 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorine magic: one new organofluorine linker leads to three new metal–organic frameworks†
K.
Peikert
,
F.
Hoffmann
and
M.
Fröba
*
Department of Chemistry, Institute of Inorganic & Applied Chemistry, University of Hamburg, Martin-Luther-King Platz 6, D-20146 Hamburg, Germany. E-mail: froeba@chemie.uni-hamburg.de; Tel: +49 40 42838 3100
First published on 21st May 2014
Abstract
We present 2-fluoro-1,3,5-benzene-tricarboxylic acid (Fbtc) as new linker for metal–organic frameworks. Interestingly, even slight variations of the synthesis conditions such as temperature or the molar ratio of metal to linker lead to three new MOFs, which we call UHM-31, UHM-32 and UHM-33. UHM-31 is isostructural to the well-known HKUST-1. The characterisation of the MOFs by single-crystal X-ray analysis and studies on their thermally induced phase change behaviour are given. Furthermore, we compare UHM-31 in terms of N2, H2, CH4 and CO2 adsorption properties with HKUST-1 and UHM-30 (the amino substituted HKUST-1).
Introduction
Over the past decade metal–organic frameworks (MOFs) have attracted much attention as they have shown excellent performance in applications like gas storage and separation,1 catalysis,2 sensing3 and drug delivery.4Compared to other microporous materials such as zeolites or activated carbon, MOFs show a great synthetic variability. Their modular construction principle is based on the combination of a great variety of metal sources and organic linkers and allows in principle tailoring of pore sizes, pore geometries and incorporation of specific functional groups. This was at the latest impressively demonstrated with the isoreticular MOF (IRMOF) series by Yaghi and coworkers in 2002.5 The pore diameter of the IRMOFs varies from 12.8 Å to 28.8 Å and functional groups such as –Br, –NH2, –OC3H7 and –C2H4 were incorporated into the prototypical cubic MOF-5-like structure. In 2011 Meek et al.6 reported a series of monohalogenated derivatives (–F, –Cl and –I) of IRMOF-1. In comparison to other functional groups, e.g. –NH2, halogens and in particular fluorine as functional groups in MOFs are rather rare in literature.
Organic fluorine compounds are known to show unusual properties and behaviour in comparison with their non-fluorinated counterparts.7 This is mainly due to the greater strength of the carbon–fluorine bond over the carbon–hydrogen bond and the large number of the non-bonding p-electrons, which function as a shield of the carbon backbone. As a result organic fluorine compounds show enhanced thermal stability and are more stable towards oxidation compared to their hydrocarbon analogues.8 Apart from that, fluorine is also known for playing a role as mineralizing agent and favours the formation of well crystalline phases.9 The group of Férey for example used these properties of fluorine in the hydrothermal synthesis of the well-known MOFs MIL-100 (ref. 10) and MIL-101.11
To the best of our knowledge, the first fluorinated MOF was published in 2004 by Pan et al.12 4,4′-(Hexafluoroisopropylidene)-bis(benzoic acid) (H2hfipbb) was reacted with Cu(NO3)2·3H2O to give a 3D interpenetrating framework with microporous 1D open channels. However, the authors did not report on special effects of the fluorination on the framework properties. One of the first groups who studied explicitly the influence of fluorine functionalization on MOF properties was the group of Omary.13 FMOF-1 was derived from 3,5-bis-(trifluoromethyl)-1,2,4-triazolate (Tz) and a silver(I) source. The authors declared: “compared to their non-fluorous counterparts, FMOFs with fluoro-lined or fluoro-coated channels or cavities are expected to possess enhanced thermal stability and catalytic activity, higher gas affinity and selectivity, and higher stability to oxidation and light”.13 FMOF-1 showed superior volumetric gas uptake and a unique hysteretic sorption of H2. The group of Cheetham14 as well as other groups15 also reported the influence of fluoro groups on H2 and CO2 storage properties. Pachfule and coworkers were the first who published a set of isostructural fluorinated and nonfluorinated MOFs and compared their H2 and CO2 storage properties.16 Isonicotinic acid (INA) and its fluorinated derivative 3-fluoroisonicotinic acid (FINA) were reacted with Co(NO3)·6H2O respectively to give two pairs (depending on the solvent) of isostructural MOFs. Comparison of the H2 and CO2 adsorption in these MOFs showed that the high H2 uptake in F-MOFs is not a universal phenomenon. The authors claim, that “factors, such as pore size, curvature, and open metal sites, can also play crucial roles in the high gas uptakes in these F-MOFs”.16
In this contribution, we present 2-fluoro-1,3,5-benzene-tricarboxylic acid (Fbtc) as new linker for MOFs. The reaction of Fbtc with copper nitrate in DMA leads to three different MOF structures, which we call UHM-31, UHM-32 and UHM-33 (see Fig. 1, UHM = University of Hamburg Materials).
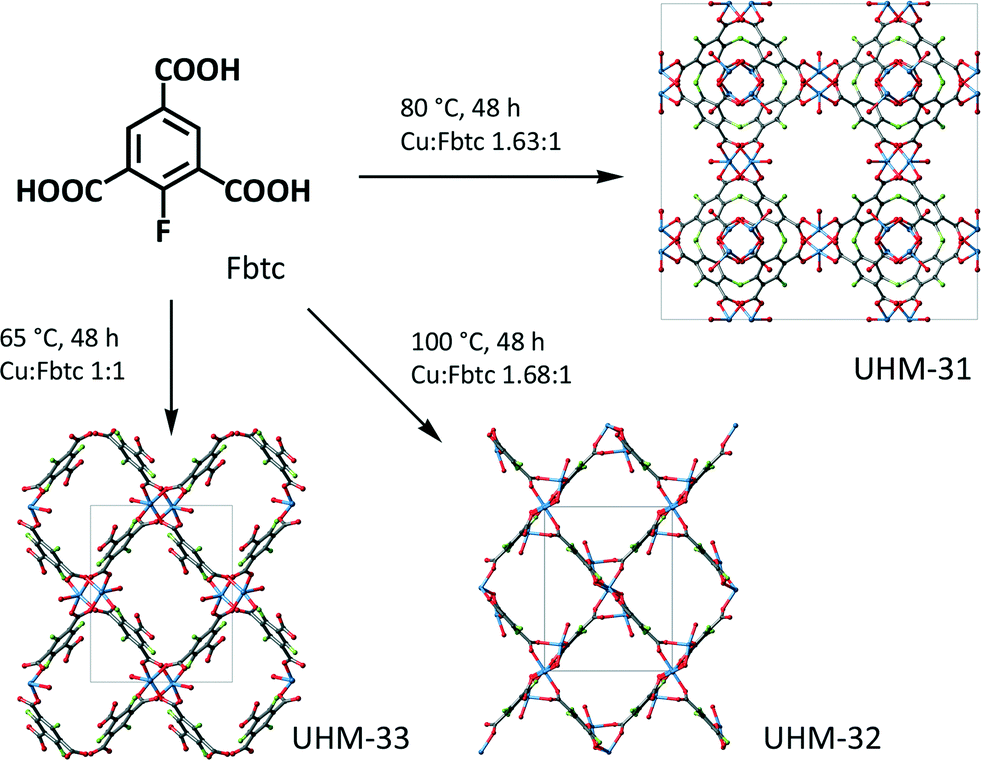 | ||
| Fig. 1 Visualisation and reaction conditions of the three new MOFs that could be synthesised from Fbtc; grey: carbon, red: oxygen, blue: copper, green: fluorine, hydrogen atoms and solvent molecules are omitted for clarity. | ||
It is a common phenomenon that one linker in combination with a certain metal can lead to more than one single MOF structure.16,17 In most cases, this is due to different solvents used in the synthesis. Here, small variations of the synthesis temperature and the molar ratio of metal to linker lead to the different structures, which is a quite rare example of this kind of ‘polymorphism’ in the area of MOFs.
As UHM-31 is the fluorinated isostructural variant of HKUST-1,18 this creates the possibility to compare fluorinated and non-fluorinated MOF counterparts concerning their gas storage properties. In this work we compare the adsorption of N2, H2, CO2 and CH4 in UHM-31 to those in HKUST-1 and UHM-30,19 the amino substituted HKUST-1, which was published by our group in 2012. Furthermore, we discuss the three new MOFs in terms of structure, topology and stability.
Results and discussion
MOF synthesis
The solvothermal reaction of Cu(NO3)2·3H2O with Fbtc (1.63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in DMA at 80 °C for 48 h leads to UHM-31, which is isostructural to HKUST-1 (ref. 18) and UHM-30.19 The reaction of Cu(NO3)2·3H2O with Fbtc (1.68:1) in DMA at 100 °C for 48 h leads to UHM-32, a three dimensional structure with an interesting new secondary building unit (SBU), see below. And the reaction of Cu(NO3)2·3H2O with Fbtc (1:1) in DMA at 65 °C for 48 h leads to UHM-33, a two-dimensional structure with paddle wheels as building unit. The synthesis of the three MOFs is strongly dependent on the conditions, in particular on the applied temperature. Reactions carried out at temperatures which are only very slightly different to the ones that lead to the pure phases easily lead to mixtures of either UHM-31 and UHM-32 (T = 70–75 °C) or UHM-31 and UHM-33 (T = 85–95 °C).
:1) in DMA at 80 °C for 48 h leads to UHM-31, which is isostructural to HKUST-1 (ref. 18) and UHM-30.19 The reaction of Cu(NO3)2·3H2O with Fbtc (1.68:1) in DMA at 100 °C for 48 h leads to UHM-32, a three dimensional structure with an interesting new secondary building unit (SBU), see below. And the reaction of Cu(NO3)2·3H2O with Fbtc (1:1) in DMA at 65 °C for 48 h leads to UHM-33, a two-dimensional structure with paddle wheels as building unit. The synthesis of the three MOFs is strongly dependent on the conditions, in particular on the applied temperature. Reactions carried out at temperatures which are only very slightly different to the ones that lead to the pure phases easily lead to mixtures of either UHM-31 and UHM-32 (T = 70–75 °C) or UHM-31 and UHM-33 (T = 85–95 °C).
Interestingly, if we applied the same conditions of the synthesis of the three different F-MOFs UHM-31, -32 or -33 to reaction mixtures of either 1,3,5-benzenetricarboxylate (btc) and Cu(NO3)2·3H2O or 2-amino-1,3,5-benzenetricarboxylate (NH2btc) and Cu(NO3)2·3H2O in DMA, we always observed phases with HKUST-1 structure. Although btc, NH2btc, and Fbtc are topologically identical only Fbtc leads to the newly observed MOF structures. Obviously, fluorine as a substituent is able to stabilize crystalline phases which are not favoured under usual conditions. However, the specific reason for that is still unclear.
Structural description
219.70 Å3) with the space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. SQUEEZE/PLATON20 was used in order to account for the electron density from disordered DMA solvent molecules, which could not be resolved (2334 electrons per unit cell, which is consistent of 4 DMA solvent molecules per formula unit). According to the three possible orientations of the linker during the incorporation into the framework the fluoro substituent shows a positional disorder over three sites, with a site occupation factor (s.o.f.) of 0.16667 (for details, see ESI†).
m. SQUEEZE/PLATON20 was used in order to account for the electron density from disordered DMA solvent molecules, which could not be resolved (2334 electrons per unit cell, which is consistent of 4 DMA solvent molecules per formula unit). According to the three possible orientations of the linker during the incorporation into the framework the fluoro substituent shows a positional disorder over three sites, with a site occupation factor (s.o.f.) of 0.16667 (for details, see ESI†).
The general description of the HKUST-1 structure can be found in the literature.18,21
In UHM-32 the coordination sphere of the copper ions leads to an untypical SBU. As Fig. 2 shows, four copper ions are linked by six carboxylic groups and two μ3-hydroxido ligands. All copper ions are surrounded in a distorted square pyramidal fashion by five oxygen atoms. Two pyramids are edge-connected by the μ3-hydroxido ligands, while the other two square pyramids are only corner connected to the tetranuclear cluster. They complete their coordination sphere by two oxygen atoms of the solvent molecules DMA (see Fig. 2a). Taking the carbon atoms of the carboxyl groups as points of extension the SBU can be described as a distorted octahedron (see Fig. 2c).
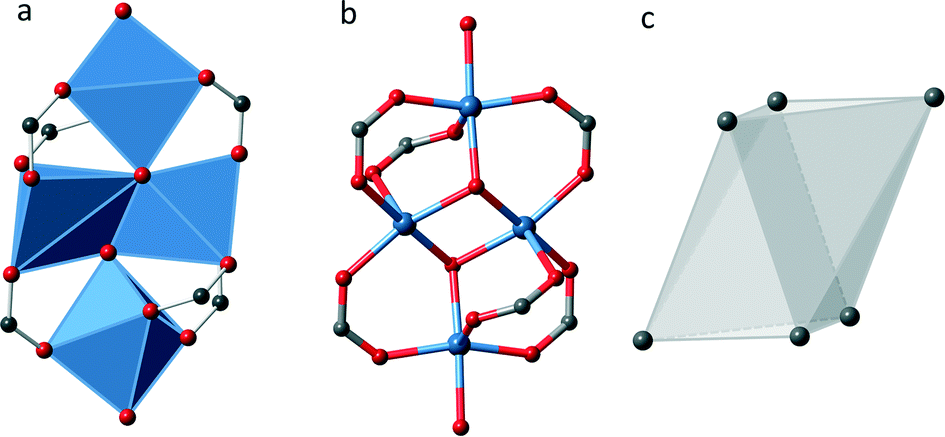 | ||
| Fig. 2 Illustration of the inorganic SBU of UHM-32; grey: carbon, red: oxygen, blue: copper. a: Illustration of the copper coordination sphere in form of the resulting polyhedra, b: illustration of the connectivity between copper ions and oxygen ions, c: resulting overall polyhedron built by the points of extension, i.e. the carbon atoms of the carboxyl groups. | ||
Fig. 3 shows an extended unit cell of UHM-32 displayed along all three directions. Crystallographically there are two SBUs at different locations, one (SBU I) at the centre of the b/c-plane (0 1/2 1/2) and the second (SBU II) at the centre of the a-axis (1/2 0 0). In addition both SBU types have different orientations with respect to each other (see Fig. 3a). Each SBU is connected to ten other SBUs, to two SBUs of the same kind along the a-axis and to eight SBUs of the other kind in form of a distorted cuboid, giving rise to a 2-fold capped cuboid as overall coordination polyhedron of the SBUs. The 3D framework contains 1D rhombic channels running along the a-axis, which show dimensions of approximately 7 × 9 Å. In the other two directions the structure shows a rather dense network.
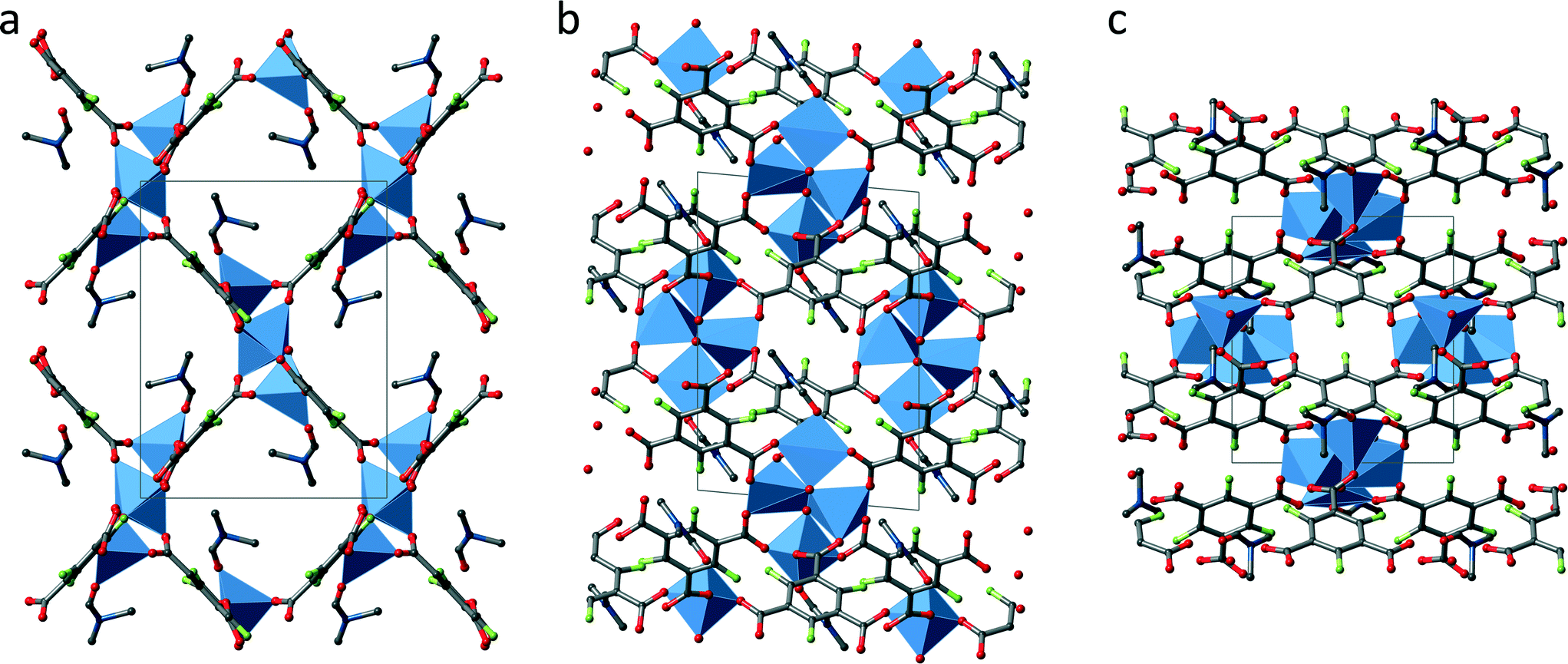 | ||
| Fig. 3 Extended unit cell of UHM-32 along the a-axis (a), the b-axis (b) and the c-axis (c); grey: carbon, red: oxygen, blue: copper, green: fluorine, hydrogen atoms and solvent molecules (apart from the DMA molecules coordinated to the SBU) are omitted for clarity. The coordination spheres of the copper ions are illustrated by the blue polyhedra. | ||
The topology of UHM-32 was analysed using TOPOS 4.0.22 The tetranuclear copper–oxygen cluster constitutes an 6-connected node, while the linker is a 3-connecting node. Accordingly, the net was classified as the binodal (3,6)-c net rtl.
262 Å, c = 14.5010 Å, α = γ = 90°; β = 91.671°, V = 2003.80 Å3). As in UHM-31 and UHM-32 the fluoro substituent shows a positional disorder over three sites (for details, see ESI†).
Fig. 4 shows an extended unit cell of UHM-33 displayed along all three directions. UHM-33 is based on the well-known copper paddle wheel motif as SBU, to which two DMA molecules are coordinated on its axial positions. These paddle wheels are placed at the centre of both the a/b- (1/2 1/2 0) and the a/c-plane (1/2 0 1/2), see Fig. 4b and c. The paddle wheels are connected to each other by only two of the three carboxylic groups of the Fbtc-linker. Surprisingly, the third carboxylic acid group remains uncoordinated. The authors are not aware of any other copper-based MOF in which this tritopic linker btc is involved in only two of the three possible bonds. This connection principle leads to 2D layers in the b/c-plane, which build rhombic meshes with channels running along the a-axis. Topologically UHM-33 belongs to the 2-dimensional uninodal 4-c layer sql.
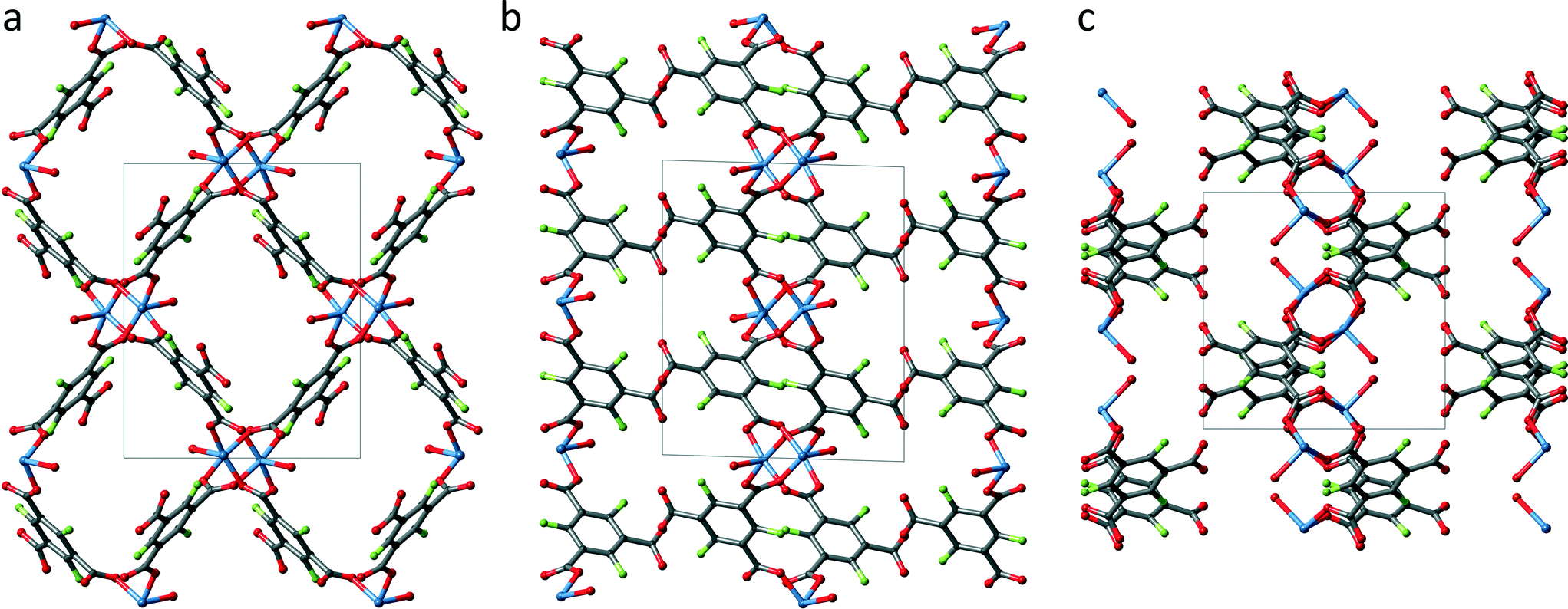 | ||
| Fig. 4 Extended unit cell of UHM-33 along the a-axis (a), the b-axis (b) and the c-axis (c); grey: carbon, red: oxygen, blue: copper, green: fluorine, hydrogen atoms and solvent molecules are omitted for clarity. | ||
Studies on thermally induced phase change behaviour
Due to the interesting new SBU of UHM-32 and the uncommon occurrence of an uncoordinated carboxylic acid function in the 2D framework UHM-33 we decided to carry out studies on probable phase changes. These studies were carried out by using temperature-dependent powder X-ray diffraction on microcrystalline phases (the phase purity/identity of all bulk samples were confirmed via comparison of the PXRD patterns with the simulated patterns of the single-crystal data sets, Fig. S4, ESI†).In Fig. 5 variable temperature powder XRD patterns recorded from 28 to 350 °C under argon atmosphere for UHM-31, -32, and -33 are shown. UHM-31 is stable up to approx. 220 °C and decomposes at higher temperatures to CuO, which is reduced to Cu2O and finally to elementary Cu. UHM-32 and UHM-33 show a quite different behaviour. Fig. 5b shows that the original phase of UHM-32 is stable up to 110 °C followed by the formation of a new phase, which is stable up to 180 °C. UHM-33 also changes its structure (Fig. 5c). The transition starts at 120 °C and the new phase is stable up to 230 °C. UHM-32 as well as UHM-33 finally decomposes to CuO.
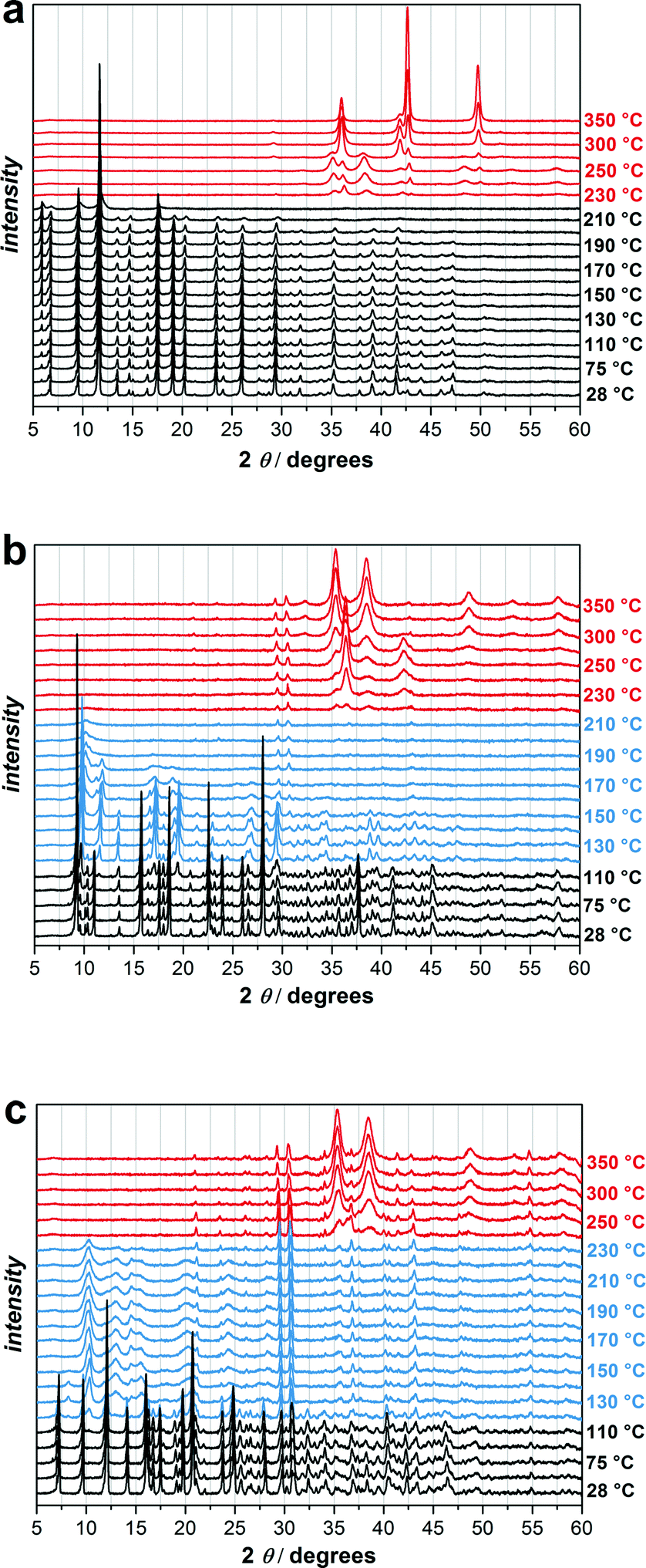 | ||
| Fig. 5 Variable temperature powder XRD pattern of UMH-31 (a), UHM-32 (b) and UHM-33 (c). For each experiment 23 diffractograms were measured, the first three ones at 28, 50 and 75 °C, followed by temperature steps of 10 °C from 100 up to 250 °C and steps of 25 °C up to 350 °C. Black represents the original MOF phase, blue the new MOF phases and red the decomposition products. The two reflections at 2θ = 29.4 and 2θ = 30.5 are probably caused by the sample holder. | ||
In order to check if the phase changes of UHM-32 and -33 are reversible, we carried out the variable temperature powder XRD experiment for another time. This time the samples were heated up to 150 °C and cooled down to room temperature again (Fig. 6). It turned out that the phase transition of UHM-32 is completely reversible, whereas the newly out of UHM-33 generated phase remains intact even after cooling down to room temperature again, meaning that the phase transition of UHM-33 is irreversible. The new phase of UMH-33 is far less crystalline than the original phase. Because of the 2D structure and the uncoordinated carboxylic acid group of the linker it is conceivable that the structure change is due to the formation of new bonds. However, this remains speculative as it was not possible to identify the structure of the new phase. The comparison of the pattern of the ‘high-temperature’ phase with that of the room-temperature phase of UHM-32 reveals that the reflections below 2θ = 12.5° do not change significantly and that the main changes occur in the region above 2θ = 12.5°. One can tentatively assume – in particular due to the fact that the phase transition is reversible – that this transition takes place without any bond breaking. It is not unlikely that the transition is a sort of ‘breathing’ mode preserving the complete connectivity of the initial phase. However, as in the case of UHM-33, we were unfortunately unable to identify or solve the structure of the high-temperature phase.
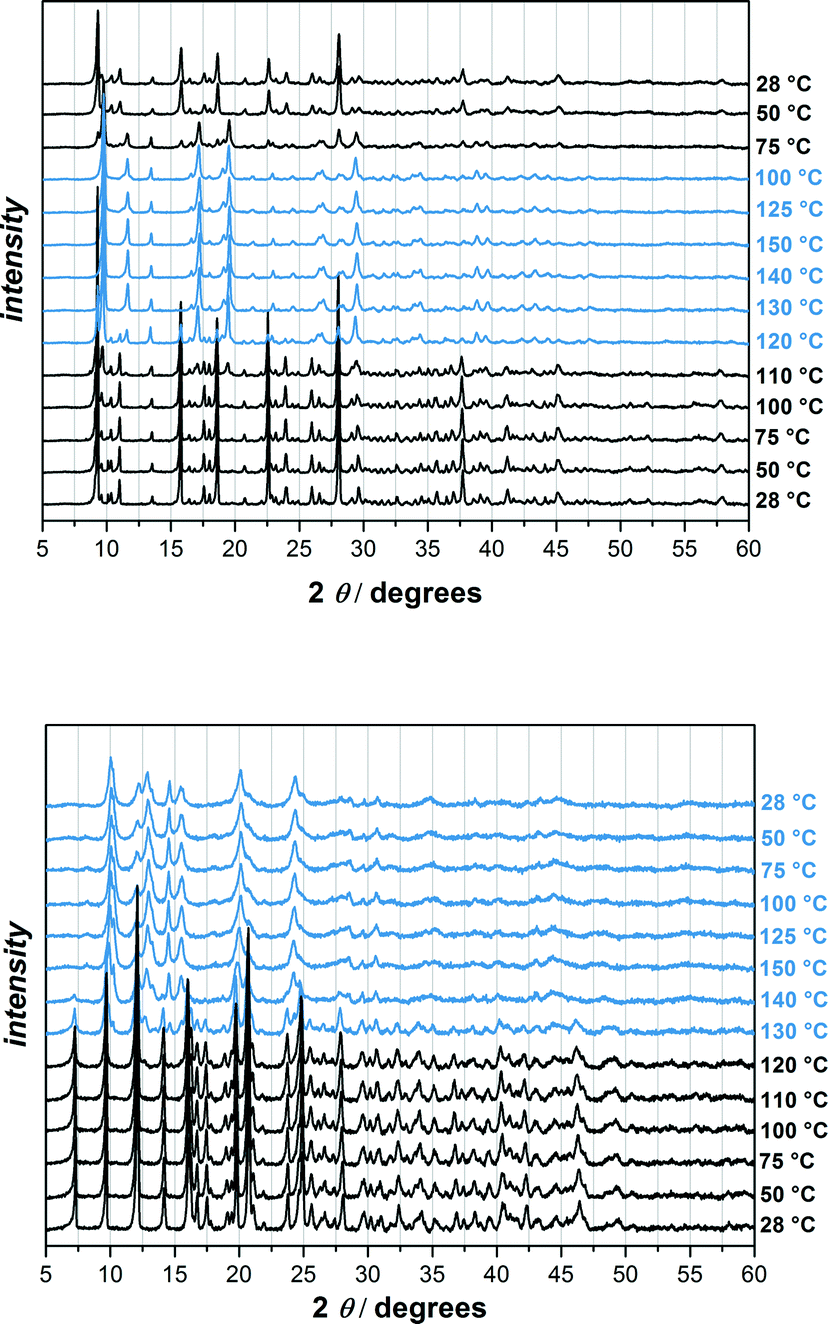 | ||
| Fig. 6 Variable temperature powder XRD patterns of UHM-32 (top) and UHM-33 (bottom). The temperature was increased from 28 °C to 50 °C and 75 °C, than in steps of 10 °C up to 150 °C. The samples were than cooled down to room temperature in steps of 25 °C. Blue represents the new built phases. | ||
Gas adsorption studies
To test the permanent porosity of UHM-31, UHM-32 and UHM-33, the MOFs were tried to activate via solvent exchange with ethanol and thermal treatment at 120 °C in vacuum and nitrogen physisorption isotherms were measured at 77 K. As already anticipated by the results of thermo-gravimetric and PXRD measurements after the solvent exchange (see ESI†) UHM-32 and UHM-33 did not show any porosity. It is probably not possible to activate UHM-32 and UHM-33 under these conditions without a significant loss of structure.The nitrogen physisorption measurement for UHM-31 shows a typical type-I isotherm (see Fig. 7). The analysis of the isotherm reveals a specific surface area of SBET = 1802 m2 g−1 (calculated from the adsorption branch and in the relative pressure interval from 0.001 to 0.024) and a micropore volume of Vpore = 0.71 cm3 g−1 (calculated at p/p0 = 0.18). These values are comparable to those known for HKUST-1 (ref. 23) and UHM-30.19
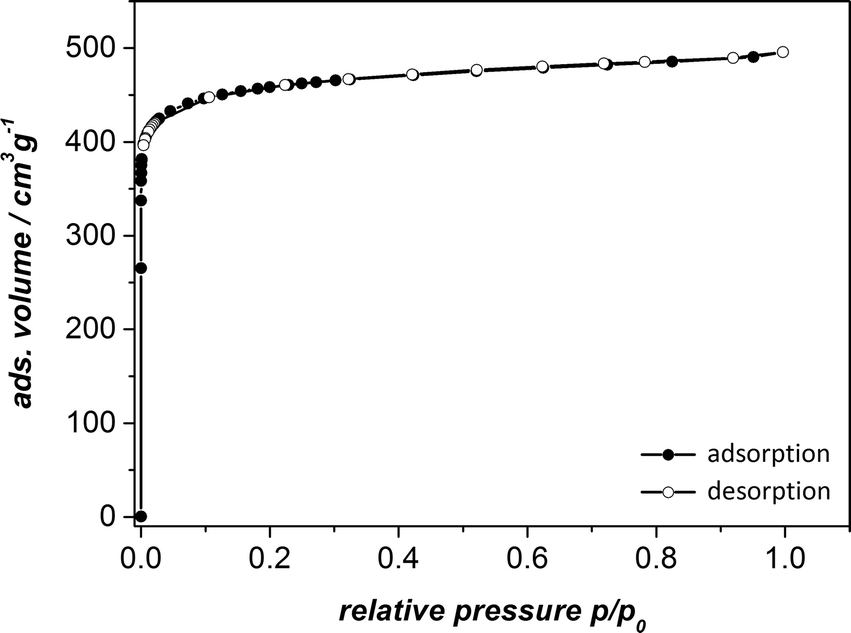 | ||
| Fig. 7 Nitrogen physisorption isotherms of activated UHM-31 measured at 77 K. | ||
Furthermore, we carried out hydrogen adsorption at 1 bar and 77 K and methane and carbon dioxide adsorption at the same pressure and 298 K. The isotherms are shown in Fig. 8 and the results are summarised in Table 1. The hydrogen uptake is 2.32 wt.%, the methane and carbon dioxide adsorption capacities amount to 1.04 mmol g−1 and 6.56 mmol g−1, respectively. Whereas the values for hydrogen and methane storage of UHM-31 are in the same range as those of HKUST-1 and UHM-30, the value for carbon dioxide adsorption is increased by around 2 mmol g−1 as compared to the non-fluorinated counterpart HKUST-1.23b In absolute terms the values are rather on the average when compared to other MOFs.24 However, to the best of our knowledge, the values of the H2 and CO2 uptake capacities (at 1 bar) are the highest values of all fluorinated MOFs so far reported.25 According to our results, the question whether the fluorination of a MOF leads to higher gas uptake can be answered with ‘it depends on the nature of the gas’! The recorded H2 uptake of 2.32 wt.% is only very slightly higher than the uptake reported for HKUST-1, so that the thesis of Pachfule et al.16 can be supported. Factors, such as pore size, curvature and open metal sites play the main role for the hydrogen uptake in this example and a special interaction between the fluorine atoms and the hydrogen molecules cannot be assumed. However, according to the results for the CO2 adsorption capacity, an increase of 2 mmol g−1 for the fluorinated version of HKUST-1 in comparison with the unfluorinated one is quite remarkable and might be indeed the result of a special interaction between the fluorine atoms and CO2, probably mediated by the relatively high quadrupole moment of CO2.
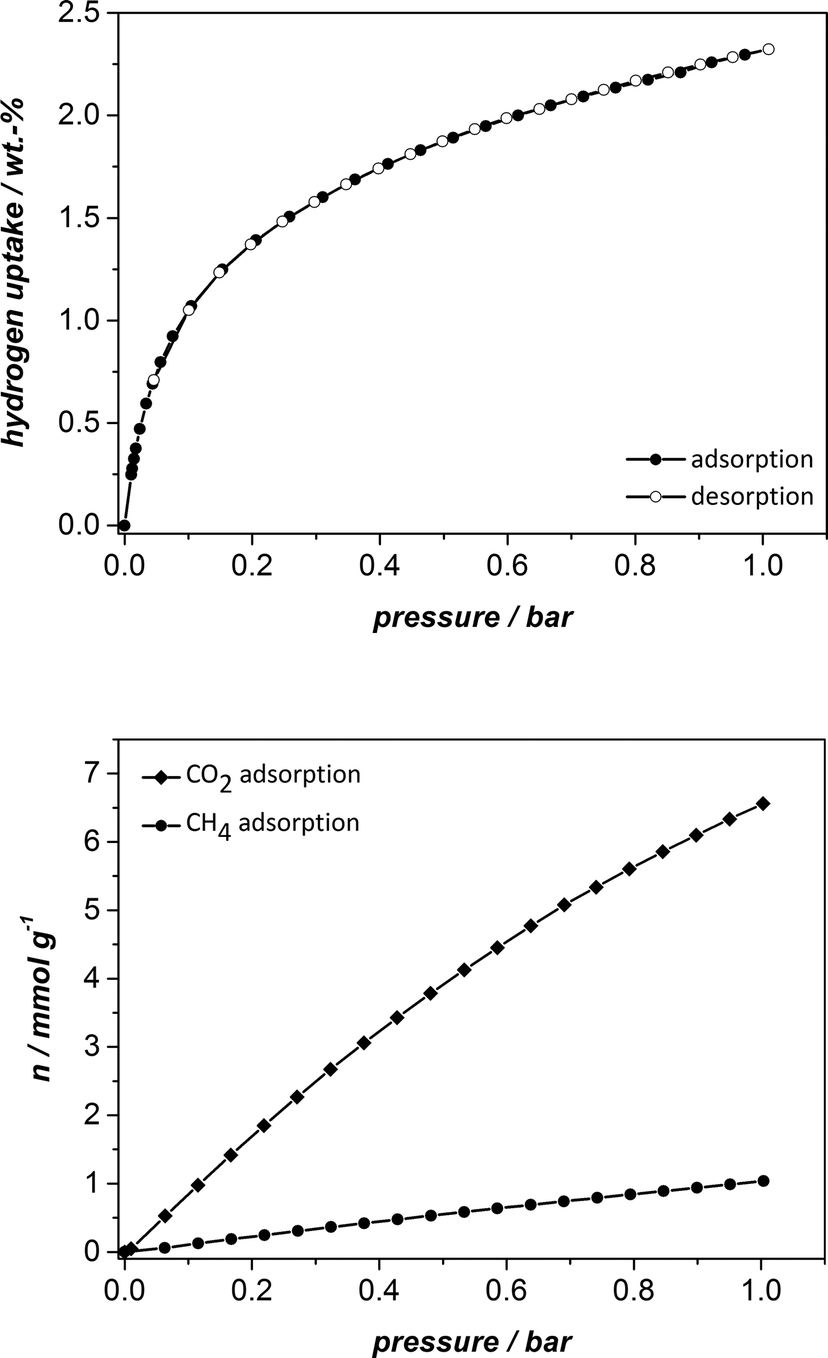 | ||
| Fig. 8 Hydrogen physisorption isotherms measured at 77 K (top) and methane and carbon dioxide physisorption isotherms at 298 K (bottom). | ||
Experimental
Chemicals
Starting materials and solvents were used without further purification. 2-Flouro-1,3,5-trimethylbenzene (Alpha Aesar, 98%), potassium permanganate (Grüssing, 99%), magnesium sulphate heptahydrate (Fluka, ≥99.0%), sulphuric acid (Grüssing, 95–97%).Methods
NMR spectra were acquired using a Bruker Fourier 300 spectrometer. Infrared spectra were acquired using a Bruker Vertex FT-IR spectrometer. Powder X-ray diffraction patterns were recorded at room temperature with a PANalytical X'Pert Pro PW3040/60 reflexion-powder diffractometer using Cu Kα radiation (45 kV, 40 mA; counting time: 74 s; steps: 0.013° (2θ)). The variable temperature XRD experiments were measured with the same instrument using the reactor chamber XRK 900 and an argon atmosphere. We used a sample holder made of glass ceramic (Macor) and Cu Kα radiation (45 kV, 40 mA; counting time: 600 s; steps: 0.013° (2θ), heating rate: 10 K min−1). Thermal analysis (thermogravimetry (TG)/mass spectrometry (MS)) was conducted under argon flow (20 mL min−1) with a NETZSCH STA 449 F3 Jupiter thermobalance coupled by capillary with an Aëolos QMS 403 mass spectrometer. The heating rate was 5 K min−1 up to 800 °C.Nitrogen physisorption data were recorded with a Quantachrome QUADRASORB-SI-MP at 77 K. The specific surface area was calculated from the adsorption branch in the relative pressure interval from 0.001 to 0.024 using the Brunauer–Emmett–Teller (BET) method. The micro pore volume was estimated from the quantity of gas adsorbed at a relative pressure of 0.18. Volumetric hydrogen physisorption data were recorded at 77 K on a Quantachrome Autosorb 1-MP (purity of helium and hydrogen: 99.999%). Carbon dioxide and methane physisorption data were recorded on a Quantachrome Autosorb iQ (purity of carbon dioxide: 99.5%, purity of methane: 99.9995%).
Synthesis of the linker
The synthesis of 2-fluoro-1,3,5-benzenetricarboxylic acid (Fbtc) was carried out according to a literature procedure.2614.0 g (101 mmol) of 2-flouro-1,3,5-trimethylbenzene was emulsified in 740 mL boiling water. 143 g (903 mmol) of potassium permanganate was mixed with 114 g (461 mmol) of magnesium sulphate heptahydrate and added to the refluxing solution in twelve portions over a period of one week. The manganese(IV) oxide was removed by filtration and treated twice with 400 mL boiling water. The combined aqueous solutions were concentrated in vacuum to 400 mL and stored over night at 6 °C. The resulting inorganic precipitate was removed by filtration and the filtrate was concentrated in vacuum to 300 mL. The solution was then acidified with sulphuric acid (50%) up to pH = 3.5 to give 7.06 g (31.0 mmol, yield: 42%) of a colourless powder. 1H-NMR (DMSO-d6, 300 MHz): δ (ppm) = 8.47 (d, 4JH,F = 6.2 Hz). 13C-NMR (DMSO-d6, 70 MHz): δ (ppm) = 165.4; 164.1 (d, 3JC,F = 2.3 Hz); 163.2 (164.6/161.0, d, 1JC,F = 275 Hz); 136.9 (d, 3J = 2.5 Hz); 127.0 (d, 4JC,F = 3.9 Hz); 121.6 (d, 2JC,F = 11.9 Hz). IR (FT-IR, KBr disk): ν (cm−1) = 3421, 3096, 2361, 2342, 1701, 1607, 1559, 1471, 1431, 1398, 1272, 1233, 1214, 1181, 1112, 983, 939, 910, 812, 791, 768, 691, 664, 619.
Synthesis of the MOFs
Conclusions
In summary, the use of 2-fluoro-1,3,5-benzenetricarboxylic acid as new linker leads to three new MOF structures: UHM-31, UHM-32 and UHM-33. Only small variations of the synthesis temperature and the ratio of metal source to linker lead to the different structures. UHM-31 is isostructural to HKUST-1. Under the same variations of synthesis conditions the use of btc or NH2btc as linker always leads to the HKUST-1 structure. Therefore, fluorine definitely has an – unfortunately unknown – influence on the formation of the different crystalline phases. Both, UHM-32 and UHM-33 show a thermally induced phase transition at around 120 °C, which is reversible for UHM-32 but irreversible for UHM-33. The low pressure gas storage capacity of UHM-31 for hydrogen and methane is only slightly higher than for HKUST-1. In contrast, the CO2 adsorption capacity of UHM-31 with 6.56 mmol g−1 is approximately 2 mmol g−1 higher than the one of HKUST-1. This is particularly interesting as it is known that fluorinated phases show a modified wetting behaviour in aqueous environments due to their hydrophobicity. With regard to possible gas storage applications – carbon capture and storage (CCS), for instance – this could be advantageous because many MOFs with high CO2 storage capacities show only a limited water stability.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG: Fr 1372/18-2) for supporting this work as part of the priority program 1362 (porous metal–organic frameworks).Notes and references
- (a) R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966 CrossRef CAS PubMed; (b) J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- (a) M. D. Allendorf, R. J. T. Houk, L. Andruszkiewicz, A. A. Talin, J. Pikarsky, A. Choudhury, K. A. Gall and P. J. J. Hesketh, J. Am. Chem. Soc., 2008, 130, 14404 CrossRef CAS PubMed; (b) L. E. Kreno, K. Leong, O. K. Farha, M. D. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2011, 112, 1105 CrossRef PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed.
- S. T. Meek, J. J. Perry IV, S. L. Teich-McGoldrick, J. A. Greathouse and M. D. Allendorf, Cryst. Growth Des., 2011, 11, 4309 CAS.
- K. Uneyama, Organofluorine chemistry, Blackwell Publishing Ltd., Oxford, 2006 Search PubMed.
- R. D. Chambers, Fluorine in Organic Chemistry, Blackwell Publishing Ltd., Oxford, 2004 Search PubMed.
- (a) J. L. Guth, H. Kessler, J. H. Higel, J. M. Lamblin, J. Patarin, A. Seive, J. M. Chezeau and R. Wey, ACS Symp. Ser., 1989, 398, 176 CrossRef CAS PubMed; (b) J. L. Guth, H. Kessler and R. Wey, Stud. Surf. Sci. Catal., 1986, 28, 121 CrossRef CAS; (c) T. Loiseau and G. Férey, J. Fluorine Chem., 2007, 128, 413 CrossRef CAS PubMed.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296 CrossRef PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Sublé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef PubMed.
- L. Pan, M. B. Sander, X. Huang, J. Li, M. Smith, E. Bittner, B. Bockrath and J. K. Johnson, J. Am. Chem. Soc., 2004, 126, 1308 CrossRef CAS PubMed.
- C. Yang, X. Wang and M. A. Omary, J. Am. Chem. Soc., 2007, 129, 15454 CrossRef CAS PubMed.
- (a) Z. Hulvey, E. H. L. Falcao, J. Eckert and A. K. Cheetham, J. Mater. Chem., 2009, 19, 4307 RSC; (b) Z. Hulvey, D. A. Sava, J. Eckert and A. K. Cheetham, Inorg. Chem., 2011, 50, 403 CrossRef CAS PubMed.
- (a) C. A. Fernandez, P.-K. Thallapally, R. K. Motkuri, S. K. Nune, J. C. Sumrak, J. Tian and J. Liu, Cryst. Growth Des., 2010, 10, 1037 CrossRef CAS; (b) W. Yang, X. Lin, A. J. Blake, C. Wilson, P. Hubberstey, N. R. Champness and M. Schröder, Inorg. Chem., 2009, 48, 11067 CrossRef CAS PubMed; (c) L. Hou, W.-J. Si, Y.-Y. Wang, Y. Guo, C. Jin and Q.-Z. Shi, Chem. Commun., 2011, 47, 5464 RSC.
- P. Pachfule, Y. Chen, J. Jiang and R. Banerjee, Chem. – Eur. J., 2012, 18, 688 CrossRef CAS PubMed.
- X.-S. Wang, S. Ma, P.-M. Forster, D. Yuan, J. Eckert, J. J. López, B. J. Murphy, J. B. Parise and H.-C. Zhou, Angew. Chem., 2008, 120, 7373 CrossRef.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS.
- K. Peikert, F. Hoffmann and M. Fröba, Chem. Commun., 2012, 48, 11196 RSC.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- (a) P. Krawiec, M. Kramer, M. Sabo, R. Kunschke, H. Fröde and S. Kaskel, Adv. Eng. Mater., 2006, 8, 293 CrossRef CAS; (b) A. Vishnyakov, P. I. Ravikovitch, A. V. Neimark, M. Bulow and Q. M. Wang, Nano Lett., 2003, 3, 713 CrossRef CAS.
- A. Blatov, Struct. Chem., 2012, 23, 955 CrossRef PubMed.
- (a) B. Xiao, P. S. Wheatley, X. B. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, S. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203 CrossRef CAS PubMed; (b) Q. M. Wang, D. M. Shen, M. Bulow, M. L. Lau, S. G. Deng, F. R. Fitch, N. O. Lemcoff and J. Semanscin, Microporous Mesoporous Mater., 2002, 55, 217 CrossRef CAS.
- (a) A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784 CrossRef CAS PubMed; (b) S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870 CrossRef CAS PubMed; (c) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS PubMed.
- P. Pachfule, R. Das, P. Poddar and R. Banerjee, Inorg. Chem., 2011, 50, 3855 CrossRef CAS PubMed.
- F. Micheel and W. Busse, Chem. Ber., 1957, 90, 2049 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details, powder XRD patterns, thermal analysis, NMR and ESI-MS studies on UHM-31. CCDC 986518–986520 contains the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00408f |
| This journal is © The Royal Society of Chemistry 2015 |