
Functional carbon-based covalent bridging bonds unlocking superior sodium-ion storage
Jinliang
Zhu
 *a,
Manchuan
Guo
a,
Miao
Hu
a,
Fang
Fu
a,
Kerou
Qiu
a,
Shijian
Wang
*b,
Guoxiu
Wang
b and
Bing
Sun
*b
*a,
Manchuan
Guo
a,
Miao
Hu
a,
Fang
Fu
a,
Kerou
Qiu
a,
Shijian
Wang
*b,
Guoxiu
Wang
b and
Bing
Sun
*b
aSchool of Resources, Environment and Materials, MOE Key Laboratory of New Processing Technology for Nonferrous Metals and Materials, Guangxi University, Nanning 530004, PR China. E-mail: jlzhu85@163.com
bCentre for Clean Energy Technology, School of Mathematical and Physical Sciences, Faculty of Science, University of Technology Sydney, Ultimo, NSW 2007, Australia. E-mail: shijian.wang@uts.edu.au; bing.sun@uts.edu.au
First published on 21st December 2024
Abstract
The development of sodium-ion batteries has gained significant momentum as a promising alternative to lithium-ion batteries, particularly for large-scale energy storage. However, the advancement of sodium-ion batteries is impeded by challenges associated with the performance of electrode materials, especially conversion-type materials such as transition metal oxides and dichalcogenides. These materials often suffer from severe volume expansion during cycling, poor electronic conductivity, and instability at the electrode/electrolyte interface. Surface modification with carbonous materials has been demonstrated to be an effective strategy to solve these challenges. This review explores the transformative role of interfacial chemical bridge bonds, particularly C–X–M bonds (where C represents carbon; X represents elements like S, O, N, P and Se; and M represents transition metals) for performance enhancement. By forming strong covalent connections between carbon materials and transition metal compounds, the carbon-coated conversion-type anode materials show enhanced structural stability, improved electronic conductivity and reduced charge transfer resistance. This review also covers advanced characterisation techniques applied to characterise and analyse these bonds, offering a detailed understanding of their contributions to sodium-ion storage. Additionally, challenges and prospects in this field are discussed for optimising electrode materials through the strategic implementation of chemical bridge bonds, providing valuable insights for advancing the next-generation high-performance sodium-ion batteries.
1. Introduction
Sodium-ion batteries (SIBs) are increasingly recognised as a sustainable and cost-effective alternative to lithium-ion batteries, particularly for large-scale energy storage applications. The abundant availability of sodium and enhanced safety profile make SIBs a promising candidate for meeting the growing global demand for energy storage solutions.1–5 Hard carbon is currently the widely used anode material among other anode materials used in SIBs due to its availability and chemical stability. However, its inherent limitations, such as low energy density and limited rate capability, restrict its effectiveness in high-performance applications.3,6–8 Therefore, some researchers have focused on developing conversion-type anode materials like transition metal oxides (TMOs) and transition metal dichalcogenides (TMDs) to address these shortcomings.9,10 These materials offer significantly higher theoretical specific capacities by undergoing reversible conversion reactions with sodium ions, thereby enhancing the overall energy density of SIBs.11–16Despite their promising advantages, conversion-type anode materials face several significant challenges. The most critical issue is the large volume expansion that occurs during cycling, leading to mechanical degradation and poor cycling stability. This expansion results from the substantial structural changes as they transform between different oxidation states.17–19 Additionally, conversion-type materials often suffer from poor electronic conductivity, which limits their rate capability and overall efficiency. They are also prone to irreversible phase transitions and side reactions with the electrolyte, further compromising their long-term stability and performance.20–22 Various strategies have been developed to mitigate these issues, including nanostructuring the materials to accommodate volume changes,17,18 hybridising them with conductive matrices such as carbon to enhance electronic conductivity,14,23–25 and applying surface coatings and electrolyte additives to stabilise the electrode/electrolyte interface.26–28
Among these strategies, carbonaceous coatings have emerged as particularly effective. Coating conversion-type anode materials with carbon improves electronic conductivity and acts as a buffer against volume expansion, creating a more stable electrode structure and facilitating better ion transport. This enhanced stability and performance can be attributed to the formation of chemical bridge bonds between the carbon coating and the conversion-type anode materials. Chemical bridge bonds, in terms of C–X–M bonds (where M represents a metal), are crucial in addressing the inherent challenges of TMOs and TMDs. These bonds mitigate severe volume changes during cycling by absorbing and distributing mechanical stress, thereby maintaining the structural integrity of the electrode.29–31 Additionally, they enhance electronic conductivity by creating continuous electron pathways at the interface, facilitating efficient charge transfer and reducing internal resistance.32–34 Chemical bridge bonds also improve ionic transport by stabilising the electrode structure and minimising defect formation, ensuring smoother ion diffusion pathways.35–37 Furthermore, these bonds stabilise the electrode/electrolyte interface by forming a chemically robust barrier, reducing the exposure of reactive sites to the electrolyte and preventing unwanted side reactions, such as the formation of an unstable solid-electrolyte interphase (SEI).16,21 Through these mechanisms, chemical bridge bonds significantly optimise the performance and durability of SIBs, utilising TMOs and TMDs as anode materials. Recently, significant progress has been made in the design, characterization, and application of C–X–M bonds in SIB anode materials; however, a comprehensive review that thoroughly analyses these aspects is still lacking.
This review provides a comprehensive analysis of the critical role that interfacial chemical bridge bonds play in SIBs, particularly in enhancing the structural stability, electronic conductivity, ion diffusion, charge transfer, electrode reversibility, and initial coulombic efficiency of composites involving transition metal compounds and carbon materials, as shown in Scheme 1. The review systematically examines the characterisation techniques commonly employed to detect and study these bonds, offering detailed insights into their specific contributions to improving sodium-ion storage performance. Additionally, it explores the challenges associated with forming and stabilising these bonds and evaluates the future prospects for developing chemical bridge bonds in SIB applications. The paper concludes by providing strategic guidance on optimising electrode materials through intelligent design and implementation of chemical bridge bonds, ultimately advancing the overall performance and commercial viability of sodium-ion batteries.
 | ||
| Scheme 1 Schematic illustration of advantages of C–X–M bonds of carbon coated anode materials. | ||
2. Formation and physical characterisation of C–X–M bonds
C–X–M bonds (X represents elements such as O, S, N, P, or Se) are typically formed via covalent bonding between the central metal (M) and the carbon framework (C) through the coordinating ligand (X). Coordinating ligands with stronger bonding capabilities, such as O, S, Se, N, and P, are prone to forming chemical bridge bonds with carbon framework and transition metals.18,31,34,40 The strong electronegativity of these elements allows them to form stable C–X–M covalent bridges through orbital hybridization. Notably, the incorporation of transition metals, as well as Bi, Sn, and Sb, can significantly enhance the electrical conductivity and structural stability of electrode materials, thereby exhibiting excellent performance in promoting sodium ion insertion/extraction within the battery and, substantially, accelerating the kinetics of battery reactions. Furthermore, these metals possess unfilled d orbitals or p orbitals, which enable them to form stable and strong coordinate covalent bonds with coordinating atoms X. Consequently, in the context of sodium-ion batteries, the formation of C–X–M bonds should preferentially involve the selection of O, S, Se, N, and P as coordinating elements, in conjunction with transition metals or Bi, Sn, and Sb featuring available orbitals as metal centers, as this synergistic combination is prone to yield stable and reliable C–X–M bonds.The approaches to induce the formation of interfacial chemical bridge bonds can be classified as: (i) introducing reactive groups enables electrostatic gravitational or van der Waals interactions between metal and carbon atoms, resulting in interfacial chemical bonding.38,39 For instance, by utilizing acid-treated carbon nanotubes (CNTs) with multiple functional groups (carbonyl, hydroxy, carboxyl, etc.) and in situ integrating them with VS4, VS4-CNT composites with a large number of C–O–V bonds were successfully prepared.30 (ii) During the material synthesis process, special reagents are used in combination with precursor materials and subjected to thermal treatment, guiding the transfer of electrons between molecules, thereby forming C–X–M bonds. A typical example is N, S co-doped carbon-encapsulated nanocube with a core–shell structure, in which the strong C–S–Mn bonds between the core and shell were introduced by vulcanization of MnCO3 nanocube-wrapped polydopamine (PDA) at a high temperature.40
Specifically, the mechanism of interfacial chemical bridge bond formation is that the central metal M acts as a Lewis acid, possessing partially filled d or p orbitals that can accept electrons. Conversely, the ligand X functions as a Lewis base, with lone pair of electrons that can participate in covalent bonding with the central metal M and the carbon materials. When the X-doped carbon materials interact with metal compounds, the metal's d or p orbitals overlap with the X element's p orbitals, forming a head-on σ bond, while simultaneously overlapping with the p orbitals of C and X to form a side-by-side π bond. This leads to a large accumulation of electrons on the C–X–M bonds, resulting in strong and stable chemical bridges (Scheme 2).
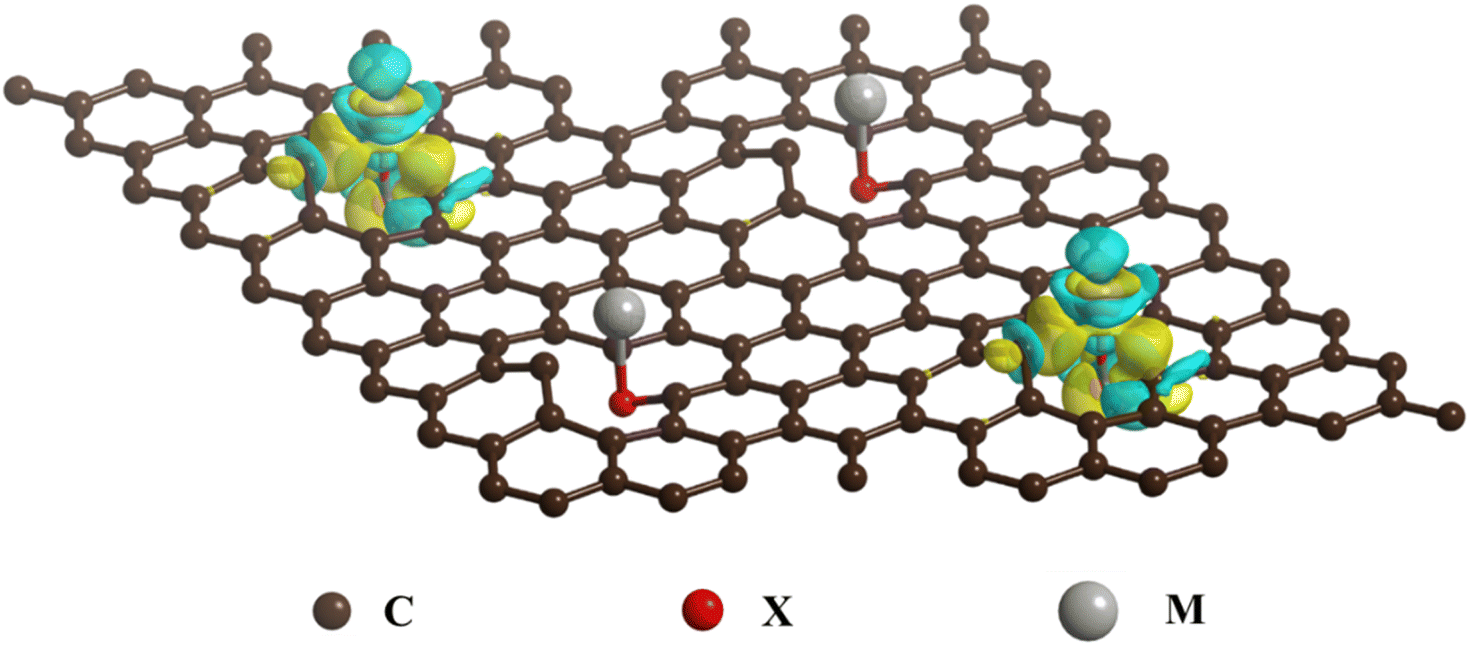 | ||
| Scheme 2 Schematic illustration of formation mechanism of C–X–M bonds of carbon coated anode materials. | ||
Generally, in order to verify the existence of C–X–M complexes and analyse their electronic properties and bonding states, the following physical characterisation techniques are commonly employed (Table 1): Fourier-transform infrared spectroscopy (FT-IR), X-ray absorption fine structure spectroscopy (XAFS),22,41,42 X-ray photoelectron spectroscopy (XPS),18,38,43 Raman spectroscopy,44–46 and electron paramagnetic resonance (EPR) spectroscopy.47,48 Additionally, the C–X–M bonds can be analysed using first-principles calculations49,50 and the electrochemical properties.
| Physical characterisation techniques | Detection content | Relevance to C–X–M bonds |
|---|---|---|
| Fourier-transform infrared spectroscopy (FT-IR) | Vibrational modes and molecular conformational analysis | Investigates molecular structure and bonding configuration |
| Raman spectroscopy | Molecular vibrational modes and phonon dynamics | Complements FT-IR, providing additional molecular structure information |
| X-ray photoelectron spectroscopy (XPS) | Elemental composition, chemical speciation, and electronic structure | Elucidates chemical bonding and electronic structure |
| X-ray absorption fine structure spectroscopy (XAFS) | Local atomic coordination and bonding environments | Evaluate the local chemical structure |
| Electron paramagnetic resonance (EPR) spectroscopy | Unpaired electron spin states and magnetic properties | Probes the spin number of unpaired electrons |
2.1 Fourier-transform infrared spectroscopy (FT-IR)
FT-IR spectra are produced when the molecules of a compound absorb specific wavelengths of infrared light, causing them to vibrate. The atoms that make up a bond or functional group are in a constant state of vibration, with each bond or functional group absorbing at a particular frequency and will be in a different position on the infrared spectrum, thus allowing information on the specific bond or functional group in the molecule to be obtained.51,52 Zhu et al.31 prepared one-dimensional core–shell structured N, S co-doped carbon-coated FeS nanowire arrays (FeS@NSC) via a simple chemical vapour deposition-like strategy and utilised FTIR measurements further to verify the possibility of the existence of C–S–Fe bonds. As shown in Fig. 1a, FTIR spectra of the samples investigated in the wavelength of 250–2000 cm−1. Near 1100 cm−1, a significant absorption peak was attributed to the stretching vibration of the C–S/C–S–Fe bond, which further illustrates the interaction between FeS and the outer carbon. A small absorption peak appears near 607 cm−1, which was caused by the stretching vibrations of the Fe–S bond. Peng et al.53 constructed MnS/Mxene heterostructure composites on carbon nanofibers by an electrostatic spinning method (Fig. 1b). In the synthesised MnS@MXene@CNF composite (MMC, where CNFs are carbon nanofibers), the presence of C–S–Mn bonds in the material was confirmed by analysing the changes in peak position and intensity of other chemical bonds in the FT-IR spectra (Fig. 1c). Compared to the MnS@CNF (MC) material, a significant increase in the adsorption peaks due to C–S bond stretching vibrations can be attributed to the newly formed C–S–Mn bond at the interface between MXene and MnS. On the other hand, the absorption peaks due to Mn–S bond stretching vibrations are more subdued in the MMC than in the MC material owing to the presence of C–S–Mn chemical bridges during the interfacial reconstruction process, which weakens the Mn–S bonds in the MnS crystals and leads to a reduction in the adsorption of Mn–S bonds in the IR.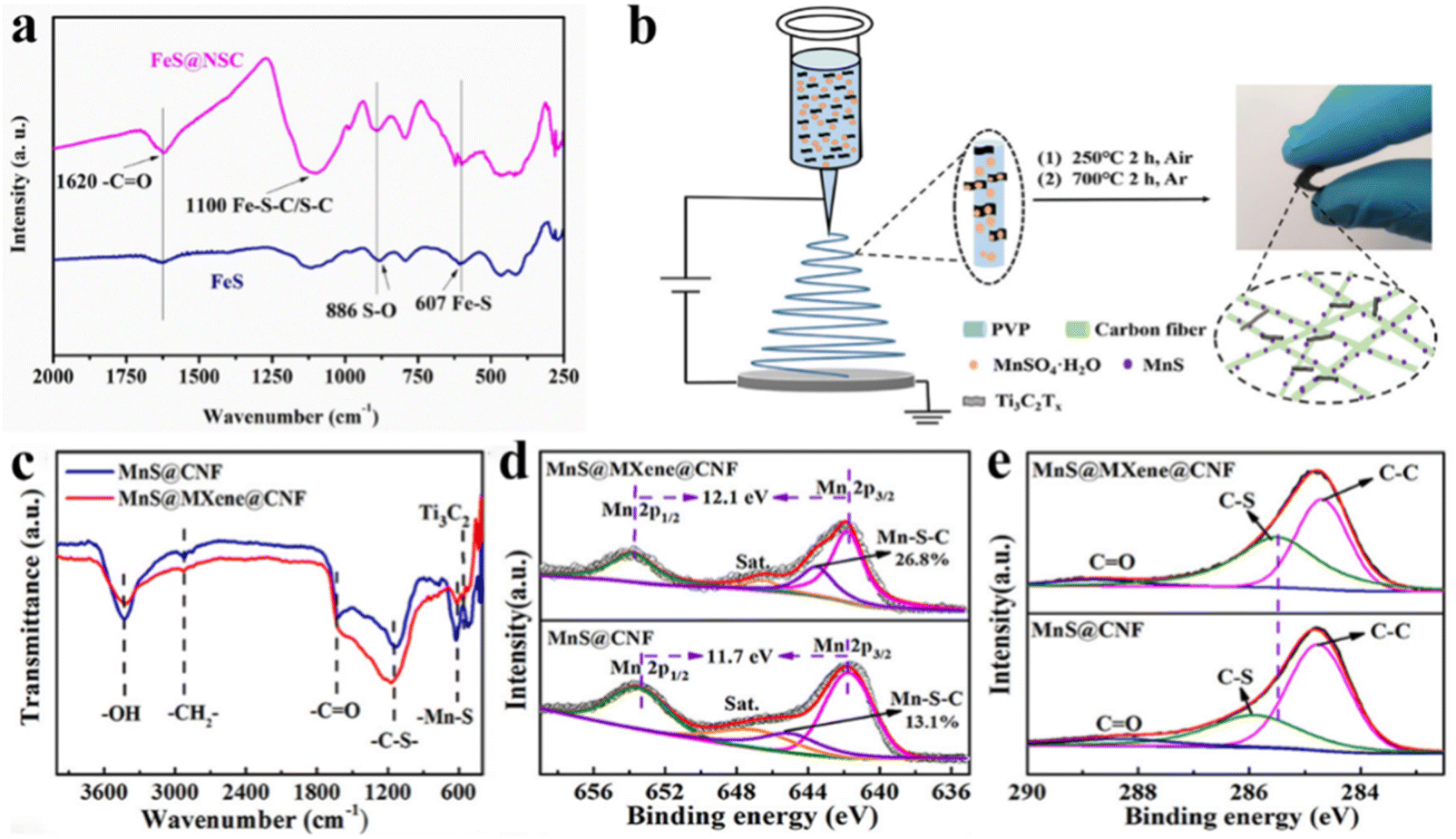 | ||
| Fig. 1 FT-IR and XPS identify the specific bond. (a) FT-IR of FeS@NSC samples. Reproduced with permission.31 Copyright 2021, Royal Society of Chemistry. (b) Synthesis process. (c) FT-IR spectra, (d) Mn 2p and (e) C 1s of MnS@MXene@CNF. Reproduced with permission.53 Copyright 2022, American Chemical Society. | ||
2.2 X-ray photoelectron spectroscopy (XPS)
As one of the most widespread means of characterising the chemical bonding information of samples, XPS serves an indispensable role. The chemical bonding information of a sample can be obtained by precisely measuring the chemical shift of the inner electron binding energy of the sample.32,54 In the high-resolution XPS spectra of the Mn 2p (Fig. 1d), a small peak at 643.5 eV was found to be caused by the formation of a C–S–Mn bond, and its chemical content (26.8%) was higher compared to the MnS@CNF (MC) material. In addition, the spin–orbit splitting value of Mn 2p in the MnS@MXene@CNF (MMC) was about 0.4 eV higher compared to the MC material, which was attributed to the movement of the outer electron Mn2+ from the MnS to the MnS/Mxene interface, resulting in a reduced shielding effect. In the high-resolution C 1s spectra (Fig. 1e), there is a slight redshift in the peak positions belonging to the C–S bond compared to the MC material, which is caused by a shift in electron density from Mn through the heterogeneous interface between MnS and MXene to the surrounding C, resulting in an increase in the electron cloud density of C and thus a reduction in the binding energy required for C excitation. Another example directly confirmed the presence of C–S–Mn bonds directly.40 In the high-resolution Mn 2p spectra, the overall peak position of the nitrogen–sulphur co-doped carbon-coated MnS nanocube (MnS@NSC) sample underwent a redshift of ∼1.45 eV compared to the commercial MnS and mechanically mixed MnS/NSC, with the appearance of an additional peak centred at 643 eV, attributable to the C–S–Mn bond formed by MnS with the outer carbon shell. The additional peak that appeared in the S 2p XPS spectrum of N, S co-doped carbon-coated MnS nanoparticles (MnS@NSC) was assigned to C–S–Mn bonds. This result was also observed in the C 1s spectra. In addition, Chen et al.55 also verified the presence of C–S–Zn bonds in their analysis of the S 2p spectra of the N, S co-doped carbon-coated ZnS composites prepared. It was apparent that the peak area ratio of S 2p3/2 and S 2p1/2 in S 2p was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for pure ZnS materials, while the area ratio of S 2p3/2 (161.5 eV) and S 2p1/2 (162.5 eV) in A-ZnS/NSC composites is lower compared to ZnS/NSC composites, which was mainly attributed to the ZnS/NSC composites with strong interactions between ZnS and carbon (C–S–Zn bond).
:1 for pure ZnS materials, while the area ratio of S 2p3/2 (161.5 eV) and S 2p1/2 (162.5 eV) in A-ZnS/NSC composites is lower compared to ZnS/NSC composites, which was mainly attributed to the ZnS/NSC composites with strong interactions between ZnS and carbon (C–S–Zn bond).
2.3 Raman spectroscopy
Raman spectroscopy measurement is a conventional means of detecting chemical bonding information on the surface of a sample. Only elastic collisions and no energy exchange occurred when a sample was excited by light at a specific frequency. The scattering spectrum was analysed when the frequency of the incident light was not equal to that of the excited light to obtain information such as the vibrational and rotational energy levels of molecules and thus determine the molecular structure and chemical bonding of the sample.56,57 Wei et al.39 also used Raman spectroscopy as an important tool for detecting C–O–V bonds. As shown in Fig. 2a, the peak observed at 872 cm−1 was ascribed to the vibrations of the C–V bond, whereas the peaks observed at 830 and 920 cm−1 were assigned to the stretching vibrations of the O–V bond, indicating that the VS4-HC composite contained both bonds. In addition, Mai et al.43 synthesised VOx clusters loaded on graphene oxide to obtain heterostructures with C–O–V bonds (Fig. 2b) and provided further clarification in determining the existence of an interfacial chemical bond (C–O–V) between VOx and graphene oxide in the VOx-G heterostructure employing Raman analysis. As shown in Fig. 2c, in the VOx-G sample with a heterogeneous structure, the peak positions exhibited by the stretching vibrations due to the V3–O and V2–O bonds are shifted to the right, indicating that a change in the electronic state of O has occurred in the VOx cluster, which may be attributed to the formation of the C–O–V bond.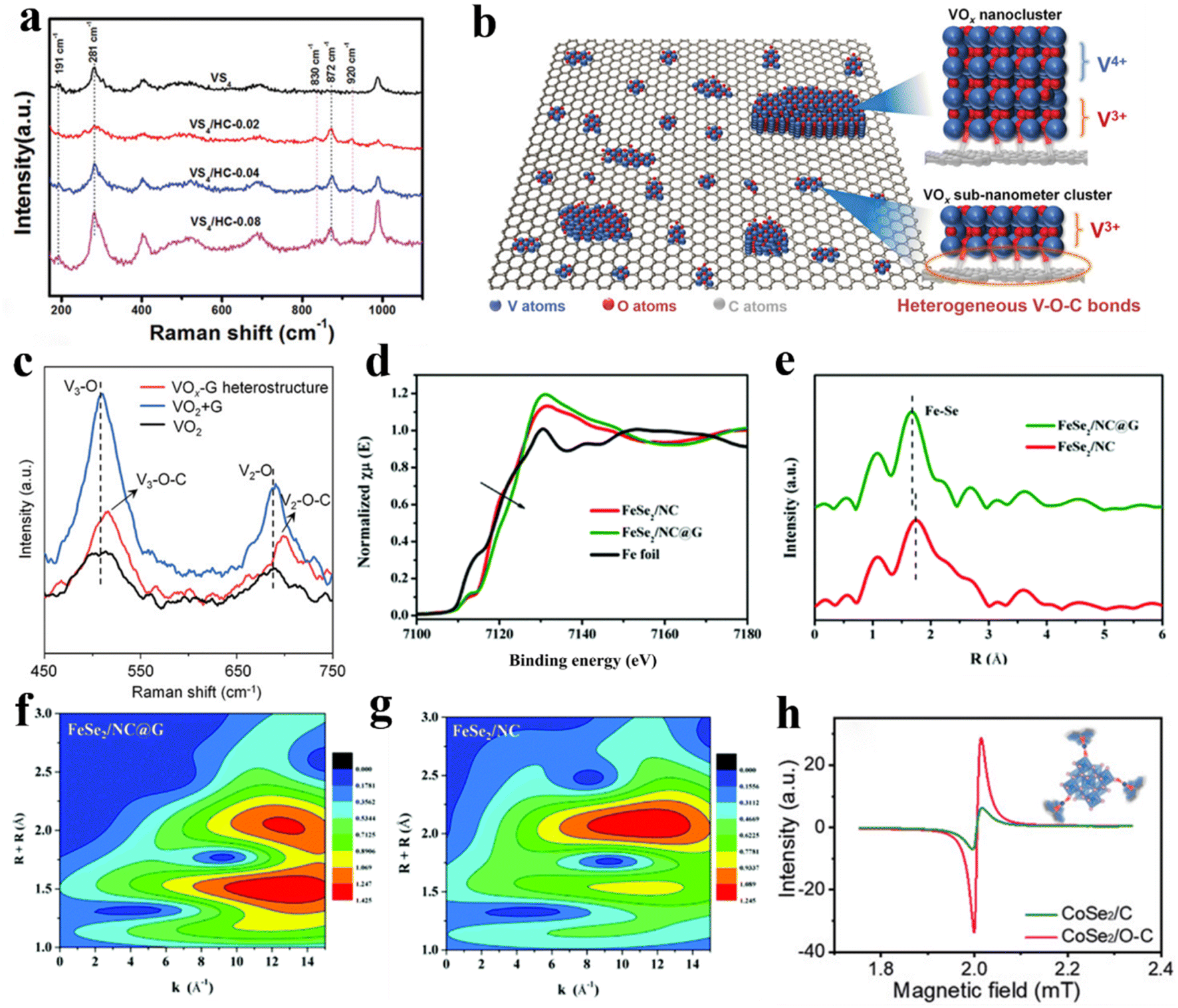 | ||
| Fig. 2 Raman spectroscopy, XAFS, and EPR verify the existence of C–X–M. (a) Raman spectra of VS4-HC materials. Reproduced with permission.39 Copyright 2020, Wiley. (b) Schematic diagram of VOx-G heterostructure with C–O–V bonds. (c) Comparative Raman spectra of VOx-based materials. Reproduced with permission.43 Copyright 2021, Wiley. (d) XANES spectra of FeSe2/NC@G composites. (e) Corresponding k3-weighted FT-EXAFS spectra. (f) Morlet WT of the k3-weighted EXAFS data of FeSe2/NC@G. (g) Morlet WT of the k3-weighted EXAFS data of FeSe2/NC/G. Reproduced with permission.58 Copyright 2020, Royal Society of Chemistry. (h) EPR electron paramagnetic resonance spectra. Reproduced with permission.60 Copyright 2021, Wiley. | ||
2.4 X-ray absorption fine structure spectroscopy (XAFS)
Notably, X-ray absorption spectroscopy (XAS) can be used to evaluate the local chemical structure of electrode materials. When the spectral region is extended to 30–50 or 50–1000 eV, it can be subdivided into X-ray absorption near-edge structure (XANES) spectroscopy and X-ray absorption fine structure (XAFS) spectroscopy. Among them, XANES can provide information about the oxidation states, while XAFS is a powerful tool for researching the local environmental chemical bonding of samples.21,42 Tao et al.58 further determined the formation of chemical bridge bonds in FeSe2/NC@G samples by XAFS testing. As shown in Fig. 2d, the FeSe2/NC@G sample has a higher position of the absorption edge compared to the Fe foil and the mixed material FeSe2/NC, which indicates that the resulting C–O–Fe chemical bridge bond makes Fe positively charged. In addition, the main peak position attributed to the Fe–Se bond (at approximately 1.88 Å) is shifted to the left compared to FeSe2/NC evident from the FT-EXAFS spectrum (Fig. 2e), indicating the presence of more C–O–Fe negatively charged coordination sites. Furthermore, the plausibility of the C–O–Fe chemical bridge bond was further confirmed by wavelet transform (WT)-EXAFS analysis (Fig. 2f and g). The intensity of the peaks attributed to Fe–Se bonds is weakened, and the intensity of the peaks attributed to Fe–O bonds is enhanced in the FeSe2/NC@G sample compared to FeSe2/NC, which is consistent with the previous. Similarly, Li et al.12 confirmed the presence of C–O–Ti bonds in the N/S co-doped TiO2–C nanofiber composites prepared by synchrotron XAS analysis.2.5 Electron paramagnetic resonance (EPR) spectroscopy
EPR technique can be used to directly probe the spin number of unpaired electrons of the sample.27,59 In recent years, researchers have analysed XPS profiles to obtain the hypothesis of the presence of chemical bridge bonds, followed by EPR profiles further to determine the plausibility of the presence of chemical bonds. Ge et al.60 compared the EPR curves of both CoSe2/C and CoSe2/O–C samples, with the results showing a stronger peak for the CoSe2/O–C sample, confirming that CoSe2/O–C has more defects and interfacial chemical bonds (Fig. 2h). Similarly, the introduction of C–O–V bonds caused a change in the spin state of the lone electron of vanadium. In the EPR image, the difference between the g value of NVO-reduced graphene oxide (rGO) samples and the g value of NVO/rGO and NVO samples is observed.61Typically, a single characterisation technique cannot robustly support the evidence for the presence of chemical bridge bonds; instead, several characterisation tools are required. For example, to detect the presence of C–S–Fe bonds, Chou et al.62 first observed two peaks near 510 and 1100 cm−1 by FT-IR spectroscopy (Fig. 3a), which were caused by vibrations of the C–S bond. This was further confirmed in the C 1s and S 2p XPS spectra (Fig. 3d and e). In addition, combined with the EDS results shown in Fig. 3b, the proportion of S atoms used for C–S bonds in S 2p was much higher than that of doped S atoms, indicating that the S portion in the C–S bonds originated from FeS2. Moreover, a strong peak was observed at 1.4 Å in the Fourier transformed-extended X-ray absorption fine structure (FT-EXAFS) spectrum (Fig. 3c), which was attributed to Fe–S bonding. Therefore, the existence of the C–S–Fe bond was confirmed.
 | ||
Fig. 3 Multiple characterization methods combined to identify C–X–M. (a) FT-IR spectra, (b) EDS patterns, (c) FT-EXAFS of FeS2/N–C and FeS2/S–C. XPS spectra of (d) C 1s and (e) S 2p. Reproduced with permission.62 Copyright 2021, Wiley. (f) Absorption energies of the various functional groups of VS4 (order from left to right: C, defective C, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –OH, and –COOH). (g) DOS curves at different states. (h) Mechanical properties of pure VS4 and the VS4-CNT heterojunction after being modified with interfacial bonds (V–O–C and V–C). Reproduced with permission.30 Copyright 2022, Wiley. O, –OH, and –COOH). (g) DOS curves at different states. (h) Mechanical properties of pure VS4 and the VS4-CNT heterojunction after being modified with interfacial bonds (V–O–C and V–C). Reproduced with permission.30 Copyright 2022, Wiley. | ||
In addition to the above methods, theoretical calculations have provided convincing evidence to confirm the presence of C–X–M bonds.63–65 Zou et al.30 anchored VS4 nanoparticles on carbon nanotubes (CNTs) with multiple functional groups to construct strong C–O–V bonds. Absorption energies of the various functional groups of VS4 calculations (Fig. 3f) and density of states (DOS) curves (Fig. 3g) were performed to corroborate the viability of C–O–V bonding, and the results indicated that certain functional groups induced a decrease in adsorption energy, which implied that the doped functional groups were beneficial for coupling interfacial chemical bonds. Moreover, through the study of the mechanical properties of VS4-CNT heterostructures with different interfacial chemical bonds (Fig. 3h), the results demonstrate that the introduction of C–O–V bonds can enhance the mechanical strength of the material more effectively, which will help to suppress the volume expansion of the material. The agreement between the density functional theory (DFT) calculations and molecular dynamics (MD) simulation results and the electrochemical performance results further confirmed the existence of strong C–O–V bonds, which will be discussed in detail later.
Thus, various physical characterisation methods were used to detect C–X–M bonds. Among these, XPS and FTIR are the most used. However, the strength and content of these bonds have not been thoroughly evaluated thus far. Therefore, exploring new characterisation techniques for detecting the bond content and strength is essential to investigate further the effects of chemical bridging bonds on composite materials.
3. The role of C–X–M bonds in sodium-ion storage
In battery systems, the C–X–M chemical bridge bond as a connection between metal compounds and carbon materials tends to cause the evolution of the physicochemical properties of the material. The study of interfacial chemical bridging bonds in SIBs has attracted much attention recently. They perform a non-negligible role in SIBs, including facilitating sodium ion diffusion, enhancing the mechanical integrity and structural stability, improving the electron transport in electrode materials, enhancing sodium ion insertion/extraction reversibility and improving initial coulombic efficiency (ICE).66–70 Therefore, the presence of C–X–M bonds confers exceptional performance on the electrode materials listed in Table 2.| Materials | Bonds | Cycle number | Initial/cycling capacity (mA h g−1) | Decay ratio per cycle (%) | References |
|---|---|---|---|---|---|
| MnS@NSC | C–S–Mn | 200 | 594.2/388.7 | 0.173 | 40 |
| FeS@NSC | C–S–Fe | 150 | 611.6/575 | 0.040 | 31 |
| FeS2/N, S–C | C–S–Fe | 5500 | 543.1/501.3 | 0.001 | 62 |
| MoS2/N-CNT | C–S/O–Mo | 100 | 555.8/486 | 0.126 | 73 |
| MoS2@rGO | C–S/O–Mo | 100 | 743.3/654.6 | 0.119 | 85 |
| A-SnS2@G | C–S–Sn | 200 | ∼1250/597 | 0.261 | 74 |
| ZnS/N, S | C–S–Zn | 100 | 517/500.7 | 0.032 | 55 |
| Cu9S5/NSC | C–S–Cu | 200 | 412/344.3 | 0.082 | 86 |
| MnS/SC | C–S–Mn | 90 | 379.5/600 | −0.646 | 70 |
| VS4–V2CTx | C–S–V | 100 | 801/416 | 0.481 | 87 |
| CoSe2/O–C | C–O–Co | 70 | 634/373.3 | 0.587 | 60 |
| FeSe2/NC@G | C–O–Fe | 50 | 529/477 | 0.197 | 58 |
| FHG | C–O–Fe | 100 | ∼318/214 | 0.327 | 34 |
| MoS2/C | C–O/N–Mo | 100 | 458.6/419.5 | 0.085 | 88 |
| MoSe2@5%GO | C–O–Mo | 50 | 458/383.6 | 0.325 | 66 |
| MoSe2/N–C | C–O–Mo | 120 | ∼498/552.1 | −0.091 | 89 |
| Sb2Se3/CNT | C–O–Sb | 200 | 428/265.3 | 0.190 | 90 |
| SS/H@C | C–O–Sb | 100 | 691.9/451.2 | 0.348 | 91 |
| SnO2/C composites | C–O–Sn | 350 | 311/270.7 | 0.037 | 18 |
| SnO2/G–S | C–O–Sn | 90 | 770/650 | 0.173 | 92 |
| SnS2/rGO | C–O–Sn | 200 | 620/575 | 0.036 | 93 |
| Mxene/TiO2 | C–O–Ti | 100 | ∼800/660 | 0.175 | 94 |
| Ti3C2/CoS2@NPC | C–O–Ti | 100 | ∼536/438.4 | 0.182 | 95 |
| Bi2Se3@rGO@NC | C–O–Bi | 300 | 288.4/160.1 | 0.148 | 96 |
| Bi2Se3–C/G-1 | C–O–Bi | 100 | 377/— | — | 33 |
| ZnO/RGO | C–O–Zn | 150 | ∼207/188.6 | 0.059 | 97 |
| VS4-CNT | C–O–V | 100 | 476.1/∼470 | 0.013 | 30 |
| FeS@C NSRs | C–S–Fe | 120 | 810.3/614.2 | 0.051 | 83 |
| SSCCM | C–O–Mo | 180 | 354.7/345.6 | 0.014 | 28 |
| NVO–rGO | C–O–V | 1000 | 168/∼164 | 0.002 | 61 |
| MoO2/N–C | C–N–Mo | 200 | ∼136/134 | 0.02 | 75 |
| FeSe2@NDC NBs | C–N–Fe | 2000 | 403.3/— | — | 79 |
| (Sb2Se3@rGO@NC) | C–N–Sb | 1000 | 426.9/100.4 | 0.327 | 81 |
| CoSe–rGO | C–Se–Co | 100 | 457.5/400.8 | 0.567 | 29 |
| SnSe2@Se–C | C–Se–Sn | 2000 | ∼600/441 | 0.08 | 78 |
| Sn4P3/RGO | C–P–Sn | 1000 | ∼710/362 | 0.348 | 98 |
| Fe1−xS@Sb@C | C–S–Sb | 100 | ∼460/397.4 | 0.13 | 99 |
3.1 Facilitating sodium-ion diffusion
In sodium-ion batteries, the diffusion kinetics of sodium ions are crucial factors of battery performance. Chemical bridge bonds create a more intimate contact between compounds and carbon materials, serve as facilitators for sodium-ion diffusion and significantly improve sodium-ion diffusion kinetics, leading to enhanced battery performance.71,72 Liu et al.73 obtained low-crystalline MoS2 nanoflakes embedded on N-doped CNTs (MoS2/N-CNT) with more defects, causing many C–S–Mo bonds between MoS2 and N-CNTs, which effectively facilitated sodium-ion diffusion. Comparing the Warburg impedance analysis of pure MoS2 and MoS2/N-CNT (Fig. 4a and b), the MoS2/N-CNT electrode containing C–S–Mo bonds exhibited smaller ion diffusion impedance, which indicated that the diffusion rate of sodium ions is faster in MoS2/N-CNT, attributed to the C–S–Mo bond. Consequently, the MoS2/N-CNT electrodes exhibited favourable electrochemical performance for application in SIBs. Wang et al.74 prepared SnS2/graphene heterostructures (A-SnS2@G) with high contact surfaces and strong interfacial C–S–Sn chemical bridge bonds. Raman spectroscopy showed a higher degree of disorder (ID/IG) in the A-SnS2@G samples with C–S–Sn bonds than in the mechanically mixed SnS2/C samples (Fig. 4c), indicating that the covalent anchoring of ultrathin SnS2 nanosheets increases the disorderliness and inhibits the stacking of graphene layers, which facilitated sodium ion diffusion. In addition, DFT calculations proved the plausibility of accelerated sodium ion diffusion after the formation of chemical bridge bonds. As shown in Fig. 4d, for the A-SnS2@G sample, charge transfer was able to occur spontaneously at the interface between graphene and SnS2 in the presence of the C–S–Sn chemical bond, indicating that a more intimate contact between SnS2 and graphene was created at the interface. Thereby, the diffusion distance of sodium ion is reduced, and the effective contact area is increased. The energy band structure diagram (Fig. 4e) of A-SnS2@G shows a downward shift of the conduction band. Moreover, the calculated DOS of A-SnS2@G near the Fermi energy level was significantly higher than that of the SnS2/C sample (Fig. 4f), indicating the bonds can facilitate the formation of a more conductive pathway for sodium ions. The facilitated sodium ion diffusion rate is also evident in the improved cell multiplicity performance. Compared with other SnS2-based materials (Fig. 4g), the multiplicative performance of the A-SnS2@G electrode has a significant advantage due to the improved sodium ion diffusion by the interfacial chemical bridge bonding. Cao et al.75 prepared a MoO2/N-doped carbon (MoO2/N–C) composite composed of MoO2 nanocrystals embedded in a carbon matrix with a C–N–Mo chemical bond by controlling annealing temperature using a carbonation-induced topochemical transformation route. Chemically bridging MoO2 with N-doped carbon substrate provided pores pathway, which facilitated fast sodium ion diffusion (Fig. 4h). Thus, the prepared sample exhibits excellent rate performance. Ge et al.60 prepared CoSe2/O–C composites with intrinsic C–O–Co bonds. Comparing the first charge–discharge platforms of other CoSe2-based materials (Fig. 5a), CoSe2/O–C exhibited smaller polarisation because of the capability of C–O–Co bonds to promote ions mobility and enhance diffusion reversibility. Additionally, the ion diffusion rates calculated from the cyclic voltammetry (CV) curves at different scan rates (Fig. 5b) confirmed that including bonds effectively increases ion mobility. By calculating the pseudocapacitance (Fig. 5c) and analysing the linear relationship between log(i) and log(v) (Fig. 5d), it was observed that the capacitance-controlled behaviour was enhanced in the low-pressure region, which was attributed to the migration of ions along the C–O–Co bond, enhancing the reaction of CoSe2. Li et al.76 found that C–O–Sn bonds could shorten the Na+ transport path. Hou et al.77 revealed that due to the construction of the Sb–O–C bond, the antimonite Sb2S3 electrode exhibited a higher Na+ diffusion rate. Yan et al.78 prepared SnSe2@Se–C, in which Se is doped into the carbon shell with a tuneable amount and form Se–C bonds, and this modification resulted in the substantial strengthening of the C–Se–Sn bond, consequently enhancing the sodium ion diffusion properties. | ||
| Fig. 4 C–X–M efficiently contribute to sodium ion diffusion kinetics. (a) Impedance plots of MoS2/N-CNT and MoS2 samples after 100 cycles at 0.1 A g−1. (b) Corresponding Z′ vs. ω−1/2 plots. Reproduced with permission.73 Copyright 2019, Royal Society of Chemistry. (c) Comparative Raman patterns of A-SnS2@G and SnS2/G samples. (d) Charge density difference of the optimised A-SnS2@G model and (e) band structure (green/pink clouds represent accumulation/depletion of electrons). (f) DOS for A-SnS2@G and SnS2/G near the Fermi energy level. (g) Comparison of the multiplicative performance with the previously reported SIB-based SnS2 anode. Reproduced with permission.74 Copyright 2022, Springer. (h) Illustration of electron and ionic transport properties in MoO2/N–C. Reproduced with permission.75 Copyright 2021, Springer. | ||
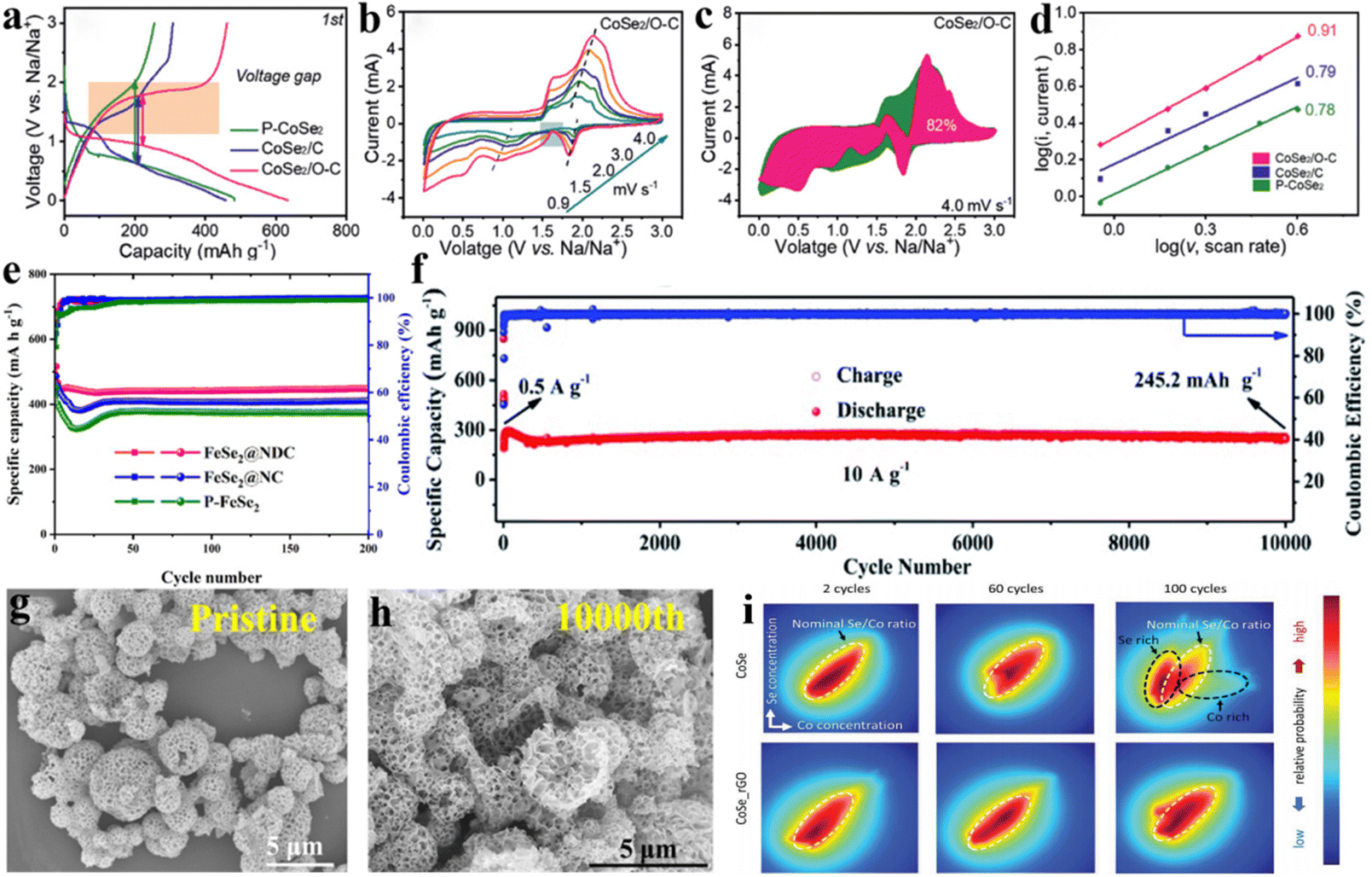 | ||
| Fig. 5 The incorporation of C–X–M bonds enhances the mechanical integrity of electrode materials. CoSe2/O–C and other CoSe2-based samples: (a) initial charge/discharge curves, (b) CV curves, (c) relative capacitance-controlled contributions, and (d) linear relationship of log(i) versus log(v). Reproduced with permission.60 Copyright 2021, Wiley. (e) Cycling performance of FeSe2@NDC, FeSe2@NC and P–FeSe2 at 0.2 A g−1. Reproduced with permission.79 Copyright 2021, American Chemical Society. (f) Cycling performance of Sb2O3@Sb@C electrodes at 10 A g−1. SEM images of (g) pristine Sb2O3@Sb@C and (h) the electrode after 10000 cycles at 10 A g−1. Reproduced with permission.80 Copyright 2021, Royal Society of Chemistry. (i) The correlation analysis of the Se and Co. Severe segregation of Se and Co is observed in the CoSe electrode after 100 cycles.29 Copyright 2018, John Wiley and Sons. | ||
3.2 Enhancing the mechanical integrity
In sodium-ion batteries, the prolonged cycling of Na+ ions in the material can trigger volume expansion, which can compromise the material's mechanical integrity and lead to capacity fading. Therefore, enhancing the mechanical integrity of electrode materials is crucial for unlocking the full potential and ensuring the long-term reliability of sodium-ion batteries. The strong C–X–M chemical bond formed between the carbon-based material and the metal compound effectively restrains the volume expansion that occurs during the long-term insertion/extraction of Na+ in the material, maintaining the mechanical integrity of the composite.26,69 The interface C–N–Fe bonds derived from the cyano group of Prussian blues (PBs)79 could contribute to the integrity of the electrode architecture. When used as an anode material for SIBs, FeSe2@NDC NBs show much better cycling stability compared with FeSe2 and FeSe2/NC (Fig. 5e). Yan et al.80 prepared Sb2O3@Sb nanoparticle composites impregnated in N-doped carbon microcages and evidenced that their strong C–O–Sb bonds were beneficial to the overall mechanical integrity of the material. Compared to the Sb2O3@NC composite without C–O–Sb bonds, the Sb2O3@Sb@C sample exhibited excellent multiplicative performance and stable long-cycle performance (Fig. 5f) at a current density of 10 A g−1, Sb2O3@Sb@C electrode obtained a higher reversible capacity of 245.2 mA h g−1 after 10000 cycles (the first five cycles being the activation process). SEM and TEM observations of the electrode after 10000 cycling (Fig. 5h) revealed a slight difference between the cycled electrode material and the original material (Fig. 5g), verifying that the heterogeneous structure with strong C–O–Sb bonds has a positive effect on protecting the mechanical integrity and enhancing the cycling performance of the material. Liang et al.28 prepared C@MoO2 composite materials with core–shell structure by introducing C–O–Mo bonds between biomass carbon and MoO2 and used as anode materials for SIBs, exhibiting excellent long-term cycling performance performance.
Similarly, Wu et al.29 Synthesised CoSe–rGO hybrid material and conducted a systematic study of the CoSe electrode and CoSe–rGO hybrid electrode using elemental sensitive transmission X-ray microscopy (TXM) (Fig. 5i) confirmed that the formation of a stable bond between CoSe and graphene, enhancing the mechanical integrity of the electrode material. Consequently, the dissolution and volume expansion of Se during Na/Li intercalation and deintercalation processes are significantly suppressed. The distinctive microstructural advantages of this hybrid material confer stable and exceptional sodium/lithium storage performance.
3.3 Improving the electron transport in electrode materials
Boosting the electrical conductivity of electrode materials is a crucial approach to enhance the performance of sodium-ion batteries. It is noteworthy that the introduction of the C–X–M bond had a positive effect on the structural electrical conductivity of sodium-ion batteries. The underlying reasons for this phenomenon are twofold: firstly, the carbon material boasts exceptional electronic conductivity, and secondly, the C–X–M chemical bridge bonds exhibit enhanced metal–ligand coordination, which creates a more efficient electron transport pathway and grants the electrode material exceptional electron transport capabilities.11,34 Zou et al.30 introduced acid-CNTs to prepare VS4-CNT heterostructure, in which VS4 nanoparticles are uniformly dispersed and tightly anchored on acid-CNT bundles with the assistance of massive C–O–V bonds. By DFT calculations (Fig. 3f and g), they demonstrated that the C–O–V bond reduced the band gap and energy barrier of the VS4-CNT heterostructure and exhibited enhanced conductivity. It was concluded from the electrochemical impedance spectroscopy (EIS) (Fig. 6a and b) pattern analysis that VS4-CNT has a smaller electron transport resistance (Rct) impedance compared to the untreated VS4/CNT composite without covalent bridge bonds, indicating that C–O–V bonded VS4-CNT has better electronic conductivity. Additionally, they Evaluated the sodium storage behaviour of VS4-CNT and untreated VS4/CNT by galvanostatic charge/discharge (GCD) (Fig. 6c and d), which indicated that the discharge capacity of VS4-CNT (530.5 mA h g−1) was significantly higher than that of VS4/CNT (444.7 mA h g−1). This could be attributed to the strong interfacial bonding of C–O–V, which enhanced the electronic conductivity of the VS4-CNT material. Ex situ XPS images (Fig. 6e–g) exhibited that the C–O–V bond and the chemical valence of the V species reversibly changed throughout the electrochemical process, allowing the C–O–V bond to maintain excellent electronic conductivity with high stability throughout the entire life cycle of the sodium-ion battery. In addition, a free-standing Na5V12O32–rGO (NVO–rGO) heterostructure with a controllable C–O–V bond was used as the cathode with highly electronic conductivity and ultralong cycling stability.61 Calculations of the band structure and density of states reveal that the introduction of a large number of C–O–V bonds in NVO–rGO leads to the disappearance of the original bandgap of NVO, resulting in an accumulation of electronic states near the Fermi level, thereby enhancing electron transport performance. The Sb2Se3@rGO@NC composite, synthesised by Chong et al.,81 demonstrated an outstanding cycle performance. The introduction of rGO and NC coatings led to the emergence of an overlapped area across the Fermi level rather than distinct gaps, as seen in the cases of Sb2Se3 and Sb2Se3@rGO (Fig. 6h). This phenomenon indicates a significant enhancement in conductivity upon the incorporation of rGO and NC. To further explore its microscopic mechanism, XPS high-resolution spectra analysis (Fig. 6i) revealed the formation of strong C–N–Sb chemical interactions during the sodiation/desodiation process of Sb2Se3@rGO@NC, and analysis of the calculated structure and charge difference picture (Fig. 6j) revealed the presence of charge transfer between Sb and N atoms. This confirms that the introduction of rGO and NC coatings leads to the formation of a large number of C–N–Sb bonds in the Sb2Se3@rGO@NC composite material, establishing electronic transport pathways and significantly enhancing the electronic conductivity of the electrode material.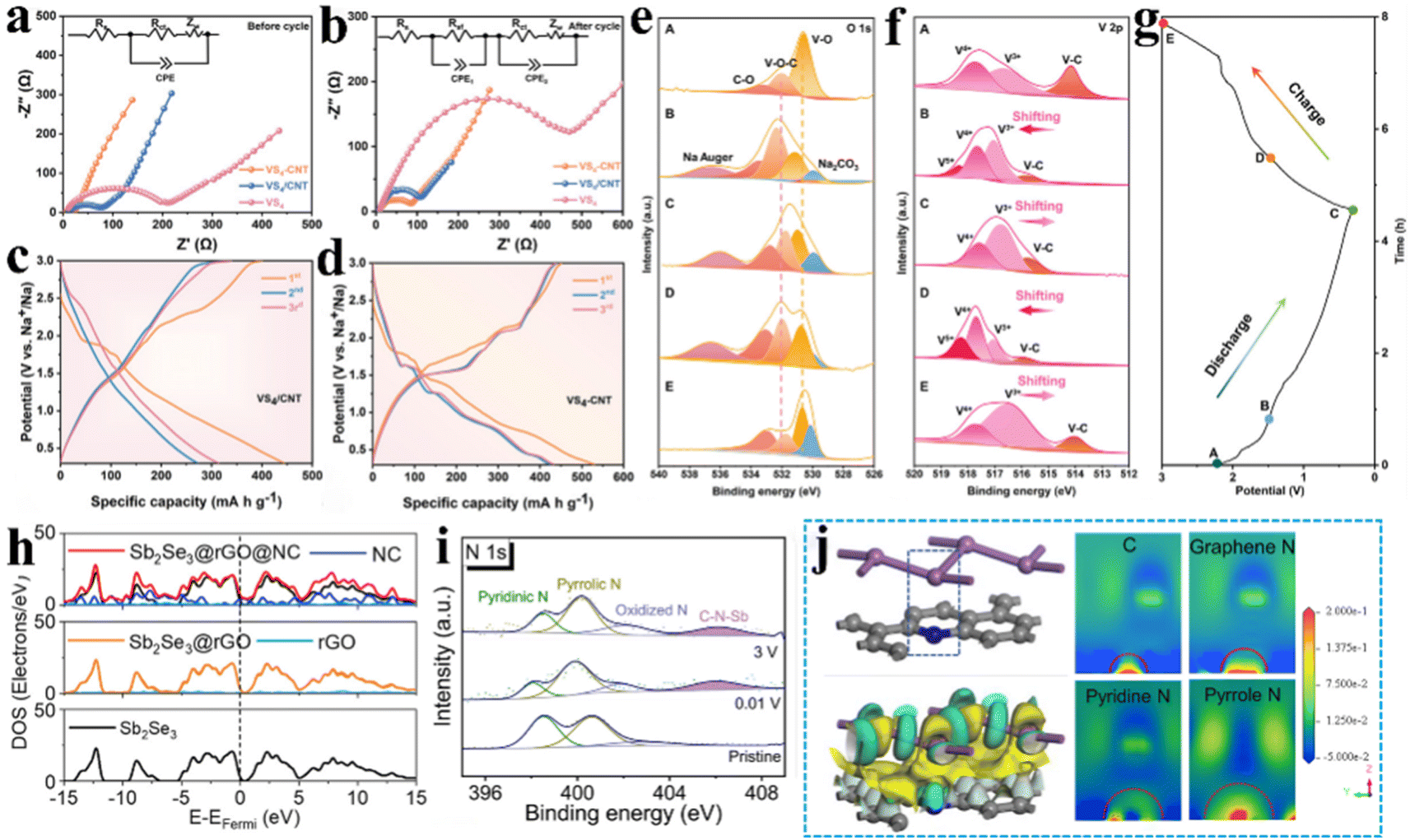 | ||
| Fig. 6 Electrochemical characterization demonstrates a significant enhancement of the electron transport in electrode materials. EIS curves of VS4-CNT electrodes (a) before and (b) after cycling; GCD curves of (c) VS4/CNT electrodes and (d) VS4-CNT electrodes. (e–g) Ex situ XPS of VS4-CNT electrodes. Reproduced with permission.30 Copyright 2022, Wiley. (h) DOS overlapping curves for Sb2Se3, Sb2Se3@rGO and Sb2Se3@rGO@NC; (i) XPS high-resolution spectra of N 1s for Sb2Se3@rGO@NC operated at full charge/discharge states. (j) Charge difference and corresponding pictures of slices between Sb and NC. Reproduced with permission.81 Copyright 2023, Wiley. | ||
3.4 Enhancing sodium ion insertion/extraction reversibility
As research on the mechanisms of sodium-ion batteries advances, the reversibility of sodium-ion insertion and extraction in electrode materials has emerged as a critical factor governing the overall performance and reliability of sodium-ion batteries. Notably, in the research on lithium-ion batteries, chemical bridge bonds have long been recognized as a crucial linking bridge for composite materials, significantly enhancing the reversibility of lithium-ion insertion and extraction in electrode materials.44,71,82 Recently, this critical role has also been identified in SIBs. The electrode material achieves sodiation/desodiation reactions via the formation of chemical bonds, and the formation and breaking of these bonds must be highly reversible to guarantee the stable performance of sodium-ion batteries. Zhu et al.40 prepared a cube-like core/shell composite of single MnS nanocubes (≈50 nm) encapsulated in N, S co-doped carbon (MnS@NSC) with strong C–S–Mn bond interactions. A real-time analysis of the structural changes of MnS@NSC/FeS@NSC using in situ TEM (Fig. 7a and b) revealed the synergistic effect of the encapsulation of carbon materials and the C–S–Mn/Fe bridge bonds, this architecture is beneficial for the sodiation/desodiation reaction at the electrode, and restricts the movement of the active substance, ensures that the active substance can still recover to its original state after the reaction. In situ Raman spectroscopy was used to investigate the intensity change of the peaks associated with the C–S–Mn bridge bonds during the charging and discharging processes. At the open-circuit voltage (2.4 V), a well-resolved strong peak was observed at approximately 654 cm−1, caused by the vibration of the C–S–Mn bond. As the discharging process proceeded, the peak strength decreased gradually. Thus, the content of the C–S–Mn bond progressively decreased until it disappeared, whereas it gradually increased as the charging progressed. This indicates that the C–S–Mn bond was reversibly broken and formed during the charging and discharging processes, which further confirms that the C–S–Mn bond may promote the reversible conversion between MnS and Na2S. Consequently, the MnS@NSC electrode exhibited a high reversible capacity (594.2 mA h g−1 at 100 mA g−1) and excellent cycling stability (329.1 mA h g−1 at 1 A g−1 after 3000 cycles). Similarly, Zhu et al.83 used in situ Raman techniques to demonstrate that during repeated transfer reaction between Na+ and FeS@C NSRs, the interfacial chemical bonds were able to break and form stably, confirming the reversibility of the redox reaction. C–S–Fe bonds can promote the reversibility of FeS@C electrode, thereby obtaining excellent reversible electrochemical performance.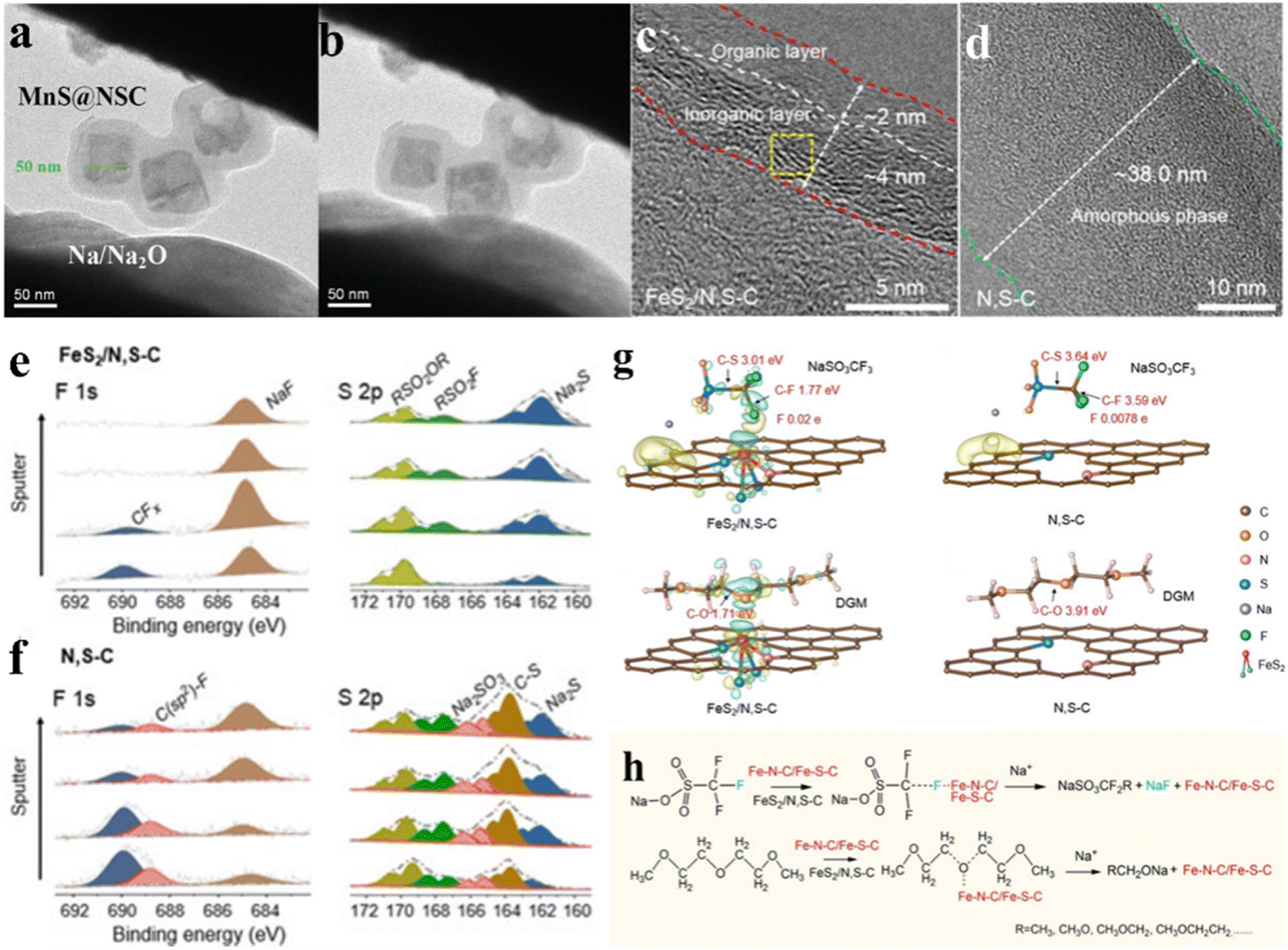 | ||
| Fig. 7 C–X–M bonds enhance the reversibility of the sodiation and desodiation processes and improve the initial coulombic efficiency (ICE) of the material. In situ TEM images of MnS@NSC during the (a) initial state and (b) sodiation. Reproduced with permission.40 Copyright 2020, Wiley. HRTEM of SEI on (c) FeS2/N, S–C and (d) N, S–C, and (e-f) their high-resolution XPS spectra at different sputtering times. (g) Differential charge densities of NaSO3CF3–FeS2/N, S–C, DGM-FeS2/N, S–C, NaSO3CF3–N, S–C, DGM-N, S–C. (h) The proposed mechanism for the decomposition process of NaSO3CF3 and DGM on FeS2/N, S–C and N, S–C electrode interface. Reproduced with permission.62 Copyright 2021, Wiley. | ||
3.5 Improving initial coulombic efficiency (ICE)
The initial coulombic efficiency of sodium-ion batteries is a key performance indicator. To improve this critical metric, researchers have employed electrode materials containing C–X–M bonds to achieve higher sodium storage capacities and facilitate the full utilization of active materials in the reactions while minimizing side reactions.9,10 This strategy can significantly enhance the initial coulombic efficiency of sodium-ion batteries.62,77,84,100–102 Chou et al.62 synthesized a high-capacity sodium storage material with FeS2 nanoclusters embedded in N, S doped carbon matrix (FeS2/N, S–C), and found that the C–S–Fe/C–N–Fe bonds with a point-matrix distribution effectively promoted the decomposition of the NaSO3CF3 and diethylene glycol dimethyl ether electrolytes, thereby forming a homogeneous and ultrathin solid electrolyte interface (SEI) layer, which enabled a significant improvement in the ICE by 92.23%. When discharged to 0.3 V, the high resolution-TEM (HRTEM) pattern (Fig. 7c and d) indicates that FeS2/N, S–C containing C–S–Fe bonds formed a thinner SEI film than N, S–C. Analysing the XPS results (Fig. 7e and f) using different sputtering times during the discharge process to 0.3 V revealed that inorganic NaF was a dominant product in the SEI film composition formed by FeS2/N, S–C. This suggests that the electrode materials containing C–X–M bonds have an excellent promoting effect on the formation of stable SEI layers. Such ultrathin and inorganic-rich SEI could prevent continuous electrolyte decomposition and minimize the undesirable reaction, enabling high ICE. Furthermore, DFT calculations were performed for NaSO3CF3–FeS2/N, S–C and NaSO3CF3–N, S–C (Fig. 7g), respectively, indicating that the C–F bonds of the former exhibited lower dissociation energies compared with those of the latter. The C–F bond was more easily broken by the C–S–Fe bond and combined with Na+ to form inorganic NaF. In addition, the ultra-small size of the FeS2 nanoclusters and the bonded rivets of C–S–Fe prevented the accumulation of reaction by-products. Fig. 7h clearly displays that Fe–N–C/Fe–S–C bonds selectively promoted the breaking of C@F and C@O bonds in NaSO3CF3 and DGM. Consequently, the FeS2/N, S–C electrodes achieved an extremely high capacity (749.6 mA h g−1 at 0.1 A g−1) and excellent rate performance in sodium-ion storage. Ji et al.84 acquired NiSe2 microsphere composites with N-doped bilateral carbon layer confinement. NiSe2 microspheres were anchored to the carbon layer film, and Ni–O–C bonds were constructed with the carbon layer. An ultra-high ICE of 93% was obtained at 0.2 A g−1, which was attributed to the introduction of Ni–O–C chemical bridging bonds leading to more sodium ions participating in the reactions and preventing side reactions from occurring.4. Conclusions and outlook
This review has provided a comprehensive analysis of the pivotal role that interfacial chemical bridge bonds play in enhancing the performance of SIBs, particularly in composites involving transition metal compounds and carbon materials. These bonds significantly improve the structural stability, electronic conductivity, ion diffusion, charge transfer, and overall electrochemical performance of SIB electrodes. By forming robust covalent linkages, such as C–X–M bonds (where X represents elements like S, O, N, P, Se, and M represents metals), these chemical bridge bonds address critical issues of SIB electrodes such as volume expansion, poor conductivity, and interfacial instability, thereby optimising the long-term cycling stability, electrode reversibility and initial coulombic efficiency of the electrodes. Through the application of advanced characterisation techniques, this review has shed light on the mechanisms by which these bonds enhance the electrochemical properties of SIBs, providing valuable insights for developing next-generation high-performance batteries.From this perspective, the construction of chemical bridge bonds is an excellent strategy for improving the performance of SIB anodes, and continued exploration and optimization of these bonds provide a promising pathway for further advancing SIB technology. However, future research on chemical bridge bonds should not be limited to their performance-enhancing effects; instead, it focuses on the detailed quantification and characterization of these bonds, as well as a deeper investigation into their microscopic mechanisms for improving battery performance. This will enable a more comprehensive understanding of the dynamic behaviour of these bonds under various cycling conditions and facilitate the development of new bond-forming strategies to further enhance the electrochemical performance of SIBs.
The integration of more advanced in situ and operando characterization techniques is particularly necessary to achieve this goal. On the one hand, these techniques can provide real-time insights into the formation process of these bonds, which will help identify controllable design schemes at the microscopic level, providing more scientific guidance for the selection and design of more stable and durable electrode materials. On the other hand, they can also provide real-time insights into the evolution process of these bonds during their operation, which is crucial for further understanding the mechanisms by which chemical bridge bonds enhance battery performance. They may even provide new perspectives and controllable solutions for addressing the inherent issues of SIBs, such as sodium-ion diffusion and interfacial resistance, ultimately guiding the design of more efficient electrode materials.
Furthermore, to achieve the commercialization of SIBs on a large scale, the design and preparation of anode materials also require further research. The strategy of using carbon-based chemical bridge bonds can significantly enhance the stability and scalability of commercial SIB electrodes. However, more in-depth research is needed on cost-effective and environmentally sustainable synthesis methods, particularly considering the challenges posed by low-temperature and high-rate conditions in practical applications. By addressing these challenges, the strategic implementation of chemical bridge bonds can play a key role in realizing the full potential of SIBs as a leading solution for large-scale energy storage.
In conclusion, this review offers novel perspectives and insights into the mechanisms by which chemical bridge bonds enhance SIB performance, informing the development of next-generation high-performance batteries.
Data availability
No data was used for the research described in the article.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Jinliang Zhu thanks the support from National Natural Science Foundation of China (52462027) and Natural Science Foundation of Guangxi (2022GXNSFAA035463). Guoxiu Wang acknowledges the funding support from Australia Research Council Discovery Project (DP240102176).References
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. L. Notten, Adv. Energy Mater., 2018, 8, 1800079 CrossRef.
- B. Liu, L. Wang, Y. Zhu, H. Peng, C. Du, X. Yang, Q. Zhao, J. Hou and C. Cao, ACS Nano, 2022, 16, 12900–12909 CrossRef CAS.
- Z. Xu, J. Wang, Z. Guo, F. Xie, H. Liu, H. Yadegari, M. Tebyetekerwa, M. P. Ryan, Y.-S. Hu and M.-M. Titirici, Adv. Energy Mater., 2022, 12, 2200208 CrossRef CAS.
- C. Yang, S. Xin, L. Mai and Y. You, Adv. Energy Mater., 2021, 11, 2000974 CrossRef CAS.
- W. Zhang, J. Lu and Z. Guo, Mater. Today, 2021, 50, 400–417 CrossRef CAS.
- P. Lu, Y. Sun, H. Xiang, X. Liang and Y. Yu, Adv. Energy Mater., 2018, 8, 1702434 CrossRef.
- Y. Yuan, Z. Chen, H. Yu, X. Zhang, T. Liu, M. Xia, R. Zheng, M. Shui and J. Shu, Energy Storage Mater., 2020, 32, 65–90 CrossRef.
- L. Zhang, W. Wang, S. Lu and Y. Xiang, Adv. Energy Mater., 2021, 11, 2003640 CrossRef CAS.
- Z. Cao, J. Cui, D. Yu, Y. Wang, J. Liu, J. Zhang, J. Yan, Y. Zhang, S. Sun and Y. Wu, Adv. Funct. Mater., 2023, 33, 2306862 CrossRef CAS.
- J. Lin, S. Lu, Y. Zhang, L. Zeng, Y. Zhang and H. Fan, J. Colloid Interface Sci., 2023, 645, 654–662 CrossRef CAS PubMed.
- Y. Qiu, F. Fu, M. Hu, P. K. Shen and J. Zhu, Chem. Eng. J., 2023, 454, 140402 CrossRef CAS.
- W. Song, H. Zhao, J. Ye, M. Kang, S. Miao and Z. Li, ACS Appl. Mater. Interfaces, 2019, 11, 17416–17424 CrossRef CAS PubMed.
- H. Sun, J. Wang, W. Li, F. Yuan, Q. Wang, D. Zhang, B. Wang and Y. A. Wu, Electrochim. Acta, 2021, 388, 138628 CrossRef CAS.
- Y. Sun, Y. Yang, X.-L. Shi, G. Suo, H. Chen, X. Hou, S. Lu and Z.-G. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 28359–28368 CrossRef CAS PubMed.
- Q. Wang, Y. Ma, L. Liu, S. Yao, W. Wu, Z. Wang, P. Lv, J. Zheng, K. Yu, W. Wei and K. Ostrikov, Nanomaterials, 2020, 10, 782 CrossRef CAS PubMed.
- J. Zhang, Z. Meng, D. Yang, K. Song, L. Mi, Y. Zhai, X. Guan and W. Chen, J. Energy Chem., 2022, 68, 27–34 CrossRef CAS.
- Y. Gao and L. Zhao, Chem. Eng. J., 2022, 430, 132745 CrossRef CAS.
- Y. Li, R. Liu, C. Wang and Y. Zhou, Energy Fuels, 2021, 35, 9029–9037 CrossRef CAS.
- J. Wang, X. Yue, Z. Xie, A. Abudula and G. Guan, Energy Storage Mater., 2021, 41, 404–426 CrossRef.
- A. Y. Maulana, J. M. Song, C. M. Futalan and J. Kim, Chem. Eng. J., 2022, 434, 134727 CrossRef.
- D. Liu, Z. Shadike, R. Lin, K. Qian, H. Li, K. Li, S. Wang, Q. Yu, M. Liu, S. Ganapathy, X. Qin, Q.-H. Yang, M. Wagemaker, F. Kang, X.-Q. Yang and B. Li, Adv. Mater., 2019, 31, 1806620 CrossRef PubMed.
- J. R. Rodriguez, S. B. Aguirre and V. G. Pol, Electrochim. Acta, 2019, 319, 791–800 CrossRef CAS.
- Z. Jiang, C. Liu, L. Zhang, T. Wei, H. Jiang, J. Zhou, M. Shi, S. Liang, Z. Su and Z. Fan, J. Power Sources, 2019, 441, 227182 CrossRef CAS.
- N. Wei, J. Cai, R. Wang, M. Wang, W. Lv, H. Ci, J. Sun and Z. Liu, Nano Energy, 2019, 66, 104190 CrossRef CAS.
- Y. Zhang, Y. Huang, V. Srot, P. A. van Aken, J. Maier and Y. Yu, Nano-Micro Lett., 2020, 12, 165 CrossRef PubMed.
- Q. Wang, T. Meng, Y. Li, J. Yang, B. Huang, S. Ou, C. Meng, S. Zhang and Y. Tong, Energy Storage Mater., 2021, 39, 354–364 CrossRef.
- R. Wei, Y. Lu and Y. Xu, Sci. China:Chem., 2021, 64, 1826–1853 CrossRef CAS.
- Q. Zhao, K. Yu, S. Wang and C. Liang, J. Alloys Compd., 2022, 927, 167004 CrossRef CAS.
- Y. Li, Y. Xu, Z. Wang, Y. Bai, K. Zhang, R. Dong, Y. Gao, Q. Ni, F. Wu, Y. Liu and C. Wu, Adv. Energy Mater., 2018, 8, 1800927 CrossRef.
- Z. Song, G. Zhang, X. Deng, Y. Tian, X. Xiao, W. Deng, H. Hou, G. Zou and X. Ji, Adv. Funct. Mater., 2022, 32, 2205453 CrossRef CAS.
- P. Wei, J. Zhu, Y. Qiu, G. Wang, X. Xu, S. Ma, P. K. Shen, X.-L. Wu and Y. Yamauchi, Chem. Sci., 2021, 12, 15054–15060 RSC.
- H. Fu, Z.-j. Du, W. Zou, H.-q. Li and C. Zhang, Carbon, 2013, 65, 112–123 CrossRef CAS.
- D. Li, J. Hu, C. Wang, L. Guo and J. Zhou, J. Power Sources, 2023, 555, 232387 CrossRef CAS.
- C. Zheng, X. Xu, Q. Lin, Y. Chen, Z. Guo, B. Jian, N. Li, H. Zhang and W. Lv, Carbon, 2021, 176, 31–38 CrossRef CAS.
- G. Suo, J. Zhang, D. Li, Q. Yu, W. Wan, M. He, L. Feng, X. Hou, Y. Yang, X. Ye and L. Zhang, Chem. Eng. J., 2020, 388, 124396 CrossRef CAS.
- H. Wang, X. Song, M. Lv, S. Jin, J. Xu, X. Kong, X. Li, Z. Liu, X. Chang, W. Sun, J. Zheng and X. Li, Small, 2022, 18, 2104293 CrossRef CAS.
- X. Zhao, W. Cai, Y. Yang, X. Song, Z. Neale, H.-E. Wang, J. Sui and G. Cao, Nano Energy, 2018, 47, 224–234 CrossRef CAS.
- Y. Teng, H. Zhao, Z. Zhang, Z. Li, Q. Xia, Y. Zhang, L. Zhao, X. Du, Z. Du, P. Lv and K. Swierczek, ACS Nano, 2016, 10, 8526–8535 CrossRef CAS PubMed.
- L.-q. Yu, S.-X. Zhao, Q.-l. Wu, J.-W. Zhao and G.-d. Wei, Adv. Funct. Mater., 2020, 30, 2000427 CrossRef CAS.
- J. Zhu, P. Wei, Q. Zeng, G. Wang, K. Wu, S. Ma, P. K. Shen and X.-L. Wu, Small, 2020, 16, 2003001 CrossRef CAS PubMed.
- M. Chen, S.-L. Chou and S.-X. Dou, Batteries Supercaps, 2019, 2, 842–851 CrossRef.
- C. Lu, R. Fang and X. Chen, Adv. Mater., 2020, 32, 1906548 CrossRef CAS.
- Y. Dai, X. Liao, R. Yu, J. Li, J. Li, S. Tan, P. He, Q. An, Q. Wei, L. Chen, X. Hong, K. Zhao, Y. Ren, J. Wu, Y. Zhao and L. Mai, Adv. Mater., 2021, 33, 2100359 CrossRef CAS PubMed.
- L. Fan, Y. Zhang, H. Zhou, Z. Guo, Y. Feng and N. Zhang, J. Mater. Sci. Technol., 2021, 88, 250–257 CrossRef CAS.
- Y. Li, L. Wang, W. Ma, B. Yang, R. Wei and Y. Yao, Int. J. Hydrogen Energy, 2021, 46, 34184–34193 CrossRef CAS.
- J. Qiu, C. Lai, Y. Wang, S. Li and S. Zhang, Chem. Eng. J., 2014, 256, 247–254 CrossRef CAS.
- I. Elizabeth, B. P. Singh, S. Trikha and S. Gopukumar, J. Power Sources, 2016, 329, 412–421 CrossRef CAS.
- C. Li, M. Shen and B. Hu, Acta Phys.-Chim. Sin., 2020, 36, 1902019 Search PubMed.
- S. Bao, T. Hou, Q. Tan, X. Kong, H. Cao, M. He, G. Wu and B. Xu, Mater. Chem. Phys., 2020, 245, 122766 CrossRef CAS.
- C. Zhao, R. Fan, T. He, J. Ma, S. Fang, D. Jin, J. Gao and Y. Cai, J. Alloys Compd., 2022, 910, 164875 CrossRef CAS.
- Y. Deng, S. Dong, Z. Li, H. Jiang, X. Zhang and X. Ji, Small Methods, 2018, 2, 1700332 CrossRef.
- Z. Shadike, E. Zhao, Y.-N. Zhou, X. Yu, Y. Yang, E. Hu, S. Bak, L. Gu and X.-Q. Yang, Adv. Energy Mater., 2018, 8, 1702588 CrossRef.
- Q. Zeng, S. Tian, G. Liu, H. Yang, X. Sun, D. Wang, J. Huang, D. Yan and S. Peng, ACS Appl. Mater. Interfaces, 2022, 14, 6958–6966 CrossRef CAS PubMed.
- I. Kallquist, R. Le Ruyet, H. Liu, R. Mogensen, M.-T. Lee, K. Edstrom and A. J. Naylor, J. Mater. Chem. A, 2022, 10, 19466–19505 RSC.
- M. Jing, Z. Chen, Z. Li, F. Li, M. Chen, M. Zhou, B. He, L. Chen, Z. Hou and X. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 704–712 CrossRef CAS PubMed.
- R. R. Jones, D. C. Hooper, L. Zhang, D. Wolverson and V. K. Valev, Nanoscale Res. Lett., 2019, 14, 231 CrossRef PubMed.
- Y. Wang and D. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 23033–23055 CrossRef CAS PubMed.
- S. Jiang, M. Xiang, J. Zhang, S. Chu, A. Marcelli, W. Chu, D. Wu, B. Qian, S. Tao and L. Song, Nanoscale, 2020, 12, 22210–22216 RSC.
- Q. Song, S. Zhou, S. Wang, S. Li, L. Xu and J. Qiu, Chem. Eng. J., 2023, 461, 142033 CrossRef CAS.
- W. Sun, W. Zhao, S. Yuan, L. Zhang, Y. Yang, P. Ge and X. Ji, Adv. Funct. Mater., 2021, 31, 2100156 CrossRef CAS.
- X. Song, X. Li, H. Shan, J. Wang, W. Li, K. Xu, K. Zhang, H. M. K. Sari, L. Lei, W. Xiao, J. Qin, C. Xie and X. Sun, Adv. Funct. Mater., 2024, 34, 2303211 CrossRef CAS.
- Y. Wan, K. Song, W. Chen, C. Qin, X. Zhang, J. Zhang, H. Dai, Z. Hu, P. Yan, C. Liu, S. Sun, S.-L. Chou and C. Shen, Angew. Chem., Int. Ed., 2021, 60, 11481–11486 CrossRef CAS PubMed.
- G. Li, H. Chen, B. Zhang, H. Guo, S. Chen, X. Chang, X. Wu, J. Zheng and X. Li, Appl. Surf. Sci., 2022, 582, 152404 CrossRef CAS.
- Z.-Z. Wang, J. Qu, S.-M. Hao, Y.-J. Zhang, F.-Q. Kong, D. Yang and Z.-Z. Yu, Chemelectrochem, 2018, 5, 2653–2659 CrossRef CAS.
- K. Zhu, S. Wei, Q. Zhou, S. Chen, Y. Lin, P. Zhang, Y. Cao, C. Wang, Y. Wang, Y. Xia, D. Cao, Z. Mohamed, X. Guo, X. Yang, X. Wu and L. Song, Nano Res., 2023, 16, 2421–2427 CrossRef CAS.
- S. Chong, X. Wei, Y. Wu, L. Sun, C. Shu, Q. Lu, Y. Hu, G. Cao and W. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 13158–13169 CrossRef CAS PubMed.
- P. Gao, H. Shi, T. Ma, S. Liang, Y. Xia, Z. Xu, S. Wang, C. Min and L. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 51028–51038 CrossRef CAS PubMed.
- T. Guo, H. Liao, P. Ge, Y. Zhang, Y. Tian, W. Hong, Z. Shi, C. Shao, H. Hou and X. Ji, Mater. Chem. Phys., 2018, 216, 58–63 CrossRef CAS.
- F. Wang, Z. Liu, H. Feng, Y. Wang, C. Zhang, Z. Quan, L. Xue, Z. Wang, S. Feng, C. Ye, J. Tan and J. Liu, Small, 2023, 19, 2302200 CrossRef CAS PubMed.
- H. Zhang, B. Liu, Z. Lu, J. Hu, J. Xie, A. Hao and Y. Cao, Small, 2023, 19, 2207214 CrossRef CAS PubMed.
- Y. Cheng, J. Huang, H. Qi, L. Cao, J. Yang, Q. Xi, X. Luo, K. Yanagisawa and J. Li, Small, 2017, 13, 1700656 CrossRef PubMed.
- X. Xie, Z. Yin, Y. Li, R. Tu, Y. Liu, H. Tong, X. Ma, Z. Ding and L. Zhou, J. Mater. Chem. A, 2022, 10, 3732–3742 RSC.
- T. Wu, M. Jing, Y. Liu and X. Ji, J. Mater. Chem. A, 2019, 7, 6439–6449 RSC.
- X. Xu, F. Xu, X. Zhang, C. Qu, J. Zhang, Y. Qiu, R. Zhuang and H. Wang, Nano-Micro Lett., 2022, 14, 91 CrossRef CAS PubMed.
- B. Huang, S. Liu, X. Zhao, Y. Li, J. Yang, Q. Chen, S. Xiao, W. Zhang, H.-E. Wang and G. Cao, Sci. China Mater., 2021, 64, 85–95 CrossRef CAS.
- C. Li, K. Pfeifer, X. Luo, G. Melinte, J. Wang, Z. Zhang, Y. Zhang, P. Dong, A. Sarapulova, H. Ehrenberg and S. Dsoke, Chemsuschem, 2023, 16, e202202281 CrossRef CAS PubMed.
- Y. Xiang, L. Xu, L. Yang, Y. Ye, Z. Ge, J. Wu, W. Deng, G. Zou, H. Hou and X. Ji, Nano-Micro Lett., 2022, 14, 136 CrossRef CAS PubMed.
- S. Xiao, Z. Li, J. Liu, Y. Song, T. Li, Y. Xiang, J. S. Chen and Q. Yan, Small, 2020, 16, 2002486 CrossRef CAS PubMed.
- J. Feng, S.-H. Luo, Y. Zhan, S.-X. Yan, P.-W. Li, L. Zhang, Q. Wang, Y.-H. Zhang and X. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 51095–51106 CrossRef CAS PubMed.
- A. Xu, C. Huang, G. Li, K. Zou, H. Sun, L. Fu, J. Ju, Y. Song, S. Wu, Z. Xu and Y. Yan, J. Mater. Chem. A, 2021, 9, 12169–12178 RSC.
- S. Chong, M. Ma, L. Yuan, S. Qiao, S. Dong, H. Liu and S. Dou, Energy Environ. Mater., 2023, 6, e12458 CrossRef CAS.
- B. Lan, X. Zhang, L. Zhao, P. Wang, C. Wei, Y. Wang and G. Wen, Electrochim. Acta, 2023, 442, 141910 CrossRef CAS.
- F. Fu, M. Hu, J. Key, P. K. Shen and J. Zhu, Carbon, 2023, 215, 118469 CrossRef CAS.
- P. Ge, S. Li, L. Xu, K. Zou, X. Gao, X. Cao, G. Zou, H. Hou and X. Ji, Adv. Energy Mater., 2019, 9, 1803035 CrossRef.
- M. Guo, H. Zhang, L. Qi, S. Zhang, Y. Qin and B. Deng, Nanotechnology, 2023, 34, 505703 CrossRef CAS PubMed.
- M. Jing, F. Li, M. Chen, J. Zhang, F. Long, L. Jing, X. Lv, X. Ji and T. Wu, J. Alloys Compd., 2018, 762, 473–479 CrossRef CAS.
- L. Chen, M. Wang, E. Li, X. Hu, S. Huang, Z. Yang, D. Zhou, H. Xie, J. Chen, Z. Ma, B. Guo, B. Yu and X. Li, Inorg. Chem. Front., 2023, 10, 4087–4101 RSC.
- M. Han, Z. Lin and J. Yu, J. Mater. Chem. A, 2019, 7, 4804–4812 RSC.
- P. Ge, H. Hou, C. E. Banks, C. W. Foster, S. Li, Y. Zhang, J. He, C. Zhang and X. Ji, Energy Storage Mater., 2018, 12, 310–323 CrossRef.
- M. Ihsan-Ul-Haq, H. Huang, J. Wu, N. Mubarak, A. Susca, Z. Luo, B. Huang and J.-K. Kim, Carbon, 2021, 171, 119–129 CrossRef CAS.
- W. Zhao, S. Yuan, S. Lei, Z. Zeng, J. Dong, F. Jiang, Y. Yang, W. Sun, X. Ji and P. Ge, Adv. Funct. Mater., 2023, 33, 2211542 CrossRef CAS.
- W. Chen, K. Song, L. Mi, X. Feng, J. Zhang, S. Cui and C. Liu, J. Mater. Chem. A, 2017, 5, 10027–10038 RSC.
- Z. Zhang, H. Zhao, J. Fang, X. Chang, Z. Li and L. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 28533–28540 CrossRef CAS PubMed.
- S. Liang, X. Wang, R. Qi, Y.-J. Cheng, Y. Xia, P. Mueller-Buschbaum and X. Hu, Adv. Funct. Mater., 2022, 32, 2201675 CrossRef CAS.
- P. Huang, H. Ying, S. Zhang, Z. Zhang and W.-Q. Han, Chem. Eng. J., 2022, 429, 132396 CrossRef CAS.
- S. Chong, L. Yuan, S. Qiao, M. Ma, T. Li, X. L. Huang, Q. Zhou, Y. Wang and W. Huang, Sci. China Mater., 2023, 66, 2641–2651 CrossRef CAS.
- M. Jing, F. Li, M. Chen, F. Long and T. Wu, Appl. Surf. Sci., 2019, 463, 986–993 CrossRef CAS.
- Q. Li, Z. Li, Z. Zhang, C. Li, J. Ma, C. Wang, X. Ge, S. Dong and L. Yin, Adv. Energy Mater., 2016, 6, 1600376 CrossRef.
- S. Yuan, J. Li, W. Xia, Z. Li, W. Tian, Y. Yang, X. Ji and P. Ge, Adv. Funct. Mater., 2024, 34, 2409295 CrossRef CAS.
- D. Yu, K. Guo, F. Hou, Y. Zhang, X. Ye, Y. Zhang, P. Ji, U. Khalilov, G. Wang, X. Zhang, K. Wang, Y. Song, X. Zhong, H. Sun, J. Zhu, J. Liang and H. Wang, Small, 2024, 20, 2312167 CrossRef CAS PubMed.
- M. Tang, X. Yin, Y. Cao, H. Ma, X. Zhang and D. Jia, Small, 2024, 20, 2406547 CrossRef CAS PubMed.
- D. Cheng, L. Lei, L. Li, A. Wei, C. Ouyang, L. Tan, Q. Huang and Y. Chen, Adv. Funct. Mater., 2024, 34, 2417253 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |