
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Third-generation solar cells: a review and comparison of polymer:fullerene, hybrid polymer and perovskite solar cells
Junfeng Yan
and
Brian R. Saunders
*
Polymer Science and Technology Group, School of Materials, The University of Manchester, Grosvenor Street, Manchester, M13 9PL, UK. E-mail: Brian.Saunders@manchester.ac.uk
First published on 26th August 2014
Abstract
The need for large scale low carbon solar electricity production has become increasingly urgent for reasons of energy security and climate change mitigation. Third-generation solar cells (SCs) are solution processed SCs based on semiconducting organic macromolecules, inorganic nanoparticles or hybrids. This review considers and compares three types of promising 3rd-generation SCs: polymer:fullerene, hybrid polymer and perovskite SCs. The review considers work reported since an earlier review (Saunders et al., Adv. Colloid Interface Sci., 2008, 138, 1) and highlights the great progress that has been made in each area. We consider the operation principles for each SC type and also review the state-of-the-art devices. The polymer:fullerene and hybrid polymer SC open circuit voltages are compared to values predicted from the well-known Scharber equation and similarities and differences discussed. The perovskite SCs are also considered and their remarkable rate of power conversion efficiency performance increase is discussed. The review considers the requirements for large-scale deployment in the contexts of semiconducting polymer and hole transport matrix synthesis and materials selection. It is concluded that the 3rd-generation SC technologies discussed here are well placed for major contribution to large scale energy production. (This has already been partially demonstrated for polymer:fullerene SCs.) Looking further ahead we propose that several of the 3rd-generation SCs considered here have excellent potential to provide the low cost large-scale deployment needed to meet the terawatt challenge for solar electricity generation.
 Junfeng Yan | Dr Junfeng Yan is currently a postdoctoral research associate in School of Materials, University of Manchester. He obtained his PhD in July 2013 in Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences. His PhD concerned fabrication of polymer brush/nanocrystal composite materials and their photoelectric performance. Afterwards he joined Prof. Brian R. Saunders group at University of Manchester as a postdoctoral researcher. His current research interest is directed assembly of nanocrystals for semiconducting polymer composites to establish solar cells. |
 Brian R. Saunders | Brian R. Saunders obtained his PhD at Monash University and worked as a postdoc with Prof. Brian Vincent at the University of Bristol between 1994 and 1997. He is a Professor of Polymer and Colloid Chemistry at the School of Materials, University of Manchester. His research career at Manchester began in 2002 and involves the application of polymer and colloid chemistry principles to solar energy and healthcare research. Current research interests involve the use of colloid and polymer chemistry principles to improve the morphology of the photoactive layers of hybrid polymer solar cells. |
1. Introduction
Third-generation solar cells (SCs) are solution processable SCs with excellent potential for large-scale solar electricity generation. This review updates and greatly extends an earlier review by one of us in 2008.1 We consider three families of 3rd-generation SCs technologies and discuss their operational principles. The review focusses on the strong progress in both their understanding and improvements in their power conversion efficiencies (PCEs) achieved in the past 6 years. The 3rd-generation SCs considered are polymer:fullerene, hybrid polymer and perovskite SCs. The first two SC types allow us to compare and assess SC types where the principle difference is the nature of the acceptor phase. Inclusion of perovskite SCs enables comparison of mixed molecular (perovskite) and colloidal organic/inorganic (hybrid polymer) SC technologies. In each case we focus on systems which have potential for large scale deployment in the future. Whilst excellent progress has been made for dye-sensitised SCs2 (DSSCs) and small molecule SCs3 a comprehensive discussion of DSSCs falls outside the scope of this review. Perovskite SCs are a disruptive SC technology that has recently captured great attention due to their outstanding power conversion efficiency (PCE) values.4 For the polymer:fullerene and hybrid polymer SCs we compare the performance of a key SC parameters to those predicted from energy level theory. The relative performance of each SC family is also compared and the potential deployment of the SCs for large scale, cost-effective, energy generation is considered. Because each of the 3rd-generation SCs considered are solution processable with PCE values that range from respectable to very high, each of the SC families are considered to have excellent potential for large-scale deployment via roll-to-roll (R2R) production. We propose that polymer:fullerene and perovskite SCs have reached points in their technological evolutions where large scale deployment is possible.1.1 Current and future global solar electricity generation
More than 80% of the world's energy mix is derived from fossil fuels,5 which in turn produce CO2. Coal, oil and gas accounted for 81.6% of the world's total primary energy supply in 2011.6 Looking ahead, the world's energy demand is forecast to double and electricity demand quadruple by 2050.7 The projected future increase in energy use is expected to result in an 80% increase in CO2 emissions without determined action to incorporate low carbon energy generation alternatives.7 Smalley estimated that 60 TW (6 × 1013 W) of power would be required to provide the anticipated future population of 10 billion people with the standard of living enjoyed by the developed world in 2004.8 Solar energy is the only renewable energy source capable of providing all of the energy needs of man in a sustainable manner. More energy arrives at the earth's surface in one hour than is used by man in one year.9 Whilst impressive gains in solar energy production have been achieved with parity reached with fossil fuel derived electricity already in several countries (below), a major expansion of SC manufacture and deployment is urgently required to answer Smalley's terawatt challenge.The Inter-governmental Panel on Climate Change (IPCC) recently concluded that (a) the human influence on the climate system is clear; (b) that atmospheric concentration of CO2 has increased to levels unprecedented in at least the last 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years and (c) that the largest contribution to total radiative forcing is caused by the increase in CO2 concentration.10 The report shows the direct relationship between global temperature and atmospheric CO2 concentration and also how future global temperature increases can be modulated by reducing CO2 output. The IPCC's Impacts, Adaption and Vulnerability report11 concluded that there will be many more negative impacts than positive impacts due to climate change in the future. The key risks involving food insecurity, fresh water access, flooding, extreme weather events, loss of marine and costal ecosystems can be reduced by limiting the rate and magnitude of climate change. Clearly, the sustainability of ecosystems that support everyday life requires future atmospheric CO2 increases to be minimised.
000 years and (c) that the largest contribution to total radiative forcing is caused by the increase in CO2 concentration.10 The report shows the direct relationship between global temperature and atmospheric CO2 concentration and also how future global temperature increases can be modulated by reducing CO2 output. The IPCC's Impacts, Adaption and Vulnerability report11 concluded that there will be many more negative impacts than positive impacts due to climate change in the future. The key risks involving food insecurity, fresh water access, flooding, extreme weather events, loss of marine and costal ecosystems can be reduced by limiting the rate and magnitude of climate change. Clearly, the sustainability of ecosystems that support everyday life requires future atmospheric CO2 increases to be minimised.
Unfortunately, progress toward reducing CO2 output has been slow to date. The International Energy Agency (IEA) estimates that the long term global temperature increase is on track for12 3.6 °C, which is well above the internationally-agreed increase climate target of 2 °C (relative to pre-industrial levels). A global temperature increase of 6 °C is expected if the business-as-usual case persists.13 A rapid transition to an energy mix containing high proportions of renewable energy is urgently required. The IPCC's mitigation document called for a tripling-to-quadrupling of the share of zero to low-carbon energy supply by 2050 to keep the temperature increase to below14 2 °C. Electricity generation currently emits about 40% of energy-related CO2 emissions.15 Hence, solar energy, a low-carbon energy source, has a key role to play in mitigating climate change.
Solar energy currently provides 2.6% of the electricity demand and 5.2% of the peak electricity demand in Europe.16 Global installed photovoltaic capacity has been growing exponentially since 2000 (see Fig. 1) and surpassed 100 GW in16 2012. The rate of installation (Fig. 1) is highest when installation is government policy driven. The impressive growth and adoption of the technology in economies such as Europe and China (with strong growth in the US17) coupled with the falling solar module prices provides good reason for optimism about the potential for solar electricity generation to become a major source of low carbon energy in the future. Indeed, solar electricity has achieved price parity with the electrical grid since 2012 in Denmark, Germany and Italy and many other European countries are expected to follow this trend.18 The IEA predicts that by 2050 solar power could generate 22% of the world's electricity.19 However, the percentage of electricity generated could be even higher if low cost, large-scale production of SCs were to occur as a result of new, disruptive, technologies. Here, we discuss new 3rd-generation SC technologies that offer realistic potential to enable this much-needed low cost electricity.
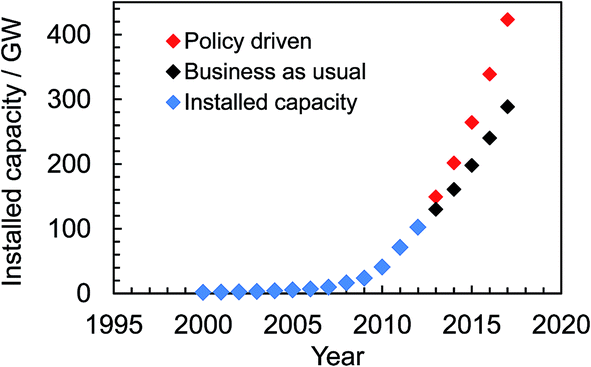 | ||
| Fig. 1 Global cumulative solar cell capacity installation scenarios until 2017. The data were taken from ref. 16. | ||
1.2 Solar cell operational fundamentals
The principles of SC operation have been described in detail elsewhere.20 A brief review is given here as a prelude to discussion for the 3rd-generation SCs. 1st-generation SCs are based on crystalline Si20 (c-Si); whilst, 2nd-generation SCs are based on thin film technology which has often involved vapour deposited semiconductors. By contrast, 3rd-generation SCs are solution processed SCs based on organics, hybrids, inorganic semiconductors21 and include nanostructured SCs.22 All SCs harvest solar radiation and convert this energy into electricity. SCs achieve power conversion using the photovoltaic effect. For the latter, photons that have energies greater than the band gap energy (Eg) are absorbed and this process excites an electron from the valence band to the conduction band. SCs have a built in asymmetry so that an electrical potential causes the electrons to reach the external circuit.20SC performance is characterised by the measurement of the current density as the voltage across the device is biased with variable load during device irradiation by light (Fig. 2(a)). The PCE is related to the short-circuit current density (Jsc) and the open circuit voltage (Voc) by:
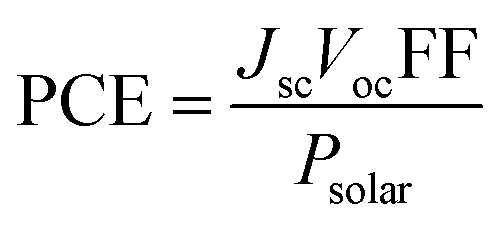 | (1) |
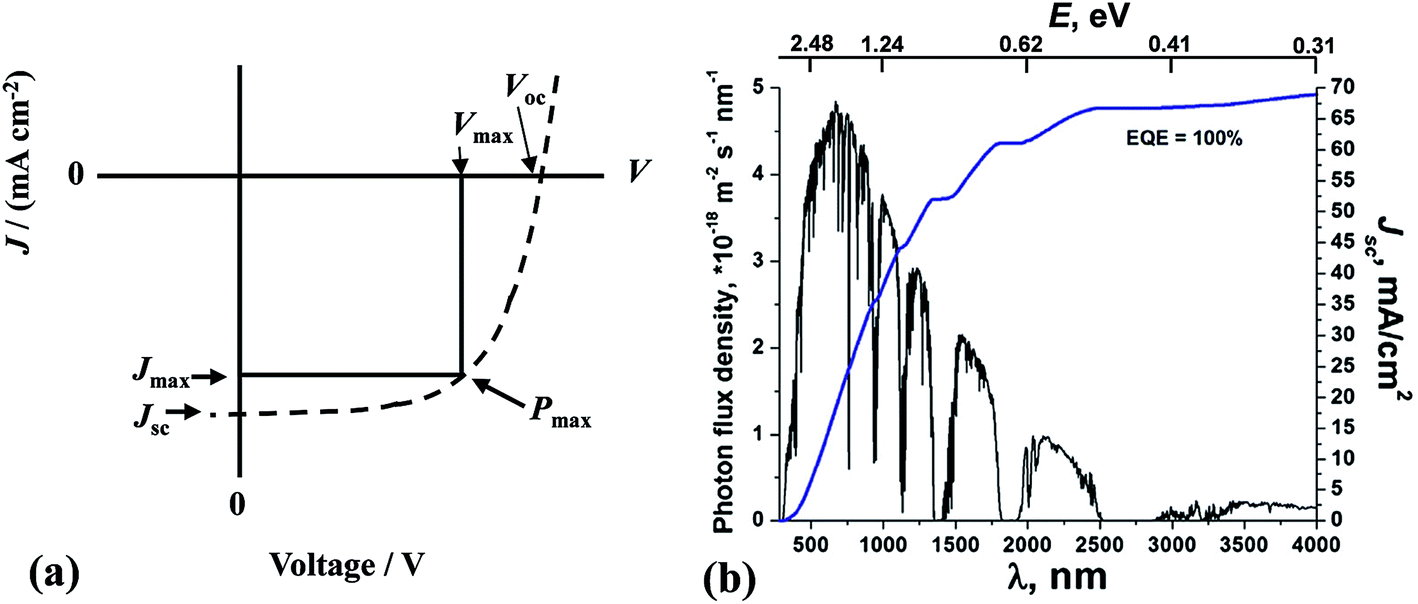 | ||
| Fig. 2 Solar cell performance and solar radiation. (a) Example of a current density vs. voltage profile for a SC. (b) The global total solar photon flux and maximum achievable integrated short-circuit current density. (b) reproduced with permission from ref. 24. The values for E have been added to the original figure here. | ||
The Jsc value for a SC depends strongly on the value for Eg. Because the photon energy (E) is inversely proportional to the wavelength (E(eV) = 1240/λ(nm)), Jsc generally increases with increasing λ across the visible and infra-red regions of the solar spectrum provided that E is greater than Eg (Fig. 2(b)). Although the PCE increases with Jsc, there is a trade-off involving Voc which means that an optimum Eg value exists for PCE. Generally, as the value for Eg decreases the values for Voc decrease. The Eg value for achieving optimum PCE values for c-Si SCs20 is between 1.0 and 1.6 eV (1240–775 nm). The maximum efficiency for a SC with an Eg of 1.1 eV was calculated by Shockley and Queisser to be 30%.25
The light intensity absorbed in a SC decreases exponentially with film thickness.20 Consequently, a key parameter governing the PCE for a SC is the thickness of the photoactive layer compared to the absorption length (1/α, where α is the absorption coefficient in cm−1). The latter is the distance over which the 63% of the incident (non-reflected) light is absorbed. It is because Si SCs have relatively low α values that the thickness of their photoactive layers must be of the order of hundreds of micrometres to millimetres. The latter adds significantly to the material and production costs for c-Si SCs.
2. Polymer:fullerene solar cells
The first two SC families discussed are polymer:fullerene and hybrid polymer SCs. Here, we focus on polymer:fullerene SCs. However, many of the principles discussed also apply to hybrid polymer SCs. Polymer:fullerene SCs have photoactive layers comprising a semiconducting polymer and a fullerene and have been the subject of several reviews.1,26–32 In the case of hybrid polymer SCs the fullerene is replaced by a semiconducting inorganic nanoparticle. Both of these SC families contain bulk heterojunctions (BHJs). A heterojunction is an interface between two different semiconducting materials. In 1986 Tang reported an excitonic device consisting of two semiconductors in a bilayer and achieved a PCE of about 1%.33 He recognised that the performance depended on the nature of the interface between the two semiconductors. After establishing photoinduced electron transfer between a semiconducting polymer and a fullerene in 1992,34 Heeger et al. mixed MEH-PPV with PCBM to give the first SC containing a dispersed heterojunction in 1995. They referred to the mixed polymer donor and fullerene composite as a BHJ.35 A key to the success of their SCs was the greatly increased Jsc which improved efficiency in collection of the photogenerated charges. In 1995 Halls et al. reported the first example of a polymer:polymer BHJ.36 BHJs can be considered as interpenetrating bicontinuous polymer/nanoparticle network. The connected nanoparticle segments are separated by a curved polymer phase. There is increasing evidence that the polymer and fullerene phases may contain a degree of molecular mixing (below).Fig. 3(a) shows the components of the most widely studied polymer:fullerene SC, poly(3-hexylthiophene):phenyl C61-butyric acid methylester (P3HT:PCBM). Between 2002 and 2010 a total of 579 publications reported the PCE of P3HT:PCBM SCs.37 The average PCE value reported for these SCs in 2010 was 3.0%. The use of vertically aligned fullerenes via a template assisted construction strategy, combined with fullerene energy level modification, has enabled a PCE value of 7.3% for P3HT:fullerene SCs to be achieved.38 The energy level diagram (Fig. 3(b)) shows that photoexcited electrons can hop from the LUMO of the P3HT donor to the LUMO of the PCBM acceptor. P3HT and PCBM act as the hole and electron transfer phases, respectively. The phase separated photoactive layer was formed during solvent evaporation and is a non-equilibrium, kinetically trapped structure. The complex morphology of the photoactive layer (Fig. 3(c)) consists of P3HT crystallites, PCBM clusters as well as amorphous P3HT and intercalated (molecularly mixed) PCBM molecules.39 Liao et al. showed that annealing promoted PCBM cluster formation and charge transport which enhanced the PCE.39 They demonstrated that the nanostructures could evolve with time using moderate heating (150 °C for 15 min); whereby, PCBM changed from well-separated to a partial attachment regime. Molecular mixing of polymer and fullerene parts is recognised to be present for many polymer:fullerene SCs and this plays an important role in both charge separation and recombination.26 The end-stage morphology for thermally annealed P3HT:PCBM films is phase separated domains of P3HT and PCBM crystals.40 The latter morphology does not enable efficient charge transport to the external circuit. (We discuss polymer:fullerene morphology further in Section 2.5.) The search for lower band gap semiconducting polymers (e.g., PTB7 (ref. 41)) and the use of processing aids for morphology enhancement (e.g., 1,8-diiodoctane, DIO42–44) have resulted in major improvements of the PCE of research grade polymer:fullerene SCs, which are approaching 10% for single junction devices (later).
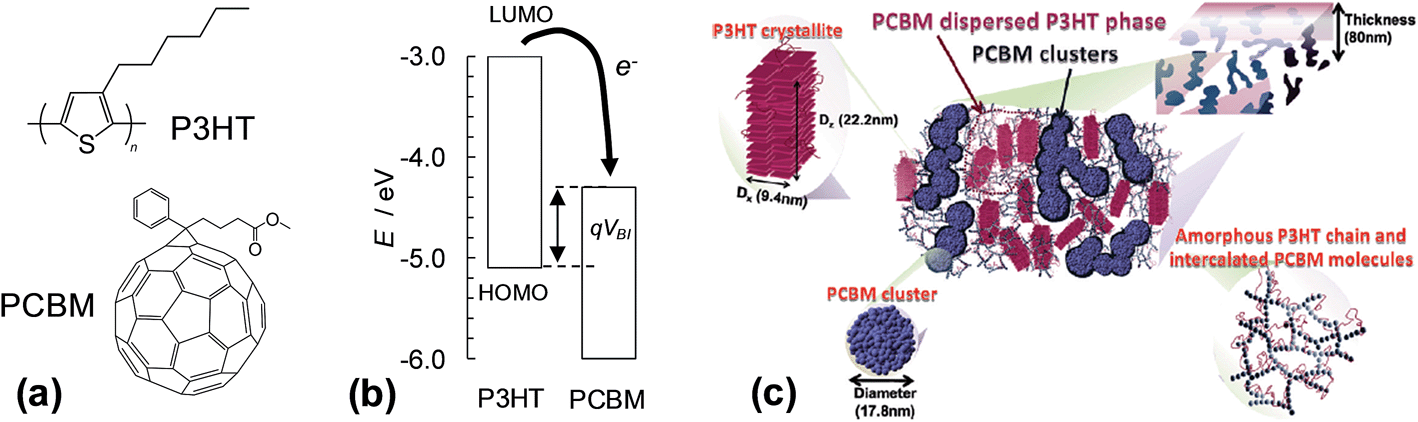 | ||
| Fig. 3 Structures, energy levels and morphologies for polymer:fullerene composites. (a) shows the structures of P3HT and PCBM. (b) Energy level diagram for P3HT:PCBM. VBI is the built-in voltage (see text) and q is the electron charge. The energy levels for P3HT and PCBM are from ref. 45 and 46, respectively. (c) Schematic of the phase-separated morphology for P3HT:PCBM film comprising PCBM-dispersed P3HT phase, P3HT crystallites, an amorphous P3HT chain region and a network of intercalated PCBM molecules and PCBM clusters. (c) reproduced with permission from ref. 39. | ||
2.1 Charge generation and transport processes within polymer:fullerene solar cells
The process of photo-generation and charge transport to the external circuit within a photoactive layer consisting of a donor and acceptor47 is depicted in Fig. 4 and can be separated into five steps. The first step is light absorption and photogeneration of an exciton (Fig. 4(a)). An exciton is an electrostatically bound electron–hole pair. In the depiction given the geminate (initially formed) exciton is created within the donor polymer phase. The efficiency of the initial exciton generation is given by the photon absorption efficiency, ηA. This parameter depends on factors such as the film thickness compared to the absorption length as well as Eg.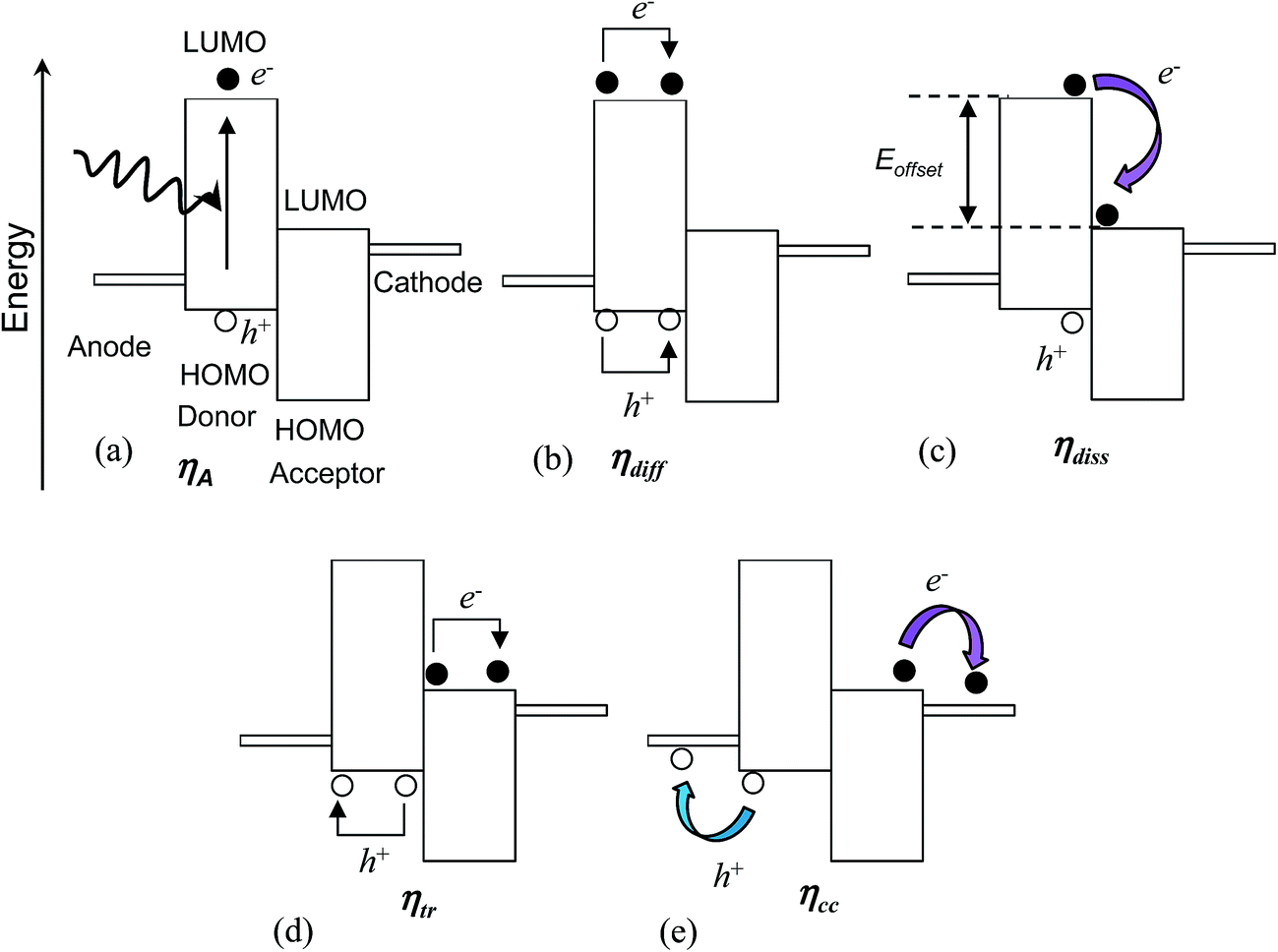 | ||
| Fig. 4 Processes responsible for photocurrent within photoactive donor/acceptor composites. The processes involve light absorption and photogeneration of an exciton (a). The latter is followed by exciton diffusion to the donor/acceptor interface (b), exciton dissociation (c), transport to the photoactive layer/electrode interface (d) and collection of the charges by the electrodes (e) which transfers the photogenerated charges to the external circuit. Note that e− and h+ represent an electron and hole, respectively. | ||
Excitons in semiconducting polymers are short-lived species that tend to rapidly recombine (annihilate). Because most semiconducting donor polymers have low dielectric constants (of about48 3), excitons are tightly bound due to coulombic attraction and have an exciton diffusion length (Lex) less than about 10 nm.26 Consequently, the polymer phase domains should be less than about 20 nm in size in order for an interface with an acceptor to be close enough to permit exciton dissociation prior to geminate recombination.29 The efficiency of the excitons reaching this heterojunction without geminate recombination is given by the exciton diffusion efficiency (ηdiff) (Fig. 4(b)).
Having reached the donor/acceptor interface (Fig. 4(c)), exciton dissociation may occur if the energy difference (offset) between the LUMOs for the donor and acceptor exceeds the exciton binding energy. The latter can be as low as 0.12 eV.49 Charge generation (from dissociation) for polymer-based excitons involves electron transfer (rather than hole transfer).26 The efficiency of the dissociation process can be described in terms of the exciton dissociation efficiency, ηdiss. The value for ηdiss approaches zero as the energy offset between the LUMOs approaches zero.
Once the exciton has dissociated the free electron and hole migrate to the photocathode and photoanode, respectively. There are a number of processes that may direct the flow of charge carriers. The migration may occur due to the driving force from the gradient in chemical potentials of the electrons and holes at the donor–acceptor junction.27 The potential energy gradient originates from the difference between the donor HOMO and acceptor LUMO. Furthermore, charge concentration gradients can produce diffusion currents.27 Electron- and hole-blocking layers are often included in BHJ SCs to direct the charge migration to the desired electrodes in order to decrease recombination.
The photo-generated holes and electrons within BHJ photoactive layers migrate (via hopping) through interconnected donor and acceptor phases, respectively. In order to reach the respective electrode holes and electrons must avoid coming into contact at an interface, and undergoing non-geminate (bi-molecular) recombination. Unfortunately, a high interfacial area favours non-geminate recombination, even though it reduces geminate recombination. The efficiency of the separated charges reaching the electrodes is given by the charge carrier transport efficiency (ηtr) (Fig. 4(d)).
Finally, the separated charges must cross the photoactive layer/electrode interfaces to reach the external circuit (Fig. 4(e)). The efficiency of charge transfer across this interface is given by the charge collection efficiency47 (ηcc). This parameter is sensitive to the nature of the electrical contact between the photoactive layer and the electrodes and decreases if these interfacial connections are highly resistive. It is also sensitive to the energy levels of each phase.47
The product of the above efficiencies gives the external quantum efficiency (EQE):
| EQE = ηAηdiffηdissηtrηcc | (2) |
The EQE is the ratio of the number of electrons collected in the external circuit to the number of incident photons. Eqn (2) illustrates the difficulty of achieving high panchromatic EQE values for BHJ SCs because each of the five efficiency parameters must be optimised to give values as close as possible to unity. The parameters ηdiff and ηtr are highly dependent on morphology. For some polymer:fullerene SCs EQE values of 100% have been reported over a limited spectral range.50 However, the EQE is usually significantly less than 100% over the whole solar wavelength range. Whilst the PCE generally increases with EQE, a high EQE is not sufficient to guarantee a high PCE. This caveat originates from the dependence of the PCE on Voc and FF (eqn (1)). Conditions that favour high EQE values (and high Jsc values) may lead to non-optimal Voc values.
Whilst the majority of polymer:fullerene SCs have a conventional design,1 some of the more recent, highest PCE devices, have adopted an inverted architecture.51,52 An example inverted device is shown in Fig. 5(a). Inverted structures can provide a PCE benefit because of vertical phase segregation and increased FF values.51 Inverted architectures are also more compatible with R2R processing and their design avoids the necessity of using reactive cathode materials (e.g., Al), which can be detrimental to device stability.53 An inverted geometry enables the use of Ag which can be screen printed and is compatible with R2R processing.54 For the research grade device architecture shown in Fig. 5(a), ZnO and MoOx acted as the electron transport and hole transport matrix, respectively.51 Those layers prevented contact of the polymer and PCBM phases with the electrodes. ITO and Ag were the cathode and anode, respectively. For a conventional polymer:fullerene SC architecture, ITO is usually the photoanode.1
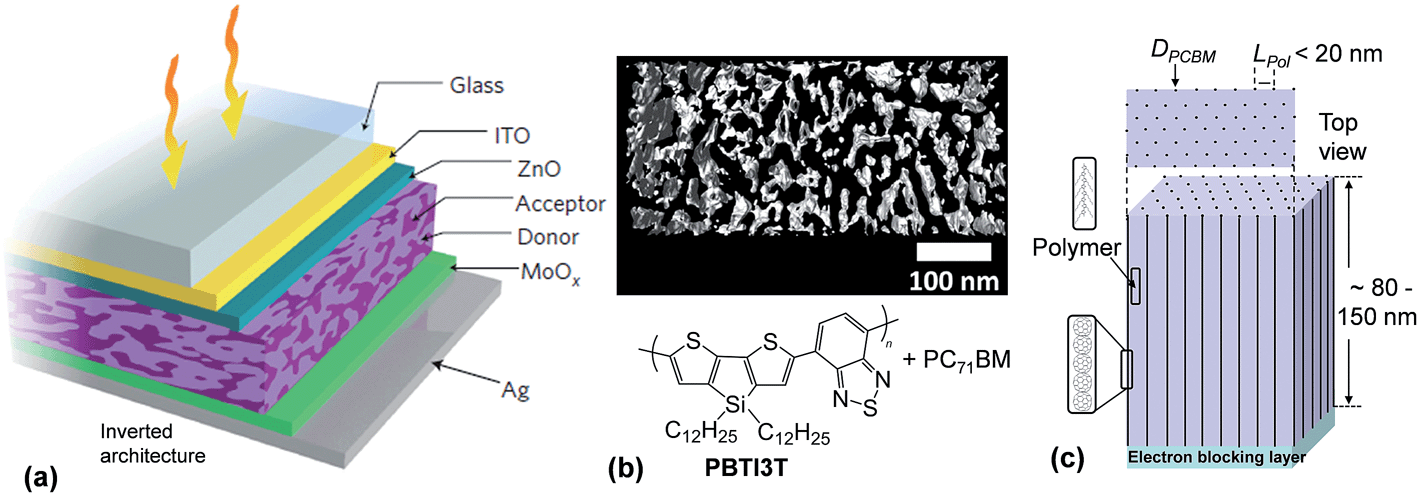 | ||
| Fig. 5 High performance polymer:fullerene solar cell architectures and morphologies. (a) Schematic of an inverted PBTI3T:PC71BM SC. (b) Electron tomography image showing morphology for a polymer:PC71BM photoactive layer. The black and white areas are the PC71BM and polymer, respectively. (c) Depiction of one ideal morphology for a polymer:PCBM layer. DPCBM and LPol are the PCBM column diameter and polymer domain length, respectively. Figures reproduced with permission from (a) ref. 51 and (b) ref. 29. | ||
The overwhelming majority of BHJ SCs reported in the literature comprise small area devices and have usually been prepared using spin coating. Typically, a low concentration polymer solution and a fullerene solution are prepared separately and then mixed. Shortly after mixing spin-coating of the mixed solution onto substrate is conducted. Spin coating results in rapid solvent evaporation and phase separation. Solution processing additives such as DIO51 may be added to the organic solvent (e.g., chloroform51) prior to spin-coating to enhance the PCE. The composite films are then thermally annealed.51,52 The aim of these treatments is to increase the structural order present within the photoactive layers so as to reduce recombination. It is generally believed that establishing more direct pathways to the electrodes decreases the probability of bimolecular recombination. The photoactive layer thickness for BHJ SCs is often 80 to 200 nm.1,52
Fig 5(b) shows a high resolution electron tomography image of a polymer:PC71BM film.29 The latter diffracts electrons more strongly than the polymer phase and appears black. The length scale of the polymer phase separated domains (Lpol) within these films is of the order of ∼20 nm which is about twice the value for Lex and this morphology is considered ideal in terms of achieving high ηdiff values. Whilst the morphology shown in Fig. 5(b) is isotropic, the optimum morphology for a BHJ SC to achieve maximum ηdiff and ηtr values is believed to be anisotropic and is depicted in Fig. 5(c). This morphology would consist of columns of vertically aligned acceptor phase (with electrically connected acceptor species and for aggregates within each column). The columns would be located within and separated by the polymer donor phase. Ideally, the columns should be equally spaced. The columns are depicted to be separated by Lpol ≃ 20 nm. An ultimate goal of BHJ SC research is to devise methods whereby this anisotropic, vertically oriented, morphology spontaneously forms both in research grade SCs and R2R-processed modules. Progress towards achieving this goal is discussed in Section 2.5.
2.2 Factors controlling polymer:fullerene solar cell performance parameters
We next consider factors controlling the three parameters that determine the PCE in eqn (1). Although the discussion given below concentrates on polymer:fullerene SCs, it also applies to the hybrid polymer SCs that are discussed in detail later.The value for Jsc is determined by the EQE. Indeed, the integrated area of an EQE vs. wavelength plot is often used to test the validity of Jsc measured from J–V plots.55 Therefore, the Jsc value is controlled by the optical properties of the light absorbing phase (Eg and α) as well as the blend morphology. In addition, a key factor controlling Jsc is the charge mobility within the polymer phase. Photoactive layers with thicknesses that are small compared to the absorption length have low ηA values. Although thicker films will have higher ηA values, the relatively low mobility of the polymer phase (typically 10−3 to 10−4 cm2 V−1 s−1 (ref. 56)) means that higher series resistance decreases FF and, hence, PCE. A low charge mobility also increases recombination. Polymer-based SCs require a thickness trade-off to be made between ηA and ηtr.
According to eqn (1) the PCE is proportional to the Voc value. In a seminal study, Scharber et al.45 compared the Voc values for 26 polymer:PCBM SCs to the HOMO positions of the polymer donors. They showed that the following relationship applied:
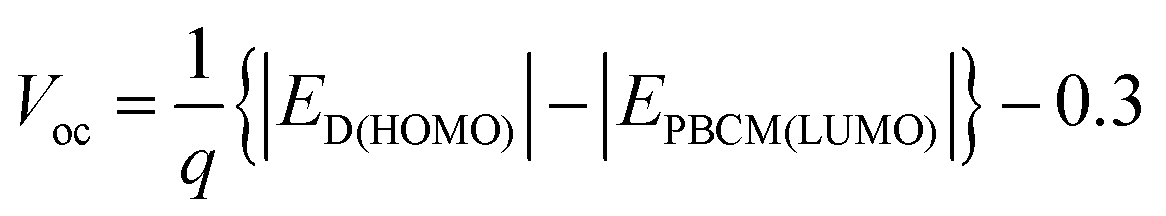 | (3) |
Eqn (3) was (and remains) very important for the design of polymer:fullerene SCs because it showed that Voc could be maximised by using polymer donors with deep HOMO energy levels. Scharber et al.45 also established a quantitative theoretical contour plot to guide polymer design to achieve improved PCE values. Their work showed that achieving a single polymer:fullerene with a PCE of 10% was theoretically possible. It followed from their study that to achieve a PCE of 10% for a polymer:PCBM SC the value for ED(LUMO) of the polymer donor should be less than ∼−3.9 eV and Eg should be less than45 1.74 eV. A suitable ED(HOMO) value would be ∼−5.7 eV. Fig. 6 shows PCE data from plotted as a function of polymer Eg for 60 polymer:fullerene SCs. It can be seen that the devices with the top 7 PCE values contained polymers with Eg values of 1.7 ± 0.2 eV, which supports the recommendation regarding Eg. We consider the other parameters later.
 | ||
| Fig. 6 Variation of PCE with Eg for polymer:fullerene SCs. The vertical line corresponds to 1.74 eV (see text). These data were taken from Table 1. The identities of the acceptors are shown in Fig. 8. | ||
The FF value is determined by competition between sweep-out of the photogenerated charge carriers and their recombination.29 The sweep-out of charge carriers is driven by the internal electric field, Vint, which is given by:29
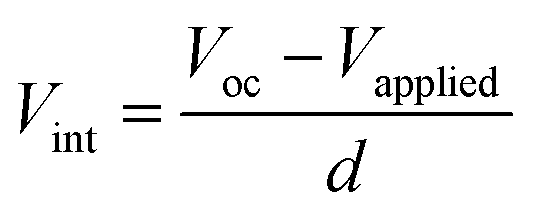 | (4) |
2.3 Polymer donors
Semiconducting polymers are the electron donors of the BHJ SCs discussed here and have been the subject of comprehensive reviews.24,56,58 Here, we focus on the key aspects of the polymers from the viewpoints of Voc, PCE and potential scale up. The overall aim of semiconducting polymer backbone design is to decrease and optimise Eg and ED(HOMO), respectively, to maximise Jsc and Voc. Strategies for decreasing Eg include58 using polymer backbones with donor–acceptor units;59,60 stabilising the quinoid structure and incorporation of electron withdrawing groups.Fig. 7 shows the structures and reported ED(HOMO) and ED(LUMO) energy levels for polymers that have been frequently been used for polymer:fullerene SCs. The polymers are classified as homopolymers (P3HT, MEH-PPV and MDMO-PPV), donor–acceptor (PCPDTBT, PDTPBT)61 and quinoid type (PTB7 and PBDTTPD).58 D–A polymers contain alternating electron-rich (donor) and electron deficient (acceptor) units. Internal charge transfer (ICT) from the donor to acceptor promotes backbone copolarity and is responsible for the decreased Eg values. One of the key advantages of D–A polymers is the ability to individually tune the ED(HOMO) and ED(LUMO),56 which results from the ability to independently adjust the electron donating ability of the donor and electron affinity of the acceptor.58 Fig. 7 illustrates donor and acceptor units. Whilst a weak donor decreases ED(HOMO) a strong acceptor decreases Eg by enhancing ICT.58
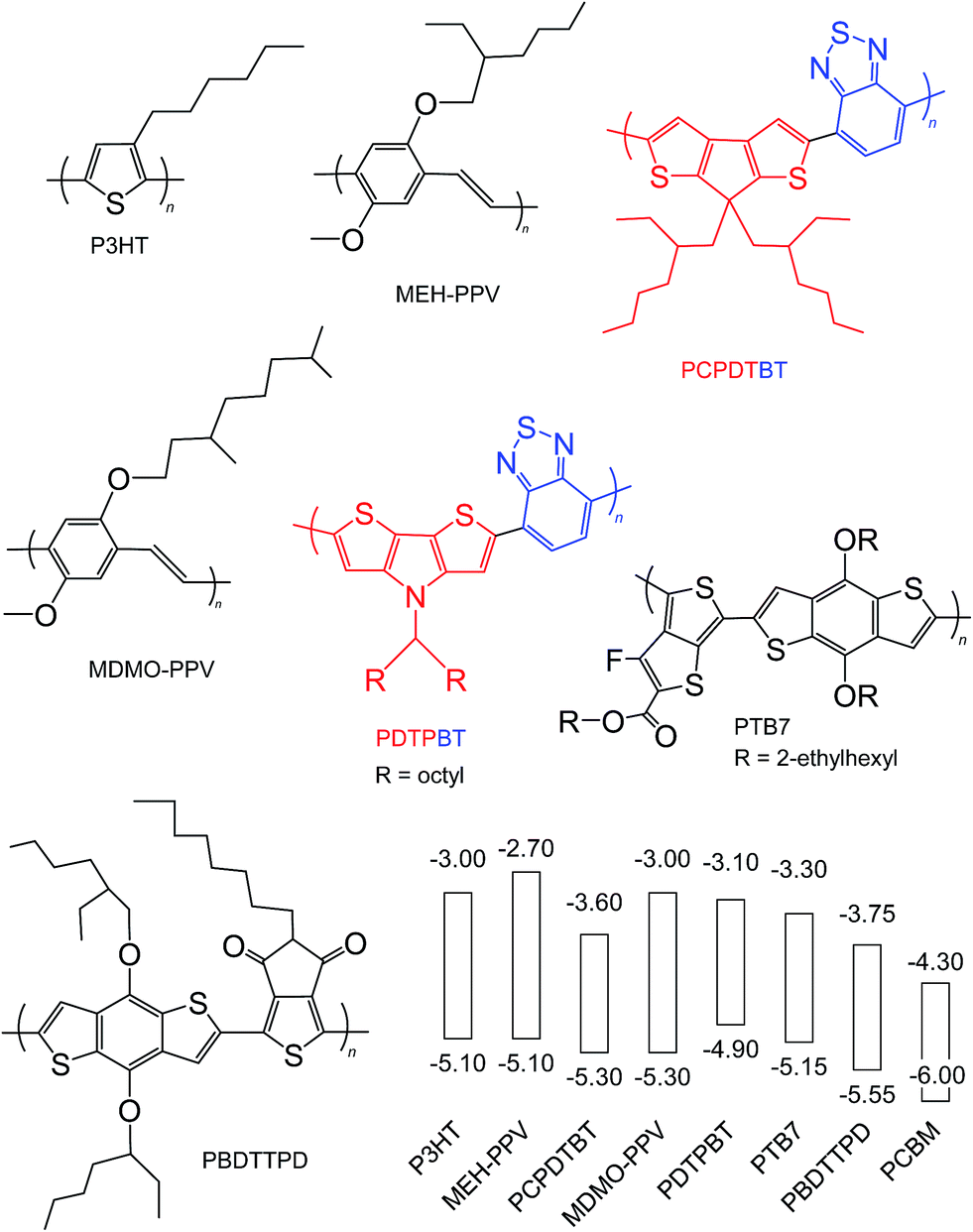 | ||
| Fig. 7 Structures and energy levels for selected polymer donors. The energy levels for P3HT are taken from ref. 45 and 46. The energy levels for MEH-PPV, PCPDTBT, MDMO-PPV, PDTPBT, PTB7 and PBDTTPD are taken from ref. 62, 63, 64, 65, 41 and 66. The energy levels for PCBM are shown for comparison and are from ref. 45. The red and blue units are the donor and acceptor units, respectively. | ||
Because the quinoid form has a lower resonance energy than the aromatic, polymer backbone design to favour quinoid formation across the repeat unit has been an effective strategy to decrease Eg.67 This approach uses two aromatic units fused together whereby one of the units has a larger resonance energy than the other.58 Substituent engineering can be used to decrease ED(HOMO)68 for semiconducting polymers. Fluorine is the smallest electron withdrawing group and has a high Pauling electronegativity and decreases the ED(HOMO).41,67 Fluorine also has a strong effect on polymer interactions and physical properties. Conversely, inclusion of electron donating groups increases ED(HOMO).69 In addition to altering the electronic properties, substituents can also affect the overall crystallinity of photoactive layers. Meager et al. showed that improved crystallinity and SC performance were obtained for diketopyrrolopyrrole polymers where the alkyl chain branching point was moved further from the backbone.70
The electrochemical Eg values for PCPDTBT, PDTPBT, PTB7 and PBDDTTPD are 1.7, 1.8, 1.85 and 1.8 eV, respectively. In this respect, these polymers (especially PBDDTTPD) most closely match the Eg design criteria from Scharber et al.45 discussed above (Eg < 1.74 eV). It can be seen from Fig. 7 (and elsewhere56) that there are now a wide range of polymers available for maximising the difference between EPBCM(LUMO) and ED(HOMO) as well as minimising Eg. Many of these are commercially available; however, their cost generally increases strongly with repeat unit complexity.
A limitation of the D–A polymers is that they generally have poor spectral coverage at low wavelengths. SCs containing D–A polymers often rely on the acceptor (usually PC71BM) to absorb photons from the lower wavelength region (to give complementary absorption) and contribute photocurrent.24 In this case excitons are created within the PC71BM phase and dissociation may involve hole charge transfer to the polymer phase.26 By tuning the Eg to the region of 1.74 eV the new D–A and quinoid-type polymers have enabled major increases in Jsc to be achieved.
An important property that has favoured success of polymer:fullerene SCs is the tendency of substituted fullerenes (e.g., PCBM and PC71BM) to have good compatibility with semiconducting polymers. The fullerenes are small enough and are sufficiently structurally similar to polymers such as P3HT that they can diffuse through the polymer matrix during annealing which improves the morphology. Cates et al.71 showed that intercalation of fullerenes could occur (i.e., molecular mixing) between the polymer side chains provided the latter were sufficiently well spaced to allow fullerene entry into this region. In that work intercalation increased the PCE due to an increased Jsc.71 The structure of the polymer can strongly influence (and even direct) the packing of the fullerene acceptors within polymer:fullerene SCs.
Whilst D–A and quinoid polymers have enabled preparation of BHJ SCs with very high PCE values, their complicated synthesis and high expense severely limits their potential for large scale use. Synthesis for the monomers usually involves more than 6 steps. In the case of PTB7 a total of 16 synthetic steps is required.24 Whilst multistep synthesis and expensive materials may be acceptable for the pharmaceutical industry, this is not likely to be the case for large scale deployment of polymer:fullerene SCs. For example a recent calculation for P3HT:PCBM SCs indicated that a solar array with an area of 137 km2 of SC foil would be required for production of 1 GWpeak. (The required area will of course decrease with increasing PCE.) The P3HT:PCBM SC foil in that study had a PCE of 1.5%.72 If we assume a hypothetical PTB7:fullerene SC foil efficiency of 3.0% it can be shown that about 7 tons of D–A polymer would be required to achieve 1 GWpeak! The masses of starting materials for a 16 step synthesis would be enormous. (Our PCE value used for this calculation may be optimistic because the PCE of scaled-up polymer:fullerene SCs does not always follow that of research grade devices.72) Clearly, semiconducting polymers with low numbers of high yield (low cost) synthetic steps are essential for achieving low-cost large-scale deployment of polymer:fullerene SCs with short energy payback times.
2.4 Fullerene acceptors
Fullerenes are both n-type semiconductors and good electron acceptors. They have been reviewed extensively elsewhere.73 We focus on the key aspects of fullerenes here. PCBM (i.e., PC61BM) was first synthesised by Hummelen and Wudl et al.74 and can be synthesised in four steps.73 PCBM has good solubility in many organic solvents. Unfortunately, PCBM has a weak absorption in the visible region (Fig. 8) and relatively high absorption lengths (below). Consequently, PCBM does not contribute substantially to Jsc within polymer:fullerene SCs. Because of this deficiency fullerenes which absorb more strongly in the solar spectrum have been developed. | ||
| Fig. 8 Structures and energy levels for selected fullerenes. The energy levels for PCBM, PC71BM, ICBA, C60-bp-CN and bis-PCBM are from ref. 45, 52 and 75, 76 and 77, respectively. The energy levels for P3HT are shown for comparison. The UV-visible spectrum is reproduced with permission from ref. 73. | ||
PC71BM was synthesised from C70 in order to increase the acceptor α value and spectral range.78 PC71BM is used in polymer:fullerene SCs to extend the absorption to 380–500 nm and is reported to give a PCE increase for polymer:fullerene SCs of about 10% compared to PC61BM.73 This success led to the synthesis of fullerenes with larger sizes (and more extensive conjugation) in order to further increase the light harvesting contribution from the acceptor. An increase in the size of the fullerenes to PC84BM increased α further and the absorption in the red part of the visible spectrum (Fig. 8). Unfortunately, the solubility of PC84BM decreased compared to PCBM. MDMO-PPV:PC84BM SCs had relatively low Voc and PCE values79 compared to MDMO-PPV:PCBM.
Considerable effort has been applied to synthesising substituted fullerenes with HOMO and LUMO levels that can be tuned by the substituents. Substituted PCBM derivatives have been used to adjust the Voc values of the SCs through modification of the EA(LUMO) levels (Fig. 8.). Although relatively high Voc values have been achieved,75,77 the PCE values for these polymer:fullerene SCs have not yet surpassed those of the highest reported values for the leading polymer:PC71BM SCs to our knowledge.
2.5 Polymer:fullerene morphology
The morphology of polymer:fullerene photoactive layers is a major factor affecting SC performance.80–83 It is widely accepted that efficient polymer:fullerene SCs require a BHJ morphology that achieves a compromise between charge dissociation and transport.82 Perfect dispersion of the acceptor within the donor matrix would maximise dissociation but not provide a pathway for the electrons to the photocathode. A morphology that provided perfect connectivity of an acceptor phase for electron transport would minimise dissociation. The best morphology for achieving high PCE values is believed to have a significant fraction of molecularly mixed polymer:fullerene phase next to phase separated pure material82 (polymer and fullerene domains). Indeed, as the PCBM concentration is increased within the photoactive layer it can be considered to fill the mixed phase until saturated and excess PCBM phase separates.80The morphology of polymer fullerene SCs has a significant effect on Eg and Voc. For example, aggregation of P3HT can cause a red shift of up to 0.5 eV.84 It is for these reasons that annealing of P3HT:PCBM, which promotes phase separation, usually results in a decrease of Voc and an increase in Jsc.82 PCBM aggregates are proposed to have a deeper EA(LUMO) compared to dispersed PCBM.85 This morphology is depicted in Fig. 3(c) and provides a mechanism whereby dissociated electrons can be transported from the mixed phase to the better inter-connected aggregated PCBM phase. Accordingly, the morphology of P3HT:PCBM is considered as a ternary blend of pure P3HT and PCBM phases with a P3HT:PCBM mixed phase.80 Annealing or the use of solvent additives or polymer structural modification alter the proportions of each of the phases. Generally, solvent additives (such as DIO) provide increased fullerene solvency and facilitate domain development82 in the phase separated regions.
One exciting area of polymer:fullerene morphology study involves vertically aligning the morphologies to achieve the type shown in Fig. 5(c). The highest PCE example of this general morphology for a polymer:fullerene SC to date involves the use of anodised alumina template-assisted nanoimprint lithography.38 Although much progress has been made using this nanoimprint lithography86,87 which is a potentially scalable technique, there have yet to be any studies reported that have demonstrated large-scale vertically aligned BHJ SCs. More work is urgently required with a view to applying the nanoimprinting stage to R2R production methods. Indeed, this could be an area where the PCE of large-scale R2R modulus could be greatly improved with major energy payback time reductions.
2.6 Comparing Voc values for polymer:fullerene solar cells with the Scharber equation
The Scharber equation (eqn (3)) has been widely used in the literature to guide rational design of polymer:fullerene SCs. Fig. 9(a) shows Voc data from 60 polymer:fullerene SCs plotted as a function of ED(HOMO) − EA(LUMO). The data shown do not include inverted SCs.51,52 The values used for ED(HOMO) and EA(LUMO) are shown in Table 1. Whilst the data are scattered they are generally consistent with the values calculated from eqn (3). The polymer:bis-PCBM (blue circle) and polymer:ICBA (yellow circle) SCs were designed to provide increased Voc as discussed above. (These acceptors are shown in Fig. 8.). Although high Voc values were obtained (close to 1.0 V), they were well below that predicted from eqn (3). It appears from the data shown in Fig. 9(a) that the maximum Voc values that can be readily achieved for polymer:fullerene SCs is in the vicinity of 1.0 V. This conclusion is further supported by the recent study of Casey et al.88 There may be an intrinsic maximum Voc value limitation in the region of 1.0 V for polymer:fullerene SCs.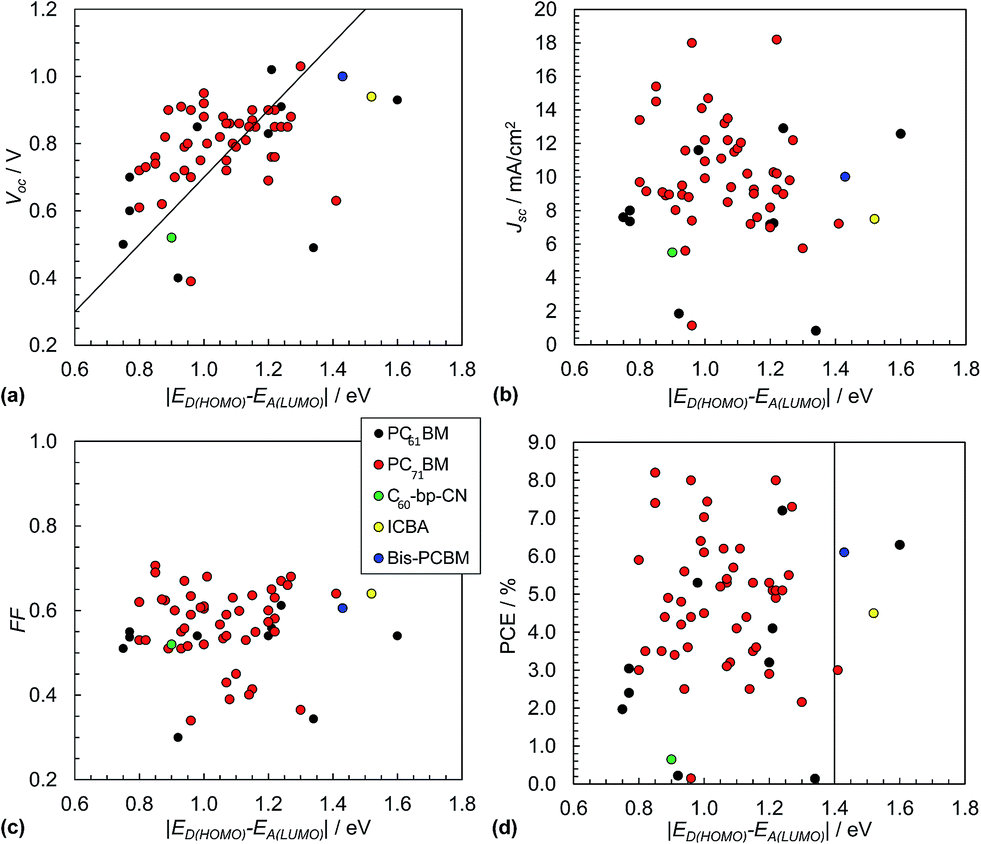 | ||
| Fig. 9 Polymer:fullerene solar cell performance characteristics. Literature data for Voc (a), Jsc (b), FF (c) and PCE (d) from 60 selected polymer:fullerene SCs plotted as a function of ED(HOMO) − EA(LUMO). The latter energy levels are depicted in Fig. 4(a). The full list of data appears in Table 1. Eqn (3) is plotted as the diagonal line in (a). The vertical line in (d) corresponds to ED(HOMO) = −5.7 eV. The legend applies to all of the graphs. The x-axis for these figures is equal to qVBI and has the same numerical value as VBI. | ||
| Entry | System | Voc/V | Jsc/mA cm−2 | FF | PCE/% | Ref. |
|---|---|---|---|---|---|---|
| a Certified PCE values indicated with an asterisk. | ||||||
| 1 | PDEBOC8/PC61BM | 0.60 | 7.35 | 0.55 | 2.4 | 90 |
| 2 | PTQTI-F/PC61BM | 0.93 | 12.58 | 0.54 | 6.3 | 91 |
| 3 | PDHF-TBT/PCBM | 0.40 | 1.85 | 0.30 | 0.2 | 92 |
| 4 | PBBTzBT-DT/PC61BM | 0.83 | 7.17 | 0.54 | 3.2 | 93 |
| 5 | PBnDT-DTffBT/PC61BM | 0.91 | 12.91 | 0.612 | 7.2 | 94 |
| 6 | P1/PC61BM | 0.49 | 0.83 | 0.344 | 0.1 | 95 |
| 7 | PTTTBO/PC61BM | 0.85 | 11.6 | 0.54 | 5.3 | 96 |
| 8 | PQTVTDPP/PCBM | 0.50 | 7.6 | 0.51 | 2.0 | 97 |
| 9 | P4/PCBM | 0.70 | 8.0 | 0.537 | 3.0 | 98 |
| 10 | PF12-TBT/PCBM | 1.02 | 7.24 | 0.559 | 4.1 | 99 |
| 11 | PTTBDT-FFT/PC71BM | 0.80 | 14.7 | 0.68 | 7.4 | 100 |
| 12 | PDPP2TBP/PC71BM | 0.80 | 11.5 | 0.63 | 5.7 | 101 |
| 13 | PBDT-TBTF/PC71BM | 0.88 | 13.21 | 0.534 | 6.2 | 102 |
| 14 | P1/PC71BM | 0.90 | 9.26 | 0.636 | 5.3 | 103 |
| 15 | PQT12oBT/PC71BM | 0.82 | 8.9 | 0.624 | 4.4 | 104 |
| 16 | PNDT-TET/PC71BM | 0.62 | 9.1 | 0.626 | 3.5 | 105 |
| 17 | PTB7/PC71BM | 0.76 | 15.4 | 0.706 | 8.2 | 52 |
| 18 | PC-TBT-TQ/PC71BM | 0.87 | 9.0 | 0.414 | 3.5 | 106 |
| 19 | P1/PC71BM | 0.69 | 7.0 | 0.6 | 2.9 | 107 |
| 20 | PBDTTPD/PC71BM | 0.92 | 10.94 | 0.604 | 6.1 | 108 |
| 21 | P(4)-4T-SiDT/PC71BM | 0.85 | 9.25 | 0.63 | 4.9 | 109 |
| 22 | P1/PC71BM | 0.86 | 9.4 | 0.39 | 3.2 | 110 |
| 23 | P-C10/PC71BM | 0.72 | 13.4 | 0.62 | 5.9 | 111 |
| 24 | IsoDPPT/PC71BM | 0.76 | 10.28 | 0.65 | 5.1 | 112 |
| 25 | PIDT-C12NT/PC71BM | 0.90 | 10.21 | 0.55 | 5.1 | 113 |
| 26 | PEBDTPD/PC71BM | 0.72 | 13.5 | 0.54 | 5.3 | 114 |
| 27 | PDTP-DFBT/PC71BM | 0.70 | 18 | 0.634 | 8.0 | 115 |
| 28 | PQCTQx/PC71BM | 0.85 | 7.6 | 0.549 | 3.6 | 116 |
| 29 | PDFCDTBT/PC71BM | 0.91 | 9.5 | 0.55 | 4.8 | 117 |
| 30 | DPP-DINI/PC71BM | 0.61 | 9.7 | 0.53 | 3.0 | 118 |
| 31 | PBTT4BT/PC71BM | 0.72 | 11.58 | 0.67 | 5.6 | 119 |
| 32 | PBDT-TFQ/PC71BM | 0.76 | 18.2 | 0.581 | 8.0* | 120 |
| 33 | BDT-TTBTT/PC71BM | 0.73 | 9.15 | 0.53 | 3.5 | 121 |
| 34 | PDPTT/PC71BM | 0.70 | 8.03 | 0.6 | 3.4 | 122 |
| 35 | PCDSeBT/PC71BM | 0.79 | 11.7 | 0.45 | 4.1 | 123 |
| 36 | PIDTT-DFBT/PC71BM | 0.95 | 12.21 | 0.61 | 7.0 | 124 |
| 37 | PQCDTB/PC71BM | 0.79 | 5.6 | 0.558 | 2.5 | 125 |
| 38 | PCZDTB/PC71BM | 0.85 | 7.2 | 0.401 | 2.5 | 125 |
| 39 | DTS-C6/PC71BM | 0.90 | 8.96 | 0.51 | 4.9 | 126 |
| 40 | P1/PC71BM | 0.86 | 8.5 | 0.43 | 3.1 | 127 |
| 41 | PDTSCBT/PC71BM | 0.82 | 11.1 | 0.567 | 5.2 | 128 |
| 42 | PBDT-FBT/PC71BM | 0.86 | 12.05 | 0.599 | 6.2 | 129 |
| 43 | PDTSTPD/PC71BM | 0.88 | 12.2 | 0.68 | 7.3 | 130 |
| 44 | PTB7/PC71BM | 0.74 | 14.5 | 0.6897 | 7.4 | 41 |
| 45 | PHCPDTMBI/PC71BM | 0.39 | 1.14 | 0.34 | 0.2 | 131 |
| 46 | PCzTh-TVDCN/PC71BM | 1.03 | 5.75 | 0.365 | 2.2 | 132 |
| 47 | PBTHDDT/PC71BM | 0.63 | 7.22 | 0.64 | 3.0 | 133 |
| 48 | PDTTTPD/PC71BM | 0.85 | 8.99 | 0.67 | 5.1 | 134 |
| 49 | P2/PC71BM | 0.81 | 10.2 | 0.53 | 4.4 | 135 |
| 50 | EH-PCzDCN/PC71BM | 0.91 | 8.94 | 0.51 | 4.2 | 136 |
| 51 | PCPDTTPD/PC71BM | 0.75 | 14.1 | 0.607 | 6.4 | 137 |
| 52 | PCPDTTTz/PC71BM | 0.75 | 12.2 | 0.59 | 5.4 | 138 |
| 53 | SilDT-BT/PC71BM | 0.88 | 9.93 | 0.52 | 4.5 | 139 |
| 54 | PBSTDTBT/PC71BM | 0.80 | 8.8 | 0.516 | 3.6 | 140 |
| 55 | PCDTBT/PC71BM | 0.90 | 8.18 | 0.573 | 5.3 | 141 |
| 56 | PFTTQx/PC71BM | 0.90 | 7.4 | 0.59 | 4.4 | 142 |
| 57 | PBDTTPD/PC71BM | 0.85 | 9.81 | 0.66 | 5.5 | 143 |
| 58 | P3HT/C60-bp-CN | 0.52 | 5.5 | 0.23 | 0.7 | 76 |
| 59 | PTTTz/ICPA | 0.94 | 7.5 | 0.64 | 4.5 | 75 |
| 60 | PBDTBDD/bis-PCBM | 1.00 | 10.02 | 0.6054 | 6.1 | 77 |
The variation of Jsc with ED(HOMO) − EA(LUMO) is shown in Fig. 9(b). The average Jsc values for these polymer:fullerene SCs containing PC71BM and PCBM were, respectively, 10.3 mA cm−2 (SD = 3.1 mA cm−2) and 7.7 mA cm−2 (SD = 4.1). The variation of the FF values with ED(HOMO) − EA(LUMO) is shown in Fig. 9(c). The average FF for the polymer:PC71BM and polymer:PCBM SCs are 0.57 (SD = 0.09) and 0.51 (SD = 0.1), respectively. The average Jsc and FF values are much less than the maximum values possible (e.g., Fig. 2(b)) which is due to considerable recombination. A recent study has shown that the value for Jsc is sensitive to polymer orientation and increases as the polymer chains become increasingly face-on with respect to the polymer/fullerene interface.89
Fig. 9(d) shows the variation of the PCE values with ED(HOMO) − EA(LUMO). The vertical line corresponds to a ED(HOMO) of −5.7 eV for SCs containing PCBM and PC71BM. The latter was reported by Scharber45 as being one factor that should provide SCs with a PCE of 10%. Accordingly, SCs with values of ED(HOMO) − EA(LUMO) to the left of the vertical line in Fig. 7(d) (and ED(HOMO) values greater than −5.7 eV) are expected to have high PCE values (depending on other factors such as Eg and ED(LUMO) values). It is noticeable that many of the PCE values obtained for SCs near the middle of this region are relatively high.
For the data shown in Fig. 9(d) the average PCE values for the polymer:fullerene SCs containing PC71BM and PCBM were 4.8% (SD = 1.7%) and 3.4% (SD = 2.4%). These values are statistically different based on a student's t-test. Whilst the data shown indicate significant improvements in the PCE for polymer:PC71BM SCs compared to polymer:PCBM SCs, this must be tensioned against the fact that the former devices generally contained more advanced polymers with lower Eg values. Nevertheless, the view that the PCE for polymer:PC71BM SCs is higher than that of polymer:PCBM SCs73 is supported from the present limited analysis.
Table 2 shows data for the polymer:fullerene SCs published in the open literature that had the highest PCE values between 2008 and the time of writing (May, 2014). Polymer:PC71BM SCs had the highest PCE values for each years between 2010 and May 2014. Whilst we have searched the literature as best we could, it is not possible to be certain that we have captured the record SCs for each of the years considered. However, the trend is certainly clear. The PCE and Jsc values have been increasing steadily over the past 6 years. We return to this point later.
| Polymer | Acceptor | PCE/% | Voc/V | Jsc/mA cm−2 | FF | Year | Ref. |
|---|---|---|---|---|---|---|---|
| a Certified PCE values indicated with an asterisk. | |||||||
| PTB7 | PC71BM | 8.2 | 0.76 | 16.4 | 0.658 | 2014 | 144 |
| PBTI3T | PC71BM | 8.7 | 0.85 | 12.8 | 0.763 | 2013 | 51 |
| PTB7 | PC71BM | 9.2* | 0.75 | 17.5 | 0.70 | 2012 | 52 |
| PBTTPD | PC71BM | 7.3 | 0.92 | 13.1 | 0.61 | 2011 | 145 |
| PTB7 | PC71BM | 7.4 | 0.74 | 14.5 | 0.69 | 2010 | 41 |
| PBDTTT-CF | PCBM | 6.8* | 0.76 | 13.4 | 0.664 | 2009 | 67 |
| PSiF-DBT | PCBM | 5.4 | 0.9 | 9.5 | 0.507 | 2008 | 146 |
There have been several announcements from companies concerning polymer SCs with efficiencies greater than 10%. Mitsubishi Chemical Corporation announced PCE values of 11.0 (ref. 147) and 11.7% (ref. 148) in 2012 and 2013. Heliatek announced a PCE of 12.0% in 2013.149 At least one or more of these SCs may have two or more junctions. It is hoped that their processes are able to maintain high PCEs upon scale-up and their technologies translate to large scale, low cost, SC deployment in the near future.
3. Hybrid polymer solar cells
Hybrid polymer SCs contain photoactive layers comprising at least two components each with a distinct chemical composition. For this discussion one component is an organic semiconducting polymer and the other is an inorganic semiconducting nanoparticle (NP). A number of reviews have been published concerning hybrid polymer SCs.1,150–153 The blending of the NPs with the polymer matrix can be achieved either by mixing,154 linking both components together155 or by in situ NP growth.156,157 One-pot preparation methods have also been used.158 The term nanocrystal (NC) is used here to describe nanometre-sized crystals; whereas, nanorods (NRs) represents rod-shaped nanometre-sized particles. NP is used to encompass NC and NR or nanometre-sized particles of arbitrary shape. Semiconducting NCs and NRs are also known as quantum dots and quantum rods, respectively.The first demonstration of a hybrid polymer SC was by Alivisatos et al.159 A design aim for hybrid polymer SCs is to use the high α values for semiconducting NPs to provide acceptors that strongly contribute to Jsc. Semiconductor NPs typically have α values of about160 105 cm−1, which gives absorption lengths of about 100 nm. The latter value is within the thickness range of hybrid polymer SC photoactive layers.1 The high α values for NPs can extend to near-IR wavelengths, which is an additional advantage for hybrid SCs.
Semiconducting NPs can be prepared using solution based methods (i.e., solvothermal methods). Their synthesis procedures allow fine control of the NP size and this, in turn, allows fine control of their α values, energy levels161,162 and Eg values. Moreover, the NP synthesis can often be simply modified to enable preparation of NRs163 and other geometries164 which means that there is design flexibility afforded by NPs that is not present for fullerenes. Whilst NCs (Fig. 10(a)) and NRs (Fig. 10(b)) are common other morphologies have been used in hybrid polymer SCs. CdSe is one of the most versatile in this regard and can be prepared as NCs, NRs, tetrapods (Fig. 10(c)) and hyperbranched forms (Fig. 10(d)). Semiconducting NPs usually have high crystallinity and mobilities, which are qualities that are advantageous for charge transport.
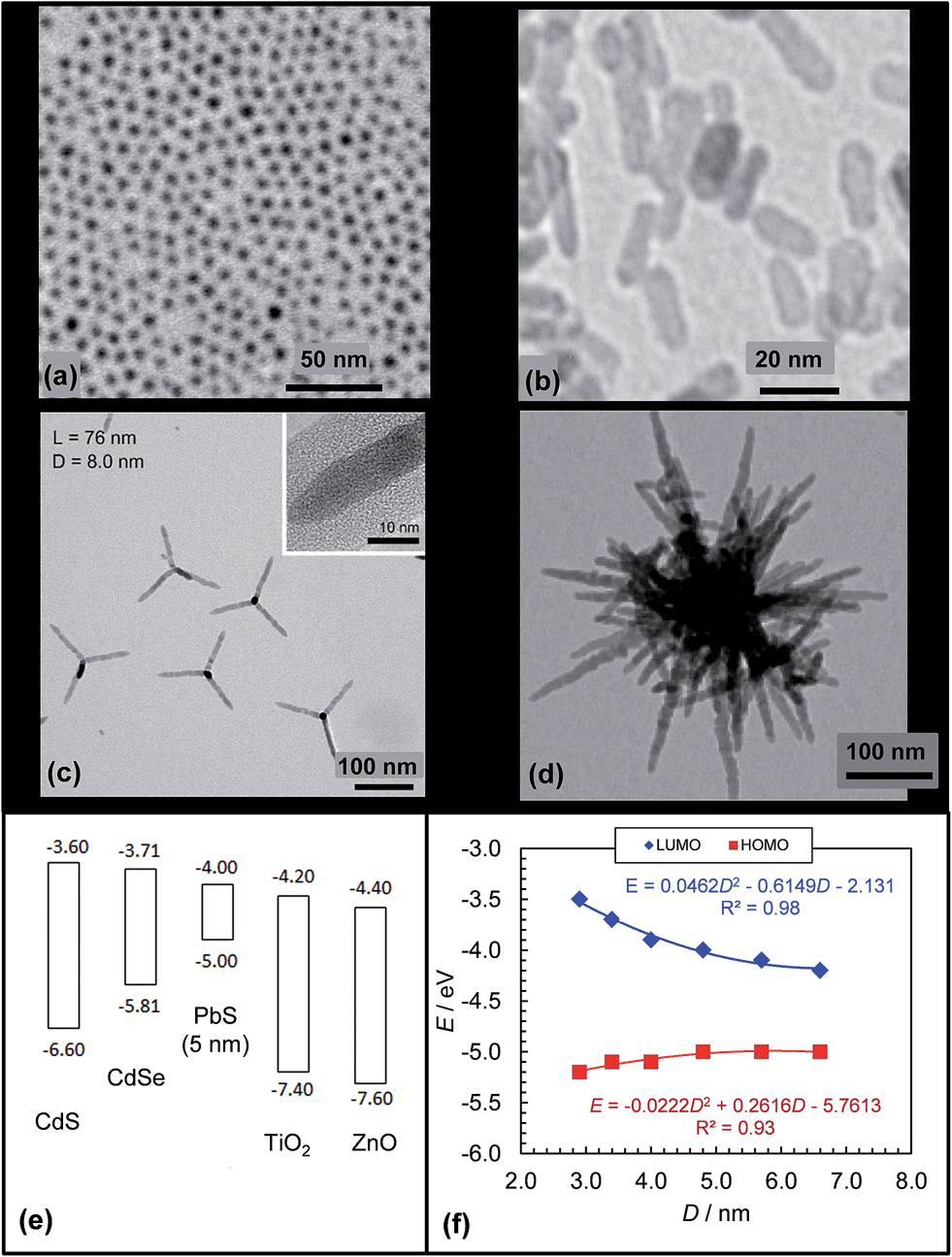 | ||
| Fig. 10 Architectures and energy levels for selected inorganic acceptors. TEM images of PbS NCs (a), ZnO NRs (b), CdSe tetrapods (c) and hyperbranched CdSe (d). (e) Energy levels for various NCs. The energy levels for CdS, CdSe, PbS, TiO2 and ZnO were obtained from ref. 62, 155, 165, 166 and 167. (f) Size dependence of PbS NC energy levels plotted from the data reported in ref. 165. Figures reproduced with permission from: (b) ref. 168, (c) ref. 163 and (d) ref. 164. | ||
Semiconducting NPs usually aggregate when dispersed in the organic solvents used to prepare hybrid polymer SCs (such as chlorobenzene or dichlorobenzene). An exception to this rule is ZnO NCs which have good dispersion stability due to surface acetate groups that are produced during synthesis.163 The dispersibility of semiconducting NPs can be improved greatly by coating the NPs with ligands that comprise binding groups and hydrophobic sequences. The ligands also increase the compatibility of the NPs with the hydrophobic semiconducting polymers during film deposition. Unfortunately, the ligands are usually insulating, which obstructs charge transport involving the NPs within photoactive films. Consequently, the ligands must be removed in order to enable efficient charge separation and transport.
The NP dispersion stability decreases upon ligand removal which has adverse effects on photoactive layer morphology if the ligand is removed prior to solvent evaporation. An early approach to removing bulky ligands used ligand exchange with pyridine154 for CdSe NPs. Pyridine was subsequently removed from the CdSe surface by heating. Although this approach was successful in increasing the PCE of the SCs the colloidal stability of the dispersions was compromised and NP aggregation was evident in the composite films.154 The morphology of films prepared from colloidally unstable dispersions is very sensitive to the mixing procedures used and can adversely affect reproducibility. Achieving reproducible, controlled, morphologies is especially important in the context of future translation of hybrid SCs to large scale deployment.
Whilst it is understood that valence and conduction bands are strictly more appropriate for bulk semiconductors, many of the inorganic semiconducting NPs discussed here are subject to quantum confinement. The latter causes the energy levels to become discrete. Consequently, HOMO and LUMO will be used throughout this discussion for NP energy levels, as is often used for semiconducting NCs such as PbS.165 When the size of semiconducting NCs is comparable to their Bohr radius, size-tuneable energy levels and also Voc and Eg values result. This size-tuneability of the energy levels is unique to semiconducting NCs and NRs and is readily achieved by varying the reaction time.169
There are a range of NPs that have been used for hybrid polymer SCs and the energy levels for selected systems are shown in Fig. 10(e). In order to provide suitable energy offsets to act as efficient acceptors the relative order of the NP HOMO and LUMOs compared to the polymer donors should be the same as those for the P3HT:PCBM SCs (see Fig. 3(b)). In addition to size-tuneable energy levels, NPs also provide the ability to tune the energy levels and Eg values by alloying. A recent hybrid polymer SC used PbSxSe1−x NC alloys.170 A key advantage of alloying is that energy level tuning can be conducted at constant NC size.
PbS NCs have recently been shown to provide relatively high PCEs (of up to 3.8%) for hybrid polymer SCs.161 This improvement became possible due to identification of the energy levels for PbS NCs. Hyun et al.165 were the first to report the variation of the HOMO and LUMO energy levels for PbS NCs (Fig. 10(f)). Those data enabled the rational design and selection of a polymer donor with a HOMO that was less deep than that of PbS161 (energy levels shown in Fig. 11(b)).
 | ||
| Fig. 11 Hybrid polymer solar cell construction and energy levels. (a) Assembly of EDT-treated PDTPBT:PbS SC. The SEM image shows a side view of the SC photoactive layer.161 The structure of PDTPBT is shown in Fig. 7. OA and EDT are oleic acid and 1,2-ethanedithiol. (b) Energy level diagram for the SC constructed using values in ref. 161. The arrows used here depict photogeneration of charges from both PDTPBT and PbS. The excitons depicted in (b) were created in the polymer phase; whereas, those depicted in (c) were created in the NC phase. (a) reproduced with permission from ref. 161. | ||
NCs subject to quantum confinement have the potential to provide more than one electron per photon within SCs.171 This process, termed multi-exciton generation (MEG) could provide SCs with efficiencies that exceed the Shockley–Queisser theoretical limit25 of 30%. The thermodynamic limit for the PCE from a MEG-based cell is 66%.171 MEG has been reviewed in detail elsewhere.171–173 When the energy levels within NCs are quantised the rate of thermal energy loss from a photoexcited electron decreases. A photoexcited electron may give up some of its energy to the lattice through collisions and excite another electron across the band gap. In order for MEG to occur the incident photon energy must be at least integer multiples of Eg. Multiple electrons have been extracted from PbS NCs within photosensitised dye sensitised solar cells.174 However, multiple electron collection required an incident photon energy that was at least 2.5Eg. MEG has recently been successfully used to increase photocurrent generation by about 4% in a solid state PbSe NC based SC.175 However, more work is needed to push the MEG-onset threshold closer to 2Eg, where a 30% increase in photocurrent is predicted.175 MEG would seem to have good potential for increasing the PCE for hybrid polymer SCs. In order to fully benefit from MEG the donor and acceptor should have complementary absorptions and the Jsc contribution due to the acceptor should be maximised. A high NP content within the hybrid polymer SC would also be needed.
3.1 Overcoming the ligand and morphology challenges for hybrid polymer solar cells
Two key challenges that adversely affect the PCE of hybrid polymer SCs are the presence of residual insulating ligand and poor morphology. Dispersed semiconducting NPs have significant inter-particle attraction due van der Waals attraction, depletion interactions and dipole–dipole interactions (for CdSe NRs).1 These attractive interactions cause NP aggregation and polymer phase separation during solvent evaporation and hybrid polymer film formation. This feature is pronounced for inorganic NPs (cf. fullerenes) because of their larger size. Consequently, there are strong driving forces that promote uncontrolled NP aggregation within hybrid polymer films prepared by blending NP dispersions and semiconducting polymer solutions.1 Furthermore, because of their large size the NPs are less able to migrate when hybrid polymer films are annealed. Consequently, the kinetically trapped NP phase is locked in place. NP aggregation results in low Jsc values due to geminate recombination which occurs because a high proportion of excitons formed in the relatively large polymer domains do note reach a NP interface before recombining. NP aggregation increases the average polymer domain size to beyond about 20 nm. A number of workers have developed innovative alternative approaches for avoiding NP aggregation as well as insulating problems from residual insulating ligand and these will be discussed below.Fig. 11(a) shows a novel hybrid polymer SC fabrication approach used for PDTPBT:PbS SCs. Seo et al.161 established a method for avoiding the colloidal stability problems associated with ligand displacement prior to hybrid film deposition. After spin coating PDTPBT:PbS films, they added 1,2-ethanedithiol (EDT) to the top of the films using another spin coating step. EDT was able to permeate through the PDTPBT:PbS layer and displaced the oleic acid (OA) ligand. Moreover, EDT acted as a (short) bridging ligand that promoted electrical contact between neighbouring PbS NCs. Their hybrid polymer SCs had photocurrent contributions from both the PDTPBT and Pb NCs. Although the NC sizes were not reported,161 when the HOMO and LUMO energy levels given in that study are compared to those reported by Hyun et al.165 (see Fig. 10(f)) an average PbS NC diameter was between about 3 and 4 nm can be estimated. Their hybrid polymer SCs had a maximum PCE of 3.8%, which was a record for hybrid polymer SCs containing PbS.
Fig. 11(b) and (c) depict the proposed exciton formation and transport pathway for the hybrid:polymer SC where both the polymer and NC produce excitons when irradiated. For illustration purposes we use the energy levels that apply to the SC reported by Seo et al.161 The general charge transport pathway for hybrid polymer SCs depicted in Fig. 11(b) and (c) has been discussed by Reiss et al.152 The diagram shows how judicious selection and control of the energy levels within hybrid polymer SCs is required to provide exciton dissociation from the polymer and NC phase and maximise contributions to Jsc. The PDTPBT:PbS SCs of Seo et al.161 contained polymer and NCs that absorbed photons in complementary regions of the spectrum, and gave two sources of photocurrent that contributed to the relatively high PCE values obtained.
Another method for reducing the effects of poor morphology control within hybrid polymer SCs is to use anisotropic NPs. Recently, scanning transmission electron microscopy using high-angle annular dark-field imaging (STEM-HAADF) was used to visualise the morphologies of P3HT:TiO2 NC and NR films176 (Fig. 12(a)–(d)). Those workers found that the NRs provided fewer network junctions and reduced inter-NP hopping which increased the charge transport efficiency. The conclusion that the use of high aspect ratio nanoparticles (i.e., NRs) increases the PCE of hybrid polymer:nanoparticle films confirmed the earlier conclusion reached by Alivisatos et al. in their study of P3HT:CdSe SCs.177 Although the values for Voc for the P3HT:TiO2 SCs were respectable (0.60 to 0.69 V) the Jsc values were low (less than or equal to 3.10 mA cm−2). The impressive high resolution topography images (Fig. 12(a)–(d)) reveal significant nanometre scale inhomogeneity of the NC and NR distributions within the composite films, which suggests that aggregation occurred. It appears that randomly oriented NRs do not seem capable of providing a solution to the aggregation problem on their own. Several groups have blended NRs with NCs in an attempt to use the NCs to fill the voids between NRs168,170 and beneficial increases in PCE occurred.
 | ||
| Fig. 12 Nanomorophologies of P3HT:TiO2 films. STEM-HAADF electron tomography images of P3HT:TiO2 NC (a and c) and NR (b and d) hybrid thin films viewed from different angles. The insets for (c) and (d) show connective networks along the film direction. (e) and (f) show TEM images of P3HT:CdS films prepared (e) without grafting and (f) with grafting using by a solvent exchange method. The images from (a)–(d) and (e) to (f) are from ref. 176 and 155, respectively. Figures reproduced with permission from: (a)–(d), ref. 176; (e) and (f), ref. 155. | ||
Ren et al.155 used added non-solvent to form P3HT nanowires and grafted CdS NCs onto the nanowire surfaces via a solvent exchange method. They suggested that addition of specific solvents changed the NC deposition from a non-grafted (Fig. 12(e)) to a chemically grafted state (Fig. 12(f)) which used the heterocyclic S on the P3HT as a ligand for the CdS NCs. Good electrical contact between neighbouring CdS NCs was achieved using a post film-deposition EDT treatment. Although important questions remain concerning the design rules for driving the heterocyclic S–CdS interaction using added non-solvent, the study is arguably the best example of achieving morphology control for hybrid polymer SCs. The PCE of 4.1%, was a record value for a hybrid polymer SC.
An innovative approach for circumventing both the ligand and morphology problems for hybrid polymer:NP films involves in situ NP formation within semiconducting polymer films.156,157 The advantages of an in situ approach are that the initial state for the NP precursors is a molecularly dispersed solution and ligand is not required. This approach has been used for P3HT:ZnO (ref. 157) and CdS:P3HT (ref. 156) films. The maximum PCE value achieved for these NC-based SCs were 2.0 (ref. 157) and 2.2%,156 respectively. The limitations of in situ NC preparation approaches are that it is difficult to achieve very high NC loadings and control over the NP particle size polydispersity. In both studies discussed above there was evidence of NP aggregation.156,157
3.2 Comparing Voc values for hybrid polymer solar cells with the Scharber equation
It is often assumed that the Voc value for hybrid polymer SCs is proportional to the magnitude of ED(HOMO) − EA(LUMO).152 This was tested using polymer:NC SC data from 26 systems (see Fig. 13(a)). The polymer:ZnO systems were exemplary because they had relatively high Voc values with all values greater than those predicted from eqn (3). ZnO appears to behave as an exceptional acceptor for hybrid polymer SCs in this regard. The Voc behaviour for the polymer:ZnO SCs contrasts to the other polymer:NC SCs which had Voc values that were mostly much lower than those expected from eqn (3). This trend contrasts to the behaviour observed for the polymer:fullerene SCs (Fig. 9(a)). There is also some evidence of linearity for the data from the hybrid polymer SCs not containing ZnO shown in Fig. 13(a) with a gradient that is lower than that predicted from eqn (3) if the P3HT nanowire/CdSe system155 is neglected (red open square).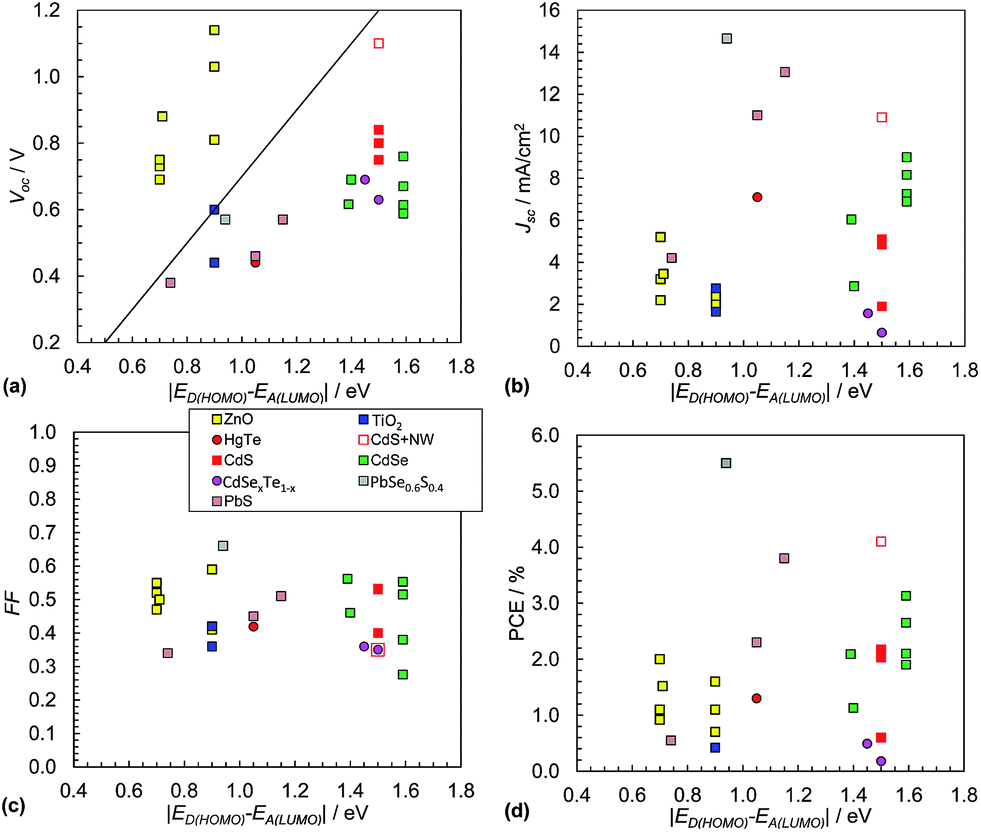 | ||
| Fig. 13 Hybrid polymer:nanocrystal solar cell performance characteristics. Literature data for Voc (a) and Jsc (b), FF (c) and PCE (d). The data are from 26 selected polymer:NC SCs. The data appear in Table 3. Eqn (3) is plotted as the diagonal line in (a). Data for hybrid polymer SCs containing NRs or tetrapods are not included. The legend for (c) applies to all figures. The x-axis for these figures is equal to qVBI and has the same numerical value as VBI. | ||
Whilst it is not possible to be certain about the origin of the differences in the trends for the Voc values between the hybrid polymer SCs containing ZnO NCs and the others shown in Fig. 13(a) there appears to be one general feature that distinguishes the data sets, which is the absence of added ligand during NC preparation. ZnO dispersions do not require added ligand for colloidal stability.163 By contrast the other (non-ZnO) NC dispersions required added ligand, which was often tri-octylphosphine oxide (TOPO) or OA, or else the NCs were generated in situ. It is known that coordinating ligands can affect Voc through modification of EA(LUMO).178 It can be seen from Fig. 13(a) that a major challenge for the majority of the non-ZnO containing hybrid polymer SCs is to increase the Voc values to closer to that predicted by the Scharber equation. A recent study has shown that NC size is important in determining Voc for polymer:CdSe SCs. It was found that Voc decreased due to traps, which became increasingly significant as the NC size decreased.179
The hybrid polymer SCs that have given the highest Jsc values to date are those containing PbS or PbSe0.6Se0.4 alloys (Fig. 13(b)). The CdS/P3HT nanowire SC155 is an exception to this trend and demonstrates how increasing order of the NCs and polymer can increase Jsc. The average Jsc value for the hybrid polymer SCs (5.4 mA cm−2, SD = 3.8 mA cm−2) is about half that for the polymer:PC71BM SCs (Fig. 9(b)). The lower Jsc for the hybrid polymer SCs is attributed to NC aggregation and/or the presence of residual ligand.
The FF values for the hybrid SCs (Fig. 13(c)) have an average value of 0.46 (SD = 0.09), with the highest being 0.67 for the SCs containing PbSe0.6S0.4. These are relatively low FF values compared to the average FF for the polymer:PC71BM SCs above. This difference is probably due to the greater extent of recombination that occurs within hybrid polymer SCs.
Whilst it is difficult to discern a clear trend for the PCE values (Fig. 13(d)) the highest values occurred when |ED(HOMO) − EA(LUMO)| is 0.9 to 1.5 eV, which is broadly in line with the results for the polymer:fullerene SCs (Fig. 9(d)). The average PCE value was 1.8% (SD = 1.3%) which is about 40% of the average value for the polymer:PC71BM films determined from the data shown in Fig. 9(d). The lower PCE is mostly due to relatively low average Jsc and FF values (above). The hybrid polymer SCs that achieved the highest PCE values are those containing PbS, CdS or CdSe. For each of these SCs the NCs can contribute photocurrent to the overall Jsc value.
Table 4 shows device performance data for hybrid polymer SCs that achieved the highest PCE value (that we were able to find) in the literature each year since 2008. The best hybrid polymer SC to date in terms of PCE is193 PDTBT:PbS0.4Se0.6 which achieved an impressive PCE of 5.5%. In order to achieve that PCE a NC layer was required on top of the PDTBT:PbS0.4Se0.6 layer which acted as a hole blocking layer. The PDTBT:PbS0.4Se0.6 SC currently holds the world record PCE for hybrid polymer:NP SCs to the best of our knowledge.
| Entry | System | Voc/V | Jsc/mA cm−2 | FF | PCE/% | Ref. |
|---|---|---|---|---|---|---|
| a Certified PCE values shown with an asterisk. | ||||||
| 1 | P3HT/CdS | 0.84 | 4.85 | 0.532 | 2.17 | 156 |
| 2 | P3HTcopol/CdS | 0.75 | 5.1 | 0.53 | 2.03 | 180 |
| 3 | P3HT/CdS | 0.8 | 1.9 | 0.4 | 0.6 | 155 |
| 4 | P3HT/CdS | 1.1 | 10.9 | 0.35 | 4.1 | 155 |
| 5 | MEH-PPV/CdSe | 0.69 | 2.86 | 0.46 | 1.13 | 62 |
| 6 | P3HT/CdSe | 0.616 | 6.04 | 0.562 | 2.09 | 181 |
| 7 | PCPDTBT/CdSe | 0.76 | 7.25 | 0.38 | 2.1 | 182 |
| 8 | PCPDTBT/CdSe | 0.67 | 9.0 | 0.515 | 3.13* | 183 |
| 9 | PCPDTBT/CdSe | 0.614 | 6.89 | 0.276 | 1.9 | 184 |
| 10 | PCPDTBT/CdSe | 0.588 | 8.16 | 0.553 | 2.65 | 181 |
| 11 | MEH-PPV/CdSe0.53Te0.47 | 0.63 | 0.65 | 0.35 | 0.18 | 62 |
| 12 | MEH-PPV/CdSe0.78Te0.22 | 0.69 | 1.57 | 0.36 | 0.49 | 62 |
| 13 | P3HT/HgTe | 0.44 | 7.1 | 0.419 | 1.3 | 185 |
| 14 | PDPPTPT/PbS | 0.46 | 11 | 0.45 | 2.3 | 186 |
| 15 | PDTPBT/PbS | 0.57 | 13.06 | 0.51 | 3.8 | 161 |
| 16 | PDTPQx/PbS | 0.38 | 4.2 | 0.34 | 0.55 | 187 |
| 17 | P3HT/TiO2 | 0.6 | 1.65 | 0.42 | 0.42 | 176 |
| 18 | P3HT/TiO2 | 0.44 | 2.76 | 0.36 | 0.42 | 166 |
| 19 | MDMO-PPV/ZnO | 1.14 | 2.3 | 0.42 | 1.1 | 188 |
| 20 | MDMO-PPV/ZnO | 1.03 | 2.0 | 0.41 | 0.7 | 189 |
| 21 | P3HT/ZnO | 0.73 | 3.2 | 0.47 | 1.1 | 189 |
| 22 | P3HT/ZnO | 0.69 | 2.19 | 0.55 | 0.92 | 190 |
| 23 | P3HT/ZnO | 0.75 | 5.2 | 0.52 | 2.0 | 157 |
| 24 | MDMO-PPV/ZnO | 0.81 | 2.4 | 0.59 | 1.6 | 191 |
| 25 | MEH-PPV/ZnO | 0.88 | 3.45 | 0.5 | 1.52 | 192 |
| 26 | PDTPBT/PbSe0.4S0.6 | 0.57 | 14.66 | 0.66 | 5.5 | 193 |
| Polymer | Acceptor | NP type | PCE/% | Voc/V | Jsc/mA cm−2 | FF | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Certified PCE values shown with an asterisk.b NC BHJ and NC bilayer present. | ||||||||
| P3HT | CdSe | Tetrapods | 2.2 | 0.63 | 7.56 | 0.471 | 2014 | 194 |
| PDTPBT | PbS0.4Se0.6 | NC & bilayerb | 5.5 | 0.57 | 14.66 | 0.66 | 2013 | 193 |
| PCPDTBT | CdSe | NR and NC | 3.6 | 0.48 | 13.86 | 0.51 | 2012 | 168 |
| P3HT | CdSe | NC | 4.1 | 1.10 | 10.9 | 0.35 | 2011 | 155 |
| PCPDTBT | CdSe | Tetrapods | 3.1* | 0.67 | 9.02 | 0.515 | 2010 | 183 |
| P3HT | CdS | NR | 2.9 | 0.65 | 9.0 | 0.48 | 2009 | 195 |
| P3HT | TiO2 | NR | 0.98 | 0.64 | 2.73 | 0.56 | 2008 | 196 |
When compared to the state-of-the-art for hybrid polymer SCs in 2008,1 remarkable improvements in the PCE values have occurred in the past 6 years. However, more progress is required if the full potential of hybrid polymer SCs is to be reached. The morphology and ligand challenge discussed in the earlier review1 still seem to plague hybrid polymer SCs although very good progress has been made in minimising their effects.155,161,193 What is still lacking are ligand-free methods to improve the compatibility of the NPs with the conjugated polymers and a method to control NP morphology to optimise Jsc. Part of the reason why hybrid polymer SCs are lagging behind polymer:fullerene SCs is because fewer researchers are working on hybrid polymer SCs. Also, there appears to be fewer (if any) major industrial research programmes for the hybrid polymer SCs as far as the authors are aware. Hopefully, the good progress reviewed here will attract more workers (and industries) into this field to accelerate the rate of PCE improvement.
4. Perovskite solar cells
Remarkable increases in the PCE of perovskite SCs has occurred since 2008. The latter are a new family of SC. The principles governing their operation are increasingly becoming clear.4,197,198 Perovskite SCs are new 3rd-generation SCs that appear to have a very good chance of contributing to large scale solar energy production based on their high PCE and compatibility with scalable processes199 and are therefore included in this review. Perovskite SCs warrant discussion because never before in the history of SC research has such rapid progress in increasing the PCE been witnessed as that which has occurred for these SCs.We briefly introduce DSSCs here because they led to the development of perovskite SCs. DSSCs are third-generation SCs and consist of several major components.200 These are a transparent conductive substrate, a high surface area n-type semiconductor (usually TiO2), a dye (sensitiser) which absorbs light and an electrolyte containing a redox mediator and a counter electrode. The dye is usually strongly bound to the surface of the n-type semiconductor. DSSCs have reached impressive PCE values of ∼11% (ref. 201) and have also been studied in solid state form.202 It was the replacement of dyes with CH3NH3PbI3 and CH3NH3PbBr3 NCs203 within DSSCs that provided the first demonstration of the ability of perovskite to act as light harvesting materials within SCs and was a key step that led to the explosion of interest of perovskite-based SCs. The reader is directed to several very good reviews to learn more about DSSCs.2,200,204,205
Perovskites have the general formula of ABX3 where A and B are monovalent and divalent ions, respectively. X is either O, C, N or a halogen.22 They are named after L. A. Perovski, a Russian mineralogist and have a cubic structure.206 The earliest perovskite example is CaTiO3. The most common perovskites currently used for SC applications are CH3NH3PbI3, CH3NH3PbBr3 and the mixed halide system, CH3NH3PbI3−xClx. Both materials are semiconductors. The structures for CH3NH3PbI3 and CH3NH3PbBr3 are shown in Fig. 14(a). Part of the success of perovskites is due to their ability to form crystals of very high quality rapidly using solution processing methods and moderate temperatures. Temperatures in the range of room temperature to 150 °C have given crystalline perovskite material.207 The energy levels for the key perovskites are shown in Fig. 14(b). Whilst the HOMO position is similar for all three systems, it can be seen that the LUMO energy is sensitive to the nature of the halogen. This feature allows energy level and Eg tuning and has been found to be particularly effective for CH3NH3Pb(I3−xBrx)3 perovskites.208 For the latter the Eg value varied from 1.55 to 2.3 eV following a quadratic relationship as x was changed from 0 to 1.0 (see Fig. 14(c)).
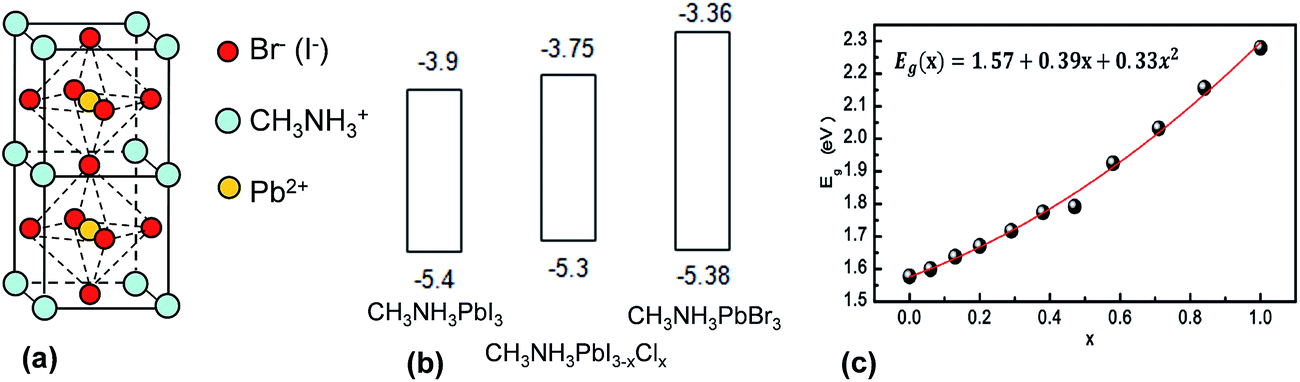 | ||
| Fig. 14 Structure and energy levels for perovskites used in solar cells. (a) Unit cell for CH3NH3PbI3 and CH3NH3PbBr3 perovskites. (b) Energy levels for CH3NH3PbI3, CH3NH3PBI3−xClx and CH3NH3PbBr3. The energy levels for the latter were taken from ref. 209, 210 and 211 respectively. (c) Variation of Eg with Br composition for CH3NH3Pb(I3−xBrx)3. (c) reproduced with permission from ref. 208. | ||
One of the earliest studies of CH3NH3PbI3 and CH3NH3PbBr3 was reported by Tanaka et al.212 They investigated optical absorption and magnetoabsorption spectra for these systems. Table 5 shows selected PCE values that exemplify the rapid progress made by perovskite SCs since 2008. In 2008 Kojima et al.203 measured a PCE of 3.8% for a NC-sensitised SC that contained CH3NH3PbI3 and CH3NH3PbBr3 NCs deposited on TiO2. Im et al.213 conducted a systematic study of perovskite NC-sensitised SCs in 2011 and reported a PCE value of 6.5%. In 2012 the first solid-state mesoscopic SC employing CH3NH3PbI3 and spiro-MeOTAD as the hole transporting was reported214 and an impressive PCE of 9.7% was reported. The rate of PCE improvement accelerated well past 10% during this period. Lee et al. reported meso-superstructured perovskite SCs in 2012 where mesoporous alumina was used as an inert scaffold for perovskite photoactive.215 That paper established that the mixed halide perovskites (CH3NH3PbI3−xClx) behaved both as a charge generator and transporter and a PCE of 10.9% was reported. Burschka et al. prepared CH3NH3PbI3 perovskite SCs using a sequential deposition method and achieved PCEs of about 15% in 2013. Wang et al.216 reported mixed halide perovskite SCs containing graphene which enhanced charge collection and gave a PCE of 15.6% (Table 5). The entire device was prepared using solution based approach with temperature less than 150 °C.216 It would seem to be ready for R2R processing if graphene can be prepared at large scale, cost-effectively, and a low cost HTM could be used. Liu et al.217 reported a PCE of 15.7% from CH3NH3PbI3 perovskite SCs prepared using ZnO NCs as the photocathode. This value has also been equalled in recent work by the Snaith group using meso-superstructured SCs and halogen bond passivation.218 The fact that three different groups achieved very high PCEs (greater than or equal to 15%) with a variety of SC architectures shows that the preparation of high quality perovskite photoactive layers is robust. This preparation robustness differs markedly from the BHJ SCs discussed above where the preparation method employed has a major influence on the morphology obtained as well as the PCE values achieved.
| Perovskite | SC typea | SC components | PCE/% | Voc/V | Jsc/mA cm−2 | FF | Yearc | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Perovskite-sensitised SC, meso-TiO2 = mesoscopic TiO2 infiltrated with perovskite, meso-super = meso superstructured photoactive layer.b Spiro is spiro-OMeTAD (see Fig. 15(d)).c Year of submission of work. | ||||||||
| CH3NH3PbI3−xClx | Meso-super | Graphene/TiO2/CH3NH3PbI3−xClx/Spirob | 15.6 | 1.04 | 21.9 | 0.73 | 2013 | 216 |
| CH3NH3PbI3 | Planar | ZnO/CH3NH3PbI3/Spiro | 15.7 | 1.03 | 20.4 | 0.75 | 2013 | 217 |
| CH3NH3PbI2Cl | Meso-TiO2 | TiO2/CH3NH3PbI2Cl/Spiro | 10.9 | 0.98 | 17.8 | 0.63 | 2012 | 215 |
| CH3NH3PbI3 | Meso-TiO2 | TiO2/(CH3NH3)PbI3/Spiro | 9.7 | 0.89 | 17.6 | 0.62 | 2012 | 214 |
| CH3NH3PbI3 | Sensitised | TiO2/CH3NH3PbI3/DSSC | 6.5 | 0.71 | 15.8 | 0.59 | 2011 | 213 |
| CH3NH3PbI3 | Sensitised | TiO2/CH3NH3PbI3/DSSC | 3.8 | 0.61 | 11.0 | 0.57 | 2008 | 203 |
Table 6 lists selected physical, spectroscopic and electronic data for the key perovskites used for SCs. The exciton binding energy is comparable to the thermal energy and this gives rise to large diffusion lengths for perovskites. Because of their relatively low exciton binding energies both free charge carriers and weakly bound excitons are believed to coexist.21
| Perovskite property | CH3NH3PbI3 | Ref. | CH3NH3PbI3−xClx | Ref. | CH3NH3PbBr3 | Ref. |
|---|---|---|---|---|---|---|
| a Diffusion length. | ||||||
| Molecular weight/(g mol−1) | 620.0 | — | 528.5 (x = 1) | 479.0 | — | |
| Wt% Pb | 33.4 | — | 39.2 | — | 43.3 | — |
| Density (g ml−1) | 4.1 | 219 | — | — | — | — |
| Mobility (cm2 V−1 s−1) | 66 | 219 | — | — | — | — |
| Dielectric constant | 6.5 | 220 | — | — | 4.8 | 212 |
| Electron LDa/nm | 129 ± 41 | 21 | 1069 ± 204 | 21 | — | — |
| Hole LDa/nm | 105 ± 32 | 21 | 1213 ± 243 | 21 | — | — |
| Exciton binding energy/meV kT | 37 (1.5) − 50 (2.0) | 220 and 221 | — | — | 2.9 | 212 |
| Absorption length (1/α)/nm | ∼100 | 21 | 100–200 | 21 | — | — |
| ELUMO/eV | −3.9 | 209 | −3.75 | 210 | −3.36 | 211 |
| EHOMO/eV | −5.4 | 209 | −5.3 | 210 | −5.38 | 211 |
| Eg/eV | 1.5 | — | 1.55 | — | 2.02 | — |
Perovskites have direct band gaps, panchromatic light absorption and high α values. The latter values were initially reported as 1.5 × 104 cm−1 at 550 nm (1/α = 665 nm) by Im et al.213 who studied perovskite NCs deposited onto TiO2. Later, Stranks et al.21 reported absorption lengths of 100–200 nm (Table 6).
The average diffusion length (LD) for CH3NH3PbI3−xClx was determined21 using 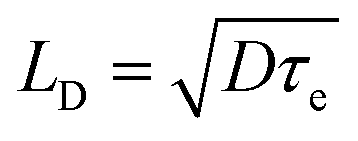 where D and τe are the diffusion coefficient and recombination lifetime in the absence of a quenching species, respectively. The values for D and τe, and hence LD, were measured using photoluminescence quenching measurements.21 The authors noted that the possibility that the LD values for the holes and electrons corresponded to diffusion of a weakly bound exciton could not be excluded.21 They reported LD values of more than 1 μm (Table 6). Edri et al. have also reported that the LD values for holes and electrons within mixed halide perovskite SCs are more than 1 μm.222 The diffusion lengths for CH3NH3PbI3−xClx are 5–10 times greater than21 1/α. This difference is in striking contrast to the BHJ systems discussed above (when Lex ≪ 1/α). As a consequence the requirement of nanostructuring the photoactive layer is relaxed for CH3NH3PbI3−xClx SCs.
where D and τe are the diffusion coefficient and recombination lifetime in the absence of a quenching species, respectively. The values for D and τe, and hence LD, were measured using photoluminescence quenching measurements.21 The authors noted that the possibility that the LD values for the holes and electrons corresponded to diffusion of a weakly bound exciton could not be excluded.21 They reported LD values of more than 1 μm (Table 6). Edri et al. have also reported that the LD values for holes and electrons within mixed halide perovskite SCs are more than 1 μm.222 The diffusion lengths for CH3NH3PbI3−xClx are 5–10 times greater than21 1/α. This difference is in striking contrast to the BHJ systems discussed above (when Lex ≪ 1/α). As a consequence the requirement of nanostructuring the photoactive layer is relaxed for CH3NH3PbI3−xClx SCs.
4.1 Preparation of perovskite solar cells
A number of different methods have been used to prepare perovskite SCs and methods are depicted in Fig. 15. They illustrate the inherent robustness of perovskite SCs because the three different SCs provided high (6.0%) to very high (15.4%) PCE values. Other designs have also been used which give very high PCEs.217 Each device depicted in Fig. 15 has the formation of a TiO2 (hole) blocking layer on FTO coated glass as the first step. The latter should be scrupulously cleaned and a variety of cleaning methods have been reported.9,21,223 The method reported by Christians et al. for hole blocking layer formation9 is the most straightforward because it does not require specialist equipment. The hole blocking layer can be formed either by spin casting213 or spray pyrolysis.223 The method reported by Im et al.213 only requires a spin coater and an oven. The hole blocking layer thickness is typically about 100 nm.9 | ||
| Fig. 15 Assembly of different perovskite solar cells. (a) A device for which a TiO2 NP network interpenetrates the perovskite phase. The example PCE and design for this type of SC are taken from ref. 9. (b) A meso-superstructured perovskite SC. This SC design and PCE are taken from ref. 224. (c) A planar perovskite SC design which used a vapour deposited mixed perovskite. This SC design and PCE are taken from ref. 225. The energy level diagram for this device is assumed to be the same as that shown in (b). (d) Structure of spiro-OMeTAD. The energy levels for TiO2, CH3NH3PbI3, CuI, Au, CH3NH3Pb3−xClx, spiro-OMETAD, Ag, and FTO are taken from ref. 166, 209, 9, 216, 210, 217, 217 and 216, respectively. | ||
After depositing the blocking layer, the SC fabrication processes shown in Fig. 15 diverge. The first design considered here in more detail is that shown in Fig. 15(a). This SC was designed with a sintered mesoporous TiO2 NP network as the medium to transfer photoexcited electrons to the photocathode (FTO). The TiO2 NP network was essential because the LD values of CH3NH3PbI3 were less than 1/α (Table 5). This layer is typically prepared by spin coating a commercial TiO2 dispersion9,223 followed by washing and sintering at 500 °C to give good electrical contact between neighbouring TiO2 NPs.
The next step is the formation of the CH3NH3PbI3 phase within the mesoporous TiO2 network. This step can be conducted using pre-mixed solutions of CH3NH3I and PbI2 (ref. 9) or sequentially by first infiltrating and drying PbI2 and subsequently CH3NH3I.223 The latter work resulted in SCs with a PCE of 15.0%. In the case of a one-step infiltration9 high concentrations of CH3NH3PbI3 solutions are used, e.g., 40 wt%.9
Ball et al.224 replaced the TiO2 with an insulating mesoporous Al2O3 network. Because the Al2O3 did not play a direct role in charge transport, the SCs were termed meso-superstructured thin film perovskite SCs.224 CH3NH3PbI3−xClx was used for that system (Fig. 15(b)). Remarkably, those SCs showed very high PCE values (e.g., 12.3%). That work demonstrated that mixed halide perovskites can fulfil the three key SC operations of light absorption, free charge carrier generation and efficient ambipolar charge transport. The mesoporous Al2O3 scaffold decreased the perovskite crystal size to less than 100 nm (ref. 224) (as determined from X-ray diffraction data and the Scherrer equation) and was believed to act as a buffering layer that inhibited leakage of current between the electrodes.224 The landmark study of Ball et al. was also highly significant for future large-scale production of perovskite SCs because the scaffold was prepared using temperatures that did not exceed 150 °C. Hence, low temperature solution processing of highly efficient perovskite SCs was demonstrated. The same group has recently demonstrated that the thermal annealing protocol used during perovskite phase crystallisation is critical for the performance of mixed halide SCs.226
Fig. 15(c) shows a planar mixed halide perovskite that was also established by the Snaith group.225 In that important study, which used vapour deposition, it was demonstrated that nanostructuring of the perovskite was not required to achieve very high PCE values (15.4%). The work demonstrated that simplified planar architectures could be used, which brought the SC architecture closer to traditional Si SCs. A simple, planar, SC device architecture offers major production benefits. However, for low cost R2R production to be realised using this geometry, a key step that will be required is the demonstration of highly efficient planar pervoskite SCs processed using low temperature (and low energy), solution, methods.
The penultimate step in SC preparation for all of the device geometries after perovskite layer formation is deposition of a hole transporting matrix (HTM). A good HTM should have high hole mobility, thermal and UV stability as well as a HOMO energy level that is well matched to that of the perovskite.227 Furthermore, it should infiltrate the mesoporous phase efficiently to optimise device efficiency.228 Spiro-OMeTAD (Fig. 15(d)) has been used as the HTM in perovskite SCs that have given the highest PCE values to date.217,223,225 However, the complexity of the spiro-OMeTAD synthesis and this materials high cost (greater than 10 times the cost of Au9) imply that it is not likely to be a viable multi-ton scale, commodity, HTM for the preparation of large scale, low cost perovskite SCs. Alternative, lower cost, HTMs are therefore urgently required. A promising alternative HTM to Spiro-OMeTAD appears to be poly(triarylamine) because of its higher hole mobility and high work function.227 CuI has also been investigated as a low cost HTM (see Fig. 15(a)). This family of SCs was shown to give a good PCE9 (6.0%) and the SC was prepared using a solution deposition method. The primary reason for the lower PCE compared to perovskite SCs prepared using spiro-OMeTAD was a reduced Voc due to an enhanced recombination rate.9 However, the good potential for low cost scale up will surely warrant further study on this type of HTM. Furthermore, the initial investigations of stability appeared promising.9 Recently, it has been reported that perovskite SCs can be prepared with an efficiency of 10.5% without a HTM.229 The latter were considered to be heterojunction SCs and offer considerable potential cost saving if the high efficiencies can be maintained upon scale up. The final step of perovskite SC construction is evaporation of the photocathode (Ag or Au). Ag can be applied using low temperature methods.
4.2 Perovskite solar cell operation principles
Perovskite SCs have been identified as a new type of SC197 with unique operation principles. Because of their low exciton binding energies (Table 5), the photoexcited charges exist as Wannier-type excitons.212 These excitons can dissociate in the bulk of a perovskite layer at room temperature.224 For CH3NH3PbI3−xClx SCs cast onto an inert scaffold (e.g., Al2O3) a built-in electric field is induced by the two selective (asymmetric) contacts which can drive charge separation throughout the photoactive layer.230 In cases where the electrodes result in a built-in voltage that is small, the hole-blocking and hole transporting layers (electron blocking) direct electron and hole flow.209 The energy offset between the perovskite valence band and hole-transporter valence band is responsible for selective charge transfer.4 It has been noted that exciton diffusion can occur both within the bulk and at interfaces.209Ponseca et al. have recently attributed the nearly ideal perovskite solar properties to highly mobile electrons and holes that form rapidly (within picoseconds) and mobilities for both species that are balanced and remain high for timescales of microseconds.198 In a recent SEM-based study Edri et al.222 reported the first direct evidence that perovskite SCs operate as a p–i–n device. An addition beneficial feature of perovskite SCs is that there is no requirement for a BHJ due to the low exciton binding energies. Consequently energy losses due energy level offsets required for BHJ SCs are not present and the ratio of Voc to Eg is very high,4 which, in turn, increases the PCE (eqn (1)). The main factor that is believed to limit perovskite SC performance is the equilibrium between the series and shunt resistance.227 Whilst a relatively thick HTM layer is required to prevent leakage through pinholes, it also increases the series resistance. Hence, HTM thickness optimisation is required.
The value of Voc for perovskite SCs is in part determined by deep level defects which act as non-radiative recombination centres. Yin et al. have shown231 that perovskite defects have low formation energies and shallow trap levels. This behaviour, which provides low recombination rates and higher Voc values, results from strong Pb lone pair s orbital and I p orbital antibonding coupling and high ionicity of CH3NH3PbI3. It follows that Pb plays a critical role in the excellent device performance of Pb-containing perovskite SCs. Accordingly, it may not be possible to achieve the high PCE values for perovskite SCs without Pb being present. The presence of Pb should not need to be a “show stopper” for widespread use of this technology provided appropriate safeguards are built into the modules to prevent environmental contamination as discussed below.
4.3 Perovskite solar cell performance and implications for scale-up
Given the very high PCEs achieved with research grade perovskite SCs (Table 5), low cost versions of perovskite SCs may already be ready for mass production. Furthermore, perovskite SCs have been demonstrated on flexible substrates (e.g., PET232) which also implies compatibility with R2R processes. Because of the enormous potential for cost effective production of perovskite SCs it is highly likely that low cost alternatives to spiro-OMeTAD will be found. The potential for large scale production of perovskite SCs appears to be excellent because (a) the photoactive materials are relatively cheap and abundant (below), (b) the PCE values are already above 15% for R2R-friendly solution processed systems216 and (c) PCE efficiency increases to beyond 20% are considered likely.227,233 However, about 33 to 43 wt% of the perovskite photoactive layer contains Pb (Table 5). An important (and perhaps critical) environmental concern is the possible release of water-soluble Pb species in the event of rainfall on modules where the encapsulation has ruptured.207 The presence of soluble Pb in drinking water can cause diseases such as anaemia.234 The EU limit for Pb in drinking water is 0.05 mg l−1.235A potentially important question is what the maximum potential release of Pb(II) is from perovskite SCs. To answer this question we assume a CH3NH3PbI3/TiO2 photoactive layer with a porosity of 0.6 (ref. 236) that is fully infiltrated with CH3NH3PbI3. Using a layer thickness of 1 μm and a perovskite density of 4.1 g cm−3 (Table 6) it can be shown that the Pb(II) content per unit area of active SC top surface corresponds to 23 Pb(II) ions per Å2. (The value per unit module area would be less than this value because the geometric fill factor would less than unity.) This value corresponds to the nominal maximum concentration of Pb(II) that could be released at the surface of this SC if all the release were to occur at once and the Pb(II) was to be placed at the top surface.
There are a wide range of materials that remove Pb(II) from water and these include functional polymers.237–239 It can be shown that a surface containing a sufficiently high surface density of polymer chains with repeat units that bind Pb(II) could be produced that could bind all of the Pb(II) present within the SC photoactive layer (equivalent to 23 Pb(II) ions per Å2). It should be possible to build in an efficient, transparent (and automatic) lead binding/absorption system within the encapsulating layer for perovskite SCs. A fail-safe design should be capable of mitigating potential contamination. Containment appears to be the key to this issue. We already live in close proximity to Pb(II), which can be contained safely in car batteries. Of course, non-Pb containing perovskites that provide high PCE values are an obvious solution to the Pb concern and are being investigated240 with Sn (ref. 241) and Cu (ref. 227) based perovskites attracting interest as possible replacements. A recent study has reported lead-free CH3NH3SnI3 SCs with a PCE of 5.73%.242 If the improvements of PCE for the latter system can follow a similar path for the Pb-based perovskite SCs, then there will be much excitement generated by these SCs in the future. To be truly scalable, Pb-free replacement perovskites should comprise abundant elements that have relatively low toxicity. Alternatively, the SC modules should be designed so as to prevent release of toxic elements in the event of rupture.
5. Comparisons of the different third-generation solar cells
Comparison of the data shown in Tables 2, 4 and 5 show that the PCE's for 2013–2014 decreased in the order: perovskite > polymer:fullerene > hybrid polymer SCs. Table 7 presents a qualitative comparison of the three different SC types discussed here. Whilst the polymer:fullerene and hybrid SCs require BHJs for maximising PCEs, the perovskite SCs do not. This difference is because of the excitons for the former two systems are Frenkel type with higher binding energies than the weakly bound Wannier excitons present within perovskites. The principle difference between the polymer:fullerene and hybrid polymer SCs is that the acceptors are inorganic. The perovskite SCs can also be considered as an hybrid organic/inorganic blend on the molecular level (Fig. 14(a)). Each of these SC types can be constructed using solution processing methods, which is one criteria that enable them to be capable of R2R processing. The latter has been demonstrated for P3HT:PCBM SCs.72 For R2R processing, the films must also have mechanical stability when strained. The strain which a thin film (e.g., 100 nm) can withstand upon bending before fracturing is strongly determined by its thickness.243 P3HT:PCBM films have a crack onset strain of 9%.243 Although similar data are not available for polymer:hybrid or perovskite SCs to our knowledge, flexible perovskite SCs on PET that withstood repeated strain cycling have been reported.244 P3HT:PCBM films have been shown to have sufficient mechanical stability for R2R processing.72| Property | Polymer:fullerene | Hybrid polymer | Perovskite |
|---|---|---|---|
| a See text for details concerning the assessments. | |||
| Bulk heterojunction | Yes | Yes | No |
| Exciton type | Frenkel | Frenkel | Wannier |
| Hybrid organic/inorganic | No | Yes | Yes |
| Solution processable | Yes | Yes | Yes |
| Roll-to-roll friendly | Yes | Yes | Yes |
| Levelised electricity cost | Low | Low | Not known |
| Environmental compatibility | Best | Good if recycled | Not known |
| Mechanical stability | Good | Not known | Good |
| Environmental stability | Good | Not known | In progress |
Cost analyses have been conducted for polymer:fullerene245–247 and hybrid polymer245 SCs. In a comprehensive study Azzopardi et al.246 calculated a levelised electricity cost (LEC) of between 0.19 and 0.50 € per kW h for a 1 kWp system with an efficiency of 7% and a 5 year module lifetime. An equivalent analysis has not been reported for polymer hybrid or perovskite SCs to our knowledge. From a related study that considered the energy payback time for hybrid polymer SCs245 it is reasonable to conclude that these SCs should have an LEC cost comparable to that of the polymer:fullerene SCs because the NCs should be solution processable.
An important question concerns the compatibility (or benignity) of each of the SC types with the environment. The latter is a particularly important issue considering the very large scale manufacture that will be required in the future for mass produced modules to significantly contribute to CO2 mitigation. The work of Azzopardi et al.245 indicated a lower CO2-eq per kW h for hybrid polymer SCs compared to polymer:fullerene SCs. Equivalent data for perovskite SCs are not yet available. The Pb-containing hybrid polymer SCs161,193 and perovskites share the potential problem of Pb contamination discussed above. However, the Pb content is lower for the hybrid polymer SCs compared to most perovskites. The organic polymer:fullerene SCs have a potential advantage in terms of environmental compatibility because they do not contain significant quantities of heavy metal ions.
6. Towards large scale deployment of third-generation solar cells
R2R processing is generally considered to offer a viable route to mass production of polymer:fullerene modules cost effectively.248 Polymer:fullerene SCs have recently been argued to be the only PV technology that enables fast manufacture of an energy producing system with a thin outline using abundant elements.72 The recent, rapid, progress for perovskite SCs implies that these new SCs may soon challenge this claim, especially since R2R-friendly processing methods have been established.199,216For large scale production to be feasible simple synthesis and processing procedures are essential. Small research grade SCs based on components which involve (costly and low yield) multiple-step synthesis are not consistent with the needs of large scale, cost-effective, processing and mass deployment even if the PCE of the SC is high.72 Cost-effective photoactive layer components are required. Scale-up using mass production processes (such as R2R) results in substantial efficiency decreases for modules compared to small area research grade SCs.72 Two sources that contribute to the PCE decrease for SC modules are ohmic losses due to relatively low conductivity of transparent electrodes and also aperture loss (or area loss).249 A key parameter for SC modules is the geometric fill factor, which is the ratio of the photoactive area to the total module area. This factor is usually significantly lower than 1.0. However, innovations in SC architecture such as formation of metal-filamentary nanoelectrodes within photoactive layers249 may offer a means to reduce aperture and ohmic losses on scale up, and increase the geometric fill factor.
The best candidate for large scale preparation of polymer:fullerene SC is currently considered to be P3HT:PCBM.72 Because the PCE values of these systems are modest, large areas are needed which, in turn, requires fast, efficient, deployment processes. An innovative Infinity concept for simultaneously installing and removing P3HT:PCBM SC foil at rates of 100 m min−1 has been demonstrated.72 Furthermore, the energy pay back times have decreased to 0.5 years in Spain. The technology appears viable for low carbon energy generation. The SCs had a constant PCE of about 1.6 to 1.8% (ref. 72) (14.7 m2 active area) on flexible ITO-free substrates. These SC foils require relatively large areas for energy generation. However, even modest improvements in PCE will greatly reduce the area requirement.
Krebs et al. have proposed that the materials developed for SCs should fit the process (R2R) if realistic large-scale deployment is to be viable.72 Their pioneering work defines a clear direction for 3rd-generation SC scientists committed to designing new SCs with realistic potential for scale-up. It follows that scalable SC systems should comprise materials which can be prepared with relatively few synthetic steps and are prepared from abundant raw materials that are cheap. The device architectures should be compatible with flexible (polymeric) substrates (and low temperature processing) to enable R2R processing with layers that can be deposited and solidified rapidly. A number of other design criteria for polymer fullerene SCs destined for R2R manufacture have been identified. The polymers (or other SC materials) should be accessible as pure materials250 and be cost-effective to manufacture.72 The photoactive material should also be sufficiently stable in solution to give ink formulations that can be deposited. These general criteria would also apply for hybrid polymer and perovskite SCs.
Because the NCs used to prepare hybrid polymer SCs can be prepared by solution methods they are also scalable. For example the PDTPBT/PbS0.4Se0.6 SC has the highest reported PCE for a hybrid polymer SC of 5.5% (Table 4). For that system the NCs were prepared using a solution based solvothermal method and a maximum temperature of 150 °C.193 The preparation method used is scalable in principle. Moreover, the fact that there are companies that focus on the scalable manufacture of semiconducting NCs251 provides greater opportunity for large scale NC preparation.
6.1 Raw material supply constraints for large-scale production of third-generation solar cells
Material supply for SCs is a key factor for future large scale deployment. Wadia et al. investigated 23 different semiconductor materials for potential large scale SC deployment.252 Of those materials, 9 were identified as having the capacity to meet or exceed the annual worldwide electricity consumption (17000 TW h) whilst being more cost effective than c-Si (see red and yellow diamonds in Fig. 16). A number of well-known semiconductors appear in the lower left quadrant which indicates that they were not considered to be viable alternatives for generating a large proportion of the World's future energy demands.
 | ||
| Fig. 16 Indexed materials extraction costs and annual potential energy production relative to crystalline Si. All values were calculated as the natural logarithm of the calculated values divided by the result for crystalline silicon (c-Si). Materials that are most attractive for large-scale future deployment appear in the upper right quadrant (in red). Materials in yellow were considered as attractive but have lower performance limits. The inset in the upper left quadrant shows energy band diagrams for selected “red diamond” systems. The main graph has been redrawn from ref. 252. The energy levels for FeS2, CZTS, Cu2O, CuO, Cu2S and Zn3P2 were obtained from ref. 253, 254, 255, 256, 257 and 258, respectively. | ||
Considering the hybrid polymer SCs discussed above in the context of Fig. 16 it is suggested that polymer:PbS SCs161 have good potential for meeting future worldwide energy production requirements in principle. A number of the other NPs have energy levels that may enable useful hybrid P3HT SCs to be prepared and these are shown in the inset of Fig. 16. These NPs include FeS2, CuO and Cu2O. Unfortunately, a study of P3HT:FeS2 SCs did not show high PCE values.259 Further work is warranted for polymer:FeS2 SCs given the potential for large scale production of these hybrid SCs. Perovskite semiconductors were not considered by Wadia et al.252 The perovskites contain Pb and I. Whilst Pb is abundant, I is less abundant. Iodine occurs naturally in the world's oceans. The Sn- and Cu-based perovskites mentioned in Section 4.3 are compositionally related to CZTS and Cu species shown in the top right quadrant of Fig. 16, which implies good potential for mass deployment. Research into hybrid polymer SCs using the nine semiconductors in the upper right quadrant is encouraged because overcoming technical barriers for their use in SCs should provide realistic opportunities for large scale SC deployment.
6.2 Solar cell stability
Long term SC PCE stability is an important issue affecting the potential for deployment. Methods for measuring polymer SC stability have been explained in detail by Gevorgyan et al.260 The different test protocols have been detailed.53 The latter includes thermal cycling between −40 and 85 °C. An aim of SC research is to prepare SCs which show little degradation in performance with time. The gold standard for stability performance is Si SCs. The latter can have operational lifetimes of about 25 years.Organic polymers undergo reaction when illuminated by light via photolytic and photochemical reactions.53 They also react to oxygen and water. The susceptibility of conjugated polymers to chemical degradation is linked to their backbone structure, side chains and substituents. The stability tends to decrease with inclusion of side chains for donor groups.53 For polymer SCs the use of inverted architectures has positively contributed to device stability54 because high work function metals (e.g., Ag) can be used in place of more reactive metals (e.g., Al).
Polymer SCs also suffer from morphological instability when heated at temperatures consistent with operation (up to 80 °C under full sun illumination). Heating enables thermally assisted reorganisation of their morphology (i.e., annealing). This process occurs because of the low glass transition temperature of the polymer phase and can lead to macroscopic phase separation.53 A realistic balance between stability and cost effectiveness has been achieved for large scale (solar park) deployment of P3HT:PCBM SCs with recycling forming a key part of the overall deployment strategy72. Impressive stabilities have been reported by Peters et al.261 for encapsulated PCDTBT:PC71BM SCs with lifetimes approaching 7 years.
Hybrid polymer SCs have been less studied in terms of stability, which is probably due to the major emphasis for this family of SCs still residing on PCE improvement. Meanwhile, perovskite SCs have been the subject of stability studies and the initial results appear promising. Leijtens et al.262 reported that their SCs retained a PCE of 6% after 1000 h exposure to white light after undergoing an initial PCE decrease. Matteocci et al.263 reported the first solar modules based on perovskites very recently and found that those prepared using spiro-OMeTAD as the HTM retained 60% of their initial PCE in air after 170 h. In each of these cases262,263 there was an initial decrease in PCE which implies some form of instability was present. However, the subsequent PCE stabilisation is encouraging and more work will undoubtedly be performed on this aspect. The sources of perovskite SC instability include UV-photodegradation caused by TiO2 (ref. 262) and also decomposition under humid conditions.208,264 The stability of polymer:fullerene SCs can be good compared to the other systems considered here (Table 7) if encapsulated. The stability for hybrid polymer SCs is less clear. For perovskite SCs, the treatments/encapsulation required to achieve long term stability are still being optimised. Clearly, a barrier to water vapour will be required for the latter.
7. Conclusions and outlook
There are a number of reasons to believe the third-generation SCs have the potential to meet the Smalley TW challenge.8 Polymer:fullerene SCs are already at the point where they can be provide the most rapid power supply deployment of any SC72 and there is good potential for PCE improvement within that process. Furthermore, high efficiency perovskite SCs are an exciting new SC technology that have now been produced using low temperature R2R-friendly processes216 and modules.263 Moreover, the tremendous achievements for the published PCE's of perovskite SCs do not appear to have benefited (yet) from industrial collaborations – which are in progress. Positive results from these collaborations are eagerly anticipated. All three 3rd-generation technologies continue to improve in terms of their PCEs (Tables 2, 4 and 5) and many researchers are mindful of future large scale deployment processes (e.g., R2R). Extrapolations of global installed SC capacity are shown in Fig. 17 based on exponential relationships that are already established for current modules. If cost-effective 3rd-generation modules can be manufactured via R2R processing and deployed using concepts such as Infinity (Section 6) then the exponential growth in the installation capacity could be maintained and the Smalley 60 TW figure could be achieved well before 2050. Of course this is a very optimistic scenario; however, it is one that 3rd-generation solar scientists and industrialists should strive to achieve. Indeed, there is good reason to be critical of the major emphasis that is currently given by most research groups and funding organisations to obtaining record breaking PCE values of laboratory-scale devices fabricated using processes and materials that have little realistic chance of being mass produced. The taxpayer, who ultimately fund most this research, may well expect a greater proportion of their research investment to be spent on truly transformative (and scalable) solar electricity generation research.265 That research will more closely relate to low cost electricity generation with significant CO2 mitigation. There is a strong argument that it is this direction that the 3rd-generation SC community and funders should move towards in order to provide realistic solutions to the urgent energy issues discussed above that challenge mankind.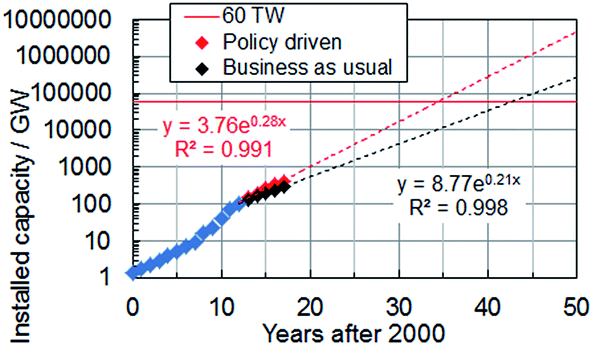 | ||
| Fig. 17 Extrapolation of global installed solar cell capacity. The graph shows fits of the data between 2012 and 2017 from Fig. 1 using years since 2000 as the time axis. The horizontal line corresponds to Smalley's 60 TW figure required for a global population of 10 billion (see text). | ||
There are several areas that warrant further work. For polymer:fullerene SCs more research is required on improving the PCE of P3HT:PCBM modules prepared by the R2R process. Methods that enable control of the morphology (e.g., Fig. 5(c)), perhaps using scaled up versions of nanoimprint lithography,87 and fit that process should provide important PCE increases and greatly reduce the area required for GW electricity production. Of course, efforts to prepare perovskite modules are important263 and their fabrication using R2R processes should be accelerated. Hybrid polymer SCs continue to hold a great deal of promise; however, they need more investment to increase the gradient of the PCE evolution. For these SCs new approaches to addressing the ligand and morphology challenges are needed. For perovskite SCs replacing the spiro-OMeTAD HTM with a cheap (sustainable) alternative is clearly required. Of course, replacing Pb with one of the abundant (less toxic) elements from Fig. 16 is desirable and may be critical to mass deployment. However, research to build in Pb(II) trapping systems into the perovskite modules would be strategic as this may help enable their larger scale deployment.
The 3rd-generation SC research reviewed here has shown very impressive progress. At least two of the technologies are well placed to contribute new SC types that could enable SCs to contribute substantially to the low CO2 energy required for the future that is urgently needed now. Indeed, it is through low cost module production (and R2R) that the future “Smalley” target of 60 TW shown in Fig. 17 might be achievable by 2050 (or earlier with policy driving) through enabling the current rate of exponential deployment to be maintained.
Acknowledgements
The authors would like to thank the EPSRC for funding our 3rd-generation solar energy research. We would also like to thank Alois Mispelon for proof-reading the manuscript. We would like to thank the referees for their helpful comments.References
- B. R. Saunders and M. L. Turner, Adv. Colloid Interface Sci., 2008, 138, 1 CrossRef CAS PubMed.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed.
- Y. Chen, X. Wan and G. Long, Acc. Chem. Res., 2013, 46, 2645–2655 CrossRef CAS PubMed.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS.
- World Energy Outlook 2013 Fact sheet, International Energy Agency, http://www.worldenergyoutlook.org.
- Key World Energy Statistics, 2013, International Energy Agency, http://www.iea.org.
- World Energy Technology Outlook – 2050, WETO-H2, European Commission, http://www.europa.eu/research/energy/.
- R. E. Smalley, Mater. Matters, 2005, 30, 412–417 Search PubMed.
- J. A. Christians, R. C. Fung and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 758–764 CrossRef CAS PubMed.
- L. Alexander, S. Allen, N. L. Bindoff, F.-M. Breon and J. Church. Working Group I Contribution to the IPCC Fifth Assessment Report Climate Change 2013: The Physical Science Basis; Summary for Policymakers 2013 Search PubMed.
- C. B. Field, V. R. Barros, M. D. Mastrandrea, K. J. Mach and M. A.-K. Abdrabo. IPCC WGII AR5: Climate Change 2014: Impacts, Adaptation, and Vulnerability; Summary for Policy Makers 2014 Search PubMed.
- Press Release from World Energy Outlook (WEO-2013), International Energy Agency, http://www.worldenergyoutlook.org/, Downloaded Feb 2014.
- IEA – Scenarios and Projections, International Energy Agency, http://www.iea.org/publications/scenariosandprojections/, Downloaded Feb 2014.
- O. Edenhofer, R. Pichs-Madruga, Y. Sokona, S. Agrawala and I. A. Bahmakov, IPCC WGIII AR5: Climate Change 2014: Mitigation; Summary for Policy Makers Search PubMed.
- Secure and efficient electricity supply during the transitions to low carbon power systems, International Energy Agency, http://www.iea.org, 2013.
- Global market outlook for photovoltaics 2013–2017, European Photovoltaic Industry Association, http://www.epia.org, Downloaded Feb 2014.
- Renewable energy technologies: solar energy perspectives, International Energy Agency, http://www.iea.org, Downloaded Feb 2014.
- Solar photovoltaics competing in the energy sector, On the road to competitiveness (September 11), European Photovoltaic Industry Association, http://www.epia.org.
- J. Kirkland, Scientific Amer., 2010 Search PubMed.
- J. Nelson, The physics of solar cells, Imperial College Press, London, 2007 Search PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- H.-S. Kim, S. H. Im and N.-G. Park, J. Phys. Chem. C, 2014, 118, 5615–5625 CAS.
- G. P. Smestad, F. C. Krebs, C. M. Lampert, C. G. Granqvist, K. L. Chopra, X. Mathew and H. Takakura, Sol. Energy Mater. Sol. Cells, 2008, 92, 371–373 CrossRef CAS PubMed.
- P. P. Khlyabich, B. Burkhart, A. E. Rudenko and B. C. Thompson, Polymer, 2013, 54, 5267–5298 CrossRef CAS PubMed.
- W. Schokley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef PubMed.
- S. D. Dimitrov and J. R. Durrant, Chem. Mater., 2014, 26, 616–630 CrossRef CAS.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed.
- J. Nelson, Mater. Today, 2011, 14, 462–470 CrossRef CAS.
- T. Wang, A. J. Pearson and D. G. Lidzey, J. Mater. Chem. C., 2013, 1, 7266–7293 RSC.
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642–6671 CrossRef CAS PubMed.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183 CrossRef CAS PubMed.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudi and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS.
- M. T. Dang, L. Hirsch and G. Wantz, Adv. Mater., 2011, 23, 3597–3602 CrossRef CAS PubMed.
- C.-Y. Chang, C.-E. Wu, S.-Y. Chen, C. Cui, Y.-J. Cheng, C.-S. Hsu, Y.-L. Wang and Y. Li, Angew. Chem., Int. Ed., 2011, 50, 9386–9390 CrossRef CAS PubMed.
- H. C. Liao, C. S. Tsao, T. H. Lin, C. M. Chuang, C. Y. Chen, U. S. Jeng, C. H. Su, Y. F. Chen and W. F. Su, J. Am. Chem. Soc., 2011, 133, 13064–13073 CrossRef CAS PubMed.
- X. Yang, A. Alexeev, M. A. J. Michels and J. Loos, Macromolecules, 2005, 38, 4289–4295 CrossRef CAS.
- Y. Liang, Z. Xu, J. Xia, S. T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619–3623 CrossRef CAS PubMed.
- D. H. Kim, A. L. Ayzner, A. L. Appleton, K. Schmidt, J. Mei, M. F. Toney and Z. Bao, Chem. Mater., 2012, 25, 431–440 CrossRef.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS PubMed.
- Y. Kim, S. A. Choulis, J. Nelson, D. D. C. Bradley, S. Cook and J. R. Durrant, Appl. Phys. Lett., 2005, 86, 063502 CrossRef PubMed.
- A. Moliton and J.-M. Nunzi, Polym. Int., 2006, 55, 583–600 CrossRef CAS PubMed.
- C. Tanase, E. J. Meijer, P. W. M. Blom and D. M. de Leeuw, Phys. Rev. Lett., 2003, 91, 216601 CrossRef CAS.
- X. Gong, M. Tong, F. G. Brunetti, J. Seo, Y. Sun, D. Moses, F. Wudl and A. J. Heeger, Adv. Mater., 2011, 23, 2272–2277 CrossRef CAS PubMed.
- S. H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS.
- X. Guo, Z. Zhou, S. J. Lou, J. Smith and D. B. Tice, Nat. Photonics, 2013, 7, 825–833 CrossRef CAS.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591–595 Search PubMed.
- M. Jorgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef PubMed.
- F. C. Krebs, S. A. Gevorgyan and J. Alstrup, J. Mater. Chem., 2009, 19, 5442–5451 RSC.
- S. Sista, M.-H. Park, Z. Hong, Y. Wu and J. Hou, Adv. Mater., 2010, 22, 380–383 CrossRef CAS PubMed.
- L. Bian, E. Zhu, J. Tang, W. Tang and F. Zhang, Prog. Polym. Sci., 2012, 37, 1292–1331 CrossRef CAS PubMed.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- S. Chen, C. E. Small, C. M. Amb, J. Subbiah, T.-h. Lai, S.-W. Tsang, J. R. Manders, J. R. Reynolds and F. So, Adv. Energy Mater., 2012, 2, 1333–1337 CrossRef CAS PubMed.
- C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S.-W. Tsang, T.-H. Lai, J. R. Reynolds and F. So, Nat. Photonics, 2012, 6, 115–120 CrossRef CAS.
- L. Pandey, C. Risko, J. E. Norton and J.-L. Brédas, Macromolecules, 2012, 45, 6405–6414 CrossRef CAS.
- Y. Zhou, Y. Li, H. Zhong, J. Hou, Y. Ding, C. Yang and Y. Li, Nanotechnology, 2006, 17, 4041–4047 CrossRef CAS PubMed.
- D. Mühlbacher, M. Scharber, M. Morana, Z. Zhu, D. Waller, R. Gaudiana and C. Brabec, Adv. Mater., 2006, 18, 2884–2889 CrossRef PubMed.
- I. H. Campbell, T. W. Hagler, D. L. Smith and J. P. Ferrais, Phys. Rev. Lett., 1996, 76, 1900–1903 CrossRef CAS.
- W. Yue, Y. Zhao, S. Shao, H. Tian, Z. Xie, Y. Geng and F. Wang, J. Mater. Chem., 2009, 19, 2199–2206 RSC.
- Y. Zou, A. Najan, P. Berrouard, S. Beaupre and B. R. Aich, J. Am. Chem. Soc., 2010, 132, 5330 CrossRef CAS PubMed.
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS PubMed.
- J. L. Brédas and A. J. Heeger, Chem. Phys. Lett., 1994, 217, 507–512 CrossRef.
- I. Meager, R. S. Ashraf, S. Mollinger, B. C. Schroeder, H. Bronstein, D. Beatrup, M. S. Vezie, T. Kirchartz, A. Salleo, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2013, 135, 11537–11540 CrossRef CAS PubMed.
- N. C. Cates, R. Gysel, Z. Beiley, C. E. Miller and M. F. Toney, Nano Lett., 2009, 9, 4153–4157 CrossRef CAS PubMed.
- F. C. Krebs, N. Espinosa, M. Hösel, R. R. Søndergaard and M. Jørgensen, Adv. Mater., 2014, 26, 29–39 CrossRef CAS PubMed.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- J. C. Hummelen, B. W. Knight, K. F. LePeq, F. Wudl and J. Yao, J. Org. Chem., 1995, 60, 532–538 CrossRef CAS.
- H. Bronstein, M. Hurhangee, E. C. Fregoso, D. Beatrup, Y. W. Soon, Z. Huang, A. Hadipour, P. S. Tuladhar, S. Rossbauer, E.-H. Sohn, S. Shoaee, S. D. Dimitrov, J. M. Frost, R. S. Ashraf, T. Kirchartz, S. E. Watkins, K. Song, T. Anthopoulos, J. Nelson, B. P. Rand, J. R. Durrant and I. McCulloch, Chem. Mater., 2013, 25, 4239–4249 CrossRef CAS.
- P. Wang, K. Yao, L. Chen, Y. Chen, F. Li, H. Wang and S. Yu, Sol. Energy Mater. Sol. Cells, 2012, 97, 34–42 CrossRef CAS PubMed.
- L. Ye, S. Zhang, D. Qian, Q. Wang and J. Hou, J. Phys. Chem. C, 2013, 117, 25360–25366 CAS.
- M. M. Wienk, J. M. Kroon, W. J. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. Janssen, Angew. Chem., 2003, 42, 3371–3375 CrossRef CAS PubMed.
- F. B. Kooistra, V. D. Mihailetchi, L. M. Popescu, D. Kronholm and P. W. Blom, Chem. Mater., 2006, 18, 3068–3073 CrossRef CAS.
- M. A. Brady, G. M. Su and M. L. Chabinyc, Soft Matter, 2011, 7, 11065–11077 RSC.
- F. Liu, Y. Gu, X. Shen, S. Ferdous, H.-W. Wang and T. P. Russell, Prog. Polym. Sci., 2013, 38, 1990–2052 CrossRef CAS PubMed.
- A. J. Moulé and K. Meerholz, Adv. Funct. Mater., 2009, 19, 3028–3036 CrossRef PubMed.
- K. Vandewal, S. Himmelberger and A. Salleo, Macromolecules, 2013, 46, 6379–6387 CrossRef CAS.
- P. J. Brown, D. S. Thomas, A. Köhler, J. S. Wilson, J.-S. Kim, C. M. Ramsdale, H. Sirringhaus and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 064203 CrossRef.
- S. Shoaee, S. Subramaniyan, H. Xin, C. Keiderling, P. S. Tuladhar, F. Jamieson, S. A. Jenekhe and J. R. Durrant, Adv. Funct. Mater., 2013, 23, 3286–3298 CrossRef CAS PubMed.
- M. Aryal, K. Trivedi and W. Hu, ACS Nano, 2009, 3, 3085–3090 CrossRef CAS PubMed.
- D. Chen, W. Zhao and T. P. Russell, ACS Nano, 2012, 6, 1479–1485 CrossRef CAS PubMed.
- A. Casey, R. S. Ashraf, Z. Fei and M. Heeney, Macromolecules, 2014, 47, 2279–2288 CrossRef CAS.
- J. R. Tumbleston, B. A. Collins, L. Yang, A. C. Stuart, E. Gann, W. Ma, W. You and H. Ade, Nat. Photonics, 2014, 8, 385–391 CrossRef CAS.
- H. Li, F. Liu, X. Wang, C. Gu, P. Wang and H. Fu, Macromolecules, 2013, 46, 9211–9219 CrossRef CAS.
- D. Dang, W. Chen, R. Yang, W. Zhu, W. Mammo and E. Wang, Chem. Commun., 2013, 49, 9335–9337 RSC.
- M. Y. Jo, S. J. Park, T. Park, Y. S. Won and J. H. Kim, Org. Electron., 2012, 13, 2185–2191 CrossRef CAS PubMed.
- M. Tsuji, A. Saeki, Y. Koizumi, N. Matsuyama, C. Vijayakumar and S. Seki, Adv. Funct. Mater., 2014, 24, 28–36 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- S. C. Cevher, N. A. Unlu, A. C. Ozelcaglayan, D. H. Apaydin, Y. A. Udum, L. Toppare and A. Cirpan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1933–1941 CrossRef CAS PubMed.
- J.-M. Jiang, P.-A. Yang, T.-H. Hsieh and K.-H. Wei, Macromolecules, 2011, 44, 9155–9163 CrossRef CAS.
- S. K. Son, B. S. Kim, C.-Y. Lee, J. S. Lee, J. H. Cho, M. J. Ko, D.-K. Lee, H. Kim, D. H. Choi and K. Kim, Sol. Energy Mater. Sol. Cells, 2012, 104, 185–192 CrossRef CAS PubMed.
- K.-C. Li, J.-H. Huang, Y.-C. Hsu, P.-J. Huang, C.-W. Chu, J.-T. Lin, K.-C. Ho, K.-H. Wei and H.-C. Lin, Macromolecules, 2009, 42, 3681–3693 CrossRef CAS.
- J.-F. Lee, S. L.-C. Hsu, P.-I. Lee, H.-Y. Chuang, J.-S. Chen and W.-Y. Chou, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4618–4625 CrossRef CAS PubMed.
- J.-H. Kim, C. E. Song, B. Kim, I.-N. Kang, W. S. Shin and D.-H. Hwang, Chem. Mater., 2014, 26, 1234–1242 CrossRef CAS.
- W. Li, A. Furlan, W. S. Roelofs, K. H. Hendriks, G. W. van Pruissen, M. M. Wienk and R. A. Janssen, Chem. Commun., 2014, 50, 679–681 RSC.
- P. Shen, H. Bin, L. Xiao and Y. Li, Macromolecules, 2013, 46, 9575–9586 CrossRef CAS.
- Q. Liao, J. Cao, Z. Xiao, J. Liao and L. Ding, Phys. Chem. Chem. Phys., 2013, 15, 19990–19993 RSC.
- K. W. Song, M. H. Choi, H. J. Song, S. W. Heo, J. Y. Lee and D. K. Moon, Sol. Energy Mater. Sol. Cells, 2014, 120, 303–309 CrossRef CAS PubMed.
- Y. J. Kim, Y.-J. Lee, J.-W. Jang, H. Cha, Y.-H. Kim, S.-K. Kwon and C. E. Park, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4742–4751 CrossRef CAS PubMed.
- D. H. Kim, H. J. Song, S. W. Heo, K. W. Song and D. K. Moon, Sol. Energy Mater. Sol. Cells, 2014, 120, 94–101 CrossRef CAS PubMed.
- B. M. Kobilka, B. J. Hale, M. D. Ewan, A. V. Dubrovskiy, T. L. Nelson, V. Duzhko and M. Jeffries-El, Polym. Chem., 2013, 4, 5329–5336 RSC.
- K. Lu, J. Fang, H. Yan, X. Zhu, Y. Yi and Z. Wei, Org. Electron., 2013, 14, 2652–2661 CrossRef CAS PubMed.
- M. Seri, M. Bolognesi, Z. Chen, S. Lu, W. Koopman, A. Facchetti and M. Muccini, Macromolecules, 2013, 46, 6419–6430 CrossRef CAS.
- K. Nie, H. Tan, X. Deng, Y. Wang, Q. Chen, Y. Huang, Y. Liu, C. Yang, Z. Huang, M. Zhu and W. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4103–4110 CrossRef CAS PubMed.
- Y. Deng, Y. Chen, J. Liu, L. Liu, H. Tian, Z. Xie, Y. Geng and F. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 5741–5747 CAS.
- S. Lu, M. Drees, Y. Yao, D. Boudinet, H. Yan, H. Pan, J. Wang, Y. Li, H. Usta and A. Facchetti, Macromolecules, 2013, 46, 3895–3906 CrossRef CAS.
- M. Wang, X. Hu, L. Liu, C. Duan, P. Liu, L. Ying, F. Huang and Y. Cao, Macromolecules, 2013, 46, 3950–3958 CrossRef CAS.
- S.-O. Kim, Y.-S. Kim, H.-J. Yun, I. Kang, Y. Yoon, N. Shin, H. J. Son, H. Kim, M. J. Ko, B. Kim, K. Kim, Y.-H. Kim and S.-K. Kwon, Macromolecules, 2013, 46, 3861–3869 CrossRef CAS.
- L. Dou, C.-C. Chen, K. Yoshimura, K. Ohya, W.-H. Chang, J. Gao, Y. Liu, E. Richard and Y. Yang, Macromolecules, 2013, 46, 3384–3390 CrossRef CAS.
- H.-J. Song, D.-H. Kim, E.-J. Lee and D.-K. Moon, J. Mater. Chem. A, 2013, 1, 6010 CAS.
- C. Du, W. Li, Y. Duan, C. Li, H. Dong, J. Zhu, W. Hu and Z. Bo, Polym. Chem., 2013, 4, 2773 RSC.
- Z. R. Owczarczyk, W. A. Braunecker, A. Garcia, R. Larsen, A. M. Nardes, N. Kopidakis, D. S. Ginley and D. C. Olson, Macromolecules, 2013, 46, 1350–1360 CrossRef CAS.
- S.-C. Lan, P.-A. Yang, M.-J. Zhu, C.-M. Yu, J.-M. Jiang and K.-H. Wei, Polym. Chem., 2013, 4, 1132–1140 RSC.
- H.-C. Chen, Y.-H. Chen, C.-C. Liu, Y.-C. Chien, S.-W. Chou and P.-T. Chou, Chem. Mater., 2012, 24, 4766–4772 CrossRef CAS.
- L. Yaowen, L. Chen, Y. Chen, C. Li, P. Zhang, L. Gao, X. Yang, Y. Tu and X. Zhu, Sol. Energy Mater. Sol. Cells, 2013, 108, 136–145 CrossRef PubMed.
- S. Subramaniyan, F. S. Kim, G. Ren, H. Li and S. A. Jenekhe, Macromolecules, 2012, 45, 9029–9037 CrossRef CAS.
- B. Kim, H. R. Yeom, M. H. Yun, J. Y. Kim and C. Yang, Macromolecules, 2012, 45, 8658–8664 CrossRef CAS.
- Y. X. Xu, C. C. Chueh, H. L. Yip, F. Z. Ding, Y. X. Li, C. Z. Li, X. Li, W. C. Chen and A. K. Jen, Adv. Mater., 2012, 24, 6356–6361 CrossRef CAS PubMed.
- H.-J. Song, D.-H. Kim, E.-J. Lee, S.-W. Heo, J.-Y. Lee and D.-K. Moon, Macromolecules, 2012, 45, 7815–7822 CrossRef CAS.
- Y. Ie, J. Huang, Y. Uetani, M. Karakawa and Y. Aso, Macromolecules, 2012, 45, 4564–4571 CrossRef CAS.
- Y.-R. Hong, J. Y. Ng, H. K. Wong, L. C. H. Moh, Y. J. Yip, Z.-K. Chen and T. B. Norsten, Sol. Energy Mater. Sol. Cells, 2012, 102, 58–65 CrossRef CAS PubMed.
- J.-S. Wu, Y.-J. Cheng, T.-Y. Lin, C.-Y. Chang, P.-I. Shih and C.-S. Hsu, Adv. Funct. Mater., 2012, 22, 1711–1722 CrossRef CAS PubMed.
- Q. Peng, X. Liu, D. Su, G. Fu, J. Xu and L. Dai, Adv. Mater., 2011, 23, 4554–4558 CrossRef CAS PubMed.
- T. Y. Chu, J. Lu, S. Beaupre, Y. Zhang, J. R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed.
- S.-H. Song, S.-J. Park, S.-C. Kwon, J.-Y. Shim, Y.-E. Jin, S.-H. Park, I. Kim, K.-H. Lee and H.-S. Suh, Bull. Korean Chem. Soc., 2012, 33, 1861–1866 CrossRef CAS.
- Z.-G. Zhang, H. Fan, J. Min, S. Zhang, J. Zhang, M. Zhang, X. Guo, X. Zhan and Y. Li, Polym. Chem., 2011, 2, 1678–1687 RSC.
- E. Ahmed, S. Subramaniyan, F. S. Kim, H. Xin and S. A. Jenekhe, Macromolecules, 2011, 44, 7207–7219 CrossRef CAS.
- G. Y. Chen, Y. H. Cheng, Y. J. Chou, M. S. Su, C. M. Chen and K. H. Wei, Chem. Commun., 2011, 47, 5064–5066 RSC.
- C.-P. Chen, S.-H. Chan, T.-C. Chao, C. Ting and B.-T. Ko, J. Am. Chem. Soc., 2008, 130, 12828–12833 CrossRef CAS PubMed.
- C. Duan, K.-S. Chen, F. Huang, H.-L. Yip, S. Liu, J. Zhang, A. K.-Y. Jen and Y. Cao, Chem. Mater., 2010, 22, 6444–6452 CrossRef CAS.
- Z. Li, S.-W. Tsang, X. Du, L. Scoles, G. Robertson, Y. Zhang, F. Toll, Y. Tao, J. Lu and J. Ding, Adv. Funct. Mater., 2011, 21, 3331–3336 CrossRef CAS PubMed.
- Z. Li, J. Ding, N. Song, X. Du, J. Zhou, J. Lu and Y. Tao, Chem. Mater., 2011, 23, 1977–1984 CrossRef CAS.
- B. C. Schroeder, Z. Huang, R. S. Ashraf, J. Smith, P. D'Angelo, S. E. Watkins, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Adv. Funct. Mater., 2012, 22, 1663–1670 CrossRef CAS PubMed.
- J.-Y. Wang, S. K. Hau, H.-L. Yip, J. A. Davies, K.-S. Chen, Y. Zhang, Y. Sun and A. K. Y. Jen, Chem. Mater., 2011, 23, 765–767 CrossRef.
- X. Xu, Y. Zhu, L. Zhang, J. Sun, J. Huang, J. Chen and Y. Cao, J. Mater. Chem., 2012, 22, 4329–4336 RSC.
- J. Zhang, W. Cai, F. Huang, E. Wang, C. Zhong, S. Liu, M. Wang, C. Duan, T. Yang and Y. Cao, Macromolecules, 2011, 44, 894–901 CrossRef CAS.
- Y. Zou, A. Najari, P. Berrouard, S. Beaupré, B. R. d. Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS PubMed.
- J. C. Yu, J. I. Jang, B. R. Lee, G. W. Lee, J. T. Han and M. H. Song, ACS Appl. Mater. Interfaces, 2014, 6, 2067–2073 CAS.
- M. S. Su, C. Y. Kuo, M. C. Yuan, U. S. Jeng, C. J. Su and K. H. Wei, Adv. Mater., 2011, 23, 3315–3319 CrossRef CAS PubMed.
- E. Wang, L. Wang, L. Lan, J. Chen, J. Peng and Y. Cao, Proc. SPIE, 2008, 7052, 70520W CrossRef PubMed.
- T. Nozawa. Techon Nikkei, http://techon.nikkeibp.co.jp/english/NEWS_EN/20120601/221131/, Downloaded Feb 2014, 2012.
- Mitsubishi Chemical, Research & Development, http://www.m-kagaku.co.jp/english/r_td/strategy/technology/topics/opv/, Dowloaded Feb 2014.
- S. Rohr. Heliatek Press Release, http://www.heliatek.com.
- M. He, F. Qiu and Z. Lin, J. Phys. Chem. Lett., 2013, 4, 1788–1796 CrossRef CAS.
- S.-S. Li and C.-W. Chen, J. Mater. Chem. A, 2013, 1, 10574–10591 CAS.
- P. Reiss, E. Couderc, J. De Girolamo and A. Pron, Nanoscale, 2011, 3, 446–489 RSC.
- J. N. Freitas, A. S. Goncalves and A. F. Nogueira, Nanoscale, 2014, 6, 6371–6397 RSC.
- W. U. Huynh, J. J. Dittmer, W. C. Libby, G. L. Whiting and P. Alivisatos, Adv. Funct. Mater., 2003, 13, 73–79 CrossRef CAS PubMed.
- S. Ren, L. Y. Chang, S. K. Lim, J. Zhao, M. Smith, N. Zhao, V. Bulovic, M. Bawendi and S. Gradecak, Nano Lett., 2011, 11, 3998–4002 CrossRef CAS PubMed.
- S. Dowland, T. Lutz, A. Ward, S. P. King, A. Sudlow, M. S. Hill, K. C. Molloy and S. A. Haque, Adv. Mater., 2011, 23, 2739–2744 CrossRef CAS PubMed.
- S. D. Oosterhout, M. M. Wienk, S. S. van Bavel, R. Thiedmann, L. J. Koster, J. Gilot, J. Loos, V. Schmidt and R. A. Janssen, Nat. Mater., 2009, 8, 818–824 CrossRef CAS PubMed.
- A. A. R. Watt, D. Blake, J. H. Warner, E. A. Thomsen, E. L. Tavenner, H. Rubinsztein-Dunlop and P. Meredith, J. Phys. D: Appl. Phys., 2005, 38, 2006–2012 CrossRef CAS.
- N. C. Greenham, Z. Peng and A. P. Alivisatos, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17628–17637 CrossRef CAS.
- Z. Hens and I. Moreels, J. Mater. Chem., 2012, 22, 10406–10415 RSC.
- J. Seo, M. J. Cho, D. Lee, A. N. Cartwright and P. N. Prasad, Adv. Mater., 2011, 23, 3984–3988 CrossRef CAS PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- R. Rhodes, M. Horie, H. Chen, Z. Wang, M. L. Turner and B. R. Saunders, J. Colloid Interface Sci., 2010, 344, 261–271 CrossRef CAS PubMed.
- I. Gur, N. A. Fromer, C.-P. Chen, A. G. Kanaras and A. P. Alivisatos, Nano Lett., 2007, 7, 409–414 CrossRef CAS PubMed.
- B.-R. Hyun, Y.-W. Zhong, A. C. Bartnik, L. Sun and H. D. Abruna, ACS Nano, 2008, 2206–2212 CrossRef CAS PubMed.
- C. Y. Kwong, A. B. Djurišić, P. C. Chui, K. W. Cheng and W. K. Chan, Chem. Phys. Lett., 2004, 384, 372–375 CrossRef CAS PubMed.
- W. J. E. Beek, M. M. Wienk and R. A. Janssen, Adv. Mater., 2004, 16, 1009–1013 CrossRef CAS PubMed.
- K. F. Jeltsch, M. Schädel, J.-B. Bonekamp, P. Niyamakom, F. Rauscher, H. W. A. Lademann, I. Dumsch, S. Allard, U. Scherf and K. Meerholz, Adv. Funct. Mater., 2012, 22, 397–404 CrossRef CAS PubMed.
- C. M. Evans, A. M. Love and E. A. Weiss, J. Am. Chem. Soc., 2012, 134, 17298–17305 CrossRef CAS PubMed.
- M. Nam, S. Kim, S. Kim, S. W. Kim and K. Lee, Nanoscale, 2013, 5, 8202–8209 RSC.
- A. J. Nozik, Inorg. Chem., 2005, 44, 6893–6899 CrossRef CAS PubMed.
- L. A. Padilha, J. T. Stewart, R. L. Sandberg, W. K. Bae and W.-K. Koh, Acc. Chem. Res., 2013, 46, 1261–1269 CrossRef CAS PubMed.
- C. Smith and D. Binks, Nanomaterials, 2013, 4, 19–45 CrossRef PubMed.
- J. B. Sambur, T. Novet and B. A. Parkinson, Science, 2010, 330, 63–66 CrossRef CAS PubMed.
- O. E. Semonin, J. M. Luther, S. Choi, H. Y. Chen, J. Gao, A. J. Nozik and M. C. Beard, Science, 2011, 334, 1530–1533 CrossRef CAS PubMed.
- S. S. Li, C. P. Chang, C. C. Lin, Y. Y. Lin, C. H. Chang, J. R. Yang, M. W. Chu and C. W. Chen, J. Am. Chem. Soc., 2011, 133, 11614–11620 CrossRef CAS PubMed.
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425–2427 CrossRef CAS PubMed.
- M. J. Greaney, S. Das, D. H. Webber, S. E. Bradforth and R. Brutchey, ACS Nano, 2012, 6, 4222–4230 CrossRef CAS PubMed.
- F. Gao, Z. Li, J. Wang, A. Rao, I. A. Howard, A. Abrusci, S. Massip, C. R. McNeill and N. C. Greenham, ACS Nano, 2014, 8, 3213–3221 CrossRef CAS PubMed.
- Y. Shi, F. Li, L. Tan and Y. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 11692–11702 CAS.
- Y. Zhou, M. Eck, C. Veit, B. Zimmermann, F. Rauscher, P. Niyamakom, S. Yilmaz, I. Dumsch, S. Allard and U. Scherf, Sol. Energy Mater. Sol. Cells, 2011, 95, 1232–1237 CrossRef CAS PubMed.
- E. Couderc, M. J. Greaney, R. L. Brutchey and S. E. Bradforth, J. Am. Chem. Soc., 2013, 135, 18418–18426 CrossRef CAS PubMed.
- S. Dayal, N. Kopidakis, D. C. Olson, D. S. Ginley and G. Rumbles, Nano Lett., 2010, 10, 239–242 CrossRef CAS PubMed.
- J. Albero, P. Riente, J. N. Clifford, M. A. Pericàs and E. Palomares, J. Phys. Chem. C, 2013, 117, 13374–13381 CAS.
- S. H. Im, H. J. Kim, S. W. Kim, S. W. Kim and S. I. Seok, Nanoscale, 2012, 4, 1581–1584 RSC.
- C. Piliego, M. Manca, R. Kroon, M. Yarema, K. Szendrei, M. R. Andersson, W. Heiss and M. A. Loi, J. Mater. Chem., 2012, 22, 24411 RSC.
- K. M. Noone, E. Strein, N. C. Anderson, P. T. Wu, S. A. Jenekhe and D. S. Ginger, Nano Lett., 2010, 10, 2635–2639 CrossRef CAS PubMed.
- W. J. E. Beek, L. H. Slooff, M. M. Wienk, J. M. Kroon and R. A. J. Janssen, Adv. Funct. Mater., 2005, 15, 1703–1707 CrossRef CAS PubMed.
- J. D. Moet, L. J. A. Koster, B. de Boer and P. W. M. Blom, Chem. Mater., 2007, 5856–5861 CrossRef.
- W. J. E. Beek, M. M. Wienk and R. A. J. Janssen, Adv. Funct. Mater., 2006, 16, 1112–1116 CrossRef CAS PubMed.
- W. J. E. Beek, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2004, 16, 1009–1013 CrossRef CAS PubMed.
- S. Shao, F. Liu, G. Fang, B. Zhang, Z. Xie and L. Wang, Org. Electron., 2011, 12, 641–647 CrossRef CAS PubMed.
- Z. Liu, Y. Sun, J. Yuan, H. Wei, X. Huang, L. Han, W. Wang, H. Wang and W. Ma, Adv. Mater., 2013, 25, 5772–5778 CrossRef CAS PubMed.
- J. Lim, D. Lee, M. Park, J. Song, S. Lee and M. S. Kang, J. Phys. Chem. C, 2014, 118, 3942–3952 CAS.
- H.-C. Liao, S.-Y. Chen and D.-M. Liu, Macromolecules, 2009, 42, 6558–6563 CrossRef CAS.
- M.-C. Wu, C.-H. Chang, H.-H. Lo, Y.-S. Lin, Y.-Y. Lin, W.-C. Yen, W.-F. Su, Y.-F. Chen and C.-W. Chen, J. Mater. Chem., 2008, 18, 4097–4102 RSC.
- H. S. Kim, I. Mora-Sero, V. Gonzalez-Pedro, F. Fabregat-Santiago, E. J. Juarez-Perez, N. G. Park and J. Bisquert, Nat. Commun., 2013, 4, 2242 Search PubMed.
- C. S. Ponseca, T. J. Savenije, M. Abdellah, K. Zheng, A. Yartsev, T. Pascher, T. Harlang, P. Chabera, T. Pullerits, A. Stepanov, J.-P. Wolf and V. Sundstrom, J. Am. Chem. Soc., 2014, 136, 5189–5192 CrossRef CAS PubMed.
- P. Docampo, J. M. Ball, M. Darwich, G. E. Eperon and H. J. Snaith, Nat. Commun., 2013, 4, 2761 Search PubMed.
- K. G. Reddy, T. G. Deepak, G. S. Anjusree, S. Thomas, S. Vadukumpully, K. R. V. Subramanian, S. V. Nair and A. S. Nair, Phys. Chem. Chem. Phys., 2014, 16, 6838–6858 RSC.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Prog. Photovolt.: Res. Appl., 2012, 20, 12–20 CrossRef PubMed.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissortel, J. Salbeck, H. Spreitzer and M. Gratzel, Nature, 1998, 395, 583–585 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- P. Docampo, S. Guldin, T. Leijtens, N. K. Noel, U. Steiner and H. J. Snaith, Adv. Mater., 2014, 26, 4013–4030 CrossRef CAS PubMed.
- A. Hinsch, W. Veurman, H. Brandt, K. Flarup Jensen and S. Mastroianni, ChemPhysChem, 2014, 15, 1076–1087 CrossRef CAS PubMed.
- M. A. Loi and J. C. Hummelen, Nat. Mater., 2013, 12, 1087–1089 CAS.
- G. Hodes and D. Cahen, Nat. Photonics, 2014, 8, 87–88 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CAS.
- O. Malinkiewicz, A. Yella, Y. H. Lee, G. M. n. Espallargas, M. Graetzel, M. K. Nazeeruddin and H. J. Bolink, Nat. Photonics, 2014, 8, 128–132 CrossRef CAS.
- A. Abrusci, S. D. Stranks, P. Docampo, H. L. Yip, A. K. Jen and H. J. Snaith, Nano Lett., 2013, 13, 3124–3128 CrossRef CAS PubMed.
- B. Cai, Y. Xing, Z. Yang, W.-H. Zhang and J. Qiu, Energy Environ. Sci., 2013, 6, 1480–1485 CAS.
- K. Tanaka, T. Takahashi, T. Ban, T. Kondo, K. Uchida and N. Miura, Solid State Commun., 2003, 127, 619–623 CrossRef CAS.
- J. H. Im, C. R. Lee, J. W. Lee, S. W. Park and N. G. Park, Nanoscale, 2011, 3, 4088–4093 RSC.
- H. S. Kim, C. R. Lee, J. H. Im, K. B. Lee, T. Moehl, A. Marchioro, S. J. Moon, R. Humphry-Baker, J. H. Yum, J. E. Moser, M. Gratzel and N. G. Park, Sci. Rep., 2012, 2, 591 Search PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- J. T. Wang, J. M. Ball, E. M. Barea, A. Abate, J. A. Alexander-Webber, J. Huang, M. Saliba, I. Mora-Sero, J. Bisquert, H. J. Snaith and R. J. Nicholas, Nano Lett., 2014, 14, 724–730 CrossRef CAS PubMed.
- D. Liu and T. L. Kelly, Nat. Photonics, 2013, 8, 133–138 CrossRef.
- A. Abate, M. Saliba, D. J. Hollman, S. D. Stranks, K. Wojciechowski, R. Avolio, G. Grancini, A. Petrozza and H. J. Snaith, Nano Lett., 2014, 14, 3247–3254 CrossRef CAS PubMed.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- M. Hirasawa, T. Ishihara, T. Goto, K. Uchida and N. Miura, Phys. B, 1994, 201, 427–430 CrossRef CAS.
- T. Ishihara, J. Lumin., 1994, 60–61, 269–274 CrossRef CAS.
- E. Edri, S. Kirmayer, S. Mukopadhyay, K. Gartsman, G. Hodes and D. Cahen, Nat. Commun., 2014, 5, 3461 Search PubMed.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- J. M. Ball, M. M. Lee, A. Hey and H. J. Snaith, Energy Environ. Sci., 2013, 6, 1739–1743 CAS.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- M. Saliba, K. W. Tan, H. Sai, D. T. Moore, T. Scott, W. Zhang, L. A. Estroff, U. Wiesner and H. J. Snaith, J. Phys. Chem. C, 2014, 118, 17171–17177 CAS.
- S. Kazim, M. K. Nazeeruddin, M. Gratzel and S. Ahmad, Angew. Chem., 2014, 53, 2812–2824 CrossRef CAS PubMed.
- D. Nanova, A. K. Kast, M. Pfannmoller, C. Muller, L. Veith, I. Wacker, M. Agari, W. Hermes, P. Erk, W. Kowalsky, R. R. Schroder and R. Lovrincic, Nano Lett., 2014, 14, 2735–2740 CrossRef CAS PubMed.
- J. Shi, J. Dong, S. Lv, Y. Xu, L. Zhu, J. Xiao, X. Xu, H. Wu, D. Li, Y. Luo and Q. Meng, Appl. Phys. Lett., 2014, 104, 063901 CrossRef PubMed.
- E. Edri, S. Kirmayer, A. Henning, S. Mukhopadhyay, K. Gartsman, Y. Rosenwaks, G. Hodes and D. Cahen, Nano Lett., 2014, 14, 1000–1004 CrossRef CAS PubMed.
- W.-J. Yin, T. Shi and Y. Yan, Appl. Phys. Lett., 2014, 104, 063903 CrossRef PubMed.
- J. You, Y. M. Yang, Q. Chen, M. Cai, T.-B. Song, C.-C. Chen, S. Lu, Y. Liu, H. Zhou and Y. Yang, ACS Nano, 2014, 8, 1674–1680 CrossRef CAS PubMed.
- N.-G. Park, J. Phys. Chem. Lett., 2013, 4, 2423–2429 CrossRef CAS.
- B. Al-Rashdi, C. Somerfield and N. Hilal, Sep. Purif. Rev., 2011, 40, 209–259 CrossRef CAS.
- V. M. Nurchi and I. Villaescusa, Coord. Chem. Rev., 2008, 252, 1178–1188 CrossRef CAS PubMed.
- P. Docampo, A. Hey, S. Guldin, R. Gunning, U. Steiner and H. J. Snaith, Adv. Funct. Mater., 2012, 22, 5010–5019 CrossRef CAS PubMed.
- I. G. Dakova, I. B. Karadjova, V. T. Georgieva and G. S. Georgiev, Microchim. Acta, 2008, 164, 55–61 CrossRef.
- B. Liu, X. Lv, X. Meng, G. Yu and D. Wang, Chem. Eng. J., 2013, 220, 412–419 CrossRef CAS PubMed.
- M. Zhang, Z. Zhang, Y. Liu, X. Yang, L. Luo, J. Chen and S. Yao, Chem. Eng. J., 2011, 178, 443–450 CrossRef CAS PubMed.
- M. D. McGehee, Nature, 2013, 501, 323–325 CrossRef CAS PubMed.
- G. Hodes, Science, 2013, 342, 317–318 CrossRef CAS PubMed.
- F. Hao, C. C. Stoumpos, D. H. Cao, R. P. H. Chang and M. G. Kanatzidis, Nat. Photon., 2014, 8, 489–494 CrossRef CAS.
- S. Savagatrup, A. S. Makaram, D. J. Burke and D. J. Lipomi, Adv. Funct. Mater., 2014, 24, 1169–1181 CrossRef CAS PubMed.
- C. Roldan-Carmona, O. Malinkiewicz, A. Soriano, G. Minguez Espallargas, A. Garcia, P. Reinecke, T. Kroyer, M. I. Dar, M. K. Nazeeruddin and H. J. Bolink, Energy Environ. Sci., 2014, 7, 994–997 CAS.
- B. Azzopardi and J. Mutale, Renewable Sustainable Energy Rev., 2010, 14, 1130–1134 CrossRef CAS PubMed.
- B. Azzopardi, C. J. M. Emmott, A. Urbina, F. C. Krebs, J. Mutale and J. Nelson, Energy Environ. Sci., 2011, 4, 3741–3753 Search PubMed.
- A. J. Medford, M. R. Lilliedal, M. Jorgenson, D. Aaro, H. Pakalski and F. C. Krebs, Opt. Express, 2010, 18, A272–A285 CrossRef CAS PubMed.
- P. Sommer-Larsen, M. Jorgensen, R. R. Sondergaard, M. Hosel and F. C. Krebs, Energy Technol., 2013, 1, 15–19 CrossRef PubMed.
- H. Kang, S. Hong, H. Back and K. Lee, Adv. Mater., 2014, 26, 1602–1606 CrossRef CAS PubMed.
- R. Po, A. Bernardi, A. Calabrese, C. Carbonera, G. Corso and A. Pellegrino, Energy Environ. Sci., 2014, 7, 925–943 CAS.
- http://www.nanocotechnologies.com, date accessed July 2014.
- C. Wadia, A. P. Alivisatos and D. M. Kammen, Environ. Sci. Technol., 2009, 43, 2072–2077 CrossRef CAS.
- Y.-Y. Lin, D.-Y. Wang, H.-C. Yen, H.-L. Chen, C.-C. Chen, C.-M. Chen, C.-Y. Tang and C.-W. Chen, Nanotechnology, 2009, 20, 405207 CrossRef PubMed.
- H. Nishi, T. Nagano, S. Kuwabata and T. Torimoto, Phys. Chem. Chem. Phys., 2014, 16, 672 RSC.
- K. K. Kasem, Surf. Interface Anal., 2011, 43, 1527–1531 CrossRef CAS PubMed.
- Y.-F. Lim, J. J. Choi and T. Hanrath, J. Nanomater., 2012, 393160 Search PubMed.
- Y. Bessekhouad, R. Brahimi, F. Hamdini and M. Trari, J. Photochem. Photobiol., A, 2012, 248, 15–23 CrossRef CAS PubMed.
- E. J. Luber, H. Mobarok and J. M. Buriak, ACS Nano, 2013, 7, 8136–8146 CrossRef CAS PubMed.
- C. Steinhagen, T. B. Harvey, C. J. Stolle, J. Harris and B. A. Korgel, J. Phys. Chem. Lett., 2012, 3, 2352–2356 CrossRef CAS.
- S. A. Gevorgyan, M. Jørgensen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 736–745 CrossRef CAS PubMed.
- C. H. Peters, I. T. Sachs-Quintana, J. P. Kastrop, S. Beaupré, M. Leclerc and M. D. McGehee, Adv. Energy Mater., 2011, 1, 491–494 CrossRef CAS PubMed.
- T. Leijtens, G. E. Eperon, S. Pathak, A. Abate, M. M. Lee and H. J. Snaith, Nat. Commun., 2013, 4, 2885 Search PubMed.
- F. Matteocci, S. Razza, F. Di Giacomo, S. Casaluci, G. Mincuzzi, T. M. Brown, A. D'Epifanio, S. Licoccia and A. Di Carlo, Phys. Chem. Chem. Phys., 2014, 16, 3918–3923 RSC.
- Y. S. Kwon, J. Lim, H.-J. Yun, Y.-H. Kim and T. Park, Energy Environ. Sci., 2014, 7, 1454–1460 CAS.
- S. Lizin, S. Van Passel, E. De Schepper, W. Maes, L. Lutsen, J. Manca and D. Vanderzande, Energy Environ. Sci., 2013, 6, 3136–3149 CAS.
| This journal is © The Royal Society of Chemistry 2014 |