
Unveiling the performance of ultrathin bimetallic CoxNi1−x(OH)2 nanosheets for pseudocapacitors and oxygen evolution reaction†
Pallavi Bhaktapralhad
Jagdale
 a,
Sayali Ashok
Patil
a,
Arupjyoti
Pathak
b,
Mukaddar
Sk
b,
Ranjit
Thapa
bc,
Amanda
Sfeir
d,
Sebastien
Royer
de,
Akshaya Kumar
Samal
a and
Manav
Saxena
*a
a,
Sayali Ashok
Patil
a,
Arupjyoti
Pathak
b,
Mukaddar
Sk
b,
Ranjit
Thapa
bc,
Amanda
Sfeir
d,
Sebastien
Royer
de,
Akshaya Kumar
Samal
a and
Manav
Saxena
*a
aCentre for Nano and Material Sciences, Jain (Deemed-to-be University), Jain Global Campus, Ramanagara, Bangalore 562112, India. E-mail: s.manav@jainuniversity.ac.in; manavsaxena19@gmail.com
bDepartment of Physics, SRM University-AP, Amaravati 522240, Andhra Pradesh, India
cCentre for Computational and Integrative Sciences, SRM University – AP, Amaravati 522240, Andhra Pradesh, India
dUniversité de Lille, CNRS, Centrale Lille, Université Artois, UMR 8181-UCCS-12 Unité de Catalyse et Chimie du Solide, Lille 59000, France
eUniversité du Littoral Côte d’Opale – Ecole d’Ingénieur du Littoral Côte d’Opale, UCEIV, UR 4492, MREI 1, 189 A, Avenue Maurice Schumann, 59140 Dunkerque, France
First published on 19th November 2024
Abstract
The rational design of highly efficient and stable electrodes is necessary for energy storage and electrocatalysis. Herein, we developed a nanometre thin bimetallic ultrathin CoxNi1−x(OH)2 nanosheet with a large lateral size by the ionic layer epitaxy (ILE) technique as an efficient bifunctional electrode material for pseudocapacitors and the oxygen evolution reaction. Its electrochemical performance was readily tuned by controlling the Co/Ni ratio. The nanosheet with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 Co:Ni ratio (termed Co1Ni3-NS) showed an excellent volumetric (areal) capacitance of 3783 F cm−3 (3 mF cm−2) at 0.3 mA cm−2 with 336 mW h cm−3 energy density at 256 W cm−3 power density and excellent stability, substantially outperforming other monometallic and bimetallic NSs. Moreover, as an electrocatalyst, Co1Ni3-NS delivered a lower overpotential (η10 = 318 mV) and Tafel slope (61 mV dec−1) in an alkaline environment. In situ Raman spectroscopy was employed to demonstrate the dynamic structural evolution of the catalyst during the OER process. Furthermore, DFT investigations further revealed that Co1Ni3-NS is a promising electrode with higher quantum capacitance and lower overpotential compared to other Co/Ni ratios. These findings pave a new way for controlled synthesis of highly efficient, bimetallic, and bifunctional electrode materials for pseudocapacitors and the OER.
:3 Co:Ni ratio (termed Co1Ni3-NS) showed an excellent volumetric (areal) capacitance of 3783 F cm−3 (3 mF cm−2) at 0.3 mA cm−2 with 336 mW h cm−3 energy density at 256 W cm−3 power density and excellent stability, substantially outperforming other monometallic and bimetallic NSs. Moreover, as an electrocatalyst, Co1Ni3-NS delivered a lower overpotential (η10 = 318 mV) and Tafel slope (61 mV dec−1) in an alkaline environment. In situ Raman spectroscopy was employed to demonstrate the dynamic structural evolution of the catalyst during the OER process. Furthermore, DFT investigations further revealed that Co1Ni3-NS is a promising electrode with higher quantum capacitance and lower overpotential compared to other Co/Ni ratios. These findings pave a new way for controlled synthesis of highly efficient, bimetallic, and bifunctional electrode materials for pseudocapacitors and the OER.
1 Introduction
Supercapacitors and electrochemical water splitting have been recognized as cutting-edge renewable and sustainable energy technologies to fulfill the next generation's energy demands, driving extensive research in developing advanced materials.1,2 Substantial research is underway, and various electrode materials have been reported separately for energy storage and conversion purposes, where each material demonstrates either of the functionalities.3 In this regard, developing cost-effective, bifunctional electrode materials would be incredibly fascinating. However, the issue is to achieve dual functionality in a single material. Bifunctional materials stand out due to their unique features, unlike monofunctional materials. Supercapacitors have high power density and are mainly classified into two categories based on the charge-storage mechanisms, i.e., electric double-layer capacitors (EDLCs) and pseudocapacitors dominated by non-faradaic processes (charge stored electrostatically at the electrode/electrolyte interface) and surface faradaic redox reactions, respectively.4–6 The electrochemical characteristics of pseudocapacitive materials are neither purely capacitive nor bulk faradaic. Instead, they bridge the gap between EDLCs and batteries, offering simultaneous high energy and power.7,8 On the other hand, water electrolysis (or splitting) is one of the clean and sustainable ways to produce hydrogen, ideally with zero carbon emissions. The anodic oxygen evolution reaction (OER) is an important but sluggish reaction step in water electrolysis due to the involvement of a complex four-electron transfer mechanism.9 This requires high overpotential, significantly reducing the overall energy conversion efficiency. To tackle this challenge, electrocatalysts with excellent catalytic activity are needed to minimize the kinetic barrier and boost the reaction rates. RuO2 and IrO2 have been recognized as state-of-the-art electrocatalysts due to their high catalytic activity and stability.10,11 However, their scarcity and high cost have driven immense research on the development of alternative abundant, cost-effective, highly efficient, and sustainable materials.To date, transition-metal-based oxides (TMOs)/hydroxides (TMHs), including CoOx, MnO2,12 NiOx,13 Co(OH)2,14–16 and Ni(OH)2 (ref. 17–19), have been widely explored to improve supercapacitor performance due to their high theoretical capacitance, excellent electrochemical redox activity, high abundance, low toxicity, high conductivity, and high stability.20 Furthermore, it has been reported that various bimetallic Co–Ni hydroxides show improved electrochemical properties compared to solitary hydroxides due to a synergistic effect of double cations and multi-redox reactions.21,22 Moreover, TMHs have been extensively used as electrocatalysts due to their high earth abundance and potentially high catalytic activity. Co(OH)2 and Ni(OH)2 exhibit high electrocatalytic activity, especially in alkaline media.16,19,23,24 However, recent reports state that, compared to monometallic systems, bimetallic CoxNi1−x(OH)2 systems offer synergistic effects by optimizing the electronic structure, enhancing electrical conductivity, and lowering reaction energy barriers, which substantially boosts catalytic activity, enabling more abundant redox reactions across multiple valence states.2,25 The morphology of the electrode materials plays a crucial role in overall electrochemical performance.
Ultrathin two-dimensional (2D) nanomaterials with large surface areas have shown promising performance in advancing supercapacitors and OER activity.26 The high surface area to volume ratio facilitates ion adsorption, and faradaic reactions improve the catalysis rate due to the higher number of exposed active sites. Owing to the short diffusion path, ultrathin 2D materials enable fast charge/discharge, ensuring high power density and offering abundant active sites and rapid mass transport.27–29 The final composition of a 2D bimetallic hydroxide predominantly depends on the ratio and reaction kinetics of the metal center under given reaction conditions. The electrochemical properties of bimetallic CoxNi1−x(OH)2 can be rationally tuned by controlling the Co and Ni atomic ratio. However, attaining precise control over material morphology and composition is one of the key synthesis challenges. The ionic layer epitaxy (ILE)30,31 approach promises controlled synthesis of nanometre-thin bimetallic CoxNi1−x(OH)2 nanosheets at the water–air interface, in which an amphiphilic molecule monolayer is employed to direct and control the growth of nanosheets with microscopic continuity.
Based on the above considerations, herein, we report the development of an ultrathin and large area, bimetallic CoxNi1−x(OH)2 nanosheet (NS) as an efficient bifunctional electrode material for supercapacitors and the OER. The CoxNi1−x(OH)2 NSs with different Co and Ni atomic ratios (i.e., Co:Ni = 1:0, 3:1, 1:1, 1:3, and 0:1 denoted as Co-NS, Co3Ni1-NS, Co1Ni1-NS, Co1Ni3-NS, and Ni-NS, respectively) have been synthesized via the ILE method with a thickness of 3.6 ± 0.5 nm. Co1Ni3-NS with an optimized Co:Ni ratio (i.e., 1:3) achieved superior performance toward the pseudocapacitor and OER process. The symmetric supercapacitor device based on Co1Ni3-NS could achieve a capacitance of 3783 F cm−3 (3 mF cm−2) at a current density of 0.3 mA cm−2 and a maximum energy density and power density of 336 mW h cm−3 and 256 W cm−3 and 66% initial capacitance retention after 10000 GCD cycles. As an electrocatalyst, the Co1Ni3-NS electrode exhibited a lower overpotential of 318 mV to reach a current density of 10 mA cm−2 showing a Tafel slope of 61 mV dec−1. Furthermore, high mass activity (1056 A g−1) and long-term durability of OER operation were achieved by Co1Ni3-NS. In situ Raman spectroscopy emphasized the structural insights into the dynamically reconstructed surface of the Co1Ni3-NS electrode during oxygen evolution.
2 Materials and methods
2.1 Materials
Cobalt nitrate hexahydrate (Co(NO3)2·6H2O, ≥99.0%), nickel nitrate hexahydrate (Co(NO3)2·6H2O, ≥99.0%), hexamethylene tetramine (HMTA) and sodium dodecyl sulphate (NaC12H25SO4, ≥99% purity) were purchased from SD Fine Ltd India. RuO2 (99.99% purity) and Nafion solution (5 wt% in lower aliphatic alcohols and water) were purchased from Sigma Aldrich. All chemicals were used without any further purification.2.2 Synthesis of CoxNi1−x(OH)2 NSs
The CoxNi1−x(OH)2 NSs were was synthesized using the ILE method. In the typical synthesis technique, 50 ml aqueous nutrient solution of 5 mM Co(NO3)2·6H2O, 5 mM Ni(NO3)2·6H2O, and 10 mM of HMTA was taken in a Petri dish. 10 μl of 0.1% SDS solution was gently spread on the surface of the reaction mixture. Afterward, the Petri dish was covered with a lid and placed in a hot air oven at 60 °C for 2 h. The reaction mixture was allowed to cool to room temperature. The Co1Ni1-NS nanosheets grown at the water–air interface were transferred onto the desired substrate for characterization and applications. The total amount of cobalt and nickel was maintained at 10 mM and the ratios of Co/Ni were adjusted to 1:0, 3:1, 1:1, 1:3, and 0:1. The synthesis details are mentioned in Table S1.†
The details of the preparation of the RuO2 electrode, electrochemical measurements, DFT calculations, and supporting figures are also provided in the ESI.†
2.3 Characterization
The surface morphology of the CoxNi1−x(OH)2 NSs was observed using a field emission scanning electron microscope (FE-SEM, JEOL, JSM-7100). The crystal structure was studied using X-ray diffraction (XRD, Rigaku SmartLab 1.54 Å Cu-Kα source) and transmission electron microscope (TEM, JEOL, JEM-2100 Plus, Japan). The elemental distribution in CoxNi1−x(OH)2 NSs was analyzed using high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and STEM energy dispersive X-ray spectroscopy (STEM-EDS) elemental mapping (FEI Titan STEM). A NX20 atomic force microscope (AFM) from Park Systems was used to determine the thickness of the NS. X-ray photoelectron spectroscopy (XPS) spectra were recorded on a Kratos Analytical AXIS Ultra DLD spectrometer with a monochromatic Al Ka X-ray radiation source (1486.6 eV) and an electron analyzer operating at a fixed pass energy of 20 eV. All the XPS results were C-corrected (285.0 eV) and fitted using CasaXPS V2.3.24 software. TEM and XPS analyses were performed on a 300 mesh carbon TEM grid and Pt (or FTO-coated glass) substrates. The rest of the characterization experiments were carried out on a Si/SiO2 wafer. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was employed to quantify the actual composition of Co and Ni in the synthesized materials. The ICP-OES sample preparation method is discussed in the ESI.†In situ Raman spectroscopy was performed using a Renishaw Raman spectrophotometer at a 532 nm excitation wavelength using a 2400 grating.3 Results and discussion
The bimetallic CoxNi1−x(OH)2 NSs were synthesized by the ILE method, in which the SDS monolayer serves as a soft template to guide the nucleation and growth of the nanosheets (see the materials and methods for synthesis details). At 60 °C, HMTA decomposes into NH3 and HCHO yielding an alkaline environment. Ammonia, a weak base in water, undergoes hydrolysis and forms ammonium and hydroxide ions. The Co2+ and Ni2+ cations in the reaction solution hydrolyze to form metal hydroxides as a stern layer guided by the SDS layer on the surface. As shown in Fig. 1a, the SDS anions self-assemble to form a closely packed amphiphilic molecule monolayer at the water–air interface. This planar soft template has strong electrostatic interaction with both the cations and helps to grow a metal hydroxide film as a stern layer at the interface. The Co and Ni cations in the aqueous solution accelerate the diffusion of hydroxide ions toward the surface, where they react with cations and form a large-area and ultrathin bimetallic cobalt-nickel hydroxide nanosheet at the water–air interface. The chemical reactions are shown below.| (CH2)6N4 + 6H2O ↔ 4NH3 + 6HCHO | (1) |
| NH3 + H2O ↔ NH3·H2O | (2) |
| NH3·H2O ↔ NH4+ + OH− | (3) |
| M2+ + 2OH− ↔ M(OH)2 | (4) |
 | ||
| Fig. 1 Synthesis of bimetallic CoxNi1−x(OH)2 NSs. (a) Schematic of ILE growth of CoxNi1−x(OH)2 NSs and (b) low-magnification FESEM image of Co1Ni3-NS. (c) AFM image of Co1Ni3-NS. Inset shows the height profile, indicating a thickness of 3.6 nm. (d) XRD pattern of bimetallic Co1Ni3-NS and monometallic Co-NS and Ni-NS, (e) TEM image of Co1Ni3-NS, (f) HRTEM image, (g) SAED pattern, (h–k) EDS elemental mapping of Co1Ni3-NS showing the uniform distribution of (i) Co, (j) Ni, and (k) O elements, and (l) TEM EDS line scanning of Co and Ni. | ||
The surface morphology of CoxNi1−x(OH)2 NSs supported on a SiO2/Si wafer was investigated using the FESEM image as shown in Fig. 1b. The nanosheet covering the entire substrate area suggests the uniform and continuous nature of the nanosheets, resulting in slight contrast (Fig. S1†). No significant overlaps or wrinkles were observed. The morphology of the CoxNi1−x(OH)2 NSs remains well maintained at different ratios of Co:Ni, indicating an identical growth mechanism and crystal structure of CoxNi1−x(OH)2 NSs under given experimental conditions (Fig. S1†). A few small cracks on the nanosheets originating during the transfer and drying process provide a good observation area to determine the thickness of the nanosheets using AFM. The AFM image (Fig. 1c) reveals that the surface of the nanosheet is smooth and flat with an average surface roughness factor (Ra) of 0.9 nm. The line profile along the crack area in Fig. 1c shows that the thickness of the nanosheet is 3.6 ± 0.2 nm. Fig. 1d demonstrates the XRD pattern of CoxNi1−x(OH)2 NSs with various ratios of Co and Ni such as 1:0, 0:1, and 1:3, denoted as Co-NS, Ni-NS, and Co1Ni3-NS, respectively. The strong diffraction peaks of Co-NS correspond to (003), (006), and (018) planes for the hexagonal phase of Co(OH)2 (JCPDS no. 046-0605), and Ni-NS can be indexed to the (003), (006), and (107) planes of Ni(OH)2 (JCPDS no. 038-715). The diffraction peak of the Co1Ni3-NS sample is located between those of Co(OH)2 and Ni(OH)2, and the intense peak resembles the exposed (003) surface of the nanosheet. These results are in good agreement with previous reports.32,33 The crystal structure of Co1Ni3-NS was investigated using the TEM image. Fig. 1e exhibits a nanosheet covered on a TEM grid. The presence of some wrinkles or overlaps is ascribed to the flexibility of the nanosheet. Further investigation of the nanosheet at high-resolution TEM (HRTEM) reveals the crystalline feature of the nanosheet, as shown in Fig. 1f. The clear lattice fringes in the HRTEM image were measured by the inverse fast Fourier transform (IFFT) method and showed an interplanar spacing of 2.7 Å corresponding to the (101) and (202) planes of Ni(OH)2 (JCPDS no. 014-0117) and the (100) and (106) planes of Co(OH)2 (JCPDS no. 046-0605), as shown in (Fig. S2†). Ni(OH)2 and Co(OH)2 have almost identical hexagonal crystal structures and parameters; hence, distinguishing the individual interplanar spacing is challenging. The hexagonal crystalline nature exhibited by selected area electron diffraction (SAED) patterns supports this conclusion. As shown in Fig. 1g, the bright diffraction spots from the SAED pattern represent the (100), (110), and (103) planes of Ni(OH)2 and Co(OH)2. Fig. 1h–k display a uniform distribution of Co and Ni throughout the nanosheet. The TEM energy diffraction spectrum (EDS) and line scanning of Co1Ni3-NS show a high ratio of Ni compared to Co (Fig. 1l and S3†), further supported by mapping from the high-angle annular dark field-scanning TEM (HAADF-STEM) image.
Furthermore, detailed information about the surface chemical composition and the valence states of the as-synthesized bimetallic cobalt-nickel hydroxide nanosheets was investigated by XPS, and the respective results are displayed in Fig. 2. The Co1Ni3-NS survey spectrum exhibited characteristic C, O, Co, Ni, and Pt peaks. The C and Pt signals originated from the atmospheric adsorbed carbon/trace of the surfactant and Pt substrate, respectively (Fig. 2a). The high-resolution Co 2p spectrum exhibited two main peaks at binding energies (BEs) of 780.7 and 796.3 eV, which are associated with the 2p3/2 and 2p1/2 spin–orbit components of Co2+ having a slit-orbit energy of 15.5 eV, as shown in Fig. 2b. The Co 2p3/2 band is curve fitted using the Shirley background and deconvoluted into two intense peaks at BE of 780.7 and 782.4, and two satellite peaks at BE 785.9 and 790.38 eV representing the characteristic peaks of Co(OH)2 as reported previously.34 The Ni 2p spectrum in Fig. 2c demonstrates two prominent peaks at BE 855.5 and 872.8 eV corresponding to 2p3/2 and 2p1/2 components with a slit-orbit energy of 17.3 eV, resembling the Ni2+ oxidation state of Ni(OH)2. The Ni 2p3/2 region is deconvoluted into peaks at BEs of 854.5, 855.5, 857.6, 860.4, 861.4, and 866.4 eV, showing characteristic peaks of Ni2+. The core level O 1s spectrum exhibited a broad peak at 531 eV associated with oxygen in –OH bonded to Co and Ni, validating the successful formation of Co1Ni3-NS (Fig. 2d). The additional peak at 532.4 eV is ascribed to the adsorbed molecular water.33 The survey spectrum of CoxNi1−x(OH)2 (Fig. S4†) depicted the atomic percentage of Co:Ni as 100:0, 59.1:40.9, 51.5:48.5, 25.2:74.8, and 0:100 for Co:Ni ratios 1:0, 3:1, 1:1, 1:3 and 0:1, respectively. Additionally, ICPS-OES was used to quantify the weight percentage of Co and Ni in CoxNi1−x(OH)2 NSs synthesized under various Co-to-Ni ratios, as shown in Table 1. The weight percentage of Co:Ni elements in CoxNi1−x(OH)2 samples was found to be 100:0, 71:29, 44:56, 27:73, and 0:100 for Co-NS, Co3Ni1-NS, Co1Ni1-NS, Co1Ni3-NS, and Ni-NS, respectively, which is further supported by the EDS spectrum and elemental mapping. These results suggest that the Co:Ni ratio in the bimetallic nanosheets can be readily tuned by adjusting the precursor concentration using the ILE strategy.
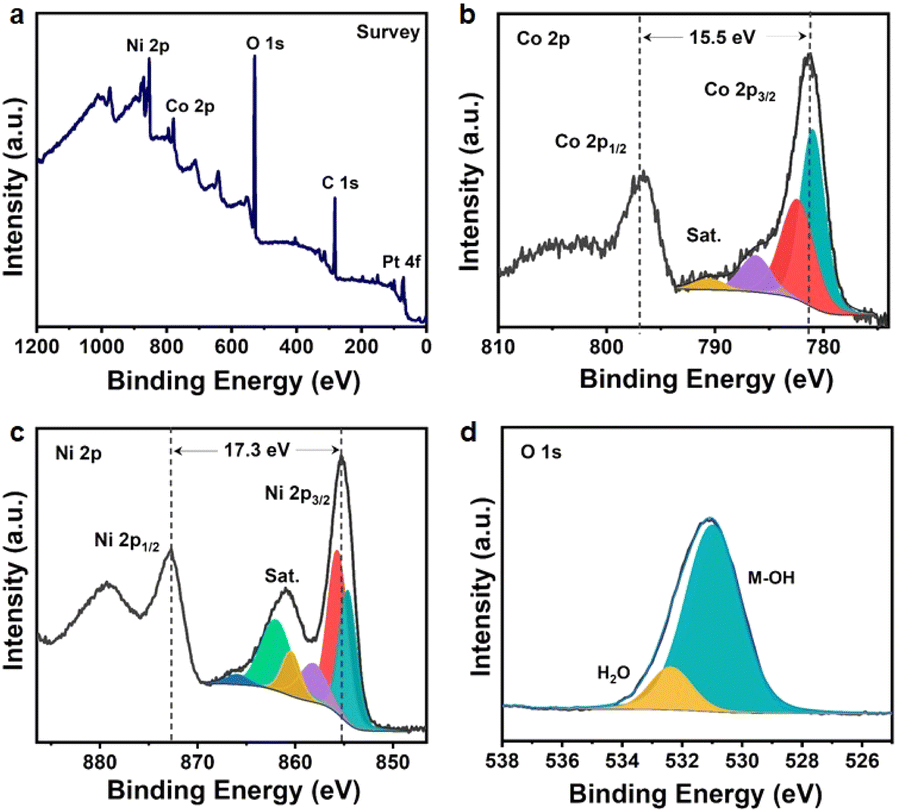 | ||
| Fig. 2 Elemental characteristics and bonding states of Co1Ni3-NS. (a) XPS survey spectrum and the high-resolution spectrum of (b) Co 2p, (c) Ni 2p, and (d) O 1s. | ||
| Sample | Ratio | Precursor concentration (mM) | Weight (ppm) from ICP | Atomic percentage (%) from XPS | |||
|---|---|---|---|---|---|---|---|
| Co:Ni |
Co(NO3)2·6H2O | Ni(NO3)2·6H2O | Co | Ni | Co | Ni | |
| Co3Ni1-NS | 3:1 |
7.5 | 2.5 | 0.56 | 0.23 | 59.1 | 40.9 |
| Co1Ni1-NS | 1:1 |
5.0 | 5.0 | 0.23 | 0.29 | 51.5 | 48.5 |
| Co1Ni3-NS | 1:3 |
2.5 | 7.5 | 0.21 | 0.57 | 25.2 | 74.8 |
3.1 Electrochemical properties of the CoxNi1−x(OH)2 NS electrode
CV and GCD measurements were carried out to investigate the electrochemical characteristics of CoxNi1−x(OH)2 NSs. A typical CV of the as-synthesized CoxNi1−x(OH)2 NSs using various Co:Ni ratios at a scan rate of 5 mV s−1 and an optimized potential window of 0 to 0.5 V is presented in Fig. 3a. The presence of one or two pairs of characteristic redox peaks in all the voltammograms represents the pseudocapacitive behavior due to the combined contribution from the following faradaic reactions of Co(OH)2 and Ni(OH)2.21| Co(OH)2 + OH− ↔ CoOOH + H2O + e− | (5) |
| CoOOH + OH− ↔ CoO2 + H2O + e− | (6) |
| Ni(OH)2 + OH− ↔ NiOOH + H2O + e− | (7) |
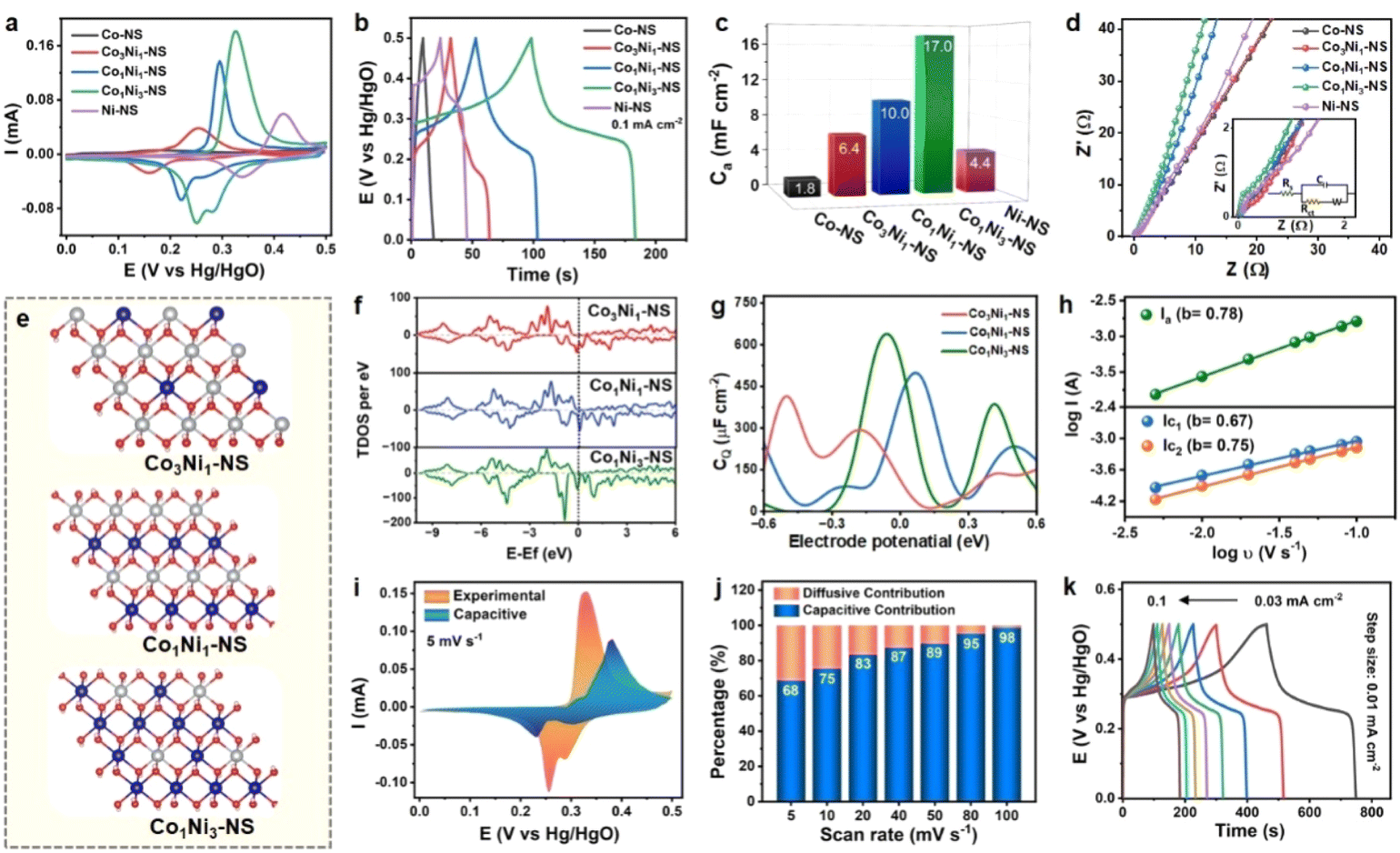 | ||
| Fig. 3 Comparison of the electrochemical performance of CoxNi1−x(OH)2 NSs synthesized using various ratios of Co:Ni: (a) CV at 5 mV s−1, (b) GCD at 0.1 mA cm−2, (c) bar graph showing areal capacitance merits, (d) Nyquist plot, (e) optimized structures of CoxNi1−x(OH)2, (f) total density of states of CoxNi1−x(OH)2 NSs with various ratios of Co:Ni, and (g) variation of quantum capacitance as a function of applied voltage for CoxNi1−x(OH)2; kinetic investigations of the Co1Ni3-NS electrode: (h) log(current density) vs. log(scan rate) plot, (i) relative contribution of the surface controlled process to the total current response of Co1Ni3-NS measured by Dunn's method, (j) capacitive and diffusive contributions of Co1Ni3-NS as a function of scan rats, and (k) GCD profile of Co1Ni3-NS at different current densities from 0.03 to 0.1 mA cm−2. | ||
The area under the curve and the redox peak position of voltammetric curves change for different ratios of Co:Ni in the electrodes. The oxidation peak of Co2+/Co3+ was observed at 0.17 V, whereas the Ni2+/Ni3+ peak was located at 0.41 V. The peak positions of the CoxNi1−x(OH)2 NS electrode potential were between those of the two monometallic (Co and Ni-) hydroxides as working electrodes. The oxidation and reduction potentials gradually shifted toward positive values with increasing Ni/Co feed ratio in CoxNi1−x(OH)2 NSs, which is attributed to the relatively higher potential of Ni2+ → Ni3+ in eqn (7). The Co/Ni ratio significantly influences the redox peak position and intensities of CoxNi1−x(OH)2 NSs, indicating the feasibility of tuning the electrochemical characteristics of bimetallic nanosheets by rational modulation of the composition. The Co1Ni3-NS (Co:Ni = 1:3) displayed a maximum integral area compared to all other optimized Co:Ni ratios. Hence, Co1Ni3-NS has optimal electrochemical performance. Notably, the contribution from the Pt substrate and SDS toward charge storage was negligible (Fig. S5†). The measured capacitance for all the CV curves mainly arises from the quasi-reversible electron transfer process from the redox process. GCD measurements analyzed the charging–discharging performance of CoxNi1−x(OH)2 NS electrodes at a constant current density of 0.1 mA cm−2. The non-linear GCD profile of CoxNi1−x(OH)2 NS electrodes in Fig. 3b is associated with multiple redox reactions, similar to the voltammetric curves. The discharging time GCD curve follows the same trend as CV confirming that the best-performing electrode is Co1Ni3-NS. Fig. 3c shows the areal capacitance of CoxNi1−x(OH)2 NS electrodes at various current densities. Co-NS has shown lower capacitance than Ni-NS, which improves significantly while introducing Ni content. Co1Ni3-NS was found to be the most appropriate combination, delivering the maximum capacitance (and capacity) values of 17.0 mF cm−2 (2.36 μA h cm−2) at 0.1 mA cm−2, followed by Co1Ni1-NS (10.0 mF cm−2, 1.38 μA h cm−2), Co3Ni1-NS (6.4 mF cm−2, 0.88 μA h cm−2), Ni-NS (4.4 mF cm−2, 0.61 μA h cm−2), and Co-NS (1.8 mF cm−2, 0.25 μA h cm−2). The areal capacitance of CoxNi1−x(OH)2 NS electrodes with various ratios of Co:Ni is shown in Fig. S6,† demonstrating that the Ni atoms in the electrode are the major source of the capacitance. The correlation between the chemical composition and electron transport behavior of CoxNi1−x(OH)2 NS electrodes was investigated by EIS (Fig. 3d). The Nyquist plot and the fitted equivalent circuit (the inset of Fig. 3d) of the five CoxNi1−x(OH)2 NS electrodes (0 ≤ x ≤ 1) with different Co:Ni ratios showed that the charge transfer resistance of Co-NS (Rct: 0.09 Ω) is relatively lower than that of Ni-NS (Rct = 0.88 Ω). The Rct values of Co3Ni1-NS, Co1Ni1-NS, and Co1Ni3-NS were measured to be 0.20, 0.27, and 0.23 Ω, respectively. The Rct and Rs values obtained for CoxNi1−x(OH)2 NS electrodes with various ratios of Co:Ni are summarized in Table S3.† Combining the abovementioned electrochemical findings and previous reports,21,32,33 cobalt atoms offer conductivity enhancement, and the nickel elements are the major source of capacitance. Consequently, the more significant straight-line tilt in a lower frequency region towards the imaginary axis signifies the superior capacitive behavior of Co1Ni3-NS, surpassing the other optimized ratios. The high electrochemical performance of Co1Ni3-NS is likely due to the following attributes: enhanced charging efficiency derived from improved electrical conductivity, improved electroactive sites due to feasible valence interchange or charge transfer between Co and Ni, and multiple redox reactions associated with the electrode material.
Additionally, theoretical studies were carried out to understand the experimental outcomes in terms of the density of states. An in-depth analysis of the quantum capacitance for CoxNi1−x(OH)2 with three distinct Co:Ni ratios 1:1, 2:1, and 3:1 was conducted. Fig. 3e shows the DFT-optimised crystal structure of CoxNi1−x(OH)2. We have determined the spin-polarized density of states for the three samples as shown in Fig. 3f. Noticeably, the total density of states (TDOS) of all these three samples shows an asymmetric nature, demonstrating their magnetic nature. Furthermore, a significantly large density of states was observed near the Fermi level of the down channel of their systems. Notably, the Co and Ni ratio of 1:3 has the relatively highest density of states near the Fermi level among all three systems.
Further, we calculated the quantum capacitance as a function of applied voltage at room temperature using the equation 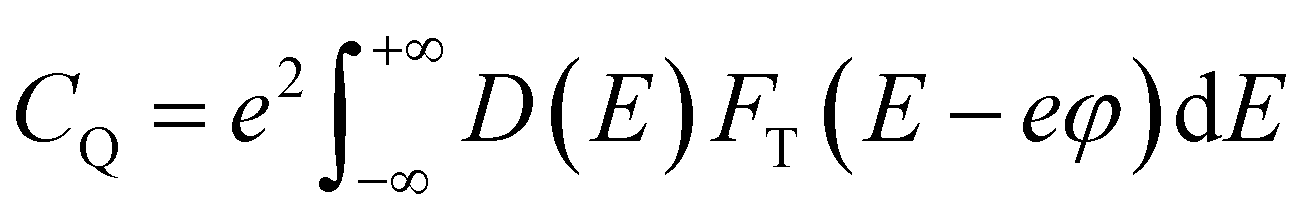 ,35 where D(E) denotes the density of states and ϕ represents the electrode potential of the system. The electronic charge is represented by e, while FT(E) illustrates the thermal broadening function, which accounts for temperature effects and can be expressed as FT(E) = (4KT)−1sech2(E/2KT). Fig. 3g showcases the dynamic changes in quantum capacitance across three distinct Co:Ni ratios 1:1, 1:3, and 3:1. For Co1Ni3-NS, the quantum capacitance peaks impressively at 624.3 μF cm−2 at −0.1 V and then decreases to 0.0 μF cm−2 at −0.4 V. Meanwhile, Co1Ni1-NS exhibits a range from a minimum of 15.3 μF cm−2 at −0.4 V to a maximum of 459.0 μF cm−2 at −0.1 V. For Co3Ni1-NS, the capacitance spans from 415.0 μF cm−2 at −0.5 V to 4.4 μF cm−2 at −0.1 V. The standout result is Co1Ni3-NS, which demonstrates the highest quantum capacitance, perfectly aligning with the experimental data. This superior capacitance is likely due to its elevated density of states in the tested range.
,35 where D(E) denotes the density of states and ϕ represents the electrode potential of the system. The electronic charge is represented by e, while FT(E) illustrates the thermal broadening function, which accounts for temperature effects and can be expressed as FT(E) = (4KT)−1sech2(E/2KT). Fig. 3g showcases the dynamic changes in quantum capacitance across three distinct Co:Ni ratios 1:1, 1:3, and 3:1. For Co1Ni3-NS, the quantum capacitance peaks impressively at 624.3 μF cm−2 at −0.1 V and then decreases to 0.0 μF cm−2 at −0.4 V. Meanwhile, Co1Ni1-NS exhibits a range from a minimum of 15.3 μF cm−2 at −0.4 V to a maximum of 459.0 μF cm−2 at −0.1 V. For Co3Ni1-NS, the capacitance spans from 415.0 μF cm−2 at −0.5 V to 4.4 μF cm−2 at −0.1 V. The standout result is Co1Ni3-NS, which demonstrates the highest quantum capacitance, perfectly aligning with the experimental data. This superior capacitance is likely due to its elevated density of states in the tested range.
Based on these primary electrochemical analyses and computational insights, Co1Ni3-NS was estimated to be the best-performing electrode. However, its electrochemical performance under high current conditions must also be investigated for practical applications. The voltammetric responses of the Co1Ni3-NS electrode at different scan rates ranging from 5 to 100 mV s−1 are exhibited in Fig. S7.† The gradual increase in the intensity and well-retained CV curve with increasing scan rate indicates excellent reversibility and capacitive behavior. A slight shift in oxidation and reduction peaks toward higher positive and negative potentials was observed due to electrode polarization. The kinetic behavior of Co1Ni3-NS based on the CV curve was investigated using the Power law,36i = aυb. The relationship between the scan rate (υ) and peak current (i) can be represented as logi = blogυ + loga, where ‘i’ is the current (A), υ is the scan rate (V s−1), and a and b are constant parameters. The value of ‘b’ is determined from the slope obtained by the linear fitting of the plot of log(i) vs. log(υ). Typically, the b value is crucial for estimating the charge-storage mechanism. The b value for the surface-controlled capacitive (or pseudocapacitive) behavior of the electrode material is typically 1.0, and that of the diffusion-controlled faradaic process is 0.5. As shown in Fig. 3h, the b values of the Co1Ni3-NS electrode were found to be 0.78 (oxidation peak), 0.67, and 0.75 (reduction peaks), indicating that the involved electrochemical processes are predominantly capacitive (or pseudocapacitive) with some influence of the diffusion-controlled process. To validate this statement, the relative contribution of the capacitive and diffusion-controlled reactions was explicitly quantified using Dunn's method,4 as shown below.
| i(V) = k1υ + k2υ1/2 | (8) |
| i(V)/υ1/2 = k1υ1/2 + k2 | (9) |
Finally, we assembled a symmetric supercapacitor device using Co1Ni3-NS as both positive and negative electrodes. Initially, CV and GCD studies were performed to optimize the optimal working potential window of the symmetric device. Fig. S8a† shows the CV response of the device at a scan rate of 100 mV s−1 in different voltage windows, where the higher cut-off potential was varied from 0.6 to 0.9 V. Notably, there was no major increment in the CV current responses after 0.8 V. Similarly, GCD in various potential ranges was recorded at a current density of 0.8 mA cm−2 as shown in Fig. S8b,† reflecting the major alteration in the quasi-linear shape of the GCD curve. Therefore, 0.8 V was considered the optimum working window for the safe and stable operation of the supercapacitor. The CV of the symmetric device was recorded at different scan rates from 5 to 100 mV s−1, as shown in Fig. 4a. The CV curve shows a quasi-rectangular shape, which is maintained even at high scan rates. Also, the GCD curve recorded at different current densities ranging from 0.3 to 0.5 mA cm−2 suggests a well-maintained symmetrical and reversible profile, as shown in Fig. 4b. These profiles did not exhibit prominent distortion with increasing scan rates or current densities, indicating the excellent rate capability of the fabricated device. The areal and volumetric capacitance values were quantified based on discharge time and are depicted in Fig. 4c. The device could deliver areal (and volumetric) capacitance values of 3.0 (3783), 2.65 (3399), 2.48 (3179), 2.36 (3028), and 2.28 mF cm−2 (2924 F cm−3) at the current densities of 0.3, 0.35, 0.4, 0.45, and 0.5 mA cm−2. Based on these findings, the symmetric device's energy and power densities were calculated at different current densities and represented in the Ragone plot, as depicted in Fig. 4d. The Co1Ni3-NS-based symmetric device delivered the highest energy density of 336 mW h cm−3 (0.26 μW h cm−2) at a power density of 153 W cm−3 (120 μW h cm−2) and a maximum power density of 256 W cm−3 (200 μW h cm−2) at 259 mW h cm−3 (0.20 μW h cm−2), implying an exceptional energy-to-power ratio compared to recently reported Co–Ni based materials such as Ni@CoNi-MOF,38 Zn–Ni–Co THO,39 NiCo-LDH,40 A-NiCo-LDH/NF,41 Co(OH)2/np NiOxHy@Ni,42 NiCoO2/Ni(OH)2/Co(OH)2,43 Ni–Co oxyhydroxides,44 MoCoFe-based hydroxides,45 CoZnNi oxyphosphide nanoarrays,46 and CoZnNiS@CNTs/rGO film.47 Table S4† compares the energy density and power density of the Co1Ni3-NS-based symmetric device with those of recently reported Co–Ni-based 2D materials. Supercapacitors often experience the self-discharge process, leading to a reduction in voltage and simultaneously affecting the device's energy density. The self-discharge rate was investigated by charging the device to 0.8 V and measuring the open-circuit potential. Fig. S9† displays the device voltage decay over time. The supercapacitor device maintained 75% potential retention even after 60 s. After 860 s under open circuit conditions, the device could hold 50% voltage making it an appropriate choice for many miniature devices. As we know, cycling stability is an essential parameter for the long-term operation of a supercapacitor device. Fig. 4e demonstrates the capacitance retention and coulombic efficiency over 10000 GCD cycles at a constant current density of 0.4 mA cm−2. During the initial 800 cycles, the increase in capacitance was observed due to the activation of the electrode material. The capacitance retention after 10000 continuous GCD cycles was 75% with an excellent coulombic efficiency of 96% indicating good cycling performance of the symmetric device. The capacitance decay is mainly due to the loss of the electrode material over long-term operation. The electrolyte's reduced resistance and enriched electrode material penetrability promote ion transportation and thus increase coulombic efficiency. The inset of Fig. 4e represents the initial and last two GCD cycles, signifying the well-maintained symmetric shapes. The EIS studies were carried out before and after cycling stability testing, as shown in Fig. 4f. The charge transfer resistance was found to increase from 205 to 210 Ω after 10000 cycles. Also, the low-frequency region of the Nyquist plot deflected towards the real axis after the stability testing, indicating resistance to ion transport, which resulted in supercapacitor performance decay over time. Post-stability XPS investigation was performed for insights into the modifications in the chemical composition and the surface chemical oxidation states of Co1Ni3-NS nanosheets. Notably, after 10000 charge–discharge cycles, considerable surface oxidation in the high-resolution Ni 2p and Co 2p spectra has been observed, as shown in Fig. S10.† The new peaks at 861.0 eV in the Ni 2p spectrum indicate the presence of Ni3+ in NiOOH. Similarly, additional peaks at 780.1 and 781.4 eV in the Co 2p scan are found due to the presence of trivalent Co ions. These results indicate that the Co3+ area has increased, and some portion of Co(OH)2 is transformed into CoOOH during the charge–discharge process. This can be further validated by the core level O 1s XPS spectra, where the peak of O–H bonds has shifted to lower BE, and a new peak of lattice oxygen has appeared at 531.2 eV, suggesting increased oxidation in the Co1Ni3-NS electrode after cycling stability testing. The exceptional electrochemical charge storage performance of the Co1Ni3-NS-based electrode is achieved due to the shortened diffusion length, and the highly exposed area of ultrathin, large-area, and uniform nanosheets offers a high surface area to volume ratio, which facilitates the electrolyte ion transport.
 | ||
| Fig. 4 Electrochemical testing of the symmetric device: (a) CV profiles at different scan rates, (b) the charge–discharge curve at various current densities, (c) areal and volumetric capacitance achieved at different current densities, (d) Ragone plot comparing the energy density and power density of the Co1Ni3-NS symmetric device with recently reported Co–Ni based electrodes, and (e) long-term cycling stability showing capacitance retention and coulombic efficiency. Inset shows the 1st and last two GCD curves. (f) EIS plot before and after cycling stability testing. | ||
3.2 Electrocatalytic OER activity of the CoxNi1−x(OH)2 NS electrode
To analyze the electrocatalytic OER performance, CoxNi1−x(OH)2 NSs synthesized with different feeding ratios of Co:Ni (1:0, 3:1, 1:1, 1:3, and 0:1) were transferred on fluorine-doped tin oxide (FTO) coated glass substrates and the electrodes were evaluated in 1 M KOH in a three-electrode system (see experimental details for more information). The FTO-coated glass substrate was chosen due to its essential merits, such as high conductivity, stability in harsh pH environments and under high oxidative conditions, better surface adhesion, and, most importantly, the FTO is electrochemically inert for the OER. The linear sweep voltammetry (LSV) curves of CoxNi1−x(OH)2 NSs at a constant scan rate of 5 mV s−1 are shown in Fig. 5a. The nearly horizontal polarization curve of the FTO-coated glass substrate indicates that the substrate does not contribute to the electrocatalytic activity in the OER process. The CoxNi1−x(OH)2 NS with a 1:3 ratio of Co:Ni, i.e., Co1Ni3-NS, exhibits outstanding OER activity substantively surpassing other monometallic and bimetallic hydroxide nanosheets and even the benchmark RuO2 catalyst. The Co1Ni3-NS electrode required the lowest overpotential of 318 mV to achieve a current density of 10 mA cm−2 compared to Co-NS (481 mV), Co3Ni1-NS (401 mV), Co1Ni1-NS (377 mV), Ni-NS (409 mV), and the benchmark RuO2 catalyst (349 mV). The overpotential value of Co1Ni3-NS is comparable to those of recently reported OER electrocatalysts, as shown in Table S5.† Moreover, the LSV in Fig. S11† reflects that SDS does not have an apparent role in the OER process. At a current density of 10 mA cm−2, RuO2 has a lower overpotential relative to Co1Ni1-NS; however, at 50 mA cm−2, Co1Ni1-NS catalytic activity became comparable to that of the state-of-the-art RuO2 catalyst. Interestingly, Co1Ni3-NS and Co1Ni1-NS could attain current densities of 157 and 93 mA cm−2 respectively at a potential 1.8 V, which are 2.3 and 1.4-fold higher than that of RuO2. These results indicate the suitability of the bimetallic Co–Ni-hydroxide nanosheets for a highly efficient OER. An interruption in mass diffusion by the RuO2 catalyst at a higher overpotential could be triggered due to the accumulation of bubbles under increased voltage bias. The Tafel plot was investigated to gain in-depth insights into the OER kinetics of CoxNi1−x(OH)2 NSs, as demonstrated in Fig. 5b. Consistent with the LSV results, Co1Ni3-NS exhibited the lowest Tafel slope of 61 mV dec−1, followed by RuO2 (66 mV dec−1), Co1Ni1-NS (71 mV dec−1), Co3Ni1-NS (73 mV dec−1), Ni-NS (79 mV dec−1), and Co-NS (81 mV dec−1), confirming the faster oxygen evolution rate of the Co1Ni3-NS electrode. The bar graph in Fig. 5c shows the overpotential and Tafel slopes of various electrocatalysts under study. Remarkably, increasing the Ni content positively influenced the OER activity, and the bimetallic nanosheets exhibited higher catalytic activity than monometallic nanosheets. This might be due to the enhanced strain offered by minimal Co atoms in the Ni lattice. The considerably high overpotential of Co3Ni1-NS could be ascribed to the additional hindrance in charge transportation driven by the double-layer structure. The onset potential is an essential parameter to determine the efficiency of the electrocatalyst. It is the least potential required to initiate the oxygen evolution at the electrode surface and it was measured from the voltammetry curves at a current density of 0.1 mA cm−2 (Fig. S12†). The rapid OER kinetics of Co1Ni3-NS were further validated by its lower charge transfer resistance (Rct).
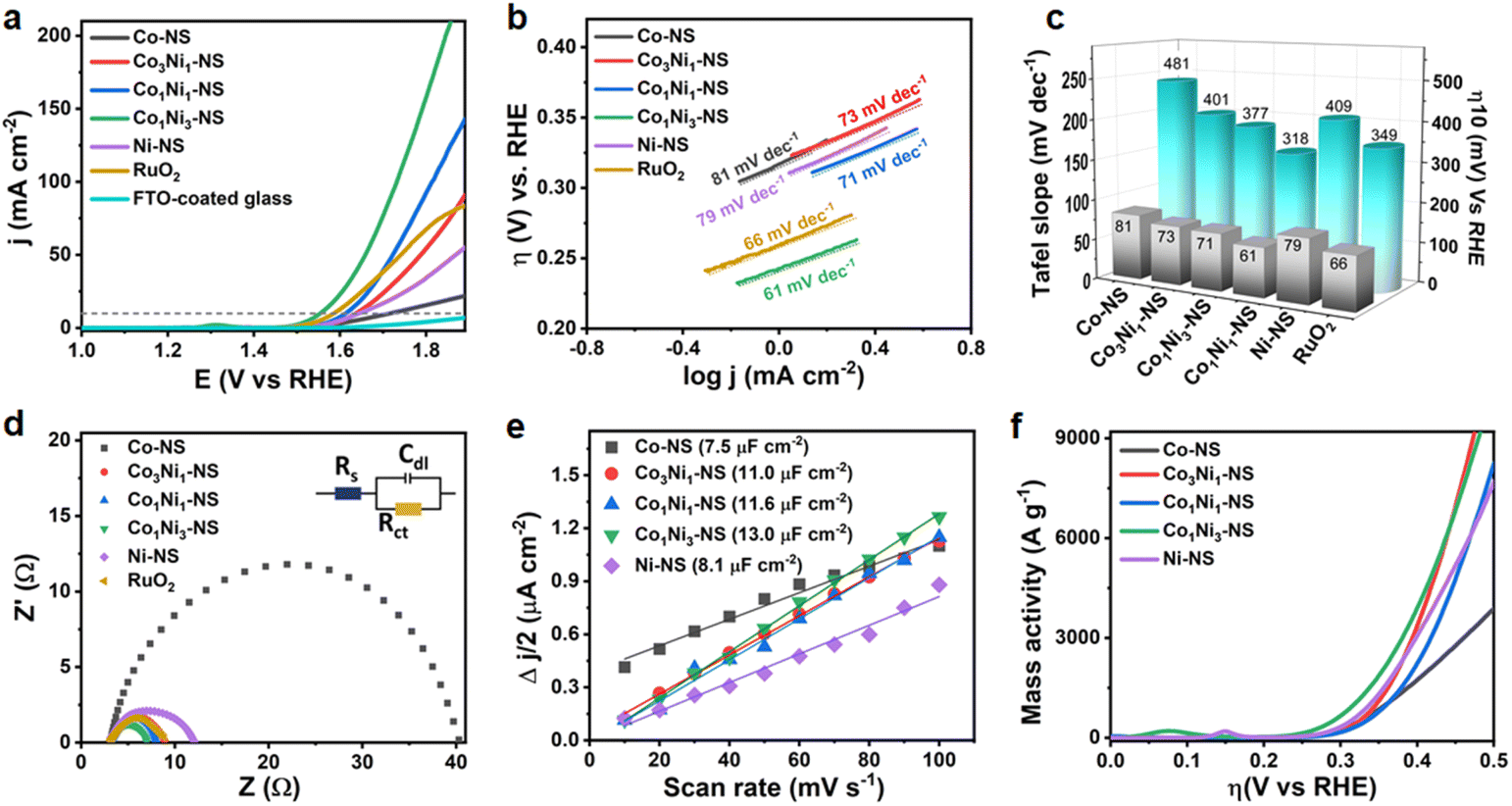 | ||
| Fig. 5 Electrocatalytic OER performance of CoxNi1−x(OH)2 NSs: (a) LSV curves at 5 mV s−1, (b) corresponding Tafel slope, (c) comparison of overpotential and Tafel slopes at 10 mA cm−2, (d) Nyquist plot recorded at a constant potential of 1.55 V (vs. RHE), inset shows an equivalent fitted circuit, (e) half of the current density difference (Δj) plot with respect to scan rates, and (f) mass activity plot against overpotential. | ||
The Co1Ni3-NS electrode showed the smallest Rct of 3.73 Ω compared to other bimetallic and monometallic hydroxide catalysts based on variations in Co:Ni ratios under the same experimental conditions (Fig. 5d). As shown in Fig. S13,† the FTO-coated glass substrate showed notably higher Rct (107 Ω), which states that the electrocatalytic activity was acquired due to a thin layer of bimetallic Co–Ni-hydroxide nanosheets. The large proportion of catalytically active sites per unit surface area is another unique feature of ultrathin nanosheets. The electrocatalytically active surface area (ECSA) defines the number of active sites involved and the intrinsic OER activity of the catalyst. The electric double-layer capacitance (Cdl) directly correlates with the ECSA, as expressed in ESI eqn (8).† The Cdl can be readily modified by tuning the ratio of Co:Ni, as shown in Fig. 5e. Co1Ni3-NS exhibited the highest Cdl of 13.0 μF cm−2 compared to the other nanosheets, revealing that more active sites of the Co1Ni3-NS surface are accessible for the OER process owing to its enhanced surface electronic structure, which is further discussed by using theoretical aspects. The control samples based on variations in the Co:Ni ratio in CoxNi1−x(OH)2 nanosheets further validated the superior OER activity of Co1Ni3-NS with a turnover frequency (TOF) of 0.054 O2 s−1 (Fig. S16†) and a mass activity of 1054 A g−1 at 318 mV vs. RHE (Fig. 5f). Such high mass activity of Co1Ni3-NS confirms the exceptional catalytic efficiency of ultrathin bimetallic nanosheets, significantly lowering the catalyst consumption. Table S6† provides the electrocatalytic activity results of CoxNi1−x(OH)2 NS, showcasing the performance metrics and comparisons across different samples. Furthermore, the electrocatalytic activity of CoxNi1−x(OH)2 with different Co to Ni ratios (1:3, 1:1, and 3:1) was theoretically investigated using density functional theory (DFT) calculations.16,48,49
Based on DFT studies, Co was identified as a potential active site for the OER, and the optimized crystal structure of CoxNi1−x(OH)2 NSs is shown in Fig. S17.† The OER typically proceeds through a four-electron transfer pathway with proton-coupled electron transfer, following the sequence * + 4OH− → OH* → O* → OOH* → O2 + 2H2O.50 Our findings indicate that cobalt (Co) serves as the active site for the OER, consistent with previous reports. Specifically, with the active site being Co (Fig. 6a), we found that the overpotential is 1.62 V for Co1Ni1-NS and 1.17 V for Co3Ni1-NS, and the lowest overpotential of 0.7 V is observed for Co1Ni3-NS, with the third step identified as the potential-determining step (PDS), as shown in Fig. 6b–d. Previous reports indicate that for bimetallic CoNi-hydroxide with Co ratios of 1:0 and 0:1, the overpotentials are 0.98 V and 1.43 V, respectively.16,48 Our findings demonstrate that Co1Ni3-NS exhibits superior activity for the oxygen evolution reaction. Further, we conducted Bader charge analysis and charge density calculations to understand the interaction between the active Co site and the intermediate molecules. These studies reveal that the charge transfer from the Co atom to the O, OH, and OOH intermediates is 0.759e, 1.199e, and 0.524e, respectively (Fig. 6e, Table S8†). This significant charge transfer suggests strong interactions between the intermediates and the surface, likely contributing to the enhanced catalytic activity. The charge density difference plots further support this, showing charge accumulation in the intermediate molecules, where cyan represents charge depletion and yellow indicates charge accumulation (Fig. 6e and S18†).51,52 These theoretical results strongly indicate that Co1Ni3-NS is an excellent electrocatalyst for the oxygen evolution reaction, aligning well with experimental findings. In addition, the 1:3 Co:Ni ratio in Co1Ni3-NS shows a unique electronic behavior based on our partial density of states (PDOS) analysis. Specifically, at the active Co site, we observe a decrease in d-band occupancy (Od) after intermediate adsorption (Fig. 6f), which suggests that charge transfer occurs during the reaction. This change in the electronic structure indicates that the 1:3 ratio creates a favorable environment for catalytic activity by allowing an optimal interaction with intermediates. Further, the shift of the d-band center (εd) to lower energy upon adsorption further points to a better balance in binding strength strong enough to hold intermediates effectively, but not so strong that it hinders the reaction. This balance is ideal for catalytic efficiency.
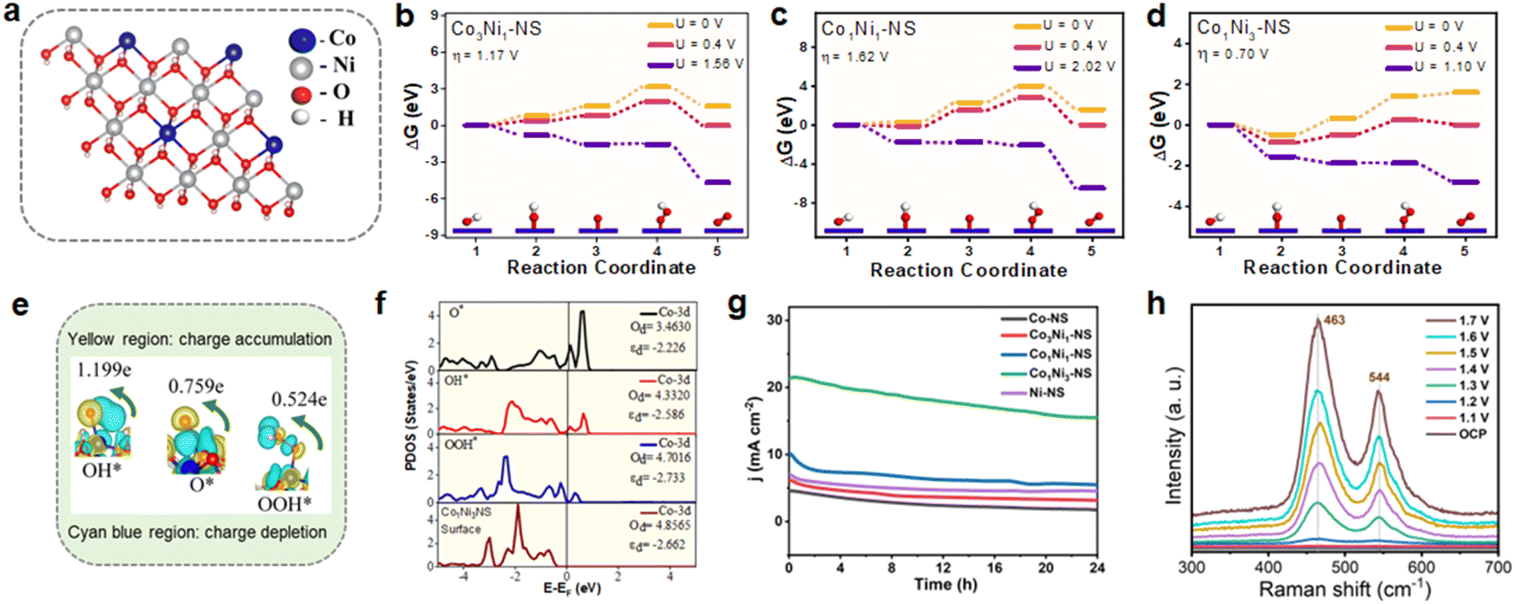 | ||
| Fig. 6 (a) Optimized structure of Co1Ni3-NS, with the Co active site highlighted and (b–d) free energy profiles of the OER at the active Co site for the CoxNi1−x(OH)2 catalyst with various ratios of Co:Ni. (e) Charge density difference plots for the intermediates O*, OH*, and OOH*, showing Bader charge values on the oxygen atom bonded to the active site, with yellow representing charge accumulation and cyan blue indicating charge depletion. (f) Partial density of states (PDOS) of the Co d-orbital at the active site before and after adsorption, comparing the values of d-band occupancy (Od) and the d-band center (εd), (g) CA curves of various catalysts at 1.59 V (vs. RHE) over 24 h, and (h) in situ Raman analysis of Co1Ni3-NS achieved during chronoamperometry measurements. | ||
The combined insights from XPS data and PDOS analysis suggest that adjusting the Co ratio not only affects the elemental makeup of the catalyst but also fine-tunes its electronic properties.53,54
The long-term durability of the electrocatalyst is an essential parameter for its practical applications. The OER performance of the CoxNi1−x(OH)2 NS electrode was tested by chronoamperometry (CA) at a fixed potential of 1.59 V versus RHE. As shown in Fig. 6g, Co1Ni3-NS exhibited exceptional electrocatalytic stability for the OER in comparison to other bimetallic and monometallic nanosheets, for as long as 24 h. The initial current density for Co1Ni3-NS could reach 20 mA cm−2 retaining 77% of its initial current density after 24 h of OER operation. The slight change in the polarization curve after continuous OER testing (Fig. S19†) indicates the superior stability of Co1Ni3-NS. Moreover, the surface morphology of the nanosheet was also least altered without peeling off from the substrate even after long-term operation, as shown in the SEM image (Fig. S20†). The sporadic bright spots on the nanosheet surface are possibly the KOH lumps that originate from the precipitation of the electrolyte during the OER, resulting in inefficient catalytic performance. The chemical bonding states and contents of the bimetallic cobalt-nickel hydroxide nanosheet after the long-term OER test were investigated by XPS. After 24 h, Co1Ni3-NS exhibited the presence of characteristic peaks of Co 2p, Ni 2p, and O 1s, as shown in Fig. S21.† The deconvoluted Co 2p and Ni 2p high-resolution XPS spectrum showed a slight positive shift in BE, as shown in Fig. S21b and c.† Also, the appearance of additional peaks revealed that the partial Co2+ and Ni2+ surface species converted into Co3+ (CoOOH) and Ni3+ (NiOOH) species. This oxidation is observed due to the OER process over long-term continuous operations. The post-stability O 1s spectrum exhibited a shift of the O–H peak to a lower BE, confirming oxyhydroxide formation in the nanosheet (Fig. S21d†).
In situ Raman spectroscopy was employed to gain better insights into the associated OER mechanism, key species, and surface self-reconstruction of Co1Ni3-NS at various potentials. As displayed in Fig. 6h, the Raman peaks at open circuit potential were observed at 463 and 526 cm−1 which could be assigned to the metal–oxygen vibrations of bimetallic Co–Ni hydroxide.55 With increasing potential (1.1 V vs. RHE), low intensity Raman bands at 465 and 544 cm−1 were observed. The intensity enhanced when the potential was increased from 1.3 to 1.7 V. The presence of two well-defined peaks around 465 and 544 cm−1 indicates the formation of the γ-NiOOH phase with Eg Ni–O bending vibration mode and A1g Ni–O stretching vibration, respectively. The blue shift in the Raman band indicates the formation of an oxyhydroxide surface. Relative to Ni, the concentration of Co is lower leading to weak Raman bands. These findings are consistent with the ex situ XPS data of Co1Ni3-NS, which was recorded after long-term stability testing. Combining the aforementioned electrochemistry results, it is believed that the bimetallic CoxNi1−x(OH)2 NS is a better electrocatalyst for the OER.
4 Conclusions
In summary, we have demonstrated the facile synthesis of nanometre-scale thin bimetallic CoxNi1−x(OH)2 NSs having a large area by the ILE strategy at the water–air interface as an efficient bifunctional electrode for supercapacitors and the OER. The synthesized nanosheets exhibited a uniform thickness of 3.6 ± 0.5 nm. Based on the electrochemical investigations, the electrochemical and electrocatalytic performance can be rationally tuned by controlling the Co/Ni ratio. Co1Ni3-NS with a Co:Ni ratio of 1:3 achieved superior activity for supercapacitors as well as OER electrocatalysis compared to that of monometallic and bimetallic CoxNi1−x(OH)2 NSs synthesized using different ratios of Co:Ni. The Co1Ni3-NS symmetric device showed an outstanding capacitance of 3783 F cm−3 (3 mF cm−2) at a current density of 0.3 mA cm−2 and maximum energy density and power density of 336 mW h cm−3 and 256 W cm−3, respectively, outperforming recently reported Co–Ni based 2D materials. Moreover, the device exhibited long-term stability, retaining 66% of its performance after 10000 continuous GCD cycles. Furthermore, as an electrocatalyst, Co1Ni3-NS exhibited superior activity for oxygen evolution in terms of low overpotential (318 mV at 10 mA cm−2), favorable reaction kinetics (Tafel slope of 61 mV dec−1), and exceptional long-term stability over 24 h of chronoamperometric experimentation in 1 M KOH. Moreover, it showed an excellent mass activity of 1056 Ag−1 and a TOF of 0.054 O2 s−1 at 318 mV, suggesting better OER activity. The overall OER performance of bimetallic Co1Ni3-NS is comparable to that of recently reported transition metal-based 2D electrocatalysts. We illustrated the structural evolution of the catalyst surface using in situ Raman spectroscopy. The DFT findings indicate that the high quantum capacitance (CQ = 624.3 μF cm−2) of Co1Ni3-NS is due to the large density of states near the Fermi level. The DFT results further indicate that Co1Ni3-NS has a comparatively lower potential barrier than other bimetallic and respective monometallic hydroxide nanosheets. In general, the supercapacitor and OER performance of bimetallic Co1Ni3-NS was substantially promoted owing to the strong interaction between Co and Ni at systematic composition, optimal surface electronic structure, and adsorption energy. The present work paves a new way to develop highly efficient and dual-functional electrode materials for supercapacitors and the OER.
Data availability
Data will be made available upon reasonable request.Author contributions
PBJ: conceptualization, experimental work, writing – original draft, investigations, experiments, and data acquisition. SAP: scientific discussion, assistance with experiments and data analysis, review and editing. AP: software and theoretical studies for the OER. MSK: software and theoretical studies for supercapacitors. RT: data curation and supervision of theoretical studies. AS: investigation and formal analysis of XPS and TEM data. SR: supervision. AKS: visualization and resources. MS: conceptualization, supervision, funding acquisition, review & editing, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PBJ acknowledges the Chhatrapati Shahu Maharaj Research Training and Human Development Institute, Pune, for a research fellowship (CSMNRF-2021/2021-22/896). MS is thankful to SERB, New Delhi, India, for research funding (EMR/2017/003368, CRG/2022/006471). The authors acknowledge the support of the National Supercomputing Mission (NSM) for providing access to PARAM Porul for carrying out the computational work, and the SRM University-AP, Andhra Pradesh, for providing the central computational facilities. RT thanks the Science and Engineering Research Board (SERB), India, for financial support (Grant No. CRG/2022/005423). The CNRS, the Chevreul Institute (FR 2638), the Ministère de l'Enseignement Supérieur et de la Recherche, the Région Hauts-de-France, the FEDER, and the MEL are acknowledged for supporting this work. The authors also thank Maya Marinova for TEM facilities and Pardis Simon for XPS resources.Notes and references
- D. Wang, Y. Wang, Z. Fu, Y. Xu, L.-X. Yang, F. Wang, X. Guo, W. Sun and Z.-L. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 34507–34517 CrossRef CAS.
- W. Wang, Y. Lu, M. Zhao, R. Luo, Y. Yang, T. Peng, H. Yan, X. Liu and Y. Luo, ACS Nano, 2019, 13, 12206–12218 CrossRef CAS.
- L. Kumar, M. Chauhan, P. K. Boruah, M. R. Das, S. A. Hashmi and S. Deka, ACS Appl. Energy Mater., 2020, 3, 6793–6804 CrossRef CAS.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- L. Zhang, X. Hu, Z. Wang, F. Sun and D. G. Dorrell, Renewable Sustainable Energy Rev., 2018, 81, 1868–1878 CrossRef.
- A. Patra, N. Kuniyil, J. R. Jose, S. Sahoo, B. Chakraborty and C. S. Rout, J. Mater. Chem. A, 2021, 9, 25852–25891 RSC.
- L. Wang, Z. H. Dong, Z. G. Wang, F. X. Zhang and J. Jin, Adv. Funct. Mater., 2013, 23, 2758–2764 CrossRef CAS.
- N. R. Chodankar, H. D. Pham, A. K. Nanjundan, J. F. S. Fernando, K. Jayaramulu, D. Golberg, Y.-K. Han and D. P. Dubal, Small, 2020, 16, 2002806 CrossRef CAS.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS.
- H. Lu, J. Tournet, K. Dastafkan, Y. Liu, Y. H. Ng, S. K. Karuturi, C. Zhao and Z. Yin, Chem. Rev., 2021, 121, 10271–10366 CrossRef CAS.
- S. J. Uke, V. P. Akhare, D. R. Bambole, A. B. Bodade and G. N. Chaudhari, Front. Mater. Sci., 2017, 4, 21 CrossRef.
- U. K. Chime, A. C. Nkele, S. Ezugwu, A. C. Nwanya, N. M. Shinde, M. Kebede, P. M. Ejikeme, M. Maaza and F. I. Ezema, Curr. Opin. Electrochem., 2020, 21, 175–181 CrossRef CAS.
- A. D. Jagadale, V. S. Kumbhar, D. S. Dhawale and C. D. Lokhande, Electrochim. Acta, 2013, 98, 32–38 CrossRef CAS.
- R. Wang, X. Yan, J. Lang, Z. Zheng and P. Zhang, J. Mater. Chem. A, 2014, 2, 12724–12732 RSC.
- P. B. Jagdale, S. A. Patil, A. Sfeir, N. Barman, A. Iqbal, S. Royer, R. Thapa, A. K. Samal, D. Ghosh and M. Saxena, Mater. Today Energy, 2024, 44, 101608 CrossRef CAS.
- S. Natarajan, M. Ulaganathan and V. Aravindan, J. Mater. Chem. A, 2021, 9, 15542–15585 RSC.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- S. Ashok Patil, P. B. Jagdale, N. Barman, A. Iqbal, A. Sfeir, S. Royer, R. Thapa, A. Kumar Samal and M. Saxena, J. Colloid Interface Sci., 2024, 674, 587–602 CrossRef CAS PubMed.
- T. Wang, H. C. Chen, F. Yu, X. S. Zhao and H. Wang, Energy Storage Mater., 2019, 16, 545–573 CrossRef.
- J.-H. Cha, S.-J. Kim, S. Jung and D.-Y. Jung, ACS Appl. Energy Mater., 2020, 3, 3854–3862 CrossRef CAS.
- L. Su, L. Gong and J. Gao, J. Power Sources, 2012, 209, 141–146 CrossRef CAS.
- S. Sriram, S. Mathi, B. Vishnu and J. Jayabharathi, Energy Fuels, 2022, 36, 7006–7016 CrossRef CAS.
- S.-Y. Lee, I.-S. Kim, H.-S. Cho, C.-H. Kim and Y.-K. Lee, Appl. Catal., B, 2021, 284, 119729 CrossRef CAS.
- B. Wang, C. Tang, H.-F. Wang, X. Chen, R. Cao and Q. Zhang, Adv. Mater., 2019, 31, 1805658 CrossRef PubMed.
- Z. Lu, G. P. Neupane, G. Jia, H. Zhao, D. Qi, Y. Du, Y. Lu and Z. Yin, Adv. Funct. Mater., 2020, 30, 2001127 CrossRef CAS.
- H. Zhang, ACS Nano, 2015, 9, 9451–9469 CrossRef CAS PubMed.
- Y. Dou, L. Zhang, X. Xu, Z. Sun, T. Liao and S. X. Dou, Chem. Soc. Rev., 2017, 46, 7338–7373 RSC.
- S. Parvin, N. Bothra, S. Dutta, M. Maji, M. Mura, A. Kumar, D. K. Chaudhary, P. Rajput, M. Kumar, S. K. Pati and S. Bhattacharyya, Chem. Sci., 2023, 14, 3056–3069 RSC.
- Y. Zhao, Y. Wang, Y. Dong, C. Carlos, J. Li, Z. Zhang, T. Li, Y. Shao, S. Yan, L. Gu, J. Wang and X. Wang, ACS Energy Lett., 2021, 6, 3367–3375 CrossRef CAS.
- F. Wang, J.-H. Seo, G. Luo, M. B. Starr, Z. Li, D. Geng, X. Yin, S. Wang, D. G. Fraser, D. Morgan, Z. Ma and X. Wang, Nat. Commun., 2016, 7, 10444 CrossRef CAS PubMed.
- L. Jia, H. Wan, X. Liu, G. Chen, N. Zhang, J. Li, W. Zhou, Y. Cao, R. Ma and G. Qiu, ChemSusChem, 2019, 12, 5274–5281 CrossRef CAS PubMed.
- U. M. Patil, J. S. Sohn, S. B. Kulkarni, S. C. Lee, H. G. Park, K. V. Gurav, J. H. Kim and S. C. Jun, ACS Appl. Mater. Interfaces, 2014, 6, 2450–2458 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- S. Kapse, B. Benny, P. Mandal and R. Thapa, J. Energy Storage, 2021, 44, 103476 CrossRef.
- P. Bhol, P. B. Jagdale, N. Barman, R. Thapa, M. Saxena and A. K. Samal, J. Energy Storage, 2023, 65, 107286 CrossRef.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185 CrossRef CAS.
- M. Hong, C. Zhou, S. Xu, X. Ye, Z. Yang, L. Zhang, Z. Zhou, N. Hu and Y. Zhang, J. Power Sources, 2019, 423, 80–89 CrossRef CAS.
- Z.-H. Huang, F.-F. Sun, M. Batmunkh, W.-H. Li, H. Li, Y. Sun, Q. Zhao, X. Liu and T.-Y. Ma, J. Mater. Chem. A, 2019, 7, 11826–11835 RSC.
- L. Zhi, W. Zhang, L. Dang, J. Sun, F. Shi, H. Xu, Z. Liu and Z. Lei, J. Power Sources, 2018, 387, 108–116 CrossRef CAS.
- D. Zha, Y. Fu, L. Zhang, J. Zhu and X. Wang, J. Power Sources, 2018, 378, 31–39 CrossRef CAS.
- F. Zhao, D. Zheng, Y. Liu, F. Pan, Q. Deng, C. Qin, Y. Li and Z. Wang, Chem. Eng. J., 2021, 415, 128871 CrossRef CAS.
- T. Qin, H. Li, R. Ren, J. Hao, Y. Wen, Z. Wang, J. Huang, D. He, G. Cao and S. Peng, CrystEngComm, 2018, 20, 6519–6528 RSC.
- X. Ren, M. Li, L. Qiu, X. Guo, F. Tian, G. Han, W. Yang and Y. Yu, J. Mater. Chem. A, 2023, 11, 5754–5765 RSC.
- Q. T. Nguyen, U. T. Nakate, J. Chen, D. T. Tran and S. Park, Composites, Part B, 2023, 252, 110528 CrossRef CAS.
- X. Chen, Y. Liu, Q. Yang, L. Li, Y. Ying and W. Shi, J. Colloid Interface Sci., 2022, 610, 427–437 CrossRef CAS PubMed.
- Y. Liu, N. Xin, Q. Yang and W. Shi, J. Colloid Interface Sci., 2021, 583, 288–298 CrossRef CAS PubMed.
- X. Zhang, B. Gao, R. Rao, F. Bi, C. Li, K. Yue, Y. Wang, J. Xu, X. Feng and Y. Yang, J. Colloid Interface Sci., 2024, 660, 423–439 CrossRef CAS PubMed.
- N. Ullah, W. Zhao, X. Lu, C. J. Oluigbo, S. A. Shah, M. Zhang, J. Xie and Y. Xu, Electrochim. Acta, 2019, 298, 163–171 CrossRef CAS.
- E. E. Siddharthan, S. Ghosh and R. Thapa, J. Mater. Chem. A, 2024, 12, 19176–19186 RSC.
- S. Das, A. Pathak, U. Phadikar, C. Kuila, A. Maji, T. Kuila, N. C. Murmu, R. Thapa and A. Kundu, Adv. Funct. Mater., 2024, 2407078 CrossRef CAS.
- S. Sarkar, A. Biswas, E. E. Siddharthan, R. Thapa and R. S. Dey, ACS Nano, 2022, 16, 7890–7903 CrossRef CAS PubMed.
- M. Andersen, Nat. Catal., 2023, 6, 460–461 CrossRef.
- L. G. M. Pettersson and A. Nilsson, Top. Catal., 2014, 57, 2–13 CrossRef CAS.
- D. Wang, Y. Zhang, L. Yang, G. Fan, Y. Lin and F. Li, J. Mater. Sci.: Mater. Electron., 2020, 31, 6467–6478 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06846g |
| This journal is © The Royal Society of Chemistry 2025 |