
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A biomass-derived nickel-based nanomaterial as a sustainable and reusable catalyst for hydrogenation of arenes and heteroarenes†
Vishakha
Goyal
ab,
Tarun
Bhatt
c,
Anshid
Kuttasseri
c,
Arup
Mahata
*c,
Radek
Zbořil
 *ad,
Kishore
Natte
*cd and
Rajenahally V.
Jagadeesh
*ab
*ad,
Kishore
Natte
*cd and
Rajenahally V.
Jagadeesh
*ab
aVSB - Technical University of Ostrava, Centre for Energy and Environmental Technologies, Nanotechnology Centre, 17. listopadu 2172/15, Ostrava 70800, Czech Republic. E-mail: radek.zboril@upol.cz; jagadeesh.rajenahally@catalysis.de
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany
cDepartment of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, 502 284, Telangana, India. E-mail: arup@chy.iith.ac.in; kishore.natte@chy.iith.ac.in
dRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute (CATRIN), Palacký University Olomouc, Šlechtitelů 27, 77 900 Olomouc, Czech Republic
First published on 31st March 2025
Abstract
Selective hydrogenation of functionalized aromatic- and hetero-aromatic hydrocarbons is an essential research area in synthetic chemistry, which gives straightforward access to an array of saturated carbo- and heterocyclic compounds. To accomplish this hydrogenation process in a more resourceful and cost-effective manner, the development and applicability of potential catalytic materials, particularly based on earth-abundant metals, are crucial. From the viewpoint of sustainability and circular economy, such catalytic systems should be derived from waste biomass. Here, we report the preparation and application of plant-based waste biomass such as pine needle-derived Ni-nanoparticles as an efficient catalyst for the hydrogenation of (hetero)arenes. The immobilization of Ni-nitrate on pine needles and subsequent pyrolysis generates zero-valent Ni-nanoparticles (5–8 nm), which are embedded in a highly mesoporous N-doped graphitic matrix. The resulting nickel nanoparticles exhibited high activity and selectivity as well as stability and reusability for the hydrogenation of functionalized arenes as well as nitrogen and oxygen-containing heteroarenes to obtain various cyclo-aliphatic compounds including tetrahydroquinolines, tetrahydroquinoxalines and dihydrobenzofurans as well as key starting materials of pharmaceutical agents. DFT calculations have been made for this Ni-catalytic (hetero)arene hydrogenation process, which revealed favorable reaction thermodynamic and kinetic as well as mechanistic feasibility for selective ring reduction.
Sustainability spotlightThe valorization of biomass to produce valuable chemicals, fuels and materials is of central importance for achieving sustainability and enabling circular economy. In this context, the preparation of nanostructured materials from waste-biomass as sustainable and reusable catalysts for chemical synthesis, energy and environmental technologies is of increasing importance. This work is related to the preparation and application of plant-based waste biomass derived nanostructured catalytic materials for hydrogenation reactions, which represent widely applied cost-effective and economically viable industrial processes. Particularly, we demonstrated the conversion of pine needles, which constitute a significant plant-based waste-material, to nitrogen doped biocarbon supported Ni-nanoparticles as a potential and sustainable catalyst for the hydrogenation of functionalized arenes as well as nitrogen and oxygen-containing heteroarenes to obtain various cyclo-aliphatic compounds including tetrahydroquinolines, tetrahydroquinoxalines and dihydrobenzofurans as well as key starting materials of pharmaceutical agents. |
Introduction
Catalytic hydrogenation of readily available arenes and heteroarenes is regarded as a powerful and appropriate strategy for preparing saturated carbo- and hetero-cyclic compounds.1–3 Particularly, the hydrogenation of heteroarenes, including quinolines, pyridines, indoles, quinoxalines, and benzofurans to produce N- and O-containing saturated heterocyclic compounds has garnered considerable importance in synthetic organic chemistry owing to their widespread applications in advanced chemicals, polymers, pharmaceuticals, and agrochemicals.4–10 In addition, the hydrogenated products of aromatic hydrocarbons such as cyclohexanes are finding increasing importance in fine and bulk chemicals as well as life science applications.4–10 To perform the hydrogenation of (hetero)arenes in a general and selective manner, the design and applicability of suitable catalysts is crucial.11–16 In this regard, from the past several years, both homogeneous17–25 and heterogeneous catalysts based on noble15 and non-noble15,16 metals have been successfully applied for these hydrogenation processes. Compared to homogeneous complexes,12,19,20,22,24,25 heterogeneous materials are appropriate due to their high stability and reusability as well as high robustness and cost-operational advantage.8,23,26–43 However, tuning their high activity and selectivity is a formidable task to perform such challenging hydrogenations under mild conditions. Nevertheless, precious metal-based heterogeneous materials13,28–36 are known to catalyze (hetero)arenes under mild conditions, but their higher price and less availability limit practical applicability. Hence the development of heterogeneous catalysts, preferably based on earth abundant metals is crucial for the advancement of (hetero)arene hydrogenation chemistry.37–43 In this regard, a few materials based on Co,10,11,15,16,42,44 Fe,39 and Ni37,41 have been reported for the hydrogenation of arenes or heteroarenes. Despite these materials, still the development of more sustainable and cost-effective heterogeneous catalysts based on 3d-metals, which should exhibit more generality as well as high selectivity for the hydrogenation of both aromatic and hetero-aromatic hydrocarbons, particularly for the chemoselective ring hydrogenation in functionalized substrates is highly desired. For the creation of sustainable and renewable catalytic materials, utilization of biomass has become the subject of intensive research.45 In addition to the production of chemicals, biomass can also be conveniently used for the preparation of carbon materials as well as potential catalytic systems.46 Noteworthy, the utilization of waste-biomass for making catalytic materials is of importance with respect to sustainability and circular economy.45 In this regard, different biomass derived biochar supported nanocatalysts have been explored for organic transformations.46–48 Among waste-biomass, pine needles represent abundantly available plant-based biomass, which can be converted to biocarbon- or biochar-based supports for nanoparticles or single atoms by pyrolysis methods.48 Over the last several years, pine needle derived biocarbon supported metal nanoparticles are prepared and applied for organic transformations including hydrogenations.8,44,49,50 This biocarbon contains various surface functional groups (C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, COOH, OH, etc.) and heteroatoms (N, S, P, etc.),50,51 which are essential moieties to create a microenvironment of supported metal nanoparticles with desired metal–heteroatom interactions. As an example, our group8,44,50,52 and others53,54 showed that metal-nanoparticles supported on N-doped carbon displayed excellent activity and selectivity for hydrogenation reactions. In these materials, metal–nitrogen interactions ensure excellent catalytic performance of metal nanoparticles. Notably, the intrinsic nitrogen atoms generated by biomass itself make it more effective since nitrogen can be easily incorporated into the carbon layers that can enhance stability, and activity as well as selectivity of metal nanoparticles.55–57 Based on the importance and applications of the (hetero)arene hydrogenation process as well as the valorization of waste-biomass from the viewpoint of sustainable and circular chemistry, here we report the preparation of pine needle derived biocarbon supported Ni-nanoparticles as a stable and reusable catalytic material for the hydrogenation of various aromatic and heteroaromatic compounds to saturated carbo- and hetero-cyclic compounds. With respect to Ni-heterogeneous catalysts, previously a few systems are reported for the hydrogenation of heteroarenes41,58–60 or arenes.61–65 However, these Ni-catalysts are applied for a few simple and non-functionalized substrates; especially in the case of arene hydrogenation they are restricted to benzene and phenol.61–65 For more general applications, it is imperative to develop Ni-based nanocatalysts that should be effective for the hydrogenation of both arenes and heteroarenes with wide substrate-scope including functionalized and structurally diverse ones. On the other hand, until now, there has been no biomass derived Ni-catalyst reported for the hydrogenation of arenes or heteroarenes. In this work, the in situ preparation of Ni-nanoparticles supported on biomass-waste pine needle-derived N-doped biocarbon (NBC) via a simple pyrolysis method without the addition of any external nitrogen ligands is demonstrated. Advantageously these pine needles serve as a self-scarifying template for the creation of biocarbon to support nanoparticles as well as for the generation of N-doped carbon. The prepared Ni-nanocatalyst is highly efficient as well as regio- and chemoselective for the hydrogenation of various heteroarenes such as quinolines, pyridines, quinoxalines, benzofurans, indoles, pyrroles, and furans as well as phenols and other arenes to produce cyclo-aliphatic compounds including tetrahydroquinolines, tetrahydroquinoxalines and dihydrobenzofurans. We also highlight the utility of this Ni-catalyzed hydrogenation protocol for the preparation of the selected pharmaceutical intermediates.
O, COOH, OH, etc.) and heteroatoms (N, S, P, etc.),50,51 which are essential moieties to create a microenvironment of supported metal nanoparticles with desired metal–heteroatom interactions. As an example, our group8,44,50,52 and others53,54 showed that metal-nanoparticles supported on N-doped carbon displayed excellent activity and selectivity for hydrogenation reactions. In these materials, metal–nitrogen interactions ensure excellent catalytic performance of metal nanoparticles. Notably, the intrinsic nitrogen atoms generated by biomass itself make it more effective since nitrogen can be easily incorporated into the carbon layers that can enhance stability, and activity as well as selectivity of metal nanoparticles.55–57 Based on the importance and applications of the (hetero)arene hydrogenation process as well as the valorization of waste-biomass from the viewpoint of sustainable and circular chemistry, here we report the preparation of pine needle derived biocarbon supported Ni-nanoparticles as a stable and reusable catalytic material for the hydrogenation of various aromatic and heteroaromatic compounds to saturated carbo- and hetero-cyclic compounds. With respect to Ni-heterogeneous catalysts, previously a few systems are reported for the hydrogenation of heteroarenes41,58–60 or arenes.61–65 However, these Ni-catalysts are applied for a few simple and non-functionalized substrates; especially in the case of arene hydrogenation they are restricted to benzene and phenol.61–65 For more general applications, it is imperative to develop Ni-based nanocatalysts that should be effective for the hydrogenation of both arenes and heteroarenes with wide substrate-scope including functionalized and structurally diverse ones. On the other hand, until now, there has been no biomass derived Ni-catalyst reported for the hydrogenation of arenes or heteroarenes. In this work, the in situ preparation of Ni-nanoparticles supported on biomass-waste pine needle-derived N-doped biocarbon (NBC) via a simple pyrolysis method without the addition of any external nitrogen ligands is demonstrated. Advantageously these pine needles serve as a self-scarifying template for the creation of biocarbon to support nanoparticles as well as for the generation of N-doped carbon. The prepared Ni-nanocatalyst is highly efficient as well as regio- and chemoselective for the hydrogenation of various heteroarenes such as quinolines, pyridines, quinoxalines, benzofurans, indoles, pyrroles, and furans as well as phenols and other arenes to produce cyclo-aliphatic compounds including tetrahydroquinolines, tetrahydroquinoxalines and dihydrobenzofurans. We also highlight the utility of this Ni-catalyzed hydrogenation protocol for the preparation of the selected pharmaceutical intermediates.
Results and discussion
Synthesis of catalytic materials
N-doped biocarbon supported Ni-nanoparticles were prepared by a pyrolysis method, which is presented in Fig. 1. Typically, crushed pine straw/needle powder was added to an ethanolic solution of Ni(NO3)2·6H2O and stirred for 6 h at room temperature at 50 °C. Then the solvent was evaporated, and the solid was dried in an oven overnight at 100 °C to get a brown-colored solid material. Finally, this material was subjected to pyrolysis at 600–800 °C under a nitrogen atmosphere with a 5 °C min−1 heating rate for 3 h to obtain Ni-nanoparticles supported on N-doped biocarbon. These new materials are denoted as Ni-NPs@NBC-T, where T is the pyrolysis temperature, NBC is the nitrogen doped biocarbon and NPs is nanoparticles. | ||
| Fig. 1 Schematic representation of the synthesis of pine-needle derived N-doped biocarbon supported Ni-nanoparticles. | ||
Evaluation of Ni-materials and optimization of the model reaction
All the prepared Ni-based materials have been tested for their activities and selectivities in the partial ring hydrogenation of quinoline (1) to obtain 1,2,3,4-tetrahydroquinoline (2) as a model reaction (Table 1). As expected, the reaction did not take place in the absence of any catalysts (Table 1, entry 1). Neither pine needle (PN) nor pine needle-derived biocarbon (NBC) was found to be active for the desired transformation (Table 1, entries 2, and 3). Next, the simple Ni-salt (Ni(NO3)2·6H2O) and its immobilized materials such as Ni(NO3)2·6H2O/PN, and Ni(NO3)2·6H2O/NBC exhibited no or very less activities (Table 1, entries 4–6). Remarkably, the materials (Ni-NPs@NBC-600-800) generated by immobilizing Ni-nitrate on pine needle powder and subsequent pyrolysis at different temperatures (600 °C, 700 °C, and 800 °C) exhibited good to excellent activities (Table 1, entries 7–9). Among these, the one (Ni-NPs@NBC-700) pyrolyzed at 700 °C displayed superior activity and as a result quantitative yield of desired product 2 was obtained with this catalyst. To compare the activities, Ni-NPs on the carbon support (Ni-NPs@C-700) were prepared and tested in the model reaction under standard reaction conditions (Table 1, entry 10). This catalyst exhibited less activity compared to that of our optimal Ni-catalyst, Ni-NPs@NBC-700.|
|
|||
|---|---|---|---|
| Entry | Catalyst | Conversion 1 (%) | Yield 2 (%) |
| a Reaction conditions: 0.5 mmol quinoline, 50 mg of catalyst, 30 bar H2, 2 mL isopropanol (IPA), 120 °C, and 24 h. b Homogeneous catalysis conditions with 5–6 mol% of Ni-salt (25 mg). Conversion and yields were determined by GC using the n-hexadecane standard. | |||
| 1a | Blank | n.d. | n.d. |
| 2a | Pine needle (PN) | n.d. | n.d. |
| 3a | Pine needle-derived biocarbon (NBC) | n.d. | n.d. |
| 4b | Ni(NO3)2·6H2O | 02 | — |
| 5a | Ni(NO3)2·6H2O/PN | 10 | 8 |
| 6a | Ni(NO3)2·6H2O/NBC | 15 | 10 |
| 7a | Ni-NPs@NBC-600 | 85 | 80 |
| 8a | Ni-NPs@NBC-700 | >99 | 99 |
| 9a | Ni-NPs@NBC-800 | 70 | 65 |
| 10a | Ni-NPs/C-700 | 45 | 35 |

Effect of reaction parameters
As per the preliminary experimental data, Ni-NPs@NBC-700 exhibits remarkable activity and selectivity for the hydrogenation of quinoline to 1,2,3,4-tetrahydroquinoline (Table 1, entry 8). As a result, this Ni-NPs@NBC-700 nanocatalyst was chosen to explore the role of reaction parameters such as solvent, reaction temperature, reaction time, H2 pressure, and catalyst amount in the model reaction. The hydrogenation of quinoline was investigated in different solvents, including polar and non-polar ones. These results are summarized in Table S1 (see ESI).† Among these, non-polar solvents are not suitable for this hydrogenation (Table S1,† entries 1 and 2), while in polar aprotic solvents, such as 1,4-dioxane and THF, the reaction moderately proceeded (Table S1,† entries 3 and 4). However, the hydrogenation reaction is very facile in polar protic solvents such as tert-butanol, ethanol, isopropanol, and water (Table S1,† entries 5–8). Among these solvents, isopropanol (IPA) was found to be the best.The influence of temperature and time was investigated, and the relevant findings are presented in Fig. 2. Temperature is an important reaction parameter because it significantly influences the rate of a reaction to get better conversion and product yield. The model reaction was carried out at different temperatures ranging from 30 to 120 °C, and the results are shown in Fig. 2a. As the temperature increases, the conversion rate also increases. As expected, the reaction is very sluggish at lower reaction temperatures (30 °C), and only 3% conversion was obtained. At 60 °C, the conversion rate improved to 20%, and at 90 °C, it reached 58%. By raising the reaction temperature to 120 °C, complete conversion occurred with 99% of the desired product, 1,2,3,4-tetrahydroquinoline. Next, the effect of reaction time was studied, and the results are displayed in Fig. 2b. The quinoline conversion rate was increased with reaction time, reaching 99% at and after 24 hours of reaction time.
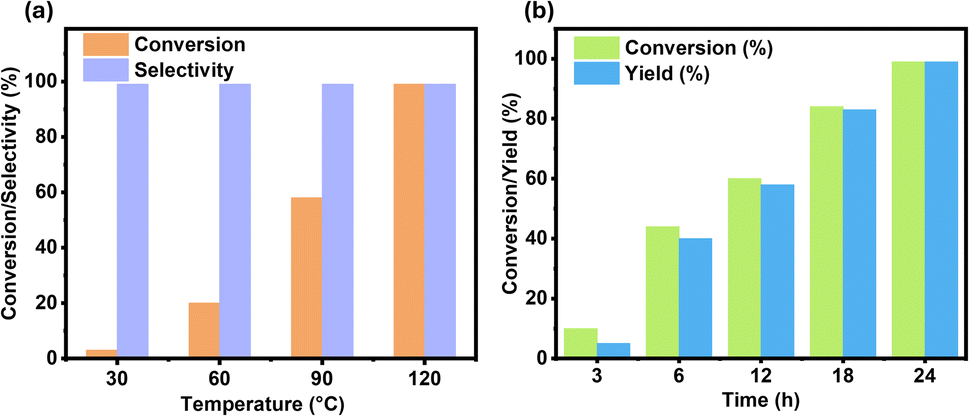 | ||
| Fig. 2 Hydrogenation of quinoline to 1,2,3,4-tetrahydroquinoline. Reaction conditions: 0.5 mmol quinoline, 50 mg of catalyst, 30 bar H2, 2 mL isopropanol (IPA), T (°C), and t (h). (a) Effect of reaction temperature (30–120 °C), and (b) effect of reaction time (3–24 h). | ||
Next, the effect of H2 pressure was measured (Fig. 3a), which is an important parameter since it is one of the reactants. The gradual increase in the conversion of quinoline was observed with increasing H2 pressure. Only 4% of conversion was obtained at 5 bar H2 pressure (Fig. 3a). The conversion was improved to 20% at 10 bar and was further enhanced to 55% at 20 bar H2 pressure. Eventually, with 30 bar of hydrogen, complete conversion of 1 and quantitative yield of 2 were achieved (Fig. 3a).
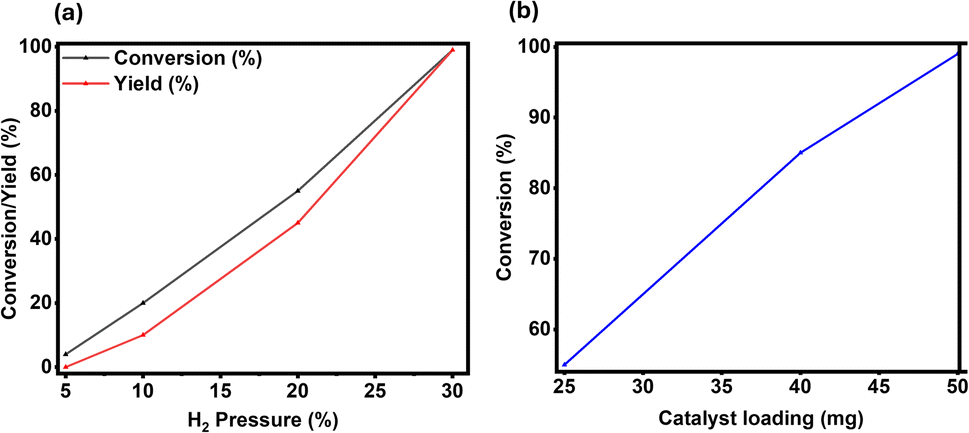 | ||
| Fig. 3 Hydrogenation of quinoline to 1,2,3,4-tetrahydroquinoline. Reaction conditions: 0.5 mmol quinoline, catalyst (x mg), H2 (x bar), 2 mL isopropanol (IPA), 120 °C, and 24 h. (a) Effect of H2 pressure (5–30 bar), and (b) effect of catalyst amount (25–50 mg). | ||
Finally, the amount of catalyst is tested, and it was found that 50 mg of catalyst is required to achieve complete conversion and obtain 99% yield of the desired product 2. All these optimization experiments confirmed that the best results for the hydrogenation of quinoline 1 to 1,2,3,4-tetrahydroquinoline 2 can be achieved with 50 mg catalyst, and 30 bar H2 pressure at 120 °C for 24 h in isopropanol solvent.
Characterization of Ni-catalysts
To know the structural features and to understand varying catalytic activities, the prepared different Ni-materials, Ni-NPs@NBC-T (Ni-NPs@NBC-600, Ni-NPs@NBC-700, Ni-NPs@NBC-800, and spent Ni-NPs@NBC-700-R), were characterized using various analytical techniques, including CHNS, ICP-AES, powder XRD (X-ray diffraction), XPS (X-ray photoelectron spectroscopy), HR-TEM (high resolution-transmission electron microscopy), N2-adsorption/desorption measurements, H2-TPR (H2-temperature programmed reduction), and Raman analyses. The XRD patterns (Fig. S3†) of Ni-NPs@NBC-600, Ni-NPs@NBC-700, and Ni-NPs@NBC-800 catalysts exhibit diffraction peaks at 44.5°, 51.6° and 76.5° on the 2θ scale, corresponding to the (111), (200), and (220) lattice planes of the metallic Ni phase with a face-centered cubic (fcc) structure (JCPDS file: no. 04-0850) (Fig. S3a–c†).66 All the pyrolyzed catalysts displayed the formation of metallic nickel with the face-centered cubic (fcc) structure, which indicates that the metallic Ni phase was intact during the pyrolysis at temperatures from 600 °C to 800 °C.66,67 Additionally, a broad peak appeared at about 25°, and is associated with the (002) lattice plane of low-crystalline graphitic carbon.67 The X-ray diffraction pattern of the recycled catalyst Ni-NPs@NBC-700-R material indicates the retention of the metallic Ni phase with a face-centered cubic (fcc) structure after recycling (Fig. S3d†).Multipoint BET nitrogen adsorption–desorption isotherms for all three catalysts, Ni-NPs@NBC-600, Ni-NPs@NBC-700, and Ni-NPs@NBC-800, were obtained to know the effect of different pyrolysis temperatures on the porosity and the surface properties. The isotherms of these catalysts show type IV characteristics with a hysteresis loop in the range of relative pressures of 0.4 to 1.0, indicating the presence of mesopores in the catalyst materials (Fig. S4†). The pore-size distribution curves of these materials are shown in Fig. S4.† Regarding pore size distribution, a sharp peak around 3–4 nm is typical for all the catalysts. However, a broad peak corresponding to the pore size of 10–35 nm and reflecting a significant number of large mesopores is well pronounced in the most active catalyst Ni-NPs@NBC-700 while the same peak is less intensive in the case of Ni-NPs@NBC-600, and Ni-NPs@NBC-800. The BET-specific surface area and pore volume of Ni-NPs@NBC-600, Ni-NPs@NBC-700, and Ni-NPs@NBC-800 are summarized in Table S3 and illustrated in Fig. S4.† High-resolution transmission electron microscopy (HR-TEM) reveals the size, distribution, and morphology of Ni-NPs@NBC-T catalysts. The representative TEM images of Ni-NPs@NBC-600, Ni-NPs@NBC-700, Ni-NPs@NBC-800, and Ni-NPs@NBC-700-R catalysts are shown in Fig. 4.
 | ||
| Fig. 4 TEM images of (a) Ni-NPs@NBC-600, (b) Ni-NPs@NBC-700, (c) Ni-NPs@NBC-800 and (d) spent Ni-NPs@NBC-700-R catalysts: (i–iii) TEM images in different magnifications, (iv) SAED pattern and (v) particle size distribution diagram. | ||
The most active Ni-NPs@NBC-700 catalyst shows that most Ni-nanoparticles are in the range of 5–8 nm size, keeping quite uniform distribution through the surface of the graphitic carbon matrix without any aggregation into larger clusters (Fig. 4b). The HR-TEM image of the Ni-NPs@NBC-700 catalyst illustrates lattice fringe d-spacing values of 0.20 nm, 0.17 nm, 0.12 nm, and 0.10 nm corresponding to the (111), (200), (220), and (222) planes of cubic metallic Ni, respectively (Fig. 4b).62 The selected-area energy diffraction (SAED) pattern of the Ni-NPs@NBC-700 nanocatalyst reveals regular diffraction rings, implying the polycrystalline nature of Ni nanoparticles. The elemental mapping of the Ni-NPs@NBC-700 catalyst confirms the presence of Ni, N, C, and O elements and the overlapping of Ni-nanoparticles with N, C, and O species (Fig. S5†). The TEM images of less active Ni-NPs@NBC-600 and Ni-NPs@NBC-800 catalysts demonstrate that Ni nanoparticles are distributed in similar size ranges with also quite homogeneous distribution, inferring that the Ni-nanoparticle size is not the key parameter contributing to the superior efficiency of the Ni-NPs@NBC-700 catalyst (Fig. 4a–c).
From this point of view, the highly mesoporous nature of the Ni-NPs@NBC-700 nanocatalyst reflecting the presence of larger pores in the catalyst structure (see Fig. S4 and Table S3†) seems to be the most distinguishing aspect compared to Ni-NPs@NBC-600 and Ni-NPs@NBC-800 catalysts. These findings suggest that 700 °C is the optimum pyrolysis temperature for obtaining small Ni nanoparticles with highly mesoporous characteristics and homogeneous dispersion in the catalyst material. Furthermore, the TEM investigation of the spent Ni-NPs@NBC-700-R nanocatalyst revealed that no aggregation or structural changes of Ni-particles occurred during/after the reaction (Fig. 4d).
The chemical composition at the surface and chemical interactions between the constituent components of Ni-NPs@NBC-T nanocatalysts were analyzed by X-ray photoelectron spectroscopy (XPS). The survey XPS spectrum of the Ni-NPs@NBC-700 catalyst confirms the presence of Ni, N, O, and C species (ESI, Fig. S1†). The N 1s spectra of all Ni-NPs@NBC-T nanocatalysts can be deconvoluted into three prominent peaks with binding energies at 398.2 eV, 399.3 eV, and 400.3 eV that correspond to pyridinic-N, Ni–Nx, and pyrrolic-N, respectively (Fig. S2i†).68 The pyridinic and pyrrolic N–C structure models could be generated by the substitution of C atoms at the edges of C6 and C5 rings during the pyrolysis.69
Interestingly, the intensity of pyridinic-N, and pyrrolic-N peaks changes with the pyrolysis temperature. In addition, the presence of the Ni–Nx moiety in all Ni-NPs@NBC-T nanocatalysts (Ni-NPs@NBC-600, Ni-NPs@NBC-700, and Ni-NPs@NBC-800) confirms the direct interaction between Ni and N atoms. However, the Ni-NPs@NBC-700 nanocatalyst has higher amounts of pyridinic-N, which might stabilize the Ni-nanoparticles by using their lone pair of electrons as a coordination site. Fig. S2ii† shows the high resolution XPS Ni 2p spectra of Ni-NPs@NBC-T nanocatalysts. All these nanocatalysts displayed binding energies at 854.2 eV, which is the characteristic Ni 2p3/2 peak of metallic Ni0.70 However, the dominant energy bands, which appeared at 856.6 eV, and 874.1 eV, along with their shake-up satellite peaks at 862.5 eV, and 880.7 eV, can be attributed to the Ni 2p3/2 and Ni 2p1/2 of Ni2+. This is evidently due to the surface oxidation of Ni0 to Ni2+ upon exposure to air during XPS measurements as XRD data are very convincing in terms of identifying Ni0 as the active phase.70 As shown in Fig. S2iii,† the C 1s XPS spectra of Ni-NPs@NBC-T nanocatalysts exhibit three binding energies at 284.2 eV, 285.3 eV, and 292.7 eV. The major energy band appearing at 284.2 eV is associated with the sp2-hybridized graphite-like carbon (C-Csp2), and the binding energy is centered at 285.3 eV, corresponding to sp3-hybridized diamond-like carbon (C-Csp3), which might be overlapped with the sp2-hybridized carbon bonded with N atoms (N-Csp2).71,72 Additionally, the weaker binding energy at 292.7 eV is ascribed to π–π* transitions.73 The O 1s core level XPS spectra of Ni-NPs@NBC nanocatalysts demonstrate two binding energies at 531.1 eV, and 532.7 eV, which can be assigned to –CO and –O–CO/C–OH, respectively.73 Furthermore, XPS analysis of the spent Ni-NPs@NBC-700-R nanocatalyst reveals no changes in the surface chemical composition or oxidation states and the binding energies of all key elements (Ni, N, C, and O) present in the catalyst material.
Next, to correlate the basic properties of the catalysts, CO2-TPD measurements of Ni-NPs@NBC-600, Ni-NPs@NBC-700, and Ni-NPs@NBC-800 nanocatalysts using CO2 as a probe molecule were carried out (Fig. S6 and Table S4†). The basic sites are classified into weak, medium, and strong, depending on the temperature desorption peaks of CO2.44 All the Ni-NPs@NBC-T nanocatalysts exhibit three major CO2 desorption peaks in the ranges of 50–150 °C, 250–490 °C, and 500–620 °C. Importantly, the most active Ni-NPs@NBC-700 nanocatalyst exhibits a broad and intense CO2 desorption peak as well as a higher amount of medium basic sites compared to Ni-NPs@NBC-600 and Ni-NPs@NBC-800 nanocatalysts (Fig. S6b and Table S4,† entry 2). This correlates well with a higher amount of pyridinic-N in the Ni-NPs@NBC-700 nanocatalyst, which is responsible for the high basicity of the catalyst,74 as corroborated by XPS analysis (Fig. S6b†)75 These findings imply that the order of the conversion (Ni-NPs@NBC-700 > Ni-NPs@NBC-600 > Ni-NPs@NBC-800) follows the content of basic sites (pyridinic nitrogen) of the catalysts (Ni-NPs@NBC-700 > Ni-NPs@NBC-600 > Ni-NPs@NBC-800). The active Ni sites keep the zero-valent state as clearly proved by XRD and magnetic measurements confirming typical superparamagnetic/ferromagnetic behavior of Ni(0) nanoparticles (Fig. S7†). The formation of zerovalent Ni-nanoparticles, the higher mesoporosity and the increased content of pyridinic nitrogen with Ni–N centers play key roles in triggering higher activity and selectivity of the Ni-NPs@NBC-700 catalyst for the hydrogenation of (hetero)arenes.
Ni-NPs@NBC-700 catalyzed hydrogenation of heteroarenes
With the set of optimized reaction conditions in hand, the substrate scope and limitations of this novel Ni-NPs@NBC-700 catalyst for the selective hydrogenation of different heteroaromatic compounds were evaluated. As shown in Scheme 1, the Ni-NPs@NBC-700 catalyst was found to be effective for the hydrogenation of several quinolines to give partially reduced products, 1,2,3,4-tetrahydroquinolines, which are an essential part of pharmaceuticals, agrochemicals, and natural products. The parent quinoline gave 1,2,3,4-tetrahydroquinolines in 99% yield (Scheme 1, product 2), which is an important precursor for (±)-galipinine, an alkaloid and other bioactive compounds.76 Next, methyl-substituted quinolines smoothly afforded excellent yields of the desired products, 2-methyl-1,2,3,4-tetrahydroquinoline and 6-methyl-1,2,3,4-tetrahydroquinoline (Scheme 1, products 3 and 4). Remarkably, in the case of halogenated quinolines, chloro-, bromo-, and fluoro-moieties are well tolerated and afforded respective halogen substituted 1,2,3,4-tetrahydroquinolines in high yields (Scheme 1, products 5–8). Interestingly, 6-fluoro-2-methyl-1,2,3,4-tetrahydroquinoline is used to produce flumequine, which is a chemotherapeutic antibiotic drug. In addition, the ester group was also tolerated under the reaction conditions (Scheme 1, product 9). Gratifyingly, sterically hindered 2-phenylquinoline also transformed into 2-phenyl-1,2,3,4-tetrahydroquinoline in excellent yield (product 10). 6-Methoxy-1,2,3,4-tetrahydroquinoline, which is a key building block for the synthesis of the tubulin polymerization inhibitor was obtained from 6-methoxy quinoline in 99% yield (Scheme 1, product 11). In the case of benzo[h]quinoline, the product 1,2,3,4-tetrahydrobenzo[h]quinoline is obtained in very poor yield (Scheme 1, product 12). Interestingly, the selective hydrogenation of acridine to 9,10-dihydroacridine was achieved in 99% yield (product 13). The obtained product derivatives have potential applications in organic light-emitting diodes.77 Next, isoquinoline was reduced to 1,2,3,4-tetrahydroisoquinoline in good yield (product 14). However, 50 bar of hydrogen is required to reduce isoquinoline to obtain 70% of the desired product. This might be due to the less reactivity of isoquinoline towards reduction compared to quinoline. Additionally, quinoxaline (product 15) and their methyl-substituted derivatives, such as 2,3-dimethylquinoxaline and 2-methyl quinoxaline, afforded corresponding tetrahydroquinoxalines in excellent yields (products 16 and 17). Notably, 1-amino-2,6-dimethylpiperidine, the key intermediate of the clopamide drug was accessed in an excellent yield by the most hindered pyridine derivative such as 2,6-lutidine (Scheme 1, product 21). Five-membered heterocycle pyrrole was also successfully transformed into pyrrolidine with excellent yield (Scheme 1, product 22). Next, hydrogenation of biomass-derived furans such as 2-methylfuran, 2,5-dimethylfuran, and furfuryl alcohols was carried out, which is of increasing interest to access bio-based O-containing cyclo-aliphatic compounds. Interestingly, 2-methyl furan afforded an excellent yield of 2-methyltetrahydrofuran (Scheme 1, product 23). However, 2,5-dimethylfuran and furfuryl alcohol gave lower conversion due to the steric hindrance (products 24 and 25). Moreover, benzofuran derivatives also afforded desired products in satisfactory yields (products 26–28).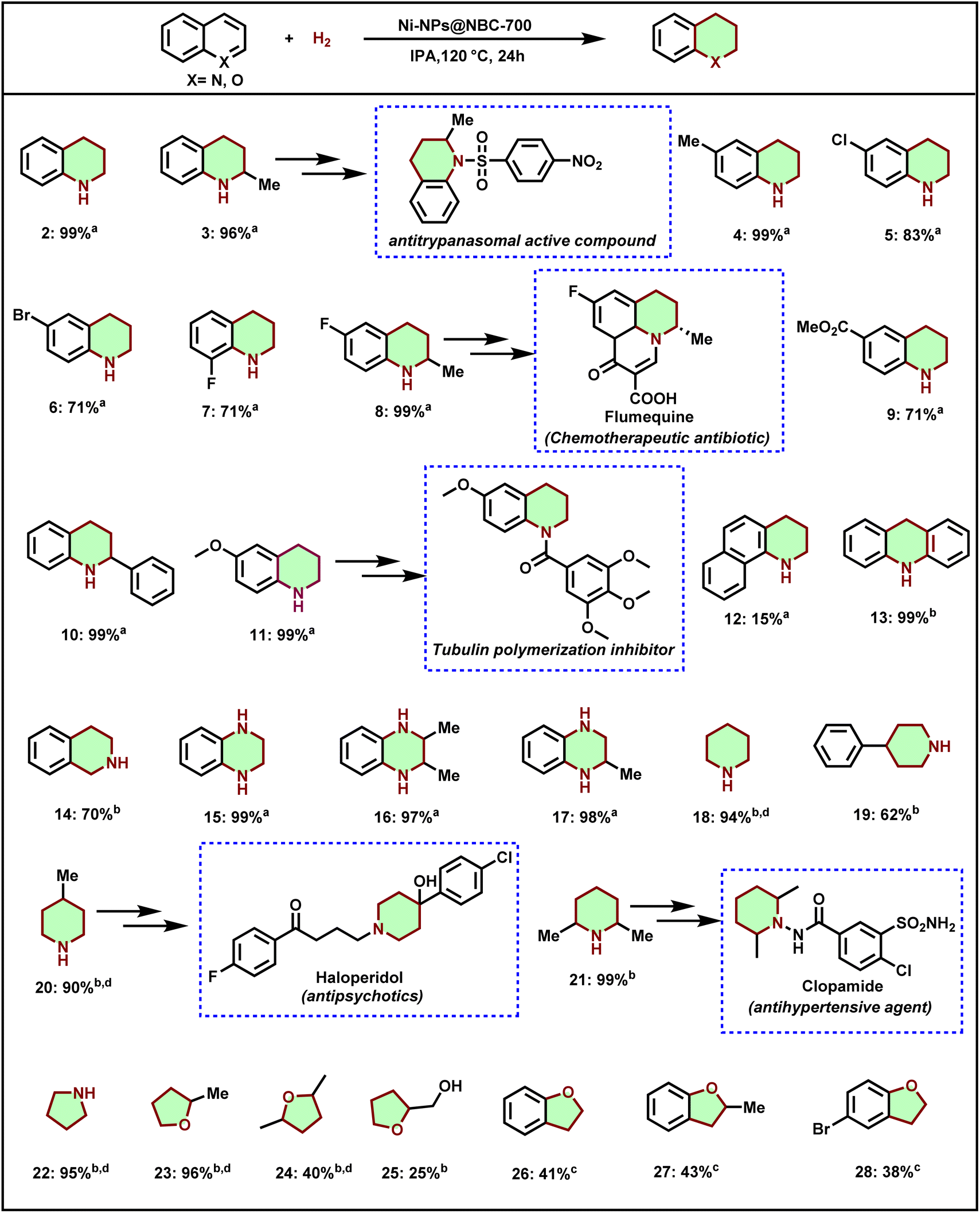 | ||
| Scheme 1 Ni-NPs@NBC-700 catalyzed hydrogenation in different heteroarenes. aReaction conditions: 0.5 mmol substrates, 50 mg catalyst, 30 bar H2, 2 mL isopropanol, 120 °C, and 24 h; isolated yield. bWith 50 bar H2 (50 bar), IPA (4 mL). cSame asb in water. dGC yields using n-hexane as the standard. | ||
In comparison to N-heterocycles, the yields of hydrogenated products of benzofuran derivatives are lower. This could be due to the high electronegativity of the oxygen atom that stabilizes the electron density within the ring system through resonance, resulting in the higher stability of the heteroarene ring. This might be the reason that the reduction of benzofurans is difficult compared to quinolines.
Ni-NPs@NBC-700 catalyzed hydrogenation of arenes
Upon being encouraged by the successful hydrogenation of N- and O-heteroarenes including sugar derived furans, we turned our interest to the hydrogenation of lignin-derived renewable phenol derivatives. For the successful hydrogenation of arenes using the optimal catalyst, a higher pressure of hydrogen (50 bar) is required compared to quinolines. Interestingly, simple phenol was transformed into cyclohexanol in 90% yield (Scheme 2, product 29). Then, substituted phenols such as 2-methylphenol, 4-methylphenol, 4-ethylphenol, 4-n-propylphenol, and 4-tertbutylphenol also afforded the corresponding saturated cyclic alcohols in excellent yields (Scheme 2, products 30–34). Such cyclohexanol-based compounds serve as key intermediates for the polymer industry allowing a straightforward and atom-economic pathway for the feedstock value addition. Similarly, benzyl alcohol and phenyl acetate also afforded cyclohexylmethanol and cyclohexyl acetate respectively in excellent yields (Scheme 2, products 35 and 36). Aniline and N-methyl aniline were hydrogenated and provided cyclohexanamine and N-methylcyclohexanamine in up to 75% yield (Scheme 2, products 37 and 38). Cyclohexylamines represent key precursors for pharmaceuticals and agrochemicals. Petrochemical-derived compounds such as benzene, toluene, o-xylene, and p-xylene were also hydrogenated and provided cyclohexanes in up to 92% yields (Scheme 2, products 39–42). Finally, N-(tert-butyl)decahydroisoquinoline-3-carboxamide (saquinavir intermediate) was prepared in good yield by the hydrogenation of N-(tert-butyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (Scheme 2, product 43).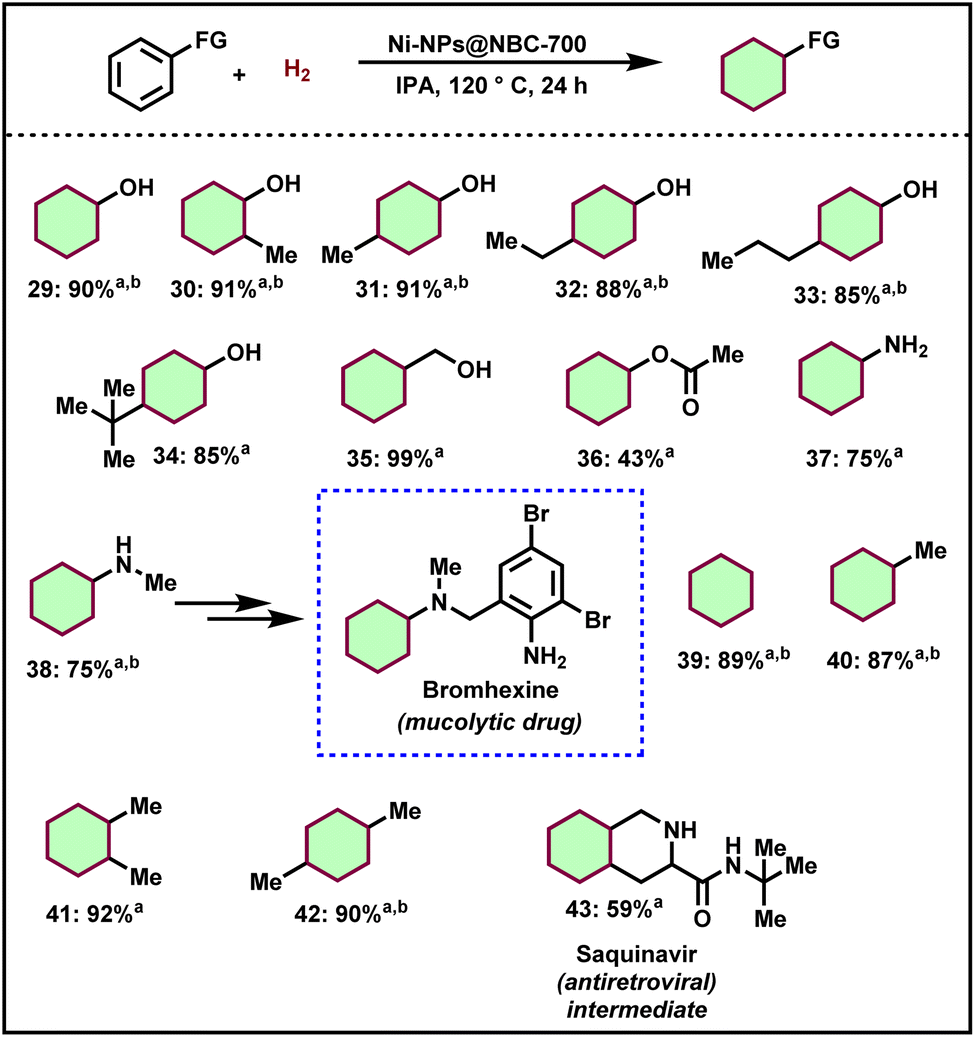 | ||
| Scheme 2 Ni-NPs@NBC-700 catalyzed ring hydrogenation of functionalized arenes. aReaction conditions: 0.5 mmol substrate, 50 mg catalyst, 50 bar H2, 2 mL isopropanol, 120 °C, and 24 h; isolated yield. bGC yields using n-hexane as the standard. | ||
Catalyst recyclability and reusability
For a given catalytic system, stability, recyclability and reusability are important features for the implementation of cost-effective processes and to facilitate easy product separation. To showcase these aspects, recycling experiments of Ni-NPs@NBC-700 have been conducted for the model reaction. As shown in Fig. 5, our Ni-nanocatalyst exhibited high stability as well as recyclability and reusability up to the 5th run without significant loss of activity or selectivity. After the 5th run, a little loss of catalytic activity, with 70% conversion of quinoline, was observed. However, the selectivity of the catalyst remained the same. Interestingly, the catalyst was easily separated from the reaction mixture using an external magnetic bar. Further, catalyst's durability/stability for the hydrogenation of quinoline (1) was tested by performing recyclability experiments with reduced reaction time. At 12 h of reaction time, the tetrahydroquinoline (2) yield was 58%, and there was no change in the yield of 2 in subsequent runs, which signifies the stability of NiNP@NBC-700 (Fig. 5). | ||
| Fig. 5 Recyclability and stability studies of the Ni-NPs@NBC-700 nanocatalyst for the hydrogenation of quinoline. Reaction conditions: 1 mmol quinoline, 100 mg catalyst, 30 bar hydrogen, 5 mL solvent (IPA), 120 °C, 24 h and 12 h. | ||
DFT calculations
Density Functional Theory (DFT) calculations have been carried out to understand the adsorption behavior of reaction intermediates, and the thermodynamic and kinetic feasibility of this Ni-catalyzed hydrogenation reaction. Since Ni(111) appears to be the most exposed surface from the experimental characterization, the reaction mechanism on the Ni(111) surface was studied, by modelling the slab considering five layers. First, different possibilities of reactant molecule adsorption on the Ni(111) surface were studied by considering the parallel and vertical adsorption of quinoline. The aromatic ring of quinoline interacts directly with the metal in parallel adsorption, whereas quinoline is adsorbed through nitrogen or one of the hydrogens of the benzene ring while adsorbing in a vertical way. Furthermore, we checked all the possibilities of adsorption considering the four possible adsorption sites on the Ni(111) surface: top site, bridge site, hexagonal close-packed (hcp) site, and face center cubic (fcc) site, as depicted in Fig. S8a and b.† As can be seen from Table S5,† the parallel adsorption of quinoline, in which the nitrogen atom is adsorbed on the bridge site has been found to be the most stable. It is worth mentioning that the most stable vertical adsorption energy (vertical top) of quinoline is 1.58 eV less stable than the least stable parallel adsorption (parallel top), indicating the strong parallel type of adsorption of aromatic species on the Ni(111) surface, and the effect of the aromatic ring on the adsorption. Such strong adsorption can be justified by investigating the average C–C bond length of the quinoline molecule which increases from 1.41 to 1.45 Å after adsorption, thus indicating the deviation from sp2 hybridization.Considering the most stable geometry of quinoline adsorption, we proceeded to study the hydrogenation pathway of quinoline on the Ni(111) surface. The possibility of competitive hydrogenation between the benzene and pyridine rings of quinolines to form 5,6,7,8-tetrahydroquinoline (5,6,7,8-THQ) and 1,2,3,4-tetrahydroquinoline (1,2,3,4-THQ), respectively has been analyzed by selectively hydrogenating the two aromatic rings. Our results show that the formation of 1,2,3,4-THQ is thermodynamically more favorable (free energy difference by ∼0.4 eV) than 5,6,7,8-THQ, thus agreeing well with the selectivity observed from the experimental results. This could be due to the basic character of nitrogen in the heterocyclic ring, which can promote the heterocyclic cleavage of hydrogen and facilitate hydrogenation.34 Furthermore, our results show that the average surface–adsorbate distance is more for 1,2,3,4-THQ (3.02 Å) compared to 5,6,7,8-THQ (2.95 Å), indicating a faster desorption of the former product.
Since 1,2,3,4-THQ has appeared to be thermodynamically more stable than 5,6,7,8-THQ, the thermodynamic and kinetic behaviors of the elementary hydrogenation reactions for the formation of 1,2,3,4-THQ were studied and all possible isomeric products in each reaction intermediate were analyzed and the most stable isomer for studying the reaction mechanism was selected. The relative energy of the isomeric products obtained for each reaction intermediate has been shown in Table S6† and the most stable geometries are shown in Fig. 6. As shown in Table S6,† the most stable isomers for the adsorbed C9H8N, C9H9N, and C9H10N are 1-hydroquinoline (1HQ), 1,4-dihydroquinoline (1,4-DHQ), and 1,3,4-trihydroquinoline (1,3,4-THQ), respectively. The detailed analysis of the adsorbed products on the surface (Table S7†) shows that the distance between the adsorbate and the surface is gradually increasing from 2.16 Å for quinoline to 2.69 Å for 1,2,3,4-THQ, respectively. This favours the smooth desorption of the product from the catalyst surface and avoids the possibility of catalyst poisoning.
 | ||
| Fig. 6 Optimized structures of the most stable reaction intermediates for the hydrogenation reaction. | ||
The thermodynamic feasibility of the elementary reactions is quantified in terms of reaction free energy (ΔG) which is calculated using the following equation, ΔG = ΔE + ΔZPE − TΔS, where ΔE, ΔZPE, and ΔS are the differences in total energy, zero-point energy, and entropy, respectively. Furthermore, the activation barriers for the elementary reaction were evaluated by identifying the transition state. As can be seen in Table S8,† the activation barrier gradually decreases (0.75 to 0.51 eV) towards higher hydrogenated intermediate formation, showing the highest activation barrier of 0.75 eV for the 1-HQ formation. However, the thermodynamic behavior shows that the 1-HQ formation is least endothermic. Such non-liner trend between thermodynamic and kinetic behaviors might be appearing due to the different nature of hydrogenation. While 1-HQ is formed by the hydrogenation at the nitrogen center, 1,4-DHQ, 1,3,4-THQ and 1,2,3,4-THQ are formed by the hydrogenation at the carbon center. Nevertheless, the later stage elementary steps gradually become kinetically favourable, and show a linear trend between thermodynamic and kinetic behavior, thus indicating kinetic feasibility of the reaction with the progress of the reaction.
According to the DFT calculations, the possible reaction pathway for the hydrogenation of quinoline is proposed (1), which is depicted in Fig. 7. The hydrogenation of quinoline includes the sequential addition of hydrogen atoms to nitrogen and carbon atoms of quinoline. Accordingly, in the first step, quinoline (1) is adsorbed to the catalyst surface (a), and the hydrogen atom is transferred to the N atom of quinoline to form the N–H bond and 1-hydroquinoline (1HQ) as shown in (b). Subsequently, H addition takes place by the hydrogenation at the carbon center to form 1,4-DHQ, followed by the formation of 1,3,4-THQ as shown in Fig. 7c and d, and finally, the formation of 1,2,3,4-THQ (2) takes place by the desorption from the catalyst surface. Interestingly, the addition of the second hydrogen atom to the partially hydrogenated quinoline takes place at the fourth place followed by the H addition at the third position due to the lower activation barrier in later stage elementary steps, thus indicating the gradual kinetic feasibility of the reaction with the progress of the reaction.
 | ||
| Fig. 7 Proposed reaction pathway for the hydrogenation of quinoline. | ||
Conclusions
We demonstrated the transformation of waste-biomass, specifically pine needles, into nitrogen-doped biocarbon supported nickel nanoparticles as an efficient and sustainable hydrogenation catalyst. This nickel-based nanostructured catalyst allows for the hydrogenation of both aromatic and heteroaromatic compounds, along with biomass-derived substrates to produce various cycloaliphatic compounds including tetrahydroquinolines, tetrahydroquinoxalines, and dihydrobenzofurans. This work holds significant relevance for sustainability as it involves the transformation of waste biomass into valuable catalytic materials, which have been utilized in important hydrogenation reactions.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the European Union under the REFRESH – Research Excellence for REgion Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 and the support from ERDF/ESF project TECHSCALE (no. CZ.02.01.01/00/22_008/0004587). We also thank Deutsche Forschungsgemeinschaft (DFG; Project number 447724917) and the State of Mecklenburg-Vorpommern for financial and general support. V. G. acknowledges the financial support of the European Union under the “MSCA Fellowships CZ at VSB-TUO 2.0” project, reg. no. CZ.02.01.01/00/22_010/0008714 within the Johannes Amos Comenius Operational Programme. T. B. is thankful to PMRF. Simulations are carried out on the Param Seva facility at the IIT Hyderabad, which is part of India's National Supercomputing Mission. A. M. thanks IIT Hyderabad (SG-145) and SERB (SRG/2023/002577) for funding.References
- M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10460–10476 CrossRef CAS PubMed.
- Z. Wei, F. Shao and J. Wang, Chin. J. Catal., 2019, 40, 980–1002 CrossRef CAS.
- A. Bordet and W. Leitner, Acc. Chem. Res., 2021, 54, 2144–2157 CrossRef CAS PubMed.
- V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS PubMed.
- J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef CAS PubMed.
- A. O'Byrne and P. Evans, Tetrahedron, 2008, 64, 8067–8072 CrossRef.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 12968–12971 CrossRef CAS PubMed.
- A. Kumar, V. Goyal, N. Sarki, B. Singh, A. Ray, T. Bhaskar, A. Bordoloi, A. Narani and K. Natte, ACS Sustain. Chem. Eng., 2020, 8, 15740–15754 CrossRef CAS.
- X. Cui, A.-E. Surkus, K. Junge, C. Topf, J. Radnik, C. Kreyenschulte and M. Beller, Nat. Commun., 2016, 7, 11326 CrossRef PubMed.
- K. Murugesan, T. Senthamarai, A. S. Alshammari, R. M. Altamimi, C. Kreyenschulte, M.-M. Pohl, H. Lund, R. V. Jagadeesh and M. Beller, ACS Catal., 2019, 9, 8581–8591 CrossRef CAS.
- K. Murugesan, V. G. Chandrashekhar, C. Kreyenschulte, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2020, 59, 17408–17412 CrossRef CAS PubMed.
- M. Wollenburg, A. Heusler, K. Bergander and F. Glorius, ACS Catal., 2020, 10, 11365–11370 CrossRef CAS PubMed.
- L. Foppa and J. Dupont, Chem. Soc. Rev., 2015, 44, 1886–1897 RSC.
- S. El Sayed, A. Bordet, C. Weidenthaler, W. Hetaba, K. L. Luska and W. Leitner, ACS Catal., 2020, 10, 2124–2130 CrossRef CAS.
- B. Zhou, V. G. Chandrashekhar, Z. Ma, C. Kreyenschulte, S. Bartling, H. Lund, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2023, 62, e202215699 CrossRef CAS PubMed.
- F. Chen, A.-E. Surkus, L. He, M.-M. Pohl, J. Radnik, C. Topf, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 11718–11724 CrossRef CAS PubMed.
- B. L. Tran, J. L. Fulton, J. C. Linehan, J. A. Lercher and R. M. Bullock, ACS Catal., 2018, 8, 8441–8449 CrossRef CAS.
- L. Ling, Y. He, X. Zhang, M. Luo and X. Zeng, Angew. Chem., Int. Ed., 2019, 58, 6554–6558 CrossRef CAS PubMed.
- X. Zhang, L. Ling, M. Luo and X. Zeng, Angew. Chem., Int. Ed., 2019, 58, 16785–16789 CrossRef CAS PubMed.
- S. Kim, F. Loose, M. J. Bezdek, X. Wang and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 17900–17908 CrossRef CAS PubMed.
- B. Chatterjee, D. Kalsi, A. Kaithal, A. Bordet, W. Leitner and C. Gunanathan, Catal. Sci. Technol., 2020, 10, 5163–5170 RSC.
- V. Papa, Y. Cao, A. Spannenberg, K. Junge and M. Beller, Nat. Catal., 2020, 3, 135–142 CrossRef CAS.
- W. Jiang, J.-P. Cao, C. Zhu, M. Zhao, Z.-H. Ni, X.-Y. Zhao, J.-X. Xie, L. Zhao, Y.-P. Zhao and H.-C. Bai, Appl. Catal., B, 2022, 307, 121137 CrossRef CAS.
- D. Moock, T. Wagener, T. Hu, T. Gallagher and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 13677–13681 CrossRef CAS PubMed.
- C. H. Schiwek, C. Jandl and T. Bach, ACS Catal., 2022, 12, 3628–3633 CrossRef CAS.
- A. Karakulina, A. Gopakumar, İ. Akçok, B. L. Roulier, T. LaGrange, S. A. Katsyuba, S. Das and P. J. Dyson, Angew. Chem., Int. Ed., 2016, 128, 300–304 CrossRef.
- A. Kulkarni, W. Zhou and B. l. Török, Org. Lett., 2011, 13, 5124–5127 CrossRef CAS PubMed.
- N. Tanaka and T. Usuki, Eur. J. Org Chem., 2020, 2020, 5514–5522 CrossRef CAS.
- Y. Duan, L. Li, M.-W. Chen, C.-B. Yu, H.-J. Fan and Y.-G. Zhou, J. Am. Chem. Soc., 2014, 136, 7688–7700 CrossRef CAS PubMed.
- C. Chaudhari, K. Sato, Y. Nishida, T. Yamamoto, T. Toriyama, S. Matsumura, Y. Ikeda, K. Terada, N. Abe, K. Kusuda, H. Kitagawa and K. Nagaoka, RSC Adv., 2020, 10, 44191–44195 RSC.
- X. Cui, Z. Huang, A. P. van Muyden, Z. Fei, T. Wang and P. J. Dyson, Sci. Adv., 2020, 6, eabb3831 CrossRef CAS PubMed.
- D. Ren, L. He, L. Yu, R.-S. Ding, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Am. Chem. Soc., 2012, 134, 17592–17598 CrossRef CAS PubMed.
- V. Colliere, M. Verelst, P. Lecante and M. R. Axet, Chem.–Eur. J., 2024, 30, e202302131 CrossRef CAS PubMed.
- Y. Motoyama, K. Ohnishi, H. Shibuya, P. Joshi and M. Yoshimura, Asian J. Org. Chem., 2023, 12, e202200666 CrossRef CAS.
- T.-N. Ye, J. Li, M. Kitano and H. Hosono, Green Chem., 2017, 19, 749–756 RSC.
- S. Kacem, M. Emondts, A. Bordet and W. Leitner, Catal. Sci. Technol., 2020, 10, 8120–8126 RSC.
- Y. Hu, M. Liu, S. Bartling, H. Lund, H. Atia, P. J. Dyson, M. Beller and R. V. Jagadeesh, Sci. Adv., 2023, 9, eadj8225 CrossRef CAS PubMed.
- N. Antil, A. Kumar, N. Akhtar, W. Begum, M. Chauhan, R. Newar, M. S. Rawat and K. Manna, Inorg. Chem., 2021, 61, 1031–1040 CrossRef PubMed.
- B. Sahoo, C. Kreyenschulte, G. Agostini, H. Lund, S. Bachmann, M. Scalone, K. Junge and M. Beller, Chem. Sci., 2018, 9, 8134–8141 RSC.
- S. Chen, W. Xue and C. Tang, ChemSusChem, 2022, 15, e202201522 CrossRef CAS PubMed.
- S. Mao, P. Ryabchuk, S. Dastgir, M. Anwar, K. Junge and M. Beller, ACS Appl. Nano Mater., 2022, 5, 5625–5630 CrossRef CAS.
- C. Bauer, F. Müller, S. Keskin, M. Zobel and R. Kempe, Chem.–Eur. J., 2023, 29, e202300561 CrossRef CAS PubMed.
- J. Zhang, C. Zheng, M. Zhang, Y. Qiu, Q. Xu, W.-C. Cheong, W. Chen, L. Zheng, L. Gu and Z. Hu, Nano Res., 2020, 13, 3082–3087 CrossRef.
- B. Singh, V. Goyal, D. Sarma, R. Kumar, T. Bhatt, A. Mahata, C. Raji Reddy, A. Narani and K. Natte, ACS Catal., 2023, 13, 9724–9744 CrossRef CAS.
- S. Sadjadi, M. Akbari, B. Léger, E. Monflier and M. M. Heravi, ACS Sustain. Chem. Eng., 2019, 7, 6720–6731 CrossRef CAS.
- P. Veerakumar, I. Panneer Muthuselvam, C.-T. Hung, K.-C. Lin, F.-C. Chou and S.-B. Liu, ACS Sustain. Chem. Eng., 2016, 4, 6772–6782 CrossRef CAS.
- T. Song, P. Ren, J. Xiao, Y. Yuan and Y. Yang, Green Chem., 2020, 22, 651–656 RSC.
- Z. Ma, T. Song, Y. Yuan and Y. Yang, Chem. Sci., 2019, 10, 10283–10289 RSC.
- F. Ferlin, F. Valentini, D. Sciosci, M. Calamante, E. Petricci and L. Vaccaro, ACS Sustain. Chem. Eng., 2021, 9, 12196–12204 CrossRef CAS.
- V. Goyal, N. Sarki, B. Singh, A. Ray, M. Poddar, A. Bordoloi, A. Narani and K. Natte, ACS Appl. Nano Mater., 2020, 3, 11070–11079 CrossRef CAS.
- S. Ahmad, F. Gao, H. Lyu, J. Ma, B. Zhao, S. Xu, C. Ri and J. Tang, Chem. Eng. J., 2022, 450, 138006 CrossRef CAS.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M.-M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- G. Hahn, P. Kunnas, N. de Jonge and R. Kempe, Nat. Catal., 2019, 2, 71–77 CrossRef CAS.
- J. L. Fiorio, M. A. Garcia, M. L. Gothe, D. Galvan, P. C. Troise, C. A. Conte-Junior, P. Vidinha, P. H. Camargo and L. M. Rossi, Coord. Chem. Rev., 2023, 481, 215053 CrossRef CAS.
- D. Li, W. Chen, J. Wu, C. Q. Jia and X. Jiang, J. Mater. Chem., 2020, 8, 24977–24995 RSC.
- Y. Zhang, L. Zhou, B. Han, L. W. Li, B. Li and L. Zhu, Chin. J. Inorg. Chem., 2022, 42, 33 CAS.
- F. Qin, W. Xue and C. Tang, Chin. J. Chem., 2025, 43, 916–924 CrossRef CAS.
- Z. Yuan, X. Li, G. Wang, Z. Zhu, Y. Liao, Z. Zhang and B. Liu, Mol. Catal., 2023, 540, 113052 CrossRef CAS.
- P. Ryabchuk, G. Agostini, M.-M. Pohl, H. Lund, A. Agapova, H. Junge, K. Junge and M. Beller, Sci. Adv., 2018, 4, eaat0761 CrossRef PubMed.
- F. Zhong, X. Ge, W. Sun, W. Du, K. Sang, C. Yao, Y. Cao, W. Chen, G. Qian, X. Duan, Z. Yang, X. Zhou and J. Zhang, Chem. Eng. J., 2023, 455, 140536 CrossRef CAS.
- T. Zhu, J. Dong, L. Niu, G. Chen, L. Ricardez-Sandoval, X. Wen and G. Bai, Appl. Clay Sci., 2021, 203, 106003 CrossRef CAS.
- Z. Zhao, X. Long, M. Millan, G. Yuan, Z. Cui, Z. Dong, Y. Cong, J. Zhang and X. Li, Carbon, 2023, 213, 118227 CrossRef CAS.
- S. Wang and L. Yang, React. Chem. Eng., 2022, 7, 170–180 RSC.
- Y. Shi, E. Xing, J. Zhang, Y. Xie, H. Zhao, Y. Sheng and H. Cao, ACS Sustain. Chem. Eng., 2019, 7, 9464–9473 CrossRef CAS.
- S. Hu, M. Xue, H. Chen and J. Shen, Chem. Eng. J., 2010, 162, 371–379 CrossRef CAS.
- P. Veerakumar, S.-M. Chen, R. Madhu, V. Veeramani, C.-T. Hung and S.-B. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 24810–24821 CrossRef CAS PubMed.
- Y. Sahoo, Y. He, M. Swihart, S. Wang, H. Luo, E. Furlani and P. Prasad, J. Appl. Phys., 2005, 98, 054308 CrossRef.
- Z. Zhuang, S. A. Giles, J. Zheng, G. R. Jenness, S. Caratzoulas, D. G. Vlachos and Y. Yan, Nat. Commun., 2016, 7, 10141 CrossRef CAS PubMed.
- X. Lu, J. He, R. Jing, P. Tao, R. Nie, D. Zhou and Q. Xia, Sci. Rep., 2017, 7, 2676 CrossRef PubMed.
- M. Jia, C. Choi, T.-S. Wu, C. Ma, P. Kang, H. Tao, Q. Fan, S. Hong, S. Liu and Y.-L. Soo, Chem. Sci., 2018, 9, 8775–8780 RSC.
- Z. Geng, Y. Lin, X. Yu, Q. Shen, L. Ma, Z. Li, N. Pan and X. Wang, J. Mater. Chem., 2012, 22, 3527–3535 RSC.
- A. Goswami, R. G. Kadam, J. Tuček, Z. Sofer, D. Bouša, R. S. Varma, M. B. Gawande and R. Zbořil, Chem. Eng. J., 2020, 382, 122469 CrossRef CAS.
- C. Zhang, J. Xiao, X. Lv, L. Qian, S. Yuan, S. Wang and P. Lei, J. Mater. Chem., 2016, 4, 16516–16523 RSC.
- M.-H. Zhang, Z.-M. Liu, G.-D. Lin and H.-B. Zhang, Appl. Catal., A, 2013, 451, 28–35 CrossRef CAS.
- X. Wang, Y. Liu, L. Zhu, Y. Li, K. Wang, K. Qiu, N. Tippayawong, P. Aggarangsi, P. Reubroycharoen and S. Wang, J. CO2 Util., 2019, 34, 733–741 CrossRef CAS.
- G. Diaz-Muñoz, R. G. Isidorio, I. L. Miranda, G. N. de Souza Dias and M. A. N. Diaz, Tetrahedron Lett., 2017, 58, 3311–3315 CrossRef.
- X. Y. Liu, Y. Y. Ma, W. Zhang, B. Song, L. Ding, M. K. Fung and J. Fan, Chem.–Eur. J., 2018, 24, 11755–11762 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5su00026b |
| This journal is © The Royal Society of Chemistry 2025 |