
The role of terminal and bridge ligands in the molecular upconversion of lanthanide(III) 1D coordination polymers†
Stefano Angerami
de Andrade
 a,
Airton Germano
Bispo-Jr
ab,
Deborah
de Alencar Simoni
a,
Javier
Ellena
c,
João
Honorato de Araujo Neto
b,
Italo Odone
Mazali
a and
Fernando Aparecido
Sigoli
*a
a,
Airton Germano
Bispo-Jr
ab,
Deborah
de Alencar Simoni
a,
Javier
Ellena
c,
João
Honorato de Araujo Neto
b,
Italo Odone
Mazali
a and
Fernando Aparecido
Sigoli
*a
aInstitute of Chemistry, State University of Campinas, Campinas, São Paulo 13083-970, Brazil. E-mail: fsigoli@unicamp.br
bInstitute of Chemistry, University of São Paulo, São Paulo, São Paulo 05508-000, Brazil
cInstitute of Physics, University of São Paulo, São Carlos, São Paulo 13566-590, Brazil
First published on 15th November 2024
Abstract
The upconversion of trivalent lanthanide ions (LnIII) has gained significant attention in recent decades due to its potential applications in energy conversion and biological imaging. Recently, the concept of LnIII molecular upconversion in coordination compounds has emerged, offering new possibilities thanks to their ease of synthesis and processing. To address the challenge of enhancing this relatively unexplored phenomenon, we investigated 1D coordination polymers containing the ErIII/YbIII pair as models to explore how structural parameters tuned by different ligands can enhance upconversion. Our approach utilized two distinct bridging ligands, [(diphenylphosphoryl)R](diphenyl)phosphine oxide, R = ethyl–dppeo or butyl–dppbo, to modulate the polymeric chain conformation and 3D packing. Additionally, three terminal ligands acac (2,4-pentanedione), tfa (1,1,1-trifluoro-2,4-pentadione), and hfa (1,1,1,5,5,5-hexafluoro-2,4-pentanedione) were employed to investigate the effects of steric and electronic factors on structural and upconversion properties. The tfa− and hfa− ligands facilitated the formation of 1D coordination polymers with the formula [Ln(μ-L)(X)3]n (Ln = ErIII/YbIII, L = dppeo or dppbo, X = tfa or hfa), while the combination of acac− and dppeo led to the dinuclear structure [Ln2(μ-dppeo)(acac)6(H2O)2]. Notably, the F⋯F and F⋯H intra/intermolecular interactions driven by the terminal ligands significantly influenced the polymer conformation, leading to more linear and loosely packed polymeric chains. Upconversion was observed exclusively in structures with the shorter bridging ligand dppeo, suggesting that reduced Ln⋯Ln distances and loosely packed polymeric chains enhance molecular upconversion. Therefore, this study lays the groundwork for understanding the structural factors that govern the molecular upconversion in polynuclear LnIII complexes.
Introduction
From the moment the first ray of sunlight graced our planet, light has been an essential force shaping life, driving innovation, and revealing the mysteries of our universe; today, spectroscopy stands at the forefront of this exploration, providing society with unparalleled insights into the natural world and technological advancements. Among the most intriguing subjects of study, the luminescence of lanthanide(III) ions (LnIII) has captivated researchers and driven forward innovations in fields ranging from medical imaging to materials science.1 This attention is not fortuitous since the luminescence of LnIII shows unique features such as narrow emission bands, high emission colour purity, long lasting emitting levels (around 10−3 s), and the possibility of achieving high emission quantum yields.2–4 These features come from 4f ↔ 4f electronic transitions involving 4f orbitals, which are protected from the outer crystal field by the external 5s and 5p orbitals. Consequently, the energy of the 4f ↔ 4f transitions barely changes depending on the ligand set around the LnIII. Moreover, 4f ↔ 4f transitions are forbidden by the Laporte transition rule considering the ion.5 Yet, this selection rule can be relaxed by removing the inversion center of the coordination sphere, leading to the mixture between levels coming from the 4f and odd-parity d orbitals. Therefore, the coordination environment of the LnIII plays a pivotal role in boosting their luminescence, seeking to drive the development of innovative technologies.6Among a broad range of opportunities to achieve luminescence in LnIII-based systems, upconversion has been considered a hotspot for key applications.7 The upconversion phenomenon is a non-linear process based on the absorption of two or more lower-energy photons and the emission of a higher energy photon.8 By converting low-energy near-infrared radiation into higher-energy visible light, lanthanide-doped materials enable advancements in bioimaging, where they enhance the visualization of biological tissues with desirable resolution and deep tissue penetration.9,10 Additionally, these materials have been investigated for use in security printing and anti-counterfeiting measures, offering specific luminescence properties that are difficult to replicate.11 Finally, in photovoltaics, LnIII upconversion could potentially improve solar cell efficiency by utilizing a broader spectrum of sunlight.12
The YbIII/ErIII pair takes the spotlight regarding the realization of upconversion.13 Upon excitation by a 980 nm laser source, YbIII works as a sensitizer to non-radiatively transfer the absorbed energy to ErIII, which then converts and emits a higher-energy photon within the visible spectral window.14 In this process, the YbIII–ErIII distance plays a crucial role in the energy transfer (ET) dynamics. That is because the ET process occurs through non-radiative mechanisms (i.e., electron exchange and/or multipolar interactions) which are strongly dependent on the donor–acceptor distance.15 Inorganic matrices, such as LnIII-doped nanoparticles have reported bright luminescence for the YbIII/ErIII pair.13 However, the possibility of utilizing coordination compounds to achieve molecular upconversion has been tempting researchers.16 These molecular-based systems offer several advantages such as easy synthesis under mild conditions and easy processability, seeking applications.17
Nowadays, some research groups have been successful in achieving LnIII molecular upconversion at low temperatures or room temperature in the solid state or in solution.18–23 As an interesting example, Murugesu and coworkers reported the room-temperature upconversion of molecular cluster-aggregate containing the YbIII/ErIII pair.24 Applying an smart strategy to achieve upconversion in solution, Sun and coworkers reported the upconversion luminescence of EuIII upon 980 nm excitation in a solution dispersions of co-crystals composed by [Yb(dbm)3bpy] and [Eu(dbm)3bpy] (dbm = dibenzoylmethane, bpy = 2,2′-bipyridine) complexes.25 In another interesting example, Charbonnière and coworkers achieved cooperative luminescence and cooperative sensitization upconversion of LnIII complexes in solution.26 Piguet and coworkers recently discussed about the importance of symmetry and rigidity for tuning the molecular upconversion of LnIII species in solution.27 In all these examples, although several advances have been made to modulate the LnIII molecular upconversion, the luminescence is still far away in performance compared to LnIII-doped nanoparticles. That is because the ions usually involved in the upconversion have their luminescence easily quenched by C–H vibrations present in the ligand scaffold. Moreover, the role of structure rigidity and Ln–Ln distances need to be better understood to boost the molecular upconversion.
There are still many questions and mysteries surrounding the LnIII molecular upconversion. To clarify some of these aspects, we focus on providing further insights to understand how structural parameters affect the molecular upconversion of the ErIII/YbIII pair. For that, herein, it is reported that the synthesis of unidimensional (1D) ErIII/YbIII coordination polymers based on dppeo or dppbo bridge ligands – [(diphenylphosphoril)(R)](diphenyl)phosphine oxide (R = ethyl or butyl, named respectively dppeo and dppbo) – and acetylacetonate (acac−), trifluoroacetylacetonate (tfa−) or hexafluoroacetylacetonate (hfa−) as terminal ligands (X). These ligands were used to synthesize the following compositions: [Ln(tfa)3(μ-dppeo)]n (1), [Ln(hfa)3(μ-dppeo)]n (2), [Ln(tfa)3(μ-dppbo)]n (3), and [Ln(hfa)3(μ-dppbo)]n (Ln = Er, Yb) (4). An alternative synthetic pathway formed a dinuclear structure with the acac− ligand, [Ln2(μ-dppeo)(acac)6(H2O)2] (5). By synthesizing these species, it is feasible to evaluate how the Ln⋯Ln distances, chain conformation and 3D packing affect the upconversion of LnIII ions. Through elegant structural analysis using single-crystal X-ray diffraction (SC-XRD) and Hirshfeld surface analysis, we identified key structural dependence correlations based on inter and intramolecular interactions. These interactions influence chain conformation and packing, thereby affecting the upconversion emission.
Experimental
A detailed experimental procedure for the precursor syntheses is described in note S1 (ESI†). The synthesis of the coordination polymers was based on a work recently reported elsewhere by Sigoli's group for the coordination polymers [Ln(tfa)3(μ-dppeo)]n and [Ln(tfa)3(μ-dppbo)]n (Ln = Eu, Tb).28 The coordination polymers 1–4 were synthesized by dissolving 0.15 mmol of [Ln(X)3(H2O)2] precursor complexes (Ln = Er or Yb) in 10 mL of ethanol followed by the addition of 0.3 mmol of the bridging ligand dissolved in 10 mL of ethanol. The solution was diluted to 50 mL with ethanol and left to crystalize in a bottle covered by pierced aluminium foil. Light-pink crystals could be observed within a few days of resting. The synthesis of 5 started with dissolving 0.3 mmol of μ-dppeo in ethanol. Aqueous solutions of YbCl3 and ErCl3, totalling 0.15 mmol each, were added dropwise into the previous mixture, followed by the addition of the Na+acac− salt dissolved in water. The solution was left to rest and pink crystals were obtained after a week. In all the syntheses, the ErIII to YbIII molar ratio was kept as 1. Finally, the apparatus used for the characterization is listed in note S1 (ESI†).Results
Synthesis pathway discussion
Coordination polymers 1–4 were synthesized by mixing [Ln(X)3(H2O)2] (Ln = Er and Yb, X = hfa− or tfa−) with the bridge ligand in ethanol at 300 K. Besides several attempts, this route was not successful in synthesizing the target [Ln(acac)3(μ-dppeo)]n and [Ln(acac)3(μ-dppeo)]n compositions. Alternatively, LnCl3 was mixed in ethanol with the bridge ligand and the deprotonated acac−, affording the dinuclear [Ln2(μ-dppeo)(acac)6(H2O)2] (5). Attempts to synthesize the analogues with the dppbo bridge were not successful. Analogous structures of 5 with EuIII, GdIII, ErIII, or DyIII were reported by Sun and coworkers,29 but starting from the [Ln(acac)3(H2O)2] and dppeo precursors dissolved in methanol under reflux.Structural characterization
1–5 were analysed by SC-XRD and the crystal structures are shown in Fig. 1. The relevant crystallographic data are contained in Table S1 (ESI†). 1–4 are 1D coordination polymers belonging to the triclinic crystal system, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, Fig. 1A–D. All LnIII ions are coordinated to 8 oxygens (coordination number of 8), 6 of them coming from the terminal ligands (tfa− or hfa−), and 2 from the bridging ligands (dppeo or dppbo). Each bridge ligand connects with the subsequent LnO8 polyhedra, forming a 1D coordination polymer chain. The coordination polyhedron of 1, 2, and 4 is better described by a distorted D4d point group (anti-square prism conformation), Fig. 1F, as determined by SHAPE analysis (Table S2, ESI†).30 Conversely, the first coordination environment of 3 fits better a distorted D2d point group (triangular dodecahedron conformation, Fig. 1F and Table S2, ESI†). 5, in turn, was obtained as a dinuclear species belonging to the triclinic crystal system, P space group, Fig. 1E. In 5, each LnIII center is bonded to three acac− bidentate ligand, one oxygen from the dppeo, and one water molecule. The coordination number of both LnIII sites is 8 and they are also described by a D4d pseudo symmetry (Fig. 1F and Table S2, ESI†).
space group, Fig. 1A–D. All LnIII ions are coordinated to 8 oxygens (coordination number of 8), 6 of them coming from the terminal ligands (tfa− or hfa−), and 2 from the bridging ligands (dppeo or dppbo). Each bridge ligand connects with the subsequent LnO8 polyhedra, forming a 1D coordination polymer chain. The coordination polyhedron of 1, 2, and 4 is better described by a distorted D4d point group (anti-square prism conformation), Fig. 1F, as determined by SHAPE analysis (Table S2, ESI†).30 Conversely, the first coordination environment of 3 fits better a distorted D2d point group (triangular dodecahedron conformation, Fig. 1F and Table S2, ESI†). 5, in turn, was obtained as a dinuclear species belonging to the triclinic crystal system, P space group, Fig. 1E. In 5, each LnIII center is bonded to three acac− bidentate ligand, one oxygen from the dppeo, and one water molecule. The coordination number of both LnIII sites is 8 and they are also described by a D4d pseudo symmetry (Fig. 1F and Table S2, ESI†).
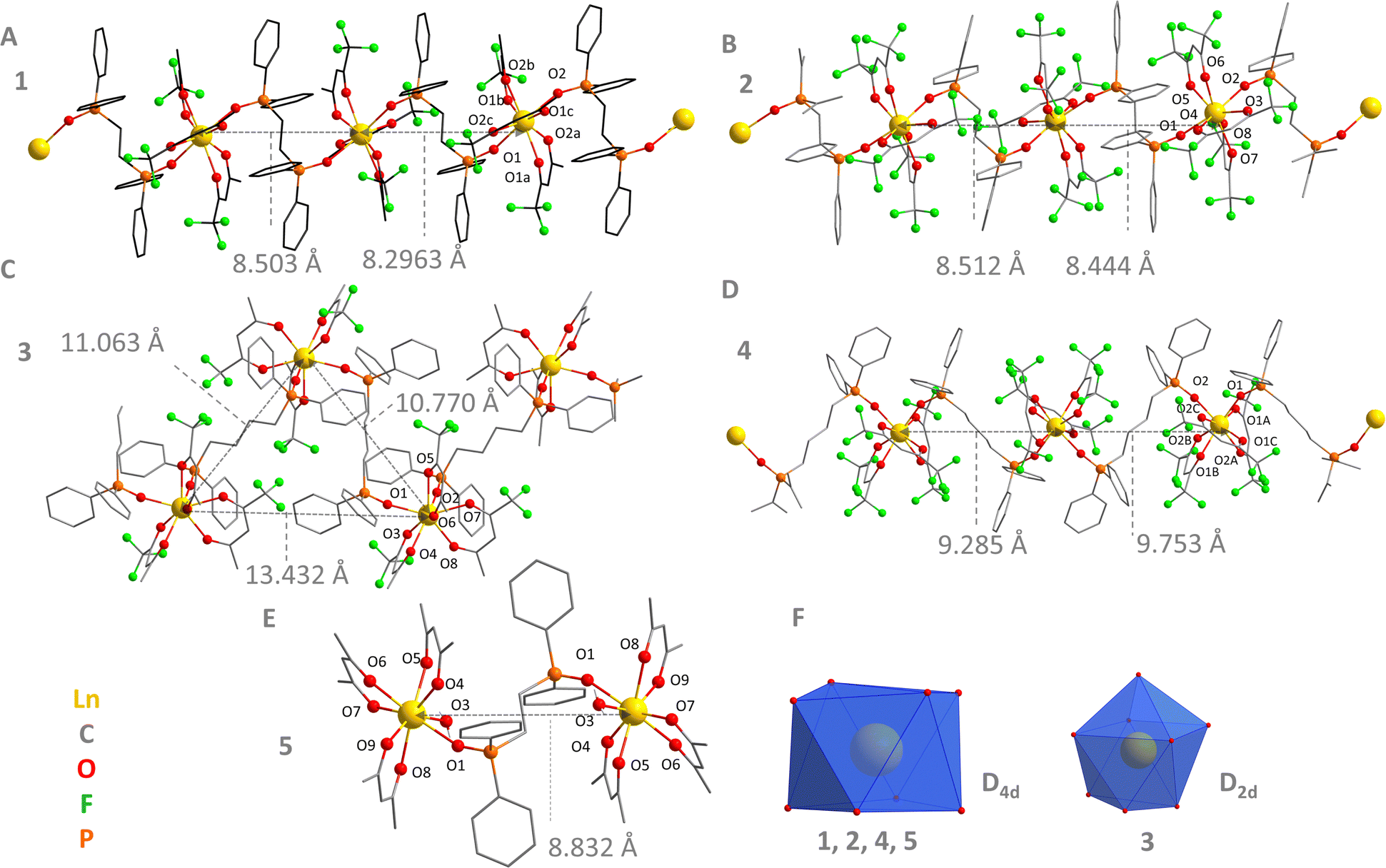 | ||
| Fig. 1 Partially-labelled crystal structures of (A) [Ln(tfa)3(μ-dppeo)]n (1), (B) [Ln(hfa)3(μ-dppeo)]n (2), (C) [Ln(tfa)3(μ-dppbo)]n (3), (D) [Ln(hfa)3(μ-dppbo)]n (4), and (E) [Ln2(μ-dppeo)(acac)6(H2O)2] (5) (Ln = Er, Yb). H atoms were removed for the sake of clarity. (F) Representative LnO8 coordination polyhedra of the compounds. | ||
To comprehend the impact of terminal and bridging ligands on the upconversion emission of the compositions, a detailed structural analysis was conducted. This analysis aimed to map local changes in the coordination environment and bond distances, as well as inter- and intramolecular interactions, including Ln⋯Ln distances, chain conformation, and molecular packing influenced by the ligands. The average Ln–O bond distances are within the 2.2 Å–2.4 Å interval (Table S3, ESI†), but the values change depending on the terminal ligand. This is not surprising considering that from acac− to tfa− and hfa−, the number of fluorine atoms increases, thus enhancing the electron-withdrawing capability of the terminal ligand.31 However, the expected trend of increasing the Ln–O bond distances from acac− to hfa− is not observed. This fact suggests that not only electronic effects control the bond conformation, but also steric interactions due to inter/intramolecular interactions. Indeed, the bite angles of the terminal ligands as well as the P–O–Ln angles also change depending on the composition, Table S4 (ESI†), confirming that steric and electronic effects lead to smooth distortions of the first coordination environment. This effect is reflected in subtle changes of the SHAPE parameter for the coordination sphere when comparing the structures (Table S2, ESI†).
The chain conformation was also mapped for the coordination polymers (Fig. 1A–D). 1, 2, and 4 form an almost linear 1D chain, as revealed by the Ln Ln–Ln angles within a single chain almost close to 180° (Table S5, ESI†). 3, in turn, presents a zig-zag chain conformation, which is in accordance with the structure previously reported for the EuIII and TbIII-based analogues.28 The shortest intramolecular Ln⋯Ln bond distances are presented in Fig. 1A–D and Table S5 (ESI†). 1 and 2 show shorter distances between sequential LnIII neighbouring ions compared with 3 and 4 (Table S5, ESI†). This is not surprising considering that the dppbo bridge ligand is longer than dppeo.
The packing arrangement of the compositions was analysed to determine the shortest Ln⋯Ln distances, Fig. 2. The coordination polymers based on dppbo (3 and 4) show a tighter packing in comparison to the dppeo equivalents (1 and 2), as demonstrated by the shortest Ln⋯Ln intermolecular distances (Table S5, ESI†). For 3, the intermolecular Ln⋯Ln distances are even shorter than the intramolecular ones due to the zig-zag chain conformation. Interestingly, 5 presented the shortest intermolecular distances among all compositions, mostly due to its reduced molecular size. For 5, the Ln⋯Ln intermolecular distance is also shorter than the intramolecular distance. By comparing 1 and 3 with 2 and 4, it is feasible to provide a clear picture on the effect of the terminal ligand on the Ln⋯Ln distances. 1 and 3 present shorter Ln⋯Ln internuclear distances when compared to 2 and 4, indicating that hfa− leads the crystals to a less packed 3D conformation. Supporting this, 2 and 4 presented an uplift in the unit cell volume of 4.3% and 8.1% respectively, in comparison to 1 and 3 (Table S1, ESI†).
 | ||
| Fig. 2 View of the packing arrangements in (A) 1, (B) 2, (C) 3, (D) 4, and (E) 5 representing the coordination polymer chains in different colors (except for 5, where each colour represents a single molecule unity). Hydrogen atoms have been omitted for the sake of clarity. The dashed lines represent the shortest Ln⋯Ln intermolecular distances of each composition. | ||
The structures were also investigated in-depth from an inter/intramolecular perspective, aiming to map the forces responsible for the packing of the crystalline structures. Several F⋯H and O⋯H hydrogen bonds are present in the compositions. The intramolecular and intermolecular interactions can be viewed in Fig. S3–S7 (ESI†), while the distances are described in Table S6–S10 (ESI†). Besides the hydrogen bonds involving the oxygen atoms in the first coordination sphere, some F⋯F interactions are detected for 1, 2, and 4, Fig. 3 and Tables S11 (ESI†).
 | ||
| Fig. 3 Shortest F⋯·F interactions (pink dashed lines) in (A) 1, (B) 2, and (C) 4. Carbon = grey; lanthanide = cyan; phosphorous = orange; fluor = light green; oxygen = red. Hydrogen atoms have been omitted for the sake of clarity. The F⋯·F interaction distances are shorter than 3 Å and the values can be found in Table S11 (ESI†). | ||
The intramolecular interactions were further investigated via Hirshfeld surface analysis obtained by using the CrystalExplorer software, Fig. 4.32 The 2D fingerprint plots of all interatomic interactions (Fig. S8–S12, ESI†) were used to compare the balance between the hydrogen bonds and F⋯F interactions in all the structures, Fig. 4F. The coordination polymers 2 and 4 (both with the hfa− terminal ligand) have overall contributions of 40–50% for F⋯H interactions, and 10–20% for F⋯F interactions. The coordination polymers 1 and 3 (both with the tfa− terminal ligand) have contributions of 30–40% of F⋯H hydrogen bonds and less than 5% of F⋯F interactions. Obviously, 5 does not have any F⋯F interaction due to the lack of fluorine in its structure. These interactions are in fact substituted by H⋯H and O⋯H contacts (70% and 15% contributions, respectively).
 | ||
| Fig. 4 Hirshfeld surfaces (HS) of (A) 1, (B) 2, (C) 3, (D) 4, and (E) 5 mapped over dnorm and shape index, S. In the dnorm HS, a red–blue–white colour scheme was used, whereas red regions represent closer contacts, blue regions represent longer ones, and white regions represent the distance of contacts which is exactly equal to the vdW separation. (F) Percentages of contacts that contribute to the total Hirshfeld surfaces area of 1–5. | ||
By mapping the intermolecular forces, it becomes feasible to propose reasonable explanations for various structural nuances that are dependent on the terminal and bridge ligands. As previously discussed, 3 deviated from the linearity of the 1D polymeric chain. Interestingly, 3 does not present F⋯F interactions at all, and only a 35% contribution of the F⋯H hydrogen bond, in contrast with 4 having about 10% (F⋯F) and 50% (F⋯H). Following, 3 has a 25% contribution of C⋯H interactions against 10% in 4. Therefore, it is possible to infer that the enhancement of fluorine interactions is a determinant factor in the linearization of the 1D polymer chain.
Finally, the shortest intermolecular distances between the LnIII ions increase in the 5 < 3 < 1 < 4 < 2 order. This sequence indicates that the use of acac− induces the shortest intermolecular distance, followed by tfa−, and hfa−, being longest. Considering that shorter intermolecular distances would lead to tighter packing of the crystals, acac− leads to a tighter packing in the structure, and hfa− a looser one. One can observe that F⋯F interactions have increasing contributions following the established sequence. As so, it is possible to determine that hfa−, and thus the presence of fluorine in higher amounts leads to a looser packing of the crystal. Finally, the lack of fluorine in 5 suggests that fluorine interactions are relevant for the formation of the polymeric structure.
After understanding the interplays between ligand characteristics and structure, we now expand the studies to the overall composition of the synthesized materials. To evaluate the bulk composition of the samples, powder X-ray diffraction (PXRD) for the crashed crystals was performed and they are presented in Fig. S13–S17 (ESI†). The experimental diffraction plans observed for the five structures agree with the ones simulated from the SC-XRD analyses. This observation indicates that the crystallinity of the sample is robust after grinding and that the composition does not present any spurious crystalline phase. Therefore, this observation characterizes the success of the synthesis process and structural purity of the samples.
The FTIR spectra for all the materials are shown in Fig. S18 and Table S12 (ESI†). The FTIR of all the compositions display C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO coupled vibrational modes peaking close to 1510 cm−1 as well as a bending mode coupled to C–H + CC vibrations at 1480 cm−1.33 The bands arising from the combination of CC + C–H vibrational modes are observed within 1290–1280 and 1280 cm−1 while the bands between 1150 and 1050 cm1 are ascribed to coupled vibrations of C–F3 + C–H bonds for 1–4. Finally, the vibrational mode assigned to the CO stretch is found close to 1630 cm−1.34 whereas the ν(PO) vibrational modes are peaking at around 1170 cm−1.
C and CO coupled vibrational modes peaking close to 1510 cm−1 as well as a bending mode coupled to C–H + CC vibrations at 1480 cm−1.33 The bands arising from the combination of CC + C–H vibrational modes are observed within 1290–1280 and 1280 cm−1 while the bands between 1150 and 1050 cm1 are ascribed to coupled vibrations of C–F3 + C–H bonds for 1–4. Finally, the vibrational mode assigned to the CO stretch is found close to 1630 cm−1.34 whereas the ν(PO) vibrational modes are peaking at around 1170 cm−1.
Luminescence features
After tailoring the structural features of the compounds, the upconversion emission at room temperature was studied under 980 nm laser excitation, Fig. 5. The emission spectra of compounds 1, 2, and 5 are a set of emission bands at around 530 nm, 545 nm and 670 nm assigned to ErIII 2H11/2 → 4I15/2, 4S3/2 → 4I15/2, and 4F9/2 → 4I15/2 transitions, respectively.35 For the three compounds, the emission band assigned to the 4S3/2 → 4I15/2 transition within the green spectral region displays the highest intensity in the emission spectrum. However, the band lying in the red spectral region also displays quite high emission intensity. This combination renders a green emission colour according to the Commission Internationale de L’éclairage CIE 1931 chromaticity diagram (Fig. S19, ESI†). | ||
| Fig. 5 Upconversion emission spectra dependent on the excitation power for (A) 1, (B) 2, and (C) 5 processed as crashed crystals. The spectra were collected at room temperature by using different powers of the 980 nm excitation laser. | ||
To guarantee that the ErIII emission occurs through the upconversion mechanism, the dependency of the emission intensity on the excitation power density was evaluated. This study is helpful since the upconversion is a non-linear process, thus, I ∝ Pn, where n is the number of photons involved in the process, I is the integrated emission intensity, and P is the laser power density.36 In the emission spectra of 1, 2 and 5, the larger the laser power, the stronger the emission intensity, Fig. 5. The plot of the integrated emission intensities versus excitation power density, Fig. S20 (ESI†), shows that the ErIII 4S3/2 → 4I15/2 transition occurs via absorption of two photons, confirming the upconversion mechanism.
In the ErIII,YbIII-based systems, the luminescence dynamics is mainly controlled by the energy transfer upconversion (ETU) mechanism, Fig. S21 (ESI†). In this process, YbIII is excited by a 980 nm photon (2F7/2 → 2F5/2) and transfers the energy directly to the ErIII 4I11/2 level. After that, two different phenomena may take place. First, the ErIII ion in the 4I11/2 level can absorb another 980 nm photon, being promoted to the 4F7/2 level. From there one, the 4F7/2 level relaxes to 2H11/2 and 4S3/2 levels giving rise to emissions at 530 nm and 545 nm, respectively, within the green spectral window. In the second case, the 4I11/2 level non-radiatively relaxes to the 4I13/2 level, which absorbs a second 980 nm photon, promoting the ion to the 4F9/2 level. From this level, the red emission at 670 nm due to the 4F9/2 → 4I15/2 transition is observed. It is worth stressing out that a high concentration of YbIII has been shown to enhance the ErIII red emission (4F9/2 → 4I15/2 transition) through the ETU mechanism.24
One should note that 3 and 4 do not show any upconversion emission, suggesting that the use of the dppbo bridge compared to dppeo is not the best strategy to induce ErIII upconversion. The lack of emission observed for 3 and 4 is better explained by the longer distance between lanthanide cores in these structures due to the extended length of μ-dppbo, as previously compared in Fig. 1. The Ln⋯Ln intramolecular distances in 1, 2 and 5 all fall in the 8.29 Å–8.52 Å interval, whilst 3 and 4 are in the 9.27 Å–11.1 Å range. This is a significant change, which affects the capacity of YbIII in sensitizing the ErIII upconversion. This occurs because the YbIII-to-ErIII non-radiative ET mechanisms are robustly dependent on the Ln–Ln distances.15 It is worth pointing out that analysis of hydrogen bonds and fluorine interactions previously showed that 2 and 4 exhibit substantial F⋯H contributions, while 1 and 3 show weaker F⋯H and minimal F⋯F interactions. This analysis highlighted that F⋯F interactions, particularly in the presence of dpeeo and/or hfa− ligands, contribute to a looser crystal packing, whereas acac− ligands lead to denser packing with shorter LnIII⋯LnIII intermolecular distances.
The tight packing and shorter intermolecular Ln⋯Ln distances of 3 and 4 could lead to an increase in the ErIII upconversion by energy transfer between LnIII centers within different chains. However, tight packing means closer polymeric chains, which could also increase the contribution of non-radiative deactivation of the LnIII emitting level due to multiphonon relaxation (molecular vibration).37 In this context, it is feasible to conclude that in the coordination polymers, the tight packing induced by the dppbo bridge ligand increases the contribution of molecular vibrations that quench the ErIII emission, shutting off any possibility of upconversion detection for 3 and 4. This packing effect seems to be less relevant for the dinuclear species 5, which is also tightly packed, but still presents upconversion emission. That is because in 5, the Ln⋯Ln intermolecular distances are way shorter than in the other structures (Fig. 2 and 3), favouring a more efficient ET and making the quenching effect due to the tight packing less relevant.
The upconversion emission intensities of 1, 2, and 5 were also compared in Fig. S22 (ESI†). Although we keep in mind that this comparison is not absolute, it suggests that 1 has the highest upconversion intensity within this series. The overall reason behind this trend lies in the role of the Ln–Ln distance and multiphonon deactivation on the luminescence behaviour, as previously discussed. Moreover, the replacement of C–H bonds by C–F in 1 and 2 could decrease the contribution of multiphonon deactivations of the ErIII emitting levels. Indeed, the literature shows that the replacement of C–H bonds by C–X bonds (X = F, Cl, and Br), which display lower vibrational mode energy, decreases the probability of multiphonon processes that quench the LnIII luminescence.38
Therefore, this work demonstrates the significant impact of the structural tailoring of compounds on their molecular upconversion. An in-depth investigation of the intermolecular and intramolecular interactions within the coordination polymers revealed significant insights into the forces governing crystal packing. Compounds 1, 2, and 5 exhibited upconversion emissions at room temperature upon 980 nm laser excitation, while the absence of upconversion in compounds 3 and 4, attributed to the detrimental role of the dppbo bridge, underscores the importance of Ln⋯Ln distances and structural hardness and packing in achieving efficient upconversion. Our findings highlight the critical role of ligand selection and structural design in optimizing the upconversion performance of lanthanide-based molecular materials, helping to contribute to the development of brighter upconverter materials for advanced photonic applications.
Conclusions
In this study, 1D coordination polymers containing the YbIII/ErIII pair were thoroughly synthesized with different ligands: dppeo or dppbo as bridging ligands, and tfa− or hfa− as terminal ligands. Additionally, the dinuclear [Ln2(μ-dppeo)(acac)6(H2O)2] species was obtained by an alternative synthetic route. The Ln⋯Ln distances and packing arrangements varied significantly among the compounds, influenced by the choice of terminal and bridging ligands. Analysis of hydrogen bonds and fluorine interactions showed that 2 and 4 exhibit substantial F⋯H contributions, while 1 and 3 show weaker F⋯H and minimal F⋯F interactions. This analysis highlighted those F⋯F interactions, particularly in the presence of dpeeo and/or hfa− ligands, contribute to a looser crystal packing, whereas acac− ligands lead to denser packing with shorter LnIII⋯LnIII intermolecular distances. The compounds with the dppeo bridge ligand exhibited robust upconversion emissions at room temperature upon 980 nm laser excitation, predominantly featuring intense green and notable red emissions, which collectively result in an overall green emission. The dppeo bridge facilitated closer Ln⋯Ln intramolecular distances and enhanced upconversion performance, whereas the dppbo bridge led to more packed 1D chains and a lack of upconversion emission. In essence, our findings elucidate the intricate relationship between structural features and upconversion performance in lanthanide-based molecular materials. This understanding provides further guidance for designing and optimizing new materials for advanced photonic applications, emphasizing the importance of precise ligand selection and structural engineering.Author contributions
S. A. A.: term, conceptualization, methodology, validation, formal analysis, investigation, data curation, and writing – original draft; A.G.B.J.: methodology, validation, investigation, data curation, formal analysis, and writing – original draft; D. A. S., J. H. A. N., and J. A. E.: methodology and formal analysis; I. O. M.: methodology, resources, writing – review & editing, and funding acquisition; F. A. S.: term, conceptualization, methodology, resources, writing – review & editing, supervision, project administration, and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to FAPESP 2021/06326-1 and INCT/INOMAT-National Institute of Science Technology in Complex Functional Materials (CNPq: 465452/2014–0 and FAPESP: 2014/50906–9). FAS thanks CNPq (304807/2022-2 and 304564/2018-4) and FAPESP 2021/08111-2. SAA thanks CNPq for the Scientific Initiation scholarship. AGBJ also thanks FAPESP (2020/02614-0) for the award of a postdoctoral scholarship. JE thanks FAPESP (2017/15850-0) and CNPq (312505/2021-3).References
- M. Hasegawa, H. Ohmagari, H. Tanaka and K. Machida, J. Photochem. Photobiol., 2022, 50, 100484 CrossRef CAS.
- A. G. Bispo-Jr, A. J. de Morais, C. M. S. Calado, I. O. Mazali and F. A. Sigoli, J. Lumin., 2022, 252, 119406 CrossRef CAS.
- S. F. N. Coelho, A. G. Bispo-Jr, N. A. Oliveira, I. O. Mazali and F. A. Sigoli, Nanoscale, 2024, 16, 7493–7503 RSC.
- A. G. Bispo-Jr, I. O. Mazali and F. A. Sigoli, Phys. Chem. Chem. Phys., 2022, 24, 13565–13570 RSC.
- P. A. Tanner, Chem. Soc. Rev., 2013, 42, 5090–5101 RSC.
- J. G. Bünzli, Trends Chem., 2019, 1, 751–762 CrossRef.
- F. Auzel, J. Lumin., 2020, 223, 116900 CrossRef CAS.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS.
- H. Dong, S. Du, X. Zheng, G. Lyu, L. Sun, L. Li, P. Zhang, C. Zhang and C. Yan, Chem. Rev., 2015, 115, 10725–10815 CrossRef CAS.
- G. Tessitore, G. A. Mandl, M. G. Brik, W. Park and J. A. Capobianco, Nanoscale, 2019, 11, 12015 RSC.
- Y. Liu, S. Liang, C. Yuan, A. Best, M. Kappl, K. Koynov, H. Butt and S. Wu, Adv. Funct. Mater., 2021, 31, 2103908 CrossRef CAS.
- A. Ghazy, M. Safdar, M. Lastusaari, H. Savin and M. Karppinen, Sol. Energy Mater. Sol. Cells, 2021, 230, 111234 CrossRef CAS.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS.
- J. A. DeLuca, J. Chem. Educ., 1980, 57, 541 CrossRef CAS.
- A. N. C. Neto, R. T. M. Jr and O. L. Malta, J. Lumin., 2019, 210, 342–347 CrossRef.
- Y. Suffren, B. Golesorkhi, D. Zare, L. Guene, H. Nozary, S. V. Eliseeva, S. Petoud, A. Hauser and C. Piguet, Inorg. Chem., 2016, 55, 9964–9972 CrossRef CAS PubMed.
- A. M. Nonat and L. J. Charbonnière, Coord. Chem. Rev., 2020, 409, 213192 CrossRef CAS.
- X. Xiao, J. P. Haushalter and G. W. Faris, Opt. Lett., 2005, 30, 1674–1676 CrossRef CAS PubMed.
- R. C. Knighton, L. K. Soro, A. Lecointre, G. Pilet, A. Fateeva, L. Pontille, L. Frances-Soriano, N. Hildebrandt and L. J. Charbonniere, Chem. Commun., 2021, 57, 53 RSC.
- G. Sun, Y. Ren, Y. Song, Y. Xie, H. Zhang and L. Sun, J. Phys. Chem., 2022, 13, 8509–8515 CAS.
- L. Aboshyan-Sorgho, M. Cantuel, S. Petoud, A. Hauser and C. Piguet, Coord. Chem. Rev., 2012, 256, 1644–1663 CrossRef CAS.
- D. A. Gálico, R. Ramdani and M. Murugesu, Nanoscale, 2022, 14, 9675–9680 RSC.
- Y. Wang, G. Sun, Q. Su, Y. Xie, F. Xing, H. Zhang and L. Sun, Chem. – Eur. J., 2024, 30, e202400911 CrossRef CAS PubMed.
- D. A. Gálico, J. S. Ovens, F. A. Sigoli and M. Murugesu, ACS Nano, 2021, 15, 5580–5585 CrossRef.
- G. Sun, Y. Xie, Y. Wang, G. A. Mandl, S. L. Maurizio, H. Zhang, X. Ottenwaelder, J. A. Capobianco and L. Sun, Angew. Chem., 2023, 62, e202304591 CrossRef CAS PubMed.
- R. C. Knighton, L. K. Soro, L. Francis-Soriano, A. Rodriguez-Rodriguez, G. Pilet, M. Lenertz, C. Platas-Iglesias, N. Hildebrandt and L. J. Charbonnière, Angew. Chem., 2022, 61, e202113114 CrossRef CAS PubMed.
- S. Naseri, I. Taarit, H. Bolvin, J. Bünzli, A. Fürstenberg, L. Guénée, G. Le-Hoang, M. Mirzakhani, H. Nozary, A. Rosspeintner and C. Piguet, Angew. Chem., 2023, 62, e202314503 CrossRef CAS PubMed.
- D. A. Lima, A. G. Bispo-Jr, D. A. Galico, S. F. N. Coelho, J. H. A. Neto, J. A. Ellena, L. Petiote, I. O. Mazali and F. A. Sigoli, Inorg. Chem., 2023, 62, 6808–6816 CrossRef.
- H. Yan, Z.-W. Che and W.-B. Sun, New J. Chem., 2023, 47, 140 RSC.
- M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575 CrossRef CAS PubMed.
- F. Habib, G. Brunet, V. Vieru, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 13242 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Cryst., 2021, 54, 1006 CrossRef CAS PubMed.
- R. Withnall and L. Andrews, J. Phys. Chem., 1987, 91, 784–797 CrossRef CAS.
- D. A. Thornton, Coord. Chem. Rev., 1990, 104, 251 CrossRef CAS.
- A. Shyichuk, S. S. Câmara, I. T. Weber, A. N. C. Neto, L. A. O. Nunes, S. Lis, R. L. Longo and O. L. Malta, J. Lumin., 2016, 170, 560–570 CrossRef CAS.
- M. Pollnau, D. R. Gamelin, S. R. Lüthi, H. U. Güdel and M. P. Hehlen, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337–3346 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., 1999, 493–504 CAS.
- Y. Ning, J. Tang, Y.-W. Liu, J. Jing, Y. Sun and J.-L. Zhang, Chem. Sci., 2018, 9, 3742 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2381906, 2381909–2381911, and 2381913. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc03823a |
| This journal is © The Royal Society of Chemistry 2025 |