Iron oxide nanoparticles induce Pseudomonas aeruginosa growth, induce biofilm formation, and inhibit antimicrobial peptide function†
Jennifer
Borcherding
a,
Jonas
Baltrusaitis
b,
Haihan
Chen
b,
Larissa
Stebounova
b,
Chia-Ming
Wu
b,
Gayan
Rubasinghege
b,
Imali A.
Mudunkotuwa
b,
Juan Carlos
Caraballo
a,
Joseph
Zabner
a,
Vicki H.
Grassian
*b and
Alejandro P.
Comellas
*a
aDepartment of Internal Medicine, University of Iowa, Iowa City, IA 52242, USA. E-mail: alejandro-comellas@uiowa.edu
bDepartment of Chemistry, University of Iowa, Iowa City, IA 52242, USA. E-mail: vicki-grassian@uiowa.edu
First published on 11th February 2014
Abstract
Given the increased use of iron-containing nanoparticles in a number of applications, it is important to understand any effects that iron-containing nanoparticles can have on the environment and human health. Since iron concentrations are extremely low in body fluids, there is potential that iron-containing nanoparticles may influence the ability of bacteria to scavenge iron for growth, affect virulence and inhibit antimicrobial peptide (AMP) function. In this study, Pseudomonas aeruginosa (PA01) and AMPs were exposed to iron oxide nanoparticles, hematite (α-Fe2O3), of different sizes ranging from 2 to 540 nm (2 ± 1, 43 ± 6, 85 ± 25 and 540 ± 90 nm) in diameter. Here we show that the greatest effect on bacterial growth, biofilm formation, and AMP function impairment is found when exposed to the smallest particles. These results are attributed in large part to enhanced dissolution observed for the smallest particles and an increase in the amount of bioavailable iron. Furthermore, AMP function can be additionally impaired by adsorption onto nanoparticle surfaces. In particular, lysozyme readily adsorbs onto the nanoparticle surface which can lead to loss of peptide activity. Thus, this current study shows that co-exposure of nanoparticles and known pathogens can impact host innate immunity. Therefore, it is important that future studies be designed to further understand these types of impacts.
Nano impactPseudomonas aeruginosa, chosen due to its prevalence and importance in human diseases, cultured with iron oxide (α-Fe2O3) nanoparticles, shows a correlation between bacterial growth and decreasing particle size. These results are attributed to an increase in soluble iron due to enhanced dissolution for the smallest nanoparticles. Antimicrobial peptide activity is also inhibited when exposed to the smallest iron oxide nanoparticles as evidenced by a decrease in bacterial killing, one of the host's first lines of defense against foreign invaders. These studies suggest that nanoparticle toxicity screening should take into consideration effects beyond direct toxicity and include co-exposures with known pathogens and their impact on host innate immunity. |
Introduction
Iron-containing nanoparticles exhibit intrinsic qualities that enhance their ability to be used in a number of industrial applications. For example, these materials are used in recording media because of their paramagnetic properties, as contrast imaging agents in biomedical applications and as catalysts in clean fuel synthesis.1 Although the properties of iron-containing nanoparticles prove beneficial in a wide-range of applications, little is known about the effects of iron-containing nanoparticles on bacterial pathogens. Through evolution, several organisms, including humans, have developed numerous mechanisms that limit iron bioavailability to microbial pathogens, keeping extremely low iron concentrations in various biological fluids (10−18 M).2 Thus, iron-containing nanoparticles can act as an exogenous iron source for bacteria, becoming potentially deleterious to human health.In our recent studies, we showed that both iron-containing particles from coal fly ash,3 a byproduct of coal combustion, and volcanic ash,4 from the eruption in Eyjafjallajökull, Iceland, impair host innate immunity and increase bacterial growth. Due to the increased use of iron-containing nanoparticles in industry, as well as its prominent natural occurrence, the potential for increased iron-containing particle exposure, as particulate matter in air, is a concern. For example, hematite, alpha-iron oxide (α-Fe2O3), is one of the most abundant polymorphs of iron oxides detected in atmospheric dust over a wide range of sizes.5 Additionally, epidemiological studies have shown that exposure to ambient air pollution is associated with increased respiratory exacerbations,6–9 pneumococcal infections,10 otitis media,11 and eye infections.12 Many of these effects are associated with exposure to particulate matter (PM), which is composed of particles of various sizes and compositions and thus very different properties.
It is known that particle size can play an important role in the chemistry of metal oxide nanoparticles and recent studies have shown that chemical and biological reactivity of iron-containing carbon nanoparticles is correlated with particle size and surface area.13 Both thermodynamic and kinetic factors can, for example, control the aqueous phase dissolution of metal oxide nanoparticles, while aggregation of nanoparticles can impact both the dissolution rate and reactivity of the nanoparticles.14 In one example, isolated nanorods of α-FeOOH (goethite) have shown an increase in iron dissolution compared to microrods due to greater surface area and chemical activity.15 In addition, nanomaterial shape and crystallinity can influence toxicological response.16
The impact of iron oxide nanoparticles on bacterial growth, virulence and innate immunity is the focus of this paper. Using well-characterized hematite (α-Fe2O3) particles of different sizes, 2 ± 1, 43 ± 6, 85 ± 25 and 540 ± 90 nm, our experimental results demonstrate that iron oxide nanoparticles increase bacterial growth with a correlation between growth and particle size and surface area. We also demonstrate that biofilm formation is found to increase in a size dependent manner that correlates with dissolved iron. Finally, we show that antimicrobial peptide (AMP) activity is inhibited when these peptides are exposed to the smallest iron oxide nanoparticles, as evidenced by a decrease in bacterial killing, one of the host's first lines of defense against foreign invaders. Several mechanisms may be operative for AMP inhibition. As discussed below, these studies suggest that the effects of co-exposures with nanoparticles and pathogens should be further studied as well as their impact on host innate immunity.
Experimental methods
Iron oxide nanoparticles
We have used iron oxide (α-Fe2O3) particles of different sizes. The details of the synthesis methods and sources of α-Fe2O3 nanoparticles used in this study are given in the ESI.†Characterization of iron oxide nanoparticles
The morphology, composition, surface area and crystalline properties of the iron oxide used in this study are provided here. Bulk crystalline phase analysis was performed using powder X-ray diffraction (XRD) utilizing Rigaku MiniFlex II spectrometer with a filtered cobalt source. Two theta angles ranged from 20° to 80° at a 0.02° step with a step dwell time of 0.6 s. JEOL 2100F TEM, operating at 200 kV accelerating voltage, was used to determine the average particle size and morphology.Iron oxide nanoparticles were prepared for TEM imaging by suspending and sonicating in methanol for 5 minutes and then depositing onto 200 mesh Cu grids (Ted Pella) and drying in air. More than 200 particles were counted in each case to determine the particles size and relevant standard deviation.
BET surface area measurements were determined using the nitrogen adsorption seven-point Brunauer–Emmett–Teller (BET) technique on a Quantochrome NOVA 4200e instrument in the P/Po range of 0.05 to 0.3. Samples were degassed at 80 °C overnight prior to the BET measurement.
Surface elemental analysis of the iron oxide nanoparticles was performed using a custom-designed Kratos Axis Ultra X-ray photoelectron spectroscopy system.17 The surface analysis chamber is equipped with an aluminum Kα X-ray gun and a 500 mm Rowland circle silicon single crystal monochromator. The X-ray gun was operated using a 15 mA emission current at an accelerating voltage of 15 kV. Low-energy electrons were used for charge compensation to neutralize the sample. High-resolution spectra were acquired in the region of interest using the following experimental parameters: 20–40 eV energy window, pass energy of 20 eV, step size of 0.1 eV, and dwell time of 1000 ms. One sweep was used to acquire a survey spectrum of all binding regions. The absolute energy scale was calibrated to the Cu 2p2/3 peak binding energy of 932.6 eV using an etched copper plate. For analysis, samples were mounted by pressing them into indium foil. All spectra were calibrated using the adventitious C1s peak at 285.0 eV. A Shirley-type background was subtracted from each spectrum to account for inelastically scattered electrons that contribute to the broad background. CasaXPS software was used to process the XPS data.18 XPS data were corrected using relative sensitivity factor values from the Kratos library for elemental quantification. The components of the peaks contain a Gaussian/Lorentzian product with 30% Lorentzian and 70% Gaussian characters. An error of ±0.2 eV is reported for all peak binding energies.
Experimental preparation and analysis of particles in M9 media
Characterized α-Fe2O3 nanoparticles were suspended in iron deficient media (M9) at pH 6.8 (BD Difco Minimal media (M9)) with 2.2 mM glucose, 0.002 M magnesium sulfate (MgSO4), 0.001 M calcium chloride (CaCl2) and 25 mM sodium succinate. Particle suspensions were sonicated for 10 minutes immediately prior to conducting experiments.Agglomeration of the nanoparticles in M9 media was monitored using a dynamic light scattering (DLS) instrument from Beckman Coulter. Iron oxide nanoparticles used in this study formed larger aggregates in M9 media of approximately the same size, ~500 nm, as measured by DLS.
Iron oxide nanoparticles were suspended in 4 mL of M9 media at a 10 μg mL−1 concentration and incubated at pH 6.8 for 4 h at 37 °C while rotating at 200 rpm. Afterwards, the samples were spun at 2950 rpm for 8 min followed by filtering through 0.2 μm filter to remove undissolved particles. A colorimetrical method using 1,10-phenanthroline which forms a complex with dissolved Fe(II) that absorbs light at 510 nm was used to measure the solution phase concentrations of iron.19 For these experiments, 40 μL of 1.5 M hydroxylamine hydrochloride was added to 1 mL of the centrifuged and filtered sample which reduces Fe(III) to Fe(II) followed by 200 μL of a 5 mM 1,10-phenanthroline solution and 200 μL of an ammonium acetate buffer. Thus, the total dissolved iron is reported here.
ATR-FTIR spectroscopy of lysozyme adsorption on α-Fe2O3 nanoparticles in M9 media
Experiments using attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) were done to determine if lysozyme, an antimicrobial peptide, readily adsorbs onto iron oxide nanoparticle surfaces. A thin layer of 2 nm α-Fe2O3 nanoparticles was deposited as a hydrosol onto the ATR crystal and allowed to dry. The same M9 media with lysozyme (600 μg mL−1) was introduced into the ATR-FTIR cell and allowed to flow over the nanoparticles. The spectra were recorded as a function of time (6 hours) and 250 scans were co-averaged at a resolution of 4 cm−1. For solution phase measurements, a spectrum of the M9 minimal media containing 600 μg mL−1 lysozyme was measured by flowing the solution over the ATR element. In another set of experiments, a blank ATR crystal spectrum was used as the background for both solution and adsorbed phase measurements. The reversibility of AMP adsorption was tested following the adsorption experiment by flowing water at ~pH 7 over the AMP adsorbed α-Fe2O3 nanoparticle thin film and collecting the spectra over a period of 6 hours.Bacterial growth experiments
Pseudomonas aeruginosa (PA01) was chosen as a model in this study due to its prevalence and importance in human diseases.6,7P. aeruginosa was grown overnight in M9 media and subcultured for 3 hours. Growth was determined by adding 10 μg mL−1 α-Fe2O3 particles to the mid-log phase growth and measuring the OD600 at 37 °C for 9 hours while correcting for particle absorbance effects at every time point. 10 μg mL−1 FeCl3 was used as a positive control and γ-Al2O3 was used as a negative control. Colony forming units per milliliter, CFU ml−1, were determined by conducting the above-mentioned experiments and sampling at four different time points to determine the changes in CFU ml−1 due to the presence of α-Fe2O3 particles in comparison to the controls.Biofilm measurements
In order to test biofilm formation in the presence of α-Fe2O3 particles, confocal imaging was used by measuring green fluorescent protein (GFP) intensity. GFP-PA01 constitutively expressing the plasmid pMRP9-1 was used and grown overnight in M9 media with 30 μg mL−1 gentamicin. The pMRP9-1 plasmid was constructed by cloning an 0.8 kp KPnI–HindIII fragment containing the Aequorea victoria GFP gene from pGFPmut2 into KpnI–HindIII digested pUCP18.20 This makes the GFP gene under control of the lac promoter and the T7 gene 10 ribosomal binding site. The overnight culture was diluted 1/100 and added to a 96 well plate with a coverslip mounted perpendicularly in order to allow the quantification of the depth of biofilm formation. After 48 hours, confocal microscopy was used to measure the depth of biofilm formation on the coverslips. Z-stack was used to create surface plots of biofilm growth and GFP intensity, as well as concavalin A–Texas red intensity. Intensities are expressed as percentage (%), i.e. the percentage of the fluorescence intensity of biofilm under each condition relative to the saturation fluorescence within the field. These percentages were measured and compared by a one-way ANOVA in order to determine the significance.Radial diffusion assay
The radial diffusion assay was conducted according to Steinberg and Lehrer.21 Briefly, PA01 was grown overnight in M9 media and subcultured. Experiments were conducted in the log phase growth; 10 μl of OD = 0.23 was added to 10 ml of underlay containing agarose and sodium phosphate buffer. An antimicrobial mixture of 600 μg of lysozyme, 200 μg of lactoferrin and 100 ng each of human neutrophil peptide (HNP) 1 & 2 were added to 5 μl wells. 10 μg mL−1 of 2 or 540 nm α-Fe2O3 particles were added to the AMP mixture and added to the 5 μl wells. Fig. S2† shows a schematic diagram of the AMP experimental protocol. First, particles were added to the antimicrobial cocktail for 1 hour at 37 °C. After 1 hour, the cocktail was spun to remove the particles. The cocktail was then added to the underlay in separate wells. After 3 hours, a nutrient filled overlay was added to the plate and allowed to incubate overnight. After overnight incubation, the diameter of the bacterial zone killing at the largest point was measured. The normalized plot of the six experiments was used and a one-way ANOVA was used to analyze the four different conditions (vide infra). In order to determine whether antimicrobial inhibition was due to the particle effects or soluble iron, these experiments were done in the absence (as stated above) as well as in the presence of nanoparticles (vide infra).Results and discussion
Iron oxide particles
The results of the characterization data for the iron oxide particles used in this study are summarized in Table 1 and discussed in this section. TEM images and XRD patterns are shown in Fig. 1 for the four different sizes of iron oxide nanoparticles used in this study. TEM images show fairly uniform particle size distributions within each sample, with evidence of particle aggregation. With the exception of 540 nm particles that were irregular in shape, the other nanoparticles have morphologies best described as spherical or rhombohedral, as is typically seen for hematite.22| Sample | Sizea (nm) | BET surface areab (m2 g−1) |
|---|---|---|
| a Particle diameter and standard deviation as determined from the TEM images. b Triplicate measurements were done for BET surface area measurements. c Al2O3 was used as a control nanoparticle that did not contain iron. | ||
| α-Fe2O3 | 2 ± 1 | 220 ± 10 |
| α-Fe2O3 | 43 ± 6 | 39 ± 1 |
| α-Fe2O3 | 85 ± 25 | 11 ± 1 |
| α-Fe2O3 | 540 ± 90 | 2.5 ± 0.1 |
| Al2O3c | 16 ± 5 | 101 ± 4 |
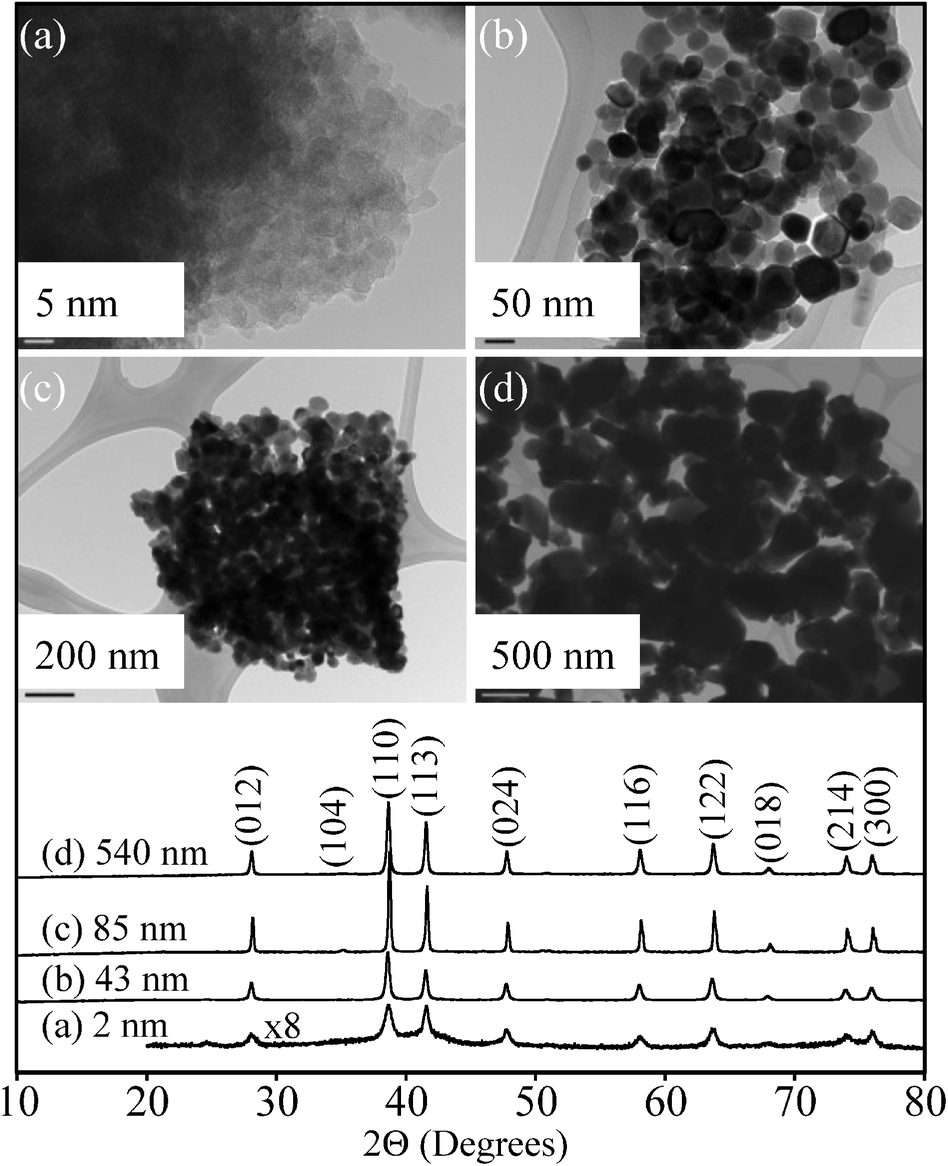 | ||
| Fig. 1 TEM images and powder X-ray diffraction (XRD) patterns (bottom curves) of α-Fe2O3 nanoparticles used in this study: (a) 2 nm, (b) 43 nm, (c) 85 nm, and (d) 540 nm. | ||
XRD data in Fig. 1 show the most intense peaks at 39° and 41° due to the (104) and (110) reflections of hematite phase, α-Fe2O3.23 Crystalline α-Fe2O3 is present in all of these different sized nanoparticles, while small peaks at 36° and 52° are due to the minor presence of other iron oxide phases.24 Additionally, there is a small amount of amorphous character as evidenced in the XRD as an underlying background in the synthesized 2 ± 1 nm iron oxide nanoparticles. This information is important in order to fully understand the biological response, in this case the growth and pathogenicity experiments (vide infra), as iron oxide and hydroxide nanoparticles can have different dissolution mechanisms depending on their crystallinity and chemistry.25 In particular, amorphous iron oxides and hydroxides have been shown to dissolve faster than their crystalline materials. Wahid and Kamalam proposed that crystalline Fe(III) oxides have to be transformed into amorphous Fe(III) oxides via microbial processing or hydration, in order to be transformed further into a bioavailable Fe(II) form.26 In fact, the Fe(III) extraction process with EDTA and ammonium oxalate from an amorphous iron oxide, hematite and goethite mixture only dissolved an amorphous fraction due to its increased reactivity.27 The dissolution rate of Fe(III) has also shown dependence on crystal size, crystalline phase, and crystallinity.28
The measured BET surface areas of these four samples increase with decreasing particle size with the smallest 2 ± 1 nm particles having the largest BET surface area of 220 ± 10 m2 g−1. Since heterogeneous solid–liquid interactions, including dissolution, are surface limited, 2 ± 1 nm particles can be expected to be the most reactive during cell growth and pathogenicity experiments. An overall summary of the properties of the particles used in this study, including the TEM derived primary particle size and BET surface areas, is provided in Table 1.
To determine the composition of the nanoparticle surfaces, XPS spectra were obtained and are shown in Fig. 2. The electrons that escape the sample during XPS analysis originate from the topmost region of surface thus making this technique very appropriate in distinguishing surface sites that may be responsible for potential nanoparticulate sample reactivity.29 The high-resolution spectra of Fe2p, O1s and C1s for nanoparticles used in this work are shown in Fig. 2. All of these have nearly the same spectral features showing very similar chemical composition of the surface. Namely, a doublet in the Fe2p region with peaks at 710.8 and 724.4 eV is due to the 2p3/2 and 2p1/2 transition in hematite.30 O1s spectrum showed a primary peak at 530.0 eV due to the surface Fe–O bonds and a broader feature at 531.7 eV, which is due to the combination of lattice and adsorbed Fe–OH bonds.30 Additionally, C1s spectrum showed a strong peak at 285.0 eV due to the adventitious carbon and 289.0 eV due to the O–C–O bonds.31
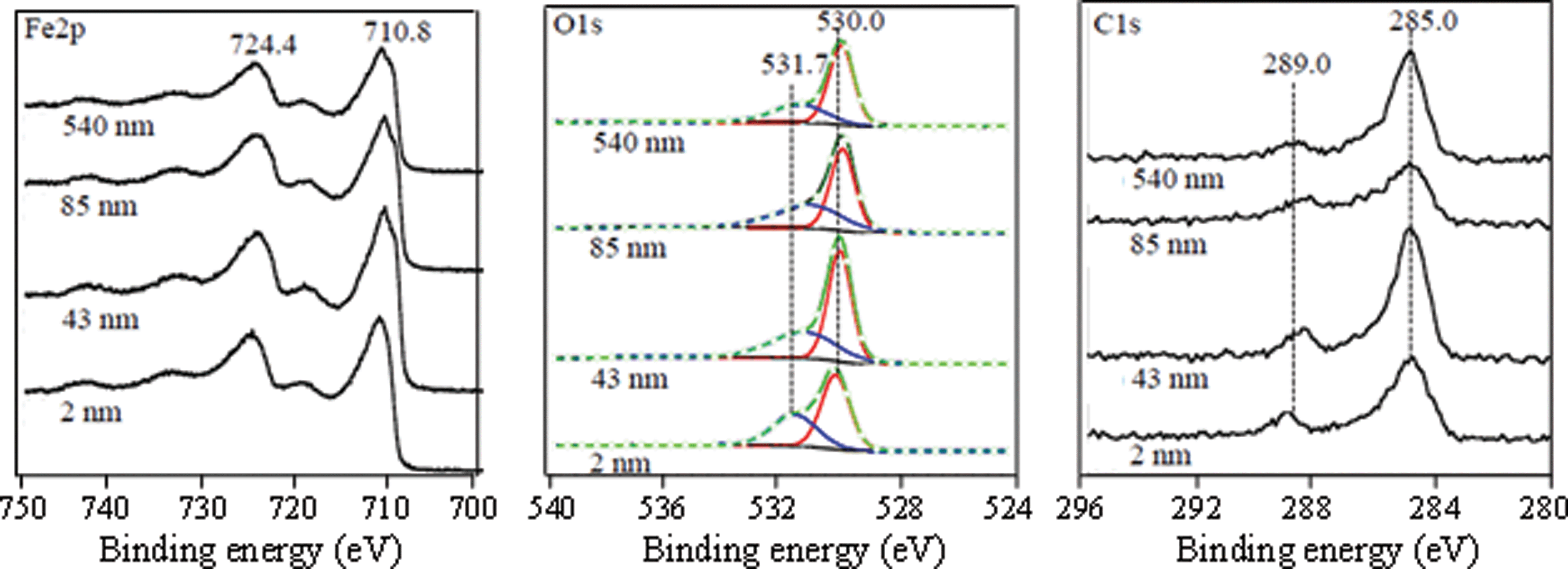 | ||
| Fig. 2 XPS high-resolution data in the Fe2p, O1s and C1s regions for α-Fe2O3 particles of different sizes (2, 43, 85 and 540 nm). The O1s region was peak-fit into two separate peaks corresponding to O and OH groups using Gaussian–Lorentzian components. The red and blue curves represent the O and OH groups at 530.0 and 530.7 eV, respectively, used to curve fit the spectra. The black solid line represents the experimental data and the green line with circular markers represents the total calculated fit. The ratio of the areas for these different O1s peaks as a function of particle size is given in Table 2. See text for further details. | ||
Collectively, these XPS data show surface species present on the hematite nanoparticles, which include structural and adsorbed hydroxyl groups and some adventitious carbon material either left over from the synthesis or from ambient air contamination. To assess the relative population of each element and corresponding species within, we performed quantification based on XPS data. The area under each peak is representative of the population of the species or functional group, allowing for the XPS spectra to be used in a quantitative way in terms of relative ratios of the elements or species. These ratios are shown in Table 2. In particular, the O1s![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe2p ratio shows a value of 1.40, close to the expected stoichiometric value of 1.5 in hematite, α-Fe2O3, in all samples except the smallest 2 ± 1 nm particles. For the smallest particles, the O1s:Fe2p ratio is found to be 1.23 suggesting a slightly reduced stoichiometry. This phenomenon can also be related to the fact that small metal oxide nanoparticles are prone to reduction in the XPS instrument under X-ray illumination under vacuum, as has been observed for 6 nm CuO particles.32 The C1s:Fe2p ratio in all samples was close to ~0.3 showing that just a little less than one-third of the surface sites are covered with carbonaceous materials. Two observed oxygen species in the O1s region, Fe–O and Fe–OH, accounted for 60% and 40% of the total oxygen, respectively. This shows that the hematite nanoparticle surface is truncated with hydroxyl groups which have been shown to be active sites for surface dissolution and adsorption.22
:Fe2p ratio shows a value of 1.40, close to the expected stoichiometric value of 1.5 in hematite, α-Fe2O3, in all samples except the smallest 2 ± 1 nm particles. For the smallest particles, the O1s:Fe2p ratio is found to be 1.23 suggesting a slightly reduced stoichiometry. This phenomenon can also be related to the fact that small metal oxide nanoparticles are prone to reduction in the XPS instrument under X-ray illumination under vacuum, as has been observed for 6 nm CuO particles.32 The C1s:Fe2p ratio in all samples was close to ~0.3 showing that just a little less than one-third of the surface sites are covered with carbonaceous materials. Two observed oxygen species in the O1s region, Fe–O and Fe–OH, accounted for 60% and 40% of the total oxygen, respectively. This shows that the hematite nanoparticle surface is truncated with hydroxyl groups which have been shown to be active sites for surface dissolution and adsorption.22
Growth of P. aeruginosa in the presence of iron oxide nanoparticles
In order to determine the effects of iron oxide nanoparticles on bacterial growth, P. aeruginosa, a known bacterial pathogen to humans,33 animals34 and plants,34,35 was grown overnight in iron deficient media (M9). To test our hypothesis of size-dependent effects of iron oxide particles on bacterial growth, 10 μg mL−1 of the four different sized α-Fe2O3 particles (2, 43, 85 and 540) were added to PA01 cultures and growth was measured. 10 μg mL−1 iron chloride (FeCl3),36 a source of soluble iron used for bacterial growth, and 10 μg mL−1 aluminum oxide (α-Al2O3), an iron-deficient control particle, were used as positive and negative controls, respectively (Fig. 3 (a)). As shown in Fig. 3 (b), the smallest nanoparticle (2 ± 1 nm) induces the greatest amount of growth when compared to larger particles. In order to understand whether the rate of growth was different between the various groups, average rates of OD600 change per hour were calculated during the log phase growth (3–7 hours). There was a size dependent rate increase (Fig. 3 (c)). Specifically, using a one-way ANOVA, the 2 nm α-Fe2O3 rate was significantly greater than the 540 nm and 85 nm rates (p < 0.05). Furthermore, a correlation between change in bacterial growth and particle BET surface area is found (R2 = 0.8153). Interestingly, this correlation is seen despite the fact that the iron oxide nanoparticles form larger agglomerates in the M9 media, near 500 nm in size as measured by DLS. However, it should be noted that there may be smaller particles or particle clusters not seen in the DLS. These smaller particles or particle clusters may be in equilibrium with the larger nearly half-micron sized aggregates.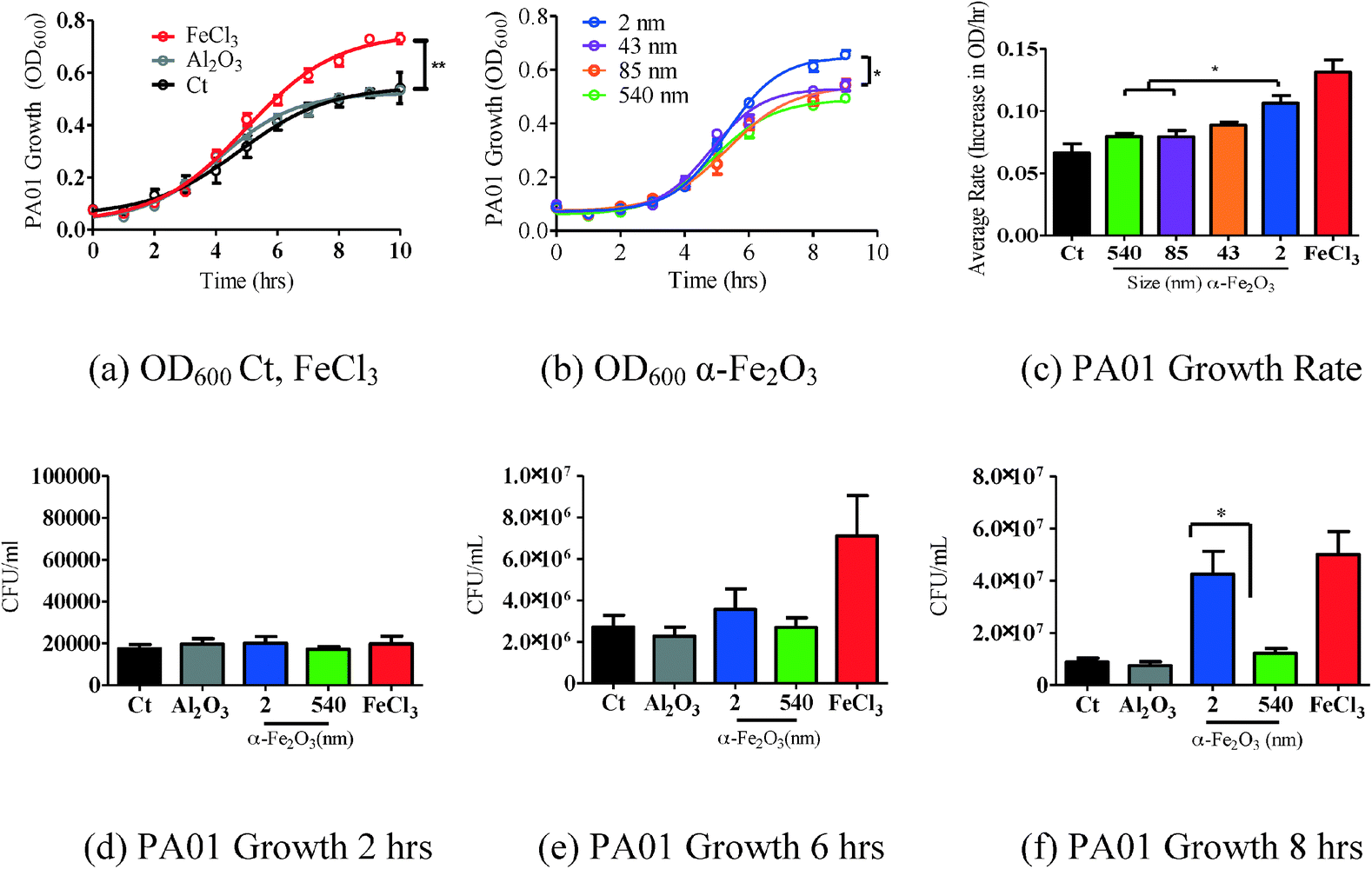 | ||
| Fig. 3 PA01 grown overnight in M9 media was subcultured and exposed to four α-Fe2O3 particles (10 μg mL−1) of different sizes and surface areas (Table 1). Growth was recorded over 9 hours. (a) FeCl3 increased OD600 significantly more than the control (Ct). (b) PA01 exposed to the smaller particles with a larger surface area had the largest growth effect. Non-linear regression (curve-fit) with a variable slope from three independent experiments was used for statistical analysis. Data were compared for all parameters of the growth curve using the extra sum of squares F-test to detect differences throughout the entire growth curve. n = 3 in triplicates. SEM reported *p < 0.0001. (c) Rate of OD600 growth was determined during the log phase growth (3–7 hours). There was a size dependent rate increase throughout the log phase. The rate of 2 nm α-Fe2O3 growth was significantly more than 540 nm and 85 nm. (d–f) CFUs mL−1 were measured at 2, 6, and 8 hours, respectively. At 8 hours, 2 nm α-Fe2O3 increased growth significantly more than 540 nm α-Fe2O3. One-way ANOVA was used to determine the significance of growth rate and CFU mL−1 experiments. SEM reported *p < 0.05. | ||
To determine whether the OD600 increase correlated with an increase in CFU mL−1 throughout the growth phase, we measured the CFUs at 2, 6 and 8 hours. At 2 hours, there was no difference in the CFU mL−1 between conditions (Fig. 3 (c)). At 6 hours, FeCl3 significantly increased growth more than the control and 540 nm particles (Fig. 3 (d)). At 8 hours, both FeCl3 and 2 nm α-Fe2O3 significantly increased growth more than the control (p < 0.05) (Fig. 3 (e)). Therefore the CFU mL−1 data correlated with the OD600 measurements of bacterial growth.
To determine whether this effect was due to different propensities of dissolved iron in the different samples, all four samples were incubated for 4 hours at 37 °C in M9 media. To test whether the iron is in the form of soluble ions, the concentration of total dissolved iron was measured colorimetrically with 1,10-phenanthroline.19 Dissolved iron was measured for 2 and 43 nm particles using this method, 9 ± 2 and 4 ± 1 μg L−1, respectively (Table 3). Blank samples were also analyzed to ensure that no significant iron was detected in the blank solution. No iron was detected after 85 and 540 nm particle dissolution at pH 6.8 in M9. Therefore, the 2 nm particle has the highest solubility and iron bioavailability for P. aeruginosa.
Furthermore, since the optimal iron requirement for bacterial growth is 0.3–1.8 μM for in vitro cultures,37 the amount of iron in the media with the other iron oxide particles could be beyond our experimental detection limits but well within the range of the iron requirement through other mechanisms such as direct iron–bacteria interactions. What is clear from these data is that iron-containing particles play a role in bacterial growth and these particles provide a source of bioavailable iron.
Iron oxide particles impact biofilm formation in a size-dependent manner due to release of soluble Fe(III) from the smaller particles
Bacteria not only exist in planktonic form but also frequently form highly structured communities called biofilms that can coat the inside of tubes such as catheters or faucets or form dense communities in the lungs of individuals with cystic fibrosis (CF) or chronic obstructive pulmonary disease (COPD).38 It has been shown that the presence of iron, specifically FeCl3, biofilm formation increases.39 Based on our growth results, we hypothesized that iron oxide particles would induce biofilm formation in P. aeruginosa cultures.Biofilms were grown on coverslips that were mounted at 90° in a 96 well plate. Mature biofilms were allowed to develop and imaged at 48 hours. In the presence of 10 μg mL−1 iron oxide particles, biofilm formation was increased more than in the presence of Al2O3, a particle of similar size used to control for particle effects (Fig. 4 (a)). FeCl3 was used as a positive control for biofilm formation and showed a significant increase in growth when comparing GFP intensity expressed as percentage and analyzed by one-way ANOVA (Fig. 4 (d)). More specifically, the mean ± standard error of the mean (SEM) percentages of GFP per group are as follows: Al2O3, 20.38 ± 2.63; 2 nm α-Fe2O3, 87.15 ± 9.11; 540 nm α-Fe2O3, 21.69 ± 6.74; and FeCl3, 91.94 ± 10.54. The smaller 2 nm α-Fe2O3 particle increased biofilm formation significantly more than the larger 540 nm α-Fe2O3 particle (Fig. 4 (b) and (c)). In order to determine whether this difference was due to increased bacterial number or matrix formation, we measured concavalin A (conA) in the biofilms in the presence of 2 nm and 540 nm α-Fe2O3 particles. The mean ± SEM for 2 nm is 41.35 ± 8.489 and 20.70 ± 50.64 for 540 nm. Therefore the matrix formation in the 2 nm biofilm was increased more than that in the 540 nm α-Fe2O3 biofilm (p = 0.0396).
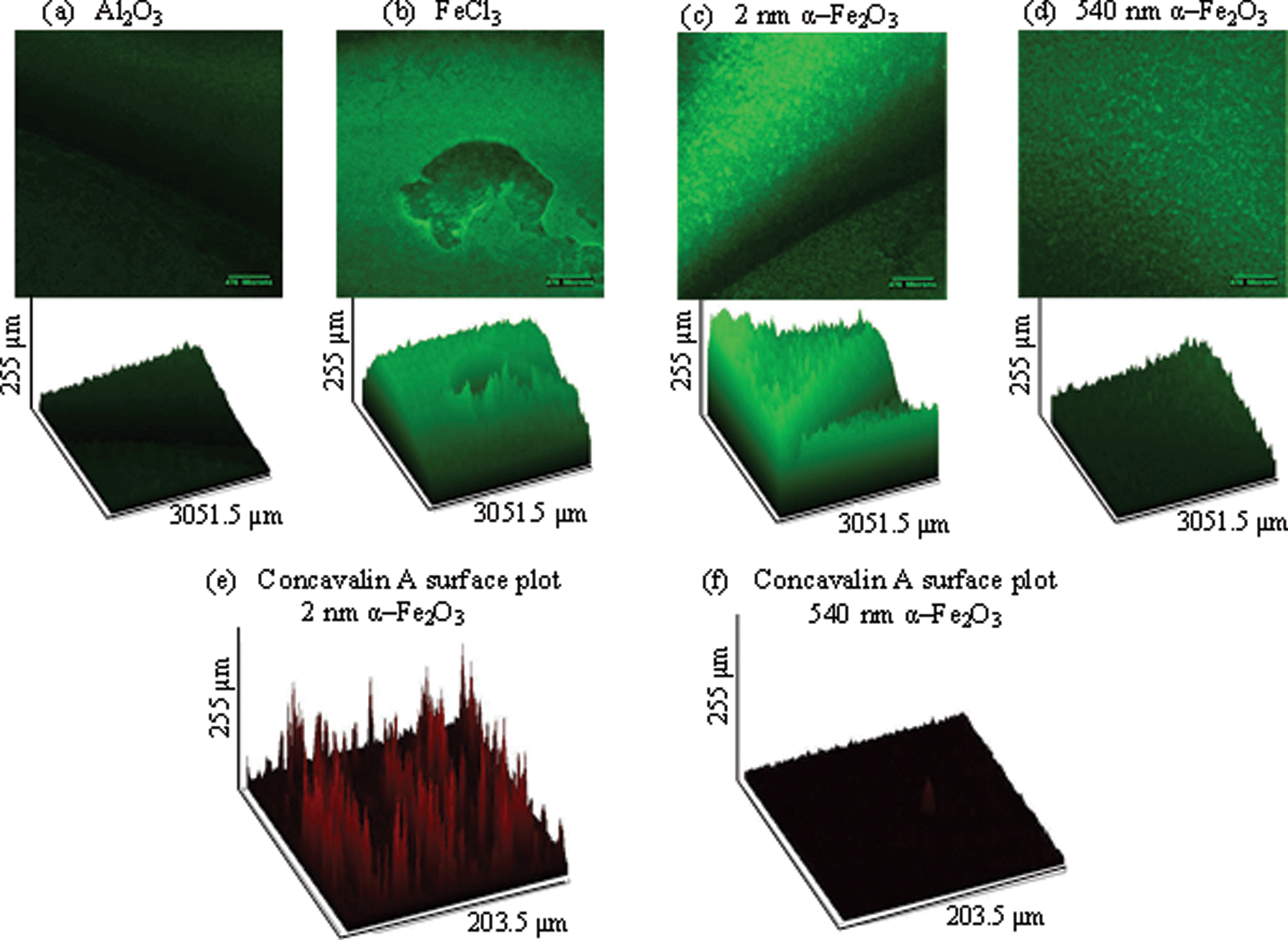 | ||
| Fig. 4 GFP-PA01 was grown overnight, diluted and exposed to α-Fe2O3 particles. Al2O3 and FeCl3 were respectively used as negative and positive controls. Biofilm was grown on a coverslip perpendicular to the bottom of a flat 24 well plate for 48 hours. (a) Al2O3 did not increase biofilm formation when exposed to GFP-PA01 for 48 hours. (b) In the presence of FeCl3, a soluble source of iron, biofilm formation significantly increased. (c) 2 nm α-Fe2O3 increased biofilm formation significantly more than (d) 540 nm α-Fe2O3. Concavalin A surface plots for (e) 2 nm α-Fe2O3 showed increased conA production than (f) 540 nm α-Fe2O3 (p = 0.0396). Significance was determined by comparing GFP or Texas Red intensity, expressed as percentage, and analyzed by one-way ANOVA. | ||
These results support our hypothesis that there is a size dependent effect of α-Fe2O3 particles on biofilm formation. Specifically, this result is consistent with planktonic growth, as a result of increased iron dissolution in the small nanoparticle samples.
Impact of iron oxide nanoparticles on antimicrobial peptide activity
The effects of iron bioavailability in iron-depleted environments such as the airway not only provide a source of iron for bacteria but can also alter host innate immunity. The airway surface liquid (ASL) is composed of a combination of antimicrobial proteins that prevent bacterial colonization. Specifically, lysozyme, lactoferrin and HNP are present all of which are part of the α-defensin subfamily and, thus, part of the host innate immunity. Each AMP has specific bactericidal activity as well as synergistic effects. Lysozyme is known to degrade the bacterial cell wall by muramidase activity.40 Lactoferrin is an iron-binding glycoprotein that sequesters iron thus inhibiting microbial respiration, and human neutrophil peptides are broad-spectrum antimicrobials that disrupt bacterial membranes.40 Lactoferrin has been shown to take up iron and inhibit both bacterial growth and biofilm formation.41 Since our results showed that α-Fe2O3 increased bacterial growth, we decided to investigate whether α-Fe2O3 particles have an effect on host innate immunity. In order to determine the effect of iron-containing particles on AMP activity, physiologically relevant concentrations of AMPs42 and iron oxide particles were incubated and the effects of bacterial killing were tested. Specifically, 600 μg mL−1 lysozyme, 200 μg mL−1 lactoferrin and 100 ng mL−1 HNP 1 & 2 were used in this mixture.In these experiments we focus on the smallest and largest particles to determine if there are any size effects associated with iron oxide particles on antimicrobial activity. In particular, the particles were incubated with the antimicrobial cocktail for 1 hour and then removed by centrifugation. The antimicrobial cocktail was added to the underlay gel containing P. aeruginosa that had been grown in M9 minimal media (Fig. S2† schematic diagram). As shown in Fig. 5 (a), both 2 nm particles and FeCl3 inhibited antimicrobial activity (p < 0.05), while 540 nm α-Fe2O3 particles did not. Since 2 nm particles have the greatest amount of dissolved iron (Table 3), we hypothesized that the smaller α-Fe2O3 provides the greatest amount of bioavailable iron and inhibits AMP activity similar to that found for FeCl3. Therefore, we incubated the particles with media for 1 hour and removed the particles by centrifugation. The soluble component of the media was then added to the antimicrobials in order to test the effect of soluble iron on antimicrobial peptide inhibition. In Fig. 5 (b), 2 nm α-Fe2O3 and FeCl3 inhibit AMP activity when compared with the control (p < 0.05), while 540 nm α-Fe2O3 is no different from the control. Therefore this result is consistent with our hypothesis that antimicrobial inhibition by iron oxide particles is due to dissolved iron and is similar to the Fe(III) effects on lysozyme antimicrobial efficacy which have been previously reported for different Escherichia coli strains, even in the presence of a chelating agent.43
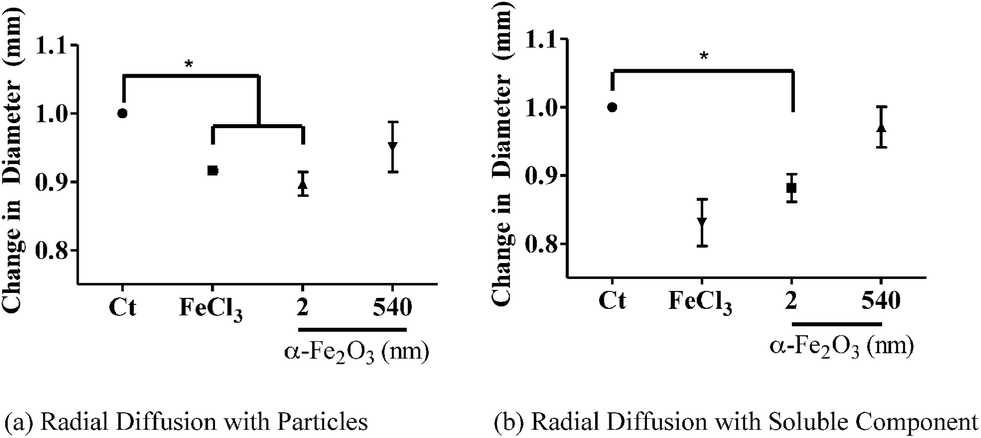 | ||
| Fig. 5 AMP activity data using the radial diffusion assay. (a) AMP activity was inhibited in the presence of FeCl3 and after exposure to 2 nm α-Fe2O3 (p < 0.05) as evidenced by the change in the zone of inhibition as measured here by a change (decrease) in diameter of bacterial killing. These data show that the positive control, FeCl3, and the smallest nanoparticles show evidence for inhibition of AMP activity. (b) Particles were incubated with M9 media for 1 hour and then removed. The soluble component of the media was added to AMPs and AMP activity was measured using radial diffusion. The ability of AMPs to kill bacteria was inhibited in the presence of the soluble component of iron in the FeCl3 sample and in the presence of the smallest particle, 2 nm α-Fe2O3 (p < 0.05). A one-way ANOVA with SEM was reported. | ||
In addition to the formation of soluble iron leading to AMP inhibition, the adsorption of AMPs on nanoparticle surfaces can also play a role in decreasing the activity of these peptides. In order to better understand this interaction, an additional experiment was done to probe the ability of AMPs to adsorb onto nanoparticle surfaces. Since it has been previously shown that lysozyme can adsorb onto nanoparticle surfaces which leads to loss of activity,44 here we investigate whether lysozyme, an important component of the α-defensin subfamily, adsorbs onto 2 nm α-Fe2O3 particle surfaces. The adsorption process is followed by ATR-FTIR spectroscopy as described in the Experimental methods section.
The collected ATR-FTIR spectra of M9 media with lysozyme in the presence of a thin film of 2 nm α-Fe2O3 particles deposited onto the ATR element as a function of time compared to the spectrum of M9 media with lysozyme in the absence of the nanoparticles are shown in Fig. 6 (a). The solution phase spectrum gives the highest intense peaks at 1077 and 989 cm−1 which correspond to HPO42− and the less intense peaks at 1156 and 936 cm−1 correspond to H2PO4−.45 The less intense peaks in the region of 1400–1800 cm−1 result from the dissolved lysozymes, succinic acid and glucose in M9 medium. A comparison of the solution phase spectrum to the adsorbed phase illustrates several distinct differences. First, it shows that the peptide readily adsorbs onto the nanoparticle surface as indicated by the absorption bands seen at 1653, 1542 and 1453 cm−1. These bands correspond to the amide I, amide II and amide III vibrational modes of lysozyme.46 Furthermore, the vibrational modes of H2PO4− and HPO42− are broadened considerably in the presence of the nanoparticles which indicate that these phosphate species are also adsorbed onto the iron oxide nanoparticle surfaces. Interestingly, as illustrated in Fig. 6 (b), adsorbed H2PO4− and HPO42− readily desorb from the surface when the M9 media are removed and pure water is introduced into the liquid cell. This behavior indicates that phosphate adsorption is weak and reversible. In contrast, absorption bands due to lysozymes remain indicating a stronger interaction between this antimicrobial peptide with the iron oxide nanoparticle surface compared to phosphate.
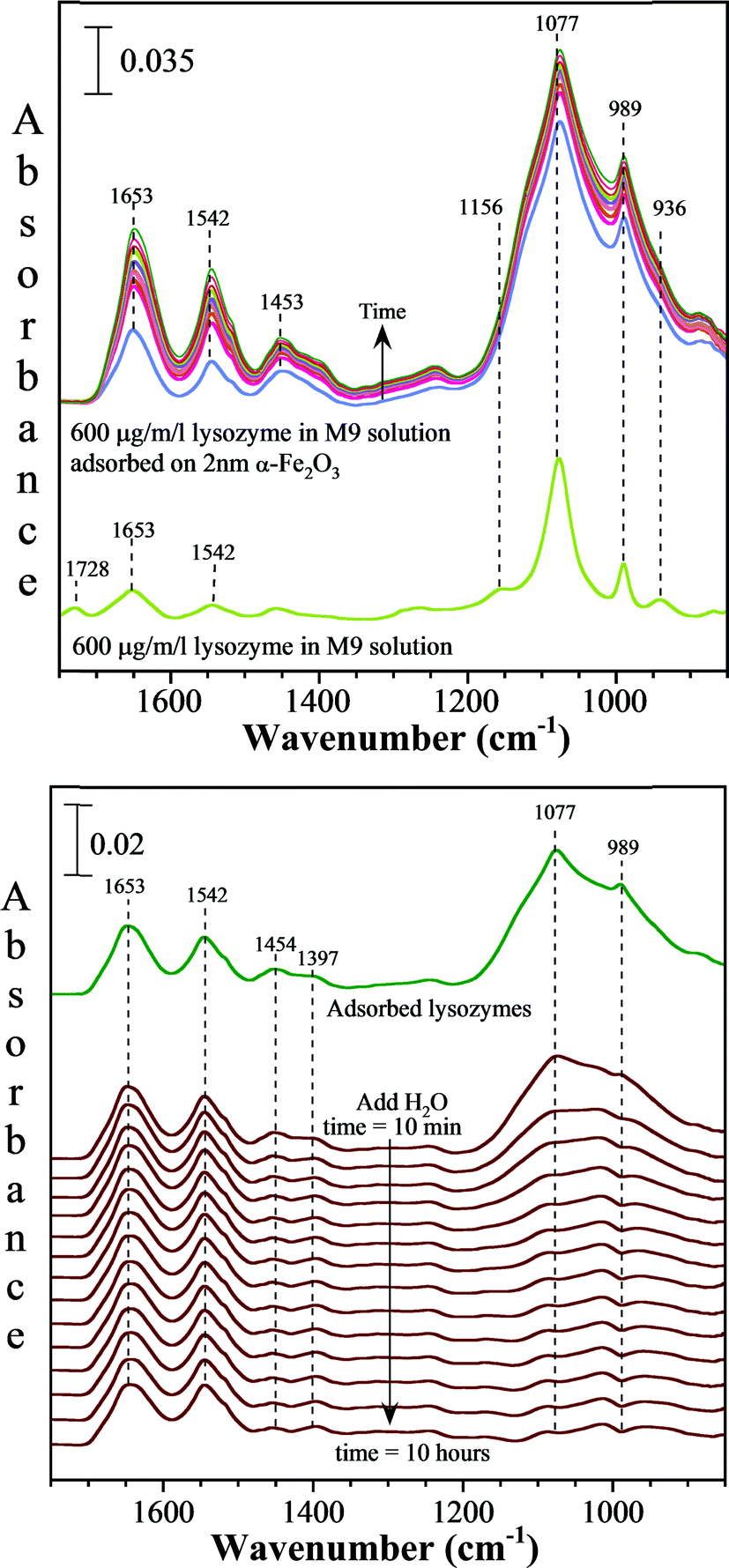 | ||
| Fig. 6 ATR-FTIR spectra of lysozyme in M9 media adsorbed on 2 nm α-Fe2O3 nanoparticles compared to the solution phase in the absence of nanoparticles (top). These data show that lysozyme as well as H2PO4− and HPO42− from the M9 media adsorb onto the nanoparticle surface. Upon removal of the M9 media containing lysozyme and replacing with water at pH 7, the spectra show that over time H2PO4− and HPO42− desorb from the surface whereas the lysozyme remains adsorbed (bottom). The spectra were collected over a period of 10 hours after introducing water. | ||
Conclusions and implications
In general, iron-containing nanoparticles are thought to be non-toxic. For example, hematite nanoparticles greater than 90 nm do not show signs of toxicity in macrophages and lung epithelial cells.47 However, the smallest nanoparticles, with a large surface area and increased dissolution, may be harmful to human health as it relates to individuals susceptible to bacterial infections and/or colonization. Our results demonstrate that iron oxide particles increased P. aeruginosa growth and biofilm formation is linked to particle size, likely explained by the increased propensity of these smaller iron particles to readily dissolve more and release Fe3+(aq) in to the aqueous medium. The current study is then in agreement with recent results showing that SPIONS (super paramagnetic iron oxide (Fe3O4) nanoparticles) also increase biofilm formation.48Although iron oxide nanoparticles themselves have no antimicrobial activity,49 our results also demonstrate that the smallest iron oxide nanoparticles inhibit AMP activity, thus impairing host innate immunity mechanisms. Recent studies in the literature have reported mechanisms of iron oxide nanoparticles in dysregulating host innate immunity.50 The data reported herein suggest that the results are correlated and related to the propensity of the smaller nanoparticles to more readily dissolve to produce Fe3+(aq) in solution affecting bacterial growth, virulence and host innate immunity. The adsorption of AMPs, in particular lysozyme, onto nanoparticle surfaces can also play a role in decreasing activity of these peptides and thus is a more general result that may be applicable to the impact of other nanoparticles. Further studies are warranted to better understand these other inhibitory impacts. This current study, along with other recently published work noted above, suggests that nanoparticle toxicity screening should take into consideration impacts beyond direct toxicity and include the effects of engineered nanomaterials on co-exposures involving known pathogens and the impact of nanomaterials on host innate immunity.
Acknowledgements
We would like to thank Dr. Michael Welsh and Dr. Liuska Pesce for their thoughtful and valuable comments. This publication was made possible by grant no. UL1RR024979 from the National Center for Research Resources (NCRR), a part of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. This publication was also supported in part by the Center for Health Effects of Environmental Contamination (CHEEC) Seed grants: FY 2010 and NIH grant: KO1HL080966 and NIH P30 ES005605.References
- D. L. Huber, Small, 2005, 1, 482–501 CrossRef CAS PubMed
.
- J. J. Bullen, H. J. Rogers, P. B. Spalding and C. G. Ward, FEMS Immunol. Med. Microbiol., 2005, 43, 325–30 CrossRef CAS PubMed
- J. A. Borcherding, H. Chen, J. C. Caraballo, J. Baltrusaitis, A. A. Pezzulo, J. Zabner, V. H. Grassian and A. P. Comellas, PLoS One, 2013, 8, e57673 CAS
- M. M. Monick, J. Baltrusaitis, L. S. Powers, J. A. Borcherding, J. C. Caraballo, I. Mudunkotuwa, D. W. Peate, K. Walters, J. M. Thompson, V. H. Grassian, G. Gudmundsson and A. P. Comellas, Environ. Health Perspect., 2013, 121, 691–698 CrossRef PubMed
- M. F. Hochella Jr., S. K. Lower, P. A. Maurice, R. L. Penn, N. Sahai, D. L. Sparks and B. S. Twining, Science, 2008, 319, 1631–1635 CrossRef PubMed
- M. A. Arbex, G. M. de Souza Conceicao, S. P. Cendon, F. F. Arbex, A. C. Lopes, E. P. Moyses, S. L. Santiago, P. H. Saldiva, L. A. Pereira and A. L. Braga, J. Epidemiol. Community Health, 2009, 63, 777–783 CrossRef CAS PubMed
- C. H. Goss, S. A. Newsom, J. S. Schildcrout, L. Sheppard and J. D. Kaufman, Am. J. Respir. Crit. Care Med., 2004, 169, 816–821 CrossRef PubMed
- S. Larrieu, A. Lefranc, G. Gault, E. Chatignoux, F. Couvy, B. Jouves and L. Filleul, Am. J. Epidemiol., 2009, 169, 1201–1208 CrossRef PubMed
- S. H. Ling and S. F. van Eeden, Int. J. Chronic Obstruct. Pulm. Dis., 2009, 4, 233–243 CrossRef CAS
- F. J. Kelly and J. C. Fussell, Clin. Exp. Allergy, 2011, 41, 1059–1071 CrossRef CAS PubMed
- E. A. MacIntyre, C. J. Karr, M. Koehoorn, P. A. Demers, L. Tamburic, C. Lencar and M. Brauer, Epidemiology, 2011, 22, 81–89 CrossRef PubMed
- R. Saxena, S. Srivastava, D. Trivedi, E. Anand, S. Joshi and S. K. Gupta, Acta Ophthalmol. Scand., 2003, 81, 491–494 CrossRef CAS
- W. J. Waldman, R. Kristovich, D. A. Knight and P. K. Dutta, Chem. Res. Toxicol., 2007, 20, 1149–1154 CrossRef CAS PubMed
- M. R. Wiesner, G. V. Lowry, E. Casman, P. M. Bertsch, C. W. Matson, R. T. Di Giulio, J. Liu and M. F. Hochella, ACS Nano, 2011, 5, 8466–8470 CrossRef CAS PubMed
- G. Rubasinghege, R. W. Lentz, M. M. Scherer and V. H. Grassian, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6628–6633 CrossRef CAS PubMed
-
C. M. Sayes and D. B. Warheit, Nanotoxicity, John Wiley & Sons, Ltd, 2009, pp. 29–39 Search PubMed
- J. Baltrusaitis, C. R. Usher and V. H. Grassian, Phys. Chem. Chem. Phys., 2007, 9, 3011–3024 RSC
-
N. Fairley and A. Carrick, The Casa Cookbook, Part I, Acolyte Science, 2005 Search PubMed
-
A. A. Schilt, Analytical Applications of 1,10-phenanthroline and Related Compounds, Pergamon Press, Oxford, 1969 Search PubMed
- B. P. Cormack, R. H. Valdivia and S. Falkow, FACS-optimized mutants of the green fluorescent protein (GFP), Gene, 1996, 173, 33–38 CrossRef CAS
- D. A. Steinberg and R. I. Lehrer, Methods Mol. Biol., 1997, 78, 169–186 CAS
-
R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, 2nd edn, 2003 Search PubMed
- R. L. Blake, R. E. Hessevick, T. Zoltai and L. W. Finger, Am. Mineral., 1966, 51, 123–129 CAS
- G. Schimanke and M. Martin, Solid State Ionics, 2000, 136–137, 1235–1240 CrossRef CAS
- G. A. Waychunas, C. S. Kim and J. F. Banfield, J. Nanopart. Res., 2005, 7, 409–433 CrossRef CAS
- P. A. Wahid and N. V. Kamalam, Biol. Fertil. Soils, 1993, 15, 144–148 CrossRef CAS
- O. K. Borggaard, J. Soil Sci., 1976, 27, 478–486 CrossRef CAS
- U. Schwertmann, Plant Soil, 1991, 130, 1–25 CrossRef CAS
- D. R. Baer and M. H. Engelhard, J. Electron Spectrosc. Relat. Phenom., 2010, 178–179, 415–432 CrossRef CAS PubMed
- J. Baltrusaitis, D. M. Cwiertny and V. H. Grassian, Phys. Chem. Chem. Phys., 2007, 9, 5542–5554 RSC
-
D. Briggs and J. T. Grant, Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy, 2003, p. 899 Search PubMed
- C.-K. Wu, M. Yin, S. O'Brien and S. J. T. Koberstein, Chem. Mater., 2006, 18, 6054–6058 CrossRef CAS
- J. R. Govan and V. Deretic, Microbiol. Rev., 1996, 60, 539–574 CAS
- L. G. Rahme, E. J. Stevens, S. F. Wolfort, J. Shao, R. G. Tompkins and F. M. Ausubel, Science, 1995, 268, 1899–1902 CAS
- T. S. Walker, H. P. Bais, E. Deziel, H. P. Schweizer, L. G. Rahme, R. Fall and J. M. Vivanco, Plant Physiol., 2004, 134, 320–331 CrossRef CAS PubMed
- Y. Kaneko, M. Thoendel, O. Olakanmi, B. E. Britigan and P. K. Singh, J. Clin. Invest., 2007, 117, 877–888 CrossRef CAS PubMed
- B. J. Kim, J. H. Park, T. H. Park, P. A. Bronstein, D. J. Schneider, S. W. Cartinhour and M. L. Shuler, Appl. Environ. Microbiol., 2009, 75, 2720–2726 CrossRef CAS PubMed
- A. S. Prince, N. Engl. J. Med., 2002, 347, 1110–1111 CrossRef PubMed
- G. M. Patriquin, E. Banin, C. Gilmour, R. Tuchman, E. P. Greenberg and K. Poole, J. Bacteriol., 2008, 190, 662–671 CrossRef CAS PubMed
- J. Wiesner and A. Vilcinskas, Virulence, 2010, 1, 440–464 CrossRef PubMed
- P. K. Singh, M. R. Parsek, E. P. Greenberg and M. J. Welsh, Nature, 2002, 417, 552–555 CrossRef CAS PubMed
- H. N. Hunter, D. B. Fulton, T. Ganz and H. J. Vogel, J. Biol. Chem., 2002, 277, 37597–37603 CrossRef CAS PubMed
- J. S. Boland, P. M. Davidson, B. Bruce and J. Weiss, J. Food Prot., 2004, 67, 285–294 CAS
- A. A. Vertegel, R. W. Siegel and J. S. Dordick, Langmuir, 2004, 20, 6800–6807 CrossRef CAS PubMed
- E. J. Elzinga and D. L. Sparks, J. Colloid Interface Sci., 2007, 308, 53–70 CrossRef CAS PubMed
- L. K. Tamm and S. A. Tatulian, Q. Rev. Biophys., 1997, 30, 365–429 CrossRef CAS
- F. S. Freyria, B. Bonelli, M. Tomatis, M. Ghiazza, E. Gazzano, D. Ghigo, E. Garrone and B. Fubini, Chem. Res. Toxicol., 2012, 25, 850–861 CrossRef CAS PubMed
- C. Haney, J. Rowe and J. Robinson, J. Biomater. Nanobiotechnol., 2012, 3, 508–518 CrossRef
- A. Amirnasr, G. Emtiazi, S. Abasi and M. Yaaghoobi, J. Biochem. Technol., 2011, 3, 280–283 CAS
- V. Kodali, M. H. Littke, S. C. Tilton, J. G. Teeguarden, L. Shi, C. W. Frevert, W. Wang, J. G. Pounds and B. D. Thrall, ACS Nano, 2013, 7, 6997–7010 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: ESI contains a detailed description of particle synthesis and sources and a schematic diagram of the antimicrobial peptide assay. See DOI: 10.1039/c3en00029j |
| This journal is © The Royal Society of Chemistry 2014 |