
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling optoelectronic properties through protonation with π-extended triphenodioxazine diimides†
Rhea
Kumar
 a,
Mario
Taddei
f,
Vasilis
Petropoulos
a,
Mattia
Russo
a,
Federico
Vernuccio
a,
Giulio
Cerullo
a,
Dario
Polli
a,
Artur
Nenov
f,
Nicola
Demitri
e,
Maurizio
Prato
*bcd,
Margherita
Maiuri
*a and
Jacopo
Dosso
*b
a,
Mario
Taddei
f,
Vasilis
Petropoulos
a,
Mattia
Russo
a,
Federico
Vernuccio
a,
Giulio
Cerullo
a,
Dario
Polli
a,
Artur
Nenov
f,
Nicola
Demitri
e,
Maurizio
Prato
*bcd,
Margherita
Maiuri
*a and
Jacopo
Dosso
*b
aDepartment of Physics, Politecnico di Milano, Piazza L. da Vinci 32, 20133 Milano, Italy. E-mail: margherita.maiuri@polimi.it
bDepartment of Chemical and Pharmaceutical Sciences, CENMAT, Centre of Excellence for Nanostructured Materials, INSTM UdR Trieste, University of Trieste, via Licio Giorgieri 1, 34127 Trieste, Italy. E-mail: prato@units.it; jacopo.dosso@units.it
cCentre for Cooperative Research in Biomaterials (CIC BiomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 194, 20014, Donostia San Sebastián, Spain
dBasque Fdn Sci, Ikerbasque, 48013 Bilbao, Spain
eElettra—Sincrotrone, Trieste S.S. 14 Km 163.5, Area Science Park, 34149 Basovizza, Trieste, Italy
fDepartment of industrial chemistry, University of Bologna, Viale del Risorgimento, 40136 Bologna, Italy
First published on 16th December 2024
Abstract
We present a combined approach integrating chemical synthesis, computational methods and advanced ultrafast spectroscopy to explore the properties of triphenodioxazine diimides (TPDODI). The TPDODI derivative shows significant redshifted absorption (λmax = 556 nm) and emission (λmax = 569 nm) compared to other non-diimide TPDO derivatives. Protonation of the TPDODI leads to further redshifts in absorption (λmax = 638 nm and 715 nm for mono- and diprotonated states, respectively), with the diprotonated form absorbing up to 800 nm. However, protonation also triggers competitive nonradiative decay processes, confirmed by transient absorption spectroscopy and linked to the Energy Gap Law via high-frequency molecular vibrations detected by two-dimensional electronic spectroscopy. Computational analysis supports these findings, particularly in highlighting the enhanced electron affinity of the monoprotonated species (LUMO = −3.61 eV vs. −4.98 eV for the neutral and monoprotonated forms, respectively). These results underscore the versatility of TPDODI for optoelectronic applications, providing key insights into the fine-tuning of n-type semiconductors, catalysts, and other advanced materials.
Introduction
Over the past two decades, studies on organic molecules for optoelectronic applications, including organic photovoltaics (OPVs) and light-emitting diodes (OLEDs), have grown rapidly. The rising interest in these materials stems from their inherent advantages, including solution processability, flexibility and the potential to mould their photophysical properties by structural fine-tuning.1,2 These characteristics make organic molecules highly attractive for use in a wide range of technologies from energy harvesting to light emission.In this context, industrially producible triphenodioxazine (TPDO)3 derivatives have emerged as promising candidates due to their remarkable versatility. These derivatives have been successfully applied across a wide range of technologies, including air- and photo-stable pigments,4,5 OPVs,4–6 dye-sensitised solar cells,4,7 air-stable organic field effect transistors,8–10 liquid crystal luminescent devices,7,11 stimuli responsive materials,12 and organic thin film transistors8,9,13 including those for toxin sensing.14,15 Of particular interest, TPDO derivatives display n-type semiconducting properties, which are relatively rare in organic molecules due to an inherent instability of organic n-type semiconductors which, among other methods, has been addressed by the incorporation of TPDO derivatives.4,8,9 This makes them attractive alternatives to traditional organic n-type semiconductors such as perylene diimides, naphthalene diimides, C60 and fluorinated oligothiophenes.16–21
Despite the promising optoelectronic properties of TPDO derivatives, there is still room to enhance their electron affinity and tuneability, which is crucial for optimizing their performance in semiconductors and other applications.22 One successful approach to achieving this is the introduction of electron-withdrawing groups on the TPDO scaffold, which lowers the LUMO levels and enhances the electron affinity.8,23 Additionally, the conversion of TPDO to its diimide form has been shown to improve both optical and electronic properties, resulting in stronger visible absorption and higher emission quantum yields.23 Recent studies on triphenodioxazine diimides (TPDODIs) have confirmed that the incorporation of the imide group enhances electron affinity, making these systems even more appealing for optoelectronic applications.23 However, despite these advances, the effects of protonation on the optical and electronic properties of TPDOs and TPDODIs remain largely unexplored.
Building on this, in this paper we report the first synthesis of a π-extended TPDODI presenting intense visible absorption and high emission quantum yield. We investigate the effect of mono- and double protonation on these derivatives, revealing significantly red-shifted absorption and emission associated with a dramatic decrease of the LUMO level of the molecule. An in-depth experimental and computational analysis reveals the optical and vibrational properties of the neutral and protonated states of the molecule, setting an important milestone for the future development of these materials.
Results and discussion
Synthesis and structural characterisation
In general, the most common route for the preparation of TPDO derivatives involves the reaction of 2,5-dihydroxy-1,4-benzoquinones with 1-aminophenols in acidic conditions, resulting in condensation reactions that lead to the formation of the heteroaromatic system. Aiming at the synthesis of TPDODI system 1, the preparation of 5-amino-6-hydroxy naphthalene imide would be required, which is however a highly challenging multi-step procedure requiring noticeable synthetic effort. To avoid this, a similar reactivity could plausibly be obtained by using a 5-amino-6-bromo naphthalene monoamide (5, Scheme 1). In fact, in presence of 2,5-dihydroxy-1,4-benzoquinone and acidic conditions, the free amine group on 5 would be able to react with the carbonyl moiety in 6, resulting in formation of an imine group. At this point, exploiting the electron-depleted nature of naphthalene imide systems and the high effective molarity arising from proximity, a nucleophilic aromatic substitution with release of HBr could take place, resulting in the formation of the final C–O bond. This would be a great advantage since it could lead to a facile synthesis of π-extended systems, avoiding the complexity related to the synthesis of amino hydroxy derivatives. Based on these assumptions, the synthesis of 5 was started by treating 2 with 2,6 diisopropyl aniline in the presence of acetic acid at 110 °C for 18 h, resulting in the formation of the corresponding naphthalene monoimide 3. Subsequently, 3 was reduced in the presence of SnCl2·2H2O in EtOAc/EtOH at 60 °C to afford the free amine derivative 4 in a 79% yield. Treatment of 4 with bromine in dioxane smoothly afforded derivative 5 in an 86% yield. At this point, derivative 5 was suspended in heptanoic acid along with 6 followed by microwave (MW) irradiation at 200 °C for 1 h, resulting in the formation of desired 1 as a dark red solid in a 50% yield (Scheme 1). Heptanoic acid was chosen as solvent since it ensures both the acid conditions for the imine formation and the high temperatures required for the nucleophilic aromatic substitution on the Br atom. | ||
| Scheme 1 (a) Synthesis of derivative 1, R = diisopropylphenyl; (b) top view and (c) side view of crystal structure of 1. Crystals obtained from slow diffusion of hexane in CHCl3. Space group: P21/n. Atom colours: O: red, N: blue, C: grey. Hydrogens omitted for clarity. CCDC 2383220.† | ||
On the other hand, when 10 was reacted to form 1Br, only traces of product could be obtained, suggesting reactivity also of the second bromine atom in the harsh reaction conditions, limiting characterization of 1Br to scXRD (Table S4 and Fig. S45, ESI†).
Derivative 1 was completely characterised via NMR and high-resolution mass spectroscopy (HRMS), confirming the formation of the desired TPDODI system. Moreover, it was possible to grow crystals of sufficient quality for single crystal X-ray diffraction (scXRD, Scheme 1b and c). From the scXRD analysis, the formation of the desired structure was confirmed. The latter proved to be almost completely planar and presented a quinoidal structure in the central ring, highlighted by marked bond alternation, in line with similar non-diimide derivatives.8,24 No significant π-stacking interactions were visible between derivatives, mostly due to the bulky nature of the diisopropyl-phenyl substituents which disrupt significantly any interaction. Also, no interactions were visible at the sp2 nitrogen atoms which is plausibly related to the steric hindrance arising from the neighbouring phenyl ring. At this point, to understand if some nucleophilic character was present on the sp2 nitrogen, an NMR experiment was performed on 1 to assess if this system could be protonated by acids resulting in the mono- and possibly doubly protonated form (Fig. 1), which are expected to present a very high electron affinity.
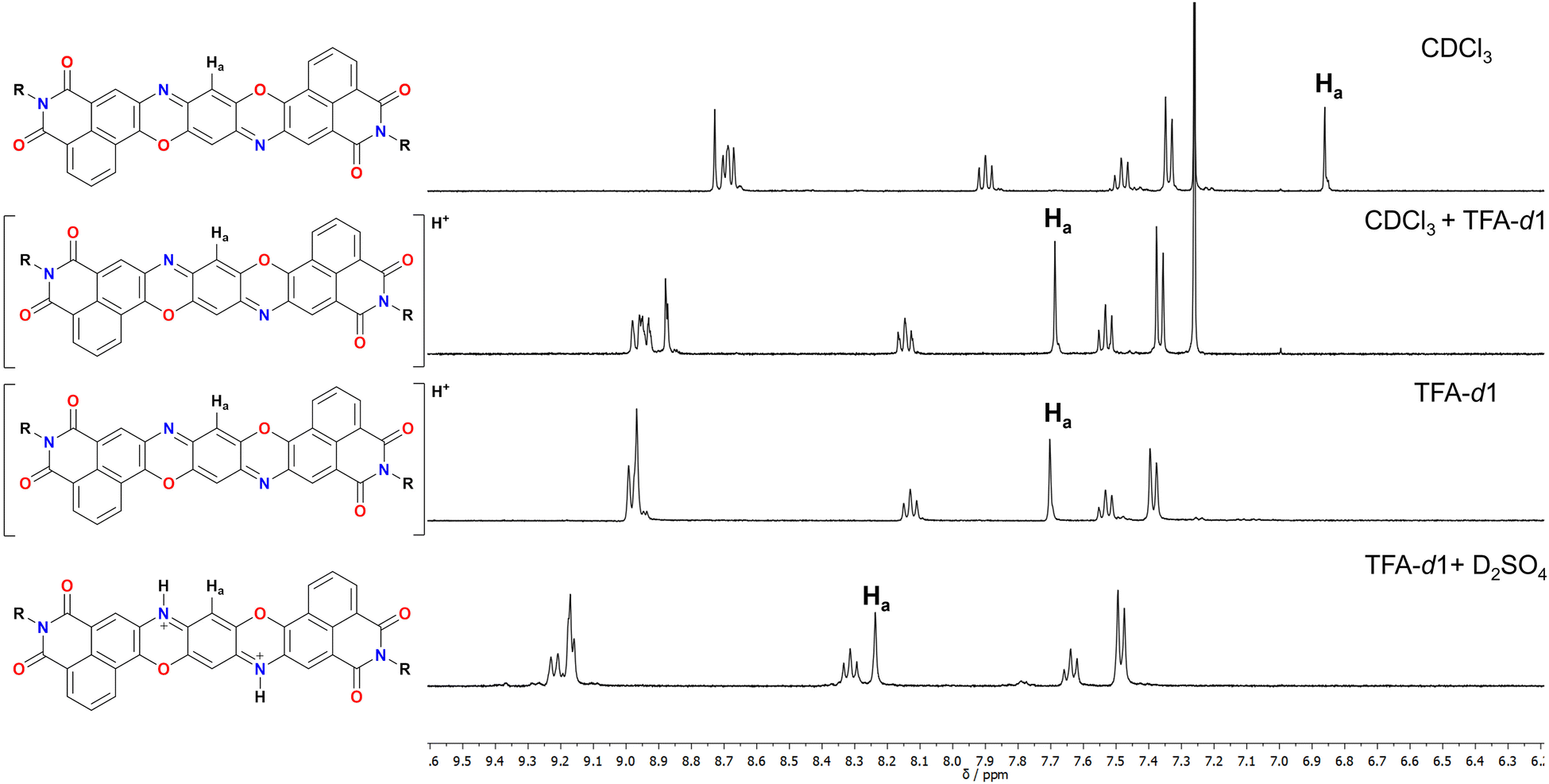 | ||
| Fig. 1 Detail of the aromatic part of 1 in different solvent systems. | ||
From these experiments it is possible to see that the diagnostic Ha proton associated with the quinoidal system undergoes a marked downfield shift upon addition of acids. Particularly, addition of TFA in CDCl3 (0.2 M) results in an immediate colour change from bright red to deep blue, associated with a downfield shift of 0.83 ppm. Recording the same spectrum in pure TFA does not significantly change the chemical shift of the peaks, suggesting quantitative protonation in both cases. The symmetric nature of the obtained spectrum however does not allow to discern between a monoprotonation with a fast interchange between the two nitrogen atoms or the doubly protonated form. Diffraction of crystals obtained in presence of TFA also did not provide satisfactory answers to this question (see ESI†). As such, to make this point clear, a further experiment was performed by adding 1 drop of D2SO4 to a TFA solution of 1. The resulting NMR spectrum presents an even more marked downfield shift for Ha (0.54 ppm), suggesting that a second protonation is occurring in this case. To support this, the absorption spectrum of 1 was measured in solvent systems of different acidity and is shown in Fig. 2a and Fig. S33 (ESI†). First, in CHCl31 presents a structured absorption with a λmax = 556 nm (ε = 6.5 × 104 M−1 cm−1) associated with an emission (λem,max = 569 nm, Fig. S28, ESI†) with a small Stokes shift consistent with its highly rigid structure. Interestingly, this emission occurs with a high quantum yield (QY) of 70%. Upon addition of increasing amounts of TFA to solutions of 1 in CH2Cl2 a new set of redshifted absorption bands appears (λmax = 638 nm, Fig. S29, ESI†), suggesting the formation of the monoprotonated form with complete conversion at ca. 0.2 M concentration of TFA. A similar titration performed with H2SO4 in THF afforded a similar result (Fig. S30 and S33, ESI†). Moreover, the absorption spectrum in pure TFA (λmax = 629 nm) is very similar to those in THF + H2SO4 (λmax = 620 nm), CH2Cl2 + TFA (λmax = 638 nm) and CHCl3 + TFA (λmax = 638 nm), thus ruling out important solvatochromic effects (Fig. S33, ESI†). Interestingly, the spectra of the neutral and monoprotonated derivatives are very similar to non-imide derivatives24 but are redshifted by ca. 36 nm corresponding to lowering of 0.16 eV in the transition energy, plausibly due to the enhanced conjugation arising from the extended diimide system. Also, compared to the non-diimide derivatives, in the case of 1, a 10-fold higher concentration of TFA is required to form quantitatively 1H+, suggesting that the electron depleted diimide systems are indeed decreasing the basic nature of the pyridinic nitrogen.24 Interestingly, a weak (QY < 1%) red emission (λem,max = 738 nm) was associated with 1H+ suggesting a great enhancement of the nonradiative decay deactivation pathways upon protonation (Fig. S31, ESI†). In line with the previously discussed NMR experiments, also in this case, dissolving 1 in a TFA solution containing 0.2 M H2SO4 resulted in the formation of a new set of redshifted absorption bands (λmax = 715 nm) consistent with the formation of 12H+.
 | ||
| Fig. 2 (a) Normalized absorption of 1 in CH2Cl2, pure TFA (11H+) and TFA with 0.2 M H2SO4 (12H+). (b) Calculated absorption spectra of 1, 1H+ and 12H+ from TD-DFT. (c) Calculated electrostatic potentials for neutral 1, 1H+ and 12H+. The colour scale indicates the full range of electrostatic potential, with the range being −0.02e to 0.02e for 1, 0.00e to 0.15e for 11H+ and 0.10e to 0.26e for 12H+. As expected, the scale is shifted to increasingly positive values with more protonation. (d)–(f) Transient absorption spectra for 1 (d), 1 in TFA (1H+, (e)) and 1 in H2SO4 (12H+, (f)) at selected timepoints within the range given by the respective colour bar for each spectrum. | ||
To estimate the HOMO and LUMO levels for derivatives 1 and 1H+, cyclic voltammetry experiments were carried out in CH2Cl2 (Fig. S36 and S37, ESI†). In the case of 1, an irreversible oxidation was visible at +0.95 V vs. Fc/Fc+, while two reversible reductions were detected at −1.19 V and −1.62 V vs. Fc/Fc+ respectively. These values are associated with a HOMO level of −5.75 eV and a LUMO of −3.61 eV vs. Fc/Fc+ which are consistent with TPDO derivatives bearing electron-withdrawing groups such as CF3,8 and differently positioned diimides.23 Interestingly, upon addition of TFA (0.2 M) the two reversible reduction peaks appear at much more positive potentials (Fig. S37, +0.18 V and −0.23 V vs. Fc/Fc+, ESI†) which are consistent with a dramatic decrease of the LUMO level (−4.98 eV vs. Fc/Fc+). This suggests that protonation results in great enhancement in the electron affinity for TPDO diimides. This result is consistent with the trend observed in calculated HOMO and LUMO levels (Fig. S38 and Table S1, ESI†), the computational details of which are discussed in detail below. The theoretical results also reveal decreasing HOMO and LUMO levels and a narrowing HOMO–LUMO gap with increasing protonation.
Electronic structure characterisation
To gain insights into the effect of protonation on the electronic structure of 1, computational studies were performed by means of density functional theory (DFT) for the ground state (GS), and time-dependent density functional theory (TD-DFT) for the first excited state employing long-range corrected CAM-B3LYP25 hybrid functional together with cc-pVDZ26 basis set as implemented in the Gaussian1627 software package. Solvent effects were treated implicitly through the polarisable continuum model (PCM).28 GS optimisation of the three systems shows that the structure is not affected upon different degrees of protonation retaining the aromatic core planar structure, in line with crystal structures obtained in presence of TFA (Fig. S44, ESI†). Calculation of vertical excitation energies reveals that the S1 bright state is characterised by the HOMO–LUMO transition (Fig. S38 and Table S2, ESI†) and, in agreement with experimental trend, we observe a progressive redshift of the absorption peak with increasing protonation: in particular, λabs = 499 nm for the neutral system, λabs = 577 nm for the monoprotonated system and λabs = 767 nm for the diprotonated system. Computed vibronic spectra of the three systems are shown in Fig. 2b. To reproduce the vibronic fine structure, we made use of the iSpectron29 code providing the GS equilibrium geometry together with its normal modes and energy gradient of the S1 excited state at the Franck–Condon point. We assume that potential energy surfaces (PESs) of the electronic states are quadratic in the coordinates with different equilibrium geometries. Moreover, different electronic states are described by the same normal modes and frequencies which are calculated only for the GS. This procedure is referred to as the vertical gradient (VG)30 approach in the literature. The simulated spectra show good agreement with experiments, although it was necessary to scale the gradients to reproduce the exact line shape (computed vibronic spectra without scaling factors are shown in Fig. S39, ESI†). We note that in the three systems, the vibronic structure is given by the activation of vibrational modes between 1300–1500 cm−1 assigned to stretching deformations of the aromatic core. In the neutral and diprotonated systems, the dominant mode is characterised by a symmetric stretching while in the monoprotonated system an anti-symmetric stretch is the dominant mode (Fig. S40, ESI†).Electrostatic potentials for 1, 1H+ and 2H+, on both S0 and S1 are shown in Fig. 2c. The neutral molecule 1 exhibits weak electron density deficiency around the central ring of the molecule in the GS. Protonation leads to delocalization of a positive charge over the central ring, asymmetrically in the case of 1H+ and symmetrically in the case of 12H+. Due to the positive charge residing on the central ring, a donor–acceptor–donor motif arises that is increasingly prevalent with increasing protonation. In all three molecules, excitation to S1 results in a partial charge displacement towards the centre (indicated by black arrows in Fig. 2c). Both 1 and 12H+ exhibit a symmetric shift on excitation due to the symmetry in their structure. Conversely, the asymmetry in 1H+ extends to asymmetric electrostatic potentials on both S0 and S1, giving rise to a non-zero dipole moment in S1.
Characterisation of excited state dynamics
To characterise the optical properties of the three systems, the photoinduced dynamics following excitation to S1 were investigated by transient absorption (TA) spectroscopy. The transient differential transmission (ΔT/T) spectra at selected probe delays are shown in Fig. 2 for 1 (Fig. 2d), 11H+ (Fig. 2e) and 12H+ (Fig. 2f). For all three systems we observe large ground state bleach (GSB) features at wavelengths corresponding to the linear absorption spectra in Fig. 2a. Due to the probe spectral range (490–710 nm), we were able to detect only the two lowest-lying vibronic peaks in the absorption spectrum for 1 (Fig. 2a), giving GSB features at 500 nm and 550 nm (Fig. 2d). At longer probe wavelengths, stimulated emission (SE) bands can be observed at 610 nm and 665 nm, though their amplitude is depleted by a superimposed negative photoinduced absorption (PA) band. The transient spectrum features isosbestic points at 570 nm and 600 nm, which lie on the ΔT/T = 0 baseline, indicating that the spectrum observed at early times evolves directly to zero. This reveals that the S1 state relaxes directly to the GS and therefore the timescale of its decay denotes the timescale of S1 → S0 relaxation. All spectral features decay slowly and persist beyond the 1.3 ns pump–probe delay available in the measurement.For 11H+ (Fig. 2e), we observe GSB features at 590 nm and 630 nm. A positive band at 710 nm comprises a superposition of GSB and SE contributions, while the remaining SE peaks appear at 805 nm and 910 nm. We identify an isosbestic point on the baseline for 11H+ at 550 nm, confirming that the S1 state of the monoprotonated molecule also relaxes directly to the ground state without branching photophysical processes. In this case, however, the ground state is entirely repopulated before 500 ps, as observed by the recovery of the signal to zero. For 12H+ (Fig. 2f), the highest-lying vibronic peak lies outside of our probe window, so we observe GSB bands at 650 nm and 725 nm. The spectrum also reveals a strongly quenched emission manifesting as only a shoulder of SE at 830 nm, otherwise masked by a PA that spans from 850 nm to longer wavelengths. Again, the isosbestic point at 850 nm demonstrates that the S1 state of 12H+ relaxes directly back to the ground state.
To examine the lifetime of the S1 state in each system, a global fit was performed for each time-resolved spectrum. The kinetic traces that show the decay of the S1 state, together with monoexponential fits to their evolution, are presented in Fig. 3. From the monoexponential fits, a lifetime that describes the relaxation from S1 to S0 was extracted and is also reported in Fig. 3. The fit for the neutral molecule 1 revealed a time constant 1.2 ± 0.13 ns. In 11H+, the S1 → S0 relaxation is accelerated to 92 ± 0.7 ps, more than an order of magnitude faster. Meanwhile, the diprotonated form 12H+ relaxes on a timescale of 27 ± 0.2 ps, accelerated a further threefold.
 | ||
| Fig. 3 Energy level diagram for 1 under neutral conditions, in TFA and in TFA + 0.2 M H2SO4 where vertical arrows depict the probed transition of relaxation from S1 to S0, alongside the dynamics of this process in each molecule and the associated lifetimes from global fitting to TA spectra in Fig. 2. | ||
The coupling of electronic transitions to high-frequency (1000–1600 cm−1) molecular vibrations, commonly carbon–carbon stretching modes, is usually responsible for accelerating the nonradiative depopulation of the fluorescent state in organic molecules.31 This generally accepted principle prompted us to investigate whether molecular vibrations that are coupled to the S1 → S0 electronic transition play a role in its altered dynamics. The ground state and excited state vibrational spectra of each molecule were therefore calculated using the same level of theory previously described. The ground state spectra can be found in the ESI† (Fig. S41) and the S1 Huang–Rhys spectra are shown in Fig. 4. All calculated spectra are generally in good agreement with one another and are also consistent with experimental ground state spectra obtained by Raman spectroscopy (Fig. S42, ESI†). All three systems show a dense manifold of high frequency vibrations in the region 1300–1500 cm−1 on both the ground and excited state, which correspond to the same stretching modes which give rise to vibronic spectra, and the frequency of the highest intensity peak is consistent for all three molecules between 1430–1450 cm−1.
 | ||
| Fig. 4 Experimental impulsively excited vibrational spectra from two-dimensional electronic spectroscopy (top) by taking the Fourier transform of kinetic traces pertaining to transitions on the S1 state and calculated Huang–Rhys spectra of each molecule on the S1 electronic excited state (bottom). | ||
To experimentally verify the observations from theoretical vibrational analysis, two-dimensional electronic spectroscopy (2DES) was also carried out. Such experiments employ sub-15-fs broadband pulses, granting a sufficiently high time resolution to probe high-frequency vibrations up to 1500 cm−1.32 Indeed, ultrashort pulses impulsively excite vibrational wavepackets, the temporal evolution of which induces an oscillating signal amplitude superimposed on the photoinduced dynamics.33 The period of the oscillation corresponds to the pump-induced nuclear motion and can be projected onto the frequency domain using the Fourier transform to resolve the vibrational spectrum of a given electronic state. In typical TA measurements, the use of broadband excitation results in a signal integrated across the entire pump wavelength range. The outcome of this experimental condition is that dynamics (especially minor oscillations in the signal due to molecular vibrations) arising from photoexcitation at different pump wavelengths may cancel to an amplitude that is within the signal-to-noise limit of the measurement. 2DES instead allows to resolve the transient ΔT/T spectra as a function of pump wavelength as well as probe wavelength, still preserving high temporal resolution, thus enabling the sensitive detection of high frequency oscillations. The resulting 2D maps can be found in the ESI† (Fig. S43).
For each molecule, the temporal evolution of a negative signal in the 2D map was extracted by plotting the signal intensity at a specific pump and probe wavelength as a function of time. As the negative signals correspond to PA transitions following resonant excitation to S1, these signals arise due to excited state absorption from S1 to a higher-lying electronic state (Sn). Therefore, by ensuring that the negative time-dependent signal is extracted from a point on the 2D map that is sufficiently far from GSB and SE features to rule out overlap with vibrational wavepackets on the ground electronic state, the oscillatory signal for a given molecule describes pump-excited vibrational wavepackets on the S1 state.34 In each case, the Fourier transform of the time-dependent signal provides a frequency-dependent spectrum that reveals the experimental vibrational spectrum of the S1 state. In Fig. 4, these S1 vibrational spectra are plotted alongside the calculated Huang–Rhys spectra, HRk = dk2ωk/2ℏ, where dk is the horizontal displacement of the S1 PES with respect to S0 along the kth normal mode with frequency ωk, obtained with iSpectron in the aforementioned VG approximation.
We note an excellent agreement between theory and experiment around 1420–1450 cm−1, arising due to the different equilibrium geometries of S1 and S0 along the high frequency stretching mode also responsible for the vibrational fine structure in the GS absorption spectra (Fig. S40, ESI†). In detail, the dominant vibrational modes from the 2DES measurement and from the theoretical approach, respectively, are 1458 ± 10 cm−1 and 1482 cm−1 for 1, 1354 ± 10 cm−1 and 1384 cm−1 for 11H+ and 1426 ± 10 cm−1 and 1428 cm−1 for 12H+ demonstrating a close agreement between theory and experiment. The discrepancies between experimental and theoretical results at lower frequencies are signatures of the different PES topology of the SN state giving rise to the PA at the wavelength chosen for the analysis with respect to S1 (Fig. S43, ESI†) and are thus less relevant for the present discussion. Overall, the similarity between the vibrational spectra for all three molecules confirms that the protonation does not have a dramatic effect on the vibrational energy levels on the S1 state, nor on the ground state.
The results in Fig. 4 reveal that the acceleration of nonradiative decay with increasing protonation is a consequence only of the decreasing S0–S1 energy gap, as the frequency of the dominant vibrational mode is largely unaffected by the protonation. We confirm this by invoking the well-accepted Energy Gap Law for calculating the rate (knr) of radiationless transitions, widely used for evaluating the competing channels of luminescence and other loss pathways that contribute to S1 decay, which states that knr accelerates as the S0–S1 energy gap decreases.35 Computing the ratio of nonradiative decay rates by varying only the S0–S1 energy gap, ΔE, and using a constant value of vibrational frequency for all three molecules, we can accurately predict the trend of accelerating nonradiative decay thus further confirming that this effect comes from the decreasing energy gap alone. In the following discussion we therefore employ an effective frequency value ω by taking an average of the three molecules’ dominant vibrational modes (Fig. S40 and Table S3, ESI†), which is 1431 cm−1 according to the peaks from calculated Haung-Rhys spectra, and 1413 cm−1 according to the results from 2DES experiments. In order to calculate the expected acceleration of nonradiative decay with narrowing ΔE, we compare approximated decay rate ratios, calculated using ΔE and ω values according to the Energy Gap Law, to the experimentally determined nonradiative decay rates. Nonradiative decay rates are calculated from the experimental S1 decay lifetimes in Fig. 3 and the fluorescence QY values (Table S3, ESI†). Because 11H+ and 12H+ both have negligible QY and are thus dominated by non-radiative decay, the knr is equal to the experimentally measured S1 decay rate, while the high (0.7) QY of 1 means that knr is three times faster than the measured S1 decay lifetime given in Fig. 3, according to k = kr + knr and QY = kr/(kr + knr).
Full treatment of the Energy Gap Law expression for a transition from S1 to S0 includes terms that consider the energy difference ΔE between the involved transition, the electronic coupling between the S1 and S0 states, and the relaxation energy related to the frequency of molecular vibrations. According to TD-DFT calculations, there is an S0–S1 conical intersection only for the deprotonated molecule that is, however, far too high in energy (ca. 2 eV from the S1 minimum) to be accessible. We therefore deduce that all three systems are in the regime of weak electronic coupling between S0 and S1 and approximate that there is no major difference in electronic coupling between the three studied molecules. This approximation allows us to derive an expression for the ratio of the rates for any given two compared molecules for which the ΔE is the only variable that is unique to a given molecule. The ratio between the rate of the S1 → S0 transition in, for example, 11H+ and 12H+ according to our approximation of the Energy Gap Law is therefore:
 | (1) |
Conclusions
In this work, we have synthesized a novel π-extended TPDO diimide derivative that exhibits intense visible absorption and high fluorescence QY. In particular, the absorption was redshifted compared to other electron depleted TPDO derivatives, plausibly due to the extended conjugation. Interestingly, this molecule demonstrates remarkable sensitivity to pH: protonation of the molecular core further shifts the absorption spectrum significantly to the near infrared (up to 800 nm for 12H+) and lowers the LUMO levels (ca. −4.98 V vs. Fc/Fc+ for 11H+). Using transient absorption spectroscopy and two-dimensional electronic spectroscopy with high temporal resolution, supported by DFT-based simulations, we have shown that protonation accelerates nonradiative decay by an order of magnitude. This behaviour is consistent with the Energy Gap Law, where the narrowing of the S0–S1 energy gap, rather than changes in the vibrational spectra, accounts for the faster decay rates. The optical properties of this new organic semiconductor molecule can therefore be finely tuned via pH, resulting in an unprecedented level of control over its absorption characteristics. This tuneability particularly in achieving low-lying LUMO levels and visible to NIR absorption has significant potential for diverse applications of organic semiconductors, especially in the photocatalysis field where red to near-infrared light absorption is highly desirable.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1Br and 1 have been deposited at the CCDC under 2383219 and 2383220-2383224 deposition codes.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. D. kindly acknowledges FRA2024 funded by the University of Trieste. J. D. acknowledges the RTDa PON ‘ricerca e innovazione’ 2014–2020. M. P. is the AXA Chair for Bionanotechnology (2016–2026). This work was supported by the University of Trieste, INSTM, and the Italian Ministry of Education MIUR (cofin Prot. 20228YFRNL). M. P. and M. M. acknowledge financial support from the European Innovation Council (PLANKT-ON, Plankton-Like Protocells for Artificial Photosynthesis Targeting Carbon-Neutral Energy Vectors, Path-finder-Open 2022, grant agreement n. 101099192). G. C. and M. M. acknowledge financial support by the European Union's NextGenerationEU Programme with the IPHOQS Infrastructure [IR0000016, ID D2B8D520, CUP B53C22001750006] “Integrated Infrastructure Initiative in Photonic and Quantum Sciences”.Notes and references
- J. Luke, E. J. Yang, C. Labanti, S. Y. Park and J.-S. Kim, Nat. Rev. Mater., 2023, 8, 839–852 CrossRef CAS.
- E. K. Solak and E. Irmak, RSC Adv., 2023, 13, 12244–12269 RSC.
- R. L. Mital and S. K. Jain, J. Chem. Soc. C, 1971, 1875–1878 RSC.
- Y. Nicolas, F. Allama, M. Lepeltier, J. Massin, F. Castet, L. Ducasse, L. Hirsch, Z. Boubegtiten, G. Jonusauskas, C. Olivier and T. Toupance, Chem. – Eur. J., 2014, 20, 3678–3688 CrossRef CAS PubMed.
- S. Yoshida, K. Kozawa and T. Uchida, Bull. Chem. Soc. Jpn., 1995, 68, 738–743 CrossRef CAS.
- F. Qiao, A. Liu, Y. Xiao, Y. P. Ou, J. Q. Zhang and Y. C. Sang, Microelectron. J., 2008, 39, 1568–1571 CrossRef CAS.
- T. Edvinsson, C. Li, N. Pschirer, J. Schöneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, J. Phys. Chem. C, 2007, 111, 15137–15140 CrossRef CAS.
- C. Di, J. Li, G. Yu, Y. Xiao, Y. Guo, Y. Liu, X. Qian and D. Zhu, Org. Lett., 2008, 10, 3025–3028 CrossRef CAS PubMed.
- Y. Nicolas, F. Castet, M. Devynck, P. Tardy, L. Hirsch, C. Labrugère, H. Allouchi and T. Toupance, Org. Electron., 2012, 13, 1392–1400 CrossRef CAS.
- L. Guo, J. Quinn, J. Wang, C. Guo, X. Li, J. Wang and Y. Li, Dyes Pigm., 2016, 132, 329–335 CrossRef CAS.
- T. Tanaka, T. Ashida and S. Matsumoto, Chem. Lett., 2011, 40, 573–575 CrossRef CAS.
- B. Huang, H. Kang, C. W. Zhang, X. L. Zhao, X. Shi and H. B. Yang, Commun. Chem., 2022, 5, 1–11 CrossRef PubMed.
- G. Gruntz, H. Lee, L. Hirsch, F. Castet, T. Toupance, A. L. Briseno and Y. Nicolas, Adv. Electron. Mater., 2015, 1, 1500072 CrossRef.
- A. Wannebroucq, G. Gruntz, J. M. Suisse, Y. Nicolas, R. Meunier-Prest, M. Mateos, T. Toupance and M. Bouvet, Sens. Actuators, B, 2018, 255, 1694–1700 CrossRef CAS.
- J. Zhu, X. Wang and H. Wang, J. Electron. Mater., 2020, 49, 4691–4696 CrossRef CAS.
- H. Li, B. C. K. Tee, J. J. Cha, Y. Cui, J. W. Chung, S. Y. Lee and Z. Bao, J. Am. Chem. Soc., 2012, 134, 2760–2765 CrossRef CAS PubMed.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS PubMed.
- H. E. Katz, J. Johnson, A. J. Lovinger and W. Li, J. Am. Chem. Soc., 2000, 122, 7787–7792 CrossRef CAS.
- H. E. Katz, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.-Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478–480 CrossRef CAS PubMed.
- H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501–510 CrossRef CAS PubMed.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- S. Jung, M. Albariqi, G. Gruntz, T. Al-Hathal, A. Peinado, E. Garcia-Caurel, Y. Nicolas, T. Toupance, Y. Bonnassieux and G. Horowitz, ACS Appl. Mater. Interfaces, 2016, 8, 14701–14708 CrossRef CAS PubMed.
- F. Sun, Y. Cui, Z. Zhao, L. Chen and Y. Xiao, Org. Lett., 2024, 26, 7875–7879 CrossRef CAS PubMed.
- E. P. Ivakhnenko, G. V. Romanenko, N. I. Makarova, A. Kovalenko, P. A. Knyazev, I. A. Rostovtseva, A. G. Starikov and V. I. Minkin, Dye. Pigm., 2020, 176, 108174 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- J. V. M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci and H. P. Hratchian, Gaussian 16, Revision A.03 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- F. Segatta, A. Nenov, D. R. Nascimento, N. Govind, S. Mukamel and M. Garavelli, J. Comput. Chem., 2021, 42, 644–659 CrossRef CAS PubMed.
- F. J. Avila Ferrer and F. Santoro, Phys. Chem. Chem. Phys., 2012, 14, 13549–13563 RSC.
- P. Ghosh, A. M. Alvertis, R. Chowdhury, P. Murto, A. J. Gillett, S. Dong, A. J. Sneyd, H. H. Cho, E. W. Evans, B. Monserrat, F. Li, C. Schnedermann, H. Bronstein, R. H. Friend and A. Rao, Nature, 2024, 629, 355–362 CrossRef CAS PubMed.
- J. Réhault, M. Maiuri, A. Oriana and G. Cerullo, Rev. Sci. Instrum., 2014, 85, 1–11 CrossRef PubMed.
- M. Liebel, C. Schnedermann, T. Wende and P. Kukura, J. Phys. Chem. A, 2015, 119, 9506–9517 CrossRef CAS PubMed.
- C. Fitzpatrick, J. H. Odhner and R. J. Levis, J. Phys. Chem. A, 2020, 124, 6856–6866 CrossRef CAS PubMed.
- Y. C. Wei, S. F. Wang, Y. Hu, L. S. Liao, D. G. Chen, K. H. Chang, C. W. Wang, S. H. Liu, W. H. Chan, J. L. Liao, W. Y. Hung, T. H. Wang, P. T. Chen, H. F. Hsu, Y. Chi and P. T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2383219–2383224. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc04626a |
| This journal is © The Royal Society of Chemistry 2025 |