
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTopological perturbation to a standard dehydrogenation catalyst, Pt3Sn†
William T. Laderera,
Xuance Jiangb,
Vojtech Vlcekb,
Harry W. T. Morgan a and
Anastassia N. Alexandrova*c
a and
Anastassia N. Alexandrova*c
aDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, USA
bDepartment of Chemistry and Biochemistry, Materials Department, University, of California, Santa Barbara, Santa Barbara, California 93106, USA
cDepartment of Materials Science and Engineering, University of California, Los Angeles, 90095, USA. E-mail: ana@chem.ucla.edu
First published on 25th June 2025
Abstract
Topological materials, which exhibit protected topological surface states (TSS) near the Fermi level, have been proposed to be good catalysts. Topological catalysis may be more prevalent than we suspect, and not limited to exotic new materials. Here we study a known dehydrogenation catalyst, Pt3Sn alloy, which happens to be a topological semimetal, and probe the participation of TSSs in catalytic dehydrogenation of methane catalyzed by this material. Through first principle modeling and detailed analysis of the electronic structure for topological and non-topological surfaces of Pt3Sn, we find that TSS get significantly altered by the binding of reaction intermediates, particularly H. However, this effect of TSS on the binding of the reagents is merely perturbative, as the majority of the adsorbate binding is achieved by not-surface-focused electronic states, located much deeper below the Fermi level. Therefore, the reaction energetics and selectivity are predominantly determined by electronic states other than TSS. The fact that TSS are available for the reagent binding does not alone guarantee that the catalysis is strongly driven by TSS. However, TSS are not to be ignored, as small changes in the energetics along the reaction profile can translate into substantial differences in the reaction rate. Hence, in our view, Pt3Sn – a topological material – is first and foremost a standard catalyst, with added topological features, and not purely a topological catalyst. Our results point at the need to carefully consider all the bonding effects at the topological material interface.
Introduction
Rarely in the field of catalysis do we have a bulk descriptor of a surface property. However, the presence of topological bulk bands in certain materials serves as an indicator of localized surface states, which have been reported to influence catalytic activity.1–4 These TSSs are characterized by their persistence against perturbations and high carrier mobility at the surface,5–8 ideal attributes of a catalytic material. Despite their potential applications, our understanding of TSS's imprint on the surface properties remains limited, particularly concerning their interactions with adsorbates. This uncertainty leads to multiple questions, for instance: if TSS are robust and unperturbed by the chemistry at the interface, then how can they participate in reagent binding? Or, if they do participate in the reagent binding, do they remain TSS? How TSSs affect electronic structure and how they might tune catalytic performance are not fully understood. The interplay between the topological properties of a material and the behavior of surface states under different reaction conditions presents an area for further research. Unraveling these mechanisms could lead to significant advancements in the design of more efficient catalysts and a deeper understanding of fundamental catalytic processes.A well-known catalytic material, Pt3Sn, has been reported to host topologically protected bulk bands.9,10 This provides us an opportunity to investigate a material that is an industrially ubiquitous dehydrogenation catalyst with many decades of research behind it.11 Understanding the topological properties of Pt3Sn can provide valuable insights into how TSSs influence its catalytic performance. The Pt3Sn(111) surface has been investigated for its catalytic properties in hydrogen evolution and the selective dehydrogenation of alkanes.12–15 Alloying Pt with Sn improves the selectivity toward the production of alkenes and reduces coke formation. This enhancement is attributed to the weakening of propene adsorption on Pt3Sn compared to pure Pt.12 Additionally, it is believed that the d-band center of Pt3Sn is closer to the Fermi level than most other Pt–Sn alloy surfaces, improving its catalytic performance.16
Concomitantly, the Pt3Sn(111) surface hosts unique topological features. The bulk topology of Pt3Sn is preserved by degeneracies enforced by time-reversal, inversion, and C3v point group symmetry.9 Consequently, surface cuts that do not preserve any of the symmetries that enforce band degeneracies will not retain TSSs, but if a subset of symmetries remain, then it is possible to observe TSSs.10,17 While we expect some differences in reactivity to come from the different surface structures and surface energies, in this study we aim to detect the role of the TSSs in catalysis, if any.
Our results are important for the search for new catalysts, with the topology of a surface facet in mind, i.e. aiming at promoting the formation of the desired surfaces if TSSs enhance the catalysis. As will be shown, topological surface cuts also need a specific computational treatment, different from the common practice, calling for the awareness of emergent surface states due to topology – a consideration that has not yet fully entered the catalysis research.
Methods
All mean-field calculations were performed using VASP 5.4 with the Perdew–Burke–Ernzerhof (PBE) functional and a density-dependent dispersion correction (dDsC).18–20 A plane-wave energy cutoff of 500 eV was used, along with an interatomic force convergence criterion of 0.01 eV Å−1 and a self-consistent field (SCF) energy convergence threshold of 1 × 10−6 eV. The PAW pseudopotentials used were from the VASP recommended PBE POTCAR set. Both bulk and slab models were converged using a uniform k-point mesh, which was optimized to ensure energy convergence with respect to k-point density. The optimized lattice parameter of the bulk was found to be 4.0113 Å, exhibiting a minimal deviation of 0.28% from the experimentally derived value.21Slab models were prepared using the publicly available python packages surfaxe22 and pymatgen.23 All slab models included a vacuum region of least 15 Å.
Electronic band structure calculations were performed using (1 × 1) surface supercells with 9 atomic layers, whereas adsorption energy calculations employed (2 × 2) supercells with 6 atomic layers. The (2 × 2) expansion corresponds to a 0.25 monolayer coverage of adsorbates. Adsorption energies were calculated using the following formula
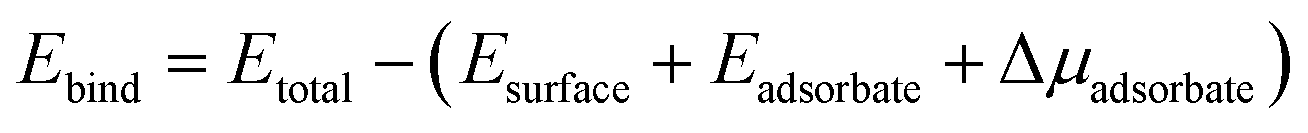
The description of individual electronic states has been based on the stochastic GW implementation using the StochasticGW code24–26 combined with an efficient random sampling of single-particle states to generate Dyson orbitals27 representing the electronic states observable e.g., via angle-resolve photoemission spectroscopy. The real-time stochastic many-body calculations employed a regular real-space grid with 128 × 128 × 64 points and the spacing of 0.41 × 0.29 × 0.34 Bohr. The electron/hole propagators were sampled by 512 random realizations of the time-dependent trajectories, and the renormalized Coulomb interactions were sampled by 10 vectors representing the density fluctuations induced by the injection of the quasiparticle, following the approaches described in ref. 24–26 and 28.
The states within interested energy window near the Fermi level are separated as a subspace.27 The quasiparticle Hamiltonians are diagonalized in the subspace with the Dyson orbitals constructed as the linear combination of DFT orbitals.
Results and discussion
Pt3Sn is a non-magnetic intermetallic compound that crystallizes in a face-centered cubic structure within the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. The unit cell contains 4 atoms, with Sn occupying the 1a = (0, 0, 0) maximal Wyckoff position and Pt occupying all
m space group. The unit cell contains 4 atoms, with Sn occupying the 1a = (0, 0, 0) maximal Wyckoff position and Pt occupying all 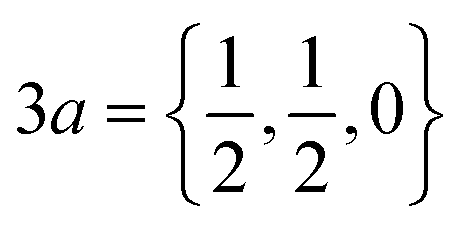 maximal Wyckoff positions. The predominant bonding interactions are of metallic nature while there is some covalency between the Pt and Sn atoms, leading to admixed valence and conductance bands with both d-type and p-type orbital character.
maximal Wyckoff positions. The predominant bonding interactions are of metallic nature while there is some covalency between the Pt and Sn atoms, leading to admixed valence and conductance bands with both d-type and p-type orbital character.
Pt3Sn is a known topological semimetal, exhibiting various topological features in its electronic structure. Band inversions in bulk Pt3Sn can occur when crystalline symmetries are broken, a hallmark of higher order topological materials. Due to the orthogonality condition of real space and reciprocal space, the breaking of real-space crystalline point group symmetries reduces the symmetry of special k-points in reciprocal space. For instance, breaking either C3v or C4v crystalline point group symmetry lifts symmetry imposed band degeneracies, Dirac points, along high-symmetry lines in reciprocal space, Λ(Γ ↔ R) and Δ(Γ ↔ X), respectively (see Fig. 1 and 2).
 | ||
| Fig. 1 (a) A Pt3Sn supercell with the (001), (111), and (110) surfaces highlighted. Sn atoms are depicted as dark blue spheres and Pt atoms are silver-grey spheres (b) depiction of the cubic reciprocal space lattice with corresponding surface projected high symmetry points. Symbols ΛD, ΔD, and TD denote the location of the Dirac points on the high symmetry lines Λ, Δ, and T, respectively. | ||
Pt3Sn is unusual in that it contains Dirac points that are sensitive to the breaking of time-reversal symmetry as well as crystalline symmetries, imbuing the material with a weak topologically insulating (WTI) phase as well as topological semimetallic behavior.9 As can be seen in Fig. 2, several essential degeneracies are broken due to the inclusion of spin–orbit coupling. For example, the conductance and valence bands meet at an intersection at Γ and R in the spinless case, which are given the irreducible representations Γ4+ and R4+. When spin–orbit is included, the k-point at Γ splits into a lower  and a higher
and a higher  state.
state.
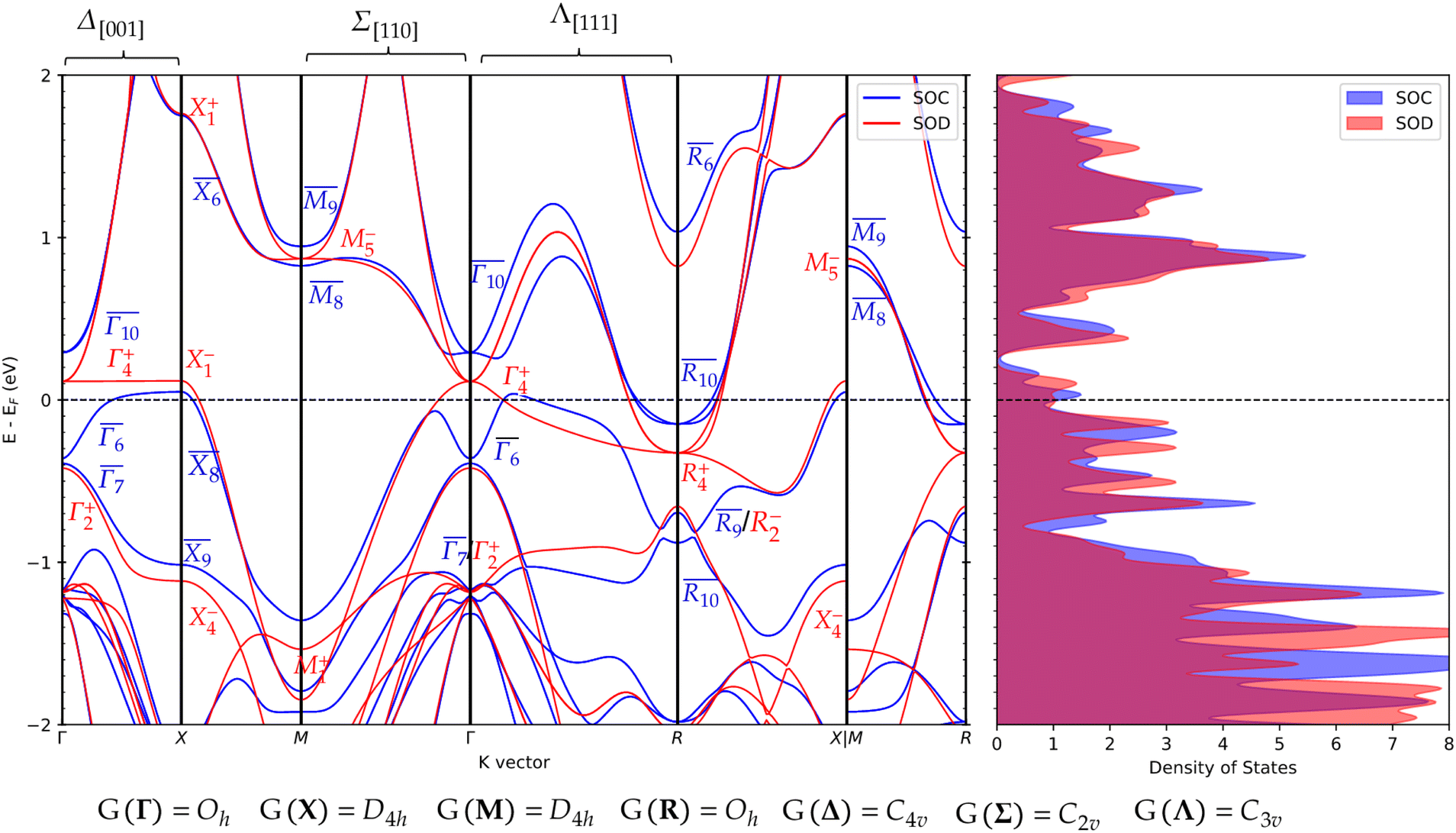 | ||
| Fig. 2 Projected band structure of bulk Pt3Sn with (in blue/SOC) and without (in red/SOD) spin orbit coupling. Bands are labeled with their irreducible representations at high symmetry points using BCS notation. Site symmetry labels for high symmetry points and lines are given at the bottom of the figure. | ||
A consequence of these splittings are TSSs – electronic states that exist on the surface of a material due to the inversion of nontrivial topological bulk bands caused by symmetry breaking. To investigate the emergence of TSSs in Pt3Sn, we construct several slab models to observe their electronic structure. We will investigate the (111), (110), and (001) terminations. Two of which, the (111) and (001), have been shown to host TSSs.9,17
Catalysts are typically used in a form of larger (supported) nanoparticles. As such, they are modeled via slab models, one facet at a time, and the most stable facet is most often used for modeling. We first determine if the of Pt3Sn surfaces in question are stable enough to occur on a nanoparticle and are thus accessible to adsorbates by creating a Wulff construction. We compute the surface energies for all unique terminations of every Miller plane investigated, and use the lowest-energy terminations in the construction. Both the (001) and the (111) have a pure Pt and mixed Pt–Sn truncation, with the latter having the lowest surface energy (see ESI Table 1†).
We find that the low index (111) and (001) surfaces are predominantly exposed, with the (111) surface comprising 96.5% of the surface area of a Pt3Sn nanoparticle, while the (001) covers the remaining exposures. Therefore, it is likely that the majority of surface chemistry will occur at the (111) surface, plus the contributions from the (001) surface and the particle edges. While the (110) surface is not competitive, we keep it in this study for comparison. (110) is a trivial cut that does not contain TSS. In this work we do not consider sites on particle edges that would correspond to higher-index surfaces, as they will be a minority. We recognize that they may be important for the overall catalytic activity. However, these states are not in focus since our goal is to find the role that TSS might play in catalysis, and pristine cuts not undergoing severe reconstructions offer the clearest identifiable TSS.
Next, we identify the TSSs on each surface using band structures and real-space visualizations of the wavefunctions. In topological insulators, the identification of TSSs can often be done by inspection due to their characteristic linear dispersion that spans the entire band-gap of the material.29,30 Identifying TSSs of topological semimetals is less straightforward and requires an understanding of the real-space location and extent of a band on a material's surface.
A surface state can be identified by using projected band structures, a method in which the local density of states (LDOS) is overlaid onto the individual bands. TSSs are highly localized surface bands and should, therefore, have a large contribution of projected density from Pt and Sn on only the first few atomic layers of the material. A distinct signature of a TSS can be seen in the projected band structure of the (001) surface. Fig. 3a presents a distinct surface (deep red) band that forms an upward-moving arc that crosses the Fermi level twice along the surface projected M–K–Γ high symmetry path.
 | ||
| Fig. 3 Projections of the slab models for the (a) Pt3Sn(001) surface band structure, (b) Pt3Sn(111) surface band structure, (c) real space representations of the surface state wavefunctions (inset solid square) with only the topmost layers of the slab shown, (d) Pt3Sn(110) surface band structure. | ||
A large projected density of surface states is only indicative of a band with high surface character, but does not address the extent of orbital overlap with bulk states or adjacent surface states which would rule out their topological origins. We can determine the localization of the surface states using real space representations of their wavefunctions. Fig. 3c depicts the wavefunctions of the (001) and (111) surface bands at Γ, with yellow and blue representing their phase. The spatial extent of the wavefunctions shows that they are entirely localized to the top four layers of the surface. This indicates that these are most likely TSSs as they have no bulk contamination.
As for the (110) surface, several bands near the Fermi level do show surface character but there is no consistent evidence in the literature that the bands are of topological origin. We also note that the (110) slab model only maintains C2v and mirror symmetry elements. Therefore, the (110) surface does not preserve the symmetries required to maintain the symmetry-enforced degeneracies that imbue Pt3Sn with topological protection.
Hence, we have established which of the three surfaces of Pt3Sn maintain TSSs. These surface bands have been claimed to be useful for catalysis because the protected states can participate in the activation of the bound small molecules, and remain robust to chemical and physical perturbations at the interface.3 However, in our view, the fact that a material is topological and is a catalyst does not yet guarantee that it is a topological catalyst, i.e. that it uses TSSs to drive the catalysis. Since Pt3Sn is a known catalyst and is apparently a topological material, we now aim to find out whether topological states play any role in its catalytic activity. We explore the influence that TSSs have on (i) hydrogen binding (a relevant descriptor for such reactions as (de)hydrogenation and hydrogen evolution), and (ii) the dehydrogenation of methane, to investigate if the catalytic mechanisms are affected.
We study the binding of hydrogen on the (1 × 1) surfaces, to understand the changes in the electronic structure of each termination upon adsorption. We systematically evaluate the hydrogen binding sites on each surface to identify the most favorable adsorption location. Next, we calculate the band structure of the surfaces, both before and after hydrogen adsorption, and project the local density of states (LDOS) onto the bands to determine the contributions of individual atoms to the overall electronic structure of the slab model.
All three surfaces have atop, bridging, and hollow binding sites for adsorbates, but the Pt3Sn(111) differs in that it possesses two inequivalent 3-fold hollow Pt sites. The two sites are commonly referred to as the hcp and fcc sites, depending on the presence or absence of a Sn atom just below the threefold arrangement of Pt atoms.
The 3-fold hollow site on Pt3Sn(111) is the most favorable hydrogen binding site, similar to the pristine Pt(111) and the Pt3Sn/Pt(111) surfaces. However, unlike Pt(111) and Pt3Sn/Pt, where hydrogen binding prefers the fcc site, on Pt3Sn(111), hydrogen binds more strongly to the hcp site. The specific hollow site preference of the Pt3Sn(111) has been claimed to be either due to the d-band center of the fcc site being lower than that of the hcp or the coverage of TSSs in the fcc site, leading to hydrogen binding with the non-surface focused bands of Pt3Sn.13,17
We observe that the addition of an adsorbate on Pt3Sn(111) significantly alters the surface band structure. After hydrogen is bound to the hcp site, the surface-like conduction band of Pt3Sn(111) appears to split along the high symmetry path M–K. The splitting causes a lowering of the conduction band energy and shows a greater surface character than before binding.
Similarly, the Pt3Sn(001) (1 × 1) surface has a strong response to the addition of an adsorbate. Upon hydrogen binding to the preferred site, the distinct surface bands shift below the Fermi level, to around −0.5 eV, as can be seen from the projected band structure (deep red in Fig. 4a). The same band also has a significant contribution of the hydrogen orbital indicating the formation of a chemical bond, between Pt-6dz2 within TSS and H 1s (Fig. 4b and c). The band remains localized to the first few surface layers. This indicates that the surface does not lose topological protection after the binding of a single hydrogen atom. However, TSSs get strongly perturbed by the adsorption.
 | ||
| Fig. 4 Comparison of the surface, hydrogen, and real space projected bands of the topological surfaces of Pt3Sn. | ||
To demonstrate that H is specifically coupling with the TSSs, we can visualize hydrogen on a (2 × 2) surface of Pt3Sn(001) using real space projections of the wavefunction. In Fig. 5, Γ shows no overlap with the Pt and H atom. In comparison, at M there is relatively good overlap between the two atoms, demonstrating this momentum dependent bonding behavior with the TSS on the Pt3Sn(001) surface. This result coincides with the band structure of the (1 × 1) supercell of the bound Pt3Sn(001) where the surface band drops below the Fermi level along the M–K path but remains unchanged at Γ. We can therefore conclude that the (2 × 2) supercell behaves similarly to that of the (1 × 1).
 | ||
| Fig. 5 Real space representation of the pristine and hydrogen adsorbed Pt3Sn(001) (2 × 2) supercell surface real-valued wavefunction at high symmetry points Γ and M. | ||
The lowering of the surface bands on both the Pt3Sn(001) and Pt3Sn(111) surfaces is due to charge transfer between the hydrogen atom and the surface bands. In addition, we observe the typical covalent Pt–H at much more negative values (see Fig. 8), which arises from hydrogen coupling to the more extended Pt d-states exposed at the surface, we also observe projected hydrogen density in the same bands that contain significant surface character near the Fermi level. This suggests multiple binding modes for hydrogen on these surfaces, with one involving the conventional, delocalized Pt d-orbitals forming the typical covalent bond, and another involving coupling to the TSSs. The hydrogen 1s orbital interacts more strongly with the lower energy Pt-d states than with the TSSs, as the latter are composed of a mixture of Pt-d and Sn-p character. While the H 1s Pt 6d interaction remains the dominant bonding mechanism, coupling to the TSSs may offer an additional, but weak, stabilizing contribution.
In comparison to the two topological surfaces, the Pt3Sn(110) surface does not display many remarkable features before or after binding. Both the projections of surface and hydrogen density show a multitude of mixed surface-bulk and hydrogen-bulk interactions, indicating that hydrogen is not interacting with highly localized surface states.
To check the many body perturbation effect on the TSSs and H coupling, we perform a large scale many-body perturbation theory calculation using the GW approximation. We employ random sampling approaches which enable treating large scale systems and specifically investigate selected states; we target the TSS state and neighboring 15 orbitals within a 0.3 eV energy window. The GW approximation indicates a significant around 1.5 eV shift down below the Fermi level (which is consistent with the typical amount of quasiparticle correction).31 The further analysis on Dyson orbital27 of this state shows that the topological surface state is robust with the many-body correction. The Dyson orbital is almost unchanged (Fig. 6a and b) compared with DFT orbitals (Fig. 5f). The z-direction line profile of charge density as shown in Fig. 6c further confirms that the Dyson orbital (red) is also localized at the surface similar to the DFT orbital (blue). Furthermore, we compare the 16 DFT orbitals with Dyson orbitals and conclude that most DFT orbitals rehybridize as the quasiparticle Hamiltonian is not diagonal in the mean-field (Kohn–Sham) basis, but we observe that TSS strongly resembles the DFT orbital (the orbital overlap is near unity).
 | ||
| Fig. 6 The side view (a) and top view (b) of the H atom and Pt3Sn surface atoms dominate Dyson orbital wavefunction from GW quasiparticle Hamiltonian diagonalization. (c) The z direction line profile of the charge density of the hybrid state. Blue: DFT orbital; red: Dyson orbital. | ||
Therefore, the many-body GW correction does not alter the topological properties and the interaction between topological states and surface molecules. The topologically protected surface states are localized at the surface even when the many-body effect is considered.
We now consider the energetics of dehydrogenation of methane on each surface of Pt3Sn. We aim to investigate the participation of surface states in dehydrogenation. Principally, we understand that the inherent activity of each termination is quite different due to their differing surface energies, and it is not possible to completely disentangle the effect of TSS from all other geometric and electronic effects influencing reaction energetics. Nevertheless, we aim to identify interactions with TSS, which may differentially affect reaction intermediates and thus participate in the catalysis.
Our computed electronic binding energies are very similar to a previous study,15 with only minor deviations. We attribute these deviations to the difference in functional and dispersion correction as there is a systematically stronger binding interaction between the adsorbates and the surfaces.32
As shown in Table 1, the electronic binding energies vary moderately across the three Pt3Sn surfaces. One might expect a large difference in the binding energies between Pt3Sn(111), the most stable facet, and the two higher energy surfaces. Yet, the binding energies for CH4, CH3, and CH2 are relatively similar. In contrast, the binding energies of CH and H differ more substantially. For instance, H binds more strongly to Pt3Sn(111) and Pt3Sn(001) than to Pt3Sn(110), with differences of −0.445 eV and −0.460 eV, respectively. These differences likely reflect changes in both the binding site geometry and the underlying electronic structure, making it difficult to disentangle geometric effects from intrinsic surface reactivity.
| Adsorbate | Pt3Sn(111) | Pt3Sn(001) | Pt3Sn(110) | |||
|---|---|---|---|---|---|---|
| Site | Ebind | Site | Ebind | Site | Ebind | |
| CH4 | Phys | −0.191 | Phys | −0.157 | Phys | −0.165 |
| CH3 | Pt atop | −2.770 | Pt atop | −2.863 | Pt atop | −2.451 |
| CH2 | Pt bridge | −4.823 | Pt–Sn bridge | −4.470 | Pt bridge | −4.524 |
| CH | hcp | −6.809 | Pt–Sn hollow | −5.704 | Pt bridge | −5.944 |
| H | hcp | −0.708 | Pt atop | −0.723 | Pt bridge | −0.263 |
Coincidentally, the preferred binding location for CH3 is the Pt-atop site on each surface. This allows us to see that the adsorbate binds more similarly to the topological (001) and (111) surfaces than the non-topological (110) surface.
It is also interesting to note that increasing Sn content at the surface tends to weaken the binding of CH. On the Pt-rich Pt3Sn(111) surface, which has a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Pt:Sn ratio, CH binds strongly at the hcp site with an electronic binding energy of −6.81 eV. In comparison, the Sn-rich Pt3Sn(110) surface, with a Pt:Sn ratio of 1:1, binds at a Pt-bridging site with a significantly weaker binding energy of −5.94 eV. This is likely due to the lack of highly coordinated Pt sites for CH on the Pt3Sn(110).
:1 Pt:Sn ratio, CH binds strongly at the hcp site with an electronic binding energy of −6.81 eV. In comparison, the Sn-rich Pt3Sn(110) surface, with a Pt:Sn ratio of 1:1, binds at a Pt-bridging site with a significantly weaker binding energy of −5.94 eV. This is likely due to the lack of highly coordinated Pt sites for CH on the Pt3Sn(110).
These differences in binding strength and site preference, governed by a combination of surface composition, geometry, and topologies, have direct implications for reaction energetics. To explore this further, we calculated the dehydrogenation pathway of methane. Fig. 7, contains the calculated reaction profile of the dehydrogenation of methane on the three surfaces of Pt3Sn. A thermodynamic correction of −0.647 eV for each hydrogen atom and −1.765 eV for each CH4 molecule was applied to account for desorbed gas phase species at operating conditions of 1 bar and 900 K.
 | ||
| Fig. 7 Reaction profile of the dehydrogenation of methane on Pt3Sn(111), Pt3Sn(001), and Pt3Sn(110) at 1 barr and 900 K. Visualizations of the global minimum structures are given on the right hand side. Asterisks above adsorbates indicate a bound species, asterisks on their own denote a bare slab. | ||
First, we follow the reaction pathway of Pt3Sn(111). Overall, the reaction is an endergonic process, with the total change in the approximate free energy being roughly 1.29 eV. Without any thermodynamic correction, this process is slightly endothermic, but at lower pressure and temperatures it becomes spontaneous (see ESI†). At first, methane becomes weakly associated to the surface near the threefold Pt-hcp hollow site. Each additionally deprotonated intermediate maintains a tetrahedral coordination environment around the central carbon atom. This requires a migration from a Pt-atop position, to a bridging Pt–Pt site, and finally a threefold Pt-hcp site, where the Pt atoms compensate for a missing hydrogen atom coordination.
The energetics of the reaction profile of the topological surfaces Pt3Sn(001) and Pt3Sn(111) follow one another closely for the first few steps. The bifurcation of the paths begins at the second deprotonation which is slightly less favorable on Pt3Sn(001). Each successive step in the profile becomes significantly less favorable for Pt3Sn(001).
In comparison, Pt3Sn(110) behaves somewhat differently than the other surfaces investigated. The first deprotonation of methane is significantly more endothermic than on other surfaces. Even after successive deprotonations, the reaction continues to be unfavorable, regardless of entropic correction, indicating that this surface is unsuitable for dehydrogenation.
The interaction of the adsorbates and each surface using density of states plots shown in Fig. 8. As established previously, the surface states must be situated near the Fermi level and therefore density of states of the adsorbate near this energetic region would indicate that they could interact with TSSs. We have also established that the energy of the surface states are modified after binding, therefore we can use the PDOS of the adsorbate at various distances away from the surface to track both the surface states and the adsorbate before and after binding.
 | ||
| Fig. 8 Comparison of the projected adsorbate density of states on the topological surfaces of Pt3Sn at different surface-adsorbate distances. | ||
In Fig. 8, the yellow peaks between −6 eV and −8 eV correspond to the bound states of the adsorbates. Each adsorbate on all surfaces binds to orbitals well below the Fermi level, as is established in the field of catalysis. Therefore, it appears that TSSs, at least in Pt based catalysts, are not the principal states to which the adsorbates bind. This does not mean that TSSs are not involved in the binding process. As mentioned previously, the binding of H on Pt3Sn(001) shows a very large energetic shift of the surface state, exhibiting coupling of the surface states and adsorbate.
When CH3 is far from both Pt3Sn(111) and Pt3Sn(001), there appears to be some adsorbate density in the energetic window in which one would expect TSSs. Yet, at equilibrium geometry, there is little to no adsorbate density. Instead, we see a large peak around −5.5 eV, indicating that CH3 does not meaningfully interact with surface states and only with lower lying bulk states on either topological surface. This behavior is almost identical for each C bearing adsorbate in this study, indicating that such species do not participate in any coupling with TSSs.
In comparison to CH3, H shows some density above and below the Fermi level before and after binding, but still within the energetic window of ± 0.5 eV in which TSSs are expected to appear. This continues to support the conjecture that H can interact with the pristine TSSs upon binding and that we can observe the energetic reorganization of the density of states due to this interaction.
We also construct the Dyson orbitals for CH and CH3 adsorbates in the energy range from −0.6 to −0.2 eV as shown in Fig. 9. To test whether an adsorbate couples to TSSs we require the corresponding wavefunction to be simultaneously localized on the adsorbate and on the topmost Pt/Sn layers. Unlike the hydrogen adsorbed Pt3Sn surface, there is no evidence of such joint localization—and hence no coupling between the CH and CH3 adsorbates and the TSSs. This is illustrated in detail in Fig. 9 showing two sets of selected single-particle states as obtained from the mean-field DFT calculation and from the diagonalization of the quasiparticle Hamiltonian (in the GW approximation). Fig. 9a illustrates a CH-dominated state that significantly couples with the bulk states when the GW many-body perturbation theory is applied (Fig. 9b). Such an observation is somewhat surprising as DFT tends to overdelocalize the single-particle states compared to the true Dyson orbitals, yet the strong dynamical renormalization here leads to even more pronounced spatial “leakage” to the substrate. Similarly, Fig. 9c and d show that the CH3 dominated states do not localize on the surface (in DFT or ion many-body perturbation theory).
 | ||
| Fig. 9 Side view of the (a) DFT and (b) dyson orbitals of CH on top of Pt3Sn(111). The side view of the (c) DFT and (d) Dyson orbitals of CH3 on top of Pt3Sn(111). The insets show enlarged views of the adsorbates. | ||
Conclusions
We have shown that TSSs will be accessible to adsorbates on the exposed surfaces of Pt3Sn. The Wulff construction demonstrates the preferential exposure of Pt3Sn(111) and Pt3Sn(001), both of which host TSSs. The exposure of TSSs provides the opportunity for chemical reactions to be influenced by TSSs.We have also demonstrated how TSSs on Pt3Sn behave in the presence of adsorbates. Principally, the effect is weak, in comparison to that of a conventional covalent bond, and would not be considered the dominant binding mode between the adsorbate and the surface. We have shown that a small number of states of H do intrude into the high lying surface bands near the Fermi level. This admixture perturbs these bands enough that they may drop below the Fermi level and create minor alterations in the energetics of binding. As a consequence, the entire dehydrogenation profile must shift to reflect this energetic alteration as each successive deprotonation contributes to the total energy of the reaction pathway.
Finally, we have shown that TSSs persist on Pt3Sn beyond the one-particle approximation before and after binding. These results show that DFT captures the topological nature of this material well.
This study raises a possibility that successful topological catalysts are first and foremost trivial catalysts, with a topological perturbation. To put these results in a broader perspective, small changes of adsorption energetics may mean quite a bit for the reaction rate, and bring the material closer to or further way from the optimal binding energy (aka in a catalytic volcano). Hence, the effect of TSS, while merely perturbative to the main bonding interaction, can still be important for finetuning of the reaction energetics. Understanding the presence and role of TSSs at specific material's facets can inform decisions regarding catalyst selection and preparation methods. Therefore, it is imperative that practitioners understand the conditions in which a particular catalyst undergoes a topological phase transition and the surfaces that can host the resultant TSSs.
Data availability
All data is included in the ESI.†Author contributions
W. T. L. performed all DFT calculations. X. J. performed GW calculations. H. W. T. M. assisted with the analysis of the electronic structure. V. V. supervised research in UCSB. A. N. A. supervised research at UCLA and is responsible for research conception and planning. The first version of the manuscript was written by W. T. L. and A. N. A. All authors contributed to manuscript writing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the and DOE BES grant DE-SC0024987 to A. N. A. and V. V., and the Brown Foundation Award 1168 to A. N. A. We thank NERSC for computational resources, and Guorong Weng for helpful discussions.References
- H. Chen, W. Zhu, D. Xiao and Z. Zhang, CO Oxidation Facilitated by Robust Surface States on Au-Covered Topological Insulators, Phys. Rev. Lett., 2011, 107, 056804, DOI:10.1103/PhysRevLett.107.056804.
- R. Xie, T. Zhang, H. Weng and G. L. Chai, Progress, Advantages, and Challenges of Topological Material Catalysts, Small Sci., 2022, 2, 202100106, DOI:10.1002/smsc.202100106.
- G. Li and C. Felser, Heterogeneous catalysis at the surface of topological materials, Appl. Phys. Lett., 2020, 116, 070501, DOI:10.1063/1.5143800.
- L. Wang, Y. Yang, J. Wang, W. Liu, Y. Liu, J. Gong, G. Liu, X. Wang, Z. Cheng and X. Zhang, Excellent catalytic performance toward the hydrogen evolution reaction in topological semimetals, EcoMat, 2023, 5, e12316, DOI:10.1002/eom2.12316.
- L. Fu, C. L. Kane and E. J. Mele, Topological Insulators in Three Dimensions, Phys. Rev. Lett., 2007, 98, 106803, DOI:10.1103/PhysRevLett.98.106803.
- J. E. Moore, The birth of topological insulators, Nature, 2010, 464, 194–198, DOI:10.1038/nature08916.
- L. Fu, Topological Crystalline Insulators, Phys. Rev. Lett., 2011, 106, 106802 CrossRef.
- J. Wang and S. C. Zhang, Topological states of condensed matter, Nat. Mater., 2017, 16, 1062–1067, DOI:10.1038/nmat5012.
- M. Kim, C. Z. Wang and K. M. Ho, Coexistence of type-II Dirac point and weak topological phase in Pt3Sn, Phys. Rev. B, 2017, 96, 205107, DOI:10.1103/PhysRevB.96.205107.
- D. Zhang, W. Jiang, H. Yun, O. J. Benally, T. Peterson, Z. Cresswell, Y. Fan, Y. Lv, G. Yu, J. G. Barriocanal, P. W. Swatek, K. A. Mkhoyan, T. Low and J. P. Wang, Robust negative longitudinal magnetoresistance and spin–orbit torque in sputtered Pt3Sn and Pt3SnxFe1-x topological semimetal, Nat. Commun., 2023, 14, 4151, DOI:10.1038/s41467-023-39408-2.
- U. Bardi, L. Pedocchi, G. Rovida, A. N. Haner and P. N. Ross, Study of the Pt3Sn(100), (111) and (110) Single Crystal Surfaces by LEISS and LEED, Fundamental Aspects of Heterogeneous Catalysis Studied by Particle Beams, ed. Brongersma HH and Santen RA van, Springer US, Boston, MA, 1991, pp. 393–397, DOI:10.1007/978-1-4684-5964-7_28.
- L. Nykanen and K. Honkala, Density Functional Theory Study on Propane and Propene Adsorption on Pt(111) and PtSn Alloy Surfaces, J. Phys. Chem. C, 2011, 9578–9586, DOI:10.1021/jp1121799.
- A. Hook, J. D. Massa and F. E. Celik, Effect of Tin Coverage on Selectivity for Ethane Dehydrogenation over Platinum–Tin Alloys, J. Phys. Chem. C, 2016, 120, 27307–27318, DOI:10.1021/acs.jpcc.6b08407.
- A. Hook and F. E. Celik, Predicting Selectivity for Ethane Dehydrogenation and Coke Formation Pathways over Model Pt–M Surface Alloys with ab Initio and Scaling Methods, J. Phys. Chem. C, 2017, 121, 17882–17892, DOI:10.1021/acs.jpcc.7b03789.
- J. Nam and F. E. Celik, Effect of Tin in the Bulk of Platinum–Tin Alloys for Ethane Dehydrogenation, Top. Catal., 2020, 63, 700–713, DOI:10.1007/s11244-020-01297-w.
- M. L. Yang, Y. A. Zhu, X. G. Zhou, Z. J. Sui and D. Chen, First-Principles Calculations of Propane Dehydrogenation over PtSn Catalysts, ACS Catal., 2012, 2, 1247–1258, DOI:10.1021/cs300031d.
- M. C. Jiang, G. Y. Guo, M. Hirayama, T. Yu, T. Nomoto and R. Arita, Efficient hydrogen evolution reaction due to topological polarization, Phys. Rev. B, 2022, 106, 165120, DOI:10.1103/PhysRevB.106.165120.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- S. N. Steinmann and C. Corminboeuf, A generalized-gradient approximation exchange hole model for dispersion coefficients, J. Chem. Phys., 2011, 134, 044117, DOI:10.1063/1.3545985.
- S. N. Steinmann and C. Corminboeuf, Comprehensive Benchmarking of a Density-Dependent Dispersion Correction, J. Chem. Theory Comput., 2011, 7, 3567–3577, DOI:10.1021/ct200602x.
- M. Ellner, Zusammenhang zwischen strukturellen und thermo-dynamischen eigenschaften bei phasen der kupfer-familie in T10-B4-systemen, J. Less-Common Met., 1981, 78, 21–32, DOI:10.1016/0022-5088(81)90140-5.
- K. Brlec, D. W. Davies and D. O. Scanlon, Surfaxe: Systematic surface calculations, J. Open Source Softw., 2021, 6, 3171, DOI:10.21105/joss.03171.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Python Materials Genomics (pymatgen): A robust, opensource python library for materials analysis, Comput. Mater. Sci., 2013, 68, 314–319, DOI:10.1016/j.commatsci.2012.10.028.
- D. Neuhauser, Y. Gao, C. Arntsen, C. Karshenas, E. Rabani and R. Baer, Breaking the Theoretical Scaling Limit for Predicting Quasiparticle Energies: The Stochastic GW Approach, Phys. Rev. Lett., 2014, 113, 076402, DOI:10.1103/PhysRevLett.113.076402.
- V. Vlček, E. Rabani, D. Neuhauser and R. Baer, StochasticGWCalculations for Molecules, J. Chem. Theory Comput., 2017, 13, 4997–5003, DOI:10.1021/acs.jctc.7b00770.
- V. Vlček, W. Li, R. Baer, E. Rabani and D. Neuhauser, Swift GW beyond 10,000 electrons using sparse stochastic compression, Phys. Rev. B, 2018, 98, 075107, DOI:10.1103/PhysRevB.98.075107.
- A. Canestraight, X. Lei, K. Z. Ibrahim and V. Vlcek, Efficient Quasiparticle Determination beyond the Diagonal Approximation via Random Compression, J. Chem. Theory Comput., 2024, 20, 551–557, DOI:10.1021/acs.jctc.3c01069.
- M. Romanova and V. Vlcek, Decomposition and embedding in the stochastic GW selfenergy, J. Chem. Phys., 2020, 153, 134103, DOI:10.1063/5.0020430.
- H. Zhang, C. X. Liu, X. L. Qi, X. Dai, Z. Fang and S. C. Zhang, Topological insulators in Bi2Se3, Bi2Te3 and Sb2Te3 with a single Dirac cone on the surface, Nat. Phys., 2009, 5, 438–442, DOI:10.1038/nphys1270.
- M. Z. Hasan and C. L. Kane, Colloquium: Topological insulators, Rev. Mod. Phys., 2010, 82, 3045–3067, DOI:10.1103/RevModPhys.82.3045.
- A. K. Watkins, D. Johrendt, V. Vlcek, S. D. Wilson and R. Seshadri, Fidelity and variability in the interlayer electronic structure of the kagome superconductor CsV3, Phys. Rev. Mater., 2024, 054204, DOI:10.1103/PhysRevMaterials.8.054204.
- S. Gautier, S. N. Steinmann, C. Michel, P. Fleurat-Lessard and P. Sautet, Molecular adsorption at Pt(111). How accurate are DFT functionals? en, Phys. Chem. Chem. Phys., 2015, 17, 28921–28930, 10.1039/C5CP04534G.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02518d |
| This journal is © The Royal Society of Chemistry 2025 |