Green synthesis and formation mechanism of cellulose nanocrystal-supported gold nanoparticles with enhanced catalytic performance
Xiaodong
Wu
a,
Canhui
Lu
a,
Zehang
Zhou
a,
Guiping
Yuan
b,
Rui
Xiong
a and
Xinxing
Zhang
*a
aState Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Chengdu 610065, China. E-mail: xxzwwh@scu.edu.cn; Fax: +86 28 85402465; Tel: +86 28 85460607
bAnalytic and Testing Center of Sichuan University, Chengdu 610065, China
First published on 13th January 2014
Abstract
Deposition of precious metal catalysts onto the surface of various supporting materials to enhance the stability and catalytic activity is highly desired. Although extensive studies have been focused on the supported metal catalysts, their preparations are mainly based on the use of reducing agents which are not environmentally benign. Herein, we report a one-pot and green synthesis of gold nanoparticles (Au NPs) deposited on cellulose nanocrystals (CNs) under hydrothermal conditions using CNs as a reducing agent and stabilizing template. Our experimental results showed that the abundant electron-rich hydroxyl groups on the surface of CNs played a key role in the reduction and immobilization of Au NPs. The obtained nanohybrid catalyst exhibited much better catalytic activity and stability than the unsupported Au NPs and other Au-containing catalysts for the reduction of 4-nitrophenol. These findings pave the way for the green synthesis of bio-supported nanohybrid catalysts and can spur advancements in nanocellulose-based nanohybrids for their application in sensors, antibacterial materials and electronic devices.
Nano impactWe report a one-step and environment-friendly synthesis of cellulose nanocrystal (CN)-supported gold nanoparticles (Au NPs) without employing any other reductants, capping or dispersing agents. CNs played a dual role as a supporting matrix and a reductant and they were used to obtain stable dispersions of Au NPs. The obtained nanohybrid catalyst exhibited much better catalytic activity and stability than the unsupported Au NPs and other Au-containing catalysts for the reduction of 4-nitrophenol. Furthermore, the sustainable and environmentally friendly characteristics of CNs, which can be derived in large quantities from various plants (from waste cotton fabrics in this study), promote the use of renewable natural resources to prepare a variety of hybrid inorganic–organic materials for the purpose of catalysis, sensors and other potential applications. |
1. Introduction
Cellulose is the most common and abundant natural polymer, which can be obtained from higher plants, tunicates, bacteria and alga. It is a renewable, biodegradable and biocompatible material which can be converted into various derivatives for versatile applications. Among the family of cellulose derivatives, cellulose nanocrystals (CNs), derived from natural cellulose fibers by controlled acid hydrolysis, have emerged as a new class of renewable nanomaterials owing to their unique properties: controlled size, morphology and surface chemistry, superior mechanical strength and self-assembling properties. The high specific surface area, nanometric dispersity and good stability of CNs make them appropriate to serve as a catalyst carrier. Bulk or microcrystalline cellulose does not possess these advantages. Hence, CNs were widely used for various applications such as polymer reinforcement,1 enzyme immobilization,2 drug delivery, as templates for mesoporous materials3 and support matrix for metallic nanoparticle synthesis.4–6Gold nanoparticles (Au NPs) have attracted great attention in the past decades due to their unique physicochemical properties7 and potential applications in optoelectronics,8 sensors9–11 and so on.12 In addition, Au NPs are one of the most promising catalysts for a variety of chemical reactions.13–15 However, Au NPs are generally unstable due to their large active surface areas which may result in self-aggregation. Thus, they are usually encapsulated in polymers16 and mesoporous matrices17 or supported on supporting materials such as graphene oxide,18 oxides,19 silica20etc. As the emphasis of science and technology is gradually shifting towards environmentally friendly and sustainable resources and processes, CNs, a “green” supporting material for precious metal NPs, are one of the best candidates for the fabrication of inorganic–organic nanohybrids.
The deposition of Au NPs on the surface of cellulose derivatives provides a new approach to hybrid heterogeneous catalysts for organic transformation. Recently, cellulose-,21 cellulose nanofiber-22 and CN-2,5 supported Au NPs have been reported. However, these methods for the preparation of Au NPs are generally based on the reduction of Au(III) using toxic or dangerous reducing agents such as sodium borohydride, hydrazine hydrate and hydrogen. In order to avoid the use of these agents, green approaches to Au NPs are necessary. To date, green syntheses of silver and platinum NPs using hydroxyl group-containing biomaterials as a reducing agent23–30 have been reported but the green reduction of Au NPs is scarce. Johnston et al.31 prepared Au NPs using lignin-containing cellulose fibers as a reducing agent. Li et al.32 synthesized Au NPs by dissolving cellulose in the ionic liquid, 1-butyl-3-methylimidazolium chloride, above 110 °C. However, to the best of our knowledge, no study reported the use of hydroxyl group-containing biomaterials as both a reducing agent and catalyst supporter for the synthesis of Au NPs.
Herein, a facile one-pot and green method to synthesize Au NPs using CNs as the reducing agent and supporter is reported for the first time. The catalytic activity of the obtained Au NPs@CNs nanohybrid was evaluated using the reduction of 4-nitrophenol (4-NP) by sodium borohydride and the result indicated that the synthesized Au NPs@CNs nanohybrid had superior catalytic performance. This approach to synthesize Au NPs@CNs nanohybrid not only avoided the use of chemical reducing agents but also improved the catalytic activity and stability of Au NPs. Furthermore, the sustainable and environmentally friendly characteristics of CNs, which can be derived in large quantities from various plants, make them attractive for large-scale applications.
2. Materials and methods
2.1 Materials
Analytical grade sulfuric acid (H2SO4, 95–98 wt%), hydrochloric acid (HCl, 36–38%), hydrogen nitrate (HNO3, 65–68%), sodium hydroxide (NaOH, ≧96%), hydrazine hydrate (N2H4·H2O, ≧80 wt%), sodium borohydride (NaBH4, ≧97%), 4-nitrophenol (4-NP, ≧98%) and other chemical reagents were all purchased from commercial sources and used without further purification. Gold(III) chloride trihydrate (HAuCl4·3H2O, 49.0 wt% Au) was purchased from J&K Scientific Ltd. Cotton fabric wastes were collected from tailoring workshops and subjected to cutting and shredding processes.2.2 Preparation of CNs
Microcrystalline cellulose (MCC) derived from waste cotton fabrics based on our previous studies33 was used to prepare CNs. The obtained MCC (4 g) was mixed with 70 mL sulfuric acid (63.5 wt%) and the mixture was stirred vigorously at 45 °C for 60 min. After the reaction was finished, the suspension was diluted ten times immediately to stop the hydrolysis. Then, the suspension was centrifuged and washed with deionized water 3 times to remove all the soluble components. Dialysis was performed to remove the residual acid in the suspension and the result was monitored by checking the neutrality of the dialyzate. The obtained CN suspension was sonicated for 30 min at room temperature.2.3 Synthesis of Au NPs@CNs nanohybrids and unsupported Au NPs
In a typical experiment, 20 mL of CNs suspension (0.2 wt%) was mixed with 20 mL of HAuCl4 solution (0.4 mM). The final mixture with the HAuCl4 concentration of 0.2 mM and the CN concentration of 0.1 wt% was stirred under magnetic stirring for 5 min to homogenize the suspension. Then, the mixture was placed in a hydrothermal synthesis reaction kettle at 120 °C for 10 h to obtain the Au NPs@CNs nanohybrid suspension.The unsupported Au NPs were synthesized by reducing HAuCl4 with N2H4·H2O under ambient conditions. 600 μL N2H4·H2O was added dropwise to a 40 mL HAuCl4 solution (0.2 mM) under magnetic stirring and then the Au NP colloid was stirred for 30 min to finish the reaction. The obtained Au NP and Au NPs@CNs suspensions were not further purified.
2.4 Catalytic performance test
In a typical experiment, an aqueous suspension of Au NPs@CNs nanohybrid (1 mL, total Au content: 0.2 μmol) was added to a solution mixture (50 mL) consisting of 4-NP (0.12 mM) and NaBH4 (38.1 mM). The reaction was performed at room temperature with continuous stirring. At intervals of 2 min, 4 mL of the mixture was taken in a quartz cuvette and analyzed via UV-vis spectroscopy. The catalytic performance test of the unsupported Au NPs was carried out as mentioned above.3. Characterization
Transmission electron microscopy (TEM) and high resolution transmission electron microscopy (HRTEM) were carried out using a transmission electron microscope (JEOL JEM-100CX, Japan) to observe the synthesized Au NPs@CNs nanohybrid and unsupported Au NPs. The selected area electron diffraction (SAED) pattern was recorded from TEM images to obtain the crystal structure. The pristine CNs and the prepared Au NPs@CNs (reacted for 10 h) were negatively stained with 1.5% aqueous uranyl acetate solution for imaging purposes. The particle sizes of the Au NPs were measured using Image-J software and at least 100 particles of the sample from different TEM images were analyzed. The X-Ray Diffraction (XRD) patterns were collected using a Philips Analytical X'Pert X-diffractometer (Philips Co., Netherlands), with Cu-Kα radiation (k = 0.1540 nm) at an accelerating voltage of 40 kV and a current of 40 mA. The data were collected from 2θ = 5–90° with a step interval of 0.03°. The X-ray photoelectron spectroscopy (XPS) experiments were performed using the Kratos XSAM 800 spectrometer using Al Kα (1486.6 eV) radiation at 12 kV × 15 mA. The fitting procedure was performed using the “XPS Peak-41” software. Zeta potential of the pristine CNs (CNs were dispersed in deionized water with concentration of 0.1 wt% and pH value of 6.8) was measured using Zetasizer nano ZEN 3600 (Malvern, UK). Fourier transform infrared spectroscopy (FTIR) studies were conducted using a Nicolet 6700 spectrophotometer (USA). The powdered samples of pristine CNs and the Au NPs@CNs nanohybrid were mixed with KBr to produce tablets for FTIR measurement. The spectra were recorded from 4000 to 400 cm−1 at a resolution of 2 cm−1. The ultraviolet visible (UV-vis) spectra were taken at room temperature using a Shimadzu UVmini-1240 spectrophotometer using a quartz cuvette with an optical path of 1 cm.4. Results and discussion
4.1 Characterization of the Au NPs@CNs nanohybrid
CNs were derived from bulk cellulose by controlled acid hydrolysis of waste cotton fabrics. A small amount of sulfate ester groups were introduced onto the surface of CNs during the sulfuric acid hydrolysis, which could electrostatically stabilize the suspension.34 After negative staining with uranyl acetate, the rod-like CNs with the length of about 100–250 nm and width of 10–20 nm could be clearly seen in Fig. 1A.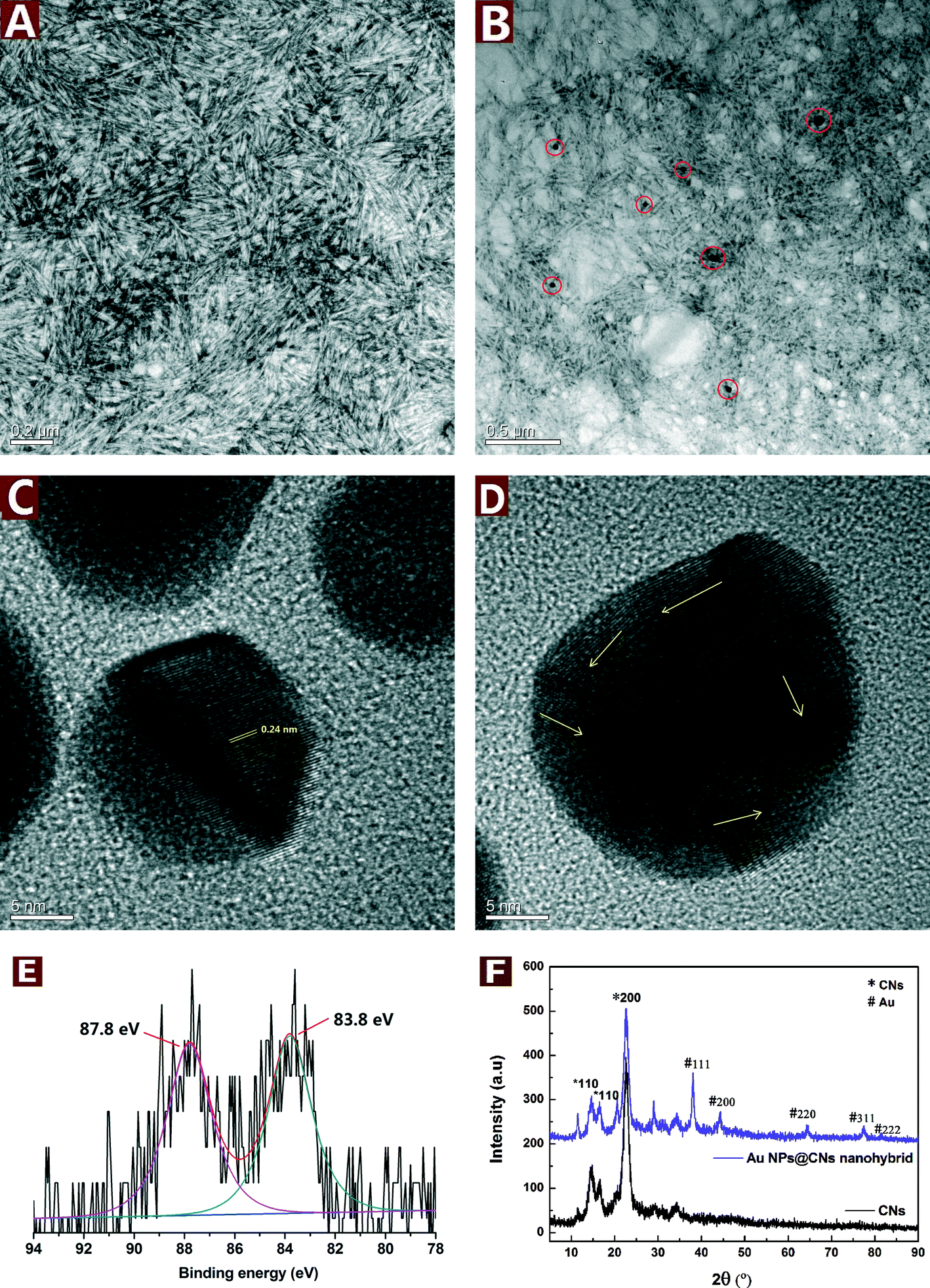 | ||
| Fig. 1 TEM images of the pristine CNs (A) and the Au NPs@CNs nanohybrid (B); HRTEM images of Au NPs in the nanohybrid (C and D); XPS spectrum of the Au NPs@CNs nanohybrid (E); XRD patterns of pristine CNs and the Au NPs@CNs nanohybrid (F). | ||
The good dispersity and hydroxyl group-rich feature of CNs make them a suitable reductant for metal precursors with high valence. During the reduction of HAuCl4 by CNs under hydrothermal conditions, the color changed from light yellow to pale purple and finally turned to dark purple after 10 h. In order to study the microstructure of the obtained Au NPs@CNs nanohybrid, the TEM observations were employed. The well-dispersed Au NPs are sparsely located on the CN surface due to the low percentage of Au in the nanohybrid (3.79 wt%). Furthermore, the morphology of CNs was well maintained after reaction with HAuCl4 as shown in Fig. 1A and B. The particle size of Au NPs was measured from at least 100 nanoparticles in several TEM images across a representative sample. The Au NPs are highly dispersed with a diameter of 30.5 ± 13.4 nm. Fig. 1C and D show the HRTEM images of the Au NPs. The lattice fringes are measured to be 0.24 nm, which matches the expected d-spacing of the [111] plane of face-centered cubic (fcc) Au.35 Besides, the atomic lattice fringes are co-directionally arranged in Fig. 1C, demonstrating a single crystalline character of these CN-supported Au NPs. Randomly packed atomic lattice fringes of CN-supported Au NPs are also observed as the arrows indicate in Fig. 1D, indicating that CN-supported Au NPs have a polycrystalline structure. The electron micrographs indicate the successful synthesis of Au NPs and their decoration onto CNs during the hydrothermal reaction.
XRD analysis was employed to investigate the crystallographic behavior of CNs and the synthesized Au NPs@CNs nanohybrid. As shown in Fig. 1E, the peaks located at 38.0°, 44.4°, 64.4° 77.2°, and 81.4° can be obviously observed, which are the characteristic peaks assigned to the diffraction of the (111), (200), (220), (311) and (222) lattice plane for Au(0) crystals,32 respectively. However, a small peak at nearly 30° was detected and could be assigned to NaCl,36 which might be introduced during the production of the sample. Both the pristine CNs and Au NPs@CNs nanohybrid show three peaks at 2θ values of 14.4°, 16.6°, and 22.6°, corresponding to the (1−10), (110), and (200) diffraction planes of cellulose I, respectively.33,37 This result indicates that the deposition of Au NPs onto CNs did not change the crystallographic property of CN scaffold. The XRD patterns further confirmed the synthesis of Au NPs in the nanohybrid as well.
The Au oxidation state in the nanohybrid was further analyzed by XPS analysis. From Fig. 1F, two peaks are observed at 83.8 and 87.8 eV, which correspond to Au(0) 4f7/2 and Au(0) 4f5/2, respectively. The results are in accordance with the literature values reported for Au(0).38 Au(III) peaks at 86.9 and 90.6 eV39 are absent in the XPS spectrum of the Au NPs@CNs nanohybrid, which could be ascribed to the small amount of Au(III) in the reaction system after hydrothermal reaction and confirmed the almost completed reduction of Au(III) to Au(0).
To conclude, the TEM, XRD and XPS results demonstrated that Au(III) was almost completely reduced to Au(0) with CNs under hydrothermal condition and highly dispersed Au NPs were successfully synthesized and decorated on the CNs. The synthesis and deposition of Au NPs onto CNs did not change the crystallographic properties and morphology of the CNs scaffold.
4.2 Effects of reaction conditions
The formation process of Au NPs can be indicated by the concomitant UV-vis spectra and color change of the reaction mixture. The UV-vis spectra of the reaction mixture had a characteristic absorption peak at 536 nm (Fig. 2A), which is attributed to the surface plasmon resonance (SPR) band of the Au NPs. At the beginning of hydrothermal reaction, no absorption peak was recorded. However, the SPR band of Au NPs appeared at about 536 nm and increased with the reaction time. The peak reached a maximum after 10 h and did not rise with longer reaction time, which suggested the end of the reaction. The plot of absorbance vs. time (Fig. 2B) indicates that the reaction proceeded fast at the beginning but slowed down over time. In addition, the color changed from light yellow to pale purple and finally turned to dark purple after reaction for 10 h (Fig. 2C). The purple color corresponds to the SPR absorption of Au NPs at about 536 nm.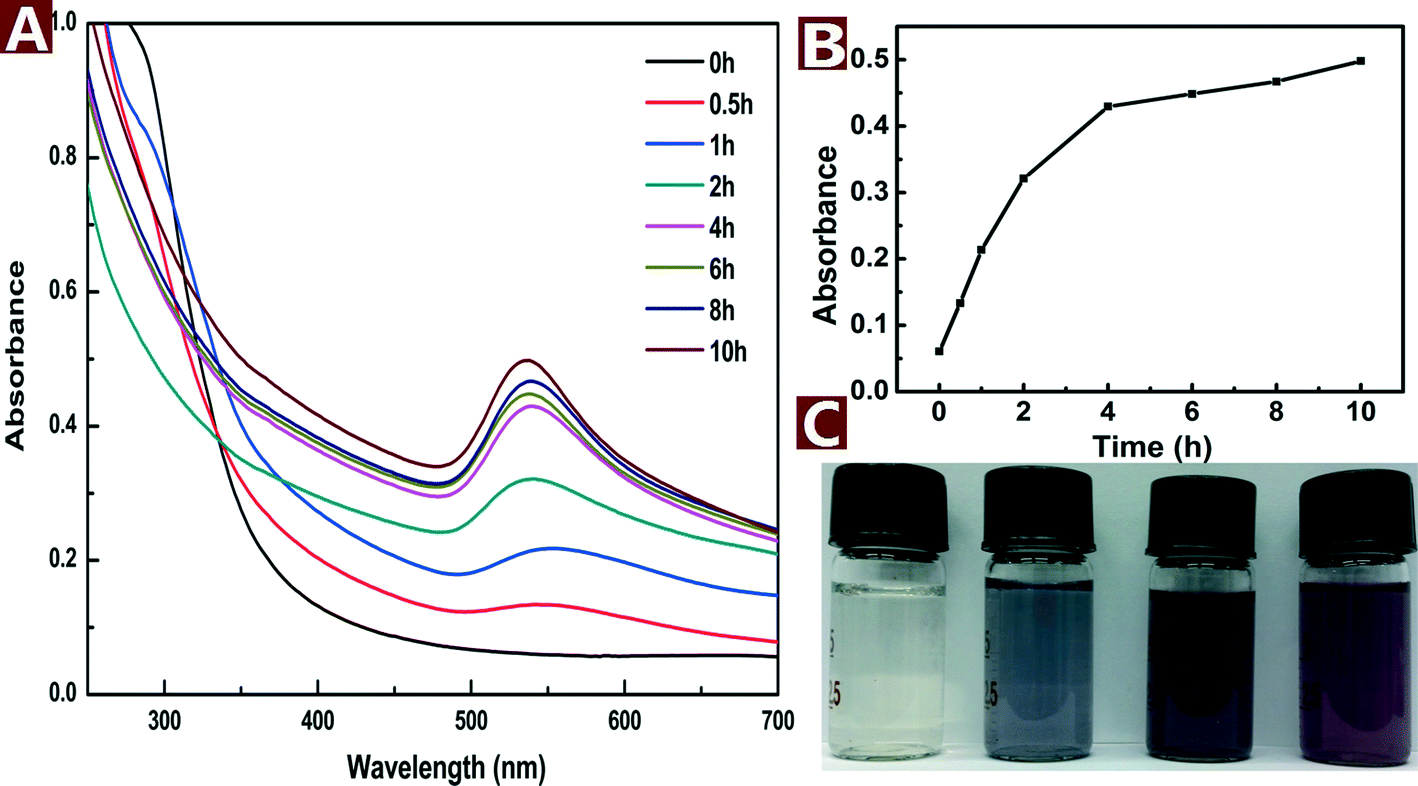 | ||
| Fig. 2 UV-vis spectra (A), peak intensity (B) and colored pictures (C) of the aqueous reaction solution with HAuCl4 (0.2 mM) and CNs (0.1 wt%) as a function of reaction time at 120 °C. | ||
The morphology of the Au NPs@CNs nanohybrid synthesized with different reaction times is studied by TEM as shown in Fig. 3. Au NPs are presented as small particles after 0.5 h of reaction in the aqueous solution at 120 °C (Fig. 3A). The average particle size is 21.8 ± 10.6 nm and several large particles can be found as well. After reaction for 2 h, the Au NPs on CNs grew to larger particles with the diameter of 23.6 ± 11.6 nm and the size of most particles is in a range of 10–35 nm (Fig. 3B). When the reaction time is extended to 10 h, the average particle size increased to 30.5 ± 13.4 nm and half of the Au NPs have a large size (>30 nm) (Fig. 3C). The growing trend of Au NPs during the reaction is clearly seen in Fig. 3A, B and C.
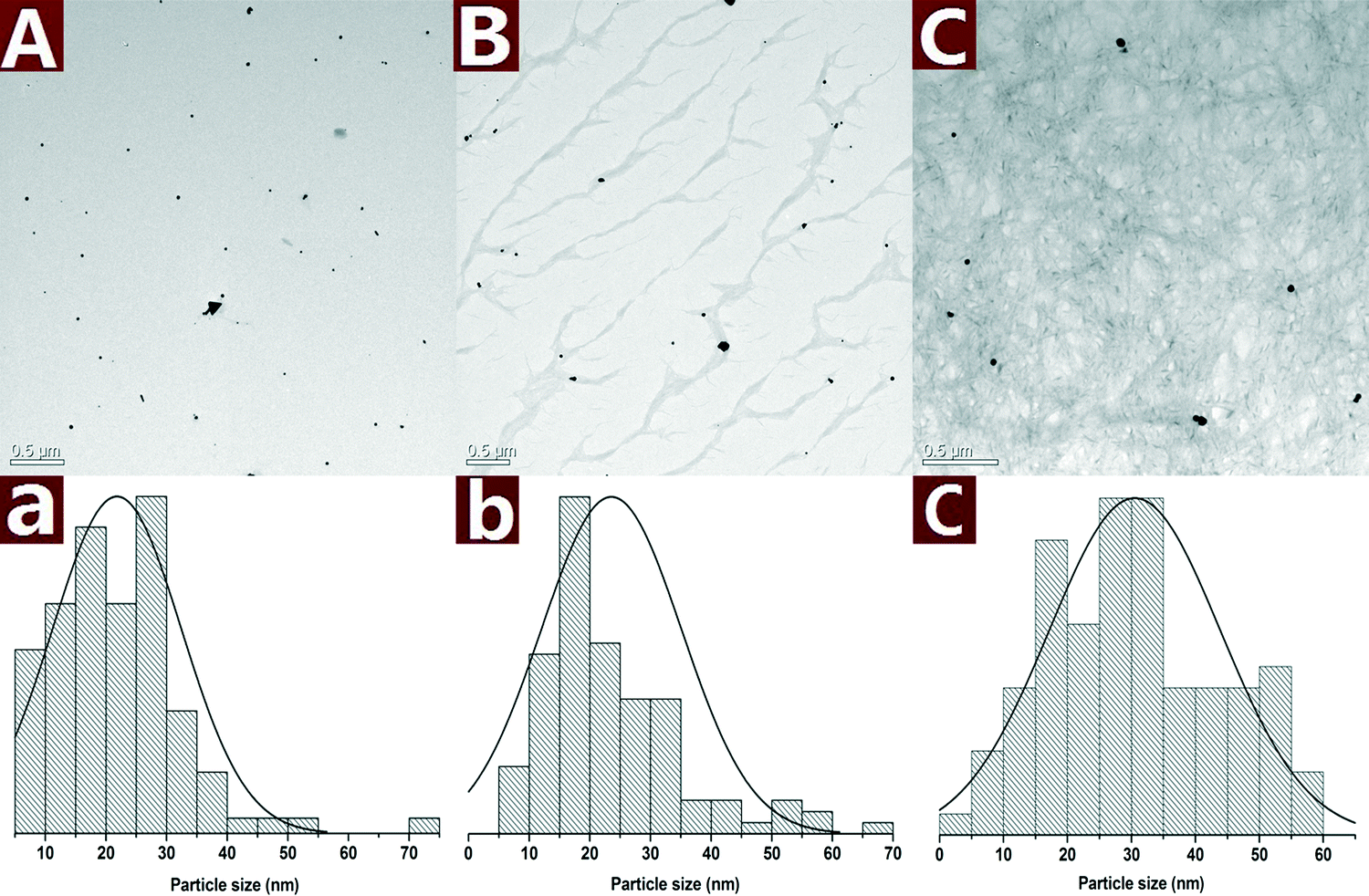 | ||
| Fig. 3 TEM images along with the statistical histograms of Au NPs obtained with different reaction times: 0.5 h (A), 2 h (B) and 10 h (C). | ||
The colored images and UV-vis spectra of Au NPs prepared with different concentrations of CNs are presented in Fig. 4A. The SPR band blue shifts from 548 to 536 nm with CN concentration increased from 0.1 to 0.5 wt%, indicating that the particle size was decreased with increasing concentration of reductive CNs. The color of the reaction mixture changed from purple to dark purple with increasing CN concentration. Fig. 4B shows the change of color and SPR band of the Au NPs under different pH values. The color change from light yellow to dark purple indicates that the pH values have a great effect on the reaction outcome. The pH of a pristine mixture of 0.2 mM HAuCl4 and 0.1 wt% CNs was 3.52. When the pH was adjusted to 2.45 with 0.5 M HCl, the appearance of UV-vis absorbance band at 312 nm assigned to [AuCl4]− indicates that the reaction did not proceed. When the pH values were adjusted from 4.46 to 12.06 with 0.5 M NaOH, the absorbance of λmax increased and the SPR band of Au NPs blue shifted, which indicates that increasing pH values result in smaller and more Au NPs in the reaction process. The results show that alkaline environment facilitates the reaction while acid environment hinders the reaction. This might be attributed to the fact that alkaline conditions can weaken the hydrogen bonding of cellulose and activate the hydroxyl groups on CNs.40 The activated hydroxyl groups have higher activity to reduce Au(III) to Au(0), thus smaller and more Au NPs were formed.
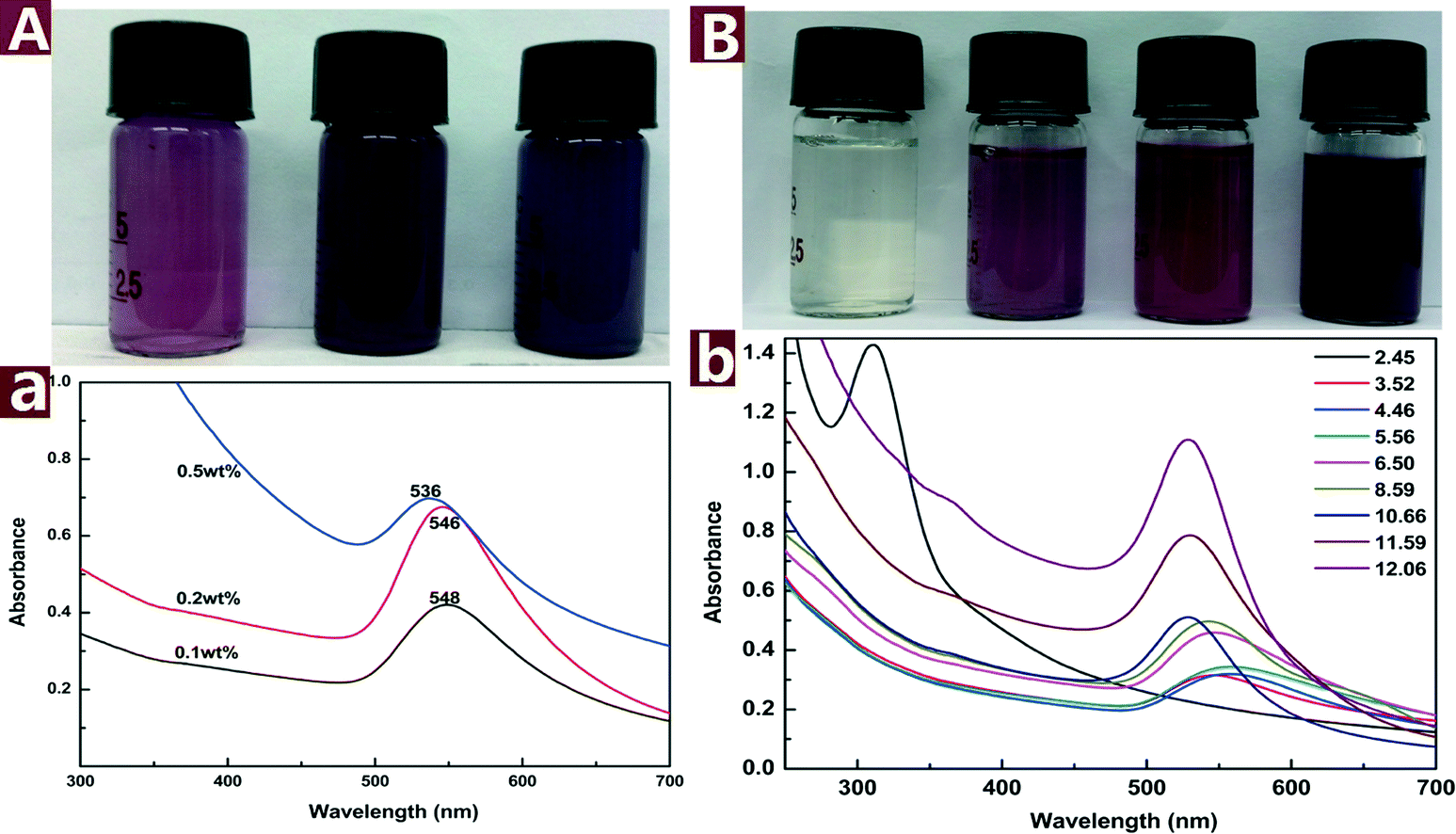 | ||
| Fig. 4 Colored pictures and UV-vis spectra of the Au NPs@CNs nanohybrid suspension: effect of CNs concentration (A) and pH values (B). | ||
4.3 Formation mechanism of the Au NPs@CNs nanohybrid
Fourier transform infrared spectroscopy (FTIR) is a useful tool to obtain rapid information about the structure changes of cellulose during various treatments. The variation of the active site of CNs during the hydrothermal reaction was investigated with FTIR. An enhanced hydrothermal reaction with the AuCl4−/CNs ratio of 5 mM/0.1 wt% at 150 °C for 10 h was used to magnify the change of related groups during the hydrothermal reaction. The FTIR spectrum of original CNs exhibits typical characteristics of cellulose as shown in Fig. 5. The representative peaks located around 3200–3500 cm−1 (O–H stretching), 2850–3000 cm−1 (C–H stretching), and 1059–1162 cm−1 (C–O and C–O–C stretching bond in cellulose)33,41 can be seen clearly. After the hydrothermal reaction, a new peak at 1733.7 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching42,43 appeared, indicating the oxidation of CNs after hydrothermal reaction. However, the C–O and C–O–C stretching bond in cellulose (1059–1162 cm−1) almost remained the same, demonstrating that the ring structure of the cellulose did not substantially change and the secondary hydroxyl groups on the ring structure remained. Thus, the oxidation of cellulose only occurred at the primary hydroxyl groups, which have higher reactivity, rather than the secondary hydroxyl group. The oxidation of cellulose with Au(III) in this hydrothermal reaction was similar to the TEMPO-oxidation of cellulose.42,44
O stretching42,43 appeared, indicating the oxidation of CNs after hydrothermal reaction. However, the C–O and C–O–C stretching bond in cellulose (1059–1162 cm−1) almost remained the same, demonstrating that the ring structure of the cellulose did not substantially change and the secondary hydroxyl groups on the ring structure remained. Thus, the oxidation of cellulose only occurred at the primary hydroxyl groups, which have higher reactivity, rather than the secondary hydroxyl group. The oxidation of cellulose with Au(III) in this hydrothermal reaction was similar to the TEMPO-oxidation of cellulose.42,44
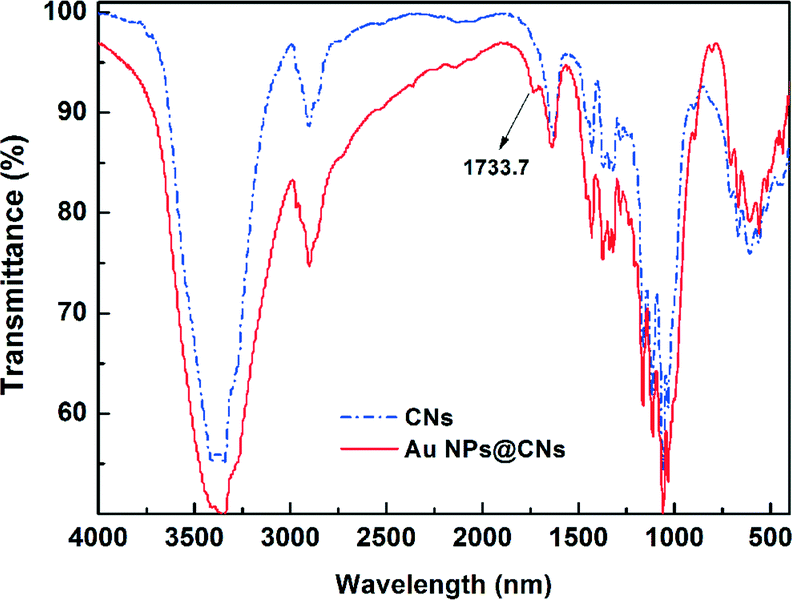 | ||
| Fig. 5 FT-IR spectra of CNs before and after the hydrothermal reaction. | ||
Based on the aforementioned results, a possible formation mechanism of the Au NPs@CNs nanohybrid is proposed in Scheme 1. There are abundant hydroxyl groups on the surface of the CNs. The electron-rich feature of hydroxyl groups makes them suitable for the reduction of Au(III). After nanometric dispersion of CNs, plenty of hydroxyl groups on the surface of CNs were exposed to Au(III). Once the required temperature was achieved, Au(III) was reduced to Au(0) by excessive hydroxyl groups, which led to the formation of Au NPs. The unsupported Au NPs aggregated due to their high surface energy, which led to the deactivation of the catalyst. On the contrary, when CNs were introduced, the oxygen-containing groups (–COO−, –OH) on the surface of CNs might act as anchor points to immobilize Au NPs through the complexing or electrostatic interaction,45–51 which prevented the aggregation of the Au NPs. In fact, the zeta potential of the pristine CNs was −19.0 mV. The negative value of the zeta potential indicated that the surface of CNs was negatively charged, while metal particles are generally positively charged without capping agents. As a result, the oppositely charged nanoparticles were combined through electrostatic force. Hence, the electrostatic or complexing interaction located the synthesized Au NPs onto the surface of oxidized CNs. Therefore, the green synthesis of CN-supported Au NP nanohybrids using CNs as both a reductant and stabilizer was realized. Due to the larger specific surface area and better dispersity of Au NPs supported on CNs, the exposed Au NPs on CNs exhibited excellent catalytic performance in the reduction of 4-NP by sodium borohydride.
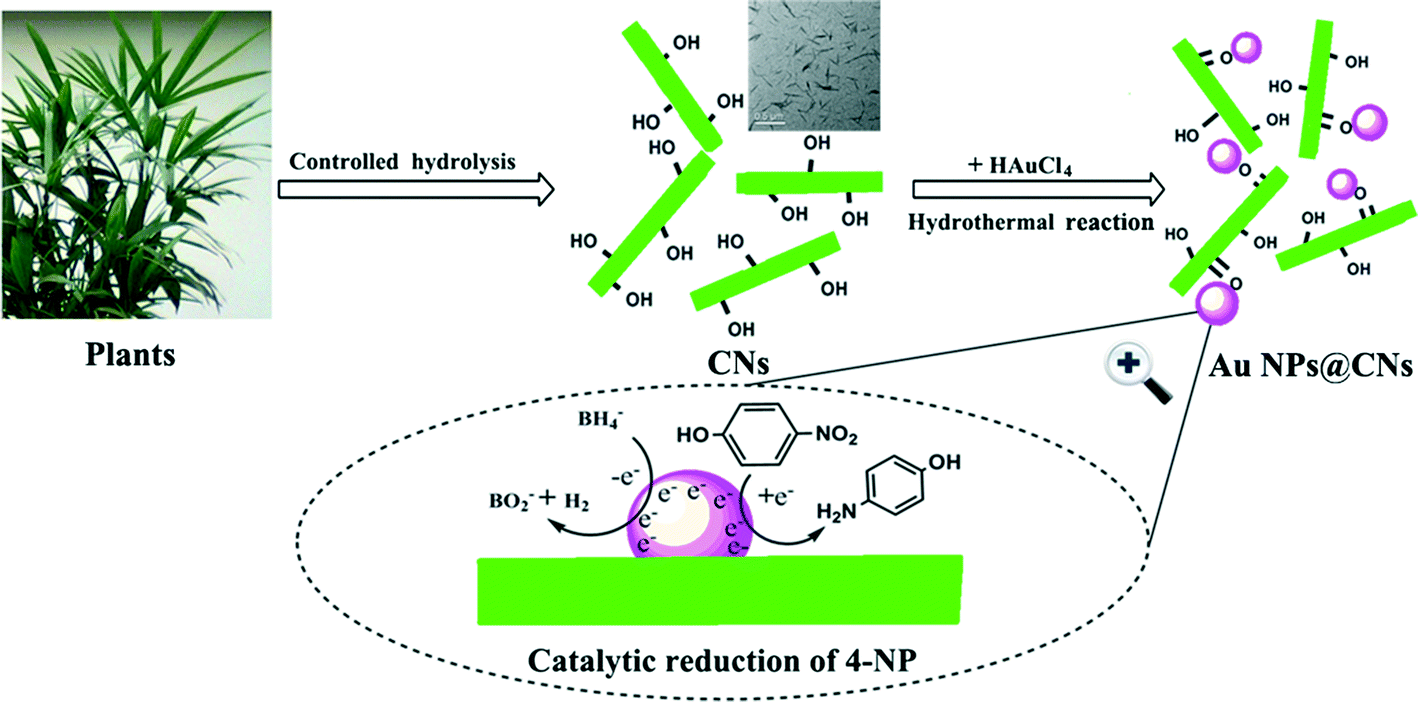 | ||
| Scheme 1 Schematic illustration of the formation mechanism of Au NPs@CNs for catalytic reduction of 4-NP. | ||
4.4 Catalytic performance studies
In order to evaluate the catalytic performance of the synthesized Au NPs@CNs nanohybrid, a model reaction of catalytic reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) with NaBH4 was employed. The reaction proceeded with the decrease of the peak absorbance at 400 nm and the emergence of a new peak at 300 nm corresponding to 4-AP. The change of the characteristic peaks as a function of time is important to understand the rate of reaction.It is necessary to point out that CNs do not have adsorption capacity or catalytic activity under the testing environment and the reduction of 4-NP with NaBH4 does not take place without catalysts.5 When the Au NPs@CNs nanohybrids were introduced, the Au NPs supported on CNs triggered the catalytic reaction by relaying electrons from the BH4− donor to the acceptor molecules of 4-NP (Scheme 1) and the reaction started.
The catalytic reaction was monitored by UV-vis spectroscopy as shown in Fig. 6A and B. As the reaction proceeded, the yellow color of 4-NP bleached with time. Meanwhile, the characteristic peak of 4-NP at 400 nm gradually dropped and a new peak at 300 nm corresponding to 4-AP appeared. However, the color bleaching catalyzed by Au NPs@CNs nanohybrid was much faster than that of the unsupported Au NPs, indicating the higher catalytic activity of the Au NPs@CNs nanohybrid. Fig. 6C provides the plot of At/Ao against time during the reduction of 4-NP, where At is the absorbance at the designated time and Ao is the initial absorbance at t = 0. For Au NPs@CNs nanohybrid catalyzed reaction, the conversion reaches 84.4% after 14 min, which is much higher than that of the unsupported Au NPs catalyzed reaction (35.1%). As shown in Fig. 6D, a good linear correlation of ln(At/Ao) versus time is obtained, indicating that the catalytic reduction proceeded with the pseudo-first-order behavior due to the use of excess NaBH4. The pseudo-first-order rate constant k at room temperature for Au NPs@CNs calculated from the slope was 2.06 × 10−3 s−1, which is nearly 4 times that of the unsupported Au NPs (0.51 × 10−3 s−1). Obviously, the catalytic performance of the Au NPs@CNs nanohybrid was superior to that of unsupported Au NPs as a result of the introduction of CNs.
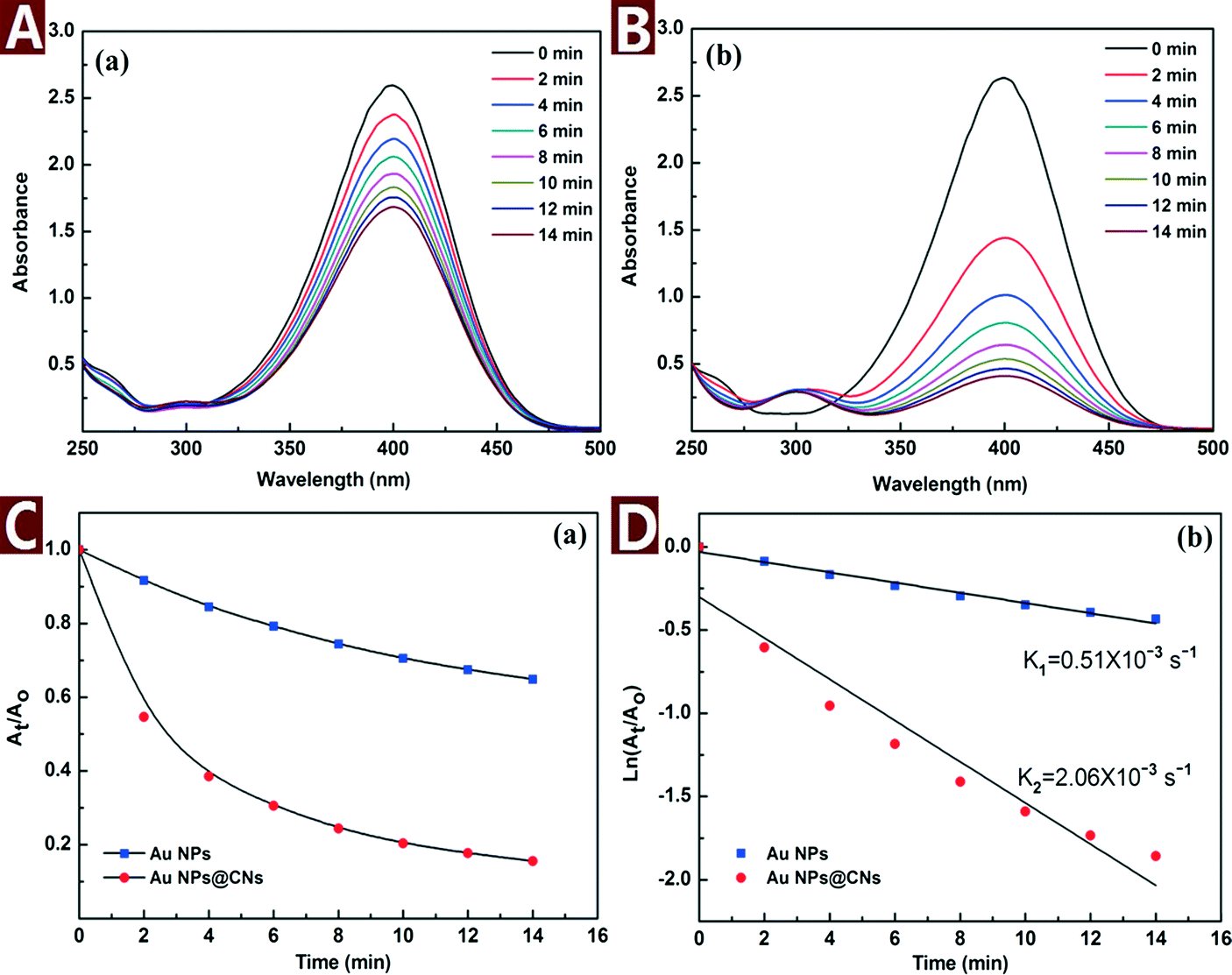 | ||
| Fig. 6 Time dependent UV-vis spectra for the reduction of 4-NP catalyzed with Au NPs (A) and Au NPs@CNs (B); plot of At/Ao (C) and ln(At/Ao) (D) against time for the reduction with different catalysts. | ||
Fig. 7 shows the TEM images and SAED pattern of the unsupported Au NPs. The particle size of the unsupported Au NPs was 18.3 ± 3.5 nm, which is smaller than Au NPs in the nanohybrid. However, the unsupported Au NPs aggregated and formed micron-sized particles (Fig. 7A and B). The SAED pattern (Fig. 7D) indicates that the aggregated Au NPs exhibit a polycrystalline structure. The aggregation of the unsupported Au NPs reduced their dispersity, stability and specific surface area, which was very important to the catalytic activity. In comparison, CNs served as a very effective catalyst supporter and avoided the aggregation of the Au NPs. The well dispersed Au NPs on the surface of CNs made full contact with the reactants which resulted in enhanced catalytic performance.
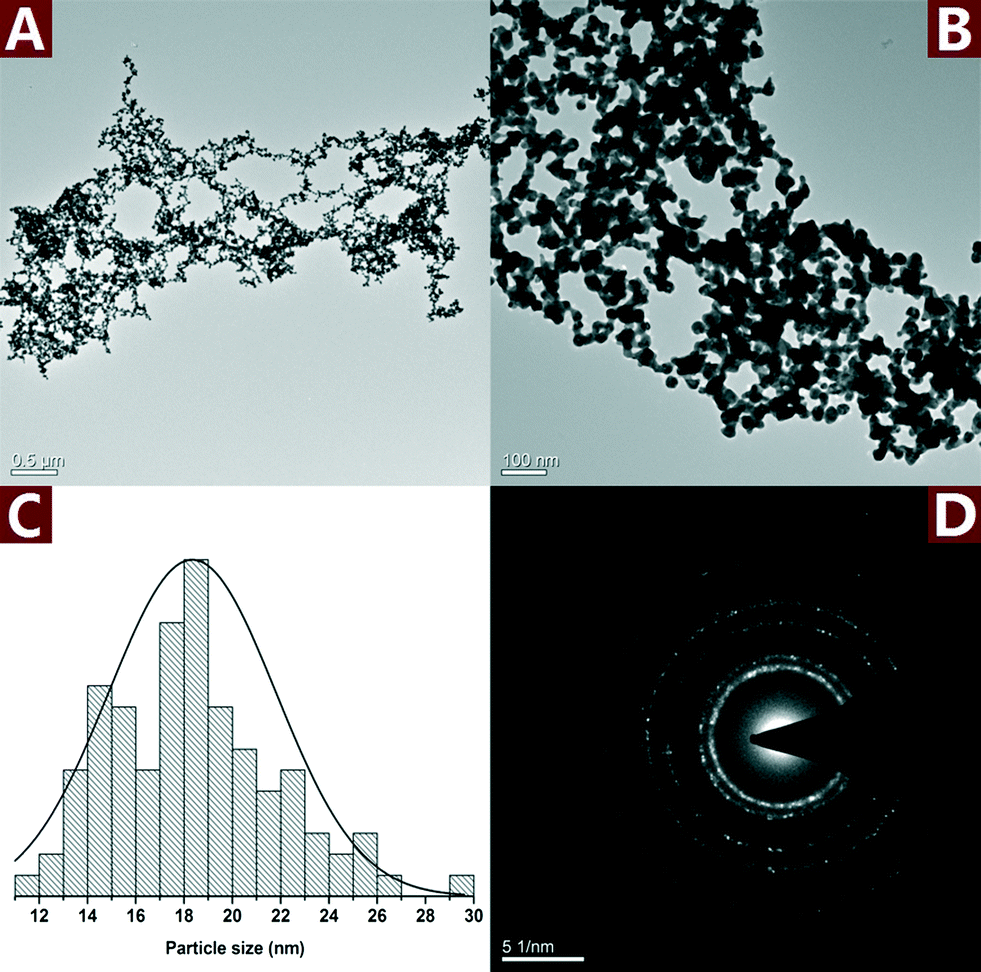 | ||
| Fig. 7 TEM images (A and B) along with the statistical histograms (C) and SAED pattern (D) of the unsupported Au NPs. | ||
To compare the catalytic performance of the Au NPs@CNs nanohybrid with other Au NP-based catalysts fairly, the turnover frequency (TOF), which is defined as moles of 4-NP reduced per mole of Au per hour, was used to evaluate the catalytic performance of the catalysts. As shown in Table 1, the TOF of the Au NPs@CNs nanohybrid is 109 h−1, which is higher than those of recently reported Au NPs-based catalysts.52–58 This result further confirms the superior catalytic performance of the Au NPs@CNs nanohybrid. In conclusion, we realized the facile synthesis of the Au NPs@CNs nanohybrid with superior catalytic performance, which is more appealing due to its one-step and green character.
| Catalyst supporter | Temp.a (K) | Size of Au NPsb (nm) | NaBH4/4-NP/Au (mol/mol/mol) | TOFc (h−1) | Reference |
|---|---|---|---|---|---|
| a Reaction temperature. b Observed by TEM. c Turnover frequency. d Poly(methyl methacrylate). e Poly(2-(dimethylamino)ethyl methacrylate) grafted on to solid polystyrene core. f Poly(N-isopropylacrylamide)-b-poly(4-vinyl pyridine). g Chitosan-coated iron oxide. h α-cyclodextrin. i Polygonal Au nanoparticles. j No data. | |||||
| PMMAd | 295 | 6.9 | 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500/15/1 500/15/1 |
89 | 52 |
| PDMAEMA-PSe | 298 | 4.2 | 28/0.14/1 | 1 | 53 |
| PNIPAP-b-P4VPf | 298 | 3.3 | 167/5/1 | 16 | 54 |
| Chitosang | 303 | 3.1 | 20/6/1 | 50 | 55 |
| α-CDh | 298 | 20 | 250/6/1 | 34 | 56 |
| Nonei | —j | 100 | 2/0.02/1 | 0.4 | 57 |
| Graphene oxide | 298 | 6–15 | 37/1.6/1 | 7 | 58 |
| CNs | 298 | 10–30 | 9720/30/1 | 109 | This work |
5. Conclusion
In this study, we have presented a one-pot and green synthesis of CN-supported Au NPs without employing any other reducing, capping or dispersing agents. CNs, which can be derived in large quantities from various plants, played a dual role as a supporting matrix and a reducing agent. The Au NPs were successfully synthesized and deposited onto CNs by hydrothermal reduction of the HAuCl4 precursor with CNs. This reduction of Au NPs was strongly affected by the CN concentration and pH value of the reaction system. The Au NPs@CNs nanohybrid showed excellent catalytic activity and stability compared to the unsupported Au NPs and other Au-containing catalysts for the reduction of 4-NP. This green synthesized catalyst has several important advantages over the traditional noble metal catalysts in terms of lower cost, absence of a toxic or dangerous reducing agent, high catalytic activity and better stability. The present work will lead to the green synthesis of new organic/inorganic nanohybrids based on nanocelluloses for application in other fields such as sensors, antibacterial materials and electronic devices.Acknowledgements
The authors would like to thank the National Science Foundation of China (51203105) for financial support.References
- A. Pei, J. M. Malho, J. Ruokolainen, Q. Zhou and L. A. Berglund, Macromolecules, 2011, 44, 4422–4427 CrossRef CAS.
- K. A. Mahmoud, K. B. Male, S. Hrapovic and J. H. T. Luong, ACS Appl. Mater. Interfaces, 2009, 1, 1383–1386 CAS.
- K. E. Shopsowitz, H. Qi, W. Y. Hamad and M. J. MacLachlan, Nature, 2010, 468, 422–426 CrossRef CAS PubMed.
- C. M. Cirtiu, A. F. Dunlop-Brière and A. Moores, Green Chem., 2011, 13, 288–291 RSC.
- E. Lam, S. Hrapovic, E. Majid, J. H. Chong and J. H. T. Luong, Nanoscale, 2012, 4, 997–1002 RSC.
- Z. H. Zhou, C. H. Lu, X. D. Wu and X. X. Zhang, RSC Adv., 2013, 3, 26066–26073 RSC.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS PubMed.
- S. A. Maier, M. L. Brongersma, P. G. Kik, S. Meltzer, A. A. G. Requicha and H. A. Atwater, Adv. Mater., 2001, 13, 1501–1505 CrossRef CAS.
- J. Zhou, K. Zhang, J. Liu, G. Song and B. X. Ye, Anal. Methods, 2012, 4, 1350–1356 RSC.
- N. Vasimalai and S. A. John, J. Mater. Chem. A, 2013, 1, 4475–4482 CAS.
- T. J. Zhang, W. Wang, D. Y. Zhang, X. X. Zhang, Y. R. Ma, Y. L. Zhou and L. M. Qi, Adv. Funct. Mater., 2010, 20, 1152–1160 CrossRef CAS.
- M. Potara, D. Maniu and S. Astilean, Nanotechnology, 2009, 20, 315602 CrossRef PubMed.
- R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2009, 11, 1366–1370 RSC.
- X. S. Tan, W. P. Deng, M. Liu, Q. H. Zhang and Y. Wang, Chem. Commun., 2009, 7179–7181 RSC.
- K. Layek, R. Chakravarti, M. L. Kantam, H. Maheswaran and A. Vinu, Green Chem., 2011, 13, 2878–2787 RSC.
- J. P. Hermes, F. Sander, T. Peterle, R. Urbani, T. Pfohl, D. Thompson and M. Mayor, Chem.–Eur. J., 2011, 17, 13473–13481 CrossRef CAS PubMed.
- A. C. Sunil Sekhar, C. J. Meera, K. V. Ziyad, C. S. Gopinath and C. P. Vinod, Catal. Sci. Technol., 2013, 3, 1190–1193 Search PubMed.
- W. B. Lu, R. Ning, X. Y. Qin, Y. W. Zhang, G. H. Chang, S. Liu, Y. L. Luo and X. P. Sun, J. Hazard. Mater., 2011, 197, 320–326 CrossRef CAS PubMed.
- J. Sa, S. F. R. T. Taylor, H. Daly, A. Goguet, R. Tiruvalam, Q. He, C. J. Kiely, G. J. Hutchings and C. Hardacre, ACS Catal., 2012, 2, 552–560 CrossRef CAS.
- S. Wang, M. Zhang and W. Zhang, ACS Catal., 2011, 1, 207–211 CrossRef CAS.
- T. Ishida, H. Watanabe, T. Bebeko, T. Akita and M. Haruta, Appl. Catal., A, 2010, 377, 42–46 CrossRef CAS PubMed.
- H. Koga, E. Tokunaga, M. Hidaka, Y. Umemura, T. Saito, A. Isogai and T. Kitaoka, Chem. Commun., 2010, 46, 8567–8569 RSC.
- L. Johnson, W. Thielemans and D. A. Walsh, J. Mater. Chem., 2010, 20, 1737–1743 RSC.
- K. Benaissi, L. Johnson, D. A. Walsh and W. Thielemans, Green Chem., 2010, 12, 220–222 RSC.
- X. B. Lin, M. Wu, D. Y. Wu, S. Kuga, T. Endo and Y. Huang, Green Chem., 2011, 13, 283–287 RSC.
- L. Johnson, W. Thielemans and D. A. Walsh, Green Chem., 2011, 13, 1686–1693 RSC.
- I. K. Sen, K. Maity and S. S. Islam, Carbohydr. Polym., 2013, 91, 518–528 CrossRef CAS PubMed.
- X. Huang, H. Wu, X. P. Liao and B. Shi, Green Chem., 2010, 12, 395–399 RSC.
- R. Xiong, C. H. Lu, Y. R. Wang, Z. H. Zhou and X. X. Zhang, J. Mater. Chem. A, 2013, 1, 14910–14918 CAS.
- J. Cai, S. Kimura, M. Wada and S. Kuga, Biomacromolecules, 2009, 10, 87–94 CrossRef CAS PubMed.
- J. H. Johnston and T. Nilsson, J. Mater. Sci., 2012, 47, 1103–1112 CrossRef CAS.
- Z. H. Li, A. Friedrich and A. Taubert, J. Mater. Chem., 2008, 18, 1008–1014 RSC.
- R. Xiong, X. X. Zhang, D. Tian, Z. H. Zhou and C. H. Lu, Cellulose, 2012, 19, 1189–1198 CrossRef CAS.
- A. F. Miller and A. M. Donald, Biomacromolecules, 2003, 4, 510–517 CrossRef CAS PubMed.
- S. K. Das, C. Dickinson, F. Lafir, D. F. Brougham and E. Marsili, Green Chem., 2012, 14, 1322–1334 RSC.
- Y. Nagaoka and S. Adachi, J. Lumin., 2014, 45, 797–802 CrossRef PubMed.
- Z. J. Cai and J. Kim, Cellulose, 2010, 17, 83–91 CrossRef CAS.
- F. Z. Zhang, X. F. Zhao, C. H. Feng, B. Li, T. Chen, W. Lu, X. D. Lei and S. L. Xu, ACS Catal., 2011, 1, 232–237 CrossRef CAS.
- L. J. Chen, H. H. Ma, K. C. Chen, H. R. Cha, Y. I. Lee, D. J. Qian, J. C. Hao and H. G. Liu, J. Colloid Interface Sci., 2011, 362, 81–88 CrossRef CAS PubMed.
- N. L. Moigne and P. Navard, Cellulose, 2010, 17, 31–45 CrossRef.
- D. Ciolacu, F. Ciolacu and V. I. Popa, Cellul. Chem. Technol., 2011, 45, 13–21 CAS.
- Y. Habibi, H. Chanzy and M. R. Vignon, Cellulose, 2006, 13, 679–687 CrossRef CAS.
- G. Biliuta, L. Fras, V. Harabagiu and S. Coseri, Dig. J. Nanomater. Bios., 2011, 6, 291–297 Search PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- D. A. Bulushev, I. Yuranov, E. I. Suvorova, P. A. Buffat and L. Kiwi-Minsker, J. Catal., 2004, 224, 8–17 CrossRef CAS PubMed.
- M. Park, B. Kim, S. Kim, D. Han, G. Kim and K. Lee, Carbon, 2011, 49, 811–818 CrossRef CAS PubMed.
- M. Song, J. Xu and C. Wu, J. Nanotechnol., 2012, 2012, 329318 CrossRef PubMed.
- D. J. Miller, L. Sun, M. J. Walzak, N. S. McIntyre, D. Chvedov and A. Rosenfeld, Surf. Interface Anal., 2005, 37, 499–508 CrossRef CAS.
- A. Kumar, S. Mandal, P. R. Selvakannan, R. Pasricha, A. B. Mandale and M. Sastry, Langmuir, 2003, 19, 6277–6282 CrossRef CAS.
- J. Manson, D. Kumar, B. J. Meen and D. Dixon, Gold Bull., 2011, 44, 99–105 CrossRef CAS PubMed.
- P. Suramanee, S. Poompradub, R. Rojanathanes and P. Thamyongkit, Catal. Lett., 2011, 141, 1677–1684 CrossRef CAS PubMed.
- K. Kuroda, T. Ishida and M. Haruta, J. Mol. Catal. A: Chem., 2009, 298, 7–11 CrossRef CAS PubMed.
- M. M. Zhang, L. Liu, C. L. Wu, G. Q. Fu, H. Y. Zhao and B. L. He, Polymer, 2007, 48, 1989–1997 CrossRef CAS PubMed.
- Y. Wang, G. W. Wei, W. Q. Zhang, X. W. Jiang, P. W. Zheng, L. Q. Shi and A. J. Dong, J. Mol. Catal. A: Chem., 2007, 266, 233–238 CrossRef CAS PubMed.
- Y. C. Chang and D. H. Chen, J. Hazard. Mater., 2009, 165, 664–669 CrossRef CAS PubMed.
- T. Huang, F. Meng and L. M. Qi, J. Phys. Chem. C, 2009, 113, 13636–13642 CAS.
- M. H. Rashid and T. K. Mandal, Adv. Funct. Mater., 2008, 18, 2261–2271 CrossRef CAS.
- W. B. Lu, R. Ning, X. Y. Qin, Y. W. Zhang, G. H. Chang, S. Liu, Y. L. Luo and X. P. Sun, J. Hazard. Mater., 2011, 197, 320–326 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |