
Structural engineering of glass for regulating chemical surroundings of dopants
Yuan
Gao
a,
Yikang
Jiang
a,
Xiaosong
Lu
 *a and
Zhiyong
Yang
*ab
*a and
Zhiyong
Yang
*ab
aJiangsu Key Laboratory of Advanced Laser Materials and Devices, School of Physics and Electronic Engineering, Jiangsu Normal University, Xuzhou, Jiangsu, China. E-mail: xingwadyx@jsnu.edu.cn; yangzhiyong@jsnu.edu.cn
bHangzhou Institute of Optics and Fine Mechanics, Hangzhou, Zhejiang 311421, China
First published on 2nd December 2024
Abstract
Optical gain materials activated by dopants are playing pivotal roles in fiber amplifiers, solid-state lasers and biological imaging. Achieving high photoluminescence (PL) efficiency in a stable matrix by regulating the chemical surroundings of dopants in an inert glass matrix remains a challenge. Here, we report a novel method for regulating the chemical surroundings of dopants by modulating the network structure of the glass matrix, achieving regulation of dopant PL efficiency. The luminescent properties of glass were adjusted not only by altering its composition, but also more importantly by controlled nanocrystallization. Furthermore, by utilizing lattice-site substitution engineering, the spectral shape and PL efficiency of active centers can be regulated by controlling the method of cation substitution at sub-nanometer scale.
Introduction
The development of optical gain materials activated by dopants is significant for many scientific technologies, such as fiber optic communication, lasers, display, lighting, and biological imaging.1–3 The luminescent properties of these gain materials are usually determined by the suitable incorporation of certain active impurities (dopants) in an inert matrix.4–6 Furthermore, the spectral characteristics of the gain materials are determined by the energy level structure of the dopant, which is closely related to the chemical surroundings in which the dopants are located. In the past few decades, significant progress has been made in doping methodology, and the dopant–host library has rapidly expanded, providing methodologies for achieving the desired optical-performance regulation in the host.7,8 However, the regulation of the chemical surroundings of dopants in an inert glass matrix, especially in chalcogenide glass, remains challenging.Gain materials activated by rare earth ions are of great importance because of their high emission efficiency and multiple photoluminescence (PL) wavebands. In recent decades, extensive research has been conducted on dopant methodologies and network structure design in order to obtain more efficient gain materials.9,10 Nevertheless, high PL efficiency and superior thermodynamic performance seem to be two contradictory aspects of gain materials, greatly limiting their practical application. It is widely acknowledged that the PL efficiency of an optical material is determined largely by the non-radiation multi-phonon relaxation (MPR) rate,11 suggesting that the phonon energy of the glass host plays a pivotal role in augmenting PL efficiency. In general, soft glass materials with extremely low phonon energy, like chalcogenide glass, have been widely used as effective gain media.12–14 However, it is widely believed that pursuing low phonon energy usually comes at the cost of sacrificing the thermodynamic property of the material.15 In comparison, a hard matrix, like silicate glass, has high resistance to damage and superior thermal stability, but its PL efficiency is relatively low due to its high phonon energy. Therefore, overcoming this traditional contradiction to obtain materials with high PL efficiency and outstanding thermodynamic property remains a challenge.
Here, we report a novel method for regulating the chemical surroundings of dopants by modulating the network structure in a glass matrix. This method regulates the distribution of doping centers in the glass matrix by adjusting the glass network structure, thereby achieving regulation of the PL intensity of doping centers. Moreover, by utilizing regulation of lattice sites, the spectral shape and PL efficiency of active centers can be regulated by controlling the method of cation substitution at the sub-nanometer scale.
Experimental
Two typical glass matrices, one with ZnSe and the other with Ga2S3, were chosen according to their unique network motifs with remarkably distinct structures. Another glass matrix with both ZnSe and Ga2S3 network units was also designed as a hybrid system. ZnSe-containing glass matrices with compositions of 60GeS2–40As2S3–xZnSe (in mol%, x = 0, 5, 10, 15) were derived from the Ge–As–S system. A glass matrix with the composition 80GeS2–20Ga2S3 (in mol%) was obtained from the Ge–Ga–S system. Hybrid glass matrices with compositions of 60GeS2–30As2S3–(10 − y)ZnSe–yGa2S3 (in mol%, y = 0, 1, 3, 10) were modified from the Ge–As–S–ZnSe system and further optimized by trial and error. For rare earth ion and transition metal ion doping, Dy2S3 and CoCl2 were selected as dopant sources. The base glass (BG) was prepared by the vacuum melt–quenching method. High-purity Ge (5 N), As (6 N), Ga (6 N), S (6 N), ZnSe (5 N), Tm2S3 (5 N) and CoCl2 (5 N) were used as the raw materials. The mixture weighed according to the composition was loaded into a silica ampoule, which was then vacuum-pumped, sealed and put into a rocking furnace. At a temperature of 900–980 °C, the ampoule was rocked for 12 h to homogenize the melt, followed by the process of quenching (in cold water) and annealing (below the glass transition temperature Tg for 5 h). The glass ceramic (GC) was obtained by heat treating the as-prepared BG at Tg + 30 °C for 5 h. The refractive indices of the samples were measured with an infrared ellipsometer. The thermodynamic stability of the glasses was analyzed by differential scanning calorimetry (DSC). The structural properties of the glass samples were analyzed by Raman spectroscopy with an excitation wavelength of 785 nm. The morphology, distribution of nanocrystals and chemical surroundings of the dopants in the glass samples were studied using transmission electron microscopy (TEM) and energy dispersion spectroscopy (EDS). The optical properties were characterized by transmission spectroscopy and PL spectroscopy. The samples doped with Dy3+ were pumped by a 1320 nm laser diode, while the samples doped with Co2+ were pumped by an 808-nm laser diode. The characterization instruments and methods were the same as those reported in ref. 16.Results and discussion
The PL spectra of 60GeS2–40As2S3–xZnSe–0.1Dy2S3 glasses with x = 0, 5, 10, and 15 are shown in Fig. 1(a). The PL decay curves (at 2.9 μm) were also measured, and a representative curve is displayed in the inset to Fig. 1(a). The lifetimes derived from the decay curves were 3.88 ms, 4.73 ms, 5.01 ms and 5.60 ms, for the glasses with x = 0, 5, 10 and 15, respectively. The emission peaks located at 1.8, 2.9 and 4.4 μm originate from 6H11/2 → 6H15/2, 6H13/2 → 6H15/2 and 6H11/2 → 6H13/2 transitions, respectively. The samples with higher ZnSe content exhibit obviously stronger emission intensity and longer lifetime. The addition of low-phonon-energy ZnSe crystals (250 cm−1)17 caused the effective phonon energy of the glass to drop off, reflected by the redshift of the longwave cut-off edge in the enlarged transmission spectra of the glass samples (Fig. 1(b)). It has been widely accepted that the non-radiation MPR rate shows exponential dependence on the number of phonons related to the transitions between 4f energy levels of rare earth ions in glasses. Hence, the probability of non-radiation MPR decreases exponentially with a reduction in the effective phonon energy of glass, leading to higher emission efficiency reflected in a stronger PL intensity and longer lifetime. On the other hand, the probability of radiative transitions of rare earth ions tends to be greatly augmented by the enhanced refractive index of the glass host.18 As shown in Fig. 1(c), an enhancement in refractive index was evidently observed in the dispersion curves with the addition of ZnSe crystals into Ge–As–S glass. Therefore, the raised refractive index of the glass host, resulting from the addition of high-refractive-index ZnSe (2.4@3 μm),17 is another factor contributing to the enhanced emission efficiency. The Tgs of as-prepared 60GeS2–40As2S3–xZnSe glasses determined from the DSC curves (see Fig. 1(d)) measured at a 10 °C ramp rate, were 265 °C, 269 °C, 281 °C and 298 °C for glasses with x = 0, 5, 10 and 15, respectively. Such an increment results from the increase in mean coordination number (MCN, 〈r〉) according to the Gibbs and DiMarzio formula.19 Previous study has shown that Zn, like Ge, tends to be quadruple coordinated in chalcogenide glass,20 exceeding the coordination number of arsenic (triple coordinated). Thus, a glass with higher Tg has a higher MCN, which means a glass network structure with a higher degree of connectivity. Based on the above results, with the addition of a low-phonon-energy substance like ZnSe, the glasses show enhanced PL efficiency and their thermodynamic stabilities are anomalously improved. In the novel glasses developed here, lower phonon energy does not run counter to better thermodynamic stability. | ||
| Fig. 1 (a) PL spectra of 60GeS2–40As2S3–xZnSe–0.1Dy2S3 glasses. The inset shows the PL decay curve measured at 2.9 μm. (b) An enlarged region near the longwave cut-off edge in the transmission spectra of these glasses. (c) Refractive index dispersion curves of these glasses. The inset shows their photographs (from left to right, x = 0, 5, 10, 15). (d) DSC curves of these glasses. | ||
High PL efficiency and superior thermodynamic stability seem to be two contradictory aspects of gain materials. But in the above case, higher emission efficiency was achieved by the addition of ZnSe on the premise that glasses maintain better thermodynamic stability. However, we found that, with the increase in dopant concentration, the prepared glass contains uncontrollable and tightly bound clusters,21 leading to severe emission quenching (concentration quenching), which limits the doping level and is a drawback of chalcogenide glasses. The addition of a certain amount of Ga is a solution to this problem, because the dopant ions can be dissolved in the glass network as charge compensators for the non-bridging sulfurs formed by the dissociation of Ge–Ge bonds and edge-shared [GaS4] tetrahedra.22 Such a scheme was proved and applied to further enhance the emission efficiency, as discussed below.
The PL spectra of 60GeS2–30As2S3–(10 − y)ZnSe–yGa2S3–0.1Dy2S3 glasses are displayed in Fig. 2, and the corresponding PL lifetimes measured at 2.9 μm were 3.40 ms, 4.23 ms, 5.20 ms and 6.58 ms, respectively. The PL intensity of the glass with 1% Ga2S3 is significantly enhanced, and the higher the Ga2S3 content, the stronger the PL intensity. It should be noted that once 1% Ga2S3 is added, the intensity enhancement factor can be as high as 8, and it reaches ∼14 when ZnSe is completely replaced by Ga2S3, while the lifetimes are significantly prolonged from 3.40 ms to 6.58 ms. These experimental results demonstrate the important role of Ga in enhancing the PL efficiencies of the glasses.
 | ||
| Fig. 2 PL spectra of 60GeS2–30As2S3–(10 − y)ZnSe–yGa2S3–0.1Dy2S3 glasses. The inset shows their photographs (from left to right, y = 0, 1, 3, 10). | ||
Fig. 3 displays PL spectra of 60GeS2–40As2S3–10ZnSe–mDy2S3 and 60GeS2–30As2S3–7ZnSe–3Ga2S3–nDy2S3 glasses. Based on the PL results, it can be confirmed that the addition of a certain amount of Ga can avoid the formation of a dopant cluster. In glass without Ga (Fig. 3(a)), the concentration quenching effect occurred at m = 0.15, whereas in sharp contrast, the quenching effect did not occur even at n = 0.5 in the glass with added Ga. Because there is one negative charge in [GaS4] tetrahedra, Dy3+ ions, acting as charge compensators, are readily dissolved into the network by dividing the [S3Ga–GaS3] unit into two [GaS4] tetrahedra with non-bridging sulfurs.22 Such a process can avoid the formation of rare earth clusters21 and greatly augment the solubility of rare earth ions in glasses. Thus, the inter-ionic distance between Dy3+ ions becomes so long that their adverse energy migration can be mitigated, leading to an increase in the concentration quenching threshold, and thus a significant enhancement in emission efficiency.
 | ||
| Fig. 3 (a) PL spectra of 60GeS2–40As2S3–10ZnSe–mDy2S3 glasses. The inset shows their photographs (from left to right, m = 0.05, 0.1, 0.15); (b) PL spectra of 60GeS2–30As2S3–7ZnSe–3Ga2S3–nDy2S3 glasses. The inset shows their photographs (from left to right, n = 0.05, 0.25, 0.5). | ||
It can be inferred that the dopant tends to accumulate around [GaS4] tetrahedra; in other words, the dopant is located in a Ga-rich region. Is the dopant in the glasses without Ga preferentially or uniformly distributed and what if Zn and Ga precipitate from their respective glass matrices? Glasses with higher PL efficiency and better thermodynamic stability can be obtained by unveiling the underlying mechanism of the above conceptions. Therefore, the chemical surroundings of the dopant are further modulated upon thermal activation after the spatial distribution of the dopant is regulated in the glasses with a distinct network structure.
Conventional wisdom has it that covalent glasses can be divided into amorphous solids and polymeric glasses, according to the structural phase transition. The chemical threshold responsible for the classification is 〈r〉 = 2.4.23 The well-known second threshold is 〈r〉 = 2.67, which governs the topological change from two-dimensional (molecular) structures to three-dimensional networks.24 Therefore, glasses with different 〈r〉 values and a tendency towards amorphous phase separation were designed and optimized: 60GeS2–40As2S3–10ZnSe–0.1Dy2S3 (ZS, 〈r〉 = ∼2.55), 60GeS2–30As2S3–7ZnSe–3Ga2S3–0.1Dy2S3 (ZGS, 〈r〉 = ∼2.57), and 80GeS2–20Ga2S3–0.5Dy2S3 (GS, 〈r〉 = ∼2.69). Upon thermal activation, crystals tend to grow from the nucleation, initially dominated by the amorphous phase separation, and self-limited by the diffusion barrier of the high-viscous glass matrix. A ZS glass ceramic (ZS GC) was obtained by heat-treating the ZS base glass (ZS BG) at 315 °C for 5 h, and a GS glass ceramic (GS GC) was obtained by heat-treating the GS base glass (GS BG) at 450 °C for 5 h.
Fig. 4(a) shows the XRD patterns of ZS BG and ZS GC samples, suggesting that pure hexagonal β-ZnS crystals were precipitated from the ZS glass matrix, as proved by the crystallization peaks of the β-ZnS crystal (P63mc, PDF no. 36-1450). It is worth mentioning that ZnSe rather than ZnS was used as the source of zinc because of the much lower melting temperature of the former. However, only ZnS crystals but no ZnSe crystals precipitated in the glass matrix upon heat treatment, mainly due to the higher bond strength of Zn–S than Zn–Se.25 The XRD patterns of the GS BG and GS GC samples are shown in Fig. 4(b). Pure cubic γ-Ga2S3 crystals were precipitated from the GS glass matrix, according to the well-matched crystallization peaks of the GS GC sample with a cubic γ-Ga2S3 crystal (F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m, PDF no. 43-0916). Fig. 4(c) shows the TEM image of ZS GC. It is found that the ZnS crystals precipitating from the glass matrix were nanorods with a diameter of about 150 nm and a length of about 500 nm. The TEM image of GS GC sample is shown in Fig. 4(d). The Ga2S3 nanocrystals have sizes of 10–30 nm, showing a uniform distribution without clusters.
3m, PDF no. 43-0916). Fig. 4(c) shows the TEM image of ZS GC. It is found that the ZnS crystals precipitating from the glass matrix were nanorods with a diameter of about 150 nm and a length of about 500 nm. The TEM image of GS GC sample is shown in Fig. 4(d). The Ga2S3 nanocrystals have sizes of 10–30 nm, showing a uniform distribution without clusters.
 | ||
| Fig. 4 XRD patterns of (a) ZS BG, ZS GC and (b) GS BG, GS GC samples. Dark field TEM of ZS GC (c) and GS GC (d) samples. | ||
It has been shown that rare earth ions tend to accumulate preferentially in the six-coordinated site of the octahedral nanocrystal structure in glass, but this is not the case when the cation of the crystal is tetrahedrally coordinated.26,27 The available TEM evidence has by now convincingly demonstrated that Dy3+ ions tend to reside in the chalcogenide glassy phase, rather than entering into the nanocrystals28 because the coordination number of Dy3+ does not match that of the cations in ZnS or Ga2S3 crystals. Fig. 5(a)–(c) show the PL spectra of the samples before and after thermal activation above Tg. The PL spectra of all GC samples show neither narrowed nor split variations, indicating that Dy3+ ions were present in the residual glass phase and did not enter the nanocrystals. The measured 2.9 μm PL lifetimes were 5.01 ms, 3.13 ms, 5.20 ms, 5.09 ms, 5.87 ms and 8.36 ms for the ZS BG, ZS GC, ZGS BG, ZGS GC, GS BG and GS GC samples, respectively. Compared with ZS BG, ZS GC showed remarkably lower PL intensity and a shorter PL lifetime (from 5.01 ms to 3.13 ms). This phenomenon should be associated with the formation of ZnS crystals in the ZS GC sample, which augments the Dy3+ concentration in residual glass, thus leading to the formation of more Dy3+ clusters. In comparison, a slightly reduced emission intensity and shortened PL lifetime (from 5.20 ms to 5.09 ms) were observed after ZGS BG underwent heat treatment. There are merely pure ZnS crystals present in the ZGS GC sample according to its similar XRD pattern to that of the ZG GC sample (Fig. 4(a)), resulting in an augmented Dy3+ concentration in the residual glass. However, the ZGS samples contain 3 mol% Ga2S3, which mitigates the formation of Dy3+ clusters, preventing a significant decrease in PL intensity and lifetime. In stark contrast, the PL intensity of the GS sample was enhanced by 3 times and the PL lifetime was prolonged from 5.87 ms to 8.36 ms after heat treatment. Similar mid-infrared PL intensity enhancements were previously observed in rare-earth-doped chalcogenide glasses after controlled crystallization.29,30 Contributing factors, including (1) doping concentration of rare earth ions; (2) local asymmetry of the field environment of the rare earths; (3) multi-phonon relaxation; (4) the refractive index of the host material; and (5) multiple scattering of light waves, were elaborated in our previous work.28 We ascribed the underlying mechanisms to the increase in dopant concentration in the glassy phase and the multiple scattering of pumped light between the nanocrystals.
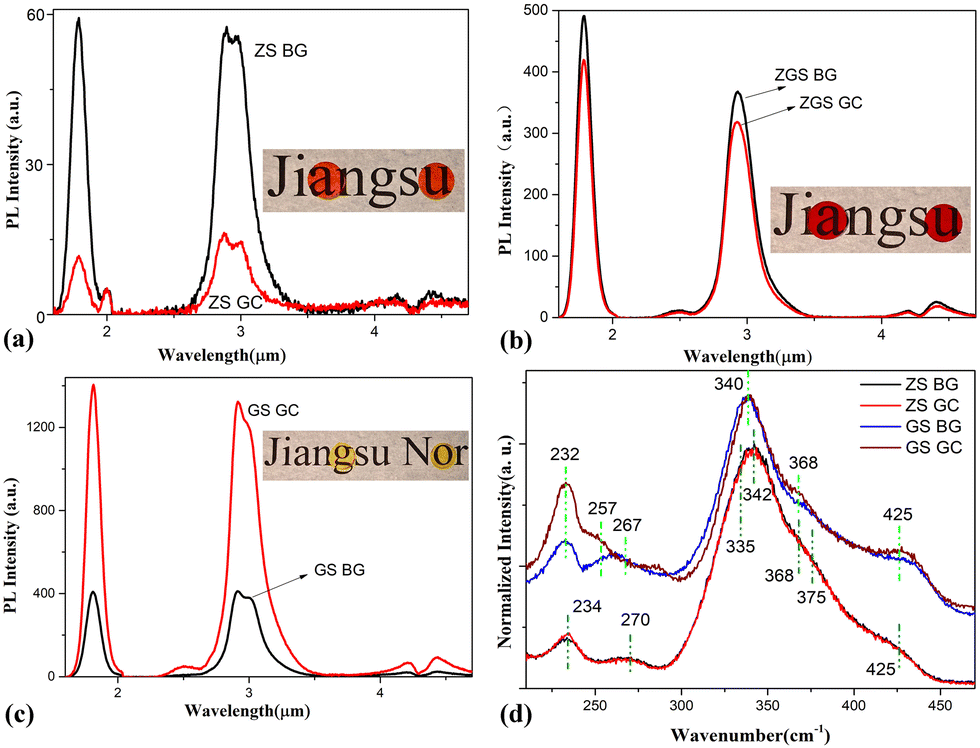 | ||
| Fig. 5 PL spectra of ZS (a), ZGS (b), and GS (c) samples before (BG) and after (GC) thermal activation above Tg. Each inset shows photographs of BG and GC samples from left to right. (d) Raman spectra of ZS and GS samples. | ||
Fig. 5(d) shows the normalized Raman spectra of the ZS and GS samples. In the ZS samples, the Raman bands are composed of vibrations of [AsS3] pyramids (∼335 cm−1, ∼375 cm−1),31 [AsSe3] pyramids (∼270 cm−1)32 and [GeS4] tetrahedra (∼234 cm−1, ∼342 cm−1, ∼368 cm−1, ∼425 cm−1).33,34 It is worth mentioning that [AsSe3] pyramids are present in the ZS sample because ZnSe was used as the raw material and some Se atoms tend to be captured by As atoms in the glass preparation process. In the GS glass samples, the curves consist mainly of vibrations of [Ge(Ga)S4] (∼232 cm−1, ∼340 cm−1, ∼368 cm−1, ∼425 cm−1) tetrahedra.33–37 The Raman band located at ∼262 cm−1 is the superposition of Ge–Ge (∼257 cm−1)38 and Ga–Ga homopolar bonds (∼267 cm−1).36 No evident difference can be observed between the Raman bands of the ZS GC and ZS BG samples, indicating no significant change in structural units takes place after heat treatment. However, there is an obvious difference between the Raman bands of the GS GC and GS BG samples. After heat treatment, the vibration of Ga–Ga bonds at ∼267 cm−1 disappears, and that of Ge–Ge bonds at ∼257 cm−1 is enhanced; meanwhile, the vibrations of [Ge(Ga)S4] tetrahedra at ∼232 cm−1, ∼368 cm−1 and ∼425 cm−1 are strengthened. These changes imply that the Ga–Ga bonds were broken and new [GaS4] units were formed by trapping the S atoms from Ge–S bonds during heat treatment. Thus, new Ge–Ge bonds were formed due to the loss of S atoms. The formation of new [GaS4] units facilitates the dissolution of Dy3+ ions in the matrix, which is beneficial for PL enhancement.
The methodology for modulation of chemical surroundings may be widely applicable to other active ions in glass. For instance, we further demonstrated it in samples activated with the transition metal ion Co2+, whose optical transitions are fairly sensitive to its chemical surroundings. The BG samples were prepared according to the compositions of 60GeS2–40As2S3–10ZnSe–0.3CoCl2 (ZC–BG) and 80GeS2–20Ga2S3–0.3CoCl2 (GC–BG). The GC samples were obtained by heat treating ZC–BG at 320 °C for 5 h (ZC–GC) and GC–BG at 460 °C for 5 h (GC–GC). Fig. 6(a) shows the visible-to-near-infrared optical transmission spectra of the ZC–GC and GC–GC samples. Three significant absorption bands can be observed in the spectrum of the GC–GC sample, namely the 0.63–0.80 μm absorption band centered at 0.73 μm, the 1.20–1.68 μm absorption band centered at 1.45 μm, and the 2.30–2.54 μm absorption band centered at 2.42 μm, which respectively originate from the 4A2(F) → 4T1(P), 4A2(F) → 4T1(F) and 4A2(F) → 4T2(F) spin-allowed transitions39 when Co2+ reside in the tetrahedral site of Ga2S3 crystals by replacing the lattice site of Ga. However, only the first two slightly different absorption bands are present in the spectrum of the ZC–GC sample. Pumped by an 808-nm diode laser source, the GC–GC sample exhibits 2.5–5.5 μm ultrabroadband and intense emission, as shown in Fig. 6(b), which consists of 2.5–4.5 μm (centered at 3.3 μm) and 3.0–5.5 μm (centered at 4.2 μm) emission bands that are respectively produced by 4T2(F) → 4A2(F) and 4T1(F) → 4T2(F) transitions39 of tetrahedrally coordinated Co2+. The full width at half maximum of the emission is ca. 2000 nm. In comparison, only the emission band of 2.5–4.5 μm (centered at 3.3 μm) can be observed in the PL spectrum of the ZC–GC sample. The much broader MIR emission band of the GC–GC sample is assumed to be associated with the multiple-coordination surroundings of Co2+. Ga atoms have several different tetrahedrally coordinated surroundings for the several sets of bond lengths in a Ga2S3 crystal (there is only one set of bond lengths in a ZnS crystal). Hence, Co2+ ions are endowed with several tetrahedrally coordinated surroundings after they replace Ga3+ ions; thus the 4F level of Co2+ tends to split in different magnitudes, leading to a wider emission band. This situation is similar to the inhomogeneous broadening of emission spectra of active ions in glass. It is worth noting that a strong absorption band at ∼4.3 μm appears in the PL spectra. This band is caused by CO2 absorption, implying that the GC–GC sample could be applied in gas sensing, as demonstrated in our previous work.40
 | ||
| Fig. 6 (a) Visible-to-near-infrared transmission spectra of ZC–GC and GC–GC samples (2 mm in thickness). The inset shows photographs of ZC–GC and GC–GC samples from left to right. (b) Featured PL spectra of ZC–GC and GC–GC samples, pumped by an 808 nm laser diode. | ||
On the other hand, the GC–GC sample exhibits much stronger PL intensity than the ZS–GC sample, as shown in Fig. 6(b). To compare their thermodynamic stabilities, the Tg of these two samples were measured. The GC–GC sample (Tg = 440 °C) has a higher Tg than the ZC–GC sample (Tg = 290 °C), suggesting better thermodynamic stability of the former. Thus, high PL efficiency and superior thermodynamic stability have been realized simultaneously by modulating the chemical surroundings of the dopant and the network structure of the glass.
To investigate the doping efficiencies that are responsible for the exclusive and intriguing optical spectroscopies of the GC–GC sample, its EDS was measured. As can be seen in Fig. 7(a–d), Ga2S3 crystals seem to disperse evenly in the amorphous solid, suggesting that the contributing mechanism underlying the nucleation is droplet phase separation. However, evident interpenetrating (complementary) elemental mappings of Ge and Ga elements were observed in Fig. 7(a) and (b), which demonstrates that the growth of Ga2S3 nucleation is self-limited by the viscous glass network, but the nucleation growth adheres to the stereotype of interpenetrating phase separation. Co2+ ions tend to accumulate preferentially in the nanocrystals, as shown in the elemental mapping similar to Ga in Fig. 7(b) and (d). With the progress of heat treatment, the Ga2S3 nanocrystals grow larger, their surface area increases and their activation barrier to impurity desorption is enhanced, enabling them to overcome the activation barrier for impurity incorporation and leading to the adsorption of Co2+ ions on the surface of the nanocrystals. Then the comparable ionic radii of Ga3+ (0.62 Å) and Co2+ (0.65 Å) allow the heterovalent substitution of Co2+–Ga3+ and the formation of tetrahedrally coordinated Co2+ ions.
 | ||
| Fig. 7 Elemental mappings of (a) Ge, (b) Ga, (c) S (d) Co in GC–GC sample. | ||
In conclusion, higher PL efficiency of Dy3+ ions can be achieved in glass with better thermodynamic stability by adding ZnSe into a Ge–As–S glass system due to the reduced effective phonon energy and augmented refractive index. The inter-ionic distance between Dy3+ ions becomes longer and the concentration quenching threshold of Dy3+ can be greatly augmented by the addition of a certain amount of Ga. As a result, the PL efficiency of Dy3+ ions in the glass network is significantly enhanced. The PL efficiency of Dy3+ ions can be further improved by regulating the glass network structure through MCN optimization and controlled nanocrystallization. Furthermore, by modulating the local chemical surroundings of Co2+, 2.5–5.5 μm ultrabroadband and intense emission has been achieved, for the first time, in the designed chalcogenide glass ceramic with new tetrahedral sites for the dopant. This ultrabroadband mid-infrared emission should be ascribed to multiple coordination surroundings of Co2+ after the lattice-site substitution of Co2+–Ga3+ in Ga2S3 crystals. All in all, it has been demonstrated that the chemical surroundings of dopants can be regulated by structural engineering of the glass network. Such a methodology may shed new light on the design and performance optimization of optical gain materials.
Data availability
Data underlying the results presented in this paper are not publicly available at this time but may be obtained from the authors upon reasonable request.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
National Natural Science Foundation of China (62305137, U21A2056); Key R&D Program of Zhejiang Province (2021C01025).References
- J. R. David, Filling the light pipe, J. Sci., 2010, 330(6002), 327–328 Search PubMed
.
- J. H. Yu, S. H. Kwon, Z. Petrášek, O. K. Park, S. W. Jun, K. Shin, M. Choi, Y. I. Park, K. Park, H. B. Na, N. Lee, D. W. Lee, J. H. Kim, P. Schwille and T. Hyeon, High-resolution three-photon biomedical imaging using doped ZnS nanocrystals, J. Nat. Mater., 2013, 12(4), 359–366 CrossRef CAS PubMed
- K. Sun, D. Tan, X. Fang, X. Xia, D. Lin, J. Song, Y. Lin, Z. Liu, M. Gu, Y. Yue and J. Qiu, Three-dimensional direct lithography of stable perovskite nanocrystals in glass, J. Sci., 2022, 375(6578), 307–310 CAS
- R. Martín-Rodríguez, R. Geitenbeek and A. Meijerink, Incorporation and luminescence of Yb3+ in CdSe nanocrystals, J. Am. Chem. Soc., 2013, 135(37), 13668–13671 CrossRef
- S. Zhou, Q. Guo, H. Inoue, Q. Ye, A. Masuno, B. Zheng, Y. Yu and J. Qiu, Topological engineering of glass for modulating chemical state of dopants, J. Adv. Mater., 2014, 26(47), 7966–7972 CrossRef CAS
- Z. Pan, Y. Y. Lu and F. Liu, Sunlight-activated long-persistent luminescence in the near-infrared from Cr3+-doped zinc gallogermanates, J. Nat. Mater., 2012, 11(1), 58–63 CrossRef CAS
- H. Zhang, Z. Yang, M. Zhou, L. Zhao, T. Jiang, H. Yang, X. Yu, J. Qiu, Y. Yang and X. Xu, Reproducible X-ray imaging with a perovskite nanocrystal scintillator embedded in a transparent amorphous network structure, J. Adv. Mater., 2021, 33(40), 2102529 CrossRef CAS PubMed
- W. B. Im, N. George, J. Kurzman, S. Brinkley, A. Mikhailovsky, J. Hu, B. F. Chmelka, S. P. DenBaars and R. Seshadri, Efficient and color-tunable oxyfluoride solid solution phosphors for solid-state white lighting, J. Adv. Mater., 2011, 23(20), 2300 CrossRef CAS
- Z. Fang, Z. Chen, W. Peng, C. Shao, S. Zheng, L. Hu, J. Qiu and B. O. Guan, Phase-separation engineering of glass for drastic enhancement of upconversion luminescence, J. Adv. Opt. Mater., 2019, 7(8), 1801572 CrossRef
- Z. Fang, J. Li, L. P. Sun, Y. Zhi, Y. Long, S. Zheng, Z. Chen, J. Qiu and B. Guan, In situ dopant-induced nano-crystallization of rare-earth-fluoride crystals in phase-separated networks for highly-efficient photoemission and photonic devices, J. Mater. Chem. C, 2021, 9(28), 9001–9010 RSC
- J. M. F. Van Dijk and M. F. H. Schuurmans, On the nonradiative and radiative decay rates and a modified exponential energy gap law for 4f–4f transitions in rare-earth ions, J. Phys. Chem., 1983, 78(9), 5317–5323 CrossRef CAS
- P. Fjodorow, M. P. Frolov and Y. V. Korostelin,
et al., Passively Q-switched 5-μm Ce3+-doped selenide glass laser using Fe: CdTe and Fe: CdSe as saturable absorbers, Opt. Lett., 2022, 47(2), 309–312 CrossRef CAS
- V. V. Koltashev, B. I. Denker and B. I. Galagan,
et al., 150 mW Tb3+ doped chalcogenide glass fiber laser emitting at λ > 5 μm, Opt. Laser Technol., 2023, 161, 109233 CrossRef CAS
- P. Fjodorow, M. P. Frolov and S. O. Leonov,
et al., Mid-infrared laser performance of Ce3+-doped selenide glass, Opt. Express, 2021, 29(17), 27674–27682 CrossRef CAS PubMed
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nanocrystals of cesium lead halide perovskites (CsPbX3, X= Cl, Br, and I): novel optoelectronic materials showing bright emission with wide color gamut, J. Nano Lett., 2015, 15(6), 3692–3693 CrossRef CAS PubMed
- X. Lu, Z. Lai, R. Zhang, H. Guo, J. Ren, L. Strizik, T. Wagner, G. Farrell and P. Wang, Ultrabroadband Mid-infrared Emission from Cr2+-doped Infrared Transparent Chalcogenide Glass Ceramics Embedded with Thermally Grown ZnS Nanorods, J. Eur. Ceram. Soc., 2019, 39(11), 3373–3379 CrossRef CAS
- S. B. Mirov, I. S. Moskalev, S. Vasilyev, V. Smolski, V. V. Fedorov, D. Martyshkin and J. Peppers, Frontiers of mid-IR lasers based on transition metal doped chalcogenides, IEEE J. Sel. Top. Quantum Electron., 2018, 24(5), 1–29 Search PubMed
- C. Lin, C. Rüssel and S. Dai, Chalcogenide glass-ceramics: Functional design and crystallization mechanism, Prog. Mater. Sci., 2018, 93, 1–44 CrossRef CAS
- A. Sreeram, D. Swiler and A. Varshneya, Gibbs-DiMarzio equation to describe the glass transition temperature trends in multicomponent chalcogenide glasses, J. Non-Cryst. Solids, 1991, 127(3), 287–297 CrossRef CAS
- J. Choi, S. J. Gurman and E. A. Davis, Structure of amorphous GexSe1−x and GexSeyZnz thin films: an EXAFS study, J. Non-Cryst. Solids, 2002, 297(2–3), 156–172 CrossRef CAS
- T. H. Lee, S. I. Simdyankin, L. Su and S. R. Elliott, Evidence of formation of tightly bound rare-earth clusters in chalcogenide glasses and their evolution with glass composition, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79(18), 180202 CrossRef
- J. Heo, J. M. Yoon and S. Y. Ryou, Raman spectroscopic analysis on the solubility mechanism of La3+ in GeS2–Ga2S3 glasses, J. Non-Cryst. Solids, 1998, 238(1–2), 115–123 CrossRef CAS
- H. He and M. F. Thorpe, Elastic properties of glasses, Phys. Rev. Lett., 1985, 54(19), 2107 CrossRef CAS
- K. Tanaka, Structural phase transitions in chalcogenide glasses, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39(2), 1270 CrossRef CAS
- X. Lu, H. Liu and K. Tian,
et al., Mid-infrared 3–4 μm emission of Ni2+ doped chalcogenide glass-ceramic fiber, Ceram. Int., 2023, 49(9), 13386–13391 CrossRef CAS
- G. Boulon, G. A. Goget, Y. Guyot, M. Guzik, T. Epicier, N. P. Blanchard, L. Chen, L. Hu and W. Chen, Conjugation of TEM-EDX and optical spectroscopy tools for the localization of Yb3+, Er3+ and Co2+ dopants in laser glass ceramics composed of MgAl2O4 spinel nano-crystals embedded in SiO2 glass, J. Mater. Chem. C, 2014, 2(44), 9385–9397 RSC
- Z. Gao, X. Lu, Y. Chu, S. Guo, L. Liu, Y. Liu, S. Sun, J. Ren and J. Yang, The distribution of rare earth ions in a γ-Ga2O3 nanocrystal-silicate glass composite and its influence on the photoluminescence properties, J. Mater. Chem. C, 2018, 6(12), 2944–9520 RSC
- X. Lu, Z. Lai, J. Ren, L. Strizik, T. Wagner, Y. Du, G. Farrell and P. Wang, Distribution of Tm3+ and Ni2+ in chalcogenide glass ceramics containing Ga2S3 nanocrystals: Influence on photoluminescence properties, J. Eur. Ceram. Soc., 2019, 39(7), 2580–2584 CrossRef CAS
- C. Lin, S. Dai, C. Liu, B. A. Song, Y. Xu, F. Chen and J. Heo, Mechanism of the enhancement of mid-infrared emission from GeS2-Ga2S3 chalcogenide glass-ceramics doped with Tm3+, Appl. Phys. Lett., 2012, 100(23), 231910 CrossRef
- R. Wang, K. Yan, M. Zhang, X. Shen, S. Dai, X. Yang, Z. Yang, A. Yang, B. Zhang and B. Luther-Davies, Chemical environment of rare earth ions in Ge28.125Ga6.25S65.625 glass-ceramics doped with Dy3+, Appl. Phys. Lett., 2015, 107(16), 161901 CrossRef
- A. Bertoluzza, C. Fagnano and P. Monti,
et al., Raman and infrared spectra of As2Sx chalcogenide glasses with x ⩽ 3, J. Non-Cryst. Solids, 1978, 29(1), 49–60 CrossRef CAS
- R. P. Wang, D. Bulla and A. Smith,
et al., Structure and physical properties of GexAsySe1−x−y glasses with the same mean coordination number of 2.5, J. Appl. Phys., 2011, 109(2), 23517 CrossRef
- M. H. Brooker, O. F. Nielsen and E. Praestgaard, Assessment of correction procedures for reduction of Raman spectra, J. Raman Spectrosc., 1988, 19(2), 71–78 CrossRef CAS
- G. Lucovsky, R. J. Nemanich and S. A. Solin,
et al., Coordination dependent vibrational properties of amorphous semiconductors alloys, Solid State Commun., 1975, 17(12), 1567–1572 CrossRef CAS
- M. Ichikawa, T. Wakasugi and K. Kadono, Glass Formation, Physico-Chemical Properties, and Structure of Glasses Based on Ga2S3-GeS2-Sb2S3 System, J. Non-Cryst. Solids, 2010, 356, 2235–2240 CrossRef CAS
- A. Tverjanovich, Y. S. Tveryanovich and S. Loheider, Raman spectra of gallium sulfide based glasses, J. Non-Cryst. Solids, 1996, 208(1–2), 49–55 CrossRef CAS
- C. Lin, L. Calvez and H. Tao,
et al., Evidence of network demixing in GeS2–Ga2S3 chalcogenide glasses: a phase transformation study, J. Solid State Chem., 2011, 184(3), 584–588 CrossRef CAS
- G. Lucovsky, F. L. Galeener and R. C. Keezer,
et al., Structural interpretation of the infrared and Raman spectra of glasses in the alloy system Ge1−xSx, Phys. Rev. B, 1974, 10(12), 5134 CrossRef CAS
- L. D. DeLoach, R. H. Page, G. D. Wilke, S. A. Payne and W. F. Krupke, Transition metal-doped zinc chalcogenides: spectroscopy and laser demonstration of a new class of gain media, IEEE J. Quantum Electron., 1996, 32(6), 885–895 CrossRef CAS
- H. Liu, Y. Gao and Y. Jiang,
et al., Ultrabroadband mid-infrared emission and gas sensing of cobalt-doped chalcogenide glass ceramics, Opt. Lett., 2024, 49(20), 5807–5810 CrossRef
| This journal is © The Royal Society of Chemistry 2025 |