 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystal structures and hydrogen bond analysis of five amino acid conjugates of terephthalic and benzene-1,2,3-tricarboxylic acids†
Anirban
Karmakar‡
*a,
Clive L.
Oliver
b,
Ana E.
Platero-Prats
c,
Elina
Laurila
a and
Lars
Öhrström
*a
aChemical and Biological Engineering, Physical Chemistry, Chalmers University of Technology, Kemivägen 10, Gothenburg, Sweden. E-mail: anirbanchem@gmail.com; ohrstrom@chalmers.se
bCentre for Supramolecular Chemistry Research, Department of Chemistry, University of Cape Town, Rondebosch, Cape Town, 7701, South Africa
cDepartment of Materials and Environmental Chemistry (MMK) and Department of Organic Chemistry (OC), Stockholm University, Sweden
First published on 27th May 2014
Abstract
Four linear connecting amino acid derived ligands, 1–4, and one potentially three connecting, 5, were prepared by the reaction of the appropriate terephthaloyl dichloride or benzene-1,3,5-tricarbonyl trichloride with the methyl ester protected amino acid. Amino acids used here were alanine (1, 5), isoleucine (2), leucine (3) and valine (4). Crystalline forms of four amino acid substituted terephthalamides (2,2′-(terephthaloylbis(azanediyl))dipropanoic acid dihydrate 1; 2,2′-(terephthaloylbis(azanediyl))bis(3-methylpentanoic acid) monohydrate 2; 2,2′-(terephthaloylbis(azanediyl))bis(4-methylpentanoic acid) dihydrate 3; 2,2′-(terephthaloylbis(azanediyl))bis(3-methylbutanoic acid) dihydrate 4) and one benzene-1,3,5-tricarboxamide molecule (2,2′,2′′-((benzene-1,3,5-tricarbonyl)tris(azanediyl))tripropionic acid hemihydrate 5) were characterised and the single crystal structures were solved. All the compounds form hydrogen bonded 2D and 3D nets. Linear connecting amino acid derivatives can be categorised into three groups depending on the hydrogen bond patterns and final structures. Compounds 1 and 2 form 3D structures but the final structure is different due to the different hydrogen bond synthons. Compounds 3 and 4 are isostructural and form 2D hydrogen bonded structures while 5 forms a hydrogen bonded pcu-net. Intermolecular interactions have been analysed with Hirshfeld surfaces and graph set symbols.
Introduction
Understanding the driving forces of the formation of extended assemblies is important for designing molecule based materials, be these for potential electronic,1 magnetic2 or porous3,4 applications. Often, the idea is to preprogram the building blocks to interact with each other in a certain way to give larger entities with the desired location and geometric relationships between them. In other words, the interaction behaviour is based on the functional groups of the molecules.Terephthalamides and benzene-1,3,5-tricarboxamides are examples of building blocks used in preparing extended assemblies.5–11 These molecules have a benzene ring core and usually contain flexible functional groups. In addition to stronger interactions such as coordination and hydrogen bonds, the role of non-covalent interactions is also important in the assembly of molecules. For example, the benzene ring in these systems offers the possibility of aromatic interactions.
The assembly process of benzene-1,3,5-tricarboxamide derivatives has been studied widely by using not only different techniques such as CD and UV-Vis but also theoretical methods.10,12,13 Terephthalamides and benzene-1,3,5-tricarboxamides have produced nanostructured materials, for instance nano-staircases and fibres are among the reported architectures.5,6,14–17 Spontaneous formation of hydrogels and their encapsulation and sorption properties of both di- and tri-substituted carboxamides have also been studied.6,18,19 In addition, terephthalamides and benzene-1,3,5-tricarboxamides derivatives have been reported to form liquid columnar crystal phases (LC), preorganized surfactants and multi-component organic cages within dynamic combinatorial libraries.20–23
Introduction of amino acids into terephthalamide and benzene-1,3,5-tricarboxamides offers the possibility of producing enantiopure chiral building blocks and structures5,7,24 with potential applications in enantioselective synthesis, separation and detection. The amino acid part can also stabilise the assemblies via hydrogen bonding and moreover provide metal coordination sites.5,11,16,17,24–26
The complexing abilities of amino acid substituted terephthalamide and benzene-1,3,5-tricarboxamides have been studied by preparing network structures with metals such as copper, cobalt, manganese, nickel, cadmium, zinc, lantanoids, and calcium.7,11,17,24–31 These studies have focused on non-chiral glycine (–H) substituted ligands, but alanine (–CH3) and phenylalanine (–CH2(C6H5)) examples are also known, although reported X-ray structures of such network compounds are rare.32,33
The choice of the ligand and metal is essential in producing such metal–organic frameworks (MOFs)34 that can have application in gas sorption, catalysis and molecular recognition.27,35–39 These properties are incorporated into the material during preparation using different building blocks. Therefore, the design of new materials with desired properties requires understanding and control of the interactions of the building blocks.
We report here the preparation and crystal structures of the amino acid derivatives 1–5 of terephthalamide and benzene-1,3,5-tricarboxamides (Fig. 1). These closely related molecules give structures with differences and similarities that are investigated not only by traditional means, but also graph set and Hirshfeld surface analyses.
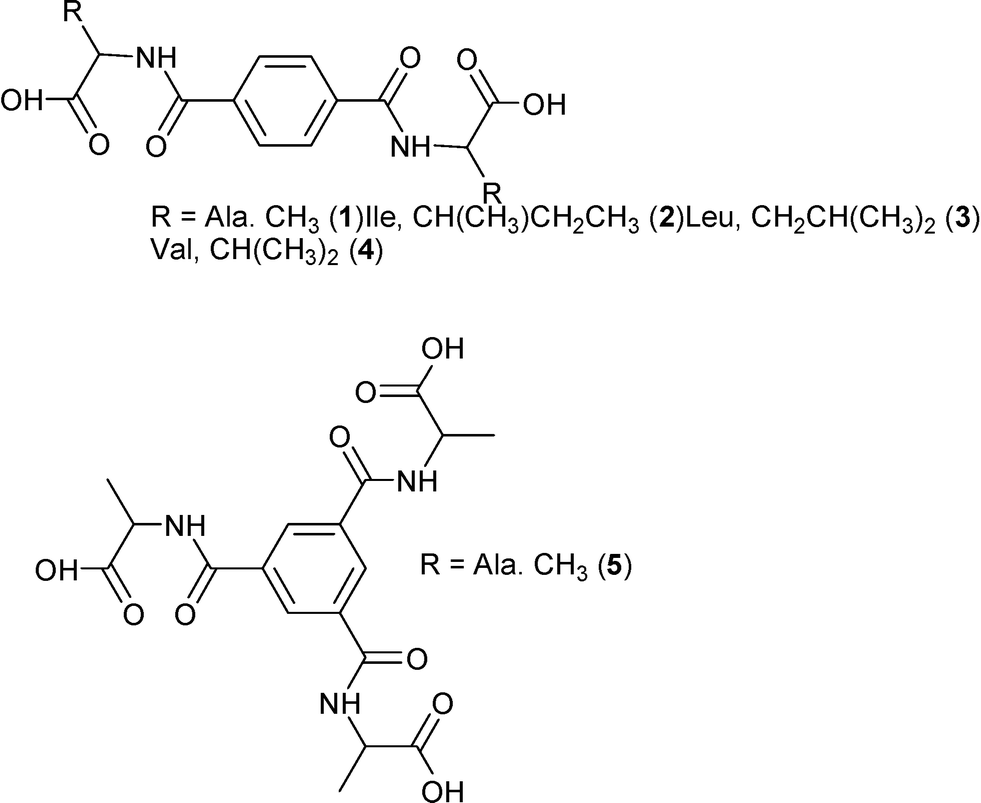 | ||
| Fig. 1 Schematic presentation of the molecules of the studied compounds 1–5. | ||
Experimental
Materials
Amino acids L-phenylalanine (≥99%), L-leucine (≥99%), L-isoleucine (≥99%) and DL-valine (≥99%) were produced by Fluka. L-alanine methyl ester hydrochloride (≥99%) was purchased from Aldrich and other amino acid methyl ester hydrochlorides were prepared according to the literature.40 Triethyl amine (99.5%), terephthaloyl dichloride (99%), benzene-1,3,5-tricarbonyl trichloride (98%) and d-chloroform (99.8%) were manufactured by Aldrich. Anhydrous magnesium sulfate (98%) and sodium hydroxide (98%) were produced by Scharlau. Hydrochloric acid (≥37%), potassium bromide (99%) and methanol (99.7%) were manufactured by Sigma Aldrich. Dichloromethane (p.a.) was produced by Merck. Dimethylsulfoxide-d6 (99.8%) was produced by Armar Chemicals.Experimental methods
The elemental analysis was performed by H. Kolbe Mikroanalytisches Laboratorium, Germany. The FTIR spectroscopy measurements were performed using a Bruker IFS-125 spectrometer in a KBr pellet. The 1H- and 13C-NMR spectra were analysed using an Agilent 400 MHz spectrometer operating at 400 and 100 MHz for proton and carbon, respectively.X-ray crystal structure determination
The crystallographic details of compounds 1–5 are summarized in Table 1. The crystals of 1–5 were immersed in cryo-oil, mounted in a Nylon loop, and measured at a temperature of 293(2) K (1, 5) and 173(2) K using an Oxford Cryostream 600 (3) and 700 (2, 4). The X-ray diffraction data were collected using an Xcalibur Sapphire3 diffractometer (1, 5), a Nonius Kappa CCD diffractometer (3) and a Bruker DUO APEX II CCD diffractometer (2, 4) with Mo Kα radiation (λ = 0.71073 Å). For 1 and 5, the CrysAlisPro programme package was used for cell refinement and data reduction,41 whilst for 3, DENZO was used, and for 2 and 4 data reduction and cell refinement were performed using SAINT-Plus.42 Space groups were determined from systematic absences using XPREP43 and confirmed using the program layer for 2–4.44 The structures were solved by direct methods using the SHELXS-97 (ref. 45) program. Semi-empirical or numerical absorption (SCALE3 ABSPACK (1), SADABS46 (2–5)) was applied to all of the data. Structural refinement was carried out using SHELXL-97 (1–5).47 The refinement procedure by full-matrix least-squares methods, based on F2 values against all reflections was performed using SHELXL-97, including the anisotropic displacement parameters for all non-H atoms. Hydrogen atoms were placed geometrically, except for O–H, N–H and water hydrogen atoms which were located in difference Fourier maps. All other hydrogen atoms were positioned geometrically and constrained to ride on their parent atoms Uiso = 1.2Ueq(parent atom). The crystals of 5 have an asymmetric carbon that is disordered over two sites with equal occupancies. It also contains electron density, interpreted as residual water molecules, that cannot be refined properly. This electron density excess was cleaned using the SQUEEZE routine of PLATON.48 CCDC numbers 993036–993040.| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. | |||||
| Empirical formula | C14H20N2O8 | C20H30N2O7 | C20H32N2O8 | C18H28N2O8 | C18H21.57N3O9.28 |
| Fw | 344.32 | 410.46 | 428.47 | 400.42 | 423.38 |
| Temp (K) | 293(2) | 173(2) | 173(2) | 173(2) | 293(2) |
| l (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Cryst. syst. | Orthorhombic | Orthorhombic | Monoclinic | Monoclinic | Trigonal |
| Space group | P212121 | P212121 | P21 | P21 |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
| a (Å) | 7.7195(5) | 6.9544(5) | 12.374(3) | 10.5665(10) | 18.3488(5) |
| b (Å) | 8.2128(5) | 11.7552(8) | 7.675(2) | 7.6596(7) | 18.3488(5) |
| c (Å) | 26.453(2) | 26.872(2) | 12.714(3) | 12.871(1) | 11.1379(6) |
| a (deg) | 90 | 90 | 90 | 90 | 90 |
| b (deg) | 90 | 90 | 107.15(3) | 106.949(2) | 90 |
| g (deg) | 90 | 90 | 90 | 90 | 120 |
| V (Å3) | 1677.1(2) | 2196.8(3) | 1153.9(4) | 996.5(2) | 3247.5(2) |
| Z | 4 | 4 | 2 | 2 | 6 |
| r calc (Mg m3) | 1.364 | 1.241 | 1.233 | 1.335 | 1.315 |
| m (Mo Kα) (mm−1) | 0.113 | 0.094 | 0.095 | 0.105 | 0.107 |
| No. reflns. | 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 431 431 |
19064 |
25132 |
9773 | 1468 |
| Unique reflns. | 5974 | 6670 | 4723 | 6055 | 1468 |
| GooF | 1.084 | 1.016 | 1.217 | 1.016 | 1.250 |
| R int | 0.0771 | 0.0605 | 0.047 (Rmerge) | 0.0286 | |
| R 1 (I3 2s) | 0.0678 | 0.0615 | 0.0489 | 0.0817 | 0.0889 |
| wR2b (I3 2s) | 0.1084 | 0.1383 | 0.1206 | 0.2048 | 0.2394 |
Synthesis of dimethyl 2,2′-(terephthaloylbis(azanediyl))dipropionate
L-Alanine methyl ester hydrochloride (1.39 g, 10 mmol) and triethylamine (2.52 g, 25 mmol) were placed in a round bottom flask and then dissolved in dry 20 ml of dichloromethane. Terephthaloyl dichloride (1.11 g, 5 mmol) in 2 ml of dry dichloromethane was added dropwise to this mixture with continuous stirring. The reaction mixture was stirred for 24 hours. Then the solvent was removed under reduced pressure and a white solid was obtained. The white solid was washed with water and then extracted with dichloromethane. The organic extracts were collected and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure and the product was obtained as a white solid. (2.4 g, 73%). νmax (KBr)/cm−1 3294 (s), 3077 (w), 3001 (w), 2954 (w), 2850 (w), 1735 (s), 1637 (s), 1546 (s), 1500 (m), 1452 (m), 1441 (m), 1372 (w), 1345 (m), 1319 (m), 1288 (m), 1230 (s), 1170 (s), 1116 (m), 1052 (m), 1017 (w), 980 (w), 928 (w), 874 (s), 836 (w), 747 (m), 678 (s), 574 (w), 501 (w). δH (400 MHz; CDCl3) 7.87 (s, 4H, Bn), 6.84 (d, 2H, –NH, J = 8Hz), 4.82 (m, 2H, –CH), 3.81 (s, 6H, –COOCH3), 1.55 (d, 6H, –CH3, J = 8Hz). δC (100 MHz; CDCl3) 173.6, 165.8, 136.7, 127.3, 52.7, 48.6, 18.6.Synthesis of 2,2′-(terephthaloylbis(azanediyl))dipropionic acid (1)
Dimethyl 2,2′-(terephthaloylbis(azanediyl))dipropionate (1.68 g, 5 mmol) and sodium hydroxide (0.6 g, 15 mmol) were dissolved in 20 mL of methanol:water mixture (4:1). The reaction mixture was stirred for eight hours at room temperature. Then the solvent was evaporated under reduced pressure. The solid product was dissolved in 10 mL of water and the solution was acidified (pH = 2) with dilute hydrochloric acid solution. The obtained white solid product was filtered and washed with water. (0.9 g, 62%). (Found: C 48.61, H 5.81, N 8.11 calc. for C14H16N2O6·2H2O C 48.83, H 5.85, N 8.14) νmax (KBr)/cm−1 3394 (bs), 3060 (w), 2982 (w), 2931 (w), 2542 (mb), 2019 (mb), 1732 (s), 1705 (s), 1639 (s), 1542 (s), 1500 (m), 1456 (m), 1406 (w), 1381 (w), 1345 (m), 1323 (m), 1289 (m), 1269 (s), 1240 (s), 1181 (s), 1039 (w), 1017 (w), 945 (w), 869 (s), 829 (m), 743 (s), 646 (m), 596 (m), 555 (w). δH (400 MHz; DMSO) 12.54 (s, 2H, –COOH), 8.77 (d, 2H, –NH, J = 8Hz), 7.95 (s, 4H, Bn), 4.42 (m, 2H, –CH), 1.39 (d, 6H, –CH3J = 8Hz). δC (100 MHz; DMSO) 174.5, 165.9, 136.7, 127.8, 48.7, 17.3.
Synthesis and analysis of dimethyl 2,2′-(terephthaloylbis(azanediyl))bis(3-methylpentanoate)
Dimethyl 2,2′-(terephthaloylbis(azanediyl))bis(3-methylpentanoate) was synthesized following the synthesis of dimethyl 2,2′-(terephthaloylbis(azanediyl))dipropionate, but using L-leucine methyl ester hydrochloride (1.8 g, 10 mmol) instead of L-alanine methyl ester hydrochloride. (2.7 mg, 64%). νmax (KBr)/cm−1 3325 (s), 3086 (w), 3043 (w), 2956 (s), 2871 (m), 1746 (s), 1635 (s), 1550 (s), 1503 (s), 1469 (w), 1437 (w), 1340 (s), 1278 (s), 1211 (s), 1163 (s), 1122 (w), 1088 (w), 1048 (w), 1018 (m), 991 (w), 872 (m), 847 (m), 748 (m), 730 (m), 642 (bs), 480 (w). δH (400 MHz; CDCl3) 7.77 (s, 4H, Bn), 6.94 (d, 2H, –NH, J = 8Hz), 4.87 (m, 2H, –CH), 3.78 (s, 6H, –COOCH3), 1.82–1.65 (m, 6H, –CH2 and –CH), 0.98 (d, 12H, –CH3, J = 8Hz). δC (100 MHz; CDCl3) 174.1, 166.1, 136.4, 127.3, 52.5, 51.1, 41.4, 25.0, 22.9, 21.8. The NMR shifts were in agreement with the literature.16Synthesis of 2,2′-(terephthaloylbis(azanediyl))bis(3-methylpentanoic acid) (2)
2,2′-(Terephthaloylbis(azanediyl))bis(3-methylpentanoic acid) was synthesized following the synthesis of 2,2′-(terephthaloylbis(azanediyl))dipropionic acid, but using dimethyl 2,2′-(terephthaloylbis(azanediyl))bis(3-methylpentanoate) (2.1 g, 5 mmol) instead of dimethyl 2,2′-(terephthaloylbis(azanediyl))dipropionate. (1.5 g, 77%). (Found: C 55.67, H 7.25, N 6.38 calc. for C20H28N2O6·2H2O C 56.06, H 7.53, N 6.54) νmax (KBr)/cm−1 3367 (bs), 3060 (w), 2962 (s), 2575 (mb), 1728 (s), 1632 (s), 1545 (s), 1499 (m), 1471 (w), 1451 (w), 1387 (w), 1338 (m), 1270 (s), 1252 (s), 1180 (m), 1169 (m), 1089 (w), 1017 (w), 967 (w), 870 (s), 854 (s), 736 (s), 600 (m). δH (400 MHz; DMSO) 12.61 (bs, 2H, –COOH), 8.71 (d, 2H, –NH, J = 8Hz), 7.95 (s, 4H, Bn), 4.44 (m, 2H, –CH), 1.80–1.55 (m, 6H, –CH2 and –CH), 0.91 (d, 6H, –CH3, J = 8Hz), 0.86 (d, 6H, –CH3, J = 8Hz). δC (100 MHz; DMSO) 174.5, 166.2, 136.7, 127.8, 51.4, 24.9, 23.4, 21.6.Synthesis of dimethyl 2,2′-(terephthaloylbis(azanediyl))bis(4-methylpentanoate)
Dimethyl 2,2′-(terephthaloylbis(azanediyl))bis(4-methylpentanoate) was synthesized following the synthesis of dimethyl 2,2′-(terephthaloylbis(azanediyl))dipropionate, but using L-isoleucine methyl ester hydrochloride (1.8 g, 10 mmol) instead of L-alanine methyl ester hydrochloride. (2.7 mg, 64%). νmax (KBr)/cm−1 3345 (s), 3051 (w), 2966 (s), 2877 (m), 1746 (s), 1638 (s), 1547 (s), 1503 (s), 1459 (m), 1434 (m), 1372 (m), 1342 (s), 1298 (s), 1254 (m), 1201 (s), 1155 (s), 1117 (w), 1089 (w), 1008 (m), 964 (w), 878 (m), 847 (m), 769 (m), 731 (s), 679 (w), 619 (bs), 589 (m), 505 (w). δH (400 MHz; CDCl3) 7.86 (s, 4H, Bn), 6.72 (d, 2H, –NH, J = 8Hz), 4.82 (q, 2H, –CH, J = 8Hz), 3.78 (s, 6H, –COOCH3), 2.03 (m, 2H, –CH), 1.53 (m, 2H, –CH2), 1.25 (m, 2H, –CH2), 0.97 (m, 12H, –CH3). δC (100 MHz; CDCl3) 172.5, 166.1, 136.9, 127.4, 56.9, 52.3, 38.2, 25.4, 15.5, 11.6. The NMR shifts were in agreement with the literature.16Synthesis of 2,2′-(terephthaloylbis(azanediyl))bis(4-methylpentanoic acid) (3)
2,2′-(Terephthaloylbis(azanediyl))bis(4-methylpentanoic acid) was synthesized following the synthesis of 2,2′-(terephthaloylbis(azanediyl))dipropionic acid, but using dimethyl 2,2′-(terephthaloylbis(azanediyl))bis(4-methylpentanoate) (2.1 g, 5 mmol) instead of dimethyl 2,2′-(terephthaloylbis(azanediyl))dipropionate. (1.6 mg, 83%). (Found: C 58.47, H 7.28, N 6.87 calc. for C20H28N2O6·H2O C 58.52, H 7.37, N 6.83) νmax (KBr)/cm−1 3463 (bs), 3411 (s), 3315 (bs), 2967 (s), 2937 (m), 2546 (w), 1725 (s), 1630 (s), 1552 (s), 1524 (s), 1495 (m), 1458 (w), 1418 (w), 1385 (w), 1348 (m), 1241 (s), 1210 (s), 1168 (m), 1083 (w), 1017 (w), 974 (w), 869 (s), 728 (s), 697 (m), 600 (w), 568 (w). δH (400 MHz; DMSO) 12.61 (bs, 2H, –COOH), 8.71 (d, 2H, –NH, J = 8Hz), 7.95 (s, 4H, Bn), 4.44 (m, 2H, –CH), 1.80–1.55 (m, 6H, –CH2 and –CH), 0.91 (d, 6H, –CH3, J = 8Hz), 0.86 (d, 6H, –CH3, J = 8Hz). 8.57 (d, 2H, –NH, J = 8Hz), 7.93 (s, 4H, Bn), 4.32 (t, 2H, –CH, J = 8Hz), 1.94 (m, 2H, –CH), 1.50 (m, 2H, –CH2), 1.26 (m, 2H, –CH2), 0.92 (d, 6H, –CH3, J = 8Hz), 0.85 (t, 6H, –CH3, J = 8Hz). δC (100 MHz; DMSO) 173.5, 166.6, 136.8, 127.9, 57.8, 36.1, 25.6, 16.1, 11.5.Synthesis of dimethyl 2,2′-(terephthaloylbis(azanediyl))bis(3-methylbutanoate)
Dimethyl 2,2′-(terephthaloylbis(azanediyl))bis(3-methylbutanoate) was synthesized following the synthesis of dimethyl 2,2′-(terephthaloylbis(azanediyl))dipropionate, but using DL-valine methyl ester hydrochloride (1.7 g, 10 mmol) instead of L-alanine methyl ester hydrochloride. (2.9 g, 75%). νmax (KBr)/cm−1 3298 (s), 3031 (w), 2965 (s), 2875 (m), 1744 (s), 1642 (s), 1537 (s), 1501 (s), 1468 (m), 1435 (m), 1347 (s), 1321 (s), 1295 (m), 1261 (s), 1201 (s), 1160 (s), 1121 (w), 1070 (m), 1020 (s), 1002 (m), 923 (w), 862 (s), 826 (w), 737 (m), 681 (bs), 577 (w). δH (400 MHz; CDCl3) 7.86 (d, 4H, Bn, J = 8Hz), 6.66 (2H, m, –NH), 4.77 (m, 2H, –CH), 3.77 (s, 6H, –COOCH3), 2.27 (m, 2H, –CH), 0.99 (m, 12H, –CH3). δC (100 MHz; CDCl3) 172.5, 166.2, 136.9, 127.4, 57.5, 52.4, 31.6, 18.9, 17.9.Synthesis of 2,2′-(terephthaloylbis(azanediyl))bis(3-methylbutanoic acid) (4)
2,2′-(Terephthaloylbis(azanediyl))bis(3-methylbutanoic acid) was synthesized following the synthesis of 2,2′-(terephthaloylbis(azanediyl))dipropionic acid, but using dimethyl 2,2′-(terephthaloylbis(azanediyl))bis(3-methylbutanoate) (1.9 g, 5 mmol) instead of dimethyl 2,2′-(terephthaloylbis(azanediyl))dipropionate. (1.5 g, 84%). (Found: C 59.15, H 6.49, N 7.55 calc. for C18H24N2O6 C 59.33, H 6.64, N 7.69) νmax (KBr)/cm−1 3444 (bs), 3325 (bs), 2967 (s), 2936 (w), 2573 (bm), 1731 (s), 1629 (s), 1537 (s), 1499 (m), 1470 (w), 1394 (w), 1375 (w), 1368 (m), 1322 (s), 1253 (w), 1236 (s), 1193 (s), 1161 (s), 1033 (w), 1017 (w), 977 (m), 915 (m), 865 (s), 815 (s), 733 (s), 636 (m), 576 (w). δH (400 MHz; DMSO) 8.56 (d, 2H, –NH, J = 8Hz), 7.94 (s, 4H, Bn), 4.28 (tr, 2H, –CH, J = 8Hz), 2.19 (m, 2H, –CH), 0.95 (tr, 12H, –CH3, J = 8Hz). δC (100 MHz; DMSO) 173.5, 166.7, 136.9, 127.9, 58.9, 29.9, 19.8, 19.3.Synthesis of trimethyl 2,2′,2′′-((benzene-1,3,5-tricarbonyl)tris(azanediyl))tripropionate
L-Alanine methyl ester hydrochloride (1.39 g, 10 mmol) and triethylamine (3.53 g, 35 mmol) were placed in a round bottom flask and then dissolved in dry 20 ml of dichloromethane. Benzene-1,3,5-tricarbonyl trichloride (0.8 g, 3 mmol) in 2 ml of dry dichloromethane was added dropwise to this mixture with continuous stirring. The reaction mixture was stirred for 24 hours. Then, the solvent was removed under reduced pressure and a white solid was obtained. The white solid was washed with water and then extracted with dichloromethane. The organic extracts were collected and dried over anhydrous magnesium sulphate. The solvent was evaporated under reduced pressure and the product was obtained. (3.2 mg, 69%). νmax (KBr)/cm−1 3235 (s), 3066 (s), 2991 (s), 2952 (s), 1751 (s), 1643 (s), 1562 (s), 1457 (s), 1437 (m), 1381 (m), 1318 (s), 1282 (m), 1212 (s), 1165 (s), 1133 (w), 1054 (s), 986 (m), 933 (w), 849 (m), 828 (w), 723 (s), 692 (s).δH (400 MHz; CDCl3) 8.13 (s, 3H, Bn), 7.72 (d, 3H, –NH, J = 8Hz), 4.73 (m, 3H, –CH), 3.79 (s, 9H, –COOCH3), 1.58 (d, 9H, –CH3, J = 8Hz). δC (100 MHz; CDCl3) 173.7, 165.9, 134.8, 128.6, 52.5, 48.8, 17.4.Synthesis of 2,2′,2′′-((benzene-1,3,5-tricarbonyl)tris(azanediyl))tripropionic acid (5)
Trimethyl 2,2′,2′′-((benzene-1,3,5-tricarbonyl)tris(azanediyl))tripropionate (2.3 g, 5 mmol) and sodium hydroxide (0.6 g, 15 mmol) were dissolved in 20 mL of methanol:water mixture (4:1). The reaction mixture was stirred for eight hours at room temperature. Then the solvent was evaporated under reduced pressure. The solid product was dissolved in 10 mL of water and the solution was acidified (pH = 2) with dilute hydrochloric acid solution. The obtained white solid product was filtered and washed with water. (1.9 mg, 91%). (Found: C 50.12, H 5.04, N 9.47 calc. for C18H21N3O9·0.5H2O C 50.00, H 5.13, N 9.72) νmax (KBr)/cm−1 3393 (bs), 3233 (m), 3066 (s), 2992 (s), 2942 (s), 1722 (s), 1644 (s), 1544 (s), 1458 (m), 1412 (w), 1382 (w), 1289 (w), 1240 (s), 1167 (s), 1053 (w), 966 (w), 922 (w), 836 (w), 741 (m), 692 (w),607 (s). δH (400 MHz; DMSO) 8.20 (s, 3H, Bn), 4.45 (m, 3H, –CH), 1.43 (d, 9H, –CH3, J = 8Hz). δC (100 MHz; DMSO) 176.5, 168.3, 134.1, 129.4, 49.4, 15.9. The NMR shifts were in agreement with the literature.24
Results
Synthesis and crystallization
Compounds 1–5 were prepared in two steps at room temperature. In the first step, the protected amino acid methyl ester is introduced to terephthaloyl dichloride (1–4) or benzene-1,3,5-tricarbonyl trichloride (5) and in the next step the amino acid is deprotected (Schemes 1 and 2). The compounds were recrystallized from methanol (1, 2), H2O (3, 4) or from D2O (5) for single crystal X-ray diffraction studies. 1–3 were obtained enantiomerically pure while racemic valine was used to prepare 4 and 5 was obtained as a racemate.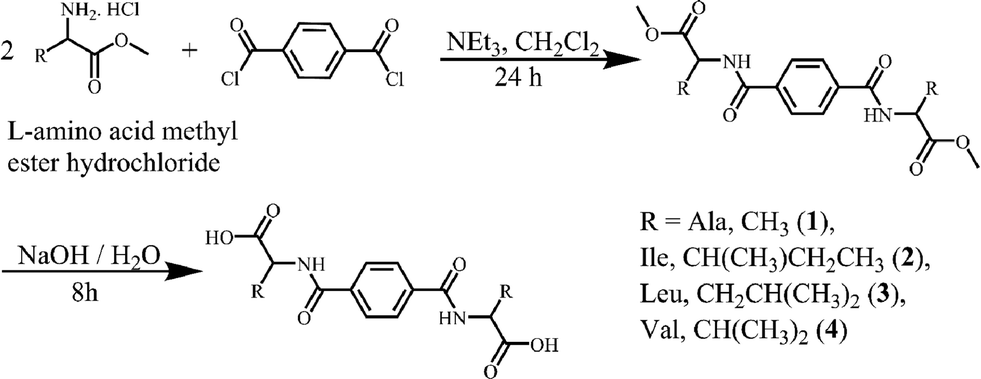 | ||
| Scheme 1 The synthesis of compounds 1–4. | ||
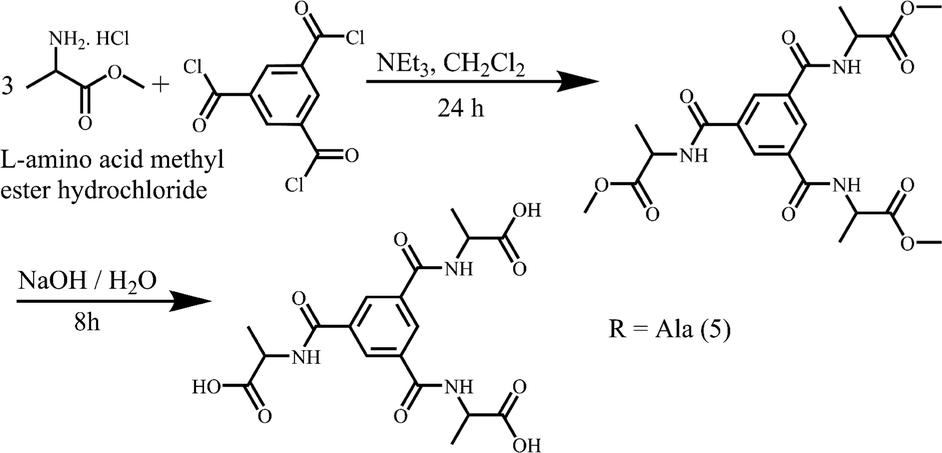 | ||
| Scheme 2 The synthesis of racemic 5. | ||
Structure descriptions
A complete set of ORTEP and packing diagrams can be found in the ESI.† | ||
| Fig. 2 Schematic presentation of dimeric units (blue and black) of 5 forming via π–π stacking and hydrogen bonding (dotted line). Graph set symbol R2,2,(16). | ||
Discussion
What we want to do here is to perform a structure comparison between our compounds using first the “big picture” methods graph set and Hirshfeld surface analyses and then, when needed, look into the structural details. For more general applicability we also include in our discussion of the compounds the earlier published structures of the glycine terephthalamide derivative 6 (ref. 32) and the glycine benzene-1,3,5-tricarboxamides derivative 7.33General hydrogen bond analysis by the graph set method
Graph set analysis is a general method to investigate the recurring hydrogen bond motifs and hydrogen bond patterns in discrete and extended assemblies.35,49,50 It does not discern the different atom types such as N or O, but once the criteria for hydrogen bonding are set, the analysis will generate the motifs such a chains (C) and rings (R) with the number of atoms directly involved in the bonding as well as the number of atoms linking these (in, for example, a ring). Table 2 shows the graph set symbols for 1–7 as calculated using Mercury. We chose to show only the ring-forming motifs as we believe these to be fairly distinct.| Comp. | Graph set symbol |
|---|---|
| Disubst. | |
| 1 | R3,3(15); R4,4(13); R4,4(18); R3,4(17); R4,4(21) |
| 2 | R4,4(13); R4,4(18); R3,4(17); R4,4(21) |
| 3 | R4,4(13); R4,4(18); R3,4(17); R4,4(18); R4,4(21); R3,4(23) |
| 4 | R4,4(13); R4,4(18); R3,4(17); R4,4(18); R4,4(21); R3,4(23) |
| 6 | R4,4(18); R2,4(28); R3,4(17); R4,4(21); R3,4(23) |
| Trisubst. | |
| 5 | R2,2(16); R2,2(22) R3,4(23); |
| 7 | R2,2(16); R2,2(22); R4,4(18); R4,4(20) |
We note first that there are no directly hydrogen bonded dimers in the 1–4 compounds, because that would have implied a 2,2 designation, (i.e. two donor atoms D and two acceptor atoms H as in the acetic acid dimer with symbol R2,2(8)). Compound 1 forms hydrogen bonds directly to another building block with a loop-back via hydrogen bonded water, the R3,3 assignment. This does not happen for compound 2 that has chains of building block–water–building block instead.
Then we can also see a number of recurring motifs in the different structures, the shorter circuits, i.e. the number in parenthesis, likely the most significant. Thus, the R4,4(13) motif is recurring in 1–4 but is absent in 6 and the R2,2(16) motif is found in both 5 and 7. An illustration of some of these motifs can be found in the ESI.†
Comparison of compounds 1–4, and 6
Before we examine in more detail the structures of 1–4 and 6, a few remarks are needed. These compounds, differing only in the amino acid chain part (R, Fig. 1), all contain three distinct entities, and each of them has different intermolecular “preferences”. Thus we have a flat benzene ring that we often find either π-stacked or with C–H σ–π interactions. Then we have aliphatic groups (except for 6) that will normally prefer to be in the presence of each other, and finally the polar hydrogen bond donors and acceptors that will generally “want” to have all free electron pairs and acidic hydrogens in bonding interactions.A convenient way to get an overall picture is to use Hirshfeld surfaces. The Hirshfeld surface reveals the intermolecular interactions, and even the weaker secondary interactions such as C–H⋯π, C⋯H and H⋯H contacts.51–53 This surface represents the volume where, inside, electron density is dominated by the atoms in the molecule in focus, while outside, electron density is dominated by all other components of the crystal. Each point on the surface has one closest atom inside and one closest atom outside the surface. We can therefore determine what percentage of the surface that originates from O⋯H, C⋯C or any other close interaction. This could be a crude first way of comparing similar structures and a diagram of these percentages for compounds 1–4§ and 6–7 are found in Fig. 3.
 | ||
| Fig. 3 The major contributions to the Hirshfeld surfaces in terms of closest atom–atom interactions for compounds 1–4 and the glycine derivative of terephthalamide 6 and benzene-1,3,5-tricarboxamide 7, a rough first way to compare intermolecular interactions. | ||
We immediately observe that the derivatives with longer aliphatic chains, 2–4, have a higher proportion of H⋯H interactions (blue), which is reasonable as these groups, as stated above, will tend to interact with each other, but also, simply, because of the higher percentage of hydrogen in these molecules.¶
Another notable feature is the higher proportion of C⋯O (purple) in the glycine derivative 6 compared especially to 1–2. This stems from the interaction of the carbonyl groups with the flat side of the benzene ring in 6, an arrangement we suggest arises because 6 lacks the CH3 groups that tend to come close to the aromatic ring in the alanine derivative 1.
One thing that is not picked up so clearly in these plots is the difference between on one side compounds 3 and 4 that form the same hydrogen bonded assemblies in unit cells that are close to identical, and compound 2.
Both structures 3 and 4 contain two lattice water molecules that connect the building blocks to form a 2D hydrogen bonded net. The interactions between the building blocks are all lattice water based. In other words there are no direct intermolecular hydrogen bonds between the actual building blocks. This is in contrast to 2 that has direct hydrogen bonds between the building blocks and only one water molecule.
It is worth noting here that 3 and 4 pack with the aliphatic groups facing each other, as shown in Fig. 4, just as intuition would suggest. This leaves the aromatic ring exposed and instead of CH3 interactions as in 1 it adopts the carbonyl interaction as in 6.
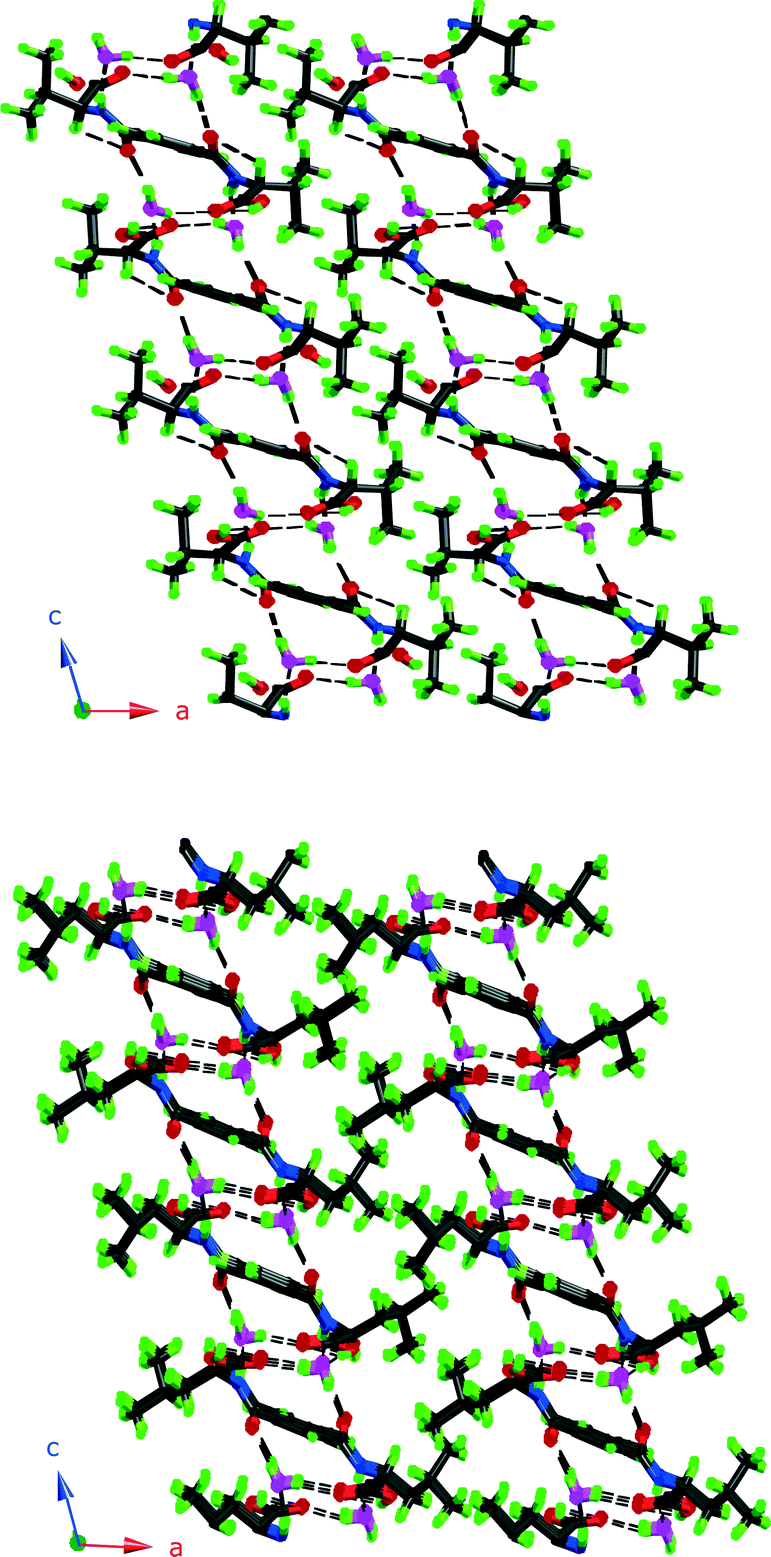 | ||
| Fig. 4 Packing diagrams of 3 and 4. Note how the aliphatic groups assemble and are kept separate from the hydrogen bonding parts. | ||
Another property of 3 and 4 that has some implications is chirality. As racemic valine was used, 4 is a racemic compound, the crystals obtained are of the R,S molecule, and a non-chirodescriptive space group would have been expected. However, solutions in P2/1c give a significantly higher R-factor than when solved and refined in the space group P21, and the apparent centre of symmetry is only pseudo. The abnormal anisotropic displacement parameters of the C8-carbon (see Fig. S18†) could be due to some minor R/S disorder present at the chiral carbon, however this could not be satisfactorily modelled.
On the other hand, 3 is enantiomerically pure (some indication of very minor isomerisation can be detected and again, solutions in P2/1c give a significantly higher R-factor) and having the chirodescriptive space group P21 is logical. Somehow there is thus a chiral arrangement of 4. This riddle is solved when one realizes first that the R-side is always facing another R-side and vice-versa and that the aliphatic groups meet around a screw axis parallel to the b-axis, thus arranging these groups in a chiral, helical way.
This observation then begs the question why compound 2 does not form the same kind of structure, being an isomer of 3. It is hard to fathom why the same packing and chiral arrangement would not be possible, but we refrain from further speculation, as this may simply be a question of crystallization conditions.
Comparing compound 5 and compound 7
The local molecular structure of 5 resembles that of the earlier published glycine derivative 2,2′,2′′-((benzene-1,3,5-tricarbonyl)tris(azanediyl))triacetic acid trihydrate337 as they both form the R2,2(19) based dimers as shown in Fig. 2. However, the absence of methyl groups makes it possible for the protruding chains to pack closer in 7. In addition, the glycine derivative has lattice water in the structure that participates in a hydrogen bond network.In contrast to 7, the dimeric units in 5 are further connected to six other dimers units via hydrogen bonding of O–H⋯O O(3)–H(3A)⋯O(1)#2 2.641(4) Å, #2 1/3 − x + y, 2/3 − x, −1/3 + z} forming a pcu-net, the most common of the six-connected nets,54 as shown in Fig. 5.
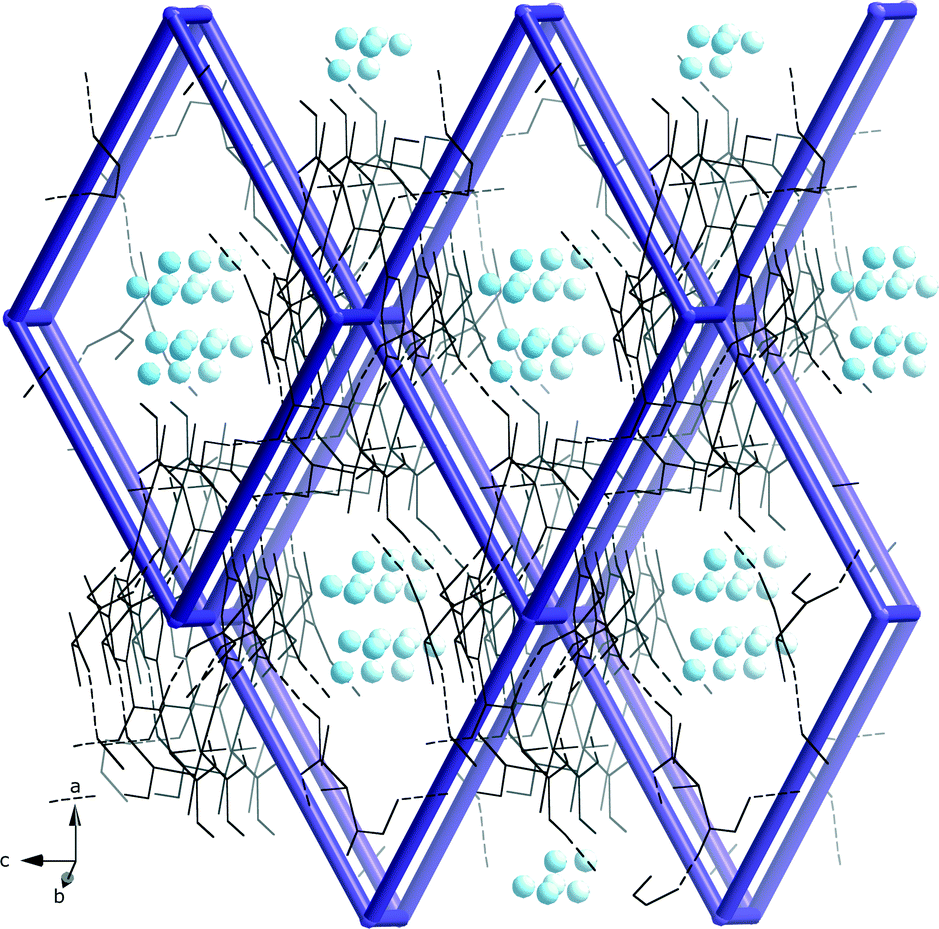 | ||
| Fig. 5 The hydrogen bonded pcu-net formed by the dimeric units of 5 with disordered water molecules (blue spheres) in the roughly 3·3 Å wide channels. | ||
Summary and conclusions
The linear disubstituted compounds can be categorized into three groups. Compound 1 and 2 form 3D hydrogen bonded nets that have some, but not all ring graph symbols identical, thus the final structure and network are not the same. Compound 1 forms chains via direct hydrogen bond of N–H⋯O between the terephthalamide derivatives. In addition two water molecules assist the chain formation and further link the chains together via hydrogen bonding. Compound 2 also contains one direct intermolecular hydrogen bond between the terephthalamide derivative molecules. However, the hydrogen bond is formed between the carboxylic COH-group and the ketone CO-group. Lattice water links the chains together. Compounds 3 and 4 form 2D hydrogen bonded structures where all the connections between terephthalamide derivatives are water mediated. Notably the graph set symbol R4,4(18) is recurring in all these substituted compounds.Compound 5 forms hydrogen bonded dimers and its comparison with 7 suggests that this motif is strong and recurring. In contrast to the glycine derivative 7, in compound 5 these dimers are connected to each other via hydrogen bonds and form a 3D pcu-net. Hirshfeld surface analyses of compounds 1–7 indicate that some of the differences between these systems and graph set symbols are also helpful in categorising and discovering differences and similarities.
From a synthesis point of view, for the further use of these ligands in building MOFs, it will be important to have complete control of the stereochemistry and less harsh conditions may be needed.
Acknowledgements
LÖ, EL and AK thank the Swedish Research Council, LÖ and CLO thank the Swedish Research Links program for Southern Africa, AK also expresses his gratitude to the Foundation for Science and Technology (FCT), Portugal, for post-doctoral fellowship (SFRH/BPD/76192/2011), AEPP was supported by the CMCSU project, the Knut and Alice Wallenberg Foundation and the MATsynCELL project, Röntgen–Ångström Cluster.Notes and references
- H. M. Heitzer, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 2013, 135, 9753–9759 CrossRef CAS PubMed.
- J. S. Miller, Chem. Soc. Rev., 2011, 40, 3266–3296 RSC.
- Metal-Organic Frameworks: Design and Application, ed. L. R. MacGillivray, Wiley, New York, 2010 Search PubMed.
- D. Farruseng, Metal-Organic Frameworks - Applications from Catalysis to Gas Storage, Wiley-VCH, Weinheim, 2011 Search PubMed.
- P. P. Bose, M. G. B. Drew, A. K. Das and A. Banerjee, Chem. Commun., 2006, 3196–3198 RSC.
- H. Cao, P. Duan, X. Zhu, J. Jiang and M. Liu, Chem. – Eur. J., 2012, 18, 5546–5550 CrossRef CAS PubMed.
- B. Wisser, A.-C. Chamayou, R. Miller, W. Scherer and C. Janiak, CrystEngComm, 2008, 10, 461–464 RSC.
- N. N. Adarsh, P. Sahoo and P. Dastidar, Cryst. Growth Des., 2010, 10, 4976–4986 CAS.
- T.-P. Tsai, Y.-T. Huang, U. Ray, Y.-J. Chang, P.-C. Cheng, C.-J. Wu, J.-D. Chen and J.-C. Wang, Polyhedron, 2010, 29, 3081–3088 CrossRef CAS PubMed.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- G. E. Kostakis, L. Casella, A. K. Boudalis, E. Monzani and J. C. Plakatouras, New J. Chem., 2011, 35, 1060–1071 RSC.
- P. J. M. Stals, J. F. Haveman, R. Martin-Rapun, C. F. C. Fitie, A. R. A. Palmans and E. W. Meijer, J. Mater. Chem., 2009, 19, 124–130 RSC.
- I. A. W. Filot, A. R. A. Palmans, P. A. J. Hilbers, R. A. van Santen, E. A. Pidko and T. F. A. de Greef, J. Phys. Chem. B, 2010, 114, 13667–13674 CrossRef CAS PubMed.
- P. Chen, P. Gao, C. Zhan and M. Liu, ChemPhysChem, 2005, 6, 1108–1113 CrossRef CAS PubMed.
- J. Roosma, T. Mes, P. Leclere, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 1120–1121 CrossRef CAS PubMed.
- S. Ray, R. P. Hegde, A. K. Das, N. Shamala and A. Banerjee, Tetrahedron, 2006, 62, 9603–9609 CrossRef CAS PubMed.
- H.-T. Zhang, Y.-Z. Li, T.-W. Wang, E. N. Nfor, H.-Q. Wang and X.-Z. You, Eur. J. Inorg. Chem., 2006, 3532–3536 CrossRef CAS.
- X. Dou, P. Li, D. Zhang and C.-L. Feng, Soft Matter, 2012, 8, 3231–3238 RSC.
- P. Li, X.-Q. Dou, Y.-T. Tang, S. Zhu, J. Gu, C.-L. Feng and D. Zhang, J. Colloid Interface Sci., 2012, 387, 115–122 CrossRef CAS PubMed.
- C. F. C. Fitie, W. S. C. Roelofs, M. Kemerink and R. P. Sijbesma, J. Am. Chem. Soc., 2010, 132, 6892–6893 CrossRef CAS PubMed.
- P. Muller, E. Weber, C. Helbig and H. Baldauf, J. Surfactants Deterg., 2001, 4, 407–414 CrossRef CAS.
- A. R. Stefankiewicz, M. R. Sambrook and J. K. M. Sanders, Chem. Sci., 2012, 3, 2326–2329 RSC.
- R. F. Ludlow and S. Otto, J. Am. Chem. Soc., 2008, 130, 12218–12219 CrossRef CAS PubMed.
- Z. Chen, X. Liu, C. Zhang, Z. Zhang and F. Liang, Dalton Trans., 2011, 40, 1911–1918 RSC.
- R. Sun, S. Wang, H. Xing, J. Bai, Y. Li, Y. Pan and X. You, Inorg. Chem., 2007, 46, 8451–8453 CrossRef CAS PubMed.
- Z. Lu, H. Xing, R. Sun, J. Bai, B. Zheng and Y. Li, Cryst. Growth Des., 2012, 12, 1081–1084 CAS.
- T. Min, B. Zheng, J. Bai, R. Sun, Y. Li and Z. Zhang, CrystEngComm, 2010, 12, 70–72 RSC.
- C. Zuo, J. Bai, R. Sun and Y. Li, Inorg. Chim. Acta, 2012, 383, 305–311 CrossRef CAS PubMed.
- G. E. Kostakis, L. Casella, N. Hadjiliadis, E. Monzani, N. Kourkoumelis and J. C. Plakatouras, Chem. Commun., 2005, 3859–3861 RSC.
- H. T. Zhang and X. Z. You, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1163–m1165 CAS.
- J. Duan, B. Zheng, J. Bai, Q. Zhang and C. Zuo, Inorg. Chim. Acta, 2010, 363, 3172–3177 CrossRef CAS PubMed.
- D. S. F. Eissmann and E. Weber, Struct. Chem. Commun., 2010, 1, 72 Search PubMed.
- B. Gong, C. Zheng and Y. Yan, J. Chem. Crystallogr., 1999, 29, 649–652 CrossRef CAS.
- S. R. Batten, N. R. Champness, X. M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- L. Öhrström, Network and graph set analysis, inSupramolecular Chemistry: From Molecules to Nanomaterials, Volume 6: Supramolecular Materials Chemistry, ed. P. A. Gale, J. W. Steed and L. Barbour, John Wiley & Sons Ltd, UK, 2012 Search PubMed.
- A. U. Czaja, N. Trukhan and U. Muller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- C. Valente, E. Choi, M. E. Belowich, C. J. Doonan, Q. Li, T. B. Gasa, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, Chem. Commun., 2010, 46, 4911–4913 RSC.
- L. Öhrström and K. Larsson, Molecule-Based Materials - The Structural Network Approach, Elsevier, Amsterdam, 2005 Search PubMed.
- A. I. Vogel, A. R. Tatchell, B. S. Furnis, A. J. Hannaford and P. W. G. Smith, Vogel's Textbook of Practical Organic Chemistry, Pearson Education, London, 5th edn, 1989 Search PubMed.
- CrysAlisPro, Oxford Diffraction Ltd., Abingdon, Oxfordshire, UK, 2005 Search PubMed.
- SAINT-Plus, Bruker AXS Inc., Madison, Wisconsin, USA., 2004 Search PubMed.
- XPREP, Bruker AXS Inc., Madison, Wisconsin, USA., 2003 Search PubMed.
- L. J. Barbour, J. Appl. Crystallogr., 1999, 351–352 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- M. C. Etter, J. C. Macdonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef.
- L. Öhrström, in Encyclopedia of supramolecular chemistry, ed. L. Barbour, Wiley Chichester, UK, 2012, vol. 8 Supramolecular materials chemistry Search PubMed.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: CIF files, additional structures and Hirshfeld plots. CCDC 993036–993040. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00605d |
| ‡ Present address: Centro de Química Estrutural, Instituto Superior Técnico, University of Lisbon, Av. Rovisco Pais, 1049-001, Lisbon, Portugal. |
| § The disorder of 5 makes a Hirshfeld surface difficult to compare and interpret. |
| ¶ Possible ways to account for the difference in composition are to weigh the contributions by atom percentages or to calculate interactions only for the part that is common to all molecules. |
| This journal is © The Royal Society of Chemistry 2014 |