
Revealing the surface oxidation mechanism and performance evolution of Nd–Fe–B sintered magnets†
Liang
Zhou
a,
Jiaying
Jin
 *ab,
Wang
Chen
ab,
Shaoqing
Ren
ab,
Mengfan
Bu
a,
Xu
Li
a,
Bo
Xin
b,
Chen
Wu
a and
Mi
Yan
*ab
*ab,
Wang
Chen
ab,
Shaoqing
Ren
ab,
Mengfan
Bu
a,
Xu
Li
a,
Bo
Xin
b,
Chen
Wu
a and
Mi
Yan
*ab
aSchool of Materials Science and Engineering, State Key Laboratory of Silicon and Advanced Semiconductor Materials, Key Laboratory of Novel Materials for Information Technology of Zhejiang Province, Zhejiang University, Hangzhou 310027, China. E-mail: jinjy@zju.edu.cn; mse_yanmi@zju.edu.cn
bState Key Laboratory of Baiyunobo Rare Earth Resource Researches and Comprehensive Utilization, Baotou Research Institution of Rare Earths, Baotou 014030, China
First published on 7th January 2025
Abstract
Achieving high magnetic properties and corrosion resistance simultaneously is a common goal for Nd–Fe–B permanent magnetic materials but remains challenging via traditional strategies. Herein, we conducted wide-range oxidation experiments to construct a tunable surface oxidation layer. Temperature-dependent and time-dependent oxidation behaviors of the typical N50 commercial-grade Nd–Fe–B sintered magnets with corresponding performance evolutions were systematically unraveled. Results showed that short-term low-temperature oxidation at 350 °C for 0.5 h or 250 °C for 3 h generated an excellent synergy of improved corrosion resistance and mechanical performance without compromising magnetic properties owing to the formation of a thin hydrophobic oxidation layer with fewer microscopic cracks under low kinetic coefficients (1.1 × 10−17 to 9.5 × 10−16 m2 s−1). High oxidation temperatures of 450–650 °C with exponentially increased kinetic coefficients (1.5 × 10−14 to 3.2 × 10−12 m2 s−1) lowered the anti-corrosion, mechanical and magnetic performance owing to the thickening of the oxidation layer with macroscopic cracks despite having superhydrophobic characteristics. With respect to the high-temperature oxidation mechanism, the formation of continuous and coarse grain boundary (GB) networks with multi-layered structures was identified in the internal oxidation zone for the first time. The multi-layered structure could be divided into four layers, with the first and second layers comprising continuous Nd/Pr/O-rich GBs with maximum oxygen concentration (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 and Imm/Ia structured Nd2O3), the third layer comprising the Fe-rich intermediate layer with extremely low concentrations of Nd/Pr and O (dominated by Imm structured α-Fe), and the fourth layer comprising a mixture of Fe, Nd/Pr and O (coexisting Imm structured α-Fe and amorphous Nd2O3). In the external oxidation zone, the single crystalline α-Fe phase without amorphous Nd2O3 was observed. Both features accelerated the inward oxygen diffusion and explained the high oxidation kinetics of the 650 °C oxidized magnet. The above correlation between the tunable oxidation behaviors and performance of the oxidized Nd–Fe–B magnets along with the temperature-dependent oxidation mechanisms provide new understandings for delicately controlling the corrosion-resistant oxidation coatings.
m1 and Imm/Ia structured Nd2O3), the third layer comprising the Fe-rich intermediate layer with extremely low concentrations of Nd/Pr and O (dominated by Imm structured α-Fe), and the fourth layer comprising a mixture of Fe, Nd/Pr and O (coexisting Imm structured α-Fe and amorphous Nd2O3). In the external oxidation zone, the single crystalline α-Fe phase without amorphous Nd2O3 was observed. Both features accelerated the inward oxygen diffusion and explained the high oxidation kinetics of the 650 °C oxidized magnet. The above correlation between the tunable oxidation behaviors and performance of the oxidized Nd–Fe–B magnets along with the temperature-dependent oxidation mechanisms provide new understandings for delicately controlling the corrosion-resistant oxidation coatings.
1. Introduction
Nd–Fe–B permanent magnets are vital in the development of renewable energy applications, such as wind and tidal power generation.1,2 Their superior maximum energy product (BH)max to other permanent magnetic materials contributes to the development of lightweight and miniaturized electronic devices.3,4 Nd–Fe–B permanent magnets possess a multiphase microstructure, including Nd2Fe14B matrix phase, Nd-rich intergranular phase and minority B-rich phase with different physicochemical properties.5,6 Among them, the Nd2Fe14B matrix phase affords the origin of high intrinsic magnetism (including saturation magnetization Ms and magnetocrystalline anisotropy field HA), while the Nd-rich intergranular phase exerts influential effects on the final extrinsic magnetic properties.7,8 On one hand, a certain fraction of the low-melting-point Nd-rich phase acts as the liquid-phase-sintering aid to permit quick densification and high remanence. On the other hand, the non-ferromagnetic Nd-rich phase is located at the continuous grain boundaries (GBs) to form a network isolating neighboring ferromagnetic Nd2Fe14B grains, which is critical for high coercivity.9–11However, apart from the desired magnetic properties, the anti-corrosion performance is also a significant indicator to guarantee the application of Nd–Fe–B permanent magnets, particularly in humid and harsh environments. Compared to other rare-earth (RE)-based permanent magnets, such as Sm2Fe17N3 prepared via the nitridation of Sm2Fe17 powders with ammonia or nitrogen gas to form a nitride layer,12,13 Nd–Fe–B with a multiphase microstructure and vulnerable Nd-rich phase exhibits poor anti-corrosion performance. Usually, the Nd-rich phase with a lower chemical potential acts as the anode, and the Nd2Fe14B matrix phase acts as the cathode to form a galvanic cell in corrosive environments. The Nd-rich intergranular phase is first eroded, followed by the loosening of the matrix phase, which leads to the pulverization of the magnets eventually.14–17 The sensitivity of Nd–Fe–B magnets to harsh conditions and poor corrosion resistance limits the lifetime of various functional applications, especially in wind turbines, generators, and hybrid electric vehicles, where the magnets need to operate for a long time under corrosive and high-temperature conditions.18–20 Therefore, the improvement of corrosion resistance is of prominent importance to ensure the long-term reliability and durability of Nd–Fe–B magnets.
From the perspective of the composition and microstructure of Nd–Fe–B magnets, two common approaches are utilized to improve corrosion resistance. The first strategy is the addition of alloying elements possessing higher electrode potential to improve the chemical stability of the GB phase and to enhance the intergranular corrosion resistance, such as Cu, Al, Ni, Nb, Dy and Tb.21–26 For instance, the addition of minor Cu to form the RE(Fe, Cu)227 and RE6Fe13Cu28 intergranular phases at the triple junctions (TJs) and GBs has been reported to yield more positive corrosion potential Ecorr and lower corrosion current density Icorr. However, this method with a limited amount of alloying elements cannot significantly alter the corrosion mechanism owing to the electrode potential difference among the multiphases while causing a high material cost owing to the introduction of expensive Dy, or deterioration of the remanence owing to the introduction of the nonmagnetic phase.23,29 The second strategy is surface anti-corrosion coating via electroplating coatings,30,31 electrolytic deposition,32 chemical vapor deposition33 and physical vapor deposition,34 which isolate the Nd–Fe–B magnets from the corrosive environment. Various coating materials have been introduced, including metal or metallic coatings such as Ni,35 Ni–P,36 Zn,37 and Al2O338 with widespread application in industrial production fields, and inorganic coatings such as Si and SiO218,34 that can reduce Icorr by three orders of magnitude to effectively block the invasion of external corrosive media. However, most electroplating or electroless plating with inferior adhesion and weak bonding force between the protective coating layer and the Nd–Fe–B substrate can facilitate crack nucleation to accelerate corrosion and increase production cost.38,39 The nonmagnetic and thick coating at dozens of micrometer scales also exerts negative effects on the magnetic properties. Consequently, the persistent exploration of new routes is still on-going towards outstanding corrosion resistance without damaging the mechanical and magnetic properties.
From the perspective of the corrosion mechanism, the corrosion of metallic materials in contact with a corrosive medium is a spontaneous electrochemical process. The root of corrosion originates from the surface. Therefore, it is feasible to improve the corrosion resistance by optimizing the microstructure and chemical composition of the material surface,40,41 catering to the requirements of stability, durability, and tunable thickness.42–44 Oxidation is one of the most common behaviors of metallic materials and provides a promising strategy for improving their anti-corrosion performance. The baking blue process in steel sectors and the hydrophobic/superhydrophobic oxide layers on the electrically conductive metal surfaces are typical examples.45–48 For Nd–Fe–B magnets, the presence of metallic Fe and Nd elements allows for the formation of a uniform oxide coating on the magnet surface via air oxidation.49 Our recent work reported the outstanding anti-corrosion performance of Nd–Fe–B permanent magnets by constructing a hydrophobic triplex surface coating,50i.e., the external outermost zone (EOZ) of the Fe2O3/Fe3O4 phase,51 the intermediate layer of amorphous Nd2O352 and the internal oxidation zone (IOZ) of amorphous Nd2O3 in a columnar α-Fe matrix.53 As reported, the nanometric Fe2O3 needles provide the hydrophobic characteristics of the outermost surface and contribute to an outstanding anti-corrosion performance. The columnar α-Fe structure, which is structurally coherent with the Nd2Fe14B matrix, ensures a stable interface and a good mechanical performance. The rapid air oxidation for 0.5 h with a limited oxide layer thickness of ∼1.5 μm ensures the well-preserved Nd–Fe–B bulk structure and high magnetic properties.50 Another advantage of the oxidization strategy is tunable oxidization coating via controlling the oxidation temperature and time, which has a significant impact on the oxidation products and microstructure of the magnet surface. The trade-off between the magnetic performance and corrosion resistance can also potentially be solved.
However, to date, few studies have been conducted on the oxidation behavior and mechanism of Nd–Fe–B magnets. Previous studies have focused on the oxidation kinetics and oxidation products,51–53 indicating a grey surface coating consisting of two oxidized layers, an EOZ of Fe2O3 and Fe3O4, and an IOZ consisting of an α-Fe matrix containing Nd-oxide particles. There is a lack of clear understanding of the oxidation mechanism and the microstructural evolution under different oxidation environments. Firdaus et al.54–56 confirmed a different microstructural evolution under low (<500 °C) and high (>700 °C) temperature conditions. However, there is still a lack of wide-range oxidation experiments to pave the way for constructing tunable surface oxidation layers. The correlation between the magnetic, mechanical and anti-corrosion performance with the microstructural evolution also remains elusive. Herein, we designed wide-range oxidation temperatures and time gradients to tune the surface oxidation coatings of typical N50 commercial-grade Nd–Fe–B sintered magnets. Temperature-dependent and time-dependent oxidation behaviors with corresponding performance evolutions were systematically investigated. A comparison between the high- and low-temperature oxidation mechanisms is also presented via detailed microstructural characterization.
2. Experimental
2.1. Material description and preparation
The N50 commercial-grade Nd–Fe–B magnets with a nominal composition of (Pr20Nd80)32FebalM1.75B0.9 (M = Cu, Al, Ga, Zr, Co, wt%) were used as the original samples in this study. The oxidation experiment was conducted in two steps. The original samples were first placed above a water bath at 75 °C for ∼30 s to obtain a uniformly coated water drop film and subsequently put into a muffle furnace for air oxidation. The oxidation temperatures increase from 250, 350, 450, 550 to 650 °C, with ranged oxidation durations from 0.5 h, 1 h. 3 h, 6 h, 12 h to 24 h.2.2. Sample characterization
5 μL water droplets were carefully placed on the surface of the original and oxidized magnets at ambient temperature to measure the static water contact angle using a KRÜSS DSA25 goniometer. Note that the averaged values after five separate measurements were obtained under the same experimental conditions to guarantee the accuracy of the results. The surface morphologies and cross-sectional microstructures of the oxidized samples were identified using scanning electron microscopy (SEM, ZEISS Sigma 300). The thickness of the oxide layer was estimated using a cross-sectional SEM image in the back-scattered electron (BSE) mode. Elemental concentration mapping was performed using an electron probe microanalyzer (EPMA, Shimadzu-1720). Phase identification of the oxidized magnet was conducted by X-ray diffraction (XRD, BRUKER D8 Advance) with a step of 0.02° and a counting time of 4 s per step. Nanoscale microstructural and chemical composition observations were conducted using transmission electron microscopy (TEM, FEI Talos F200X) equipped with an energy dispersive X-ray spectrometer (EDS). A focused ion beam (FIB, FEI Strata 400S) was used to prepare the TEM foils.Electrochemical measurements in 3.5% NaCl aqueous solution were performed by a standard three-electrode cell consisting of an Ag/AgCl reference electrode, working electrode (using the sample surface) and Pt counter electrode (AMETEK VersaSTAT 3F workstation). The open circuit potential (OCP) reached its steady state after immersion in the electrolyte for 40 minutes. Electrochemical impedance spectroscopy (EIS) was performed in frequencies ranging from 100 kHz to 0.01 Hz, at sinusoidal potential perturbation of 10 mV. The impedance data were fitted using ZView software. Polarization curves were recorded at a scan rate of 2.0 mV s−1. Magnetic measurements at room temperature were performed using an NIM-62000 hysteresigraph analyser. The thermal stability of the magnetic performance was evaluated by the irreversible loss of the open-circuit flux from 20 to 150 °C. Mechanical performance was also evaluated using Agilent G200 nanoindentation.
3. Results and discussion
Fig. 1 shows the cross-sectional BSE SEM images of the Nd–Fe–B sintered magnets processed under different oxidation durations (0.5–24 h) and oxidation temperatures (250 to 650 °C). The dark gray coating on the Nd–Fe–B magnet surface refers to the oxidation layer, which is highlighted by the white dotted lines and arrows. It can be clearly observed that the thickness of oxidation layer d increases with higher oxidation temperatures or elongating oxidation durations. Compared to the oxidation time, the oxidation temperature plays a more significant role in the growth of the oxide layer. To clarify the temperature dependence of oxidation kinetics, the measured d based on the BSE SEM images in Fig. 1 is plotted in Fig. 2a. Results confirm that d is more significantly influenced by the oxidation temperature, exhibiting the parabolic kinetics with the best-fit lines and being consistent with the references.49,51–53 According to the Wagner diffusion model, d as a function of oxidation time t can be described using d2 = k(T) × t. Based on the Arrhenius equation of k(T) = k0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−E/RT), k(T) is the parabolic rate constant related to oxidation temperature T. Here, k0 is the pre-exponential factor of diffusivity, E is the activation energy, and R is the gas constant.57 Based on the linear regression lines depicted in Fig. 2b, the detailed values of k(T) at different oxidation temperatures can be calculated and plotted in Fig. 2c. Clearly, as the oxidation temperature increases, an exponentially increased diffusion coefficient is observed, i.e., from k(250 °C) = 1.1 × 10−17 m2 s−1 to k(650 °C) = 3.2 × 10−12 m2 s−1.
exp(−E/RT), k(T) is the parabolic rate constant related to oxidation temperature T. Here, k0 is the pre-exponential factor of diffusivity, E is the activation energy, and R is the gas constant.57 Based on the linear regression lines depicted in Fig. 2b, the detailed values of k(T) at different oxidation temperatures can be calculated and plotted in Fig. 2c. Clearly, as the oxidation temperature increases, an exponentially increased diffusion coefficient is observed, i.e., from k(250 °C) = 1.1 × 10−17 m2 s−1 to k(650 °C) = 3.2 × 10−12 m2 s−1.
 | ||
| Fig. 1 Cross-sectional BSE SEM images of Nd–Fe–B sintered magnets processed under different oxidation durations (0.5 to 24 h) and oxidation temperatures (250 to 650 °C). The dark-gray coating on the Nd–Fe–B magnet surface refers to the oxidation layer (pointed out by the white dotted lines and arrows), which thickens with the increase of oxidation temperature or oxidation time. | ||
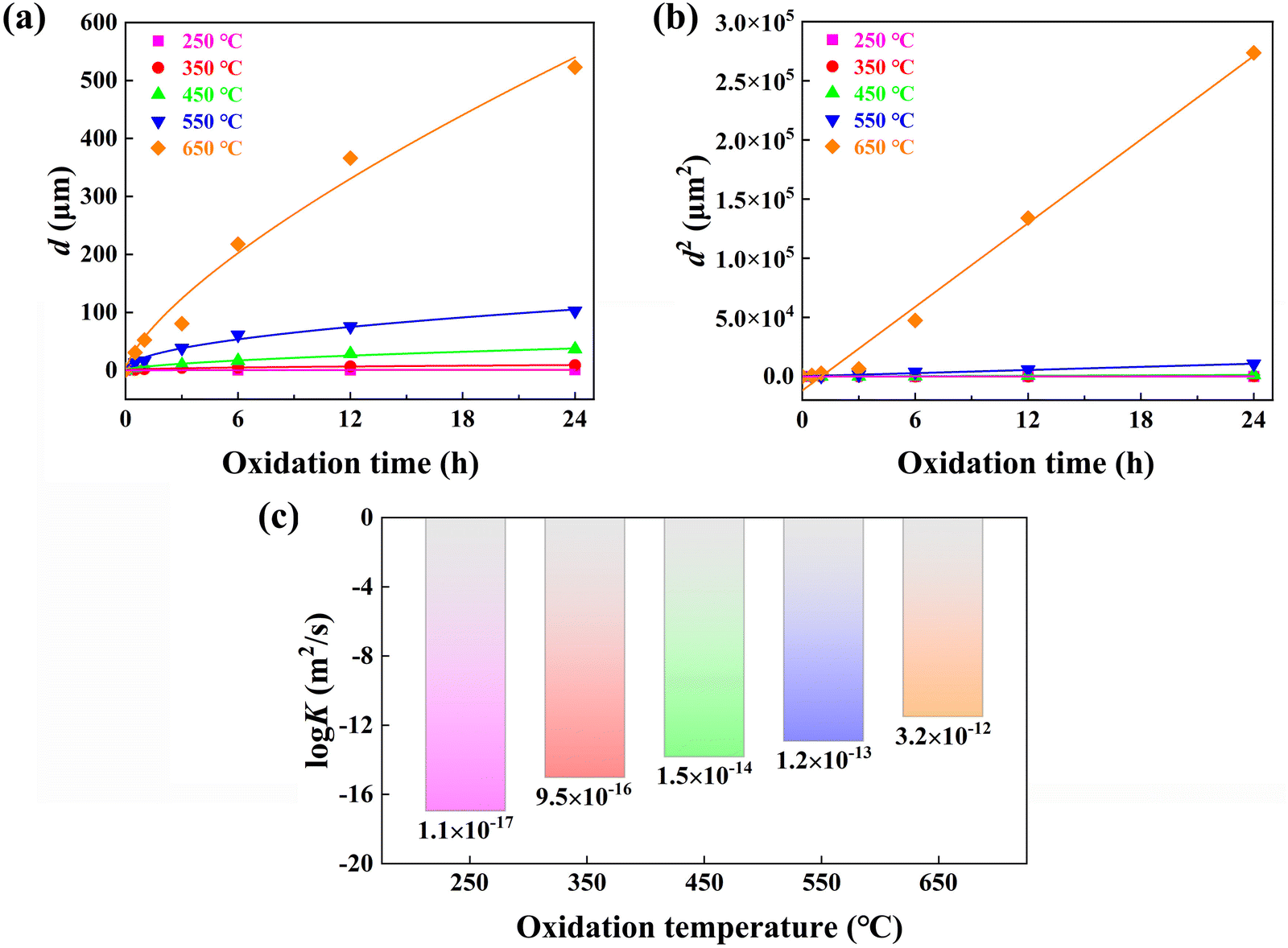 | ||
| Fig. 2 Plots of the kinetic data as a function of oxidation time for bulk Nd–Fe–B magnets oxidized at temperatures between 250 and 650 °C. (a) Time-dependent thickness (d) of the oxidation layer based on the BSE SEM measurements in Fig. 1, showing the parabolic kinetics with the best-fit lines. (b) Thickness squared against oxidation time with fitted linear regression lines. (c) Logarithmic dependences of the Arrhenius diffusion coefficient (logK) on the oxidation temperature. | ||
Note that under the low oxidation temperature of 250 °C, a short-term oxidation duration of 0.5–3 h yields a thin oxide layer that cannot be clearly identified via the BSE SEM images. However, under the high oxidation temperature of 650 °C and long-term oxidation duration of 6–24 h, large cracks appear at the magnet surface, which is possibly due to the exacerbated oxidation reaction and resultant internal stress. Additionally, being different from the magnets processed under lower oxidation temperatures, 650 °C oxidation yields the formation of continuous GBs in white contrast. In particular, the continuous GBs exhibit a coarsening trend for the 650 °C-3 h magnets, which can be the diffusion channel of inward oxygen from the surface toward the inner bulk magnet. Such evolved microstructure and oxidation kinetics dependent on the oxidation temperature and time highly affect the final anti-corrosion and magnetic performance. Previous studies on the 350 °C oxidation of Nd–Fe–B magnets revealed that 0.5–3 h duration yields the best anti-corrosion performance, lowering the Icorr by ∼80% relative to the original magnets.50 With regard to the 650 °C oxidized magnets for 6–24 h duration, the presence of huge cracks evidently fails to enhance the corrosion resistance. Therefore, two typical durations of 0.5 h and 3 h upon wide-ranged oxidation temperatures from 250 to 650 °C are chosen for detailed comparisons in the following figures.
Fig. 3 illustrates a comparison of the anti-corrosion performance of the original and oxidized Nd–Fe–B magnets in a 3.5% NaCl solution. Fig. 3a and d depict the fitted impedance spectra curves, with the corresponding electrochemical equivalent circuit shown in Fig. S1 (ESI†) and fitted electrochemical parameters listed in Tables S1 and S2 (ESI†). Taking Rct as a representative parameter for comparison, the Rct value of the 0.5 h oxidized magnet gradually increases from 1450 Ω cm2 for the original magnet to 2527 Ω cm2 after 250 °C oxidation, peaks at 4584 Ω cm2 after 350 °C oxidation, and then decreases at higher oxidation temperatures. As exemplified by the 650 °C-0.5 h oxidized magnet, the Rct value is lowered to 1475 Ω cm2, which is similar to that of the original magnet. However, for the 3 h oxidized magnet, the corrosion resistance gradually deteriorates with increasing oxidation temperatures. Specifically, the magnets subjected to 250 °C and 350 °C oxidation exhibit better anti-corrosion properties (Rct of 3356 Ω cm2 and 3273 Ω cm2), while the 550 °C and 650 °C oxidized magnets exhibit even poorer corrosion resistance (Rct of 1059 Ω cm2 and 547 Ω cm2) than that of the original magnet.
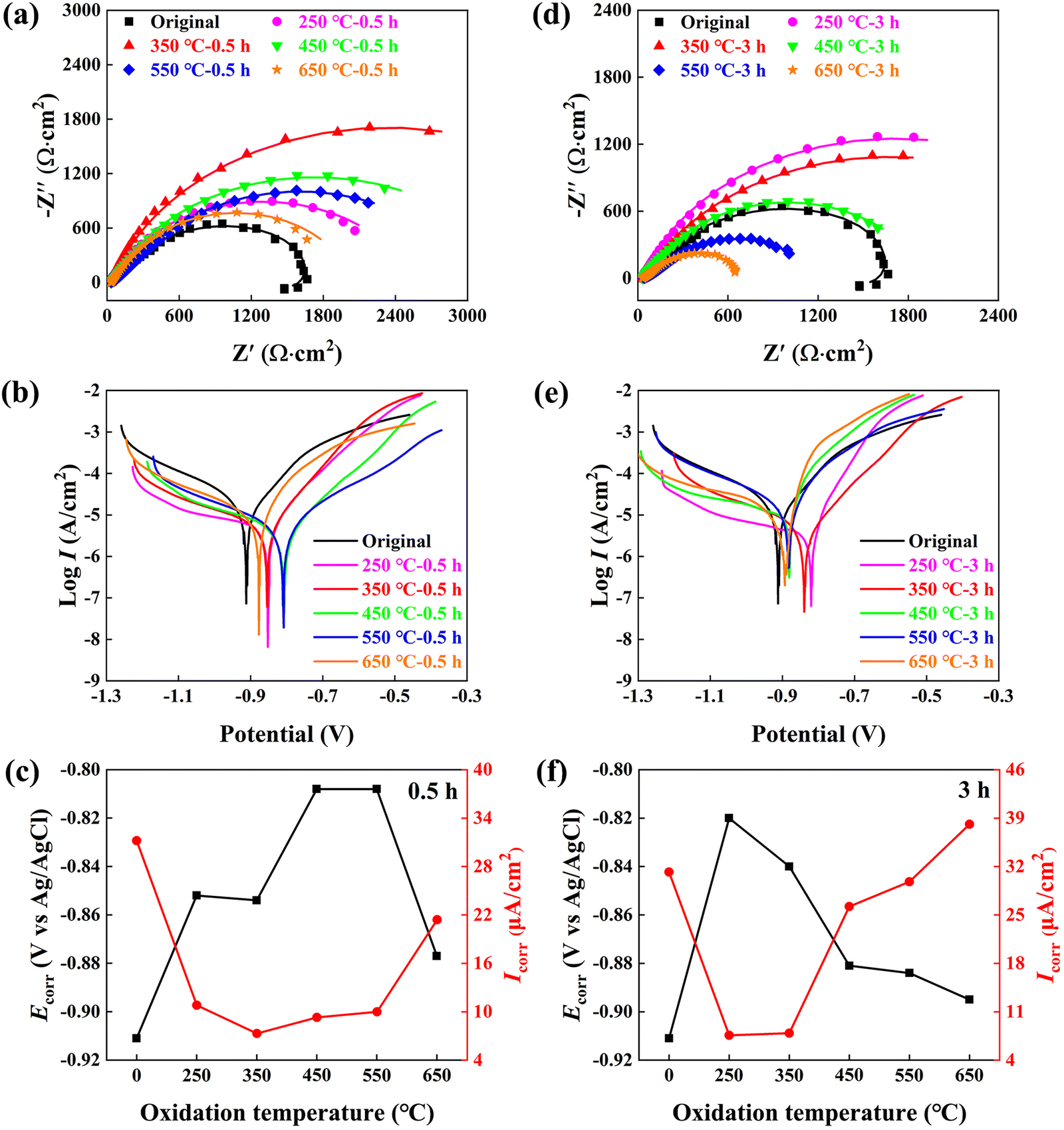 | ||
| Fig. 3 Anti-corrosion performance of 0.5 h and 3 h oxidized Nd–Fe–B magnets at different temperatures (250 to 650 °C) in 3.5% NaCl solution: (a) and (d) Nyquist plots, (b) and (e) polarization curves, (c) and (f) derived corrosion potential (Ecorr) and corrosion current density (Icorr) as a function of oxidation temperature. | ||
Based on impedance spectra, the short-term low-temperature oxidation generates excellent corrosion resistance (i.e., 350 °C-0.5 h or 250 °C-3 h), which is further confirmed by the electrochemical polarization results shown in Fig. 3b, c, e and f. The Icorr value is minimized to 7.3 μA cm−2 after 350 °C-0.5 h oxidation, or to 7.6 μA cm−2 after 250 °C-3 h oxidation, compared to the original magnet (Icorr = 31.2 μA cm−2). Furthermore, the magnets subjected to low-temperature oxidation of 250–350 °C and short-term oxidation of 0.5–3 h exhibit an approximate corrosion potential Ecorr of −0.85 to −0.82 V, compared to the original magnet (−0.91 V). However, the high-temperature oxidation of 650 °C deteriorates Icorr to 21.4 μA cm−2 after 0.5 h oxidation, and further to 38.1 μA cm−2 after 3 h oxidation. The electrochemical polarization and impedance spectra results indicate that the oxide layer gradually loses its protective effect with increasing temperature and elongation time.
Similarly, the magnetic measurement results in Fig. 4 also reveal the negative effects imposed by high-temperature oxidation. Here, two important magnetic parameters at room temperature, coercivity Hcj and remanence Br are compared, as depicted in Fig. 4a and b. Evidently, the 350 °C-0.5 h or 250 °C-3 h magnets exhibit similar Hcj and Br values to the original magnet, while a high oxidation temperature of 650 °C for 3 h severely deteriorates Hcj from 15.05 kOe to 10.92 kOe and Br from 13.88 kG to 13.54 kG. The irreversible flux loss hirr is also shown in Fig. 4c and d to evaluate the thermal stability at elevated service temperatures from 20 °C to 150 °C. Clearly, the 250 °C and 350 °C oxidized magnets exhibit similar hirr to the original magnet (typically ∼15% at 150 °C). However, at higher oxidation temperatures of 550 °C and 650 °C, the hirr values are significantly increased. For example, the hirr value reaches 52% for the 650 °C-3 h oxidized magnet at a service temperature of 150 °C, demonstrating the severely damaged magnetic properties.
 | ||
| Fig. 4 Magnetic performance of 0.5 h and 3 h oxidized Nd–Fe–B magnets as a function of oxidation temperature: (a) and (b) remanence Br (in black color) and intrinsic coercivity Hcj (in red color), (c) and (d) irreversible flux loss hirr. (c) hirr of 0.5 h oxidized Nd–Fe–B magnets at different service temperatures. (d) hirr of 3 h oxidized Nd–Fe–B magnets at a service temperature of 150 °C. | ||
The mechanical performance illustrated in Fig. S2 (ESI†) reveals the simultaneously enhanced micro hardness and modulus of the 250 °C and 350 °C oxidized magnets. The above results demonstrate that outstanding comprehensive performance, including improved anti-corrosion resistance and mechanical performance without compromising magnetic properties, can be attained after low-temperature and short-duration oxidation (250 °C-3 h and 350 °C-0.5 h). Because the performance of the oxidized magnets is critically reliant on the oxidation temperature and time, this stimulates us to investigate the microstructural evolution of the surface oxidation layer. To identify the phase components of the oxidation layer, XRD patterns of the 0.5 h and 3 h oxidized magnets are shown in Fig. 5a and b. Based on the standard PDF cards, the peaks at 29.1°, 38.1°, and 44.4° corresponding to the Nd2Fe14B matrix phase are easily observed in the original Nd–Fe–B magnet. The XRD spectrum of the 250 °C oxidized magnet is similar to that of the original one due to either low temperature or short time. New peaks of α-Fe (44.3°, 63.3°) and Nd2O3 (27.7°) phases appear by increasing the oxidation temperature to 350 °C for 3 h, representing the partial decomposition of the Nd2Fe14B matrix phase during the oxidation process. By further increasing the oxidation temperature, the intensity of the Fe-oxide peaks gradually increases, indicating a higher concentration of oxides coated at the magnet surface. Conversely, the peaks of the α-Fe phase exhibit a gradual weakening trend. The limited penetration depth of XRD detection indicates a gradual thickening of the surface layer composed of Fe-oxide, resulting in a progressive attenuation of the α-Fe signal. Moreover, a weak and broad peak at 2θ ∼ 30° appears to be the amorphous Nd2O3. The XRD results evidently demonstrate that the oxidation temperature exerts a more pronounced influence on the phase formation of the oxidation layer than the oxidation time.
 | ||
| Fig. 5 XRD patterns of (a) 0.5 h and (b) 3 h oxidized Nd–Fe–B magnets at different temperatures as well as the original magnet for comparison. The standard PDF cards for Nd2Fe14B, α-Fe, Fe2O3, Nd2O3 and Fe3O4 phases are also provided at the bottom. | ||
The evolution of surface morphology via SEM is shown in Fig. 6. No evident products appear for a low oxidation temperature of 250 °C. As the oxidation temperature increases, the nanometric needle-like precipitates appear on the surface, and their amount gradually increases to form a denser network structure. When the temperature is increased to 550 °C, a small amount of sheet-like products appear among the needle products (Fig. S3, ESI†), and the quantity of sheet products gradually increases as the temperature and time increase. However, for the 650 °C-3 h oxidized magnet, the amount of needle-like products decreases sharply. Besides, more surface cracks with larger dimensions form upon higher oxidation temperature and longer oxidation time (Fig. S4, ESI†). It is worth noting that although the number of microscopic cracks seems to be reduced in the 650 °C oxidized magnet compared to the 450 °C oxidized magnet (Fig. S4b, c, f, and g, ESI†), low-magnification SEM images in Fig. S4d and h (ESI†) indicate the formation of large cracks. This suggests that the surface cracks gradually transform from microscopic cracks to macroscopic cracks upon high-temperature or long-term oxidation.
 | ||
| Fig. 6 Surface morphologies of (a)–(e) 0.5 h and (f)–(j) 3 h oxidized Nd–Fe–B magnets at different temperatures: (a) 250 °C-0.5 h, (b) 350 °C-0.5 h, (c) 450 °C-0.5 h, (d) 550 °C-0.5 h, (e) 650 °C-0.5 h; (f) 250 °C-3 h, (g) 350 °C-3 h, (h) 450 °C-3 h, (i) 550 °C-3 h, and (j) 650 °C-3 h. Average water contact angles as a function of oxidation temperature for (k) 0.5 h and (l) 3 h oxidized Nd–Fe–B magnets after at least five separate measurements on each sample. Representative photos observed via microgoniometer are also shown in the bottom inset. | ||
The amount of needle-like products affects the surface hydrophobicity. The measured water contact angle (WCA) is shown in Fig. 6k and l, which rises drastically from ∼60° of the original magnet to ∼99° after a low-temperature oxidation of 250 °C. Elevated oxidation temperature from 250 °C to 650 °C or elongating oxidation duration from 0.5 h to 3 h further increases the average WCA. Upon 650 °C oxidation, the WCA of the 0.5 h oxidized magnet reaches the maximum value of 165°, while the WCA of the 3 h oxidized magnet decreases from 156° to 132°. This is presumably caused by the reduced amount of needle-like Fe-oxide products at the oxidation surface. Representative photographs of water droplets on the original and oxidized surfaces are also displayed in the inset in Fig. 6k and l. It is known that the surface with a WCA < 90° is hydrophilic, 90° < WCA < 150° is hydrophobic, and WCA > 150° is superhydrophobic.58,59 Therefore, once it has been oxidized, the original magnet undergoes a transition from hydrophilic to hydrophobic, with increasing hydrophobicity observed upon increasing temperature and time. In particular, the WCA of the 550 °C-3 h or 650 °C-0.5 h oxidized magnets exceeds 150° and exhibits a superhydrophobicity characteristic, which is theoretically advantageous in improving the corrosion resistance of bulk magnets. However, high-temperature oxidation at 450–650 °C induces cracks that compromise the surface integrity and provide direct access for corrosion media to penetrate the inner substance. This may account for the deteriorated corrosion resistance of high-temperature oxidized magnets.
Note that Fig. 1 also reveals the presence of continuous GBs in the 650 °C oxidized magnet, which may account for the high diffusion coefficient of k(650 °C) = 3.2 × 10−12 m2 s−1. Consequently, further characterization of the 650 °C-3 h oxidized magnet is conducted to elucidate the high-temperature oxidation mechanism. Fig. 7a illustrates a cross-sectional BSE SEM image and corresponding EPMA elemental mappings of the 650 °C-3 h oxidized magnet, revealing the multilayer structure with gradually decreased oxygen concentration from the outermost layer towards the inner layer. The external oxidation region mainly comprises Fe-oxide with the highest oxygen concentration, named area A. The second layer belongs to the internal oxidation region (area B), which primarily comprises α-Fe and serves as the main phase of the oxide layer. The oxygen concentration of area B is lower than that of area A while possessing a higher Fe concentration. Area C consists of the Nd2Fe14B matrix phase with a low oxygen concentration, indicating an inward diffusion of oxygen. Besides, the thick GBs exhibit higher oxygen concentration than the Nd2Fe14B matrix phase, emphasizing that the GB region is more easily oxidized and is one of the main channels of oxygen diffusion. Fig. 7c shows the elemental concentration gradient from area B to A, which can explain the difference between the EOZ and IOZ more clearly. It should be observed that there is an intermediate layer at the interface between areas A and B, mainly consisting of Nd/Pr and O, which is consistent with the results reported by Chen et al.50Fig. 7b and d show an enlarged view of the continuous white-contrast GB networks and corresponding line scan profile. The concentrations of O and Nd/Pr in the continuous white-contrast GB networks are significantly higher than other areas, which reveals a violent oxidation reaction occurred. The 650 °C-0.5 h oxidized magnet also exhibits a similar phenomenon (Fig. S5, ESI†). Compared to other oxidized magnets at different temperatures, the presence of a continuous GB phase in the 650 °C magnet is unique, which can increase the channels of inward oxygen diffusion and induce a large diffusion rate of k(650 °C) = 3.2 × 10−12 m2 s−1 that is five orders of magnitude larger than that of k(250 °C).
 | ||
| Fig. 7 (a) Cross-sectional BSE SEM images and corresponding EPMA elemental mappings of a 650 °C-3 h oxidized magnet. (b) Continuous white-contrast networks and corresponding EPMA elemental mappings from the black-rectangle region in (a). Corresponding line scan profiles: (c) line 1 from (a), and (d) line 2 from (b). | ||
To precisely determine the oxidation mechanism of the 650 °C-3 h oxidized magnet, an elaborate examination is performed via two site-specific TEM foils of the 650 °C-3 h oxidized magnet, with the specific location shown in Fig. S6 (ESI†). Fig. 8 shows the TEM characterization results of the region of interest (ROI 1) at the interface between area A and area B in Fig. 7a. The bright field image (BFI) in Fig. 8a provides an overview from the fragile outermost surface to the internal oxidation zone. The selected area electron diffraction (SAED) pattern and corresponding high resolution TEM (HRTEM) image obtained from zone I in Fig. 8b and c reveal the coexisting Fe2O3 and Fe3O4 phases in the outermost layer, which is consistent with the Fe-oxide EPMA results. Fig. 8d shows an enlarged image of zone II, which is divided into regions A and B. The SAED pattern and HRTEM image taken from zone II and the corresponding FFT image in Fig. 8e–g demonstrate that region B refers to the amorphous Nd2O3 phase and region A refers to the evenly distributed crystalline α-Fe phase (Imm [100]).
 | ||
| Fig. 8 TEM characterization of ROI 1 at the interface between area A and area B of the 650 °C-3 h oxidized magnet shown in Fig. 7(a). (a) Overview BFI showing the FIB sample from the fragile outermost surface to the internal oxidation zone. (b) SAED pattern and (c) corresponding HRTEM image obtained from zone I. (d) Enlarged BFI of zone II, (e)–(g) SAED pattern, HRTEM and corresponding FFT images taken from zone II. (h)–(j) SAED pattern, HRTEM and corresponding FFT images taken from zone III. (k) Enlarged BFI of zone IV, (l)–(t) SAED patterns along different zone axes of regions C–F in zone IV, with corresponding HRTEM images and FFT insets. (u) STEM-EDS mappings of zone IV. | ||
However, with the increasing depth from the outermost oxide layer, the phase distribution alters, as shown by zone III in Fig. 8h–j. The mixture of polycrystalline α-Fe and amorphous Nd2O3 is homogeneously distributed. Because the oxygen diffuses inward through GBs,54 here more emphasis is placed on the GBs, as shown by zone IV in Fig. 8k–t. Within the interior of GB, regions labeled as C–F refer to the complex Nd2O3 phases with different crystal structures. Based on the SAED patterns along two different zone axes, regions C and D can be determined as P321 and Pm1 structured Nd2O3, respectively, belonging to the trigonal crystal system. Region E possesses an Imm structure, while region F possesses an Ia structure. According to previous studies,60–62 different crystal structures of Nd2O3 originate from different oxygen concentrations, which represent the inhomogeneous oxidation of GB. From the STEM-EDS mappings demonstrated in Fig. 8u, it can be observed that the Nd2O3 GBs possess higher Nd and O concentrations than adjacent regions.
For ROI 2 of the continuous and thick GBs from area B in Fig. 7a, TEM characterization results are shown in Fig. 9 and Fig. S7 (ESI†), exhibiting a notable microstructural feature that differs from the GBs in Fig. 8. Fig. 9b is an enlarged BFI of zone V, showing that a hierarchical structure forms from the oxidized GB to the oxidized matrix phase, designated as region A–D. From Fig. 9c–j, region A refers to Ia-type Nd2O3 along the [111] zone axis. Region B corresponds to Pm1-type Nd2O3 along the [121] zone axis, containing a higher oxygen content than that of region A. Region C is α-Fe phase with Imm structure along the [111] zone axis. It is also noteworthy that region B and region C exhibit a certain crystallographic orientation relationship (OR) following [121]Pm1-Nd2O3//[111]Imm-α-Fe. In addition, both region C and region D belong to the Imm structured α-Fe phase with the same crystallographic orientation along the [111] zone axis. However, region C is the pure α-Fe phase, while region D is a mixture of crystalline α-Fe phase and amorphous Nd2O3 phase. The STEM-EDS mappings in Fig. 9l show that region B corresponds to continuous GB enriched with Nd/Pr and O, which possess the maximum oxygen concentration. Region C is the intermediate layer between the oxidized GBs and the matrix phase, which has a higher Fe concentration and extremely low concentrations of Nd/Pr/O. Region D is a mixture of Fe, O and Nd/Pr, which is consistent with zone IV in Fig. 8. Such a peculiar multi-layered structure in zone V is schematically shown in Fig. 9m. Combining the results of elemental mapping and HRTEM images, we envision a relationship between the continuous GB (regions A and B) and the intermediate layer (region C). A violent oxidation reaction occurs to form the continuous and thick GBs, which require large amounts of Nd/Pr and O in a short time. Because the elemental concentration of the original GB phase is no longer sufficient to fulfill the oxidation reaction, it is necessary to consume the Nd/Pr and O elements at the edge of the matrix phase, which leads to the formation of the intermediate α-Fe layer. This can be confirmed by the certain crystallographic OR between regions B and C, as well as by the same crystallographic orientation of the α-Fe phase between regions C and D.
 | ||
| Fig. 9 TEM characterization of ROI 2 of the 650 °C-3 h oxidized magnet within area B shown in Fig. 7(a). (a) BFI of the continuous and thick grain boundary, (b) enlarged image of zone V. (c) SAED pattern taken from region A in zone V, and (d) corresponding HRTEM image and FFT inset. (e) HRTEM image of the A/B/C interface. (f) HRTEM image of the C/D interface. (g)–(j) Corresponding FFT images of regions B–D in zone V. (k) and (l) STEM-EDS mappings of the B/C/D interface. (m) Schematic of the multi-layered structure in zone V. | ||
For another zone VI containing the continuous and thick GBs, a multi-layered structure similar to zone V also appears, designated as region E–H in Fig. 10. The SAED pattern, HRTEM and FFT images in Fig. 10b–d reveal that region G is a mixture of α-Fe phase and crystalline Nd2O3 with the same Imm structure and crystallographic orientation along the [100] zone axis, following [100]Imm-Nd2O3//[100]Imm-α-Fe. As shown in Fig. 10e–h, region E is Pm1-type Nd2O3 along the [121] zone axis and region F is an Imm-type Nd2O3 along the [100] zone axis, following [100]Imm-Nd2O3//[121]Pm1-Nd2O3. From Fig. 10i–k, region H with the coexisting α-Fe phase and amorphous Nd2O3 is analogous to region D in zone V. Through the combined microstructural and elemental distribution characterization results in Fig. 10b–l, the schematic of the multi-layered structure of zone VI is shown in Fig. 10m. It can be inferred that although the formation of the continuous and thick GBs consumes the massive Nd/Pr and O atoms at the edge of the matrix phase and yields the α-Fe intermediate layer, the residual Nd/Pr and O atoms may also facilitate the uniform distribution of crystalline Imm-type Nd2O3 within the α-Fe layer, following a certain crystallographic OR.
 | ||
| Fig. 10 TEM characterization of zone VI of the 650 °C-3 h oxidized magnet. (a) Enlarged BFI. (b) SAED pattern of region G, which is taken along the [100] zone axis of Imm structured α-Fe phase, (c) corresponding HRTEM and (d) FFT images, showing the co-existence of Imm structured α-Fe and Nd2O3 phases. (e) HRTEM image of the G/F/E interface, and (f)–(h) corresponding FFT images of region E and F, showing the co-existence of Imm structured and Pm1 structured Nd2O3. (i) HRTEM image of the G/H interface, and (j) corresponding FFT image, showing the co-existence of Imm structured α-Fe and Nd2O3 phases in the crystalline state, together with the amorphous Nd2O3. (k) FFT image of region H, showing the co-existence of Imm structured α-Fe phase and amorphous Nd2O3. (l) STEM-EDS mappings of the E/F/G/H interface. (m) Schematic of the multi-layered structure in zone VI. | ||
Fig. 11 presents a new layered structure in zone VII. Results show that region I corresponds to the Imm-structured α-Fe phase along the [111] zone axis. However, unlike zone V and zone VI, where the α-Fe and amorphous Nd2O3 phases coexist homogeneously, the amorphous Nd2O3 in region J is spherically embedded in the α-Fe phase, as clearly observed from the STEM-EDS mappings in Fig. 11f and schematically shown in Fig. 11g. It is considered that the residual Nd2O3 phase in the α-Fe intermediate layer (region I) gradually grows to form spherical amorphous Nd2O3 at the I/J interface.
 | ||
| Fig. 11 TEM characterization of zone VII of the 650 °C-3 h oxidized magnet. (a) Enlarged BFI. (b) SAED pattern of region I, and (c) HRTEM image of region I and corresponding FFT image. (d) HRTEM image of the J/I interface, (e) corresponding FFT image and (f) STEM-EDS mappings. (g) Schematic of the multi-layered structure in zone VII. | ||
For a better understanding, the 350 °C-3 h oxidized magnet is also selected for TEM characterization, as shown in Fig. 12. Compared to the 650 °C-3 h oxidized magnet, besides the absence of continuous and thick GBs, the distribution of α-Fe phase and amorphous Nd2O3 are also different. As shown by the SAED pattern, HRTEM and FFT images of zone I in Fig. 12b-e, the microcrystalline Imm α-Fe phase with multiple orientations ([100] zone axis in Fig. 12d and [111] zone axis in Fig. 12e) and amorphous Nd2O3 phase always coexist. However, for the 650 °C-3 h oxidized magnet shown in Fig. 8d and e, a single crystalline Imm α-Fe phase ([100] zone axis) without the identification of amorphous Nd2O3 is identified in the entire region A.
 | ||
| Fig. 12 TEM characterization of the 350 °C-3 h oxidized magnet. (a) BFI of the typical region from the outermost surface to the internal oxidation zone, (b) SAED pattern taken from zone I, (c) HRTEM image of zone I, corresponding FFT images of (d) region A and (e) region B. | ||
Based on the above results, it can be concluded that the oxidation temperature exerts a more pronounced impact on the thickness of the oxidation layer than that of oxidation time (Fig. 1 and 2), as well as the phase components (Fig. 5) and resultant performance (Fig. 3, 4 and Fig. S2, ESI†). The short-term low-temperature oxidation of 350 °C-0.5 h or 250 °C-3 h generates excellent corrosion resistance and magnetic and mechanical properties. A low temperature fails to guarantee the formation of the oxidation layer in a short time (250 °C-0.5 h or 250 °C-1 h), while an excessively high temperature (650 °C-3/6/12/24 h) triggers a violent oxidation reaction and substantial internal stress, leading to the formation of large macroscopic cracks and undermining structural integrity. Consequently, the 650 °C oxidized magnet exhibits the worst comprehensive performance, including lower anti-corrosion, magnetic and mechanical performance than the original magnet (Tables S1 and S2, ESI†). The WCA results show that the original hydrophilic surface transforms into a hydrophobic or superhydrophobic state due to the formation of nanometric Fe oxide needles on the outermost layer (Fig. 6). Such a hydrophobic or superhydrophobic surface helps isolate the corrosive medium and is beneficial in enhancing corrosion resistance. However, when the oxidation temperature rises to 650 °C, a superhydrophobic surface with large macroscopic cracks compromises the surface integrity, provides additional diffusion access for the corrosion medium to decompose the matrix substance, and accounts for the deteriorated corrosion resistance of high-temperature oxidized magnets. Meanwhile, the thickening of the oxidation layer (high kinetic coefficients of 3.2 × 10−12 m2 s−1 and hundreds of micrometers oxidation layer upon high-temperature oxidation of 650 °C) together with the large macroscopic cracks leads to deteriorated magnetic properties of the oxidized magnets (Br, Hcj and hirr in Fig. 4). It is also noted that being different from the monotonically reduced Hcj, Br first increases and then decreases with increasing oxidation temperature, exhibiting a slightly enhanced value for the 450 °C-3 h oxidized magnet (Fig. 4b). This can be attributed to the thickening of IOZ primarily composed of a soft-magnetic α-Fe phase, which possesses exceptionally high saturation magnetic polarization (JS = 21.5 kG).63
New understandings of the high-temperature oxidation mechanism are also grained. For the 650 °C-3 h oxidized magnet, continuous and coarse GB networks with significantly higher oxygen concentrations appear in the IOZ (Fig. 7 and Fig. S5, ESI†). Detailed TEM characterization (Fig. 8–11) reveals the overall microstructure of the 650 °C-3 h oxidized magnet, with two main features that facilitate oxygen diffusion and permit the larger k(T). First, compared to the 350 °C-3 h oxidized magnet (Fig. 12), an evenly distributed single crystalline α-Fe phase with the absence of amorphous Nd2O3 appears in the outermost layer for the 650 °C-3 h oxidized magnet (Fig. 8). The high-angle GBs between the neighboring crystalline α-Fe phase with different crystalline orientations and growth directions offer more channels for inward oxygen diffusion. Second, for the first time, we observe the continuous and thick GB regions with multi-layered structures in the IOZ (Fig. 9–11). The multi-layered structure can be divided into four layers: the first and second layers refer to continuous Nd/Pr/O-rich GBs with the maximum oxygen concentration (Pm1 and Imm/Ia structured Nd2O3), the third layer refers to a Fe-rich intermediate layer with extremely low concentrations of Nd/Pr and O (dominated by Imm structured α-Fe), and the fourth layer refers to a mixture of Fe, Nd/Pr and O (coexisting Imm structured α-Fe and amorphous Nd2O3). Certain crystallographic orientation relationships (ORs) are identified in the multi-layered GBs, including [100]Imm-Nd2O3//[121]Pm1-Nd2O3 at the interface of the first and second layers (Fig. 10h), [121]Pm1-Nd2O3//[111]Imm-α-Fe at the interface of the second and third layers (Fig. 9h), and [100]Imm-Nd2O3//[100]Imm-α-Fe within the third layer (Fig. 10j). Besides, amorphous Nd2O3 spherically embedded in the α-Fe phase is observed within the fourth layer (Fig. 11e).
Note that the first and second layers dominated by Nd2O3 with different crystal structures, the intermediate third layer dominated by α-Fe between the continuous GB phase and the oxidized matrix phase, and the fourth layer containing spherical amorphous Nd2O3 embedded in the α-Fe matrix phase are distinct from the oxidation products in the EOZ or the previous publications. These new microstructural characteristics can be explained by the progressive oxidation reaction. It is inferred that the formation of thick and continuous GBs requires a large amount of Nd/Pr and O, promoting the complete decomposition of the neighboring matrix phases to generate more Nd/Pr and Fe. The released Nd/Pr atoms participate in the oxidation reaction of the GB phase, resulting in the formation of Nd/Pr/O-rich RE-oxide with different O concentrations and crystal structures in the first and second layers, together with the Fe-rich intermediate layer. Part of the residual Nd/Pr atoms also generate the uniformly distributed crystalline Nd2O3 phase in the α-Fe intermediate layer (Fig. 10m). Furthermore, the residual Nd2O3 phase in the intermediate α-Fe layer gradually grows and moves toward the oxidized matrix phase, which results in the formation of a pure α-Fe phase in the intermediate third layer, and the partially spherical amorphous Nd2O3 was embedded in α-Fe matrix phase in the fourth layer (Fig. 11g).
4. Conclusions
Temperature-dependent and time-dependent oxidation behaviors of the Nd–Fe–B sintered magnets with the corresponding performance evolutions are systematically unraveled. The main conclusions can be summarized as follows:(1) Within the low-temperature range (250–350 °C), the low kinetic coefficients (1.1 × 10−17–9.5 × 10−16 m2 s−1) generate the thin and hydrophobic oxidation layer with fewer cracks over short durations of 0.5–3 h, ensuring the excellent anti-corrosion, magnetic and mechanical performance of the oxidized magnets.
(2) Within the high-temperature range (450–650 °C), a thickening oxidation layer with exponentially increased kinetic coefficients (1.5 × 10−14–3.2 × 10−12 m2 s−1) exhibits hydrophobic or superhydrophobic characteristics and generates large macroscopic cracks, resulting in the lowered anti-corrosion, magnetic and mechanical performance of the oxidized magnets.
(3) The high-temperature oxidation mechanism is unveiled with two main new features that differ from the low-temperature one. One is the formation of a single crystalline α-Fe phase with the absence of amorphous Nd2O3 in the EOZ, and the other is the formation of continuous and coarse GB networks with a multi-layered structure in the IOZ. Both features accelerate the inward oxygen diffusion and explain the high oxidation kinetics of the 650 °C oxidized magnet.
In summary, the above findings on the correlation between the tunable oxidation behaviors and the performance of oxidized Nd–Fe–B magnets provide new insights into designing corrosion-resistant scalable permanent magnets. New understandings of temperature-dependent oxidation mechanisms also provide guidelines for delicately controlling oxidation coatings for future industrial applications.
Author contributions
L. Zhou: formal analysis, investigation, methodology, writing – original draft, writing – review & editing. J. Jin: conceptualization, supervision, funding acquisition, project administration, methodology, writing – original draft, writing – review & editing. W. Chen: formal analysis, data curation, validation. S. Ren: data curation, validation, writing – review & editing. M. Bu: data curation, validation. X. Li: resources, validation. B. Xin: resources, software. C. Wu: resources. M. Yan: supervision, funding acquisition, project administration.Data availability
The authors declare that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key Research and Development Program of China (2022YFB3505800), “Leading Goose” R&D Program of Zhejiang Province (2022C01110), National Natural Science Foundation of China (52171192 and U21A2052), Natural Science Foundation of Inner Mongolia (2023ZD26).References
- D. C. Nababan, R. Mukhlis, Y. Durandet, M. I. Pownceby, L. Prentice and M. A. Rhamdhani, Corros. Sci., 2021, 189, 109560 CrossRef CAS.
- Z. Liu, J. He, Q. Zhou, Y. Huang and Q. Jiang, J. Mater. Sci. Technol., 2022, 98, 51–61 CrossRef CAS.
- O. Gutfleisch, M. Willard, E. Bruck, C. Chen, S. Sankar and J. Liu, Adv. Mater., 2011, 23, 821–842 CrossRef CAS.
- H. Sepehri-Amin, S. Hirosawa and K. Hono, Advances in Nd–Fe–B Based Permanent Magnets, Elsevier B. V, Amsterdam, 2018, pp. 269–372 Search PubMed.
- W. Mo, L. Zhang, Q. Liu, A. Shan, J. Wu and M. Komuro, Scr. Mater., 2008, 59, 179–182 CrossRef CAS.
- X. Tang, J. Li, H. Sepehri-Amin, A. Bolyachkin, A. Martin-Cid, S. Kobayashi, Y. Kotani, M. Suzuki, A. Terasawa, Y. Gohda, T. Ohkubo, T. Nakamura and K. Hono, NPG Asia Mater., 2023, 15, 50 CrossRef CAS.
- J. Herbst, Rev. Mod. Phys., 1991, 63, 819–898 Search PubMed.
- V. Chaudhary, S. Mantri, R. Ramanujan and R. Banerjee, Prog. Mater. Sci., 2020, 114, 10068 Search PubMed.
- G. Hrkac, T. Woodcock, K. Butler, L. Saharan, M. Bryan, T. Schrefl and O. Gutfleisch, Scr. Mater., 2014, 70, 35–38 Search PubMed.
- X. Shi, M. Zhu, X. Wang, S. Zhou, X. Wang, M. Zhang and W. Li, J. Rare Earths, 2020, 38, 735–741 CrossRef CAS.
- M. Soderznik, H. Sepehri-Amin, T. Sasaki, T. Ohkubo, Y. Takada, T. Sato, Y. Kaneko, A. Kato, T. Schrefl and K. Hono, Acta Mater., 2017, 135, 68–76 CrossRef CAS.
- J. M. D. Coey and T. Iriyama, Bonded Sm-Fe-N permanent magnets, Woodhead Publishing, 2022, pp. 305–342 Search PubMed.
- J. Xu, Y. Yi, B. Jia, H. Xue, G. Tian, Z. Ma and Y. Hou, J. Mater. Chem. C, 2024, 12, 14714–14728 RSC.
- X. Zhang, Y. Ma, B. Zhang, Y. Li, M. Lei, F. Wang, M. Zhu and X. Wang, Corros. Sci., 2014, 87, 156–166 Search PubMed.
- K. Tokuhara and S. Hirosawa, J. Appl. Phys., 1991, 69, 5521–5523 CrossRef CAS.
- R. Sueptitz, K. Tschulik, M. Uhlemann, M. Katter, L. Schultz and A. Gebert, Corros. Sci., 2011, 53, 2843–2852 CrossRef CAS.
- B. Peng, J. Jin, Y. Liu, C. Lu, L. Li and M. Yan, Corros. Sci., 2020, 177, 108972 CrossRef CAS.
- Z. Shi, H. Zeng, Y. Yuan, N. Shi, L. Wen, H. Rong, D. Zhu, L. Hu, L. Ji, L. Zhao and X. Zhang, Adv. Funct. Mater., 2023, 33, 2213042 CrossRef CAS.
- Q. Feng, Y. L. Huang, H. F. Li, J. Y. Yu, C. Y. Wang, Z. J. Wu, Y. F. Yao, Y. H. Hou, W. Li, L. Ma and H. B. Yu, J. Alloys Compd., 2023, 940, 168827 CrossRef CAS.
- J. Zheng, S. Wang, S. Zhang, W. Cai, L. Qiao, H. Chen, Y. Ying, W. Li, J. Yu and S. Che, J. Mater. Res. Technol., 2023, 22, 3020–3032 CrossRef CAS.
- Y. Wu, M. Zhu, P. Shen, Y. Fang, Q. Sun, L. Zhang, C. Wang, X. Song, M. Zheng and W. Li, J. Mater. Res. Technol., 2023, 24, 6369–6377 CrossRef CAS.
- T. Zhou, X. Yang, Q. Wang, J. Chen, W. Bao, B. Yu, R. Liu and G. Xie, J. Alloys Compd., 2023, 968, 172079 CrossRef CAS.
- J. Yan, Q. Wu, X. Hu, J. Jia, Y. Zhao, M. Zou and H. Ge, J. Magn. Magn. Mater., 2024, 600, 172129 CrossRef CAS.
- K. Wang, Y. Dai, J. Deng, J. Wang, H. Zhou, Q. Yao and W. Huang, J. Rare Earths, 2024, 42, 345–353 CrossRef CAS.
- K. Zhang, Z. Yue, Y. Ma, J. He, X. Li, W. Gong and Y. Huang, J. Alloys Compd., 2023, 969, 172391 CrossRef CAS.
- J. Yan, Q. Wu, H. Ge, Z. Xi and J. Liang, Mater. Chem. Phys., 2024, 315, 128901 CrossRef CAS.
- J. Jin, M. Yan, W. Chen, W. Zhang, Z. Zhang, L. Zhao, G. Bai and J. Greneche, Acta Mater., 2021, 204, 116529 CrossRef CAS.
- L. Liang, T. Ma, P. Zhang, J. Jin and M. Yan, J. Magn. Magn. Mater., 2014, 355, 131–135 CrossRef CAS.
- C. Huang, C. Mo and S. Ou, J. Magn. Magn. Mater., 2024, 600, 172166 CrossRef CAS.
- Y. Zhan, Y. Dai, D. Tan, Z. Yi, J. Wu, W. Jiang and Z. Cao, J. Alloys Compd., 2023, 961, 170956 CrossRef CAS.
- X. Li, Y. Ma, S. Peng and L. Tian, RSC Adv., 2024, 14, 8641–8651 RSC.
- X. Zhang, J. Xu, J. Luo, Z. Hu, J. Huang and Y. Ma, J. Alloys Compd., 2023, 969, 172442 CrossRef CAS.
- X. Zhang, J. Luo, J. Xu, J. Chen, J. Huang, Y. Ma and M. Xue, Trans. Nonferrous Met. Soc. China, 2024, 34, 1606–1617 CrossRef CAS.
- J. Tao, B. Liu, P. Zhang, G. Xu, J. Lv, J. Huang, J. Yan, W. Sun, B. Li, D. Wang and Y. Wu, J. Rare Earths, 2023, 41, 1203–1210 CrossRef CAS.
- P. Zhang, Q. Liu, J. Huang, J. Cui, W. Sun, B. Li and G. Xu, J. Alloys Compd., 2022, 922, 166206 CrossRef CAS.
- W. Zhao, L. Zhang, H. Zhao, C. Yuan, J. Yan, Z. Li, L. Xin, C. Li, F. Wu, S. Ye and X. Zhang, J. Mater. Sci., 2024, 59, 11510–11532 CrossRef CAS.
- L. Duan, J. Chen, P. Zhang, G. Xu, J. Lv, D. Wang, W. Shen and Y. Wu, J. Alloys Compd., 2023, 936, 168292 CrossRef CAS.
- Q. Zhao, S. Geng, G. Chen and F. Wang, Corros. Sci., 2021, 192, 109837 CrossRef CAS.
- X. Ding, Y. Wu, L. Yang, C. Xu, S. Mao, Y. Wang, D. Zheng and Z. Song, Vacuum, 2016, 131, 127–134 CrossRef CAS.
- R. Singh and N. B. Dahotre, J. Mater. Sci.: Mater. Med., 2007, 18, 725–751 CrossRef CAS PubMed.
- X. Liu, M. Gong, S. Deng, T. Zhao, T. Shen, J. Zhang and D. Wang, Adv. Funct. Mater., 2021, 31, 2009032 CrossRef CAS.
- G. Azimi, R. Dhiman, H. Kwon, A. Paxson and K. Varanasi, Nat. Mater., 2013, 12, 315–320 CrossRef CAS PubMed.
- J. Lee, S. Shin, Y. Jiang, C. Jeong, H. Stone and C. Choi, Adv. Funct. Mater., 2017, 27, 1606040 CrossRef.
- G. Barati Darband, M. Aliofkhazraei, S. Khorsand, S. Sokhanvar and A. Kaboli, Arab. J. Chem., 2020, 13, 1763–1802 CrossRef CAS.
- B. Singh, B. Jena, S. Bhattacharjee and L. Besra, Surf. Coat. Technol., 2013, 232, 475–481 CrossRef CAS.
- G. Chen, L. Xue, J. Wang, Z. Tang, X. Li and H. Dong, Corros. Sci., 2020, 173, 108797 CrossRef.
- Y. Ye, D. Zhang, J. Li, T. Liu, J. Pu, H. Zhao and L. Wang, Corros. Sci., 2019, 147, 9–21 CrossRef CAS.
- X. Zhang, F. Jiang, R. Chen, Y. Chen and J. Hu, Corros. Sci., 2020, 174, 108836 CrossRef.
- J. Jacobson and A. Kim, J. Appl. Phys., 1987, 61, 3763–3765 CrossRef CAS.
- W. Chen, J. Jin, J. Yu, L. Zhou, B. Peng, S. Fu, X. Liu, G. Bai and M. Yan, J. Mater. Chem. C, 2023, 11, 6884–6893 RSC.
- Y. Li, H. Evans, I. Harris and I. Jones, Oxid. Met., 2003, 59, 167–182 CrossRef CAS.
- I. Skulj, H. Evans and I. Harris, J. Mater. Sci., 2008, 43, 1324–1333 CrossRef CAS.
- J. Breton and J. Teillet, IEEE Trans. Magn., 1990, 26, 2652–2654 CrossRef.
- M. Firdaus, M. Rhamdhani, W. Rankin, M. Pownceby, N. Webster, A. D’Angelo and K. McGregor, Corros. Sci., 2018, 133, 374–385 CrossRef CAS.
- M. Firdaus, M. Rhamdhani, Y. Durandet, W. Rankin and K. McGregor, Corros. Sci., 2018, 133, 318–326 CrossRef CAS.
- M. Firdaus, M. Akbar Rhamdhani, Y. Durandet, W. John Rankin, K. McGregor and N. Webster, 4th Symposium on Rare Metal Eraction and Processing, San Diego, CA, 2017 Search PubMed.
- W. Liu, M. Yue, G. Wang, D. Zhang and J. Zhang, Corrosion, 2010, 66, 055004 CrossRef.
- E. Kobina Sam, D. Kobina Sam, X. Lv, B. Liu, X. Xiao, S. Gong, W. Yu, J. Chen and J. Liu, Chem. Eng. J., 2019, 373, 531–546 CrossRef CAS.
- A. Lafuma and D. Quere, Nat. Mater., 2003, 2, 457–460 CrossRef CAS PubMed.
- W. Mo, L. Zhang, Q. Liu, A. Shan, J. Wu and M. Komuro, Scr. Mater., 2008, 59, 179–182 CrossRef CAS.
- K. Makita and O. Yamashita, Appl. Phys. Lett., 1999, 74, 2056–2058 CrossRef CAS.
- T. Sasaki, T. Ohkubo and K. Hono, Acta Mater., 2016, 115, 269–277 CrossRef CAS.
- J. M. D. Coey, Magnetism and Magnetic Materials, Cambridge University Press, 2010, pp. 439–463 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc03843f |
| This journal is © The Royal Society of Chemistry 2025 |