
Molten salt-assisted synthesis of a nitrogen-doped biochar catalyst at low temperature for enhanced degradation of acetaminophen†
Gongyong
Feng
a,
Wenhao
Li
a,
Haibo
Ye
a,
Teng
Wang
ac,
Yang
Liu
ac,
Binghua
Jing
c,
Chunyang
Nie
*b,
Didi
Li
*a and
Zhimin
Ao
 *c
*c
aGuangdong Key Laboratory of Environmental Catalysis and Health Risk Control, Guangzhou Key Laboratory Environmental Catalysis and Pollution Control, Institute of Environmental Health and Pollution Control, School of Environmental Science and Engineering, Guangdong University of Technology, Guangzhou 510006, China. E-mail: ddli@gdut.edu.cn
bResearch Institute of Interdisciplinary Sciences (RISE) and School of Materials Science & Engineering, Dongguan University of Technology, Dongguan 523808, China. E-mail: niechunyang@dgut.edu.cn
cAdvanced Interdisciplinary Institute of Environment and Ecology, Beijing Normal University, Zhuhai 519087, China. E-mail: zhimin.ao@bnu.edu.cn
First published on 3rd December 2024
Abstract
Nitrogen (N) doping is an efficient modification route to improve the catalytic performance of biochar in peroxymonosulfate (PMS) activation. However, conventional synthetic methods for high-performance N-doped biochar catalysts often require high temperatures (>700 °C), which are energy intensive. To address this issue, a molten salt-assisted method was employed to synthesize a N-doped biochar catalyst from alkaline lignin at a relatively low temperature of 500 °C (named AL@NX). This catalyst was subsequently utilized for the catalytic oxidation of acetaminophen (APAP) through PMS activation. Results demonstrated that the molten salt treatment improved the carbonization of lignin, specific surface area and pyridine N-doping level in the biochar. Notably, pyridine N was identified as the primary active site in AL@NX for PMS activation. Accordingly, AL@NX exhibited a reaction rate constant (Kobs) for APAP oxidation that was 8 times higher than that observed for N-doped biochar synthesized without the use of molten salt. The results of electron paramagnetic resonance (EPR) spectroscopy, quenching experiments and electrochemical measurements revealed that the AL@NX/PMS system operates through an O2˙−-dominated dual activation pathway (radical-based and nonradical). The unique oxidative properties of O2˙− in the AL@NX/PMS system impart broad pH adaptability, high selectivity for APAP, and high degradation performance in real water bodies. These findings highlight the promising application prospects of the AL@NX/PMS system for water remediation applications. Furthermore, this work proposes a green, energy-efficient approach to the resource utilization of biomass waste, contributing to the achievement of carbon neutrality goal.
1. Introduction
Advanced oxidation processes (AOPs) utilizing peroxymonosulfate (PMS) have been widely applied in decontaminating aqueous refractory organic pollutants.1 PMS contains an asymmetric peroxide bond, making it easily susceptible to activation by various external stimuli, leading to the generation of diverse reactive oxygen species (ROS). The activation methods for PMS mainly include thermal, photochemical, electrochemical, ultrasonic and chemical catalyst approaches.2–4 Among these, chemical catalysts have been popularly utilized to activate PMS due to their ease of operation, high efficiency and low energy requirements.5 Transition metal- and carbon-based materials are the two mainstream types of catalysts for PMS activation, each with its own merits and demerits.6 Carbon-based materials, in particular, manifest good catalytic activity, excellent acid/alkali resistance, tunable electronic properties and environmental friendliness. However, their stability can be a limiting factor.7–11 Many kinds of carbonaceous materials, such as carbon nanotubes (CNTs), graphene (Gr), ordered mesoporous carbon (OMC) and nanodiamonds (NDs), have been applied in PMS-based AOPs for the degradation of organic pollutants.12In recent years, biochar has emerged as a promising alternative to well-designed nanocarbons (such as CNTs, Gr and NDs) as a carbon catalyst due to its potential for preparation from the ubiquitous biomass sources on Earth. Particularly, the conversion of biomass waste into biochar offers an effective pathway for carbon fixation and reducing carbon dioxide emissions, which is highly beneficial for achieving carbon neutrality goals.13 However, the catalytic activity of pristine biochar for PMS activation is generally limited. To overcome this limitation, surface engineering strategies are usually adopted to enhance the reactivity of biochar and improve its catalytic performance, breaking its inherent chemical inertness.14 Various types of surface engineering, including physical methods (e.g., grinding and ultrasonic treatment), biological methods (e.g., leveraging microbial metabolic activity) and chemical methods (e.g., heteroatoms doping and oxygenation) have been attempted for biochar.15–17 Among them, nitrogen (N) doping is a very popular approach due to its easy operation and maneuverable doping level.18–20 With the unique electronic configuration of biochar, the introduction of nitrogen atoms can alter the degree of charge delocalization, thus reducing the inertness of sp2-hybridized carbon, leading to a higher catalytic activity.21 Many N-doped biochar catalysts have been reported to exhibit excellent performance in activating PMS to degrade pollutants.22 For instance, Cai et al. synthesized BDK-900 material by utilizing a waste bean dreg precursor, and the as-prepared BDK-900 showed good catalytic activity towards PMS activation for bisphenol A degradation.23 The N-doped biochar derived from waste elm bark attained a rapid degradation of high concentrations of tetracycline.18 In our previous study, an N-doped alkaline lignin biochar catalyst (MX@N-800) was prepared, and it can rapidly eliminate APAP within 15 min by activating PMS.24
Despite these achievements, the syntheses of high-performance N-doped biochar catalysts usually require high pyrolysis temperatures (more than 700 °C), while those prepared at low pyrolysis temperatures demonstrate unsatisfactory catalytic performance. In this regard, the resource utilization of biomass waste for water remediation is relatively energy intensive, which is disadvantageous to the achievement of a low-carbon economy in the wastewater treatment industry. Therefore, it is imperative to develop simple synthetic methods at low temperatures to obtain highly efficient N-doped biochar catalysts for persulfate activation. Unfortunately, relevant studies have been infrequently found in the literature.
Molten salt-assisted methods have been widely employed for the synthesis of inorganic materials because molten salt can offer a strong polarizing force to destabilize solid chemical bonds in the precursors and thus initiate synthetic reactions.25 Meanwhile, because the diffusivity of species that have low solubility in common solvents (water and organic solvents) might be promoted in the ‘liquid’ molten salt solvent, the reaction temperature can be reduced, and reaction dynamics can be accelerated.26 Furthermore, molten salt can be easily washed with water and recycled after synthesis, rendering it an economical and environmentally benign auxiliary pyrolysis method.27 Many studies have reported the fabrication of carbonaceous materials with tunable microporosity and surface structure via molten salt-assisted pyrolysis methods.28,29 In addition, it was found that the pyrolysis of biomass in molten salt media can prevent nitrogen loss and promote graphitization.30,31 On these grounds, the utilization of molten salt can be a promising route for achieving low-temperature synthesis of high-performance N-doped biochar catalysts.
Herein, we used alkaline lignin (AL) as the biomass source and mixed NaCl/KCl salt to prepare an N-doped biochar catalyst (AL@NX) through a straightforward pyrolysis process at 500 °C. To understand the influence of molten salt on N-doped biochar, the extrinsic (e.g., morphology, textural parameters, etc.) and intrinsic (e.g. graphitization degree, doping level, contents of different N formats, etc.) properties of AL@NX and several AL-derived biochar counterparts were examined. The performances of the above biochar and previously reported MX@N-800 as catalysts to activate PMS for eliminating APAP were systematically compared, and AL@NX was demonstrated to be the best performing catalyst. Then, the effects of several operational parameters, including dosages of catalyst and oxidant, and initial pH on APAP removal by AL@NX/PMS system, were investigated. A series of scavenging experiments, electron paramagnetic resonance (EPR) analysis and electrochemical measurements were carried out to examine the activation pathway. Thereafter, the main active sites in AL@NX were identified by comparing the variation in contents of different N formats in fresh and repeatedly used catalyst samples. Finally, the stability and efficiency of the AL@NX/PMS system for removing APAP in the actual water body were assessed. The results demonstrated the good application prospects of AL@NX in practical water remediation.
2. Materials and methods
2.1 Chemicals
PMS was purchased from Alfa Aesar. Alkaline lignin and humic acid were acquired from Shanghai McLean Biochemical Co., Ltd. APAP, methanol (MeOH), tert-butanol (TBA), potassium dichromate (K2Cr2O7), urea, p-benzoquinone (PBQ), β-carotene, sodium chloride (NaCl), potassium chloride (KCl), sodium dihydrogen phosphate (Na2H2PO4), heavy water (D2O), 2,2,6,6-tetramethyl-4-piperidinol (TEMP, 98.0%), and 5,5-dimethyl-1-pyrroline N-oxide (DMPO, 97.0%) were from Aladdin Reagent Inc. (Shanghai, China). All chemicals utilized in this study were of analytical grade, and the solutions were prepared using 18.2 MΩ cm ultrapure water. Lake water samples were collected from natural water bodies in Guangzhou.2.2 Synthesis and characterizations of AL@NX
To synthesize the biochar catalyst, AL underwent a molten salt-assisted low-temperature pyrolysis process. Initially, 5 g AL was subjected to heating at 400 °C for 2 h in an argon atmosphere with a heating rate of 5 °C min−1, producing a powdery precursor named AL-400. Subsequently, AL-400 was mixed with 3 g urea, 5 g NaCl, and 5 g KCl, and the mixture was pyrolyzed at 500 °C for 4 h in an argon atmosphere at a heating rate of 1 °C min−1. Upon completion of the heating process, the resultant black powder was washed several times with ultrapure water to remove the residual salts and then dried at 60 °C for 6 h to obtain AL@NX. For comparison, AL@X, AL@N, and AL@BC were fabricated under the same conditions using precursors of AL-400 + NaCl + KCl, AL-400 + urea, and AL-400, respectively. The characterization methods of the as-prepared catalysts are shown in ESI (Text S1).†2.3 Removal experiments
All the pollutant removal experiments were conducted in a 150 mL conical flask under magnetic stirring at room temperature. First, a 5 mg catalyst was uniformly dispersed into 50 mL of APAP solution with an initial concentration of 50 mg L−1. Then, 1 mM PMS was added to initiate the degradation reaction. 0.5 mL of the reaction solution was sampled at the designed times and then filtered through a polytetrafluoroethylene filter with a size of 0.22 μm. The obtained filtrate was then loaded into a high-performance liquid chromatography (HPLC) vial. Unless otherwise specified, the pH of the solution was not adjusted. For the quenching experiments, corresponding quenching agents were added to the reactor before adding the catalyst and PMS. All experiments were conducted in triplicate, and the results are presented as mean values accompanied by their corresponding standard deviations.2.4 Analysis method
APAP concentration was monitored using the HPLC system (Elite EClassical3100). Types of reactive species were identified by EPR spectroscopy (Text S2†) with DMPO and TEMP employed as spin-trapping agents. Details of the electrochemical analysis can be found in Text S3.† The removal efficiency of APAP was calculated using eqn (1), and the corresponding reaction rate constant (Kobs, min−1) was calculated using a pseudo-first-order kinetic model (eqn (2)):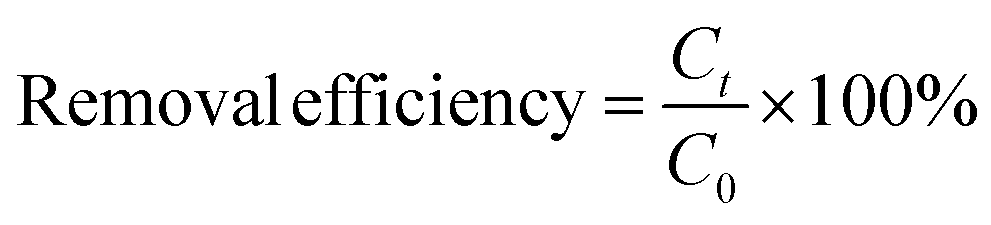 | (1) |
 | (2) |
3. Results and discussion
3.1 Characterizations
The morphologies of the biochar catalysts were observed using scanning electron microscopy (SEM). As shown in Fig. 1a, AL@BC comprises large and thick flakes containing macropores. After treatment with molten salt, smaller flakes appear in the AL@X compared to AL@BC (Fig. 1b), possibly because the decomposition of alkaline lignin was accelerated by molten salt during pyrolysis, causing the lignin structure to break down into fragments.32,33 AL@N comprises fragments with sizes ranging from micrometers to several hundreds of nanometers (Fig. 1c), suggesting that N-doping with urea can influence the morphology of AL@BC. Compared to AL@BC, AL@X and AL@N, the dimension of flakes in AL@NX is significantly reduced, and more pores are present in AL@NX (Fig. 1d). This should be ascribed to the combined action of molten salt and N-doping. | ||
| Fig. 1 SEM images of (a) AL@BC, (b) AL@X, (c) AL@N and (d) AL@NX. | ||
Fig. 2a displays the N2 adsorption–desorption isotherms of four as-prepared biochar samples. AL@BC, AL@X and AL@N possess typical type III isotherms characterized by a weak hysteresis loop that corresponds to type H3/H4. In contrast, AL@NX exhibits a typical type I isotherm with an H4 loop, indicating the main presence of micropores.34 The pore size distribution curve in Fig. S1† further supports the microporous feature of AL@NX. Table 1 lists the pore structure parameters of the four catalysts. Apparently, AL@X and AL @N possess higher specific surface area (SBET) than Al@BC, indicating that molten salt and N-doping are both useful ways to improve the porosity of AL@BC. Surprisingly, AL@NX exhibits a much higher SBET (295.174 m2 g−1) compared to the other three samples. This suggests that the synergistic effect exerted by molten salt and N-doping can significantly promote the formation of micropores in biochar. The large SBET of AL@NX is beneficial to the adsorption of organic pollutants and exposure of active sites, thereby enhancing its catalytic performance.35
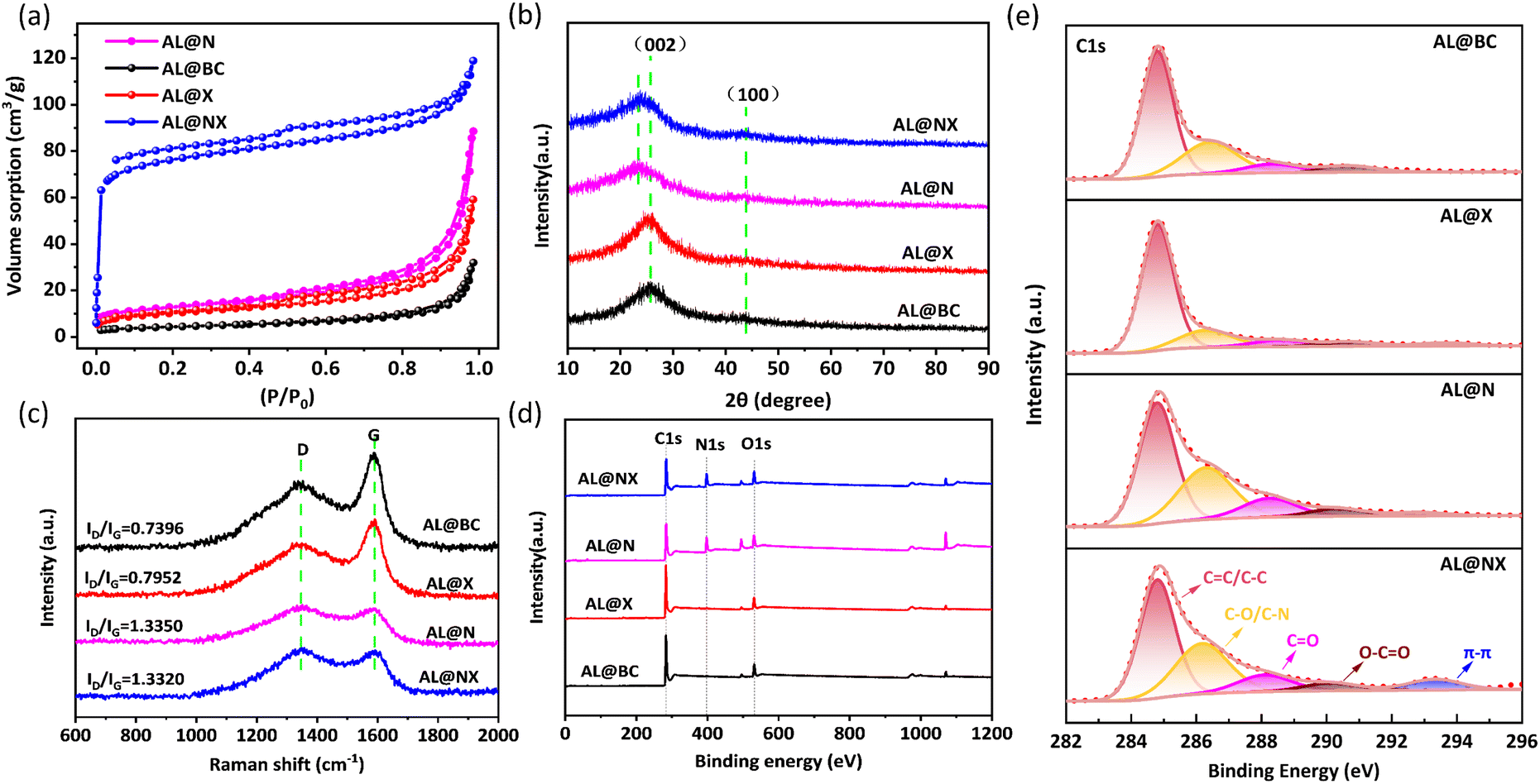 | ||
| Fig. 2 (a) N2 adsorption isotherms, (b) XRD patterns, (c) Raman spectra, (d) XPS full spectra and (e) C1s fine spectra of as-prepared biochar samples. | ||
| Samples | AL@BC | AL@X | AL@N | AL@NX |
|---|---|---|---|---|
| S BET (m2 g−1) | 15.487 | 36.573 | 45.048 | 295.174 |
| Average pore size (nm) | 12.771 | 10.011 | 12.164 | 2.493 |
| Pore volume (cm3 g−1) | 0.0494 | 0.0915 | 0.1369 | 0.1839 |
Further investigations into the structures of the prepared biochar samples were conducted using X-ray diffraction (XRD) and Raman spectroscopy analyses. As depicted in Fig. 2b, two characteristic peaks located at approximately 2θ of ∼25° and ∼44° are observed in the XRD patterns of AL@BC and AL@X, corresponding to the (002) and (100) planes of graphite, respectively.36 In addition, the acquired broad and weak diffraction peaks suggest the low graphitization degree of the as-prepared biochar samples. Compared with AL@BC and AL@X, the (002) diffraction peak of AL@N and AL@NX slightly shifts to the left, which may be attributed to the reason that N-doping expands the interlayer distance of the material, leading to a decrease in the diffraction angle.37 Notably, the absence of characteristic peaks of NaCl and KCl crystals indicates the successful removal of salts during the washing process. In the Raman spectra of four biochar samples (Fig. 2c), representative D and G bands of carbonaceous materials are present, signifying the disorder caused by vacancies or impurity doping and the E2g stretching vibration associated with the sp2-hybridized graphite structure, respectively.38 The ID/IG ratio generally serves as an index of defective levels of carbon materials.39 Our results show that the ID/IG ratio of AL@X is slightly higher than that of AL@BC, while the ID/IG ratios for AL@N and AL@NX are quite similar, both of which are much higher with regard to AL@BC. These phenomena imply that N-doping greatly increases the defective degree of carbon materials because the introduction of N atoms can expose more defect sites and edge planes.40
High-resolution X-ray photoelectron spectroscopy (XPS) was performed to further explore the chemical element compositions and proportions of functional groups on the surface of the materials. As shown in Fig. 2d, the surfaces of AL@BC and AL@X consist predominantly of carbon (C) and oxygen (O), while distinctive N peaks were detected at the surface of AL@N and AL@NX, revealing the successful incorporation of N atoms into the biochar structure. Meanwhile, the deconvolution of the C1s peak was conducted to understand the proportions of functionalities in biochar (Fig. 2e). As observed, the C1s band showed five peaks located at 284.8, 285.96, 287.29, 288.95, and 293.35 eV, assignable to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C, C–O/C–N, CO, O–CO, and π–π* shake up, respectively.35Table 2 depicts the surface elemental compositions and proportions of the functionalities of the as-prepared biochar samples. The elemental composition of AL@X resembles AL@BC, while the proportions of functional groups in the two samples are quite different. In comparison to AL@BC, the content of CC is higher, while the contents of oxygenated groups are lower in AL@X, indicating that molten salt treatment can promote the carbonization of lignin and reduce the formation of oxygenated groups.36 Upon N-doping, the contents of the O atom and CO group in AL@N are increased. The N-doping level in AL@NX is similar to that in AL@N, but the contents of the C atom and CC in the former are higher than those in the latter. This may also be attributed to the rearrangement of surface functional groups within the carbon material in the molten salt environment.21
C/C–C, C–O/C–N, CO, O–CO, and π–π* shake up, respectively.35Table 2 depicts the surface elemental compositions and proportions of the functionalities of the as-prepared biochar samples. The elemental composition of AL@X resembles AL@BC, while the proportions of functional groups in the two samples are quite different. In comparison to AL@BC, the content of CC is higher, while the contents of oxygenated groups are lower in AL@X, indicating that molten salt treatment can promote the carbonization of lignin and reduce the formation of oxygenated groups.36 Upon N-doping, the contents of the O atom and CO group in AL@N are increased. The N-doping level in AL@NX is similar to that in AL@N, but the contents of the C atom and CC in the former are higher than those in the latter. This may also be attributed to the rearrangement of surface functional groups within the carbon material in the molten salt environment.21
| Compositions (at%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| C1s | N1s | O1s | CC/C–C |
C–O/C–N | CO |
O–CO |
π–π* | |
| AL@NX | 73.42 | 16.27 | 10.31 | 50.19 | 25.13 | 10.68 | 10.08 | 3.92 |
| AL@N | 70.87 | 17.91 | 11.22 | 39.82 | 38.55 | 15.07 | 5.71 | 0.85 |
| AL@X | 90.19 | — | 9.81 | 69.65 | 12.93 | 6.60 | 9.62 | 1.19 |
| AL@BC | 89.39 | — | 10.61 | 60.05 | 17.66 | 12.29 | 6.97 | 3.03 |
Based on the above results, we proposed a plausible mechanism for the molten salt-assisted synthesis of AL@NX. Under a pyrolysis temperature of 500 °C, the eutectic mixture of NaCl/KCl melts, and the molten salts possess strong polarity and high thermal conductivity. Consequently, the covalent bonds in AL-500 became unstable via solvent interactions, and the precursor was efficiently carbonized into a porous carbon framework. Meanwhile, the urea was readily decomposed into NH3 and CO2 at a low pyrolysis temperature due to the presence of molten salt. Then, the gaseous NH3 is dissolved into the molten salt solvent and reacts with the carbon framework, producing an N-doped carbon material with a high doping level. The produced CO2 can also activate the carbon material to produce more pores.41 Additionally, the alkaline metal ions in the NaCl/KCl melt can be inserted into the carbon framework during the pyrolysis process, and they can be removed by the washing step, leaving micropores in the carbon material. However, the molten salt can also affect the formation of N species in carbon material and thus affect the catalytic activity towards PMS activation (see the following sections).
3.2 Catalytic performances of biochar catalysts
The APAP removal efficiency achieved by the as-prepared biochar catalysts by activating the PMS was evaluated. PMS alone is almost unable to oxidize APAP.24 Fig. S2† describes the APAP adsorption behaviors of biochar catalysts. As observed, all samples could only adsorb less than 5% APAP in 40 min, indicating their poor adsorption capacity for APAP. Therefore, the adsorption process was not investigated in subsequent organic removal experiments. Fig. 3a displays the APAP degradation performances of various biochar/PMS systems. The degradation efficiency attained by AL@BC/PMS and AL@X/PMS systems is less than 10%, suggesting the chemical inertness of AL-BC and AL@X towards PMS activation. In contrast, AL@N and AL@NX demonstrate good catalytic activity towards PMS activation because the degradation efficiency achieved by AL@N/PMS and AL@NX/PMS systems within 30 min is around 98% and 100%, respectively. These results show that the doped N species in biochar are critical active sites in biochar for PMS activation. In addition, the corresponding degradation rate constant Kobs of AL@NX/PMS system (0.672) is about 8 times higher than that of AL@N/PMS system (0.087) (Fig. 3b). The superior catalytic activity of AL@NX to AL-N should arise from the difference in bond configuration of N atoms because the catalytic activity of different C–N functionalities towards PMS activation is disparate,16 which is discussed later. | ||
| Fig. 3 (a) APAP removal performances and (b) the corresponding pseudo-first-order kinetics for APAP degradation in different biochar/PMS systems; effect of (c) PMS concentration, (d) catalyst dosage, (e) pH and (f) stirring speed on APAP removal by AL@NX/PMS system. Conditions: [catalyst]0 = 0.1 g L−1, [APAP]0 = 50 mg L−1, [PMS]0 = 1 mM, temperature = 25 °C. | ||
It is noteworthy that the Kobs for APAP oxidation over the AL@NX/PMS system is also higher than that for APAP oxidation over the previously reported MX@N-800/PMS system (0.5032) under the same experimental conditions.24 Meanwhile, the APAP removal performance of AL@NX is better than other documented catalysts (Table S1†), including carbonaceous materials (CM), CoFe layered double oxide/g-C3N4 composite (CoFe LDO/g-C3N4) and Enteromorpha-derived Fe–N-doped carbon matrix (Fe–N–C).42–44 The above results demonstrate that the utilization of molten salt not only reduces the synthesis temperature of N-doped biochar catalysts but also enhances catalytic performance, which is highly desirable for achieving a low-carbon economy in the coming era.
Fig. S3† displays the catalytic performance of AL@NX in cyclic APAP removal experiments. As observed, the APAP degradation rate in the 2nd cycle significantly declined compared to the 1st cycle, indicating that the catalytic activity of AL@NX-2nd is lower than that of fresh biochar. The declined catalytic activity of AL@NX can be caused by several reasons, such as the loss of active sites, adsorption of residual degradation intermediates on its surface, and minor structural changes.45 Hence, we first examined the morphology of AL@NX-2nd via SEM. As shown in Fig. S4,† the morphology of AL@NX-2nd is similar to that of fresh AL@NX. A good porosity was still observed, indicating no significant structural collapse of the biochar catalyst during the PMS activation process. The N2 adsorption–desorption isotherm of AL@NX-2nd was also acquired (Fig. S5†). Obviously, AL@NX-2nd shows the same isotherm type as the fresh sample, confirming that the porous structure of biochar is stable. The SBET of AL@NX-2nd (165 m2 g−1) decreases possibly due to the coverage of intermediates onto the biochar catalyst. Therefore, we presumed that the declined catalytic activity of AL@NX should be attributed to the adsorption of residual intermediates and loss of active sites (which is discussed in detail in Section 3.6).
3.3 Operation parameter optimization for AL-NX/PMS system
To enhance the APAP removal efficiency of the AL@NX/PMS system, we investigated the effects of catalyst and oxidant dosages, as well as the initial pH. As shown in Fig. 3c, the APAP removal efficiency increased from 63.5% to 100% as the PMS concentration increased from 0.25 to 0.5 mM while maintaining a fixed catalyst concentration of 0.1 g L−1. Further increasing the PMS concentration to 1 or 2 mM accelerated the APAP oxidation rate to a certain degree, while the attained oxidation rates under the two conditions are very close. These results indicate that AL@NX shows high utilization efficiency of PMS for eliminating APAP.Fig. 3d displays the influence of catalyst dosage on APAP removal by applying AL@NX/PMS system under a fixed PMS concentration of 0.5 mM. As observed, the complete removal of APAP within 30 min was attained with the catalyst dosage in the range of 0.05–0.2 g L−1, while a higher catalyst dosage can promote the organic oxidation rate due to the affordance of more adsorption and catalytic sites. The excellent removal performance of AL@NX under a low catalyst dosage of 0.05 g L−1 is beneficial to the reduction of catalyst cost in practical water treatment.
The impact of initial pH on the removal performance of the AL@NX/PMS system was investigated in Fig. 3e. As observed, the AL@NX/PMS system exhibited excellent APAP degradation performance across the pH range of 3–10, revealing the wide adaptability of biochar catalysts to varying pH levels.
The APAP removal performance under different stirring speeds is illustrated in Fig. 3f. APAP was totally degraded under stirring speeds ranging from 250 to 750 rpm, while the oxidation rate was remarkably accelerated when the stirring speed was increased from 250 to 500 rpm. This is possible because increasing the stirring speed can promote the mass transfer between oxidants and catalysts, as well as organics and as-generated reactive species, thus accelerating the reaction rate. However, a further increase in speed to 700 rpm only slightly improved the oxidation rate. This suggests that a moderate stirring speed is sufficient to facilitate the APAP removal by the AL@NX/PMS system.
We also examined the degradation performance of the AL@NX/PMS system on various contaminants, including p-nitrophenol, sulfamethoxazole (SMZ), bisphenol A (BPA) and phenol. As shown in Fig. S6,† the removal efficiencies of SMZ, phenol, BPA and p-nitrophenol are 95.5%, 61.5%, 53.0%, and 1.0%, respectively. Notably, the selectivity exhibited by the AL@NX/PMS system towards organic oxidation is different from the selectivity of SO4˙−, ˙OH, 1O2 and nonradical carbon–persulfate complexes.46–48 Meanwhile, the selectivity of the AL@NX/PMS system differs from that of the previously reported MX@N-800/PMS system whose primary reactive species is an activated state of electron-deficient N-doped biochar.24,49 Accordingly, the AL@NX/PMS system should be dominated by other types of reactive species, such as O2˙−, which is demonstrated in the subsequent section.
3.4 Identification of main reactive species in the AL@NX/PMS system
The activation of PMS by carbonaceous materials can generate various types of ROS, such as SO4˙−, ˙OH, and 1O2.50 To discern the types of ROS produced in the AL@NX/PMS system, we first conducted quenching experiments. TBA, β-carotene, and PBQ are selected as quenchers for ˙OH, 1O2, and O2˙−, respectively, while MeOH is used as a quencher for ˙OH and SO4˙−.51Fig. 4a depicts that the addition of 500 mM TBA or MeOH did not significantly inhibit the APAP degradation rate in the AL@NX/PMS system, while complete APAP removal was still achieved within 10 min. The addition of β-carotene exerted a similar effect to MeOH on APAP removal in the AL@NX/PMS system. These results reveal that SO4˙−, ˙OH and 1O2 may be produced from PMS activation over AL@NX but minimally contribute to APAP oxidation. K2Cr2O7 is reported to be able to eliminate the presence of homogeneous electron transfer in the solution.52 Our study shows that adding K2Cr2O7 into the AL@NX/PMS system had a minor influence on APAP removal, suggesting the absence of homogeneous electron transfer within the solution. However, the addition of PBQ significantly suppressed APAP removal in the AL@NX/PMS system, and the inhibition level increased with the PBQ concentration (Fig. 4b). This phenomenon is opposite to that observed for MX@N-800 in our previous study wherein the addition of p-BQ barely suppressed the removal of APAP in the MX@N-800/PMS system.24 Hence, O2˙− is deduced to be the main ROS produced from PMS activation over AL@NX and responsible for APAP oxidation. | ||
| Fig. 4 (a and b) Effects of various quenchers on the APAP degradation in the AL@NX/PMS system. EPR spectra of AL@NX/PMS system using (c) DMPO and (d) TEMP as trapping agents. | ||
To further confirm the types of generated ROS in the AL@NX/PMS system, we conducted an EPR analysis using DMPO and TEMP as spin traps to detect the presence of SO4˙−, ˙OH, O2˙−, and 1O2.53 As depicted in Fig. 4c, no signals were detected for the PMS alone solution, while a distinct seven-line signal (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1:2:1:2:1) attributed to DMPOX was observed for the AL@NX/PMS system. The detection of DMPOX can be attributed to the oxidation of DMPO by the strong oxidizing species generated by the AL@NX-activated PMS.54Fig. 4d displays that a weak triplet signal (1:1:1) resembling 2,2,6,6-tetramethylpiperidine oxide (TEMPO)55 was detected for the PMS alone solution, and the intensity of the TEMPO signal was significantly enhanced for the AL@NX/PMS system. These seemingly suggest the production of 1O2 in the AL@NX/PMS system, contradicting the results of quenching experiments. It has been reported in the literature that TEMP can also be oxidized by other strong oxidizing species besides 1O2 to produce a TEMPO signal and the EPR results alone cannot reliably prove the presence of 1O2.45 Therefore, we further conducted APAP degradation experiments in D2O solution to verify whether 1O2 was present in the AL@NX/PMS system based on the fact that the lifetime of 1O2 in D2O is approximately 18-fold longer compared to that in regular water.56 As shown in Fig. S7,† no difference in degradation performance between the two solvent systems was observed, indicating the absence of 1O2 in the AL@NX/PMS system. We also compared the EPR signal of TEMPO in D2O solution with that in ultrapure water. Fig. 5a illustrates that the intensity of the TEMPO signal in D2O is even lower than that in ultrapure water, further ruling out the involvement of 1O2 in the AL@NX/PMS system.
:2:1:2:1:2:1) attributed to DMPOX was observed for the AL@NX/PMS system. The detection of DMPOX can be attributed to the oxidation of DMPO by the strong oxidizing species generated by the AL@NX-activated PMS.54Fig. 4d displays that a weak triplet signal (1:1:1) resembling 2,2,6,6-tetramethylpiperidine oxide (TEMPO)55 was detected for the PMS alone solution, and the intensity of the TEMPO signal was significantly enhanced for the AL@NX/PMS system. These seemingly suggest the production of 1O2 in the AL@NX/PMS system, contradicting the results of quenching experiments. It has been reported in the literature that TEMP can also be oxidized by other strong oxidizing species besides 1O2 to produce a TEMPO signal and the EPR results alone cannot reliably prove the presence of 1O2.45 Therefore, we further conducted APAP degradation experiments in D2O solution to verify whether 1O2 was present in the AL@NX/PMS system based on the fact that the lifetime of 1O2 in D2O is approximately 18-fold longer compared to that in regular water.56 As shown in Fig. S7,† no difference in degradation performance between the two solvent systems was observed, indicating the absence of 1O2 in the AL@NX/PMS system. We also compared the EPR signal of TEMPO in D2O solution with that in ultrapure water. Fig. 5a illustrates that the intensity of the TEMPO signal in D2O is even lower than that in ultrapure water, further ruling out the involvement of 1O2 in the AL@NX/PMS system.
 | ||
| Fig. 5 (a) EPR spectra of AL@NX/PMS system in D2O and regular water using TEMP as the trapping agent. (b) EPR spectra of AL@NX/PMS system in methanol solution using DMPO as the trapping agent. (c) LSV curves. (d) Current response. (I–t curves obtained at 0.0 V vs. Ag/AgCl). | ||
The generation of O2˙− from PMS activation over AL@NX was also examined using DMPO as the spin agent and methanol as the solvent. As shown in Fig. 5b, no signals were observed in the PMS alone solution, while characteristic EPR signals corresponding to DMPO-O2˙− adducts were detected when AL@NX was added to the PMS solution, and signal intensity increased with reaction time. In particular, the introduction of APAP into the AL@NX/PMS system caused a noticeable decrease in the intensity of DMPO-O2˙− adducts, resulting from the consumption of O2˙− by APAP. These phenomena consolidated the production of O2˙− and its important contribution to organic oxidation in the AL@NX/PMS system.
It has been reported that the dissolved oxygen in water can be transformed into O2˙−via interacting with catalysts.46 To determine whether dissolved oxygen contributed to the generation of O2˙− in the AL@NX/PMS system, the solution was purged with N2 to eliminate dissolved oxygen, and the efficiency of APAP degradation was assessed. As demonstrated in Fig. S8,† the absence of O2 has minimal impact on the removal efficiency of APAP in AL@NX/PMS system. This indicates that the O2˙− in AL@NX/PMS system is primarily derived from the activation of PMS rather than dissolved oxygen. In addition, the involvement of photoirradiation in PMS activation over AL@NX was ruled out. As shown in Fig. S9,† the APAP degradation performance of the AL@NX system under the dark environment quite resembles that under the irradiation of ambient light, indicating the non-involvement of photoirradiation.
Because the addition of a high concentration of PBQ did not completely suppress the degradation of APAP in the AL@NX/PMS system, we speculated that the nonradical pathway via the electron-transfer mechanism, which is frequently found in various types of carbon/persulfate systems, also contributed to APAP oxidation in the AL@NX/PMS system.57 The LSV curves in Fig. 5c show that the current density just enhanced slightly with the addition of PMS to the AL@NX alone system, while a more pronounced increase in current density was witnessed when AL@NX, PMS and APAP co-existed. This can be explained by the fact that the presence of a catalyst mediated the electron transfer between PMS and the organic pollutant, inducing an increased electric current.58 The results of chronoamperometry analysis further support the electron-transfer mechanism. As illustrated in Fig. 5d, when PMS was introduced to the electrolyte, the intensity of current was suddenly increased but then rapidly decreased to a value close to zero, suggesting a small amount of electron transfer between AL@NX and PMS. However, subsequent injection of APAP into the electrolyte caused a noticeable current increase, and the current intensity slowly declined, indicating a considerable amount of electron transport among AL@NX, PMS and APAP. To conclude, the above measurements demonstrated the occurrence of an electron-transfer mechanism in the APAP degradation processes over AL@NX and PMS systems.
3.5 Exploration of the main active sites in AL@NX for PMS activation
For N-doped carbonaceous materials, several types of N functionalities can be formed when N atoms are bonded with C atoms, including pyridine N, pyrrole N and graphite N, and they demonstrate distinct catalytic activity towards PMS activation. For instance, Ren et al. proposed that graphite N was the main active site in N-doped carbon nanotubes for PMS activation.59 Sun and co-workers reported that pyridine N and graphite N in N-doped sludge biochar played dominant roles in PMS activation.60 To determine the dominant catalytic sites in AL@NX towards PMS, the contents of different N formats in fresh and used biochar catalysts were investigated by XPS spectroscopy (Fig. 6a–c).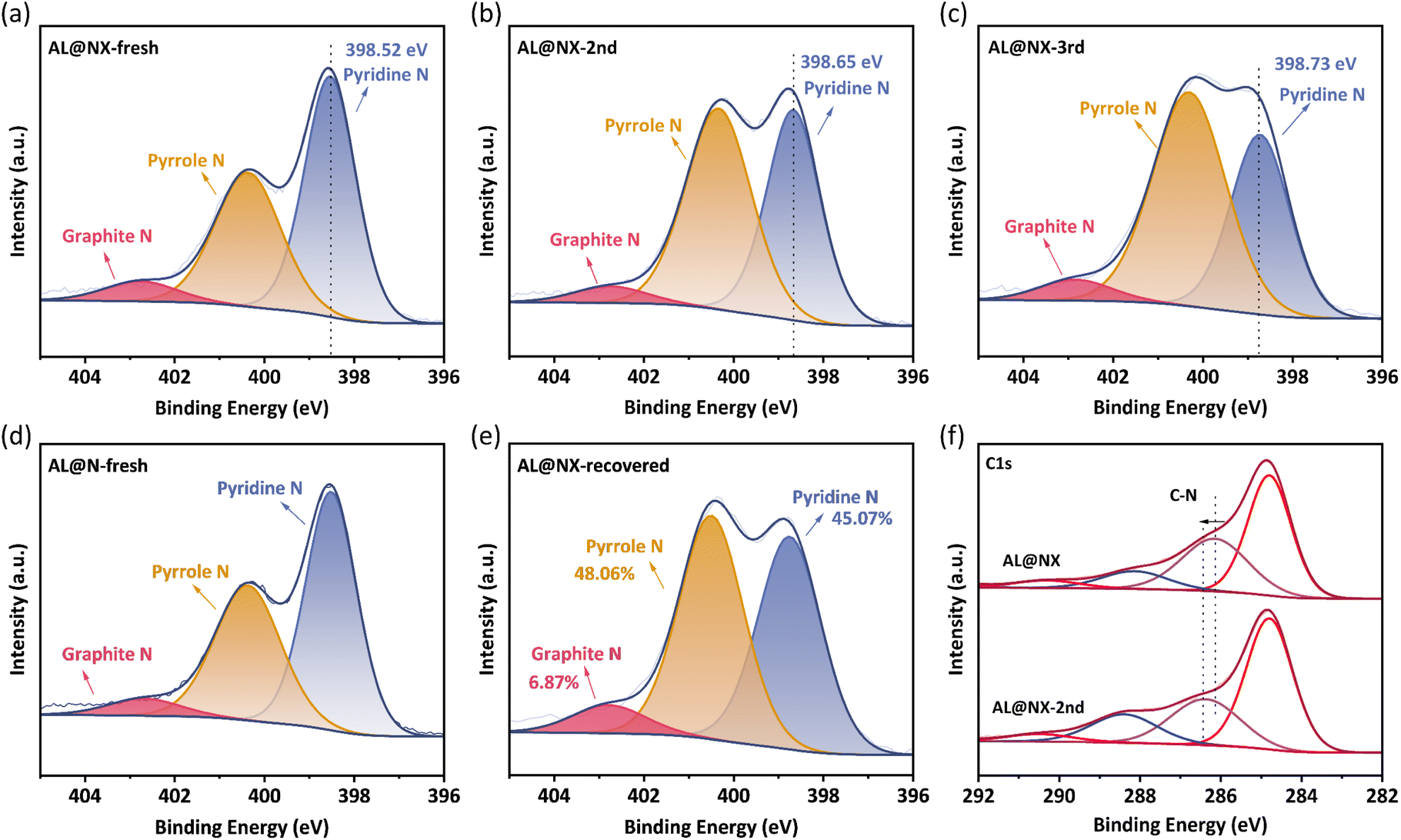 | ||
| Fig. 6 Deconvolution of N1s spectra of (a) AL@NX, (b) AL@NX-2nd, (c) AL@NX-3rd, (d) AL@N, and (e) AL@NX-recovered. (f) C1s spectra of AL@NX before and after catalytic reaction. | ||
The N1s peak profile can be deconvoluted into three components located at 398.64 ± 0.1 eV, 400.31 ± 0.1 eV, and 402.63 ± 0.1 eV, assignable to pyridine N, pyrrole N and graphite N, respectively.61 Detailed information on biochar samples is listed in Table S2.† The N species in fresh AL@NX are dominated by pyridine N (52.92%) and pyrrole N (41.52%). By comparison, the proportion of pyridine N in the AL@NX-2nd (used once) and AL@NX-3rd (used twice) samples decrease and the contents of pyrrole and graphite N increase. In addition, the peak of pyridine N in AL@NX shifts toward higher binding energy, while the pyrrole N and graphite N peaks remained basically unchanged in relation to AL@NX-2nd. Meanwhile, the binding energy of C–N species in the C 1s XPS spectrum of AL@NX was upshifted compared to AL@NX-2nd (Fig. 6f). These phenomena signify remarkable charge transfer between pyridine N and the adjacent C atoms in AL@NX and PMS during the activation process. Fig. S3† displays the catalytic performance of AL@NX in cyclic APAP removal experiments. The APAP degradation rate in the 2nd cycle significantly declined compared to the 1st cycle, indicating that the catalytic activity of AL@NX-2nd is lower than that of fresh biochar. By combining the results of XPS analysis with cyclic experiments, we deduce that pyridine N is the main active site in AL@NX for PMS activation. For AL@NX-2nd and AL@NX-3rd samples, their contents of pyridine N are lower than a fresh sample but still maintained at a relatively high level (41.04% and 33.77%), which explains the phenomenon that a high APAP removal efficiency of 90% was attained during the 2nd and 3rd runs.
Previous studies have shown that passivated carbon catalysts can recover their catalytic activity through secondary pyrolysis because the residual intermediates can be removed via volatilization and the exposure of active sites is improved.45 In this work, a thermal annealing treatment on AL@NX-2nd was carried out to regenerate the catalytic activity (see Text S4† for details). It was observed that biochar catalytic activity could be remarkably recovered after the thermal annealing process (Fig. S3†). Meanwhile, the surface chemistry of recovered AL@NX was analyzed by XPS (Fig. 6e). The elemental composition of the recovered sample is very similar to that of the fresh sample. Additionally, the relative proportion of pyridine N in recovered AL@NX (45.07%) is measured to be higher than that of AL@NX-2nd. The SBET of recovered AL@NX was measured to be 244 m2 g−1 (Fig. S5†). These results confirm the efficiency of secondary pyrolysis in regenerating biochar catalysts.
The N1s configuration of AL@N was also investigated for comparison (Fig. 6d). As listed in Table S2,† the proportions of pyridine and graphite N in AL@N are lower than those in AL@NX, indicating that treatment with molten salt can also affect the bonding configuration of N atoms in the carbon matrix. Due to the lower content of pyridine N and SBET, a smaller amount of exposed pyridine N active sites is present on the surface of AL@N relative to AL@NX, accounting for the lower catalytic activity of AL@N towards PMS activation.
3.6 Reaction mechanisms and degradation pathway of APAP in AL@NX/PMS system
Based on the aforementioned results, the catalytic mechanism for PMS activation over AL@NX is illustrated in Fig. 7. When N atoms are incorporated into the carbon frameworks, the electronic structure of biochar changes. Due to the larger atomic number of N than C, the doped N atoms deliver a higher electron density than the neighbouring C atoms regardless of the types of N species, giving rise to the formation of electron-deficient C atoms in N-doped carbon materials. For AL@NX with a high doping level of pyridine N, abundant electron-deficient C atoms adjacent to pyridine N are present, while the electron-deficient C atoms have been reported to be efficient for PMS activation.62 When PMS is adsorbed onto the electron-deficient C atoms, fast charge transfer occurs between the PMS molecule and carbon catalyst, and PMS then loses electrons to the electron-deficient carbon atoms (denoted as C+ in the equations) to form O2˙−viaeqn (3) and (4).45,60 Interestingly, Gao et al. proposed that the electron-deficient C atoms adjacent to graphite N atoms are the main catalytic sites in N-doped nanosheets to activate PMS into 1O2,63 while Wang et al. suggested that the electron-deficient C atoms in N vacancy modified g-C3N4 are responsible for the generation of O2˙− from PMS activation.64 Therefore, we speculated that the type of N-doping configuration and electrical property of the carbon catalyst significantly impact the activation pathway of PMS and perhaps the transformation of O2˙− into 1O2. Apart from doped N atoms, a certain amount of carbonyl group (CO) also exists on the surface of AL@NX. The interactions between CO and PMS can evolve metastable carbon-PMS complexes to partially oxidize APAP.65 Owing to the occurrence of the abovementioned reactions, a dual radical-based and nonradical pathway is involved in the AL@NX/PMS system, while O2˙− serves as the primary ROS.| HSO5− → H+ + SO52− | (3) |
| SO52− + H2O + C+ → O2˙− + 2H+ + SO42− | (4) |
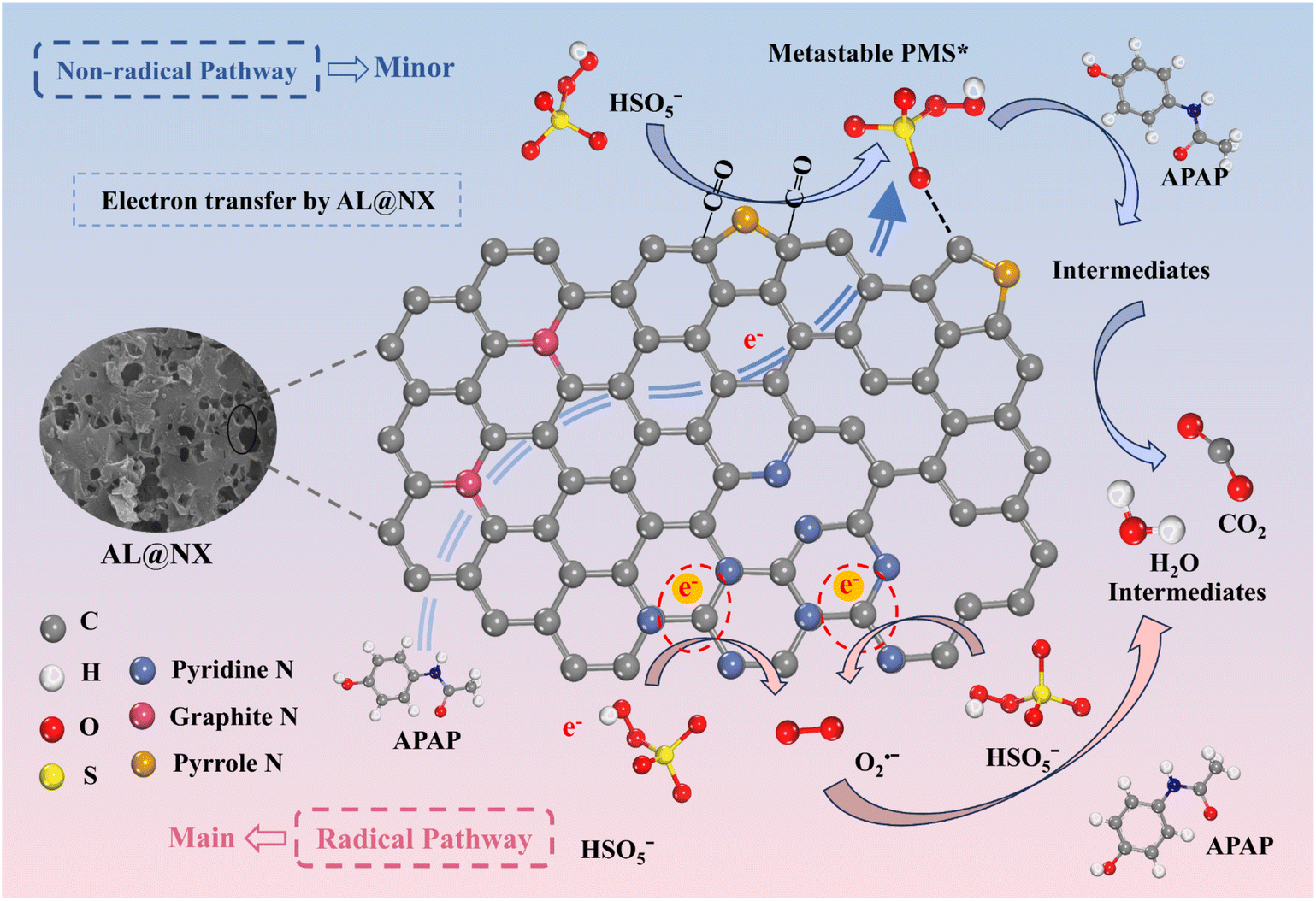 | ||
| Fig. 7 Proposed mechanism of APAP degradation in AL@NX/PMS system. | ||
As we mentioned earlier, the PMS activation pathway over AL@NX is different from that over MX@N-800, which should be related to the distinct physicochemical properties of the two biochar catalysts. Compared to AL@NX, MX@N-800 possesses a much higher SBET, higher graphitization degree and a lower amount of nitrogen doping. The nitrogen species in MX@N-800 are also dominated by pyridine N and pyrrolic N, while the percentage of pyrrolic N is higher than that of pyridine N. Because the electron density delivered by pyridine and pyrrolic N and the charge transport properties of graphitic carbon and amorphous carbon are disparate, different PMS activation pathways are induced. To fully understand the influences of various N-doping configurations and graphitization degree on the carbon-catalyzed PMS activation process, further in-depth studies combining theoretical calculations and experimental investigations are necessary.
The APAP degradation intermediates in the AL@NX/PMS/APAP system were analysed by liquid chromatography-mass spectrometry (LC-MS). Nine potential products were identified. Their detailed information is presented in Table S3.† Based on the oxidation route involving O2˙− and electron transfer, three possible degradation pathways were proposed (Fig. 8). In pathways 1 and 2, O2˙− initially attacks the benzene ring of APAP or substitutes the acetyl group connected to nitrogen, forming 4-aminophenol (m/z: 109.05) or N-(3,4-dihydroxyphenyl) formamide (m/z: 167.06).66 Subsequently, 4-aminophenol is converted to 4-aminocatechol (m/z: 125.05) via an –OH addition mechanism or is further oxidized by radicals to form benzene-1,2,4-triol (m/z 126.03) and short-chain organic acids.67 Meanwhile, N-(3,4-dihydroxyphenyl) formamide undergoes further oxidation through different pathways to form short-chain intermediates and acetamide (m/z: 59.04).68 For pathway 3, under the attack of O2˙−, the negatively charged phenoxyl ion in APAP loses an electron to form a phenoxyl radical. The unpaired electron in the phenoxyl radical delocalizes through resonance onto the conjugated positions of the benzene ring and is subsequently involved in a coupling reaction to form an acetaminophen dimer (m/z: 300.11).69 Notably, most of the intermediates in pathways 1 and 2 can be oxidized into low-molecular-weight ion fragments and further mineralized to CO2, H2O, and NH4+, while the dimer intermediates in pathway 3 are more resistant to further mineralization. It is important to point out that all intermediates involved in this proposal, except p-diphenol, are observed in the mass spectrum, even at low intensities, and most of them were also found in previous studies.
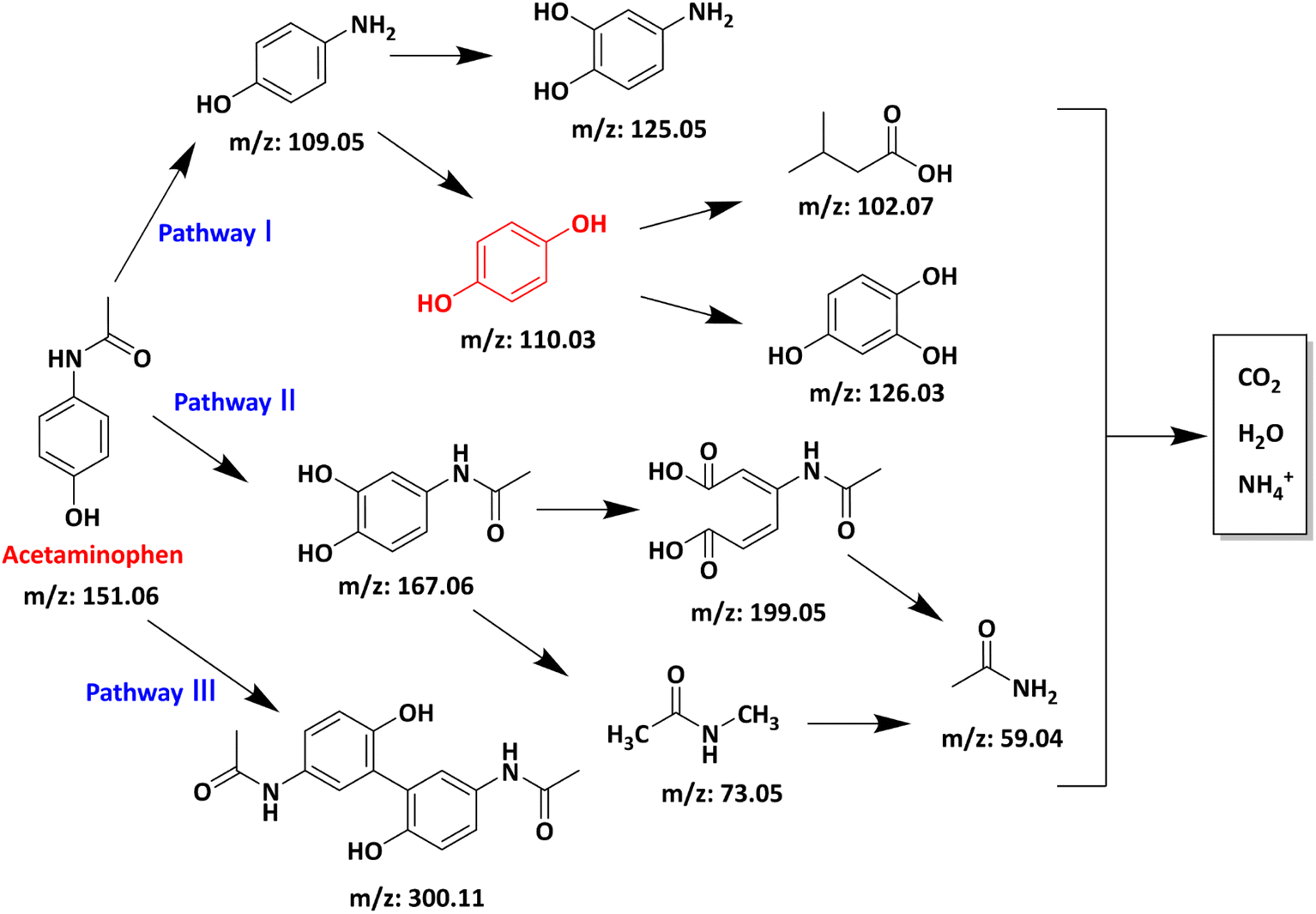 | ||
| Fig. 8 Proposed degradation pathway of APAP in AL@NX/PMS system. | ||
3.7 Performance of AL@NX/PMS system in actual water bodies
The compositions of the actual water body are quite complicated and diverse types of anions and cations, and natural organic matters (NOMs) are ubiquitously present. These substances may interfere with the reactions between catalysts and PMS differently, thus causing distinct impacts on the elimination of organic pollutants. In this study, we investigated the impacts of several types of anions involving Cl−, H2PO42−, and HCO3− and humic acid (HA) on APAP removal by applying the AL@NX/PMS system. As shown in Fig. 9a, the addition of 10 mM Cl−/H2PO42− marginally affected the APAP degradation, indicating the good tolerance of the AL@NX/PMS system to the two anions. Conversely, the presence of 10 mM HCO3− markedly hindered APAP degradation because the removal efficiency diminished to 70%. The inhibitory effect exerted by HCO3− may be ascribed to competitive reactions between HCO3− and AL@NX with PMS, leading to greater consumption of PMS. However, the activation efficiency of PMS by HCO3− is lower than that of AL@NX, thus reducing the degradation efficiency of APAP. A similar phenomenon has also been observed for other kinds of biochar/PMS systems.70–74Fig. 9b illustrates the influences of different concentrations of HA on APAP removal by the AL@NX/PMS system. Obviously, HA exerts a negative effect on APAP degradation with a concentration in the range of 1–10 mg L−1, and the inhibitory extent increases with the HA concentration. Such phenomena are likely to result from the surface covering of AL@NX by oxygen-containing groups in HA, which masks the active sites.75 Meanwhile, HA can compete with APAP to react with ROS, impeding the APAP oxidation process.76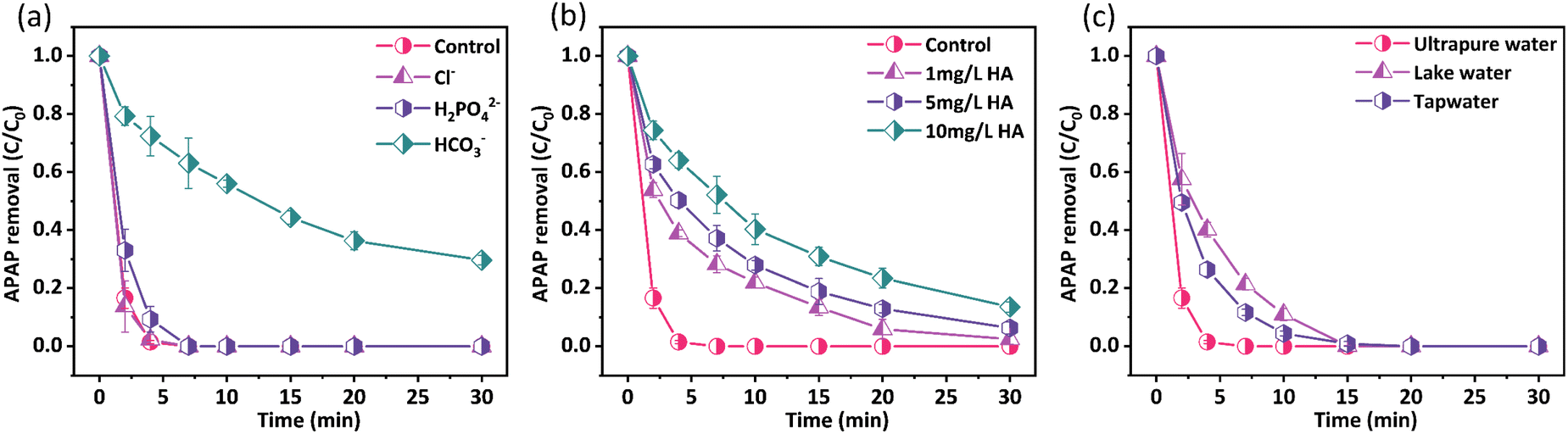 | ||
| Fig. 9 (a) Effects of inorganic anions, (b) HA concentration, (c) different water bodies on removal of APAP by AL@NX/PMS system. Conditions: [catalyst]0 = 0.1 g L−1, [APAP]0 = 50 mg L−1, [PMS]0 = 1 mM, temperature = 25 °C. | ||
To further evaluate the potential of AL@NX in actual water treatment, APAP degradation experiments in tap water and natural lake water were conducted. Fig. 9c depicts that the APAP oxidation rate in tap water and lake water was marginally reduced compared to ultrapure water, which should be due to the presence of HCO3− and HA in the actual water (detailed water quality information of lake water is presented in Table S4†), deteriorating APAP oxidation by reactive species.77 Despite the decreased oxidation rate, complete removal of APAP from both tap water and lake water was achieved within 15 min, indicating the promising potential of the AL@NX/PMS system in practical water remediation.
4. Conclusions
In this work, we prepared a high-performance N-doped biochar catalyst derived from AL for PMS activation using a simple molten NaCl/KCl salt-assisted low-temperature pyrolysis method. AL@NX demonstrated higher catalytic activity than AL@N and N-doped alkaline lignin biochar prepared at a high pyrolysis temperature of 800 °C in our previous work. O2˙− and electron-transfer mechanisms co-exist in the AL@NX/PMS system, while O2˙− made a larger contribution to APAP oxidation. Pyridine N is identified as the main active site in AL@NX, and O2˙− is assumed to evolve from the interaction between PMS and electron-deficient C atoms adjacent to pyridine N. AL@NX/PMS system is demonstrated to show good stability and high efficiency towards APAP removal in actual water bodies, indicating its promising application prospects in water remediation. Overall, we offer an energy-efficient pathway for the utilization of biomass resources in the field of water decontamination, which is helpful in achieving a low-carbon economy in the wastewater treatment industry.Data availability
The data are available from the corresponding author on reasonable request.Author contributions
Gongyong Feng: data curation, investigation, methodology, formal analysis, validation, writing-original draft. Wenhao Li: methodology, formal analysis, writing-reviewing, and editing. Haibo Ye: formal analysis, writing-reviewing, and editing. Teng Wang: methodology, writing-reviewing, and editing. Yang Liu: writing-reviewing, and editing. Binghua Jing: methodology, formal analysis. Didi Li: writing-reviewing, and editing. Chunyang Nie: methodology, formal analysis, writing-reviewing, and editing. Zhimin Ao: conceptualization, supervision, funding acquisition, resources, writing-reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 22176041, 22406011, 22166025, and T2421005), National Key R&D Program of China (no. 2022YFC3901800), Jiangxi Natural Science Foundation (grant no. 20232BAB203037), Guangzhou Science and Technology Planning Project (No. 2023A04J0918), and the “Fundamental Research Funds for the Central Universities”.Notes and references
- C. Zhou, Y. Liang, W. Xia, E. Almatrafi, B. Song, Z. Wang, Y. Zeng, Y. Yang, Y. Shang, C. Wang and G. Zeng, J. Hazard. Mater., 2023, 441, 129871 CrossRef CAS PubMed.
- Q. Du, C. Zhu, C. Yue, F. Cun, Z. Du, F. Liu and A. Li, Appl. Catal., B, 2024, 343, 123570 CrossRef CAS.
- H. Hou, L. Fang, L. Liu, B. Zhao, D. Wu, Z. Ma, B. Hu, X. Wang and S. Yu, J. Cleaner Prod., 2023, 426, 139150 CrossRef CAS.
- X. Xue, N. Xue, D. Ouyang, L. Yang, Y. Wang, H. Zhu, A. Aihemaiti and J. Yin, ACS Appl. Mater. Interfaces, 2023, 15, 57003–57014 CAS.
- Y. Liu, L. Liu and Y. Wang, Environ. Sci. Technol., 2021, 55, 9691–9710 CrossRef CAS PubMed.
- J. Lee, U. von Gunten and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
- W. D. Oh, Z. L. Dong and T. T. Lim, Appl. Catal., B, 2016, 194, 169–201 CrossRef CAS.
- H. Wang, Z. Zeng, P. Xu, L. Li, G. Zeng, R. Xiao, Z. Tang, D. Huang, L. Tang, C. Lai, D. Jiang, Y. Liu, H. Yi, L. Qin, S. Ye, X. Ren and W. Tang, Chem. Soc. Rev., 2019, 48, 488–516 RSC.
- B. Song, M. Chen, S. Ye, P. Xu, G. Zeng, J. Gong, J. Li, P. Zhang and W. Cao, Carbon, 2019, 144, 1–7 CrossRef CAS.
- P. Duan, Y. Qi, S. Feng, X. Peng, W. Wang, Y. Yue, Y. Shang, Y. Li, B. Gao and X. Xu, Appl. Catal., B, 2020, 267, 118717 CrossRef CAS.
- W.-D. Oh, G. Lisak, R. D. Webster, Y.-N. Liang, A. Veksha, A. Giannis, J. G. S. Moo, J.-W. Lim and T.-T. Lim, Appl. Catal., B, 2018, 233, 120–129 CrossRef CAS.
- W. Huang, S. Xiao, H. Zhong, M. Yan and X. Yang, Chem. Eng. J., 2021, 418, 129297 CrossRef CAS.
- B. Song, E. Almatrafi, X. Tan, S. Luo, W. Xiong, C. Zhou, M. Qin, Y. Liu, M. Cheng, G. Zeng and J. Gong, Renewable Sustainable Energy Rev., 2022, 164, 112529 CrossRef CAS.
- L. Chen, X. Jiang, S. Ma, W. Chen, B. Xu, Z. Dai, W. Jiang, Y. Peng and J. Li, Chem. Eng. J., 2023, 475, 146115 CrossRef CAS.
- W. Zhang, H. Cheng, Q. Niu, M. Fu, H. Huang and D. Ye, Environ. Sci. Technol., 2019, 53, 7632–7640 CrossRef CAS PubMed.
- S. Ye, G. Zeng, X. Tan, H. Wu, J. Liang, B. Song, N. Tang, P. Zhang, Y. Yang, Q. Chen and X. Li, Appl. Catal., B, 2020, 269, 118850 CrossRef CAS.
- J. Qu, Y. Xu, X. Zhang, M. Sun, Y. Tao, X. Zhang, G. Zhang, C. Ge and Y. Zhang, Appl. Catal., B, 2022, 316, 121639 CrossRef CAS.
- G. Fang, C. Liu, J. Gao, D. D. Dionysiou and D. Zhou, Environ. Sci. Technol., 2015, 49, 5645–5653 CrossRef CAS PubMed.
- L. He, C. Yang, J. Ding, M.-Y. Lu, C.-X. Chen, G.-Y. Wang, J.-Q. Jiang, L. Ding, G.-S. Liu, N.-Q. Ren and S.-S. Yang, Appl. Catal., B, 2022, 303, 120880 CrossRef CAS.
- M. He, P. Zhao, R. Duan, S. Xu, G. Cheng, M. Li and S. Ma, J. Hazard. Mater., 2022, 424, 127568 CrossRef CAS PubMed.
- X. Chen, W.-D. Oh, Z.-T. Hu, Y.-M. Sun, R. D. Webster, S.-Z. Li and T.-T. Lim, Appl. Catal., B, 2018, 225, 243–257 CrossRef CAS.
- S. Ahmad, L. Liu, S. Zhang and J. Tang, J. Hazard. Mater., 2023, 446, 130727 CrossRef CAS PubMed.
- S. Cai, Q. Zhang, Z. Wang, S. Hua, D. Ding, T. Cai and R. Zhang, Appl. Catal., B, 2021, 291, 120093 CrossRef CAS.
- W. Li, C. Nie, X. Wang, H. Ye, D. Li and Z. Ao, Sep. Purif. Technol., 2023, 323, 124418 CrossRef CAS.
- L. Tian, J. Li, F. Liang, J. Wang, S. Li, H. Zhang and S. Zhang, Appl. Catal., B, 2018, 225, 307–313 CrossRef CAS.
- C. Xu, H. Liu, D. Wang, D. Li, Y. Zhang, X. Liu, J. Huang, S. Wu, D. Fan, H. Liu and H. Pan, Appl. Catal., B, 2023, 334, 122835 CrossRef CAS.
- X. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237–8265 RSC.
- C. Xin, W. Shang, J. Hu, C. Zhu, J. Guo, J. Zhang, H. Dong, W. Liu and Y. Shi, Adv. Funct. Mater., 2022, 32, 2108345 CrossRef CAS.
- M. Demir, Z. Kahveci, B. Ahoy, N. K. R. Palapati, A. Subramanian, H. T. Cullinan, H. M. El-Kaderi, C. T. Harris and R. B. Gupta, Ind. Eng. Chem. Res., 2015, 54, 10731–10739 CrossRef.
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS PubMed.
- L. Huang, Z. Hu, H. Jin, J. Wu, K. Liu, Z. Xu, J. Wan, H. Zhou, J. Duan, B. Hu and J. Zhou, Adv. Funct. Mater., 2020, 30, 1908486 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- P. Li, H. Xie, Y. Liu, J. Wang, Y. Xie, W. Hu, T. Xie, Y. Wang and Y. Zhang, J. Power Sources, 2019, 439, 227096 CrossRef CAS.
- Y. Du, G. Zheng, J. Wang, L. Wang, J. Wu and H. Dai, Microporous Mesoporous Mater., 2014, 200, 27–34 CrossRef CAS.
- J. Yu, L. Tang, Y. Pang, G. Zeng, H. Feng, J. Zou, J. Wang, C. Feng, X. Zhu, X. Ouyang and J. Tan, Appl. Catal., B, 2020, 260, 118160 CrossRef CAS.
- S. Zhu, X. Huang, F. Ma, L. Wang, X. Duan and S. Wang, Environ. Sci. Technol., 2018, 52, 8649–8658 CrossRef CAS PubMed.
- K. Wang, G. Gu, S. Hu, J. Zhang, X. Sun, F. Wang, P. Li, Y. Zhao, Z. Fan and X. Zou, Chem. Eng. J., 2019, 368, 896–904 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- M. Nie, Y. Deng, S. Nie, C. Yan, M. Ding, W. Dong, Y. Dai and Y. Zhang, Chem. Eng. J., 2019, 369, 35–45 CrossRef CAS.
- Y. Long, J. Dai, S. Zhao, Y. Su, Z. Wang and Z. Zhang, Environ. Sci. Technol., 2021, 55, 5357–5370 CrossRef CAS PubMed.
- Y. Liu, M. Paskevicius, M. V. Sofianos, G. Parkinson and C.-Z. Li, Carbon, 2021, 172, 454–462 CrossRef CAS.
- S. Liu, P. Qi, S. Xu, P. Jin, B. Li, W. Yu and B. Young, J. Environ. Chem. Eng., 2022, 10, 107853 CrossRef CAS.
- Y. Shen, M. J. M. d. De Vidales, G. Gorni, A. Gomez-Herrero, F. Fernandez-Martinez and A. J. Dos santos-Garcia, Chem. Eng. J., 2022, 444, 136610 CrossRef CAS.
- L. Peng, X. Duan, Y. Shang, B. Gao and X. Xu, Appl. Catal., B, 2021, 287, 119963 CrossRef CAS.
- J. B. Zhang, C. Dai, Z. Wang, X. You, Y. Duan, X. Lai, R. Fu, Y. Zhang, M. Maimaitijiang, K. H. Leong, Y. Tu and Z. Li, Water Res., 2023, 244, 120534 CrossRef PubMed.
- Y. Xiang, X. Xie, H. Zhong, F. Xiao, R. Xie, B. Liu, H. Guo, D. Hu, P. Zhang and H. Huang, Environ. Sci. Technol., 2024, 58, 8846–8856 CrossRef CAS PubMed.
- J. Wu, J. Wang, C. Liu, C. Nie, T. Wang, X. Xie, J. Cao, J. Zhou, H. Huang, D. Li, S. Wang and Z. Ao, Environ. Sci. Technol., 2022, 56, 13996–14007 CrossRef CAS PubMed.
- R. Hao, L. Du, X. Gu and S. Li, Sep. Purif. Technol., 2023, 306, 122586 CrossRef.
- W. Ren, G. Nie, P. Zhou, H. Zhang, X. Duan and S. Wang, Environ. Sci. Technol., 2020, 54, 6438–6447 CrossRef CAS PubMed.
- Y. Zhou, X. Wang, C. Zhu, D. D. Dionysiou, G. Zhao, G. Fang and D. Zhou, Water Res., 2018, 142, 208–216 CrossRef CAS PubMed.
- X. Duan, Z. Ao, D. Li, H. Sun, L. Zhou, A. Suvorova, M. Saunders, G. Wang and S. Wang, Carbon, 2016, 103, 404–411 CrossRef CAS.
- P. Peljo, E. Smirnov and H. H. Girault, J. Electroanal. Chem., 2016, 779, 187–198 CrossRef CAS.
- M. Sun, J. Qu, T. Han, J. Xue, K. Li, Z. Jiang, G. Zhang, H. Yu and Y. Zhang, J. Cleaner Prod., 2023, 383, 135415 CrossRef CAS.
- X. Li, Y. Jia, M. Zhou, X. Su and J. Sun, J. Hazard. Mater., 2020, 397, 122708 CrossRef PubMed.
- X. Cheng, H. Guo, Y. Zhang, X. Wu and Y. Liu, Water Res., 2017, 113, 80–88 CrossRef CAS PubMed.
- A. A. Gorman and M. A. J. Rodgers, Chem. Soc. Rev., 1981, 10, 205–231 RSC.
- M. M. Wang, L.-J. Liu, J.-T. Wen, Y. Ding, J.-R. Xi, J.-C. Li, F.-Z. Lu, W.-K. Wang and J. Xu, Environ. Sci. Technol., 2022, 56, 12613–12624 CrossRef CAS PubMed.
- Y. Deng, L. Li, H. Zeng, R. Tang, Z. Zhou, Y. Sun, C. Feng, D. Gong, J. Wang and Y. Huang, Chem. Eng. J., 2023, 457, 141339 CrossRef.
- Y. Gao, Y. Zhu, L. Lyu, Q. Zeng, X. Xing and C. Hu, Environ. Sci. Technol., 2018, 52, 14371–14380 CrossRef CAS PubMed.
- H. Sun, X. Peng, S. Zhang, S. Liu, Y. Xiong, S. Tian and J. Fang, Bioresour. Technol., 2017, 241, 244–251 CrossRef CAS PubMed.
- G.-X. Yang and H. Jiang, Water Res., 2014, 48, 396–405 CrossRef CAS PubMed.
- X. Cheng, H. Guo, Y. Zhang, G. V. Korshin and B. Yang, Water Res., 2019, 157, 406–414 CrossRef CAS PubMed.
- Y. Gao, Z. Chen, Y. Zhu, T. Li and C. Hu, Environ. Sci. Technol., 2020, 54, 1232–1241 CrossRef CAS PubMed.
- G. Wang, Y. Zhao, H. Ma, C. Zhang, X. Dong and X. Zhang, Sci. Total Environ., 2021, 756, 144014 CrossRef PubMed.
- F. C. Carniel, L. Fortuna, D. Zanelli, M. Garrido, E. Vazquez, V. J. Gonzalez, M. Prato and M. Tretiach, J. Hazard. Mater., 2021, 414, 125387 CrossRef PubMed.
- L. Chen, H. Ji, J. Qi, T. Huang, C.-C. Wang and W. Liu, Chem. Eng. J., 2021, 406, 126811 CrossRef.
- M. Noorisepehr, B. Kakavandi, A. A. Isari, F. Ghanbari, E. Dehghanifard, N. Ghomi and F. Kamrani, Sep. Purif. Technol., 2020, 250, 116950 CrossRef CAS.
- S. Humayun, M. Hayyan, Y. Alias and A. Hayyan, Sep. Purif. Technol., 2021, 270, 118730 CrossRef CAS.
- S. Zhang, J. Zou, J. Li, Y. Huang, J. Wu, X. Liao, Q. Li, B. Yuan and J. Ma, Sep. Purif. Technol., 2023, 327, 124910 CrossRef CAS.
- H. Meng, C. Nie, W. Li, X. Duan, B. Lai, Z. Ao, S. Wang and T. An, J. Hazard. Mater., 2020, 399, 123043 CrossRef CAS PubMed.
- J. Liang, X. Duan, X. Xu, K. Chen, Y. Zhang, L. Zhao, H. Qiu, S. Wang and X. Cao, Environ. Sci. Technol., 2021, 55, 10077–10086 CrossRef CAS PubMed.
- W. Liu, C. Nie, W. Li, Z. Ao, S. Wang and T. An, J. Hazard. Mater., 2021, 414, 125552 CrossRef CAS PubMed.
- C. Nie, Z. Dai, W. Liu, X. Duan, C. Wang, B. Lai, Z. Ao, S. Wang and T. An, Environ. Sci.:Nano, 2020, 7, 1899–1911 RSC.
- C. Nie, Z. Dai, H. Meng, X. Duan, Y. Qin, Y. Zhou, Z. Ao, S. Wang and T. An, Water Res., 2019, 166, 115043 CrossRef CAS PubMed.
- D. He, X. Guan, J. Ma and M. Yu, Environ. Sci. Technol., 2009, 43, 8332–8337 CrossRef CAS PubMed.
- C. Zhao, B. Shao, M. Yan, Z. Liu, Q. Liang, Q. He, T. Wu, Y. Liu, Y. Pan, J. Huang, J. Wang, J. Liang and L. Tang, Chem. Eng. J., 2021, 416, 128829 CrossRef CAS.
- P. Zhang, X. Tan, S. Liu, Y. Liu, G. Zeng, S. Ye, Z. Yin, X. Hu and N. Liu, Chem. Eng. J., 2019, 378, 122141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07221a |
| This journal is © The Royal Society of Chemistry 2025 |