lines.49 Once determined, the value of the CDL, ECSA was calculated according to eqn (4) (ref. 50)
| ECSA = CDL/Cs | (4) |
Measurement of crystallite size
The Scherrer equation (eqn (5)) was employed to calculate the crystallite sizes based on the obtained diffraction data.| τ = (Kλ)/βcosθ | (5) |
Characterization
High-resolution transmission electron microscopy (HR-TEM) images were obtained using a cold field transmission electron microscope (JEM-F200, Jeol, Japan).The morphology of the thermally treated samples was investigated using a Phenom ProX Desktop SEM instrument.
The crystallographic phases were identified using a Bruker D8-Advance X-ray diffractometer (XRD) with Cu-Kα radiation (λ = 1.54186 Å).
LC-MS analyses were performed using a Waters Acquity Premier UPLC system equipped with a Xevo G3 QTOF. An ACQUITY UPLC BEH C18 Column, 130 Å, 1.7 μm, 2.1 mm × 150 mm was used. The mobile Phase A was 100% distilled water from Avidity Science, Duo System, Type 1 water +0.1% Formic acid (Waters Formic Acid P/N 186006691-1). The mobile phase B: 100% Acetonitrile from Fisher, Optima LC-MS Grade + 0.1% Formic acid (Waters Formic Acid P/N 186006691-1). The flow rate was 0.4 mL min−1 and the injection volume was 1 μL. Three repeats were performed each time for solutions prepared as follows: 0.65 g of finely ground citrus peel powder, either from lemon or orange, was suspended in 8 mL distilled water in a 10 mL microwave tube and the pH was adjusted to coincide with the pH of the synthesis solution as determined experimentally. The suspension was irradiated at 68 °C for 11 minutes under continuous stirring using a CEM Discover SP Microwave Reactor. The suspension was then pre-filtered through a 0.2 μm PTFE filter, before the solution was injected in the LC-MS.
X-ray photoelectron spectroscopy (XPS) analysis was performed on a PHI VersaProbe II (Physical Electronics) equipped with an Al Kα X-ray source (1486.6 eV) to determine the surface chemical composition of the samples. Table 6 summarises XPS fittings.
| C 1s (eV) | C1 (284.3) | C2 (285) | C3 (286.2) | C4 (288.9) |
|---|---|---|---|---|
| LS/FWHM | DS(0.1,450)/1 | DS(0.05,450)/1.2 | GL(30)/1.7 | GL(30)/1.7 |
Data availability
Data for this article are available at https://salford-repository.worktribe.com/.Author contributions
R. A. and S. A. conceived and supervised the study. FDL and PD performed the electrocatalytic test and IN synthesised the electrocatalysts. FDL and AS performed materials characterization, whereas RA contributed to the analysis of the XPS data. CI performed TEM measurements. RA and FDL drafted the manuscript. All the authors contributed to revision and finalization of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. A. wishes to acknowledge Lee Harman and Paulina Kalinowska at the University of Salford for performing LC-MS analysis.Notes and references
- S. Ye, M. Hall, H. Cao and P. He, Degradation Resistant Cathodes in Polymer Electrolyte Membrane Fuel Cells, ECS Trans., 2016, 3, 657–666 CrossRef.
- R. Arrigo, T. Sasaki, J. Callison, D. Gianolio and M. E. Schuster, Monitoring dynamics of defects and single Fe atoms in N-functionalized few-layer graphene by in situ temperature programmed scanning transmission electron microscopy, J. Energy Chem., 2021, 64, 520–530 CrossRef.
- O. A. Baturina, Q. Lu, A. Purdy, F. Xu, B. Dyatkin, X. Sang, R. Unocic, T. Brintlinger and Y. Gogotsi, Effect of Nanostructured Carbon Support on Copper Electrocatalytic Activity toward CO2 Electroreduction to Hydrocarbon Fuels, Catal. Today, 2017, 288, 2–10 CrossRef CAS.
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu, A. Epshteyn, T. Brintlinger, M. Schuette and G. E. Collins, CO2 Electroreduction to Hydrocarbons on Carbon-Supported Cu Nanoparticles, ACS Catal., 2014, 4, 3682–3695 CrossRef CAS.
- N. Gutiérrez-Guerra, L. Moreno-López, J. C. Serrano-Ruiz, J. L. Valverde and A. de Lucas-Consuegra, Gas Phase Electrocatalytic Conversion of CO2 to Syn-Fuels on Cu Based Catalysts-Electrodes, Appl. Catal., B, 2016, 188, 272–282 CrossRef.
- J. Masa, W. Xia, M. Muhler and W. Schuhmann, On the Role of Metals in Nitrogen-Doped Carbon Electrocatalysts for Oxygen Reduction, Angew. Chem., Int. Ed., 2015, 54, 10102–10120 CrossRef CAS PubMed.
- G. L. Chai and Z. X. Guo, Highly Effective Sites and Selectivity of Nitrogen-Doped Graphene/CNT Catalysts for CO2 Electrochemical Reduction, Chem. Sci., 2016, 7, 1268–1275 RSC.
- Q. Li, W. Zhu, J. Fu, H. Zhang, G. Wu and S. Sun, Controlled Assembly of Cu Nanoparticles on Pyridinic-N Rich Graphene for Electrochemical Reduction of CO2 to Ethylene, Nano Energy, 2016, 24, 1–9 CrossRef CAS.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, Achieving Highly Efficient, Selective, and Stable CO2 Reduction on Nitrogen-Doped Carbon Nanotubes, ACS Nano, 2015, 5, 5364–5371 CrossRef PubMed.
- Y. Lum, Y. Kwon, P. Lobaccaro, L. Chen, E. L. Clark, A. T. Bell and J. W. Ager, Trace Levels of Copper in Carbon Materials Show Significant Electrochemical CO2 Reduction Activity, ACS Catal., 2016, 6, 202–209 CrossRef CAS.
- M. M. Titirici, et al., Sustainable carbon materials, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- M. W. Thielke, G. Tian and J. Sobrido, Sustainable electrodes for the next generation of redox flow batteries, JPhys Mater., 2022, 5, 024004 CrossRef CAS.
- The World Counts, Wasted Food Statistics, The World Counts, 2023 Search PubMed.
- A. Budai, et al., Biochar Carbon Stability Test Method: An assessment of methods to determine biochar carbon stability, Int. Biochar Initiative, 2013, 1–10 Search PubMed.
- T. Miah, et al., Orange Peel Biomass-derived Carbon Supported Cu Electrocatalysts Active in the CO2-Reduction to Formic Acid, ChemPhysChem, 2023, 202200589 CrossRef PubMed.
- A. Loiudice, et al., Tailoring Copper Nanocrystals towards C2 Products in Electrochemical CO2 Reduction, Angew. Chem., 2016, 128, 5883–5886 CrossRef.
- B. C. Marepally, et al., Electrocatalytic reduction of CO2 over dendritic-type Cu-A and Fe-based electrodes prepared by electrodeposition, J. CO2 Util., 2020, 35, 194–204 CrossRef CAS.
- G. A. Tompsett, C. Conner and K. S. Yngvesson, Microwave Synthesis of Nanoporous Materials, ChemPhysChem, 2006, 7, 296–319 CrossRef CAS PubMed.
- R. Arrigo and M. E. Schuster, On the High Structural Heterogeneity of Fe-Impregnated Graphitic-Carbon Catalysts from Fe Nitrate Precursor, Catalysts, 2019, 9, 303 CrossRef.
- A. I. Large, S. Wahl, S. Abate, I. da Silva, J. J. Delgado Jaen, N. Pinna, G. Held and R. Arrigo, Investigations of Carbon Nitride-Supported Mn3O4 Oxide Nanoparticles for ORR, Catalysts, 2020, 10, 1289 CrossRef CAS.
- B. Kumar, et al., Nanohybrid Cu@C: synthesis, characterization and application in enhancement of lubricity, Compos. Interfaces, 2020, 27, 777–794 CrossRef CAS.
- R. Arrigo, et al., Influence of Synthesis Conditions on the Structure of Nickel Nanoparticles and their Reactivity in Selective Asymmetric Hydrogenation, ChemCatChem, 2020, 12, 1491–1503 CrossRef CAS.
- T. Ghodselahi, M. A. Vesaghi, A. Shafiekhani, A. Baghizadeh and M. Lameii, XPS study of the Cu@Cu2O core-shell nanoparticles, Appl. Surf. Sci., 2008, 255, 2730–2734 CrossRef CAS.
- M. T. Greiner, et al. , The oxidation of copper catalysts during ethylene epoxidation, Phys. Chem. Chem. Phys., 2015, 17, 25073–25089 RSC.
- R. Arrigo, et al., Tuning the acid/base properties of nanocarbons by functionalization via amination, J. Am. Chem. Soc., 2010, 132, 9616–9630 CrossRef CAS PubMed.
- R. Arrigo, et al., Dynamics at Polarized Carbon Dioxide–Iron Oxyhydroxide Interfaces Unveil the Origin of Multicarbon Product Formation, ACS Catal., 2022, 12, 411–430 CrossRef CAS.
- R. Arrigo, et al., Dynamics over a Cu–graphite electrode during the gas-phase CO2 reduction investigated by APXPS, Faraday Discuss., 2022, 236, 126–140 RSC.
- V. Kuzmenko, et al., Cellulose-derived carbon nanofibers/graphene composite electrodes for powerful compact supercapacitors, RSC Adv., 2017, 7, 45968–45977 RSC.
- G. Nieto, et al. , Valorization of Citrus Co-Products: Recovery of Bioactive Compounds and Application in Meat and Meat Products, Plants, 2021, 10, 1069 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- P. Connor, J. Schuch, B. Kaiser and W. Jaegermann, The Determination of Electrochemical Active Surface Area and Specific Capacity Revisited for the System MnOx as an Oxygen Evolution Catalyst, Z. fur Phys. Chem., 2020, 234, 979–994 CrossRef CAS.
- X. Liu, et al., Structural disorder determines capacitance in nanoporous carbons, Science, 2024, 384, 321–325 CrossRef CAS PubMed.
- C. J. Bondue, M. Graf, A. Goyal and M. T. M. Koper, Suppression of Hydrogen Evolution in Acidic Electrolytes by Electrochemical CO2 Reduction, J. Am. Chem. Soc., 2021, 143, 279–285 CrossRef CAS PubMed.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, Investigating Electrode Flooding in a Flowing Electrolyte, Gas-Fed Carbon Dioxide Electrolyzer, ChemSusChem, 2020, 13, 400–411 CrossRef CAS PubMed.
- (a) F. P. Abramo, et al., Electrocatalytic production of glycolic acid via oxalic acid reduction on titania debris supported on a TiO2 nanotube array, J. Energy Chem., 2021, 68, 669–678 CrossRef; (b) F. P. Abramo, et al., Nanostructure-performance relationships in titania-only electrodes for the selective electrocatalytic hydrogenation of oxalic acid, J. Catal., 2024, 429, 115277 CrossRef CAS.
- D. Gianolio, M. D. Higham, M. G. Quesne, M. Aramini, R. Xu, A. I. Large, G. Held, J. J. Velasco-Vélez, M. Haevecker, A. Knop-Gericke, C. Genovese, C. Ampelli, M. E. Schuster, S. Perathoner, G. Centi, C. R. A. Catlow and R. Arrigo, ACS Catal., 2023, 13, 5876–5895 CrossRef CAS PubMed.
- S. J. Raaijman, N. Arulmozhi and M. T. M. Koper, Morphological Stability of Copper Surfaces under Reducing Conditions, ACS Appl. Mater. Interfaces, 2021, 13(41), 48730–48744 CrossRef CAS PubMed.
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard, J. Yano and E. J. Crumlin, Subsurface oxide plays a critical role in CO2 activation by Cu(111) surfaces to form chemisorbed CO2, the first step in reduction of CO2, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CrossRef CAS PubMed.
- G. Liu, M. Lee, S. Kwon, J. Eichhorn, A. K. Bucley, F. D. Toste, W. A. Goddard III and F. M. Toma, CO2 reduction on pure Cu produces only H2 after subsurface O is depleted: Theory and experiment, Proc. Natl. Acad. Sci. U.S.A., 2021, 118, e2012649118 CrossRef CAS PubMed.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Tailoring Copper Nanocrystals towards C2 Products in Electrochemical CO2 Reduction, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, Grain-Boundary-Dependent CO2 Electroreduction Activity, J. Am. Chem. Soc., 2015, 137, 4606–4609 CrossRef CAS PubMed.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Controlling the selectivity of CO2 electroreduction on copper: The effect of the electrolyte concentration and the importance of the local pH, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. García de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- Y. Zhang, C. Liang, J. Wu, H. Liu, B. Zhang, Z. Jiang, S. Li and P. Xu, Recent Advances in Magnetic Field-Enhanced Electrocatalysis, ACS Appl. Energy Mater., 2020, 3, 10303–10316 CrossRef CAS.
- M. Ye, T. Shao, J. Liu, C. Li, B. Song and S. Liu, Phase engineering of Cu@Cu2O core-shell nanospheres for boosting tandem electrochemical CO2 reduction to C2+ products, Appl. Surf. Sci., 2023, 622, 156981 CrossRef CAS.
- R. Arrigo, T. Sasaki, J. Callison, D. Gianolio and M. E. Schuster, Monitoring dynamics of defects and single Fe atoms in N-functionalized few-layer graphene by in situ temperature programmed scanning transmission electron microscopy, J. Energy Chem., 2022, 64, 520 CrossRef CAS.
- R. Arrigo, et al., Pd Supported on Carbon Nitride Boosts the Direct Hydrogen Peroxide Synthesis, ACS Catal., 2016, 6, 6959–6966 CrossRef CAS.
- C. Jimenez, et al., Effect of carbon support on the catalytic activity of copper-based catalyst in CO2 electroreduction, Sep. Purif. Technol., 2020, 248, 1170831 CrossRef.
- X. Li, et al., Titanium dioxide nanotubes as model systems for electrosorption studies, Nanomaterials, 2018, 8, 404 CrossRef PubMed.
- J. Qiao, P. Jiang, J. Liu and J. Zhang, Formation of Cu nanostructured electrode surfaces by an annealing-electroreduction procedure to achieve high-efficiency CO2 electroreduction, Electrochem. Commun., 2014, 38, 8–11 CrossRef CAS.
- L. de Sousa, C. Harmoko, N. Benes and G. Mul, Optimizing the Ink Formulation for Preparation of Cu-Based Gas Diffusion Electrodes Yielding Ethylene in Electroreduction of CO2, ACS ES&T Eng., 2021, 1, 1649–1658 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00463a |
| This journal is © The Royal Society of Chemistry 2025 |