 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluation of the formation pathways of cocrystal polymorphs in liquid-assisted syntheses†
Franziska
Fischer
ab,
Gudrun
Scholz
b,
Sigrid
Benemann
a,
Klaus
Rademann
b and
Franziska
Emmerling
*a
aBAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Str. 11, 12489 Berlin, Germany. E-mail: franziska.emmerling@bam.de
bDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
First published on 27th May 2014
Abstract
The synthesis of the polymorphic cocrystal caffeine:anthranilic acid was investigated to obtain a better understanding of the processes leading to the formation of different polymorphic forms. In the case of these cocrystal polymorphs synthesized by liquid-assisted grinding a distinct influence of the dipole moment of the solvent was found. A pre-coordination between the solvent molecules and the caffeine:anthranilic acid cocrystal could be identified in the formation of form II. In the case of form II the solvent can be regarded as a catalyst. The formation pathway of each polymorph was evaluated using synchrotron X-ray diffraction.
Introduction
A key challenge in pharmaceutics is to evoke the best therapeutic effect of a drug and to improve its physicochemical properties especially water solubility and bioavailability. Polymorphs, cocrystals, and polymorphs of cocrystals of a given active pharmaceutical ingredient (API) exhibit different structures leading to different, often enhanced, physicochemical properties.1–3 The term “cocrystal” is still under debate. Here we define cocrystals as a two- or more component crystalline phase consisting of uncharged organic compounds, which interact via intermolecular forces. A pharmaceutical cocrystal consists of at least one API and a coformer, typically a small organic molecule.4–8 Since the formation of a cocrystal can change the physicochemical properties of an API, the crystallization processes, synthesis pathways and properties of these crystalline forms are of high interest in chemistry and pharmaceutics. Among the numerous studies in the field of cocrystals only a few examples of polymorphs of cocrystals are known.9–14 Examples of this phenomenon are caffeine cocrystals. Caffeine is used in the pharmaceutical treatment of migraine and primary apnea and it is a drug for all seasons.15 It represents an ideal API model for cocrystal formation.16–19 Due to the weakly basic nature of caffeine, its tendency for cocrystallization is higher than that for formation of a salt. Two polymorphic forms and one hydrate form of caffeine were characterized.20–24Trask prepared one of the first caffeine containing cocrystals with glutaric acid as a coformer using the so called liquid-assisted grinding (LAG) method.14 In this milling process a small amount of solvent is added to the solid reactant mixture to enhance the reaction rate. Many more investigations followed using other dicarboxylic acids including maleic acid, malonic acid and oxalic acid as coformers.25 Several caffeine cocrystals and cocrystal solvates with diverse coformers were described.26–32 Interest in the formation of caffeine cocrystals increased intensively and synthesis methods were developed in a targeted manner. Recently, Eddleston introduced cocrystallization by freeze-drying and a new form of a caffeine:theophylline cocrystal was identified.12 Bučar pushed forward the application of heteronuclear seeding and described the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cocrystal caffeine:benzoic acid. The cocrystal could not be obtained by common synthesis techniques such as grinding or from solution.27
:1 cocrystal caffeine:benzoic acid. The cocrystal could not be obtained by common synthesis techniques such as grinding or from solution.27
Despite the numerous known cocrystals of caffeine, there are only a few cocrystals showing the phenomenon of polymorphism. Trask reported that by changing the polarity of the solvent in solvent-drop grinding synthesis the polymorphic form of the cocrystal of caffeine with glutaric acid can be controlled.14 Moreover, Ghosh and Trask described that the caffeine cocrystals with 4-chloro-3-nitrobenzoic acid and trifluoroacetic acid are able to crystallize in two different forms.11,33 Two polymorphic forms were also found by Schultheiss during cocrystallization of caffeine with pterostilbene as a cocrystal former.34
Very recently, Jones published a caffeine:anthranilic acid (cf:ana) cocrystal revealing a high diversity of crystal forms: two polymorphs, two hydrates, and seven solvates. The polymorphic forms were prepared by LAG. Finding a mechanistic explanation for the formation of polymorphs I and in particular II still remains challenging.9 Because of its structural diversity, the equimolar cocrystal cf:ana is an ideal model system for investigations on the formation processes. In this study, we show that the polymorphic forms of the cocrystal can be synthesized by LAG in a controlled way. X-ray diffraction investigations unambiguously allow distinguishing the formation of the different polymorphs, while Raman and solid-state NMR (ssNMR) studies give detailed insight into the hydrogen bond network.
To understand the influence of the solvent in the formation of a distinct polymorph in a LAG process, different solvents were added during the milling process. Based on these experiments, the important role of the solvent could be clarified.
Results and discussion
Prior to the interpretation of liquid-assisted grinding experiments the possible salt formation has to be excluded. Therefore, Raman and ssNMR spectra of the reactants and the cocrystals were collected (Fig. 1).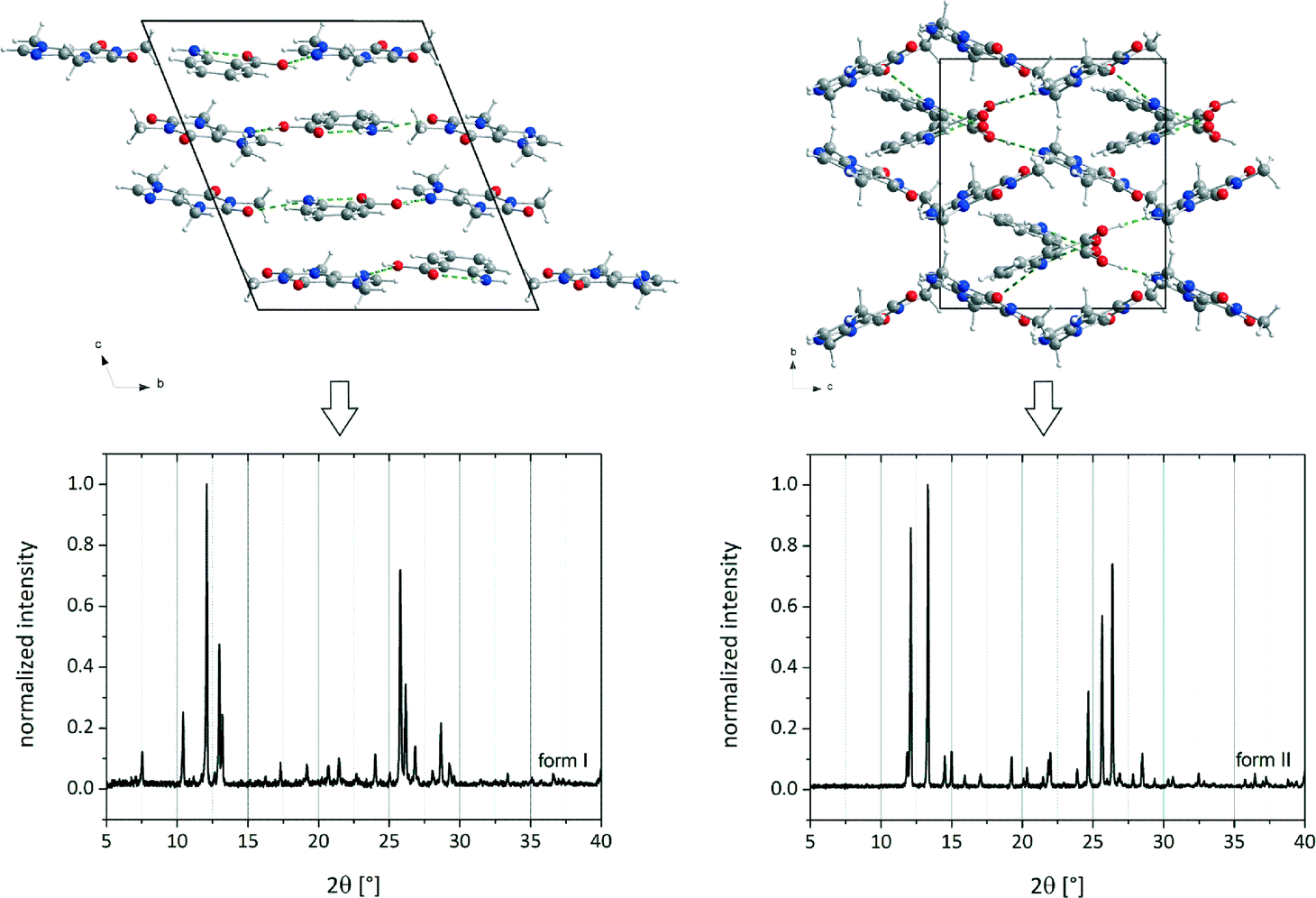 | ||
| Fig. 1 Crystal structures and PXRD patterns of cf:ana form I (left) and form II (right).9 | ||
The Raman spectra of both the polymorphic cocrystal forms and the reactants were recorded. Since the hydrogen-bonding arrangements of form I and form II are equivalent, the chemical environment of the molecules in the cocrystal polymorphs is identical, and the Raman spectra are very similar as shown in Fig. 2.
 | ||
| Fig. 2 Raman spectra of ana, the cf:ana cocrystal polymorphs, and caffeine. | ||
The Raman bands of caffeine at 1654 cm−1 and 1698 cm−1 (stretching vibration of carbonyl groups) and of anthranilic acid (ana) at 1245 cm−1 (deformation vibration of the amino group) and 1373 cm−1 (stretching vibration of the carboxylate group) were analyzed to determine if there is a complete shift of the acidic proton of ana towards caffeine.35,36 The carbonyl band of the caffeine molecule at 1654 cm−1 shifts by 12 cm−1 (form I) and 2 cm−1 (form II) to higher wavenumbers, whereas the second carbonyl band appears at 1665 cm−1 in form I and 1658 cm−1 in form II. These small Raman shifts reveal that the caffeine molecules are not highly affected in the conformation of the cocrystal. The amino group of ana is also not influenced, because the Raman deformation vibration band at 1245 cm−1 slightly shifts by 5 cm−1 (form I) and 4 cm−1 (form II). A strong shift of the symmetrical stretching band of the carboxylate group can be observed. In ana the band appears at 1373 cm−1, while in both polymorphic forms of the cocrystal the band is located at 1333 cm−1. This strong shift can be attributed to the change of the chemical environment concerning the acid group of ana. In pure ana a homosynthon between the carboxylic acid groups is formed,37 and the protons of both carboxylic acids are delocalized. In contrast, in the cocrystal the proton of the acidic group of ana is involved in the hydrogen bond to a carbonyl oxygen atom of caffeine and is not bridged in such a strong manner. It is connected to the oxygen atom of ana and the cocrystal consists only of neutral molecules.
For further characterization the polymorphs of the cocrystal were investigated via ssNMR. Fig. 3 shows the spectra of both polymorphs along with the superimposed spectra of the two reactants measured under identical conditions. The methyl groups and the tertiary carbon of caffeine have resonance absorptions at 3.5 ppm and 7.8 ppm, respectively. These signals superimpose with the ssNMR signal of ana at 6.9 ppm, which is attributed to the aromatic protons and the amino groups of ana. The proton of the carboxylic group shows a signal at 17.0 ppm. This distinct shift reveals that the acidic proton of ana is bridged in a strong manner because of the crystal packing in pure ana as shown in the Raman results.
 | ||
| Fig. 3 1H magic angle spinning (MAS) NMR spectra of the cf:ana cocrystal form I (above) and form II (middle) and the superimposed spectra of the reactants (below) without the spinning sidebands. | ||
The spectra of the cocrystal polymorphs resemble on a first view the sum of the spectra of the reactants but are not identical. More importantly, the strongly bridged acidic proton of ana is highly influenced, since its resonance position shifts ca. 3 ppm in both cocrystal forms. The other protons seem not to be highly affected in the cocrystal. Based on these results, it can be concluded that the acidic proton of ana is not as strongly bridged in the cocrystal as in pure ana.
To investigate the morphology of the cocrystal forms, SEM micrographs were taken. As shown in Fig. 4, both forms exhibit rod-shaped crystals, which are arranged locally in a parallel manner. This similarity can be traced back to the resembling crystal structures of the polymorphs and similar crystallization conditions. The main difference resides in the length of the crystals. In form I only short crystals can be obtained. In the case of form II, the crystals are elongated slightly. Despite this intensive characterization using SEM, Raman and ssNMR spectroscopy an unambiguous differentiation between the polymorphs is only possible based on the XRD data.
 | ||
| Fig. 4 SEM micrographs of the cf:ana cocrystal material form I (left) and form II (right). | ||
In order to investigate the formation of the polymorphic forms of the cf:ana cocrystal different solvents were added to the reactants caffeine and ana during the milling synthesis. In a typical experiment 1 g of the cocrystal reactants were milled together with 250 μL of the solvent at 30 Hz for 25 min. The results of the syntheses are summarized in Table 1. Mostly, polymorph I is formed. In each experiment a pure cocrystal form is obtained and no crystalline phase mixtures were found. It can be supposed that the crystallization of form I proceeds without any interaction or pre-coordination between the solvent and the reactants. This polymorph is also formed by neat grinding or by adding non-polar solvents, which cannot create any intermolecular bonds with the reactants. Both polymorphic forms show a comparable thermodynamic stability, since the melting points differ only by 2 K (Fig. S2†). This assumption is supported by temperature dependent PXRD measurements. These measurements show that the polymorphs do not convert into each other below the melting points (Fig. S3†). Slurry experiments reveal that in an 1:1 slurry of form I and form II in heptane only form I can be obtained after seven days. Therefore, form I can be considered slightly more stable compared to form II.9 Based on the results of the temperature dependent PXRD experiments the preferred formation of a distinct polymorph based on its thermodynamic stability can be revealed.
| Compound | Solubility in water [mg ml−1] | Solubility in EtOH [mg ml−1] | Compound | Solubility in water [mg ml−1] | Solubility in EtOH [mg ml−1] |
|---|---|---|---|---|---|
| Anthranilic acid | 5 | 200 | Caffeine | 16 | 15 |
| Solvent | Dipole moment [×10−30 C m] | Polymorphic form | Solvent | Dipole moment [×10−30 C m] | Polymorphic form |
| 1,4-Dioxane | 1.3 | Form I | Methanol | 5.5 | Form I |
| 2-Butanone | 9.2 | Form I | n-Heptane | 0.0 | Form I |
| Acetophenone | 9.7 | Form I | n-Hexane | 0.0 | Form I |
| Chloroform | 3.7 | Form I | n-Pentane | 0.0 | Form I |
| Cyclohexane | 0.0 | Form I | Nitromethane | 10.3 | Form I9 |
| Dichloromethane | 6.0 | Form I | Tetrahydrofuran | 5.7 | Form I |
| Diethyl ether | 4.2 | Form I | |||
| Ethanol | 5.7 | Form I | Acetone | 10.0 | Form II |
| Ethyl acetate | 6.2 | Form I | Acetonitrile | 11.7 | Form II |
| Ethylene glycol | 6.7 | Form I | Dimethylformamide | 12.7 | Form II |
The formation of the polymorphs of the cf:ana cocrystal form I or II depends decisively on the solvent which is added in the LAG synthesis. Only three of the 18 solvents lead to form II. The screening reveals that two facts play an important role for the formation of form II: the dipole moment of the solvent and the functional group of the solvent. Only if the solvent molecule has a high dipole moment, more precisely higher than 10 × 10−30 C m, can form II be formed. This includes the solvents acetone, acetonitrile, dimethylformamide and nitromethane. However, nitromethane seems to be an exception. Despite its high dipole moment, nitromethane generates form I.9 In comparison to other solvents, only nitromethane has a nitro group as a functional group. This implies that aside from the high dipole moment a carbonyl or nitrile group is also necessary for the cocrystal formation of form II. These observations suggest that the formation of form II is triggered by a pre-coordination to the solvent molecule.
Both polymorphic forms exhibit the same synthon. Each caffeine molecule is connected to two ana molecules via hydrogen bonds. One hydrogen bond is formed between the imidazole nitrogen atom from a caffeine molecule and the carboxyl oxygen atom from an ana molecule. An additional hydrogen bond is formed between the amino group of an ana molecule and the carbonyl group of a caffeine molecule, leading to a chain structure. The main difference between the polymorphic forms is the environment around the latter hydrogen bond. In form I, all molecules of the chains are in plane leading to a planar layered structure, whereas in form II the caffeine molecule is twisted at the amino–carbonyl hydrogen bond resulting in zigzag-type chains. Based on this difference, it can be assumed that the described torsion results from the pre-coordination to the solvent molecules, which takes place at the amino group of ana. Since the solvent molecule occupies too much space, the caffeine molecule cannot bind in the same planar layer and is twisted approximately 30° as shown in Fig. 5.
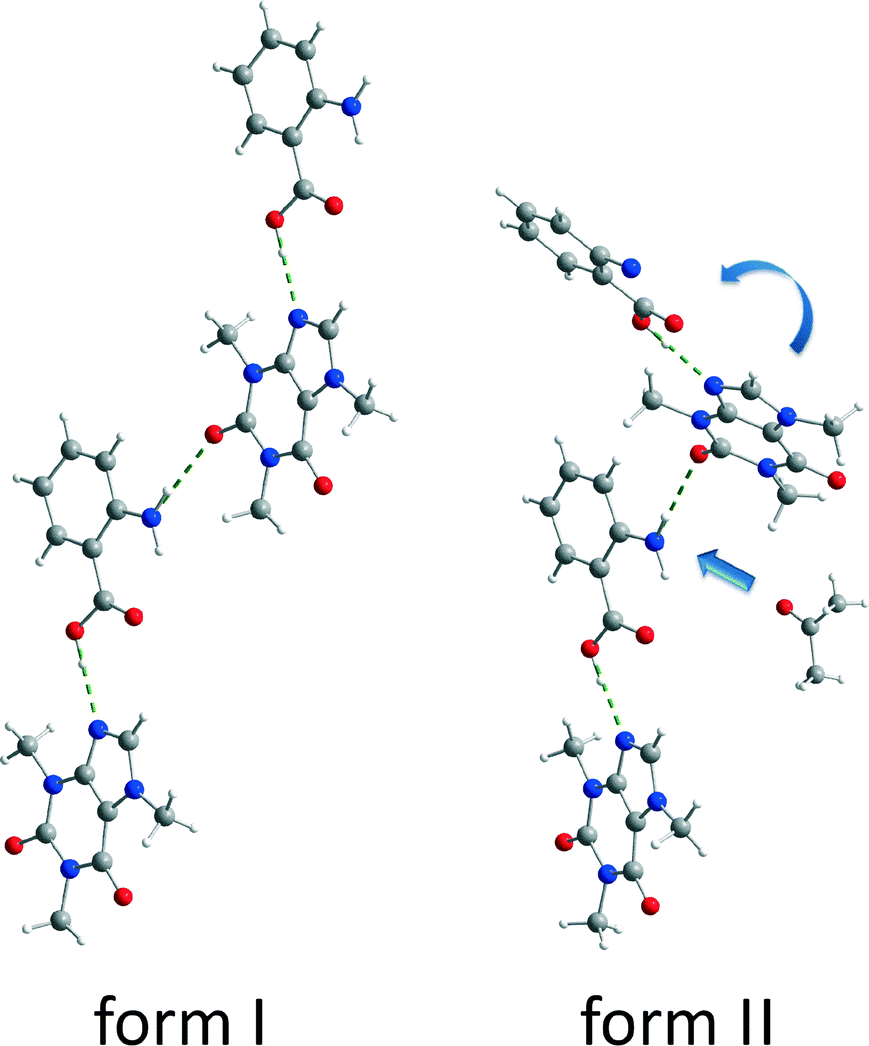 | ||
| Fig. 5 Bonding arrangement of the cf:ana form I (left) and form II (right). | ||
Systematic investigations with different volumes of acetonitrile added to the reactant mixture during the milling process were performed. Here, it can already be observed that after addition of 5 μL of acetonitrile to a total load of 1 g of cocrystal material, form II crystallizes spontaneously (Fig. 6). This observation indicates that the coordination of the reactants with only a few solvent molecules already leads to a sufficient amount of the cocrystal form II as seeds for further crystallization. Because the solvent is not embedded in the crystal system, it can be thought of as a catalyst. Fig. 6 compares the powder patterns of the cocrystals synthesized using different amounts of acetonitrile. The intensity of the reflections decreases when a smaller amount of the solvent is used during grinding. The amount of the solvent accelerates the product formation.
 | ||
| Fig. 6 PXRD patterns of cf:ana form II with different amounts of acetonitrile (5 μL–250 μL) added to 1 g of the reactant mixture. | ||
To exclude a similar pre-coordination for form I, milling experiments were conducted by adding different mixture ratios of ethanol and acetonitrile in the LAG synthesis. If both polymorphic forms were induced by pre-coordination with the solvent molecules, a transition of one form to another would appear with the corresponding increase of the solvent. Fig. 7 illustrates that at a volume mixing ratio of 49:1 (ethanol:acetonitrile) the exclusive formation of polymorph II is triggered by the presence of acetonitrile. Consequently, a pre-coordination with the solvents leading to form I can be ruled out.
 | ||
| Fig. 7 PXRD patterns of cf:ana form II with different solvents (250 μL) added to 1 g of the reactant mixture: acetonitrile (ACN), ethanol and acetonitrile (49:1, corresponding to the volumes of the solvents), and ethanol. | ||
To elucidate the catalytic influence of the solvents forming form II, the synthesis of both cocrystal polymorphs was investigated via pseudo-in situ monitoring. The grinding synthesis was stopped after different periods of time and small amounts of the material were immediately investigated using synchrotron X-ray diffraction. Fig. 8 illustrates exemplarily the formation pathway of form II, which was obtained by adding 250 μL of acetonitrile. While the reflections of ana are not observable in the powder pattern after 20 s, the first reflections of the cocrystal appear at that time. The reflections of caffeine disappear after 100 s of the LAG synthesis, and the cocrystal formation can be regarded as completed. The monitoring of the cocrystal synthesis also reveals that the formation pathway leads directly to form II without any transitional stages.
 | ||
| Fig. 8 PXRD patterns of the cf:ana cocrystal form II after different milling time intervals. | ||
In a similar experiment the formation of form I (Fig. S4†) was also observed by pseudo-in situ monitoring using 250 μL of dioxane. Here, no reflections of ana can be observed after 10 s. The formation of form I is much slower, and no caffeine reflections can be observed after 6 min. It can be postulated that the pre-coordination prior to the formation of form II causes also an acceleration of the reaction rate.
Experimental
Materials
Caffeine (C8H10N4O2) (ReagentPlus®, Sigma Aldrich, Germany), anthranilic acid (C7H7NO2) (≥98%, Acros Organics, USA), and all solvents were used without further purification.Milling synthesis
The synthesis of the two polymorphs was conducted by neat or liquid-assisted grinding (LAG) in a ball mill (MM400, Retsch, Germany) at 30 Hz for 25 min. A 10 mL steel vessel with two steel balls (10 mm) was used for a total load of 1 g of the reactants. In the case of LAG 250 μL of the solvent was added.PXRD measurements
The obtained product was measured by PXRD. All X-ray diffraction experiments were carried out using a D8 diffractometer (Bruker AXS, Karlsruhe, Germany) in transmission geometry (Cu-Kα1 radiation, λ = 1.54056 Å).Synchrotron X-ray diffraction
Pseudo-in situ measurements were performed at the micro-focus beamline μSpot (BESSY II, Helmholtz Centre Berlin for Materials and Energy, Germany) in transmission geometry. The powder patterns were collected with a wavelength of 1.0039 Å using a Si (111) double-crystal monochromator and were recorded with a two-dimensional MarMosaic CCD X-ray detector with 3072 × 3072 pixels. In a typical experiment, XRD patterns were collected for 30 s. The obtained scattering images were processed and converted into diagrams of scattered intensities versus scattering vector q (q = 4π sinθ/λ) employing an algorithm from the FIT2D software.41 For the graphical representations, q-values were converted to the diffraction angle 2θ (Cu) to provide a direct comparison to the results obtained by XRD with Cu radiation.
Raman measurements
Raman measurements were performed using a Raman RXN1™ Analyzer (Kaiser Optical Systems, France). The spectra were collected using a laser with a wavelength of λ = 785 nm and a contactless probe head (working distance 1.5 cm, spot size 1.0 mm). Raman spectra were recorded with an acquisition time of 5 s for 5 accumulations. Measurements at NIR excitation radiation (λ = 785 nm) using an irradiation of 6.6 W cm−2 were performed.ssNMR spectroscopy
1H MAS NMR spectra were recorded using a Bruker AVANCE 400 spectrometer using a 2.5 mm double-bearing MAS probe (Bruker BioSpin) and a spinning speed of 20 kHz. The 1H MAS NMR spectra were recorded with π/2 pulse lengths of 3.7 μs, a recycle delay of 5 s and an accumulation number of 256. Existing background signals were suppressed with a phase-cycled depth pulse sequence based on Cory and Ritchey.42SEM
SEM micrographs were obtained using a ZEISS Supra 40 scanning electron microscope equipped with a thermal field emission cathode (Schottky-emitter, ZrO/W cathode). The acceleration voltage was set to 10 kV and the working distance was 6.2 mm. The samples were additionally deposed with a thin layer of carbon. The micrographs were acquired with an in-lens (SE1) secondary electron detector, a (SE2) secondary electron detector of type Everhart–Thornley and a QBSD back-scatter electron detector.43Conclusions
The formation pathways of two polymorphs of the caffeine:anthranilic acid cocrystal were investigated under mechanochemical conditions. Small amounts of solvents added during the grinding process lead to different polymorphs depending on the solvent. Solvents with low dipole moments favour the crystallization of form I. Solvents with high dipole moments lead to the crystallization of form II. In these cases the solvent molecules interact with the amino group of ana molecules. Consequently, the caffeine molecules have to twist for the development of the cocrystal synthon. For solvents with lower dipole moment a pre-coordination is not observed and the caffeine molecules are arranged in parallel in the layered structure of form I.Pseudo-in situ monitoring of the synthesis pathway of both polymorphs using synchrotron radiation revealed that pre-coordination initializes not only the crystallization of the polymorphic form II but also accelerates the cocrystal formation with respect to the synthesis of form I. In addition, since only small amounts of the solvents are needed to cause the crystallization of form II, it can be seen as a catalyst.
These investigations show that in the liquid-assisted grinding synthesis the kind of solvent added during the milling process has an enormous influence and has an effect on the fate of the cocrystal product.
Acknowledgements
We are grateful to Dr. M. Feist for DTA/TG measurements.References
- B. Sarma, J. Chen, H. Y. Hsi and A. S. Myerson, Korean J. Chem. Eng., 2011, 28, 315–322 CrossRef CAS.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859–866 CrossRef CAS.
- J.-P. Brog, C.-L. Chanez, A. Crochet and K. M. Fromm, RSC Adv., 2013, 3, 16905–16931 RSC.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CAS.
- C. B. Aakeröy, M. E. Fasulo and J. Desper, Mol. Pharmaceutics, 2007, 4, 317–322 CrossRef PubMed.
- M. C. Etter and G. M. Frankenbach, Chem. Mater., 1989, 1, 10–12 CrossRef CAS.
- T. Friščić and W. Jones, J. Pharm. Pharmacol., 2010, 62, 1547–1559 CrossRef PubMed.
- M. J. Zaworotko, Cryst. Growth Des., 2006, 7, 4–9 Search PubMed.
- N. Madusanka, M. D. Eddleston, M. Arhangelskis and W. Jones, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 72–80 CAS.
- M. J. Bevill, P. I. Vlahova and J. P. Smit, Cryst. Growth Des., 2014, 14, 1438–1448 CAS.
- S. Ghosh, A. Mondal, M. S. R. N. Kiran, U. Ramamurty and C. M. Reddy, Cryst. Growth Des., 2013, 13, 4435–4441 CAS.
- M. D. Eddleston, B. Patel, G. M. Day and W. Jones, Cryst. Growth Des., 2013, 13, 4599–4606 CAS.
- N. Schultheiss, M. Roe and S. X. M. Boerrigter, CrystEngComm, 2011, 13, 611–619 RSC.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2004, 890–891 RSC.
- B. N. Cronstein, J. Hepatol., 2010, 53, 207–208 CrossRef CAS PubMed.
- B. B. Fredholm, K. Battig, J. Holmen, A. Nehlig and E. E. Zvartau, Pharmacol. Rev., 1999, 51, 83–133 CAS.
- R. B. Lipton, W. F. Stewart, R. E. Ryan, J. Saper, S. Silberstein and F. Sheftell, Arch. Neurol., 1998, 55, 210–217 CrossRef CAS.
- S. Ghosh and C. M. Reddy, CrystEngComm, 2012, 14, 2444–2453 RSC.
- E. Lu, N. Rodriguez-Hornedo and R. Suryanarayanan, CrystEngComm, 2008, 10, 665–668 RSC.
- S. S. Pinto and H. P. Diogo, J. Chem. Thermodyn., 2006, 38, 1515–1522 CrossRef CAS PubMed.
- G. D. Enright, V. V. Terskikh, D. H. Brouwer and J. A. Ripmeester, Cryst. Growth Des., 2007, 7, 1406–1410 CAS.
- T. E. Gorelik, A. Sarfraz, U. Kolb, F. Emmerling and K. Rademann, Cryst. Growth Des., 2012, 12, 3239–3242 CAS.
- A. Sarfraz, A. Simo, R. Fenger, W. Christen, K. Rademann, U. Panne and F. Emmerling, Cryst. Growth Des., 2012, 12, 583–588 CAS.
- J. Leiterer, F. Emmerling, U. Panne, W. Christen and K. Rademann, Langmuir, 2008, 24, 7970–7978 CrossRef CAS PubMed.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013–1021 CAS.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2012, 14, 2381–2385 RSC.
- D.-K. Bučar, G. M. Day, I. Halasz, G. G. Z. Zhang, J. R. G. Sander, D. G. Reid, L. R. MacGillivray, M. J. Duer and W. Jones, Chem. Sci., 2013, 4, 4417–4425 RSC.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, L. R. MacGillivray and G. G. Z. Zhang, Cryst. Growth Des., 2009, 9, 1932–1943 Search PubMed.
- D. K. Bučar, R. F. Henry, X. C. Lou, T. B. Borchardt and G. G. Z. Zhang, Chem. Commun., 2007, 525–527 Search PubMed.
- H. D. Clarke, K. K. Arora, H. Bass, P. Kavuru, T. T. Ong, T. Pujari, L. Wojtas and M. J. Zaworotko, Cryst. Growth Des., 2010, 10, 2152–2167 CAS.
- B. Das and J. B. Baruah, Cryst. Growth Des., 2010, 11, 278–286 Search PubMed.
- T. Friščić, L. Fabian, J. C. Burley, W. Jones and W. D. S. Motherwell, Chem. Commun., 2006, 5009–5011 Search PubMed.
- A. V. Trask, J. van de Streek, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 2233–2241 CAS.
- N. Schultheiss, S. Bethune and J. O. Henck, CrystEngComm, 2010, 12, 2436–2442 RSC.
- I. Pavel, A. Szeghalmi, D. Moigno, S. Cîntă and W. Kiefer, Biopolymers, 2003, 72, 25–37 CrossRef CAS PubMed.
- J. S. Suh and M. Moskovits, J. Am. Chem. Soc., 1986, 108, 4711–4718 CrossRef CAS.
- G. E. Hardy, W. C. Kaska, B. P. Chandra and J. I. Zink, J. Am. Chem. Soc., 1981, 103, 1074–1079 CrossRef CAS.
- A. L. McClellan, Tables of experimental dipole moments, WH Freeman, 1963 Search PubMed.
- A. M. Awwad and A. H. Al-Dujaili, J. Chem. Eng. Data, 2001, 46, 1349–1350 CrossRef CAS.
- Merck Index, ed. S. Budavari, 12th edn, 1996 Search PubMed.
- A. P. Hammersley, K. Brown, W. Burmeister, L. Claustre, A. Gonzalez, S. McSweeney, E. Mitchell, J.-P. Moy, S. O. Svensson and A. W. Thompson, J. Synchrotron Radiat., 1997, 4, 67–77 CrossRef CAS PubMed.
- D. G. Cory and W. M. Ritchey, J. Magn. Reson., 1988, 80, 128–132 Search PubMed.
- A. Guilherme, V. D. Hodoroaba, S. Benemann, J. Coroado and M. L. Carvalho, J. Anal. At. Spectrom., 2014, 29, 51–57 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H MAS NMR spectra of the reactants and the cocrystal. See DOI: 10.1039/c4ce00472h |
| This journal is © The Royal Society of Chemistry 2014 |