
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancement in the nonlinear optical properties of silver nanoprisms through graphene oxide anchoring†
Fadeela Chundekatt
Ummer‡
a,
Hao
Yuan‡
b,
Isabelle
Russier-Antoine
b,
Fabien
Rondepierre
b,
Pierre-François
Brevet
b,
Pierre
Mignon
b,
Nandakumar
Kalarikkal
 a and
Rodolphe
Antoine
*b
a and
Rodolphe
Antoine
*b
aInternational and Inter University Centre for Nanoscience and Nanotechnology (IIUCNN), Mahatma Gandhi University, Kottayam, Kerala-686560, India
bUniv Lyon, Université Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, 69622 Lyon, France. E-mail: rodolphe.antoine@univ-lyon1.fr
First published on 17th January 2025
Abstract
Efficient nonlinear optical materials remain a subject of significant interest in the scientific community, with ongoing efforts focused on optimizing their properties for practical applications. This paper aims at exploring how the nonlinear optical properties of silver nanoprisms are affected by the interaction with graphene oxide sheets. For this purpose, we produce nanocomposites consisting of citrate-passivated silver nanoprisms anchored both electrostatically and covalently on graphene oxide nanosheets in a cost-effective and reproducible manner. The novelty of the technique hinges on the covalent functionalization of silver nanoprisms onto graphene oxide (GO) nanosheets according to the 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride crosslinking method, using the existing carboxylic groups present both at the surfaces of the nanoprisms and the GO nanosheets. The formed hybrid nanocomposites were characterized by TEM measurements and exhibit nonlinear optical (NLO) properties, in particular a strong second harmonic scattering response as well as a multiphoton excited fluorescence spectrum characterized by a broad band in the visible range between 350 and 700 nm. In addition, the NLO response is sensitive to the nature of the interaction (electrostatic or covalent), which might be attributed to different charge transfer capabilities between covalently or electrostatically bound silver particles onto graphene oxide. Such nanocomposites are therefore promising for new applications in the areas of optoelectronics and photovoltaics.
1 Introduction
Efficient nonlinear optical silver based materials remain a subject of significant interest in the scientific community,1–7 with ongoing efforts focused on optimizing their properties for practical applications. Silver nanoparticles (AgNPs) have excelled by their exceptional conductive, electronic, and optical properties.8 However, a disadvantage when employing AgNPs as a functional material is their tendency to agglomerate, which hampers their performance. This could be avoided by using a template or supporting material to keep the AgNPs well dispersed. To enhance the dispersion of AgNPs, graphene oxide (GO) is an ideal supporting material.9 GO has both sp2 and sp3 carbon domains and it gets dispersed into water readily due to the presence of an oxygen-containing group.10 Moreover, GO's oxygen functional groups act as optimal reactive sites for the anchoring of metal nanoparticles and preventing their agglomeration. GO-based nanocomposites allow for attractive properties of multiple functional components to be combined. For instance, GO can be merged with plasmonic NPs to obtain composites with superior nonlinear optical (NLO) properties.11 For example, a drastic improvement in the nonlinear absorption of silver NP-decorated graphene oxide nanohybrids produced via pulsed laser ablation using a nanosecond laser was demonstrated by Nancy et al.12 In addition, a 4-fold increase in two-photon excitation emission intensity is observed from the GO–gold nanocrystal composites compared to pure gold nanocrystals.13 Also a strong enhancement of the nonlinear absorption and optical limiting (OL) of graphene oxide–gold nanorod hybrids over GO and gold nanorods was also reported.14 The two-photon absorption of GO sheets, nonlinear scattering of GO and plasmonic silver NPs, and charge transfer between GO and silver NPs are key parameters to enhance NLO and OL properties of nanocomposites.15–17One of the first steps in developing this silver-graphene oxide nanocomposite is optimizing the NPs’ morphology and dispersion over the GO surface to enhance their properties. Beyond spherical particles, there is a large palette of anisotropic nanomaterials, such as 1D and 2D nanomaterials (e.g. nanorods, nanowires, …) and 3D nanostructures (e.g. prisms, pyramids, stars, flowers, nanourchins, tadpole, …). Alteration of the structural symmetry of nanoparticles (indeed such particles possess asymmetry axes) leads to remarkable nonlinear optical properties. Three-fold anisotropy of prisms points to the possible enhancement of their quadratic nonlinear optical (NLO) response compared to that of centrosymmetric nanoparticles such as nanospheres and nanorods.18 In addition, it has been shown that the nanoprism shape noncentrosymmetry has a leading role in the quadratic NLO response although surface defects induce deviations from the ideal threefold symmetry.19
This paper aims at exploring how the nonlinear optical properties of silver nanoprisms are affected by the interaction with graphene oxide sheets. For this purpose, we produce nanocomposites consisting of citrate-passivated silver nanoprisms anchored both electrostatically and covalently on graphene oxide nanosheets in a cost-effective and reproducible manner. The novelty of the technique hinges on the covalent functionalization of silver nanoprisms onto graphene oxide (GO) nanosheets according to the 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride crosslinking method, using the existing carboxylic groups present both at the surfaces of the nanoprisms and the GO nanosheets. The formed hybrid nanocomposites, which were characterized by TEM measurements, exhibit nonlinear optical (NLO) properties, in particular a strong second harmonic scattering response as well as a multiphoton excited fluorescence spectrum characterized as a broad band in the visible range between 350 and 700 nm. In addition, the NLO response is sensitive to the nature of the interaction (electrostatic or covalent), whichmight be attributed to different charge transfer capabilities between covalently or electrostatically bound silver particles onto graphene oxide. Such nanocomposites are therefore promising for new applications in the areas of optoelectronics and photovoltaics.
2 Experimental
2.1 Materials
Graphite flakes, silver nitrate (AgNO3), trisodium citrate (Na3C6H5O), sulphuric acid (H2SO4), phosphoric acid (H3PO4), potassium permanganate (KMnO4), hydrogen peroxide (H2O2) and sodium borohydride (NaBH4) are used. The chemicals are of analytical grade and purchased from Merck. De-ionized water is used throughout the synthesis and purification of the samples.2.2 Preparation of silver nanoprisms
Citrate-capped triangular silver nanoprisms are synthesized using a slightly modified method used by Panzarasa20 and the protocol is summarized in Fig. S1 (ESI†). About 100 μL of 0.1 M AgNO3 is dispersed in 100 mL of deionized water taken in a round bottom flask under stirring conditions, followed by adding 1.5 mL of 0.3 M trisodium citrate. The slightly yellow-coloured solution of silver seeds thus formed is subsequently etched with 280 μL of 30% H2O2 solution. Vigorous stirring is ensured for about 10 minutes for homogeneous dispersion of the reaction mixture. After that, the stirring rate is reduced, and 1 mL of 0.1 M NaBH4 solution is quickly added. The stirring is continued until a stable turquoise blue-coloured silver nanoprism solution is formed (see Fig. S2, ESI†). The synthesized silver nanoprisms are then concentrated by centrifuging at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The concentrated prism is then stored at 4 °C for further use.
000 rpm. The concentrated prism is then stored at 4 °C for further use.
To calculate the production yield of silver nanoprisms, 10 sets of silver nanoprism solutions are prepared with the same procedure. Each set of solutions is concentrated to 20 mL by centrifuging at 15000 rpm in a HERMLE cooling centrifuge for about 1 hour. The concentrated solutions are then mixed and purified using milli Q water several times using centrifugation. The resulting solution is then concentrated by evaporating water at 80 °C for one day and finally freeze-dried in a pre-weighed PET bottle at −70 °C for three days. The final weight obtained is 0.004087 g. Thus, the production yield in one set of synthesis (using 100 μL of 0.1 M silver nitrate solution) is 37.9%.
2.3 Preparation of graphene oxide
Graphene oxide is prepared using Tour's modified method,13 as follows; about 1 g of graphite flakes were treated within a 9:1 H2SO4/H3PO4 acid mixture. Add 6 g of KMnO4 in small quantities to this reaction mixture with stirring. After the KMnO4 addition, the ice bath is removed, and the reaction vessel is kept at 50 °C for about 12 hours. The oxidation is terminated by adding 150 mL of ice containing 3 mL of 30% H2O2. This step must be performed with caution as it is exothermic. The formed product is then cooled to room temperature and filtered to remove unoxidized graphite. Finally, the graphite oxide formed is collected after washing successively with water, HCl, and ethanol to ensure the pH is neutral. The solution is then sonicated in a bath sonicator for one hour and dried at 80 °C to get graphene oxide.
2.4 Preparation of graphene oxide-silver nanoprism hybrids
The graphene oxide–silver nanoprism hybrids are prepared by two methods: (i) by physical mixing of graphene oxide solution and concentrated silver nanoprisms (GO–Ag) and (ii) by covalently linking graphene oxide and silver nanoprisms (GO–X–Ag). This step is achieved via the functionalization of the graphene oxide with the amino group by the reaction with EDC and ethylene diamine. Initially, 50 mg of graphene oxide is dispersed in 100 mL water. To prepare GO–X–Ag, 50 mL of as-prepared graphene oxide dispersion is treated with 50 mg EDC and 100 μL ethylene diamine for about 3 hours. Finally, the pH of the dispersion is adjusted to 6 with the addition of HCl. The product formed is washed several times with water and redispersed in 50 mL water. The amine-functionalized graphene oxide thus obtained is then treated with 10 mL of concentrated silver nanoprism solution by mixing with the help of a magnetic stirrer. To prepare GO–Ag, 50 mL of as-prepared graphene oxide dispersion is mixed with 10 mL of concentrated silver nanoprisms by stirring for about 1 hour. Fig. 1 shows the anticipated mechanism of covalent linking.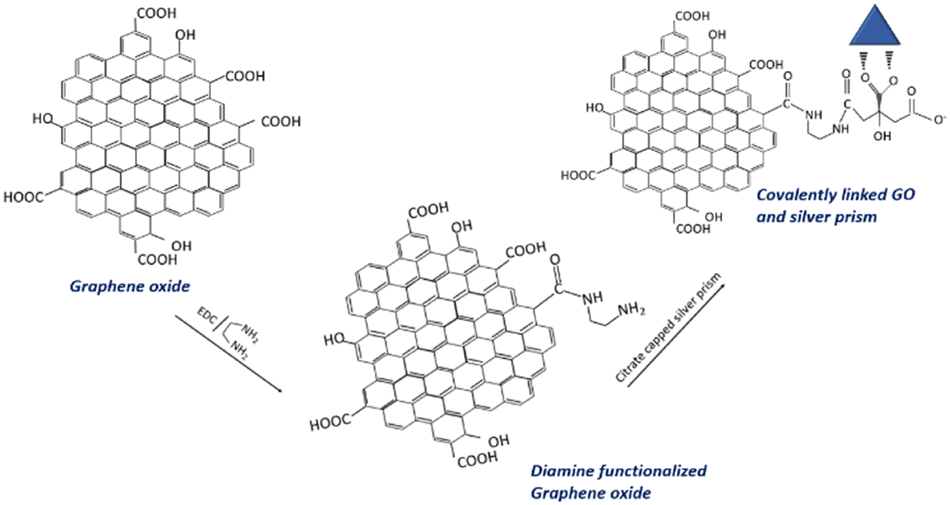 | ||
| Fig. 1 Schematic of an anticipated mechanism of covalent linking of graphene oxide and triangular silver prisms. | ||
2.5. Instrumentation
FTIR spectra are obtained using an FT-IR spectrometer ((PerkinElmer Spectrum Two) with an attenuated total reflection (ATR) contact sampling method. Raman measurements (Raman spectra and Raman images) are obtained using a Confocal Raman, WITec equipped with a x100 objective (NA = 0.5) with an excitation wavelength of 532 nm. The laser power at the samples is typically 10–20 mW, and the data acquisition time was 1 second. The Raman imaging is carried out by selecting an area of 100 μm × 100 μm and data acquisition for 30 minutes. XRD measurements are done using the Multipurpose X-ray diffractometer equipped with a D/teX Ultra 250 1D detector and integrated with the Smart Lab SE software, an Intelligence guidance system powered by Rigaku. XPS analysis is carried out using XPS, PHI 5000 Versa Probe II, ULVAC-PHI Inc., equipped with a micro-focused (100 μm, 15 kV) monochromatic Al-Kα X-Ray source (hν = 1486.6 eV). Both wide scan spectra (survey scan) and narrow scans (high-resolution spectra) were recorded. Wide scan spectra were recorded with an X-ray source power of 50 W and pass energy of 187.85 eV. Narrow scan spectra of the major elements were recorded at 46.95 eV pass energy. XPS spectra were processed using PHI's Multipak software. The binding energy was referenced to the C1s peak at 284.8 eV. The FTIR, Raman, XRD, and XPS analysis was performed using thin films of the samples by drop casting the sample dispersions on a glass substrate. TEM images were taken with a JEOL JEM 2100 transmission electron microscope (TEM) with an accelerating voltage of 200 kV.UV-vis absorption spectra were recorded on an Avantes AvaSpec-2048 spectrophotometer with an AvaLight DH-S deuterium lamp. Photoluminescence fluorescence emission spectra were recorded with a Horiba FluoroMax-4 spectrophotometer. For absorption and one-photon excited photoluminescence spectra, the silver nanoprism solution was diluted 60 times, while GO–Ag and GO–X–Ag were diluted 10 times, with the same nanoprism concentration in silver nanoprism, GO–Ag and GO–X–Ag solutions. A 10 mm light path quartz cuvette is used.
2.6. Nonlinear optics characterization
The setup for hyper-Rayleigh scattering (HRS) and two-photon excited fluorescence (TPEF) or more generally multi-photon excited fluorescence emission (MPEF) has been described in detail in previous works.21,22 Briefly, the light source for the HRS and MPEF measurements was a mode-locked femtosecond Ti: sapphire laser delivering at the fundamental wavelengths at 800 nm pulses with a duration of about 140 fs at a repetition rate of 80 MHz.23 After passing through a low-pass filter to remove any unwanted harmonic light generated before the cell, a fundamental beam of about 400 mW was focused by a low numerical aperture microscope objective into a 0.5 cm spectrophotometric cell containing the aqueous solutions. We recorded the absorption spectra before and after laser irradiation for all these samples (AgNPs, GO–Ag, GO–X–Ag). Actually, after laser irradiation for more than 20 min, we didn’t observe any change in the feature absorption peak of AgNPs, demonstrating no laser-induced damage under our measurement conditions (800 nm, 400 mW). The HRS and MPEF light were collected at an angle of 90° from the incident direction by a 2.5 cm focal length lens. The HRS light was separated from the excitation light by a high-pass filter and a monochromator set at the second harmonic wavelength. The HRS light was then detected with a photomultiplier tube working in the single-photon counting regime. The first hyperpolarizability of the nanoparticles was obtained using the internal reference methods,16 where the output scattered harmonic intensity is recorded as a function of the nanocomposite concentration in water.22 Linear plots were thus obtained, and slopes were compared to a reference. Here, we used neat water as the reference in both methods, the first hyperpolarizability of which is taken as 0.087 × 10−30 esu.24 For the MPEF signal, the scan wavelength of the spectrometer (iHR320 spectrometer) was set between 350 and 750 nm, and the same detection unit was used. The MPEF cross sections are extracted and calculated by a reference method6 according to the following equation:where σMPEF is the two-photon excited fluorescence cross-section, c is the concentration, and I is the fluorescence intensity. Fluorescein (10 μM, in ethanol) was used as a reference solution. At 800 nm,

 of the fluorescein is 33.3 GM.25
of the fluorescein is 33.3 GM.25
2.7. Computational details
The GO models have been built from a graphene sheet made of 10 × 10 × 1 unit cells, on which have been randomly attached hydroxyl and peroxide groups according to the quantitative XPS results (see Computational details in ESI†). Six models have been built and only one has been retained for both the electrostatic and covalently bound calculations because of the lower energy of the system, or because of chemically acceptable hydroxyl/epoxide groups during geometry relaxation procedures. For the covalently bound model, a defect has been included by removing a portion of C atoms, making it possible to represent the edge of GO and covalently attach the molecule. This kind of structural motif has already been identified in previous work as a structural motif of GO.26 For these models both ions and cell parameters have been relaxed. Due to the limited resources necessary to perform the present calculations, we used a cluster with seven Ag atoms. The complexes made of the citrate molecule bound to two sodium cations and Ag7 have been relaxed in the gas phase before being added to the GO models. Then it was added to the GO models at a reasonable distance between the Na+ cation and GO oxygen atoms (around 2.5–3.0 Å allowing relatively soft Na+–O interactions) before relaxing the entire system. In the case of the covalently bound model, one oxygen atom of the carboxylate group has been replaced by the –[NH–CH2]2– chain covalently bound to the GO edge. For the model including GO and molecules Na+ salts and Ag clusters, the periodic box parameters are about a = b ≈ 25 Å, c ≈ 30 Å, α = β ≈ 90°, γ ≈ 60°. A Bader charge analysis27 has been performed using a grid-based Bader analysis algorithm28–30 in order to get atomic charges. Interaction energies were computed for the electrostatic models as the difference in energy between the whole GO-Mol-Na+2-Ag7 complex minus the GO and Mol-Na+2-Ag7 isolated moieties optimized as described above. All calculations have been performed using the VASP package31,32 using the projector augmented wave (PAW) method to describe ionic cores and valence electrons through a plane wave basis.33,34 The PBE functional35 has been employed by including dispersion contributions through the Grimme D3 scheme.36 Structural relaxations were performed with a Brillouin zone sampling restricted to the Γ-point and a cut-off energy of 400 eV. For the charge, band structure and density of state analyses, K-points along the Γ–M–K–Γ sampling have been applied including 30 points between each vertex. For the energy calculation, a 6 × 6 × 6 Brillouin zone sampling has been applied for all, complex and isolated, systems. For the analyses and energy calculations, the precision has been increased to the accurate mode. All detailed information can be found in the ESI.†3 Results and discussion
3.1 Characterization of the GO and GO–Ag nanocomposites
 | ||
| Fig. 2 UV-vis extinction spectra of (a) GO, (b) silver nanoprisms and (c) GO–X–Ag and GO–Ag dispersed in water. | ||
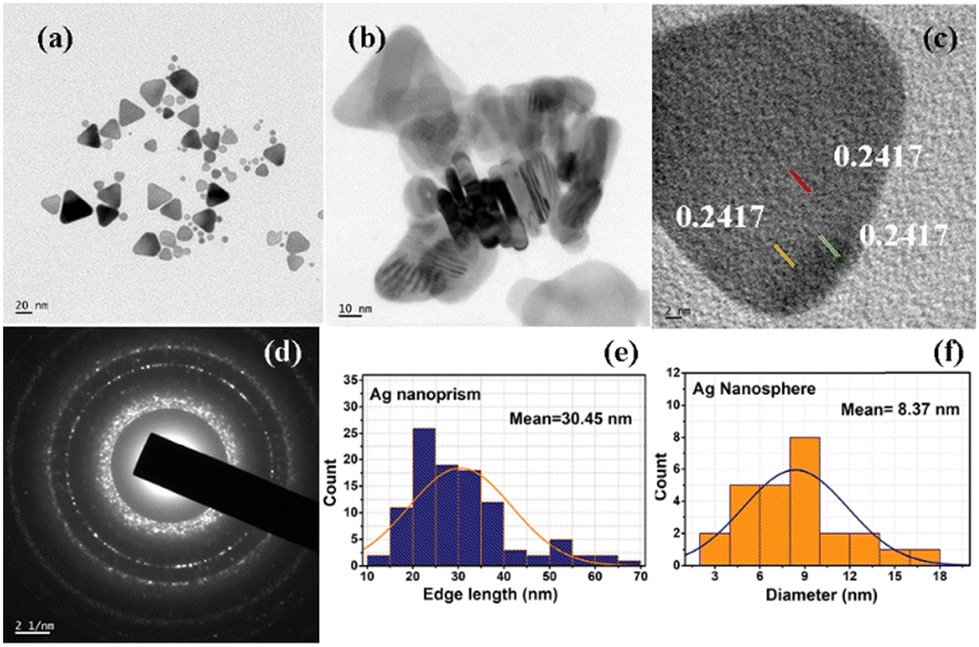 | ||
| Fig. 3 (a) TEM images of the silver nanoprisms, (b) TEM image of the stacked silver nanoprisms showing thickness of the prism, (c) high resolution TEM image of a prism showing d-spacing corresponding to the (111) plane, (d) SAED pattern of a silver nanoprism and (e) and (f) histograms of nanoprism edge (e) and nanosphere diameter (f) formed during nanoparticle synthesis. | ||
Fig. 4(A) shows the TEM images of electrostatically bound GO–Ag, and Fig. 4(B) shows the TEM image of covalently bound GO–X–Ag. As seen from the TEM images, the prisms decorated on the graphene oxide sheets in GO–Ag are intact and preserved their triangular plate-like morphology. However, in the case of GO–X–Ag, we can see that the edges of the prisms are rounding off significantly, which may be due to the complex chemistry between amine functional groups introduced on the graphene oxide and the citrate linkage around the silver nanoprisms. Briefly, according to various reports on silver nanoprisms, the carboxylic acid groups on the citrate ion play a crucial role in its shape.40 Covalent linking is achieved by making use of a reaction between the citrate ligands present on the silver prisms and the induced amino groups on the graphene oxide by EDC/diamine coupling. This will definitely make a change in ligand nature on the silver prism surface, which may cause the rounding-off of the corners. This effect is noted by researchers even in the physical linking (electrostatic) of graphene oxide and they reported that it is mainly because of the loss of the citrate ions from the surface of the silver prism during the washing stage.41,42 Since we are not adding more citrate species during the covalent linking, the change in ligand nature over the surface of the triangular silver prism will result in the rounding off of the edges, as the corners are energetically high and it will try to minimize the energy when conditions favour it.
 | ||
| Fig. 4 TEM images of (A) GO–Ag and (B) GO–X–Ag. | ||
 | ||
| Fig. 5 (a) Raman spectra of GO and GO–Ag, and (b) GO–X and GO–X–Ag. | ||
 | ||
| Fig. 6 Wide scan XPS spectra of GO–Ag and GO–X–Ag. | ||
Fig. S4(a) and (b) (ESI†) show the deconvoluted C 1s spectra of GO–Ag and GO–X–Ag, respectively. The C 1s spectra of GO–Ag (Fig. S4(b), ESI†) show characteristic peaks at 284.82 eV, 286.79 eV, and 288.31 eV, corresponding to the functional groups C–C, C–O, and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. The C 1s spectra of GO–X–Ag (Fig. S4(c), ESI†) show peaks at 284.74 eV, 286.29 eV, and 288.89 eV. The decrease in the intensities of the peaks corresponding to C–O and O–CO in the C 1s spectra of GO–X–Ag suggests the chemical conversion of the corresponding functional groups during covalent functionalization. This observation is consistent with the nature of the deconvoluted O 1s spectra of GO–Ag and GO–X–Ag. The considerable decrease in O 1s peak intensity of O–CO at 531.3 eV in GO–X–Ag compared to GO–Ag indicates the conversion of carboxylic acid groups present in GO into amide. The deconvoluted N 1s spectra of GO–X–Ag show two peaks at 397.6 eV and 399.9 eV, characteristic of the functional groups –CO–NH and –NH, respectively. The higher intensity of –CO–NH compared to the –NH indicates that the hybrid contains more amide groups than free amine groups. These observations further confirm the covalent integration of the individual constituents of the hybrid. The intense doublets of GO–Ag hybrid at 367.8 eV and 376.8 eV are owing to Ag 3d5/2 and Ag 3d3/2 with spin-orbit coupling energy of 6 eV, confirming the presence of silver (Ag0) in the hybrid. The presence of a Ag 4s peak in the GO–X–Ag peak indicates the presence of silver in the composite.
O, respectively. The C 1s spectra of GO–X–Ag (Fig. S4(c), ESI†) show peaks at 284.74 eV, 286.29 eV, and 288.89 eV. The decrease in the intensities of the peaks corresponding to C–O and O–CO in the C 1s spectra of GO–X–Ag suggests the chemical conversion of the corresponding functional groups during covalent functionalization. This observation is consistent with the nature of the deconvoluted O 1s spectra of GO–Ag and GO–X–Ag. The considerable decrease in O 1s peak intensity of O–CO at 531.3 eV in GO–X–Ag compared to GO–Ag indicates the conversion of carboxylic acid groups present in GO into amide. The deconvoluted N 1s spectra of GO–X–Ag show two peaks at 397.6 eV and 399.9 eV, characteristic of the functional groups –CO–NH and –NH, respectively. The higher intensity of –CO–NH compared to the –NH indicates that the hybrid contains more amide groups than free amine groups. These observations further confirm the covalent integration of the individual constituents of the hybrid. The intense doublets of GO–Ag hybrid at 367.8 eV and 376.8 eV are owing to Ag 3d5/2 and Ag 3d3/2 with spin-orbit coupling energy of 6 eV, confirming the presence of silver (Ag0) in the hybrid. The presence of a Ag 4s peak in the GO–X–Ag peak indicates the presence of silver in the composite.
Additional characterization of GO and the GO–Ag nanocomposites (FT-IR, XRD, confocal Raman mapping) is given in the ESI,† see Fig. S5–S8.
3.2 Nonlinear optical properties
Two-photon or more generally multiphoton excitation photoluminescence or fluorescence upon excitation at ∼800 nm has been scarcely reported. MPEF from metal NPs is supposed to be initiated by the excitation of electrons from the d-valence band to the sp-conduction band via two-photon absorption, followed by intraband scattering relaxation of the excited electron to the states near the Fermi surface before electrons and holes recombine radiatively.47,48 Au and Ag NPs are poor light emitters under one- or two-photon excitation; however, anisotropic Au and Ag NPs (nanorods,…)49 and coupled Au or Ag nanostructures were found to exhibit strong emission under two-photon excitation.50,51 In addition, enhancement of MPEF may be possible by the “lightning rod effect” near the sharp edge of the nanoparticles (in particular with nanoprisms).52Fig. 7 shows typical MPEF spectra measured for silver nanoprisms and silver nanocomposites excited at a wavelength of 800 nm. MPEF is suggested (both 2- (2PEF) and 3- (3PEF) photon excited fluorescence) since emission is observed at wavelengths below 400 nm, i.e. shorter than half the fundamental exciting wavelength. A broadband centered at 500 nm with an additional shoulder at 600 nm is observed for both silver nanoprisms and GO–silver nanocomposites. In addition to the broad MPEF band, a strong sharp peak was also observed at 400 nm corresponding to enhanced second harmonic generation (SHG) of the silver nanostructures from the 800 nm fundamental wavelength. The multi-photon excited photoluminescence cross sections were measured using the reference method (as described in the experimental section). The SHG response is evaluated using the hyper-Rayleigh scattering technique used to generate incoherent second harmonic scattered light from the sample cell. The HRS light response as a function of sample concentration is used to estimate the first hyperpolarizability of the different nanomaterials using the internal reference methods (with water as the reference) (see Fig. 8). NLO data are given in Table 1.
 | ||
| Fig. 7 MPEF spectra of silver nanoprisms, GO–Ag and GO–X–Ag diluted in water (same concentration of silver nanoprisms as 1.905 pM). Details on concentration calculations are given in the ESI.† | ||
 | ||
| Fig. 8 HRS response as a function of sample concentration for (A) Ag, (B) GO–Ag and (C) GO–X–Ag. Details on concentration calculations are given in the ESI.† | ||
| Samples | σ MPEF (GM unit) | 1027β (esu) |
|---|---|---|
| Silver nanoprisms | (1.5 ± 1) × 106 | 867 ± 30 |
| GO–Ag nanocomposites | (6.5 ± 1) × 106 | 1200 ± 30 |
| GO–X–Ag nanocomposites | (5 ± 1) × 106 | 890 ± 50 |
Regarding the physical origin of the observed NLO responses, with MPEF, we must consider separately odd and even phenomena, i.e. quadratic (2PEF) or cubic (3PEF) MPEF since the two are probably operating in the present case. The fact that cubic MPEF occurs is indicated by the existence of fluorescence emitted at wavelengths shorter than 400 nm, i.e. shorter than half the exciting fundamental wavelength. In MPEF, one must consider the selection rules during the exciting step. If for cubic MPEF, centrosymmetry is not an issue, this is the case in quadratic MPEF as this initial excitation step requires centrosymmetry of the molecule due to the selection rules. On the opposite side, hyperpolarizability requires the breaking of centrosymmetry, for the same reason as the selection rules as it is a three-photon process as a whole and not a two-photon step followed by a one-photon step. Hence, at the 800 nm excitation wavelength, the centrosymmetry role points to the conclusion that the GO–Ag system is less centrosymmetric than the GO–X–Ag system whereas the two systems present a similar symmetry from the point of view of their states involved in the excitation step of the MPEF process. The key feature here in this comparative study of MEPF and SHG is therefore centrosymmetry.
The MPEF cross sections of silver nanostructures are very large (about 106–107 GM) and are about 1 to 2 orders of magnitude larger than those reported for gold nanorods (average aspect ratio 3, average size 90 × 30 nm).53 Also, the influence of graphene oxide on the MPEF cross sections of silver nanoprisms is noticeable and leads to an enhancement. However, the enhancement is larger when graphene oxide interacts electrostatically with silver nanoprisms rather than covalently. Hyperpolarizabilities follow the same trend (see Table 1). Also, the hyperpolarizability value for silver nanoprisms is about 4 orders of magnitude higher than the one reported for gold nanoprisms with similar dimensions.19 This difference in hyperpolarizability between gold and silver was already evidenced with nanospheres.54
To go further in determining the origin of this nonlinearity, the SHG response of the samples was recorded as a function of the fundamental input beam polarization angle. The polar plots (displayed in Fig. S9, ESI†) exhibit a dipolar contribution as evidenced by a two-lobe pattern for the silver nanoprisms. Due to the small size of those nanoprisms, it is expected that retardation is negligible leading to simple dipolar excitations. This is further confirmed by the retardation parameter indicating the deviation from purely dipolar polar plots that never exceed 0.04. Surprisingly, this parameter reaches 0.19 for the GO–Ag system whereas it is again 0.04 for the covalently bound GO–X–Ag system. This may be attributed to some agglomeration of the silver nanoprisms once electrostatically attached to the GO flakes. This may be seen in Fig. 4(A) where silver nanoprisms appear closer to each other whereas they appear well separated on the covalently bound system. It is worth noting further that because several silver nanoprisms are present on a single GO flake, they do not behave as independent nanoprisms in solution anymore. This phenomenon is strikingly much more present for the electrostatically bound ones as they appear to aggregate more. The measured depolarization ratios for the silver nanoprisms deviate also from the pure three-fold symmetry with a value of only 0.45 instead of the expected 0.67 value. This feature underlines some deviations to the perfect shape and potentially the role of a nonperfect citrate layer in affecting the nanoprism symmetry, see Table S2 (ESI†). A striking result is also the strong influence of the way GO interacts with nanoprisms on the hyperpolarizability values. A factor of about 1.5 in enhancement is observed with nanoprisms electrostatically interacting with GO, while no significant enhancement is observed when silver nanoprisms are covalently bound to the GO surface. This result is quite unexpected and puzzling. But looking more in detail into the TEM images, it appears that the process of covalent linking leads to more rounded corners for the nanoprisms. It has been evidenced by Ledoux-Rak and co-workers that the corner sharpness of nanoprisms strongly influences the nonlinearity.18 One may therefore suggest that a similar effect is responsible for the decrease in hyperpolarizability value for GO–X–Ag as compared to the GO–Ag nanocomposites. Nevertheless, if the silver nanoprisms do not appear to be much affected in their symmetry when electrostatically interacting with the GO flakes, their symmetry parameter is unchanged, they present on the opposite a much better-defined three-fold symmetry once covalently bound to the GO flakes as seen from the symmetry parameter reaching a value of 0.65, close to the 0.67 perfect value of the three-fold symmetry.
Another striking result that could be of interest for developing multi-photon imaging contrast agents is the strong enhancement by a factor of 3 to 4 of the multi-photon brightness of silver nanoprisms when combined with graphene oxide. This enhancement may be explained by the possible efficient charge transfer of a “donor–acceptor like character” between silver nanoprisms and GO surface in the hybrid materials as evidenced by the pioneering works by Yadav et al. on hybrids of graphene oxide and gold nanoparticles.55 Interestingly, one can notice an effect of the way the GO surface interacts with silver nanoprisms. Covalent bonding leads to a smaller MPEF brightness as compared to electrostatic bonding. To understand the correlation of interfacial charge transfer with the concentration and type of the functional linkers attached to the graphene oxide sheet, we have investigated the silver—graphene oxide interface with covalent versus electrostatic configurations from first-principles calculations.
3.3 DFT calculations
Periodic DFT calculations were performed on two models of the electrostatic configuration (GO–Ag1 & GO–Ag2) and one model representing the covalently bound system (GO–X–Ag). For the two electrostatic configurations, the computed binding energies indicate an important stabilization of about 3.6 and 3.3 eV (see Table 2). In these configurations, the Na+–citrate molecule-Ag cluster complex is stabilized over the GO surface through bridging electrostatic interactions between the carboxylate citrate groups and the Na+ cation and the GO hydroxyl or epoxide O atoms. Indeed, the strong binding energies are explained by rather short Na+–O distances with the GO surface ranging from 2.3–2.5 Å with hydroxyl O atoms, 2.5 Å with the epoxide for GO–Ag1, and ∼2.3 Å with a dangling O atom belonging to an epoxy atom of the initial GO (see Table 2). The larger binding energy for GO–Ag1 may be explained by the rather short Na+–Oepoxy interaction although the GO–Ag2 complex is stabilized by 3 Na+–OH interactions instead of 2 for GO–Ag1. For the covalently bound system, the distances are similar, the only difference lies in the fact that citrate is somehow farther from the surface because of the NH–[CH2]2–NH chain and because of the hole leading to less available GO's O atoms.| GO–X–Ag | GO–Ag1 | GO–Ag2 | |
|---|---|---|---|
| Binding energies | |||
| ΔE (eV) | −3.602 | −3.258 | |
| Charges | |||
| qCit | −1.58 | −1.92 | −2.01 |
| qAg | 0.82 | 0.89 | 0.88 |
| qNa+ | 0.84 | 1.68 | 1.69 |
| qGO | −0.08 | −0.65 | −0.57 |
| qCit-Ag | −0.76 | −1.04 | −1.13 |
| Distances | |||
| Na+–citrate carboxylates O | |||
| dNa+–CO | 2.272 | 2.492 | 2.724 |
| dNa+–CO | 2.253 | 2.519 | 2.267 |
| dNa+–CO | 2.336 | 2.221 | |
| dNa+–CO | 2.245 | 2.578 | |
| 2.236 | |||
| Ag–citrate carboxylate O | |||
| dCO–Ag | 2.189 | 2.189 | 2.190 |
| dCO–Ag | 2.199 | 2.197 | 2.196 |
| Na+–GO epoxide O | |||
| d Na+–Oepox | 2.513 | ||
| Na+–GO hydroxyl O | |||
| dNa+–OH | 2.317 | 2.327 | 2.247 |
| 2.420 | 2.466 | ||
| 2.438 | |||
| Na+–GO dangling O | |||
| dNa+–O | 2.296 | 2.271 | |
| H bond CO carboxylate–hydroxyl GO | |||
| H-bond CO–HO | 1.551 | ||
The computed atomic charges on the citrate moiety, the Ag cluster and the GO sheet are shown in Table 2. One can see that the charge on the Ag cluster does not show any difference between the covalently bound system and the electrostatic configurations. This may be due to the rather small size of the Ag cluster which is not sufficient to drive the charge transfer. Instead, the charge transfer seems to be controlled by the presence of the carboxylate groups on the citrate molecule. Indeed, for the covalently bound system, there is only one COO− while there are two for the electrostatic complex. The presence of the COO− groups and their associated electron withdrawing character thus led to a larger negative charge on the citrate moiety for the electrostatic complex as compared to the covalently bound system. This remarkable aspect also suggests that a larger amount of negative charge on the citrate moiety will also allow a larger transfer to the Ag-nanoparticle. This cannot be observed from the present calculations because of the small size of the Ag cluster considered (around 4.7 Å large) here limited by the resources necessary to perform the present calculations. However, one can reasonably expect a charge transfer to the Ag-nanoparticle of the size of several nm. As such, the observed difference between the MPEF cross-section between the electrostatic and covalently bound systems can be explained by the difference in charge transfer driven by the presence of the carboxylate groups on the citrate moieties (Fig. 9).
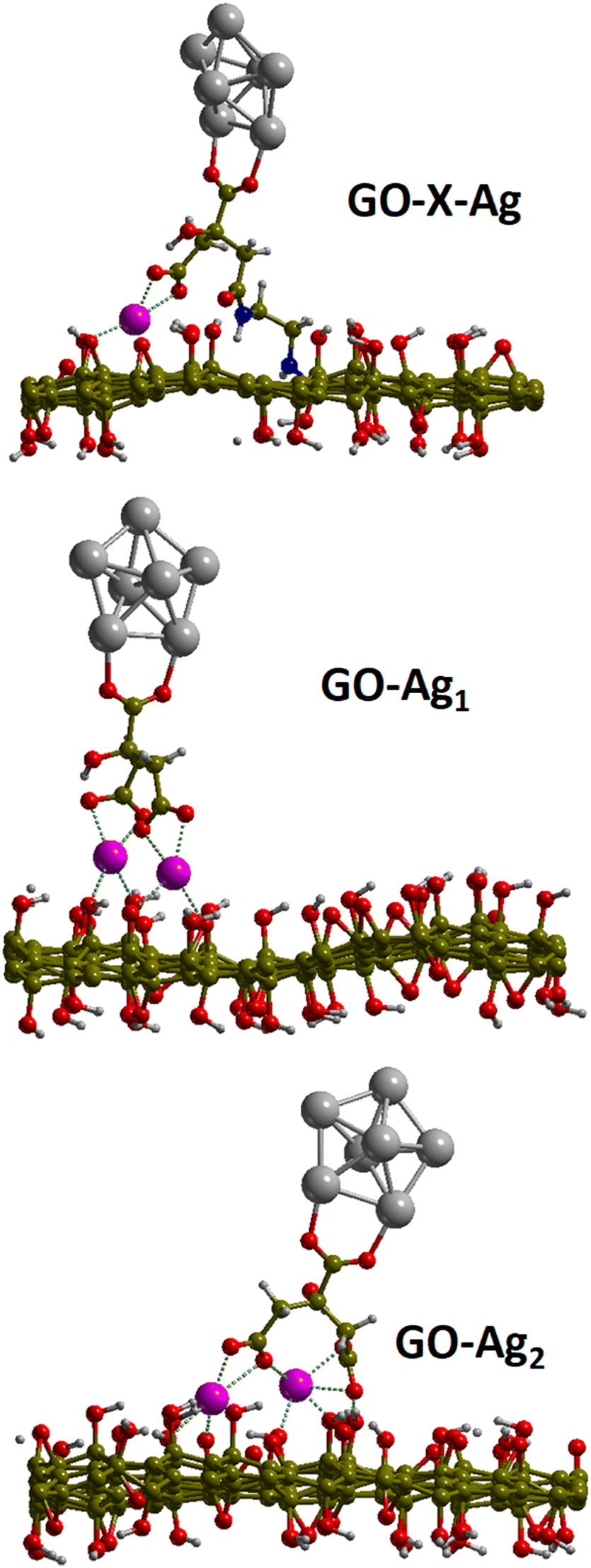 | ||
| Fig. 9 Geometries of the relaxed structures of the covalent and electrostatic configurations. | ||
Conclusions
In conclusion, we have synthesized nanocomposites consisting of citrate-passivated silver nanoprisms anchored both electrostatically and covalently on graphene oxide nanosheets in a cost-effective and reproducible manner. We report the covalent functionalization of silver nanoprisms onto graphene oxide (GO) nanosheets according to the 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride crosslinking method, using the existing carboxylic groups present both at the surfaces of the nanoclusters and the GO nanosheets. The formed hybrid nanocomposites exhibit remarkable nonlinear optical (NLO) properties upon femtosecond laser excitation at 800 nm, in particular a strong second harmonic scattering response as well as a multiphoton excited fluorescence spectrum characterized by a broad band in the visible range between 350 and 700 nm. In addition, the NLO response is sensitive to the nature of the interaction (electrostatic or covalent) and might be attributed to different charge transfer capabilities between covalently or electrostatically bound silver particles onto graphene oxide. Such nanocomposites are promising for new applications in the areas of optoelectronics since for the same excitation wavelength, a differentiated response in terms of wavelength for MPEF (broad band) and SHG (narrow band) is observed. Also, they can serve as new molecular probes for imaging due to high cross sections for MPEF and SHG.Author contributions
FCU, synthesis and characterization, writing, HY, NLO investigations, characterization, writing, IR-A, FR, P-FB, NLO investigations and analysis, PM, computations, RA and NK, fundings, RA conceived the initial idea and coordinated the work, writing. All authors provided critical feedback and helped to shape the final manuscript.Data availability
The data supporting the findings of this study are available within the article and its ESI files.† Additional data that support the findings of this study are available from the corresponding author upon reasonable request. This includes computational codes and raw data files generated during the study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge CNRS for funding through International Emerging Actions between Institut Lumière Matière, CNRS, France, and Mahatma Gandhi University, India. HY is grateful for PhD fellowships donated by the China Scholarship Council (CSC, No. 202206140023). The author FC is grateful for JRF provided by Mahatma Gandhi University, Kottayam. The author NK would like to acknowledge the financial support from the CRS program of UGC-DAE CSR, Kolkata Centre (UGC-DAE-CSR-KC/CRS/19/RC08/0421), India, SERB: CRG (Grant No. CRG/2021/001506), UGC-Govt. of India for funding through the Innovative Program and Special Assistance Program (SAP Grant No. F.530/12/DRS/2009; F.530/13/DRS II/2016), RUSA 2.0 scheme supported by Department of Higher Education, Govt. of India, UGC: Scheme for Promotion of Academic and Research Collaboration (SPARC Grant No. P930, P1400, P1429, P1460) supported by MHRD, Govt. of India, DST: Nano Mission (Grant No. SR/NM/NS-1420-2014(C)), DST: Fund for Improvement of S&T Infrastructure (FIST Grant No. SR/FST/P SI-143/2009), DAE-Board of Research in Nuclear Sciences (BRNS Grant No. 39/29/2015-BRNS/39009), and DST Promotion of University Research and Scientific Excellence (PURSE Grant No. SR/S9/Z-23/2010/22(C, G)), Government of India programs for providing the facilities for research and development in IIUCNN, MGU. The authors FC and NK express gratitude to Mr Anu A.S and Mr Jeffy Manuel, Analytical Engineers, IIUCNN, MGU, for the HRTEM imaging and Ms Anu Mathew, SAIF, MGU and NIIST, Thiruvananthapuram for different experimental support.Notes and references
- W. Zhou, J. Wu, W. Liu and S.-P. Guo, Coord. Chem. Rev., 2023, 477, 214950 CrossRef.
- B. Sahraoui, R. Czaplicki, A. Klöpperpieper, A. S. Andrushchak and A. V. Kityk, J. Appl. Phys., 2010, 107, 113526 CrossRef.
- I. Russier-Antoine, F. Bertorelle, N. Calin, Z. Sanader, M. Krstic, C. Comby-Zerbino, P. Dugourd, P.-F. Brevet, V. Bonacic-Koutecky and R. Antoine, Nanoscale, 2017, 9, 1221–1228 RSC.
- R. Allu, D. Banerjee, R. Avasarala, S. Hamad, S. V. Rao and G. K. Podagatlapalli, Opt. Mater., 2019, 96, 109305 CrossRef CAS.
- B. Binish, B. Lokesh, Y. Veer, S. Peters, M. Abith, T. C. S. Girisun and K. M. Rahulan, Sci. Rep., 2024, 14, 5642 CrossRef CAS PubMed.
- H. Fakhouri, M. Perić, F. Bertorelle, P. Dugourd, X. Dagany, I. Russier-Antoine, P.-F. Brevet, V. Bonačić-Koutecký and R. Antoine, Phys. Chem. Chem. Phys., 2019, 21, 12091–12099 RSC.
- Ž. Sanader, M. Krstić, I. Russier-Antoine, F. Bertorelle, P. Dugourd, P.-F. Brevet, R. Antoine and V. Bonačić-Koutecký, Phys. Chem. Chem. Phys., 2016, 18, 12404–12408 RSC.
- X. F. Zhang, Z. G. Liu, W. Shen and S. Gurunathan, Int. J. Mol. Sci., 2016, 17, 1534 CrossRef PubMed.
- H. Ahmad, M. Fan and D. Hui, Composites, Part B, 2018, 145, 270–280 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560–10564 CrossRef CAS.
- C. Zheng, W. Chen, Y. Huang, X. Xiao and X. Ye, RSC Adv., 2014, 4, 39697–39703 RSC.
- P. Nancy, A. K. Nair, R. Antoine, S. Thomas and N. Kalarikkal, Nanomaterials, 2019, 9, 1201 CrossRef CAS.
- Y. H. Lee, L. Polavarapu, N. Gao, P. Yuan and Q.-H. Xu, Langmuir, 2012, 28, 321–326 CrossRef CAS PubMed.
- A. Salah, S. Hassab-Elnaby and M. A. Ramadan, SN Appl. Sci., 2023, 5, 288 CrossRef CAS.
- F. Bertorelle, S. Basu, H. Fakhouri, M. Perić Bakulić, P. Mignon, I. Russier-Antoine, P.-F. Brevet, S. Thomas, N. Kalarikkal and R. Antoine, Nano Express, 2020, 1, 030005 CrossRef.
- I. Russier-Antoine, H. Fakhouri, S. Basu, F. Bertorelle, P. Dugourd, P.-F. Brevet, P. Velayudhan, S. Thomas, N. Kalarikkal and R. Antoine, Chem. Commun., 2020, 56, 3859–3862 RSC.
- J. Butet, P.-F. Brevet and O. J. F. Martin, ACS Nano, 2015, 9, 10545–10562 CrossRef CAS PubMed.
- H. Minh Ngo, E. Drobnyh, M. Sukharev, Q. Khuong Vo, J. Zyss and I. Ledoux-Rak, Isr. J. Chem., 2023, 63, e202200009 CrossRef CAS.
- K. Nadolski, E. Benichou, N. Tarnowicz-Staniak, A. Żak, C. Jonin, K. Matczyszyn and P.-F. Brevet, J. Phys. Chem. C, 2020, 124, 14797–14803 CrossRef CAS.
- G. Panzarasa, J. Chem. Educ., 2015, 92, 1918–1923 CrossRef CAS.
- F. Bertorelle, I. Russier-Antoine, N. Calin, C. Comby-Zerbino, A. Bensalah-Ledoux, S. Guy, P. Dugourd, P.-F. Brevet, Ž. Sanader, M. Krstić, V. Bonačić-Koutecký and R. Antoine, J. Phys. Chem. Lett., 2017, 8, 1979–1985 CrossRef CAS PubMed.
- I. Russier-Antoine, F. Bertorelle, M. Vojkovic, D. Rayane, E. Salmon, C. Jonin, P. Dugourd, R. Antoine and P.-F. Brevet, Nanoscale, 2014, 6, 13572–13578 RSC.
- H. Yuan, I. Russier-Antoine, C. Moulin, P.-F. Brevet, Ž. Sanader Maršić, M. Perić Bakulić, X. Kang, R. Antoine and M. Zhu, Nanoscale Horiz., 2025 10.1039/D4NH00454J.
- J. Duboisset, G. Matar, I. Russier-Antoine, E. Benichou, G. Bachelier, C. Jonin, D. Ficheux, F. Besson and P. F. Brevet, J. Phys. Chem. B, 2010, 114, 13861–13865 CrossRef CAS.
- M. A. Albota, C. Xu and W. W. Webb, Appl. Opt., 1998, 37, 7352–7356 CrossRef CAS.
- P. Feicht and S. Eigler, ChemNanoMat, 2018, 4, 244–252 CrossRef CAS.
- R. F. W. Bader, P. L. A. Popelier and T. A. Keith, Angew. Chem., Int. Ed. Engl., 1994, 33, 620–631 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- M. Yu and D. R. Trinkle, J. Chem. Phys., 2011, 134, 064111 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- D. Zhang, X. Liu and X. Wang, J. Inorg. Biochem., 2011, 105, 1181–1186 CrossRef CAS.
- J. Benalcázar, E. D. Lasso, C. M. Ibarra-Barreno, J. A. Arcos Pareja, N. S. Vispo, J. C. Chacón-Torres and S. Briceño, ACS Omega, 2022, 7, 46745–46755 CrossRef PubMed.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- N. Li, Q. Zhang, S. Quinlivan, J. Goebl, Y. Gan and Y. Yin, ChemPhysChem, 2012, 13, 2526–2530 CrossRef CAS PubMed.
- B. Tang, S. Xu, J. An, B. Zhao and W. Xu, J. Phys. Chem. C, 2009, 113, 7025–7030 CrossRef CAS.
- P. Wang, H. He and Y. Jin, Small, 2012, 8, 3438–3442 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- D. López-Díaz, J. A. Delgado-Notario, V. Clericò, E. Diez, M. D. Merchán and M. M. Velázquez, Coatings, 2020, 10, 524 CrossRef.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- C. Dai, X. Yang and H. Xie, Mater. Res. Bull., 2011, 46, 2004–2008 CrossRef CAS.
- M. R. Beversluis, A. Bouhelier and L. Novotny, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 115433 CrossRef.
- K. Imura, T. Nagahara and H. Okamoto, J. Phys. Chem. B, 2005, 109, 13214–13220 CrossRef CAS PubMed.
- H. Wang, T. B. Huff, D. A. Zweifel, W. He, P. S. Low, A. Wei and J. X. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15752–15756 CrossRef CAS.
- Z. Guan, L. Polavarapu and Q.-H. Xu, Langmuir, 2010, 26, 18020–18023 CrossRef CAS PubMed.
- F. Han, Z. Guan, T. S. Tan and Q.-H. Xu, ACS Appl. Mater. Interfaces, 2012, 4, 4746–4751 CrossRef CAS.
- K. Imura, T. Nagahara and H. Okamoto, Appl. Phys. Lett., 2006, 88, 023104 CrossRef.
- P. Zijlstra, J. W. M. Chon and M. Gu, Nature, 2009, 459, 410–413 CrossRef CAS PubMed.
- I. Russier-Antoine, E. Benichou, G. Bachelier, C. Jonin and P. F. Brevet, J. Phys. Chem. C, 2007, 111, 9044–9048 CrossRef CAS.
- R. K. Yadav, J. Aneesh, R. Sharma, P. Abhiramnath, T. K. Maji, G. J. Omar, A. K. Mishra, D. Karmakar and K. V. Adarsh, Phys. Rev. Appl., 2018, 9, 044043 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc03863k |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |