
Vanadium-doped Li2TiSiO5 anodes for boosting capacity and cycling stability of lithium-ion batteries†
Yuting
Cai
a,
Hao
Huang
a,
Zhongcheng
Song
*a,
Xinxin
Dong
b,
Mengyuan
Tong
a,
Qihu
Wu
a,
Chao
Yu
a,
Lixia
Sun
*a,
Ziqi
Sun
 c,
Ting
Liao
d and
Pingan
Song
*e
c,
Ting
Liao
d and
Pingan
Song
*e
aSchool of Chemistry and Chemical Engineering, Jiangsu University of Technology, Changzhou 213001, China. E-mail: songzhongcheng@jsut.edu.cn; sunlixia@jsut.edu.cn
bState Key Laboratory of Organic–Inorganic Composites, Center for Fire Safety Materials, Beijing University of Chemical Technology, Beijing, 100029, China
cCentre for Materials Science, School of Chemistry and Physics, Queensland University of Technology, Brisbane, QLD 4000, Australia
dSchool of Mechanical Medical and Process Engineering, Queensland University of Technology, George Street, Brisbane, QLD 4000, Australia
eCentre for Future Materials, School of Agriculture and Environmental Science, University of Southern Queensland, Springfield 4300, QLD, Australia. E-mail: pingan.song@usq.edu.au
First published on 31st January 2025
Abstract
Lithium-ion batteries (LIBs) represent one of the most ideal electrochemical energy storage devices due to their long cycle life, high specific energy, and high-power density. Li2TiSiO5 (LTSO) has been proposed as a promising anode material for LIBs, because of its favorable operating potential of 0.28 V vs. Li+/Li and desired safety and stability. However, its application has been significantly impeded by some key drawbacks, including slow Li+ transfer rates and low electrical conductivity. Herein, we proposed vanadium(V)-doping engineering for synthesizing Li2Ti1−xVxSiO5 (x = 0, 0.25, 0.5, 0.75) anode materials via a sol–gel method. Because of the partial replacement Ti4+ with V5+ ions in the structure, the as-prepared V-doped Li2Ti0.95V0.05SiO5 shows a high reversible capacity of 235 mA h g−1 after 130 cycles at a rate of 0.5 A g−1, nearly three-fold that of the pristine LTSO anode. The improved cycling stability and multiplicity performances are largely attributed to the increased conductivity, and this excellent lithium storage performance opens up new opportunities for further practical applications of novel silicon-based carbon materials as electrode materials in high-power storage devices. This study provides a simple and effective method for fabricating high-performance LTSO anode materials, thus facilitating their practical applications in rechargeable LIBs.
1. Introduction
Lithium-ion batteries (LIBs) have become central to the electronics market nowadays. LIBs are widely used across various sectors due to their exceptional specific capacity. Graphite is the predominant choice for anode materials in LIBs due to its structural properties, which facilitate the reaction between Li+ and the electrodes.1–7 However, graphite poses safety risks because its operating voltage is close to the redox potential of Li+/Li, which can easily lead to the formation of dendritic lithium on the surface of battery electrodes. The growth of lithium dendrites can result in internal short circuits, jeopardizing battery safety and potentially leading to explosion.8–12 Therefore, it has been desirable to develop safer batteries with higher redox potentials.13Recently, LTSO has been reported as a promising anode alternative to graphite because of its desirable operating potential of 0.28 V. This means that it can help avoid lithium plating without significantly compromising energy density.14–16 LTSO has demonstrated stable performance after many charge/discharge cycles, indicating its potential as a commercial anode material for LIBs.17,18 Despite its theoretical specific capacity of 315 mA h g−1, its capacity is often solely as low as 138 mA h g−1 in practical applications due to its insufficient ionic and electronic conductivity.19–22 Meanwhile, the formation of an unstable solid electrolyte interface (SEI) can lead to significant volumetric fluctuations in the battery material, resulting in capacity degradation. To address these issues, various strategies have been developed, including particle size reduction,23 preparation of nanoporous materials,24 carbon coating,4 and doping with metal ions.25,26
Vanadium(V)-doping has recently been applied to boost electrochemical performances of catalysts27,28 and electrodes.24,33 For example, the lithium extra layered catalyst (LLC) exhibited a more stable cycling performance partially because of improved electronic conductivity, upon being doped by V5+ cations. Recently, it has gained considerable attention to use heteroatom doping for improving the electrochemical performance of LIBs in the last few years.29 In addition, titanium-based compounds have been recently reported to show altered ectrochemical properties and significant morphological changes in V-doped TiO2 and Na2Ti3O7 for LIBs.24,30 Moreover, V5+ cations are considered some of the most suitable dopants for LTSO due to the similarity in ionic radii between Ti4+ (0.68 nm) and V5+ (0.59 nm). This similarity ensures unchanged lattice parameters and that the spatial structure of Li4Ti5O12 remain during the substitution process.31–33 Therefore, we hypothesise that the V-doping of LTSO by replacing some of the Ti sites is expected to improve the ion migration rate of LTSO without altering the space structure and thus to enhance its capacity and cycling stability.34,35
This work aims to dope LTSO with V5+ cations to replace some of the Ti4+ to verify our V-doping assumption. Li2Ti1−xVxSiO5 (x = 0, 0.25, 0.5, 0.75) anode materials with varying levels of V-doping were synthesized using the simple sol–gel method. The V-doped Li2Ti1−xVxSiO5 shows a reversible specific capacity as high as 235 mA h g−1 after 130 charge/discharge cycles at a rate of 0.5 A g−1 current density, nearly 300% higher than 129 mA h g−1 for LTSO. Additionally, the V-doped anode demonstrates excellent electrochemical performance in terms of rate capability. Density functional theory (DFT) calculations further validate the improved electrochemical performances of Li2Ti1−xVxSiO5. This work provides a simple and promising approach to develop high-performance LTSO anode materials, which hold significant potential as commercially viable anode materials for rechargeable LIBs.
2. Results and discussion
Li2Ti1−xVxSiO5 (x = 0, 0.25, 0.5, 0.75) anode materials were synthesized via the facile sol–gel approach (Fig. 1a), and their chemical structures are confirmed using XRD spectra (Fig. 1b). Pure LTSO exhibits a well-defined tetragonal phase, with an inevitable impurity peak of lithium appearing at the 18.4° diffraction peak.10,36,37 In comparison, as-synthesized Li2Ti1−xVxSiO5 anode materials with different V-doped concentrations show narrow and sharp diffraction peaks, indicating the higher crystallinity of Li2Ti1−xVxSiO5. Moreover, the specific diffraction peaks match the standard pattern for LTSO (PDF#130268) very well, implying that the V-doping does not change the crystal structure (Fig. 1b). | ||
| Fig. 1 (a) Schematic diagram of the synthesis of Li2Ti1−xVxSiO5 (x = 0, 0.025, 0.05 and 0.075) samples. (b) XRD plots of Li2Ti1−xVxSiO5 (x = 0, 0.025, 0.05 and 0.075) (Li2Ti0.975V0.025SiO5-LTSO-2.5%, Li2Ti0.95V0.05SiO5-LTSO-5% and Li2Ti0.925V0.075SiO5-LTSO-7.5%). (c) Enlarged XRD spectra of Li2Ti1−xVxSiO5 (x = 0, 0.025, 0.05, and 0.075). (d) Refined graphs of Li2TiSiO5. (e) Refined graphs of Li2Ti0.95V0.05SiO5. (f) Crystal structure of Li2Ti0.95V0.05SiO5. (g) 2 × 2 × 4 structure perspective view of Li2Ti0.95V0.05SiO5. | ||
According to the Bragg equation 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ = nλ. Fig. 1c shows a zoomed-in view of the strongest crystal plane, where the peak position between 28° and 29° varies with the amount of doping. The V-doping leads to such significant shifts in these two peak locations. Specifically, the diffraction peaks of the doped Li2Ti1−xVxSiO5 materials shift to the right as the doping level increases from 0% to 5%, but shifts to the left as the doping increases further from 5% to 7.5%. A minor amount of V doping causes the diffraction angle to increase and the position to move to the right because the ionic radius of V5+ is lower than the ionic radius of Ti4+. This occurs without altering the lattice. Further doping of V complicates the doping situation, though some stray peaks appear, suggesting that the doping amount reaches some sort of saturation. Additionally, excessive doping of V causes the lattice spacing to increase, which in turn reduces the diffraction angle and shifts the peak position to the left once more. The stabilization energies of the doping process initially increase slowly and then increase sharply as more V is introduced. When the V concentration reaches 5%, the stabilization energies becomes significantly higher, which can lead to the most stable LTSO structure. Therefore, 5% V doping appears to be the optimum concentration that can maintain the lattice structure and improve electrochemical properties of LTSO without compromising its stability. The diffraction peaks of LTSO-7.5% V move back to a lower value, probably because of the law of conservation of charge.38 The peaks of the doped samples first deviate to a higher angle (right shift) when the doping content increases from 0.0% to 5.0% and then move to a smaller angle (left shift) with doping from 5.0% to 7.5%. The charge neutrality is destroyed, so a charge compensation reaction occurs to comply with the charge neutrality rule.38 The most likely defects to arise are conduction electrons and oxygen vacancies, as higher energy is required for the formation of titanium vacancies.39
sinθ = nλ. Fig. 1c shows a zoomed-in view of the strongest crystal plane, where the peak position between 28° and 29° varies with the amount of doping. The V-doping leads to such significant shifts in these two peak locations. Specifically, the diffraction peaks of the doped Li2Ti1−xVxSiO5 materials shift to the right as the doping level increases from 0% to 5%, but shifts to the left as the doping increases further from 5% to 7.5%. A minor amount of V doping causes the diffraction angle to increase and the position to move to the right because the ionic radius of V5+ is lower than the ionic radius of Ti4+. This occurs without altering the lattice. Further doping of V complicates the doping situation, though some stray peaks appear, suggesting that the doping amount reaches some sort of saturation. Additionally, excessive doping of V causes the lattice spacing to increase, which in turn reduces the diffraction angle and shifts the peak position to the left once more. The stabilization energies of the doping process initially increase slowly and then increase sharply as more V is introduced. When the V concentration reaches 5%, the stabilization energies becomes significantly higher, which can lead to the most stable LTSO structure. Therefore, 5% V doping appears to be the optimum concentration that can maintain the lattice structure and improve electrochemical properties of LTSO without compromising its stability. The diffraction peaks of LTSO-7.5% V move back to a lower value, probably because of the law of conservation of charge.38 The peaks of the doped samples first deviate to a higher angle (right shift) when the doping content increases from 0.0% to 5.0% and then move to a smaller angle (left shift) with doping from 5.0% to 7.5%. The charge neutrality is destroyed, so a charge compensation reaction occurs to comply with the charge neutrality rule.38 The most likely defects to arise are conduction electrons and oxygen vacancies, as higher energy is required for the formation of titanium vacancies.39
The standard lattice parameters (x-values of 0, 0.025, 0.05 and 0.075) of the synthesized Li2Ti1−xVxSiO5 samples were refined using the FullProf software40 and the crystal structures of LTSO and Li2Ti0.95V0.05SiO5 were analysed using the VESTA41 software (Fig. 1f and g).The relevant crystal structure data are listed in Table S1.† Additionally, the refinement results for Li2Ti0.975V0.025SiO5 and Li2Ti0.925V0.075SiO5 are illustrated in Fig. S2.† It is evident that as the doping ratio increases, the volume of the unit cell initially decreases (from Li2TiSiO5 to Li2Ti0.975V0.025SiO5 and then to Li2Ti0.95V0.05SiO5) and then increases (from Li2Ti0.95V0.05SiO5 to Li2Ti0.925V0.075SiO5). Consequently, the d-spacing (distance between crystalline planes) decreases and then increases, resulting in a corresponding increase and a decrease in the diffraction angle according to the Bragg equation. The refinement results are also in very good agreement with the fact that the diffraction angles first shift to the right and then to the left in the experiment. In addition, theoretical calculations are carried out to further investigate 5% V-doped LTSO (Fig. 1f). Conductivity is critical for fast electron transfer, which strongly depends on a largely altered electronic structure.42 This indicates that the conductivity is enhanced when V is doped in appropriate proportions.43,44 In general, V5+ can chemically reduce Ti4+ to Ti3+ and coexists with Ti4+. The generated Ti4+/Ti3+ redox pairs can act at a relatively high operating voltage (typically 1.5 V, vs. Li+/Li), which has demonstrated good reversibility and been used in commercial LIBs.45 Furthermore, all doped materials seem to show increased electrical conductivity.31,46,47 Hence, V-doping can not only successfully replace Ti to improve the conductivity, but also improve the stability of the structure.
The Rp and wRp values of LTSO and Li2Ti0.95V0.05SiO5 are both lower than 10%, and the value of χ2 is less than 2, suggesting that the XRD data of the experimental results of Li2Ti0.95V0.05SiO5 are in good accordance with the theoretical model (Fig. 1f), indicating a high degree of consistency. The V atom successfully replaces the Ti atom and coexists with Ti in the structure (Fig. 1g). The V-doped LTSO decreases the lattice constant, increases the Li occupancy at tetrahedral sites, and decreases occupancy at octahedral sites, forming a stable cubic structure with high ionic conductivity. Furthermore, the V's stable oxidation state can promote a close-packed oxygen distribution, thus contributing to a higher specific energy for the cell.
Doping improves the interfacial stability of the material and reduces the structural changes of the material during charging and discharging, thus improving cycling stability.48 Generally, doping can change the electronic structure of a material and reduce its band gap width. A decrease in the band gap width means that the conductivity of the material is increased because electrons can jump from the valence band to the conduction band more easily.49 We also performed relevant DFT calculations and the results are consistent with the reduced band gap width of the doped Li2Ti0.95V0.25SiO5 compared to Li2TiSiO5, as seen in Fig. 4a and b. Doping increases the specific surface area of the material, thereby increasing the contact area between the material and the electrolyte and accelerating charge transfer.50 In this regard, a relevant BET test was also added. Compared to Li2TiSiO5, specific surface area, pore volume, and average pore size are increased for Li2Ti0.95V0.25SiO5.
Fig. 2a and b show the morphologies of both pure LTSO and as-prepared Li2T0.95V0.05SiO5. Both samples exhibit irregular shapes, further indicating that V doping does not significantly alter the structure of LTSO. As compared with LTSO (Fig. 2a and b), the high-resolution transmission electron microscopy (HRTEM) images show that there are no observed significant changes in the morphology of the doped Li2Ti0.95V0.05SiO5 (Fig. 2c and d). The lattice fringes of Li2Ti0.95V0.05SiO5 are clearly observable for the (001) and (101) planes, with spacing measurements of 0.445 nm and 0.372 nm, respectively. The diffraction points of Li2Ti0.95V0.05SiO5 (shown in Fig. 2e and f) correspond to the (001) and (101) crystal planes, in addition to their respective lattice stripes and the (221) crystal plane (Fig. 2h). Furthermore, the energy dispersive X-ray (EDX) mapping results (Fig. 2i) indicate the uniform distribution of O, Si, Ti, and V within the Li2Ti0.95V0.05SiO5 compound, representing other evidence for the successful V-doping of LTSO.
 | ||
| Fig. 2 (a) LTSO transmission electron microscopy and (b) Li2Ti0.95V0.05SiO5 transmission electron microscopy. (c and d) High resolution TEM images of Li2Ti0.95V0.05SiO5 at different magnifications. (e and f) Lattice fringes of Li2Ti0.95V0.05SiO5 (400) and (102) crystal faces. (h) Electron diffraction pattern of the crystal face (221) of the Li2Ti0.95V0.05SiO5 sample. (i) Elemental mapping of the Li2Ti0.95V0.05SiO5 sample. | ||
In addition, the binding energy peaks of Li2Ti0.95V0.05SiO5 are almost the same as those of pure LTSO. The binding energy peak of Ti 2p decreases slightly after V-doping, suggesting that the valence state of Ti is likely to be reduced due to the incorporation of V.51 The in situ X-ray photoelectronic spectroscopy (XPS) spectra show that the peaks of Ti 2p and V 2p are slightly shifted to the right and their binding energies are slightly reduced after reduction (Fig. S1e and f†). In addition, LTSO is much coarser than Li2Ti0.95V0.05SiO5, with irregular particle sizes and a compact structure (Fig. S3†).
As-synthesised V-doped Li2Ti1−xVxSiO5 (x = 0, 0.025, 0.05, and 0.075) anode materials are then used for preparing lithium half-cells, with their electrochemical performances tested. The rate performances of V-doped Li2Ti1−xVxSiO5 at different current densities are investigated (Fig. 3a). Li2Ti0.95V0.05SiO5 exhibits the best rate performance, outperforming the other V-doped samples. Compared with pure LTSO, all the V-doped samples (x = 0, 0.025, 0.05, and 0.075) show increased specific capacity at varied current densities, and Li2Ti0.95V0.05SiO5 gives rise to the most significant improvement. It delivers a specific capacity of approximately 185 mA h g−1 at a current density of 0.5 A g−1, far exceeding the capacity of its pure LTSO counterpart. The as-prepared Li2Ti0.95V0.05SiO5 cell shows a long-cycle capacity of around 235 mA h g−1 after over 130 cycles at a current density of 0.5 A g−1, achieving the best long-cycle performance at high current densities (Fig. 3b). The N2 adsorption–desorption isotherms and corresponding pore size distribution curves of LTSO and Li2Ti0.95V0.05SiO5 samples are shown in Fig. 3c and d, respectively. The specific surface areas of pure LTSO and Li2Ti0.95V0.05SiO5 were 1.7839 and 2.6124 m2 g−1, the pore volumes were 0.0021 and 0.0033 cm3 g−1, and the average pore sizes were 4.6166 and 5.0233 nm, respectively (Table 3). The V-doping of Li2Ti0.95V0.05SiO5 can increase the specific surface area of LTSO and provide more pores and active sites, and in general, an appropriate increase in the specific surface area will increase the contact area between the electrode material and the electrolyte, which is conducive to the rapid transport and reaction of lithium ions.55 More notably, the graded porosity of these samples is also supported (Fig. 3d). More porosities in the range of 1 nm, 3–10 nm, and 20–60 nm were detected, in a structure that can accelerate the ion diffusion rate for competitive ionic conductivity.56
 | ||
| Fig. 3 (a) Rate specific capacity of Li2Ti1−xVxSiO5 (x = 0, 0.025, 0.05, and 0.075) materials at different current densities. (b) Cycling specific capacity of Li2Ti1−xVxSiO5 (x = 0, 0.025, 0.05, and 0.075) materials at 0.5 A g−1 current density. (c) N2 adsorption–desorption isotherms of LTSO and Li2Ti0.95V0.05SiO5. (d) Corresponding pore size distribution curve. (e) Charge–discharge curves of Li2Ti1−xVxSiO5 (x = 0, 0.025, 0.05, and 0.075) materials for the 1st cycle at 0.5 A g−1 current. (f) Charge–discharge curves of Li2Ti1−xVxSiO5 (x = 0, 0.025, 0.05, and 0.075) materials for the 10th cycle at 0.5 A g−1 current. (g) Cyclic voltammograms of pure samples of Li2TiSiO5. (h) Cyclic voltammograms of pure samples of Li2Ti0.95V0.05 SiO5. (i) Fitted equivalent diagram of the impedance circuit of Li2Ti1−xVxSiO5 (x = 0, 0.025, 0.05, and 0.075). (j) The relationship between Z′ and ω−1/2 for LTSO and Li2Ti1−xVxSiO5 (x = 0, 0.025, 0.05, and 0.075) samples. (k) Comparison of the multiplicity properties of Li2Ti0.95V0.05 SiO5 with those of previously reported V-doped and Si-based materials. | ||
The constant current charge/discharge profiles show that after 10 cycles, the specific capacity of the Li2Ti1−xVxSiO5 samples gradually stabilizes (Fig. 3e and f). Moreover, 5% V-doped LSTO is most effective in increasing the storage capacity of lithium without compromising the electrochemical response.
The electrochemical reactions of LTSO and Li2Ti0.95V0.05SiO5 are examined through cyclic voltammetry (CV), as shown in Fig. 3g and h. The results indicate that as-synthesized Li2Ti0.95V0.05SiO5 exhibits a favorable CV curve, showing promise for its exceptional electrochemical performances. Its CV data are in good consistency with theoretical predictions. An inflection point of the discharge is observed to appear at around 1 V, probably because of the formation of a solid electrolyte interface (SEI) layer by LTSO, as expressed using the below equation (eqn (1)):
| Li2TiSiO5 + 2Li+ + 2e− → 2Li + TiO2 + SiO2 + Li2O | (1) |
The impedance diagram gives a straight line in the low-frequency region but a semicircle in the high-frequency region (Fig. 3i). The low-frequency straight line has a slope representing the ion diffusion rate. Meanwhile, the diameter of the semicircle in the high-frequency region directly corresponds to the charge transfer resistance (Rct). A smaller Rct value indicates faster ion transfer kinetics and an improved electrochemical performance.31 The electrochemical impedance fitting results, shown in Table 2, suggest that Li2Ti0.95V0.05SiO5 has the smallest Rct value, as further confirmed using the Weber factor (Fig. 3j). Hence, as-synthesized Li2Ti0.95V0.05SiO5 has a low contact and charge transfer impedance, which favors enhanced conductivity and faster ion mobility in the insertion/embedding reaction, thereby improving overall electrochemical performances.
During the discharge of Li2Ti0.95V0.05SiO5, some Ti and V ions with reduced binding energies are converted to Ti3+ and V4+ (Fig. S1†). V5+ exerts a stronger restriction effect on the anion movement, leading to a more stable electrode/electrolyte interface, which increases the material's conductivity.30 Also, the presence of V5+ generates additional free electrons, thereby enhancing the electrochemical properties of the doped anodes.33 This can facilitate the incorporation of more Li+, thus increasing the specific capacity of Li2Ti0.95V0.05SiO5.
The lithium-ion diffusion coefficients (DLi+) are calculated based on the following two equations eqn (2) and (3).57,58
| Zre = Rs + Rct + σω−1/2 | (2) |
| DLi+ = 0.5R2T2/A2n4F4C2σ2 | (3) |
485C mol−1. The concentration of lithium ions in the solid is 5.86 × 10−3 mol cm−3, denoted as C. The lithium-ion diffusion coefficient of Li2Ti0.95V0.05SiO5 is 2.491 × 10−9 cm2 s−1, which is about 1.65 times higher than that of LTSO (see Table 2) indicating that Li2Ti0.95V0.05SiO5 is more conducive to lithium-ion diffusion, which can be attributed to its higher lithium ion diffusion coefficient and shorter lithium ion transfer channel. This can explain why Li2Ti0.95V0.05SiO5 shows a better electrochemical performance than LTSO, which is consistent with the results of the charging and discharging processes.59
The rate performances of Li2Ti0.95V0.05SiO5 are compared to those of previously reported Si-based materials14,17,38,52,53 and V-doped materials30,51,54 in the range of 5–10 cycles at the same current density (Fig. 3i and Table 1). The as-synthesized Li2Ti0.95V0.05SiO5 maintains a higher specific capacity than its most anode counterparts. Upon returning to lower current densities, it exhibits the highest specific capacity and excellent capacity retention.
| Materials | Current density (A g−1) | Specific capacity (mA h g−1) | Cycle number | Ref. | |
|---|---|---|---|---|---|
| Si-based | Li2Ti0.95V0.05SiO5 | 0.2 | 264.3 | 20 | This work |
| LTSO | 0.2 | 210 | 10 | 14 | |
| LTSO | 0.2 | 215 | 20 | 17 | |
| LTSO | 0.2 | 235 | 20 | 38 | |
| LTSO | 0.2 | 225 | 5 | 52 | |
| Li2MnSiO4 | 0.2 | 210 | 20 | 53 | |
| V-doped | VNTO-15 | 0.2 | 118 | 20 | 30 |
| V0.1Ti3C2Tx-d | 0.2 | 218 | 20 | 51 | |
| V0·1TiO2 | 0.2 | 188 | 20 | 54 | |
| Sample | R s (Ω) | R ct (Ω) | δ (Ω cm2 s−1/2) | D Li+ (cm2 s−1) |
|---|---|---|---|---|
| LTSO | 1.747 | 179 | 2.63 | 1.513 × 10−9 |
| LTSO-2.5% V | 7.262 | 75.04 | 2.06 | 2.466 × 10−9 |
| LTSO-5% V | 10.92 | 63.01 | 2.05 | 2.491 × 10−9 |
| LTSO-7.5% V | 4.605 | 107 | 2.62 | 1.525 × 10−9 |
| Sample | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore size (nm) |
|---|---|---|---|
| LTSO | 1.7839 | 0.0021 | 4.6166 |
| LTSO-5% V | 2.6124 | 0.0033 | 5.0233 |
The energy band structures of LTSO and V-doped Li2Ti0.95V0.05SiO5 are presented in Fig. 4a and b, respectively. The band gap of undoped LTSO is estimated to be 3.544 eV, while the band gap of V-doped Li2Ti0.95V0.05SiO5 is reduced to 2.504 eV. This significant reduction in the band gap indicates that the V-doping leads to a significant improvement in the electrical conductivity of LTSO.60 The electron resistance of the electrode material is further analyzed using a density of states (DOS) diagram, with DOS plots shown in Fig. 4c and d. The presence of the element V and the hybridization between the 3d state of Ti and V are more pronounced (Fig. 4d), indicating stronger interatomic interactions, which is a characteristic of transition metal oxides.61
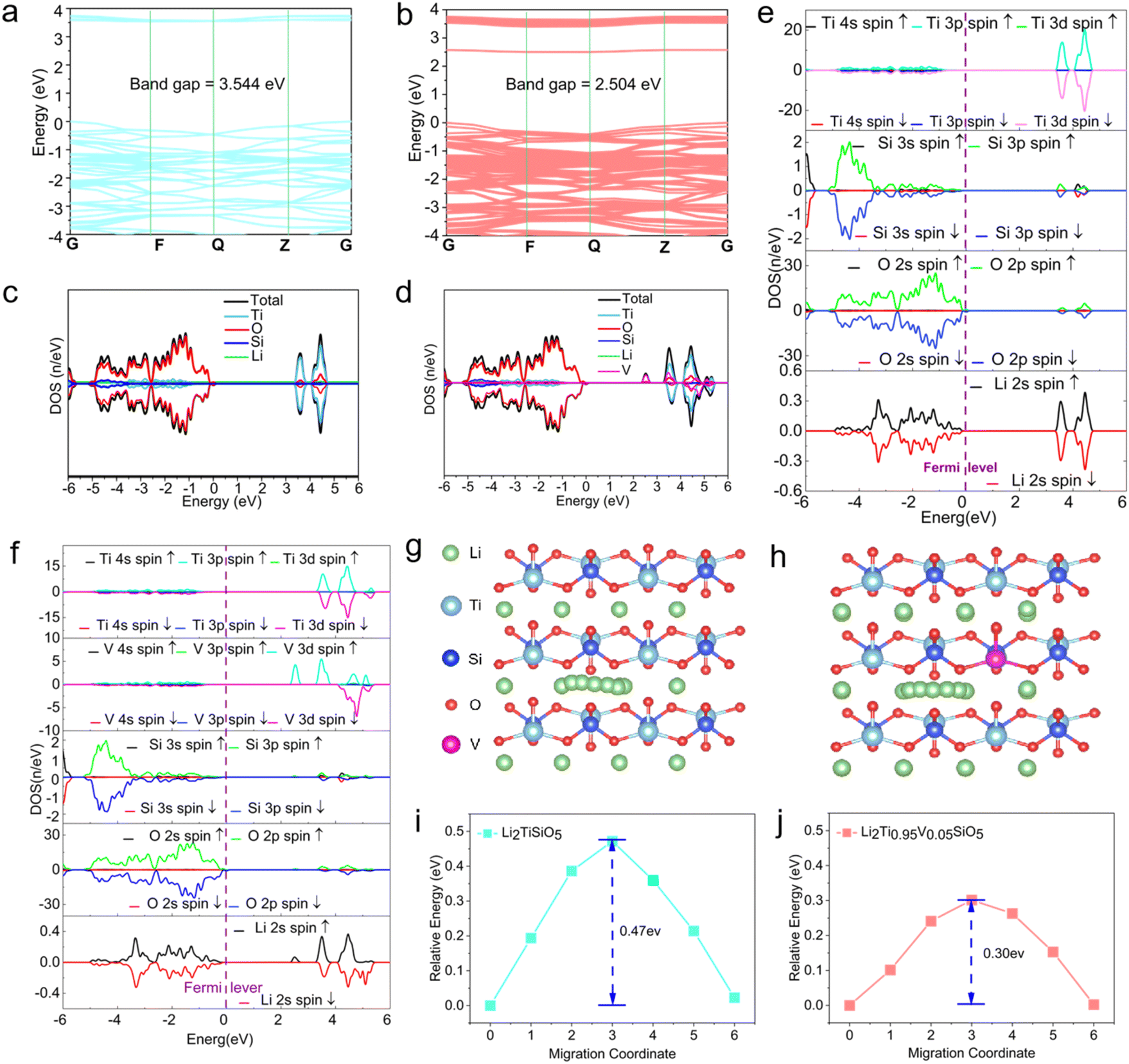 | ||
| Fig. 4 (a) Energy band diagram of Li2TiSiO5. (b) Energy band diagram of Li2Ti0.95V0.05SiO5. (c) Density of states plots (DOS) for Li2TiSiO5. (d) DOS for Li2Ti0.95V0.05SiO5. (e) Partial density of states (PDOS) of Li2TiSiO5. (f) PDOS map of Li2Ti0.95V0.05SiO5. (g) Lithium-ion migration path diagram for Li2TiSiO5 (010) crystal faces. (h) Lithium-ion migration path diagram for Li2Ti0.95V0.05SiO5 (010) crystal faces. (i) Migration energy barrier diagram for Li2TiSiO5 (010) crystal faces. (j) Migration energy barrier diagram for Li2Ti0.95V0.05SiO5 (010) crystal faces. | ||
The electron resistance of the electrode materials can be analyzed using the DOS plots. For an ideal anode material, the band gap should be as small as possible. LTSO shows the partial density of states (PDOS) in both spin-up and spin-down configurations (Fig. 4e). The occupied states of the Fermi energy levels are mainly composed of O 2p orbitals, while the Ti 3d orbitals contribute predominantly to the conduction band. In comparison, for Li2Ti0.95V0.05SiO5, the Ti 3d orbitals contribute less to the conduction band compared to LTSO, and the substitution of Ti with V results in increased interatomic interactions and greater orbital contributions (Fig. 4f). The V 3d and Li 2s orbitals are closer to the Fermi energy level, thus giving rise to a smaller band energy in Li2Ti0.95V0.05SiO5. This further confirms the improved electronic conductivity of Li2Ti0.95V0.05SiO5, which is well consistent with the energy band results.
Fig. 4g and h show schematic diagrams of Li+ diffusion in LTSO and Li2Ti0.95V0.05SiO5 along the (010) crystal plane. The doping of V ions into the crystal structure of LTSO (Fig. 4h) leads to a reduction in the migration energy barriers. The energy barriers for LTSO and Li2Ti0.95V0.05SiO5 are calculated from the path diagrams to be 0.47 eV and 0.3 eV, respectively, on the (010) crystallographic plane. The significantly reduced migration energy barrier in Li2Ti0.95V0.05SiO5 indicates that V-doping significantly lowers the Li+ energy barrier. These findings are in good consistence with the impedance fitting and Li-ion mobility calculations from previous electrochemical tests, further demonstrating that V-doping enhances the ionic conductivity of LTSO. These results clearly demonstrate that the as-designed Li2Ti0.95V0.05SiO5 shows great potential as a promising anode material for LIBs.62
3 Conclusion
In conclusion, we have successfully prepared V-doped Li2Ti1−xVxSiO5 (x = 0, 0.25, 0.5, 0.75) anode materials with varied doping ratios via V-doping engineering. The V-doped Li2Ti0.95V0.05SiO5 material can effectively substitute Ti with V in the crystal structure, achieving the optimum chemical composition. Compared with the undoped LTSO, the V-doped Li2Ti0.95V0.05SiO5 exhibits significantly enhanced electrochemical performances. After 130 charge–discharge cycles at a current density of 0.5 A g−1, Li2Ti0.95V0.05SiO5 achieves a specific capacity of approximately 235 mA h g−1, which is much higher than 62.1 mA h g−1 for undoped LTSO. These results demonstrate that Li2Ti0.95V0.05SiO5 significantly improves the electrochemical properties of LTSO, making it a promising candidate for high-performance anode materials for LIBs. This work offers a facile yet promising V-doping strategy for the design of Ti-based anode materials for LIBs, and the developed Li2Ti1−xVxSiO5 anodes show great potential as high-performance anode materials for their real-world applications in LIBs.4. Experimental
Samples of V-doped LTSO materials were prepared by the sol–gel technique. The required elements were weighed according to the stoichiometric ratios and sequentially added to a beaker containing anhydrous ethanol. The mixture was stirred for 4 hours, with ultrasonication applied for 30 minutes to ensure uniform mixing. After drying, the solution was moved to a blast drying oven and held at 80 °C for 12 hours. The dried precursor was then ground, and the V-doped LTSO material was obtained by calcining it in a tube furnace under an argon atmosphere at 870 °C for 8 hours.4.1 Material properties
The X-ray diffractometer (XRD) model X′Pert Powder was utilized to examine the crystallinity and composition of the material. During this procedure, the anode was exposed to radiation from a copper Kα target. The scanning range of the radiation was from 10° to 80°, at a rate of 5° per minute. The samples were compared with known standard reference cards to determine the purity and impurity content. Additionally, microstructures were examined using a Sigma 500 electron microscope, while the lattice structures were specifically analyzed using a Talos transmission scanning electron microscope.4.2 Electrochemical measurements
Conductive carbon black (Super P) and polyvinylidene fluoride (PVDF) were measured at a weight ratio of 80:10:10 and milled with the synthesized LTSO material for 1 hour. The milled mixture was then uniformly blended with N-methyl-2-pyrrolidine (NMP) and applied onto copper foil. The coated foil was dried in a vacuum oven at 80 °C for 12 hours. After drying, the electrode sheets were rolled and cut into a single electrode sheet with a surface area of around 1.1 cm2 and an active material loading of around 2 mg cm−2. The assembly of CR2016 coin cell batteries was conducted in an argon-filled glove box with H2O and O2 concentrations maintained below 0.1 ppm. The batteries were assembled using a standard Celgard 2400 separator and an electrolyte consisting of a 1 M LiPF6 organic solution. The cyclic voltammetry (CV) testing was performed on a Shanghai Chenhua CH1660E electrochemical workstation. Constant current charge/discharge and rate performance tests were carried out using a Blue Power system (Land CT2001A).
4.3 Theoretical calculation method
We used the VASP63 software to perform the first principles approach calculations to perform all density functional theory (DFT) calculations64 within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE)65 formulation. We chose the projected augmented wave (PAW) potentials66 to describe the ionic cores and took valence electrons into account using a plane wave basis set with a kinetic energy cutoff of 520 eV. Partial occupancies of the Kohn–Sham orbitals were allowed using the Gaussian smearing method and a width of 0.05 eV. The electronic energy was considered self-consistent when the energy change was smaller than 10−5 eV. A geometry optimization was considered convergent when the energy change was smaller than 0.05 eV Å−1. The Brillouin zone integration was performed using 2 × 2 × 1 Monkhorst–Pack k-point sampling for a structure. Finally, ion migration was determined using the nudged elastic band (NEB) method. The NEB method discretized the path between the reactant and product into a series of structural images.Data availability
All the data supporting this article have already been included in this article and no new data were generated or analysed as part of this article.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52171212 and 51972151) and the Australian Research Council (Grant No. DP240102728). This research was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), supported by the Australian Government.References
- Z. Li, J. Huang, B. Y. Liaw, V. Metzler and J.-B. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS.
- G. Wang, C. Lu, X. Zhang, B. Wan, H. Liu, M. Xia, H. Gou, G. Xin, J. Lian and Y. Zhang, Nano Energy, 2017, 36, 46–57 CrossRef CAS.
- J. Yang, Y. Wang, W. Li, L. Wang, Y. Fan, W. Jiang, W. Luo, Y. Wang, B. Kong, C. Selomulya, H. K. Liu, S. X. Dou and D. Zhao, Adv. Mater., 2017, 29, 1700523 CrossRef PubMed.
- L. Ping, Z. Zhi-An, L. Jie and L. Yan-Qing, J. Cent. South Univ. Technol., 2010, 17, 1207–1210 CrossRef.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- S. Xin, Y.-G. Guo and L.-J. Wan, Acc. Chem. Res., 2012, 45, 1759–1769 CrossRef CAS PubMed.
- F.-F. Cao, Y.-G. Guo and L.-J. Wan, Energy Environ. Sci., 2011, 4, 1634–1642 RSC.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- Z. Guo, J. Zhu, J. Feng and S. Du, RSC Adv., 2015, 5, 69514–69521 RSC.
- P. Wang, G. Zhang, J. Cheng, Y. You, Y.-K. Li, C. Ding, J.-J. Gu, X.-S. Zheng, C.-F. Zhang and F.-F. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 6138–6143 CrossRef CAS PubMed.
- Y. Liu, M. Zhang, Y. Zhang, Y. Liu, L. Wang, X. Li, M. Xue, B. Li and X. Tao, J. Supercrit. Fluids, 2019, 154, 104596 CrossRef CAS.
- J. Liu, K. Song, P. A. van Aken, J. Maier and Y. Yu, Nano Lett., 2014, 14, 2597–2603 CrossRef CAS PubMed.
- L. Jin, R. Gong, J. Zheng, C. Zhang, Y. Xia and J. P. Zheng, ChemElectroChem, 2019, 6, 3020–3029 CrossRef CAS.
- J. Liu, Y. Liu, M. Hou, Y. Wang, C. Wang and Y. Xia, Electrochim. Acta, 2018, 260, 695–702 CrossRef CAS.
- S. Wang, R. Wang, Y. Bian, D. Jin, Y. Zhang and L. Zhang, Nano Energy, 2019, 55, 173–181 CrossRef CAS.
- Y. Wu, S.-H. Bo and Y. Xia, J. Power Sources, 2020, 467, 228292 CrossRef CAS.
- J. Liu, W. K. Pang, T. Zhou, L. Chen, Y. Wang, V. K. Peterson, Z. Yang, Z. Guo and Y. Xia, Energy Environ. Sci., 2017, 10, 1456–1464 RSC.
- Y. Wu, Z. Xu, Y. Liu, J. Chen, L. Peng, O. J. Borkiewicz, H. Zhu, S.-H. Bo and Y. Xia, J. Phys. Chem. C, 2021, 125, 3733–3744 CrossRef CAS.
- Y. Mei, Y. Li, F. Li, Y. Li, Y. Jiang, X. Lan, S. Guo and X. Hu, J. Mater. Sci. Technol., 2020, 57, 18–25 CrossRef CAS.
- K. S. Ng, C.-S. Moo, Y.-P. Chen and Y.-C. Hsieh, Appl. Energy, 2009, 86, 1506–1511 CrossRef CAS.
- S. Lee, J. Kim, J. Lee and B. H. Cho, J. Power Sources, 2008, 185, 1367–1373 CrossRef CAS.
- S. Piller, M. Perrin and A. Jossen, J. Power Sources, 2001, 96, 113–120 CrossRef CAS.
- H. Raj, S. Saxena and A. Sil, Mater. Technol., 2017, 32, 196–201 CrossRef CAS.
- S. Saxena and A. Sil, IETE Tech. Rev., 2016, 33, 60–63 CrossRef.
- N. Wang, J. Yue, L. Chen, Y. Qian and J. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 10348–10355 CrossRef CAS PubMed.
- C. Wang, S. Wang, Y.-B. He, L. Tang, C. Han, C. Yang, M. Wagemaker, B. Li, Q.-H. Yang, J.-K. Kim and F. Kang, Chem. Mater., 2015, 27, 5647–5656 CrossRef CAS.
- K. Liu, J.-A. Wang, J. Yang, D. Zhao, P. Chen, J. Man, X. Yu, Z. Wen and J. Sun, Chem. Eng. J., 2021, 407, 127190 CrossRef CAS.
- Y. Lee, J. Shin, H. Kang, D. Lee, T.-H. Kim, Y.-K. Kwon and E. Cho, Adv. Sci., 2021, 8, 2003013 CrossRef CAS PubMed.
- L. Sun, J. Hu, W. Bai, W. Mao and Z. Song, Front. Chem., 2023, 11, 1189866 CrossRef CAS PubMed.
- S. Chandel, Zulkifli, J. Kim and A. K. Rai, Dalton Trans., 2022, 51, 11797–11805 RSC.
- T.-F. Yi, J. Shu, Y.-R. Zhu, X.-D. Zhu, C.-B. Yue, A.-N. Zhou and R.-S. Zhu, Electrochim. Acta, 2009, 54, 7464–7470 CrossRef CAS.
- A. Mahmoud, J. M. Amarilla, K. Lasri and I. Saadoune, Electrochim. Acta, 2013, 93, 163–172 CrossRef CAS.
- S. Saxena and A. Sil, Mater. Res. Bull., 2017, 96, 449–457 CrossRef CAS.
- D. Kong, L. Shen, R. Mo, J. Liu, R. Tao, W. Shi, S. Ma, C. Zhang and Y. Lu, Nanoscale, 2020, 12, 13918–13925 RSC.
- J. Liu, D. Su, L. Liu, Z. Liu, S. Nie, Y. Zhang, J. Xia, H. Deng and X. Wang, Nanoscale, 2020, 12, 19702–19710 RSC.
- I. Belharouak, G. M. Koenig Jr and K. Amine, J. Power Sources, 2011, 196, 10344–10350 CrossRef CAS.
- L. Noerochim, R. S. Prabowo, W. Widyastuti, D. Susanti, A. Subhan and N. H. Idris, Batteries, 2023, 9, 38 CrossRef CAS.
- Y. Li, Y. Mei, X. Lan, Y. Jiang and X. Hu, Mater. Res. Bull., 2021, 136, 111145 CrossRef CAS.
- F. Ambriz-Vargas, R. Zamorano-Ulloa, A. Romero-Serrano, J. Ortiz-Landeros, J. Crespo-Villega, D. Ramírez-Rosales and C. Gómez-Yáñez, J. Mex. Chem. Soc., 2017, 61, 317–325 CAS.
- P. Paufler, Cryst. Res. Technol., 1995, 30, 494 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- G. Qu, J. Wang, G. Liu, B. Tian, C. Su, Z. Chen, J.-P. Rueff and Z. Wang, Adv. Funct. Mater., 2019, 29, 1805227 CrossRef.
- H. Huang, G. Zhao, X. Yu, X. Shen, M. Wang, X. Bai and N. Zhang, J. Mater. Chem. A, 2022, 10, 577–584 RSC.
- C. Li, Y. Lin, X. Li, Z. Li, P. Luo, Y. Jin and Z. Li, Colloids Surf., A, 2023, 660, 130681 CrossRef CAS.
- J. C. Pérez-Flores, M. Hoelzel, F. García-Alvarado and A. Kuhn, ChemPhysChem, 2016, 17, 1062–1069 CrossRef PubMed.
- Z. Yu, X. Zhang, G. Yang, J. Liu, J. Wang, R. Wang and J. Zhang, Electrochim. Acta, 2011, 56, 8611–8617 CrossRef CAS.
- T.-F. Yi, J. Shu, Y.-R. Zhu, X.-D. Zhu, R.-S. Zhu and A.-N. Zhou, J. Power Sources, 2010, 195, 285–288 CrossRef CAS.
- K. Min and Y.-H. Shin, Phys. Chem. Chem. Phys., 2021, 23, 2038–2045 RSC.
- S. G. Kang and D. S. Sholl, J. Alloys Compd., 2017, 693, 738–743 CrossRef CAS.
- J.-S. Seo and B.-K. Na, Korean J. Chem. Eng., 2021, 38, 1826–1833 CrossRef CAS.
- T. Yao and H. Wang, J. Colloid Interface Sci., 2021, 604, 188–197 CrossRef CAS PubMed.
- Y. Mei, S. Guo, Y. Jiang, F. Li, Y. Li and X. Hu, Electrochim. Acta, 2020, 344, 136149 CrossRef CAS.
- S.-S. Liu, L.-J. Song, B.-J. Yu, C.-Y. Wang and M.-W. Li, Electrochim. Acta, 2016, 188, 145–152 CrossRef CAS.
- M. Tian and Z. Hao, Funct. Mater. Lett., 2023, 16, 2340006 CrossRef CAS.
- Y. Liu, B. Li, M. Zhang, Y. Zhang, H. Zhu, N. Xue, J. Zhuang, X. Zhao and X. Tao, Electrochim. Acta, 2021, 387, 138469 CrossRef CAS.
- L.-M. Jin, R.-Q. Gong, J.-S. Zheng, C.-M. Zhang, Y.-Y. Xia and J. P. Zheng, ChemElectroChem, 2019, 6, 3020–3029 CrossRef CAS.
- X. Wang, H. Hao, J. Liu, T. Huang and A. Yu, Electrochim. Acta, 2011, 56, 4065–4069 CrossRef CAS.
- X. Zhang, J. Wu, S. Li, Y. Li, X. Li, Z. Zhu and J. Li, Solid State Ionics, 2022, 386, 116047 CrossRef CAS.
- Y. Cai, H. Huang, W. Bai, L. Sun, Z. Song, Z. Sun, S. Huo and P. Song, J. Mater. Chem. A, 2024, 12, 21895–21904 RSC.
- T. Chakraborty, B. Monserrat, A. Tănase, R. I. Walton and B. Karasulu, J. Mater. Chem. A, 2024, 12, 10059–10071 RSC.
- S. Karimzadeh, B. Safaei, W. Huang and T.-C. Jen, J. Energy Storage, 2023, 67, 107572 CrossRef.
- R. Pulido, N. Naveas, R. J. Martin-Palma, F. Agulló-Rueda, V. R. Ferró, J. Hernández-Montelongo, G. Recio-Sánchez, I. Brito and M. Manso-Silván, Materials, 2022, 15, 6237 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- B. Dec and R. Bogdanowicz, Photonics Lett. Pol., 2018, 10, 39–41 CrossRef CAS.
- A. A. Adllan and A. Dal Corso, J. Phys.:Condens. Matter, 2011, 23, 425501 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08073d. |
| This journal is © The Royal Society of Chemistry 2025 |