
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced cycling performance of bilayered vanadium oxide cathode in Li-ion batteries via dual metal-ion preintercalation†
Xinle
Zhang
 and
Ekaterina
Pomerantseva
*
and
Ekaterina
Pomerantseva
*
Department of Materials Science and Engineering, Drexel University, Philadelphia, PA 19104, USA. E-mail: ep423@drexel.edu
First published on 10th March 2025
Abstract
Chemical preintercalation of Li+ or Mg2+ ions can improve the specific capacity or cycling stability, respectively, of bilayered vanadium oxide (BVO) electrodes in Li-ion cells. However, advancing both properties simultaneously in a single material via chemical preintercalation of both Li+ and Mg2+ ions has never been reported. Herein, we experimentally demonstrated that by simultaneously pre-intercalating electrochemically active Li+ ions and structure-stabilizing Mg2+ ions, the specific capacity and cycling stability of the BVO electrodes in Li-ion cells can be synergistically improved. Additionally, we revealed the role of the interlayer structural water in the charge storage and degradation mechanisms of dual metal-ion preintercalated BVO electrodes. With the simultaneous preintercalation of 0.19 Li+ ions and 0.10 Mg2+ ions into the interlayer structure of BVO, the LMVO electrode demonstrated a specific capacity of ∼245 mA h g−1 in a potential window of 2.0–4.0 V (vs. Li/Li+) with a capacity retention of 58% after 50 cycles. Low-temperature vacuum-annealing at 200 °C reduced the hydration degree (n in δ-Li0.19Mg0.10V2O5·nH2O) of LMVO (denoted as LMVO-200) from 0.85 to 0.67 H2O per V2O5 without phase transformation and increased the contribution of diffusion-limited process while reducing the surface-controlled fraction of the charge storage mechanism, demonstrating further enhanced capacity retention of ∼66% after 100 cycles. Additionally, GITT experiments demonstrated that the dual metal-ion preintercalation and vacuum-drying treatment facilitated the diffusion of Li+ ions. The ex situ XRD and ATR FTIR analyses of the LMVO-200 electrode revealed its reversible bulk and local structure evolution during the electrochemical operation, enabling excellent cycling stability in Li-ion cells. This work demonstrates a general strategy to synergistically enhance the specific capacity and cycling stability of layered oxide electrode materials for intercalation batteries.
1. Introduction
Rechargeable batteries are indispensable in modern society for powering portable electronics and transport electrification. However, they are experiencing a surge in demand for higher energy and power density, thus, the structure and chemistry of the cathode materials need to be optimized for achieving higher capacity and longer lifespan.1–3 Commercialized cathode materials in Li-ion batteries, such as LiFePO4 and LiMn2O4 spinel4,5 or layered LiNixCoyMnzO2 and LiNixCoyAlzO2,6 usually deliver specific capacities below 200 mA h g−1, and their capacity and cycling stability are compromised in other battery systems.7,8 Moreover, the consumption of critical elements such as nickel (Ni) and cobalt (Co) in commercial cathode materials of Li-ion batteries raises concerns regarding material sustainability.9 Cathode materials based on layered vanadium oxides have demonstrated high capacity and versatility in a wide range of electrolyte systems10–15 owing to their spacious and tunable interlayer regions that enable fast intercalation and diffusion of various charge transport ions. In a lithium-ion battery system, bilayered vanadium oxides (BVOs) can accommodate up to two Li+ ions per unit cell when vanadium is reduced from V5+ to V4+,10 enabling a high theoretical capacity of 294 mA h g−1. However, maintaining such high capacity over extended cycling requires the preintercalation of inorganic metal ions16–20 or organic species21–23 to stabilize the large interlayer regions and suppress phase transformations,24 structural degradation,11 and electrochemical side reactions25 during the electrochemical operation of BVO electrodes.Chemical preintercalation is a versatile synthesis approach that enables inserting water-soluble foreign species into the interlayer regions of layered electrodes prior to electrochemical cycling, thereby enhancing the cathodic performance in multiple battery systems.26 It has been shown that the single alkali or alkali-earth metal ion preintercalated BVOs (δ-MxV2O5·nH2O, where M stands for Li, Na, K, Mg, or Ca) enable improved cycling performance in lithium-ion,19,20 sodium-ion,17,27 potassium-ion,18 magnesium-ion,28 and calcium-ion29 batteries with non-aqueous electrolytes. To understand the mechanisms of the chemical preintercalation effect, it has been proposed that the preintercalated metal ions can be distinguished as either electrochemically active (same as the electrolyte ions) or electrochemically inactive (different from the electrolyte ions).26 Chemical preintercalation of electrochemically active ions facilitates solid-state ion diffusion, thus leading to increased specific capacity, while chemical preintercalation of electrochemically inactive ions stabilizes the structure of electrode materials, thus improving the cycling stability.26 For instance, in the non-aqueous Li-ion cells, δ-Li0.51V2O5·nH2O exhibited the highest initial specific capacity compared to other metal ion preintercalated phases, but it exhibited poor cycling stability due to the lack of structure-stabilizing ions in the interlayer regions; the δ-Mg0.37V2O5·nH2O phase demonstrated superior cycling stability in Li-ion cells due to the stabilization effect of electrochemically inactive Mg2+ ions; however, the specific capacity was compromised.19,20 Therefore, the synthesis approach must be innovated to achieve synergistically enhanced capacity and stability for layered electrode materials by simultaneously pre-intercalating both electrochemically active and stabilizing ions.
It has been demonstrated that integrating two types of metal ions in layered vanadium oxides enhances their electrochemical cycling performance, but the mechanism and the role of chemically preintercalated ions remain elusive. One perspective ascribes that chemical preintercalated metal ions act as “pillars” that induce interlayer distance expansion,19,30 thus altering the transport of the electrolyte ions from diffusion-controlled faradaic process to surface-redox-controlled pseudocapacitive process.31 Jiang et al. reported an Mn and Li “dual-ion-in-sequence” preintercalated vanadium oxide that led the interlayer distance to expand from 9.4 Å to 11 Å, which enhanced ion diffusivity and pseudocapacitive behavior that was favorable for aqueous zinc-ion energy storage.32 Another study demonstrated that a large interlayer distance of 14.1 Å, achieved through simultaneously pre-intercalating K+ and Mg2+ ions into the interlayer regions, synergistically enhanced the specific capacity and cycling stability of the vanadium oxide electrode in zinc-ion cells.33 Since then, widening the interlayer regions of layered vanadium oxides via pre-intercalating more than one type of monovalent, bivalent, or even trivalent metal ions has been demonstrated for enhancing the charge storage properties of layered vanadium oxides.14,34–38 In comparison, the dual metal-ion preintercalation also alters the confined chemical environment of the ion transport channels (interlayer regions of layered vanadium oxide), and therefore, it might bring favorable energy storage features to electrode materials, such as improved redox reactivity or electronic conductivity. For instance, compared to the single Ni2+ or K+ preintercalated layered vanadium oxide electrodes, Ni2+ and K+ co-preintercalated electrodes not only exhibited a higher capacitive charge contribution but also facilitated the electron transport and minimized the charge storage overpotential, leading to a reduced charge transfer resistance, increased diffusion coefficient, and good electrochemical reversibility.39 In addition, it has been shown that the confined Na+ ions in the interlayer region of the MXene-derived δ-NaxKyV2O5·nH2O electrode facilitated the desolvation of the neighboring electrochemically active K+ ions and facilitated their diffusion in the electrodes of non-aqueous potassium batteries.40 Despite the preintercalation of Li+ or Mg2+ ions demonstrating the capability of enhancing specific capacity or cycling stability separately, and a high capacity and cycling stability have been reported by using V2CTx MXene as a precursor for a chemical preintercalation synthesis with Li+ or Mg2+ ions,20 chemical preintercalation of layered vanadium oxide electrodes with both Li+ and Mg2+ ions to achieve high capacity and stability has never been experimentally demonstrated.
In this work, we developed an innovative synthesis approach to simultaneously chemically pre-intercalate electrochemically active Li+ ions and stabilizing Mg2+ ions within the BVO structure. The amounts of chemically preintercalated Li+, Mg2+, and structural water per V2O5 were determined using atomic absorption spectroscopy (AAS) and thermogravimetric analysis (TGA), respectively, suggesting a chemical formula of δ-Li0.19Mg0.10V2O5·0.85H2O, denoted as LMVO. Additionally, low-temperature vacuum annealing was applied to LMVO, resulting in a reduced hydration degree (δ-Li0.19Mg0.10V2O5·0.67H2O, denoted as LMVO-200) without significantly altering its layered structure. The physical properties and electrochemical mechanisms of the LMVO and LMVO-200 polymorphs were studied in comparison to the δ-Li0.33V2O5·0.88H2O (LVO) and δ-Mg0.22V2O5·0.92H2O (MVO), demonstrating that the unique structure, morphology, chemistry, and enhanced electrochemical cycling performance can be achieved by tuning the chemically preintercalated metal ions and hydration degrees. Additionally, we conducted the ex situ XRD and FTIR analyses of the post-cycled electrodes to further understand the material structural evolution and degradation during battery operation. By experimentally demonstrating the synergistically enhanced capacity and cycling stability of LMVO and LMVO-200 in Li-ion cells achieved via dual metal-ion preintercalation strategy, we propose this methodology could be extended to other hydrated layered electrode materials and beyond lithium-ion battery systems.
2. Experimental methods
2.1 Dual metal-ion chemical preintercalation synthesis
The LMVO was synthesized using a two-step approach, including a wet chemical sol–gel step,41 followed by hydrothermal treatment, as illustrated in Fig. 1. For the sol–gel step, 1.43 g of anhydrous MgCl2 (99.9%, Thermo Scientific Chemicals) was dissolved in 15 mL of deionized water (ACS Reagent, Thermo Scientific) in a 250 mL beaker, followed by the addition of 15 mL of H2O2 (30 wt% aq., Thermo Scientific) to form a transparent solution. 0.5 g of V2O5 powder (99.6%, Thermo Scientific) was slowly added to the solution over a span of 15 minutes, followed by stirring for an hour at room temperature to form a clear orange sol. Then, the temperature of the stirring plate was raised to 60 °C and kept at this value for another 4 hours to induce precipitation. After that, the beaker was covered with Parafilm in a fume hood to age the precipitate for 4 days, as our previous work showed that the aging step is necessary to obtain a single bilayered phase.17 The aged precipitate was thoroughly washed with deionized water through vacuum filtration, then transferred into a 23 mL Teflon-lined autoclave (Parr Instruments) and mixed with 15 mL of 3 M LiCl (aq.) for hydrothermal treatment at 220 °C for 24 hours. The hydrothermally treated precipitate was thoroughly washed and filtered with deionized water, air-dried in a 105 °C oven for 24 hours, and denoted as LMVO. A portion of the LMVO powder was treated in a vacuum oven at 200 °C for 24 hours and denoted as LMVO-200. For reference, the LVO phase was synthesized using a similar approach with 2.33 g of LiCl (98+%, Thermo Scientific Chemicals) added in the sol–gel step; the MVO phase was prepared via the same sol–gel step as the LMVO powder, but it was hydrothermally treated in a 15 mL 0.5 M MgCl2 (aq.) solution instead of the LiCl solution used for the LMVO synthesis. Both reference materials were air-dried in a 105 °C oven for 24 hours.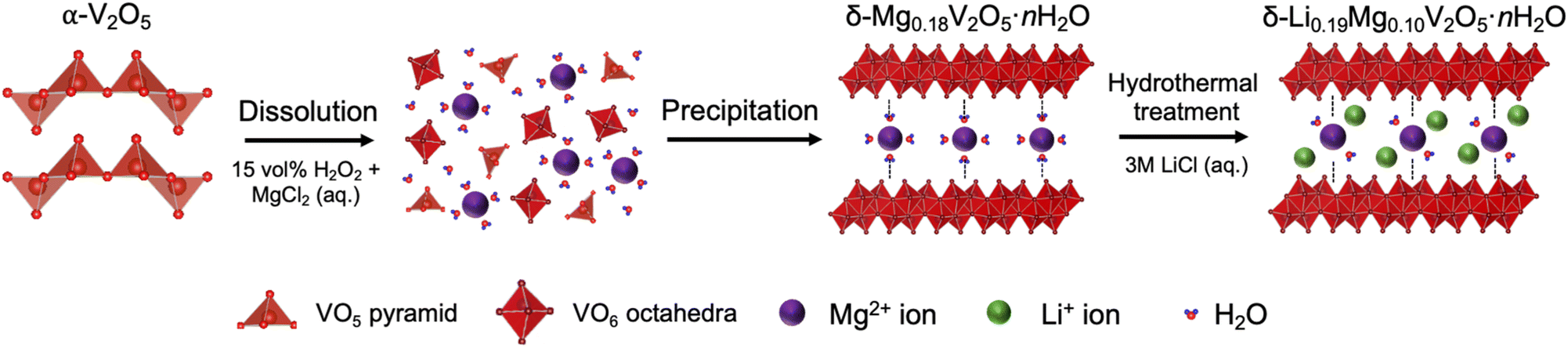 | ||
| Fig. 1 Schematic of the dual Li+ and Mg2+ ion chemical preintercalation of bilayered vanadium oxide. | ||
2.2 Physical characterizations
The X-ray diffraction (XRD) data were acquired using Rigaku MiniFlex 600 with Cu-Kα radiation (λ = 1.54 Å) from 2θ ranging from 3° to 40° with a step size of 0.02° and step duration of 0.5 s. The background of the diffraction patterns was subtracted using CrystalDiffract 6 software. The amount of structural water in each sample was evaluated using a Q50 thermogravimetric analyzer (TA instruments, USA) by measuring the weight loss from 100 °C to 1000 °C with a temperature ramp of 10 °C min−1, and the measurements were held at 100 °C for 60 min to exclude the impact of the physisorbed water. The FTIR pellets were prepared by hand-grinding 1 mg of the sample powder with 200 mg of KBr powder (ACS reagent, ≥99.0%) and pressing the powder in a 13 mm mold at 50 psi. The FTIR spectra in transmission mode were recorded using an INVENIO R Spectrometer (Bruker, U.S.A.) between 400 and 4000 cm−1. The stoichiometric formula of each sample was determined using a flame atomic absorption spectrometer (Shimadzu AA7000, Japan) equipped with an air-acetylene or nitroxide-acetylene burner for analyzing alkali-metal or transition-metal elements, respectively. The AAS calibration curves were developed using standard solutions purchased from Inorganic Ventures, USA. The scanning electron microscopy (SEM) images were captured using Apero 2S Low Vac (ThermoFisher, USA) with a 5 kV electron accelerating voltage captured using a trinity in-lens detector. XPS measurements were recorded on a Physical Electronics VersaProbe 5000 instrument using a monochromatic Al Kα source and ionized Ar for charge compensation. The high-resolution V 2p spectra were taken at a pass energy of 23.5 eV with a step size of 0.05 eV. The peak fitting and data analysis were carried out using CasaXPS software. A Shirley background was used for the V 2p spectra quantification.2.3 Electrochemical characterizations
The electrode slurry was prepared by ground mixing 70 wt% of the active material and 20 wt% carbon black (100% compressed, Thermo Scientific Chemicals), which was suspended in an NMP (99.5% 1-methyl-2-pyrrolidinone, Acros Organics) solution with 10 wt% of Kynar Flex PVDF (Arkema, USA) in a polypropylene cup using Flacktek SpeedMixer (Flacktek, USA). The electrode slurry was cast onto the aluminum current collector using an 80 μm film applicator. The electrode films were dried overnight at 105 °C in the vacuum oven and then punched into 10 mm electrode disks. The punched electrode disks were dried at 150 °C in the vacuum oven for 24 h before being transferred into the glovebox to minimize the amount of structural water in the electrodes, following the previously established procedures for hydrated oxides.42 The electrodes were tested in type 2032 coin-cells with a two-electrode configuration, where 12 mm lithium metal disks served as both counter and reference electrodes, 16 mm polymer films served as a separator (Celgard 2325, USA), and 1 M LiPF6 in EC/DEC (Gotion Inc., USA) was used as the electrolyte. The electrochemical properties were evaluated within a voltage window of 2.0–4.0 V using a multichannel potentiostat (Biologic VMP3, USA) and electrochemical workstation (Arbin Instruments, USA). All potentials in this work are reported with respect to the Li/Li+ reference electrode. The cyclic voltammetry curves were acquired using the scan rates of 0.1, 0.2, 0.4, 0.6, 0.8, and 1 mV s−1, respectively. The cycle life test was conducted by galvanostatically cycling the cells with a current density of 20 mA g−1, and the rate capabilities were assessed by cycling for 10 cycles at each of 20, 50, 100, 200, and 20 mA g−1 current densities at room temperature, respectively. To analyze the charge storage contributions due to the surface-controlled and diffusion-limited processes, cyclic voltammetry curves were acquired at multiple scan rates from 0.1 mV s−1 to 1 mV s−1. The measured peak current (i) and scan rate (v) comply with the following relationship:| i = avb |
log(i) = log(a) + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(v) log(v) |
By determining the slope of log(i) versus log(v) in the experimental data fitted to the equation above, the b-value was calculated. The b-value typically ranges from 0.5 to 1.0 and indicates the proportion of the diffusion-limited charge storage process (b = 0.5) and surface-controlled charge storage process (b = 1.0).
The GITT experiments consisted of 1800 s (30 min) of discharge periods at a current density of 10 mA g−1 and 72000 s (1200 min) of rest periods, which left the cells in an open circuit to reach the equilibrium states. The diffusion coefficients of Li+ ions (DLi+) were derived using the following equation:43,44
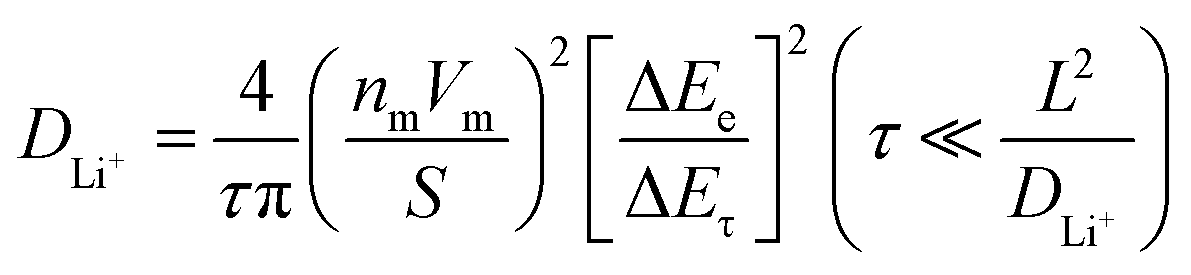
2.4 Ex situ XRD and FTIR analysis
The Li-ion cells containing the LMVO-200 electrode were cycled using a multichannel potentiostat (Biologic VMP3, USA) at the cut-off potentials of 3.6 V (open circuit potential), 2.7 V, 2.0 V (during 1st discharge), 3.0 V, and 4.0 V (during 1st charge) via linear sweep voltammetry (LSV) at a scan rate of 0.05 mV s−1. A relatively slow scan rate was chosen in order to minimize the hysteresis effect during the electrochemical operation. The electrochemically cycled cells were disassembled in an argon-filled glovebox, and the working electrodes were air-dried at 105 °C after being rinsed three times with dimethyl carbonate (99+%, Thermo Scientific Chemicals) prior to the ex situ XRD and FTIR analyses. The ex situ XRD patterns were acquired using a Rigaku MiniFlex 600 with Cu-Kα radiation (λ = 1.54 Å) over a 2θ range from 3° to 40°, with a step size of 0.02° and a step duration of 1.0 s. The attenuated total reflectance (ATR) mode was employed to non-destructively obtain the FTIR spectra of the ex situ electrodes, using an INVENIO R Spectrometer (Bruker, USA) scanned between 400 and 4000 cm−1.3. Results and discussions
The XRD patterns of LVO, MVO, LMVO, and LMVO-200, shown in Fig. 2a, confirm that the chemical preintercalation synthesis performed in this work yields nanostructured bilayered vanadium oxides with varying interlayer distances that depend on the type of the metal ions present in the sol–gel and hydrothermal treatment steps. The d-spacings of LVO and MVO, calculated from the positions of the (001) peaks, are 11.16 Å and 13.19 Å, respectively. These values are consistent with the previously reported δ-Li0.51V2O5·nH2O and δ-Mg0.37V2O5·nH2O phases and correspond to the BVO structures preintercalated with hydrated Li+ or Mg2+ ions, respectively.16,19,20 The joint analysis of TGA (Fig. 2b) and AAS (Fig. 3) was conducted to determine the stoichiometry between lithium, magnesium, vanadium, and structural water in the chemically preintercalated polymorphs in this work, as listed in Table 1. The contents of structural water in each sample were determined by evaluating the weight loss characteristics from 100 to 650 °C. The stoichiometries of the LVO and MVO reference materials in this work were determined as δ-Li0.33V2O5·0.88H2O and δ-Mg0.22V2O5·0.92H2O, respectively. The LMVO phase synthesized with both Li+ and Mg2+ ions shows an interlayer distance of 11.18 Å, similar to that of the LVO phase, but the AAS and EDS (ESI, Fig. S1†) analysis suggests that the LMVO possesses both lithium and magnesium elements with a stoichiometry of Li0.19Mg0.10V2O5·0.85H2O. The MVO obtained from the sol–gel step (denoted as MVO-aged), which is the precursor of LMVO prior to hydrothermal treatment, was determined to have a formula of δ-Mg0.18V2O5 (ESI, Figu. S2†). Comparing the magnesium content in MVO-aged (Mg0.18) and LMVO (Mg0.10) reveals that Mg2+ ions in MVO-aged were partially exchanged with Li+ ions from the LiCl solution during the hydrothermal treatment. The XRD analysis indicated that vacuum drying of LMVO at 200 °C (LMVO-200) caused the d-spacing to reduce from 11.18 Å to 10.98 Å without a phase transformation, and the TGA analysis indicates that the ∼0.2 Å decrease in d-spacing correspond to the dehydration of LMVO from 0.85 to 0.67 H2O per Li0.19Mg0.10V2O5. The XRD and TGA analyses confirmed that the interlayer distance is dominated by the hydration degree of the interlayer regions (n in δ-MxV2O5·nH2O), which can be impacted by the nature of metal ions introduced during the chemical preintercalation synthesis. Moreover, the TGA curves of LVO, LMVO, and LMVO-200 samples exhibited weight-regaining characteristics at temperatures between 650 to 680 °C, suggesting the presence of oxygen vacancies in these samples that were refilled by air oxygens during the TGA measurements.20,27,45 The FTIR spectra, shown in Fig. 2c, were acquired to understand the characteristics of structural water and the V–O in-plane structures. The normalized intensity of water bands at ∼1629 cm−1 (O–H bending) and 3429 cm−1 (O–H stretching) aligns with the hydration degrees determined via the TGA weight loss analysis: MVO exhibits the highest intensity, LVO and LMVO show comparable but lower intensities, and the most dehydrated LMVO-200 phase displays the weakest structural water characteristics (Fig. 2c). Additionally, the FTIR spectra exhibited the fingerprint region of bilayered vanadium oxide, including the V–O–V out-plane stretching, V–O–V in-plane stretching, and V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands at 522, 769, and 1012 cm−1, respectively, which confirmed the bilayered intra-layer structure of all samples in this study.27,45,46
O stretching bands at 522, 769, and 1012 cm−1, respectively, which confirmed the bilayered intra-layer structure of all samples in this study.27,45,46
 | ||
| Fig. 2 (a) XRD patterns, (b) TGA weight loss curves, and (c) FTIR spectra of the LVO, MVO, LMVO, and LMVO-200 phases. | ||
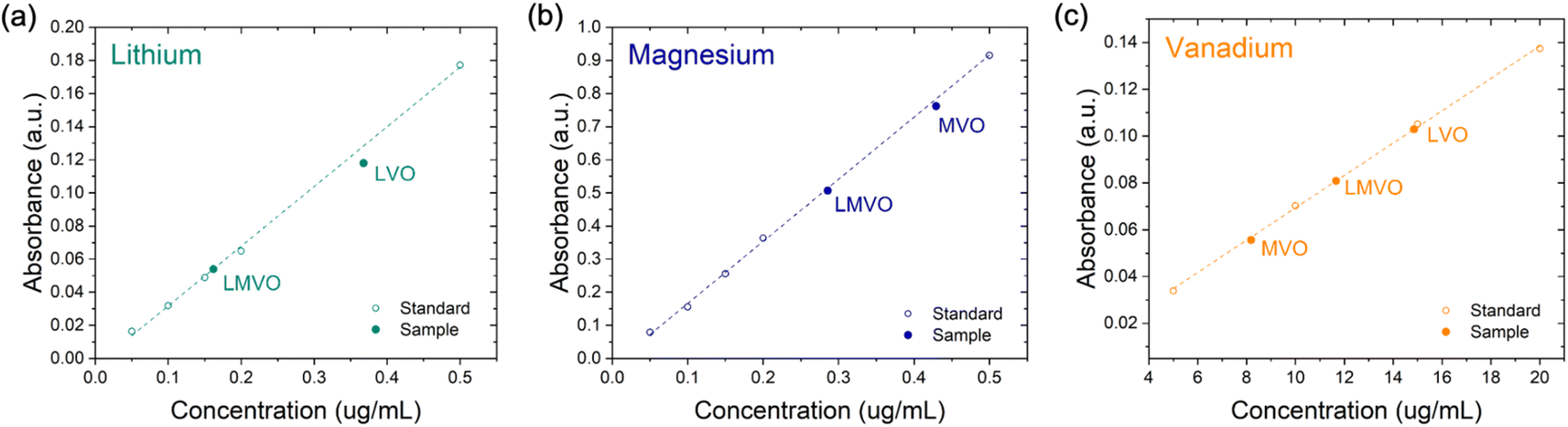 | ||
| Fig. 3 AAS calibration curves for (a) lithium, (b) magnesium, and (c) vanadium as well as the absorbances of the AAS samples prepared with LVO, MVO, and LMVO that correspond to the concentrations of each element. | ||
:V and Mg:V ratios (at%) determined via AAS, weight loss of structural water (wt%) determined via TGA, and stoichiometry of LVO, MVO, LMVO, and LMVO-200 according to the AAS and TGA analyses
| Sample name | Li:V ratio (via AAS) |
Mg:V ratio (via AAS) |
Structural water loss (wt%, via TGA) | Chemical formula |
|---|---|---|---|---|
| LVO | 0.1666 | N/A | 7.90 | δ-Li0.33V2O5·0.88H2O |
| MVO | N/A | 0.1098 | 8.12 | δ-Mg0.22V2O5·0.92H2O |
| LMVO | 0.0971 | 0.0511 | 7.60 | δ-Li0.19Mg0.10V2O5·0.85H2O |
| LMVO-200 | 0.0971 | 0.0511 | 6.12 | δ-Li0.19Mg0.10V2O5·0.67H2O |
Fig. 4 shows the SEM images of the LVO, MVO, and LMVO particles at low and high magnifications. Consistent with the literature, the SEM images indicate that LVO and MVO particles represent approximately 200 nm wide one-dimensional (1D) nanobelts.19 However, the nanobelts in the SEM images of LVO appear more disordered and entangled, while those in the SEM images of MVO are straighter, better aligned, and packed parallel to each other. The simultaneous preintercalation of lithium and magnesium ions leads to drastic changes in the morphology: the LMVO powder is composed of crumpled nanosheets. It appears that the chemical preintercalation of Li+ and Mg2+ ions simultaneously induces the transformation of 1D nanowires into two-dimensional (2D) crumpled nanosheets, and according to the literature, a 2D morphology typically offers superior electrochemical cycling stability to BVO compared to 1D nanowires with similar chemistries.20,47 The SEM images imply that the transition of morphology from 1D to 2D could be a factor that contributes to the electrochemical cycling stability of LMVO.
 | ||
| Fig. 4 (a–c) Low magnification and (d–f) high magnification SEM images of the (a and d) LVO, (b and e) MVO, and (c and f) LMVO phases. | ||
To study the influence of the chemical preintercalation of metal ions on the vanadium oxidation states, XPS spectra in the V 2p region of LVO, MVO, LMVO, and LMVO-200 were analyzed and are shown in Fig. 5. The peaks at binding energies of ∼517.2 and ∼524.5 eV correspond to the V 2p3/2 and V 2p1/2 characteristics of V5+, respectively. Due to the preintercalation of positively charged metal ions, the characteristics of V 2p3/2 and V 2p1/2 from V4+ were identified in the V 2p region of MVO and LMVO-200 at the binding energies of ∼515.8 and ∼523.2 eV. The XPS spectra suggest that no vanadium reduction occurred in LVO and LMVO, revealing the strong charge-shielding effect of structural water,48–50 which is located at the interlayer regions and surrounds the chemically preintercalated Li+ and Mg2+ ions. We believe the structural water in MVO mitigated the vanadium reduction by the same charge-shielding effect, but the charges carried by the chemically preintercalated Mg2+ ions were not fully neutralized, therefore leading to the reduction of vanadium. After vacuum-drying at 200 °C, the formation of V4+ in LMVO-200 implies the introduction of additional bonds and increased stacking order of the bilayers, which have been demonstrated to improve electrochemical stability during battery operation.27 The XPS analysis revealed that the vanadium in LVO and LMVO can be electrochemically reduced from its highest valence state during battery operation, thus enabling their relatively high specific capacity compared to MVO and LMVO-200, in which vanadium reduction occurred during synthesis. To further understand the characteristics of oxygen and oxygen vacancies, the XPS spectra in the O 1s region are shown in ESI, Fig. S3.† According to the XPS O 1s spectra, all samples show characteristics of the V–O lattice oxygen at ∼530 eV and two shoulder peaks at the binding energies of ∼531.2 and ∼532.5 eV, which correspond to the characteristics of oxygen vacancies and structural water, respectively.14,51 The presence of oxygen vacancy characteristics in the O 1s spectra of LVO, LMVO, and LMVO-200 agree with the presence of weight-regaining characteristics observed at 650 to 680 °C in the TGA curves. Additionally, other oxygen characteristics in the O 1s XPS spectra, such as water–oxygen substituted oxygen vacancies, also show significance in electrochemical applications. For example, the O 1s XPS spectrum suggests the presence of oxygen vacancies in MVO (ESI, Fig. S3b†), but no refilling of oxygen vacancies (weight regaining characteristic) is observed in its TGA weight loss curve. This observation indicates the oxygen vacancies in the V–O sublattice of MVO might be occupied by structural water oxygens, similar to the characteristic reported in layered molybdenum oxides,52,53 and leading to no weight-regaining characteristic in its TGA weight loss curve.
 | ||
| Fig. 5 XPS spectra and fittings in the V 2p region of (a) LVO, (b) MVO, (c) LMVO, and (d) LMVO-200. | ||
The CV curves at the scan rates of 0.1–1.0 mV s−1, demonstrated in Fig. 6, suggest that the four samples underwent similar two-step redox processes between 2.0–4.0 V. At a scan rate of 0.1 mV s−1, the first cathodic/anodic pair is located at ∼2.45/2.80 V (peak 2/peak 3), and another cathodic peak appears at ∼2.80 V (peak 1) with a broad shoulder anodic peak at above 3.0 V (peak 4). As the scan rate increases, all the cathodic/anodic peaks in the CV curves shift to lower/higher potentials due to the charge polarization effect. We performed the b-value analyses for peaks 2 & 3 (peaks 1 & 4 are too broad to reliably identify their potentials), shown in Fig. 6(b, d, f and h), to further understand the charge storage mechanism. Since the peaks in the CV curves of LVO broadened and shifted significantly when the scan rate increased to 0.6 mV s−1, the fitting of the b value for LVO is only taken from cycles with the scan rates of 0.1, 0.2, and 0.4 mV s−1. The peaks 2 and 3 of LVO, labeled in Fig. 6(a), were determined from the b-values of 0.893 and 0.824, respectively, indicating a pseudocapacitive characteristic dominated by the surface-controlled capacitive processes. The two anodic peaks in MVO exhibit a strong rate-dependent behavior, as peak 3 dominates the anodic process at low scan rates (0.1–0.4 mV s−1), but it was surpassed by peak 4 at higher scan rates (0.6–1.0 mV s−1). The b-value analysis reveals that the peak 2/3 in MVO is dominated by a diffusion-limited process (b = 0.707/0.606). As the scan rate increases (>0.6 mV s−1), peaks 1 and 4 in MVO broaden significantly, which suggests the surface-controlled process gradually dominates the charge storage mechanism. This observation suggests that peaks 1 and 4 in MVO possibly involve the pseudocapacitive processes mediated by structural water at high scan rates.54,55 The peaks 2 and 3 in the CV curves of MVO were suggested to have smaller b values compared to those of LVO, revealing that higher diffusion-limited kinetics could be enabled by pre-intercalating electrochemically inactive Mg2+ ions. The b values of the three peaks in LMVO are between those of LVO and MVO, suggesting that the chemically preintercalated Li+ ions are electrochemically active and effectively facilitate ion diffusion. Peaks 2 and 3 of LMVO were determined to have b values of 0.818 and 0.785, respectively. These values decreased to 0.779 and 0.752 after vacuum-drying at 200 °C (as shown by LMVO-200, Fig. 6h), indicating that the removal of structural water mitigated the charge-shielding effect of structural water in LMVO-200, therefore diminishing the surface-controlled charge storage contribution.49,50,54,55
 | ||
| Fig. 6 Sweep rate-dependent cyclic voltammetry study of (a and b) LVO, (c and d) MVO, (e and f) LMVO, and (g and h) LMVO-200 electrodes. (a, c, e and g) CV curves with the scan rates of 0.1, 0.2, 0.4, 0.6, 0.8, and 1.0 mV s−1, and (b, d, f and h) b values derived from the logi vs. logv profiles at the peak current densities of the corresponding CV curves. | ||
To further investigate the effects of the dual metal-ion preintercalation on specific capacity and cycling stability, electrochemical cycling tests were conducted in Li-ion cells containing LMVO and LMVO-200 electrodes, using LVO and MVO electrodes as references. Fig. 7(a) shows the 2nd cycle galvanostatic charge–discharge profiles of each electrode, indicating their specific capacities in the initial state. The discharge curves exhibit two plateaus at ∼2.80 and ∼2.45 V and the charge curves exhibit one plateau at ∼2.80 V, in agreement with the potentials of the redox peaks observed in the CV curves (0.1 mV s−1). The LMVO electrode exhibited an initial specific capacity of 244.55 mA h g−1, which is between that of LVO and MVO, suggesting that the specific capacity and cycling stability of the BVO electrodes are tunable by controlling the chemistry of the interlayer region. After partially removing the structural water from LMVO via vacuum annealing at 200 °C, the LMVO-200 electrode showed an initial discharge capacity of 223 mA h g−1, which decreased by ∼20 mA h g−1 compared to the LMVO electrode. This trend could be attributed to the decreased interlayer distance and surface-controlled capacitive contribution due to dehydrating the interlayer regions, implied by the XRD pattern and the above b-value analysis. Fig. 7(b) shows the comparison of the cycling stabilities within 100 discharge cycles at a fixed current density of 20 mA g−1. The LMVO electrode, containing 0.19 Li+ and 0.10 Mg2+ per V2O5, exhibited the stabilization effect of Mg2+ without significantly compromising specific capacity due to the concurrent preintercalation of Li+ ions. The LMVO-200 electrode, which was partially dehydrated compared to LMVO, demonstrated the highest electrochemical cycling stability in this work, retaining ∼66% of the specific capacity relative to its initial state after the 100th cycle. The enhanced electrochemical stability of LMVO-200 is attributed to its most dehydrated interlayer regions enabled by vacuum-drying, which suppresses the structural water-involved parasitic reactions, such as irreversibly producing electrochemically inactive LiOH during the electrochemical cycling.25
 | ||
| Fig. 7 Galvanostatic cycling results for the LVO, MVO, LMVO, and LMVO-200 electrodes: (a) charge–discharge profile of the 2nd cycle, (b) cycling stability for 100 cycles, (c) rate capabilities at current densities of 20, 50, 100, 200, and 20 mA g−1 (electrodes were cycled at each current density for 10 cycles), and (d) Li+ diffusion coefficients derived from the GITT experiments. | ||
Ex situ XRD analysis of the electrodes after 20 galvanostatic cycles at 20 mA g−1 (ESI, Fig. S4†) was conducted to understand the degradation mechanism of the BVO polymorphs in this work. Before cycling, the interlayer distances of the active material in the pristine electrodes (ESI, Fig. S4a†) vary from ∼11 to ∼13 Å, which generally agrees with the interlayer distances calculated from the powder XRD patterns (Fig. 2a). After 20 cycles, the XRD patterns of the post-cycling electrodes (ESI, Fig. S4b†) suggest that their interlayer distances declined to ∼10.8 Å regardless of the chemistry of the interlayer regions, possibly implying that the extraction of varying amounts of structural water occurred during electrochemical cycling. This finding underscores the importance of minimizing interlayer structural water without inducing phase transformation to mitigate the cathode-originated structural water shuttling effect. Additionally, the XRD patterns of the electrodes after 20 cycles maintained good shape and intensity of their (001) peaks, suggesting that all four polymorphs were able to retain their layered structure during the first 20 cycles. The ex situ XRD analysis of the post-cycling electrodes revealed that the capacity decay during the first 20 cycles, especially for the LVO electrodes, could be attributed to the shuttling of the interlayer structural water rather than a disordered layered structure or irreversible structural transformation.
The rate capabilities of the LVO, MVO, LMVO, and LMVO-200 electrodes are shown in Fig. 7c. The LVO electrode exhibited capacity degradation as the current density increased as it only retained ∼70 mA h g−1 and ∼30 mA h g−1 at current densities of 100 and 200 mA g−1, respectively. The MVO electrode delivered ∼120 mA h g−1 at 100 mA g−1 and ∼95 mA h g−1 at 200 mA g−1, performing better compared to the LVO electrode. The LMVO and LMVO-200 electrodes exhibited a similar cycling characteristic as the cycle life test in the first 10 cycles. As the current density increases, the LMVO electrode delivered ∼220 mA h g−1 at 50 mA h g−1, ∼190 mA h g−1 at 100 mA g−1, and ∼165 mA h g−1 at 200 mA g−1, suggesting the rate capability was significantly improved by the simultaneous preintercalation of Li+ and Mg2+ ions. The specific capacity of the LMVO electrode decreased by ∼17 mA h g−1, ∼15 mA h g−1, and ∼18 mA h g−1 as the current densities increased from 20 to 50 mA g−1, 50 to 100 mA g−1, and 100 to 200 mA g−1, respectively. In comparison, the specific capacity of the LMVO-200 electrode decreased by only ∼18 mA h g−1, ∼10 mA h g−1, and 12 mA h g−1 under the same conditions, suggesting further enhanced rate capabilities due to partial dehydration enabled via vacuum-drying. When the current density was set back to 20 mA g−1, the LVO electrode only retained ∼150 mA h g−1, which is significantly lower than the first 10 cycles at 20 mA g−1. The recovered specific capacities of the MVO, LMVO, and LMVO-200 electrodes at 20 mA g−1 were ∼140, ∼210, and ∼190 mA h g−1, respectively, each declining by ∼30 mA h g−1 compared to their specific capacities during the first 10 cycles.
The diffusion coefficients shown in Fig. 7d, derived from the GITT experiments (ESI, Fig. S5†), indicate that the dual metal-ion preintercalation enables higher diffusion coefficients of Li+ ions in the LMVO electrodes compared to the single metal-ion preintercalated LVO or MVO electrodes. After the vacuum drying treatment, the LMVO-200 electrode demonstrated the highest diffusion coefficient in this study, which could be attributed to the partial dehydration that further enhanced the synergistic effect of the Li+ and Mg2+ dual metal-ion preintercalation. In general, LMVO-200 demonstrated the best overall electrochemical performance in Li-ion cells, which is attributed to the simultaneous preintercalation of electrochemically active Li+ and structure-stabilizing Mg2+ ions, followed by a vacuum-drying procedure that minimizes the content of structural water without phase transformation, which led to the charge contribution from the surface-controlled capacitive partially shifting to diffusion-limited processes and suppressing the irreversible parasitic reaction between the Li+ ions and structural water.
To investigate the local and bulk structural evolution of the dual metal-ion preintercalated and partially dehydrated BVO electrodes during electrochemical cycling, the post-cycled LMVO-200 electrode at different potentials were analyzed using ex situ XRD and FTIR techniques. The CV curve of the studied electrode is shown in Fig. 8(a), indicating the cut-off potentials selected for the ex situ XRD and FTIR analysis. The (001) reflection in the XRD patterns of the pristine LMVO-200 electrode, shown in Fig. 8(b), indicates that the interlayer distance of LMVO-200 slightly increased from 10.98 Å (from its powder XRD) to 11.36 Å during the electrode preparation steps, but generally retained its layered structure. As the discharge progressed from OCP (∼3.55 V) to 2.0 V, the (001) reflection shifted to a higher 2θ angle, indicating a decrease in the interlayer distance of LMVO-200 to 8.81 Å due to the electrochemical intercalation of Li+ ions. The increase in potential up to 4.0 V on the charge cycle, corresponding to the extraction of Li+ ions, led to a low-angle shift of the (001) reflection that corresponds to the increase of the interlayer distance to 11.05 Å, revealing the “lattice breathing effect” and good stability of the layered crystal structure of LMVO-200 upon a reversible electrochemical cycle. The FTIR spectra of the LMVO-200 electrode in the BVO fingerprint region, revealing the structural changes within the V–O bilayers at various potentials, are shown in Fig. 8(c). The FTIR spectrum of the pristine LMVO-200 electrode exhibits similar characteristics to its powder FTIR spectrum, primarily featuring three bands corresponding to the V–O–V out-of-plane, V–O–V in-plane, and VO stretching at wavenumbers of ∼500 cm−1, ∼720 cm−1, and ∼1000 cm−1, respectively. The band at ∼1000 cm−1, indicating the stretching of VO, shifts to a lower wavenumber and broadens significantly upon discharge, suggesting a shortening of the atomic distance between vanadium and terminal (vanadyl) oxygen. Additionally, the intensities of the V–O–V out-of-plane and in-plane bands at ∼500 and ∼720 cm−1 attenuate significantly upon discharge, indicating a loss of bilayered ordering within the V–O 2D slabs. However, when the electrode's potential is restored to 4.0 V, the V–O slabs revert to their bilayered structure, as the characteristics of bilayered vanadium oxide reappear in the ex situ FTIR spectrum at 4.0 V. The combination of XRD and FTIR ex situ analyses revealed that the excellent cycling stability of the LMVO-200 electrode is attributed to its good structural reversibility from the aspect of bulk (inter-bilayer) and local (intra-bilayer) structures during the electrochemical operation in Li-ion cells.
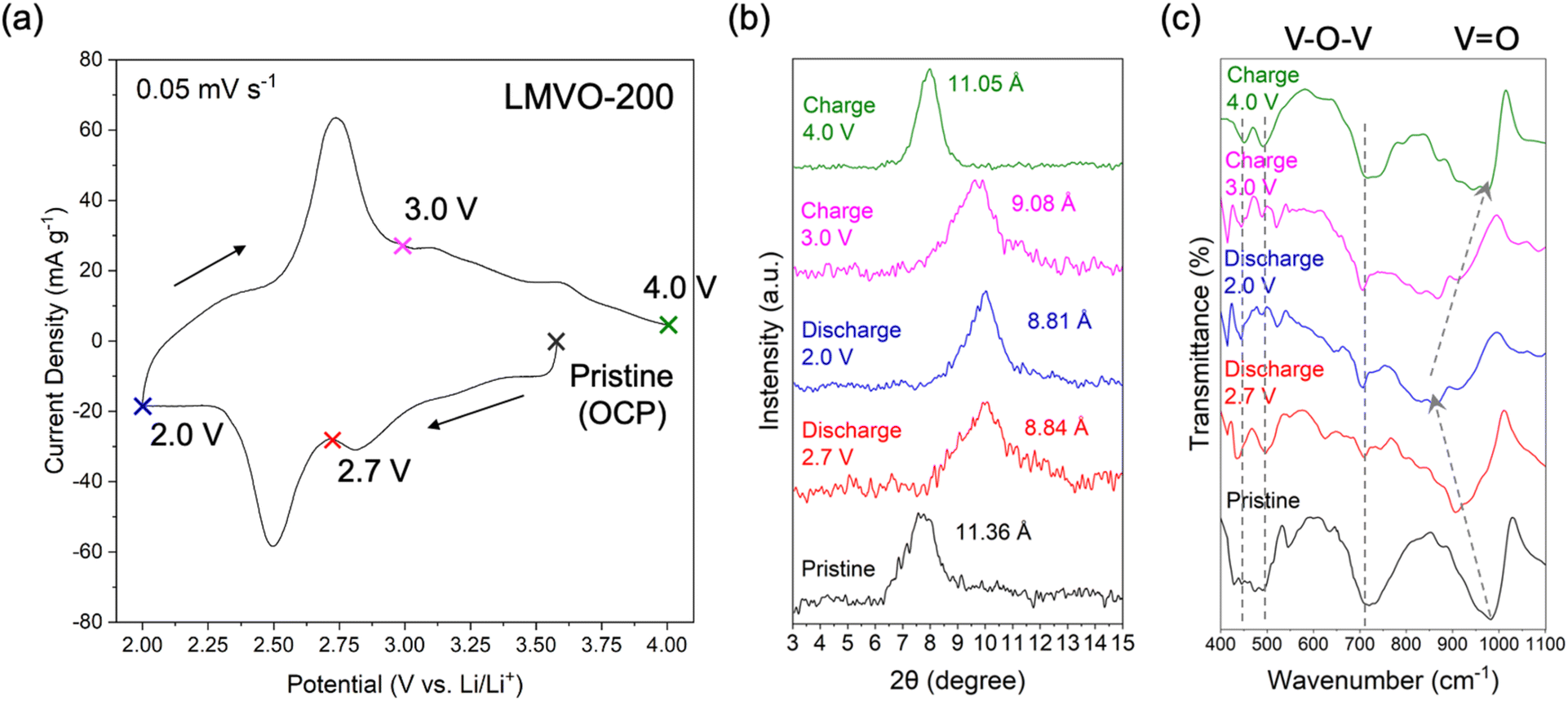 | ||
| Fig. 8 (a) CV curve of the LMVO-200 electrode scanned at 0.05 mV s−1 with the indication of the open circuit potential (OCP) and potentials at which ex situ XRD and FTIR analyses were performed: 2.7 V (discharge), 2.0 V (discharge), 3.0 V (charge), and 4.0 V (charge); (b) ex situ XRD patterns and (c) ex situ ATR FTIR analysis of the vanadium oxide fingerprint region for the LMVO-200 electrode at the selected potentials, respectively. | ||
This work not only experimentally demonstrated the non-aqueous cycling performance of layered electrode materials can be advanced via engineering the chemistries of the interlayer regions but also elucidated the role of multi-species (Li+, Mg2+, and structural H2O) from the interlayer regions of the electrode material in rechargeable Li-ion batteries. The tuning of chemistries from the interlayer regions highlighted the opportunities of advancing charge storage properties of other layered electrode materials with a large interlayer spacing (e.g., layered manganese49 and tungsten oxide54,55) for beyond non-aqueous electrolyte and beyond lithium-ion chemistries, as the electrochemically inactive ion could be identified (the electrochemical active ion is obvious). Additionally, developing new layered electrode materials using this methodology requires the innovation of new synthesis methods and processing approaches to achieve the desired structure and chemistry. The ex situ XRD and ATR FTIR analysis in this work emphasized that understanding the intra-layer structure evolution and reversibility is equally important as the widely investigated and acknowledged inter-layer structural evolution during electrochemical operation. A similar experiment protocol or methodology could be established to holistically study the structural evolution of layered electrode materials for a better understanding of the charge storage and degradation mechanisms, therefore fostering the availability of better materials for electrochemical energy storage technologies.
Conclusions
In this work, we demonstrated a feasible two-step synthesis route, including the chemical preintercalation of magnesium, followed by hydrothermal treatment in a LiCl solution, which allowed to simultaneously introduce 0.19 Li+ and 0.10 Mg2+ ions into the interlayer region of nanostructured BVO, producing a material with the chemical formula of δ-Li0.19Mg0.10V2O5·0.85H2O. Through a low-temperature vacuum drying, the hydration degree (n in δ-Li0.19Mg0.10V2O5·nH2O) was reduced from 0.85 to 0.67 without a phase transformation. By comparing the BVO polymorphs with individually preintercalated Li+ or Mg2+ ions, we experimentally demonstrated that the specific capacity and cycling stability of BVOs can be improved by the simultaneous preintercalation of Li+ and Mg2+ ions, which act as species that facilitate diffusion and stabilize the structure during electrochemical cycling in Li-ion cells, respectively. The b-value analyses revealed that the preintercalation of Li+ enhances the surface-controlled charge storage process, while the preintercalation of Mg2+ promotes the charge storage contribution from the diffusion-limited process. By comparing the b-values determined for the LMVO and LMVO-200 electrodes, we experimentally demonstrated that the surface-controlled charge transport process can be further facilitated by the interlayer structural water, enabling a higher capacity of the LMVO electrode. By pre-intercalating two types of metal ions simultaneously, δ-Li0.19Mg0.10V2O5·0.85H2O exhibited synergistic improvements in specific capacity and cycling stability, delivering 245 mA h g−1 during the initial discharge cycles and retaining ∼58% of its capacity after 50 cycles. This cycling stability is comparable to the ∼57% capacity retention of MVO (δ-Mg0.22V2O5·0.92H2O) and significantly higher than the 22% retention observed for LVO, achieved without any notable significant sacrifice in specific capacity. The dual metal ion preintercalated and vacuum-dried δ-Li0.19Mg0.10V2O5·0.67H2O electrode retained ∼66% of its initial specific capacity after 100 cycles. This improvement is attributed to the reduction of structural water, which leads to a transition from a surface-controlled process to a diffusion-limited process while simultaneously suppressing water-related parasitic reactions. The diffusion coefficients, derived from the GITT experiments, suggested that the simultaneous preintercalation of Li+ and Mg2+ ions combined with the vacuum-drying step at 200 °C effectively facilitated the diffusion of Li+ ions, compared to the single metal ion preintercalated electrodes. Through the analyses of diffusion coefficients and b-values, we conclude that the preintercalated Li+ ions define specific diffusion sites in the interlayer regions during synthesis, leading to the increased capacity due to higher Li+ diffusion coefficients and the preintercalated Mg2+ ions substantially improve the cycling stability without significantly sacrificing the specific capacity by promoting the diffusion-limited charge storage contribution. Through the post-cycled analyses of electrodes via XRD and ATR FTIR, the excellent cycling stability of the δ-Li0.19Mg0.10V2O5·0.67H2O electrode in Li-ion cells was attributed to its highly robust intra- and inter-bilayer structure reversibility during the electrochemical operation. By experimentally demonstrating that specific capacity and cycling stability can be synergistically enhanced through dual metal-ion preintercalation, this work underscores the importance of understanding the role of multiple interlayer species introduced during the synthesis and processing steps. Furthermore, it suggests that similar studies could be extended to other layered electrodes and energy storage systems beyond Li-ion batteries.Data availability
Data for this article, including XRD, TGA, FTIR, AAS, XPS, and electrochemical analyses, are available at Materials Commons 2.0 at https://doi.org/10.13011/m3-s85w-cv37.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation grant DMR-2106445. The XRD, SEM and XPS analyses were conducted using instruments in the Materials Characterization Core (MCC) at Drexel University. We thank Prof. Kevin Owens from the Department of Chemistry at Drexel University for providing us with access to the atomic absorption spectroscopy analysis in the Instrumental Analytical Laboratory.References
- A. Manthiram, A reflection on lithium-ion battery cathode chemistry, Nat. Commun., 2020, 11(1), 1550, DOI:10.1038/s41467-020-15355-0.
- K. Chen, P. Barai, O. Kahvecioglu, L. Wu, K. Z. Pupek, M. Ge, L. Ma, S. N. Ehrlich, H. Zhong and Y. Zhu, et al., Cobalt-free composite-structured cathodes with lithium-stoichiometry control for sustainable lithium-ion batteries, Nat. Commun., 2024, 15(1), 430, DOI:10.1038/s41467-023-44583-3.
- S. N. Lauro, J. N. Burrow and C. B. Mullins, Restructuring the lithium-ion battery: A perspective on electrode architectures, eScience, 2023, 3(4), 100152, DOI:10.1016/j.esci.2023.100152.
- G. Zhang, M. Li, Z. Ye, T. Chen, J. Cao, H. Yang, C. Ma, Z. Jia, J. Xie and N. Cui, et al., Lithium Iron Phosphate and Layered Transition Metal Oxide Cathode for Power Batteries: Attenuation Mechanisms and Modification Strategies, Materials, 2023, 16(17), 5769 CrossRef CAS PubMed.
- Y. Huang, Y. Dong, S. Li, J. Lee, C. Wang, Z. Zhu, W. Xue, Y. Li and J. Li, Lithium Manganese Spinel Cathodes for Lithium-Ion Batteries, Adv. Energy Mater., 2021, 11(2), 2000997, DOI:10.1002/aenm.202000997.
- M. Akhilash, P. S. Salini, B. John and T. D. Mercy, A journey through layered cathode materials for lithium ion cells – From lithium cobalt oxide to lithium-rich transition metal oxides, J. Alloys Compd., 2021, 869, 159239, DOI:10.1016/j.jallcom.2021.159239.
- K. M. Abraham, How Comparable Are Sodium-Ion Batteries to Lithium-Ion Counterparts?, ACS Energy Lett., 2020, 5(11), 3544–3547, DOI:10.1021/acsenergylett.0c02181.
- L. Peng, Y. Zhu, D. Chen, R. S. Ruoff and G. Yu, Two-Dimensional Materials for Beyond-Lithium-Ion Batteries, Adv. Energy Mater., 2016, 6(11), 1600025, DOI:10.1002/aenm.201600025.
- L. Ming, M. Ruzi, D. Anting, Z. Kai, G. Fan and X. Chengliang, Electrochemical technology to drive spent lithium-ion batteries (LIBs) recycling: recent progress, and prospects, Energy Mater., 2024, 4(6), 400070, DOI:10.20517/energymater.2024.29.
- A. Moretti and S. Passerini, Bilayered Nanostructured V2O5·nH2O for Metal Batteries, Adv. Energy Mater., 2016, 6(23), 1600868, DOI:10.1002/aenm.201600868.
- S. Tepavcevic, H. Xiong, V. R. Stamenkovic, X. Zuo, M. Balasubramanian, V. B. Prakapenka, C. S. Johnson and T. Rajh, Nanostructured Bilayered Vanadium Oxide Electrodes for Rechargeable Sodium-Ion Batteries, ACS Nano, 2012, 6(1), 530–538, DOI:10.1021/nn203869a.
- D. S. Charles, M. Feygenson, K. Page, J. Neuefeind, W. Xu and X. Teng, Structural water engaged disordered vanadium oxide nanosheets for high capacity aqueous potassium-ion storage, Nat. Commun., 2017, 8(1), 15520, DOI:10.1038/ncomms15520.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, A high-capacity and long-life aqueous rechargeable zinc battery using a metal oxide intercalation cathode, Nat. Energy, 2016, 1(10), 16119, DOI:10.1038/nenergy.2016.119.
- P. Qi, H. Wang, L. Li, X. Shen, L. Ou, M. Chen, Y. Lu, K. Hao and Y. Tang, Facile Synthesis of Oxygen-Vacancy-Rich Li–K Coinsertion Vanadate Nanoflakes for High-Performance Zn-Ion Batteries, ACS Sustain. Chem. Eng., 2023, 11(40), 14691–14700, DOI:10.1021/acssuschemeng.3c02616.
- X. Xu, F. Xiong, J. Meng, X. Wang, C. Niu, Q. An and L. Mai, Vanadium-Based Nanomaterials: A Promising Family for Emerging Metal-Ion Batteries, Adv. Funct. Mater., 2020, 30(10), 1904398, DOI:10.1002/adfm.201904398.
- F. Ming, H. Liang, Y. Lei, S. Kandambeth, M. Eddaoudi and H. N. Alshareef, Layered MgxV2O5·nH2O as Cathode Material for High-Performance Aqueous Zinc Ion Batteries, ACS Energy Lett., 2018, 3(10), 2602–2609, DOI:10.1021/acsenergylett.8b01423.
- M. Clites, B. W. Byles and E. Pomerantseva, Effect of aging and hydrothermal treatment on electrochemical performance of chemically pre-intercalated Na–V–O nanowires for Na-ion batteries, J. Mater. Chem. A, 2016, 4(20), 7754–7761, 10.1039/C6TA02917E.
- M. Clites, J. L. Hart, M. L. Taheri and E. Pomerantseva, Chemically Preintercalated Bilayered KxV2O5·nH2O Nanobelts as a High-Performing Cathode Material for K-Ion Batteries, ACS Energy Lett., 2018, 3(3), 562–567, DOI:10.1021/acsenergylett.7b01278.
- M. Clites and E. Pomerantseva, Bilayered vanadium oxides by chemical pre-intercalation of alkali and alkali-earth ions as battery electrodes, Energy Storage Mater., 2018, 11, 30–37, DOI:10.1016/j.ensm.2017.09.005.
- P. Ridley, C. Gallano, R. Andris, C. E. Shuck, Y. Gogotsi and E. Pomerantseva, MXene-Derived Bilayered Vanadium Oxides with Enhanced Stability in Li-Ion Batteries, ACS Appl. Energy Mater., 2020, 3(11), 10892–10901, DOI:10.1021/acsaem.0c01906.
- X. Zhang, R. Andris, T. Averianov, M. J. Zachman and E. Pomerantseva, Hybrid bilayered vanadium oxide electrodes with large and tunable interlayer distances in lithium-ion batteries, J. Colloid Interface Sci., 2024, 674, 612–623, DOI:10.1016/j.jcis.2024.06.164.
- Y. Zhang, L. Xu, H. Jiang, Y. Liu and C. Meng, Polyaniline-expanded the interlayer spacing of hydrated vanadium pentoxide by the interface-intercalation for aqueous rechargeable Zn-ion batteries, J. Colloid Interface Sci., 2021, 603, 641–650, DOI:10.1016/j.jcis.2021.06.141.
- Y. Tong, Y. Zang, S. Su, Y. Zhang, J. Fang, Y. Yang, X. Li, X. Wu, F. Chen and J. Hou, et al., Methylene blue intercalated vanadium oxide with synergistic energy storage mechanism for highly efficient aqueous zinc ion batteries, J. Energy Chem., 2023, 77, 269–279, DOI:10.1016/j.jechem.2022.10.040.
- C. K. Christensen, D. R. Sørensen, J. Hvam and D. B. Ravnsbæk, Structural Evolution of Disordered LixV2O5 Bronzes in V2O5 Cathodes for Li-Ion Batteries, Chem. Mater., 2019, 31(2), 512–520, DOI:10.1021/acs.chemmater.8b04558.
- L. W. Wangoh, Y. Huang, R. L. Jezorek, A. B. Kehoe, G. W. Watson, F. Omenya, N. F. Quackenbush, N. A. Chernova, M. S. Whittingham and L. F. J. Piper, Correlating Lithium Hydroxyl Accumulation with Capacity Retention in V2O5 Aerogel Cathodes, ACS Appl. Mater. Interfaces, 2016, 8(18), 11532–11538, DOI:10.1021/acsami.6b02759.
- E. Pomerantseva, Chemical Preintercalation Synthesis of Versatile Electrode Materials for Electrochemical Energy Storage, Acc. Chem. Res., 2023, 56(1), 13–24, DOI:10.1021/acs.accounts.2c00193.
- M. Clites, J. L. Hart, M. L. Taheri and E. Pomerantseva, Annealing-Assisted Enhancement of Electrochemical Stability of Na-Preintercalated Bilayered Vanadium Oxide Electrodes in Na-Ion Batteries, ACS Appl. Energy Mater., 2020, 3(1), 1063–1075, DOI:10.1021/acsaem.9b02098.
- Y. Xu, X. Deng, Q. Li, G. Zhang, F. Xiong, S. Tan, Q. Wei, J. Lu, J. Li and Q. An, et al., Vanadium Oxide Pillared by Interlayer Mg2+ Ions and Water as Ultralong-Life Cathodes for Magnesium-Ion Batteries, Chem, 2019, 5(5), 1194–1209, DOI:10.1016/j.chempr.2019.02.014.
- B. Jeon, H. H. Kwak and S.-T. Hong, Bilayered Ca0.28V2O5·H2O: High-Capacity Cathode Material for Rechargeable Ca-Ion Batteries and Its Charge Storage Mechanism, Chem. Mater., 2022, 34(4), 1491–1498, DOI:10.1021/acs.chemmater.1c02774.
- Y. Zhao, C. Han, J. Yang, J. Su, X. Xu, S. Li, L. Xu, R. Fang, H. Jiang and X. Zou, et al., Stable Alkali Metal Ion Intercalation Compounds as Optimized Metal Oxide Nanowire Cathodes for Lithium Batteries, Nano Lett., 2015, 15(3), 2180–2185, DOI:10.1021/acs.nanolett.5b00284.
- S. Fleischmann, Y. Zhang, X. Wang, P. T. Cummings, J. Wu, P. Simon, Y. Gogotsi, V. Presser and V. Augustyn, Continuous transition from double-layer to Faradaic charge storage in confined electrolytes, Nat. Energy, 2022, 7(3), 222–228, DOI:10.1038/s41560-022-00993-z.
- H. Jiang, Y. Zhang, M. Waqar, J. Yang, Y. Liu, J. Sun, Z. Feng, J. Sun, Z. Pan and C. Meng, et al., Anomalous Zn2+ Storage Behavior in Dual-Ion-In-Sequence Reconstructed Vanadium Oxides, Adv. Funct. Mater., 2023, 33(7), 2213127, DOI:10.1002/adfm.202213127.
- Z. Feng, Y. Zhang, J. Sun, Y. Liu, H. Jiang, M. Cui, T. Hu and C. Meng, Dual ions enable vanadium oxide hydration with superior Zn2+ storage for aqueous zinc-ion batteries, Chem. Eng. J., 2022, 433, 133795, DOI:10.1016/j.cej.2021.133795.
- S. Bai, X. Wang, Q. Wang, Z. Chen and Y. Zhang, Bimetallic Intercalated Vanadium Oxide As a High-Performance Cathode for Aqueous Zinc Ion Batteries, ACS Appl. Mater. Interfaces, 2024, 16(17), 22403–22410, DOI:10.1021/acsami.4c01648.
- Z. Li, L. Yang, S. Wang, K. Zhu and H. Li, Co-insertion of K+ and Ca2+ in vanadium oxide as high-performance aqueous zinc-ion battery cathode material, J. Alloys Compd., 2024, 992, 174589, DOI:10.1016/j.jallcom.2024.174589.
- C. Liu, L. Zhang, D. Chen and K. Jiang, Dual ions pre-intercalated hydrate vanadium oxide as cathode drives high-performance aqueous zinc ions storage, J. Alloys Compd., 2023, 947, 169476, DOI:10.1016/j.jallcom.2023.169476.
- W. Liu, X. Liu, F. Ning, S. Subhan, Y. Liu, Q. Li, J. Zhang, S. Lu and J. Yi, Fabrication of a heterovalent dual-cation pre-embedded hydrated vanadium oxide cathode for high-performance zinc ion storage, J. Mater. Chem. A, 2024, 12(20), 11883–11894, 10.1039/D4TA00376D.
- J. Wang, X. Zhao, J. Kang, X. Wang, H. Yu, C.-F. Du and Q. Yan, Li+, Na+ co-stabilized vanadium oxide nanobelts with a bilayer structure for boosted zinc-ion storage performance, J. Mater. Chem. A, 2022, 10(40), 21531–21539, 10.1039/D2TA05803K.
- S. Li, X. Jia, J. Liu, Z. Liu and G. Cao, Engineering hydrated vanadium oxide by K+ and Ni2+ incorporation for aqueous zinc ion batteries, Mater. Chem. Phys., 2022, 287, 126358, DOI:10.1016/j.matchemphys.2022.126358.
- H. Yang, Q. Li, L. Sun, S. Zhai, X. Chen, Y. Tan, X. Wang, C. Liu, W.-Q. Deng and H. Wu, MXene-Derived Na+-Pillared Vanadate Cathodes for Dendrite-Free Potassium Metal Batteries, Small, 2024, 20(5), 2306572, DOI:10.1002/smll.202306572.
- J. Livage, Sol-gel chemistry and electrochemical properties of vanadium oxide gels, Solid State Ionics, 1996, 86–88, 935–942, DOI:10.1016/0167-2738(96)00336-0.
- R. Zhang, T. Averianov, R. Andris, M. J. Zachman and E. Pomerantseva, Liquid Phase Exfoliation of Chemically Prelithiated Bilayered Vanadium Oxide in Aqueous Media for Li-Ion Batteries, J. Phys. Chem. C, 2023, 127(2), 919–929, DOI:10.1021/acs.jpcc.2c06875.
- Y. Yang, G. Liu, Z. Fang, M. Liu, Y. Zhao, X. Lai, J. Bi and D. Gao, Dual-Guest Intercalated Vanadium Oxides with a Robust Channel for Efficient Zn Ion Diffusion and Storage, ACS Sustain. Chem. Eng., 2024, 12(10), 3941–3950, DOI:10.1021/acssuschemeng.3c06125.
- X. H. Rui, N. Ding, J. Liu, C. Li and C. H. Chen, Analysis of the chemical diffusion coefficient of lithium ions in Li3V2(PO4)3 cathode material, Electrochim. Acta, 2010, 55(7), 2384–2390, DOI:10.1016/j.electacta.2009.11.096.
- M. Prześniak-Welenc, M. Łapiński, T. Lewandowski, B. Kościelska, L. Wicikowski and W. Sadowski, The Influence of Thermal Conditions on V2O5 Nanostructures Prepared by Sol-Gel Method, J. Nanomater., 2015, 2015(1), 418024, DOI:10.1155/2015/418024.
- C. Sanchez, J. Livage and G. Lucazeau, Infrared and Raman study of amorphous V2O5, J. Raman Spectrosc., 1982, 12(1), 68–72, DOI:10.1002/jrs.1250120110.
- T. Averianov, X. Zhang, R. Andris, D. Olds, M. Zachman and E. Pomerantseva, The Influence of MAX phase etching conditions on the synthesis and properties of V2CTx MXene-derived K-preintercalated bilayered vanadium oxides in non-aqueous K-ion batteries, ACS Appl. Nano Mater., 2025 Search PubMed.
- H. Yu, M. Aakyiir, S. Xu, J. D. Whittle, D. Losic and J. Ma, Maximized crystal water content and charge-shielding effect in layered vanadate render superior aqueous zinc-ion battery, Mater. Today Energy, 2021, 21, 100757, DOI:10.1016/j.mtener.2021.100757.
- S. Boyd, K. Ganeshan, W.-Y. Tsai, T. Wu, S. Saeed, D.-e. Jiang, N. Balke, A. C. T. van Duin and V. Augustyn, Effects of interlayer confinement and hydration on capacitive charge storage in birnessite, Nat. Mater., 2021, 20(12), 1689–1694, DOI:10.1038/s41563-021-01066-4.
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang and Y. Shao, et al., Water-Lubricated Intercalation in V2O5·nH2O for High-Capacity and High-Rate Aqueous Rechargeable Zinc Batteries, Adv. Mater., 2018, 30(1), 1703725, DOI:10.1002/adma.201703725.
- Y. Zhao, S. Liang, X. Shi, Y. Yang, Y. Tang, B. Lu and J. Zhou, Synergetic Effect of Alkali-Site Substitution and Oxygen Vacancy Boosting Vanadate Cathode for Super-Stable Potassium and Zinc Storage, Adv. Funct. Mater., 2022, 32(32), 2203819, DOI:10.1002/adfm.202203819.
- D. Omo-Lamai, X. Zhang, R. Andris, M. J. Zachman and E. Pomerantseva, Chemical preintercalation of magnesium ions into α-MoO3 structure for improved electrochemical stability in Li-ion cells, J. Alloys Compd., 2024, 1005, 175954, DOI:10.1016/j.jallcom.2024.175954.
- M. Yu, H. Shao, G. Wang, F. Yang, C. Liang, P. Rozier, C.-Z. Wang, X. Lu, P. Simon and X. Feng, Interlayer gap widened α-phase molybdenum trioxide as high-rate anodes for dual-ion-intercalation energy storage devices, Nat. Commun., 2020, 11(1), 1348, DOI:10.1038/s41467-020-15216-w.
- J. B. Mitchell, W. C. Lo, A. Genc, J. LeBeau and V. Augustyn, Transition from Battery to Pseudocapacitor Behavior via Structural Water in Tungsten Oxide, Chem. Mater., 2017, 29(9), 3928–3937, DOI:10.1021/acs.chemmater.6b05485.
- J. B. Mitchell, R. Wang, J. S. Ko, J. W. Long and V. Augustyn, Critical Role of Structural Water for Enhanced Li+ Insertion Kinetics in Crystalline Tungsten Oxides, J. Electrochem. Soc., 2022, 169(3), 030534, DOI:10.1149/1945-7111/ac58c8.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00673b |
| This journal is © The Royal Society of Chemistry 2025 |