
Chemical conversion pathways for carbohydrates
Chandrani
Chatterjee
*,
Frances
Pong
* and
Ayusman
Sen
*
Department of Chemistry, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: chandrani@psu.edu; fyp101@psu.edu; asen@psu.edu
First published on 26th September 2014
Abstract
Biomass has emerged as a potential alternative feedstock to dwindling fossil fuel reserves. Starting in the 1990s, extensive research has been directed towards the synthesis of useful platform chemicals from cellulosic biomass. Chemical conversion processes of biomass have evolved as a parallel approach to thermochemical and enzymatic synthetic routes. In this review, we summarize the recent developments in liquid phase chemical conversions of monosaccharides, disaccharides, and polysaccharides. The reaction processes explored are hydrolysis, oxidation, reduction, hydrogenation, hydrogenolysis, esterification, etherification, glycosylation, dehydration, as well as the functionalization of the polysaccharide backbone. Our review follows a “process-driven” approach where the existing carbohydrate conversion pathways are classified according to the types of chemical processes involved.
 Chandrani Chatterjee | Chandrani Chatterjee completed her B.Sc. Honours in Chemistry from Presidency College Kolkata, India in 2004 and her MS in Chemistry from IIT Bombay, India in 2006. She joined the research group of Professor Malcolm H. Chisholm, at Ohio State University for her PhD. She developed mechanistic understanding of (porphyrin)M(III) catalyst systems for copolymerization of CO2 and cyclic anhydride with epoxides. She completed her PhD in 2012 and currently is a Post-doctoral Fellow at The Institute of Energy and Environment, Pennsylvania State University. Her current research, under supervision of Professors Ayusman Sen, Rob Rioux and Chunsan Song, focuses on the development of new catalytic processes for the conversion of CO2 into chemical building blocks. |
 Frances Pong | Frances Pong graduated from Wellesley College in 2005 with a B.A. in Chemistry; while at Wellesley she studied the behavior of nanocomposite N-isopropylacrylamide hydrogels with Professor Nolan Flynn. Immediately after, she joined Cabot Corporation as a research associate and worked on aqueous and solvent particle dispersions. In 2009, she joined the Chemistry Ph.D. program at Pennsylvania State University under the supervision of Professor Ayusman Sen. Her research interests include organometallic polymerization catalysts, biomass conversion, and studying the behavior of synthetic and biological nanomotors/nanopumps. |
 Ayusman Sen | Ayusman Sen was born in Calcutta, India and holds a Ph. D. from the University of Chicago where he was first introduced to catalysis. Following a year of postdoctoral work at the California Institute of Technology, he joined the Chemistry Department of the Pennsylvania State University where is currently a Distinguished Professor. He is a Fellow of the American Association for the Advancement of Science. His research interests encompass catalysis, organometallic and polymer chemistry, and nanotechnology. He is the author of approximately 335 scientific publications and holds 24 patents. Sen's pastime centers on enological and gastronomical explorations. |
1. Introduction
Currently, a majority of essential commodity chemicals and fuels are produced from fossil fuels, which are non-renewable resources such as coal, petroleum and natural gas. The decrease in fossil fuel reserves and the resulting price hike for petrochemicals1 have thus mandated the search for alternative, inexpensive, renewable resources: abundant, ever-growing, terrestrial biomass.2,3 The conversion of waste biomass into fuels, fine chemicals, and commodity materials can potentially reduce national dependence on oil-exporting countries. The use of bio-based technology has also been universally recognized as a means to reduce the emission of CO2, a well-known greenhouse gas which is produced during the combustion of fossil fuels.4 As a result, during the last 50 years, a significant portion of research has been directed towards the production of biodiesel and biofuel to serve growing energy demands.4–7 The drive towards bio-based alternatives, which seeks to replace the conventional petrochemical processes with new technologies, must also be financially competitive, if not advantageous, for the fuel/chemical industry. Much emphasis, therefore, has been placed upon an integrated biorefinery approach, which couples the production of inexpensive, high volume biogas and biofuels with the production of low volume, high-value platform chemicals,3,6,8 to provide the economic incentive for the implementation of this new “bio-based economy”. From both economic and ethical perspectives, abundant and inexpensive carbohydrates are ideal feedstocks for production of both biofuels and platform chemicals. In our review, we define the platform chemicals as sustainable chemical intermediates which can be synthesized in either one or two steps from carbohydrates.In 2004, the Department of Energy (DOE) of the United States identified a series of valuable platform chemicals and the basic technology required to produce these chemicals from biorenewable carbohydrates following an integrated biorefinery approach.9 Further conversion of these platform chemicals provide access to a plethora of “secondary chemicals”, which can be incorporated into surfactants, polymers, fabrics, resins etc. for application in the personal care, textile, transportation, polymer, and packaging industries. Selected platform chemicals produced by chemical conversion of carbohydrate biomass are summarized in Chart 1.
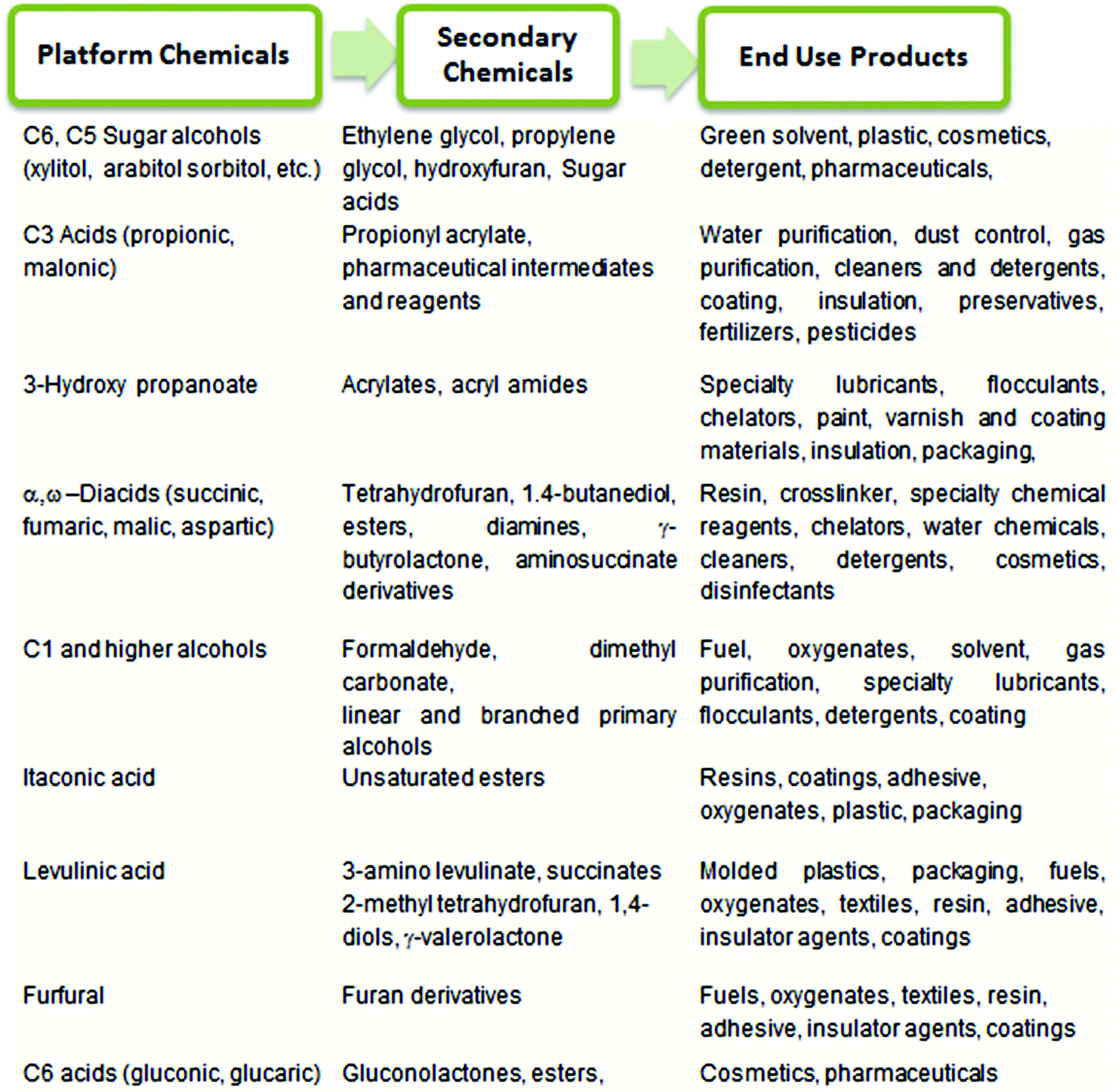 | ||
| Chart 1 A summary of platform chemicals produced by chemical conversion. | ||
A decade after the DOE report, technology development for commercially viable biomass conversion processes still remains the most significant challenge. This problem has been addressed in detail in several recent reviews.6,10–12 One, however, must also consider that biomass conversion is still in its infancy – petrochemical technologies have been thoroughly researched and developed since the early 1900s, while extensive studies of the conversion of cellulosic biomass began only during the 1990s. Hence, further development of commercially viable synthetic routes for the biomass conversion is necessary for the success of this integrated bio-refinery approach.
The current review is thus focused on the description and critical analysis of the recent developments in the liquid phase chemical conversions of biomass at temperatures of ≤300 °C for the production of platform chemicals. We will not cover enzymatic or high temperature thermochemical process (500–800 °C), which include gasification and pyrolysis. The production of biofuels is also out of the scope of this review. Most biomass conversion reviews are organized around conversion to specific platform chemicals. In contrast, we intend to organize our review around the chemical conversion routes themselves. Herein, we discuss recent advances in academic research and we hope to help researchers establish a broader understanding and a more comparative perspective of the existing chemical processes.
2. Carbohydrates as feedstocks
Carbohydrates account for 75 wt% of plant biomass, and can be categorized into four classes: sugar, starch, cellulose, and hemicellulose, as shown in Fig. 1.13 Carbohydrates of relatively high purity can be harvested from a wide variety of biomass wastes (land, agricultural, aquatic) and crops (corn, switch grass).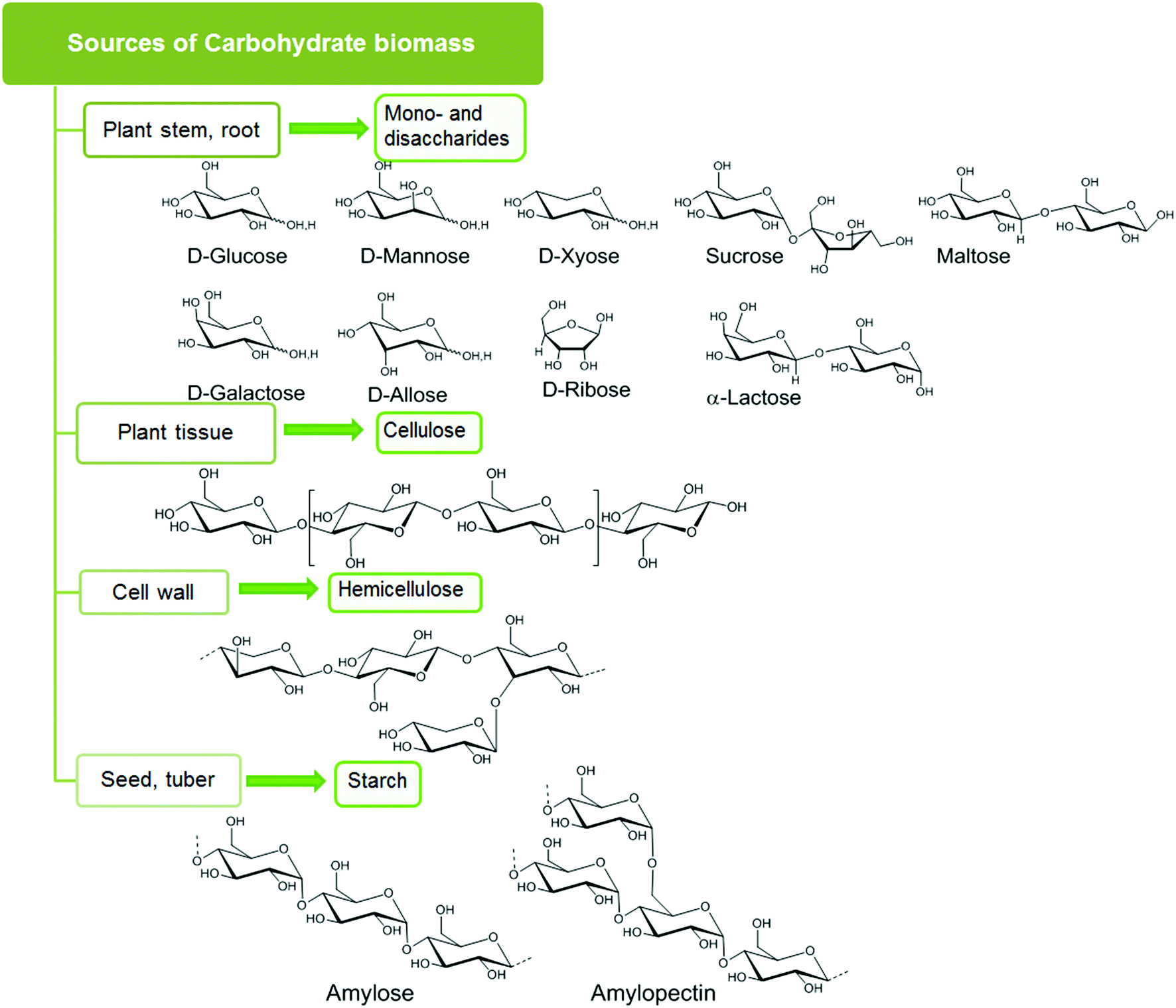 | ||
| Fig. 1 Structural types of carbohydrate. | ||
2.1. Sugars
Plants store solar energy in the form of monomeric hexose and pentose sugar units which are commonly referred to as monosaccharides. D-Glucose and D-xylose are the most abundant hexose and pentose sugars. Other rarer hexose and pentose sugars are D-galactose, D-mannose, D-fructose and D-ribose. Two monosaccharides joined together by a glycosidic linkage form a disaccharide. Common disaccharides include: sucrose, which contains a glucose and a fructose unit; lactose, which contains a glucose and a galactose unit; and maltose, comprised of two glucose units. These mono- and disaccharides serve as the basic building blocks of complex carbohydrate networks.2.2. Starch
Polysaccharide starch is mainly composed of two α-glucose polymers, amylose with a straight chain structure and amylopectin with a branched chain structure. Typically starch consists of 10–20% amylose and 80–90% amylopectin, and the actual content of these polymers varies, depending on the plant source. The monomeric glucose units in the polysaccharide are joined by α 1,4-glycoside linkages in amylose and both α 1,4-glycoside linkages and α 1,6-glycoside linkages in amylopectin. Due to low crystallinity (crystallinity index 35%), starch can be easily hydrolyzed or degraded by enzymes.2.3. Cellulose
Cellulose is the major structural component in carbohydrate biomass with typically 40–50% content by weight. Linear polymeric strands of unbranched β-glucose form the basic structure of cellulose. These glucose units are joined by β 1,4-glycoside linkages, which form both inter- and intra-strand hydrogen-bonded helices. In woody biomass, these cellulose strands are intertwined with hard, water-impermeable matrices of lignin. As a result, cellulose is rigid, crystalline, insoluble in water, and difficult to hydrolyze under mild acidic or enzymatic conditions. Crystalline cellulose can adopt different polymorphic forms (i.e. Iα, Iβ, II, III, IV) where the relative arrangement of the individual cellulose strands are different and the resulting hydrogen bonding also changes. The Iα, (algae and bacterial cellulose) and Iβ (wood, cotton) polymorphs are found in natural cellulose. These two forms can be present on the same microfibrillar network. Cellulose II is prepared from these natural forms by alkaline treatment, also known as mercerization or by solubilization followed by recrystallization. Liquid ammonia treatment of cellulose I and II produces cellulose III, which upon further pretreatment produces cellulose IV. Only polymorphs I and II are significant for use as large scale biorenewable feedstock.142.4. Hemicellulose
Hemicellulose is a branched polymer made of both hexoses (D-glucose, D-mannose, D-galactose) and pentoses (D-xylose and L-arabinose). The most abundant monomer present in hemicellulose is xylose. Oxidized sugars, e.g. mannuronic acid and galacturonic acids, comprise a small portion of the hemicellulose structure. Hemicellulose forms the linkage between cellulose and lignin fractions and must be hydrolyzed to isolate cellulose from lignin. Hemicellulose is amorphous and can be easily hydrolyzed into its’ component monomers, which can then be used for further conversion into fine chemicals.2.5. Pretreatment of carbohydrate biomass
Due to their complex structure and the presence of interpenetrating, rigid, hydrophobic lignin layers, ligninocellulosic feedstocks cannot be directly used in further conversion. A pretreatment step is necessary in biomass conversion processes to separate the carbohydrate fractions (cellulose and hemicellulose) from lignin.15 This step makes the isolated carbohydrates more susceptible towards subsequent steps in chemical conversion. Pretreatment usually involves both physical separation (milling, steam wash) and chemical degradation (acid or base hydrolysis) techniques. The pretreated carbohydrates are then subjected to further conversion (enzymatic, thermochemical, chemical).Due to the presence of H-bonded, hard crystalline segments, the dissolution of cellulose requires the presence of strongly acidic pretreatment, typically using 60–90% H2SO4 at ∼180 °C. Depending on the reaction time and acid concentration, the desired products – glucose and glucose oligomers – may undergo partial degradation. In order to improve product yield, mechanical pretreatments, such as ball milling, are often applied to weaken the H-bonding.16 This allows for the use of milder acidic and lower temperature conditions, which suppress byproduct formation and improve the overall reaction yield. It has been observed that longer ball milling time can increase the rate of cellulose hydrolysis at milder reaction conditions – lower temperature ∼100 °C and dilute acidic hydrolysis.
3. Carbohydrate conversion processes
A considerable amount of research has been dedicated to developing platform chemicals from cellulosic materials. The primary goal of biomass conversion processes is to deoxygenate the carbohydrate backbone, which generally liberates CO2 and H2O as byproducts.17,18 Instead of chemical conversion or “cracking” into small molecules, in an alternative approach, the pretreated biopolymer is chemically modified to incorporate useful functional groups and properties while keeping the ligninocellulosic backbone intact.19 These chemically modified biopolymers are used as the main components of high volume commodities such as lubricants, coatings, resins, papers, packaging materials and surfactants etc. without requiring further purification or characterization.20 In contrast to the high temperature, gas-phase transformations of petrochemicals, the transformations of carbohydrates are generally carried out in aqueous phase or biphasic systems, at moderate to high temperature (<500 °C) due to their hydrophilic nature.17,21 For better understanding, the basic biomass conversion strategies are classified herein according to the main process or technology involved in the processes. A summary of the advantages and disadvantages of thermochemical conversions, biocatalysis, and chemical conversion are listed in Chart 2.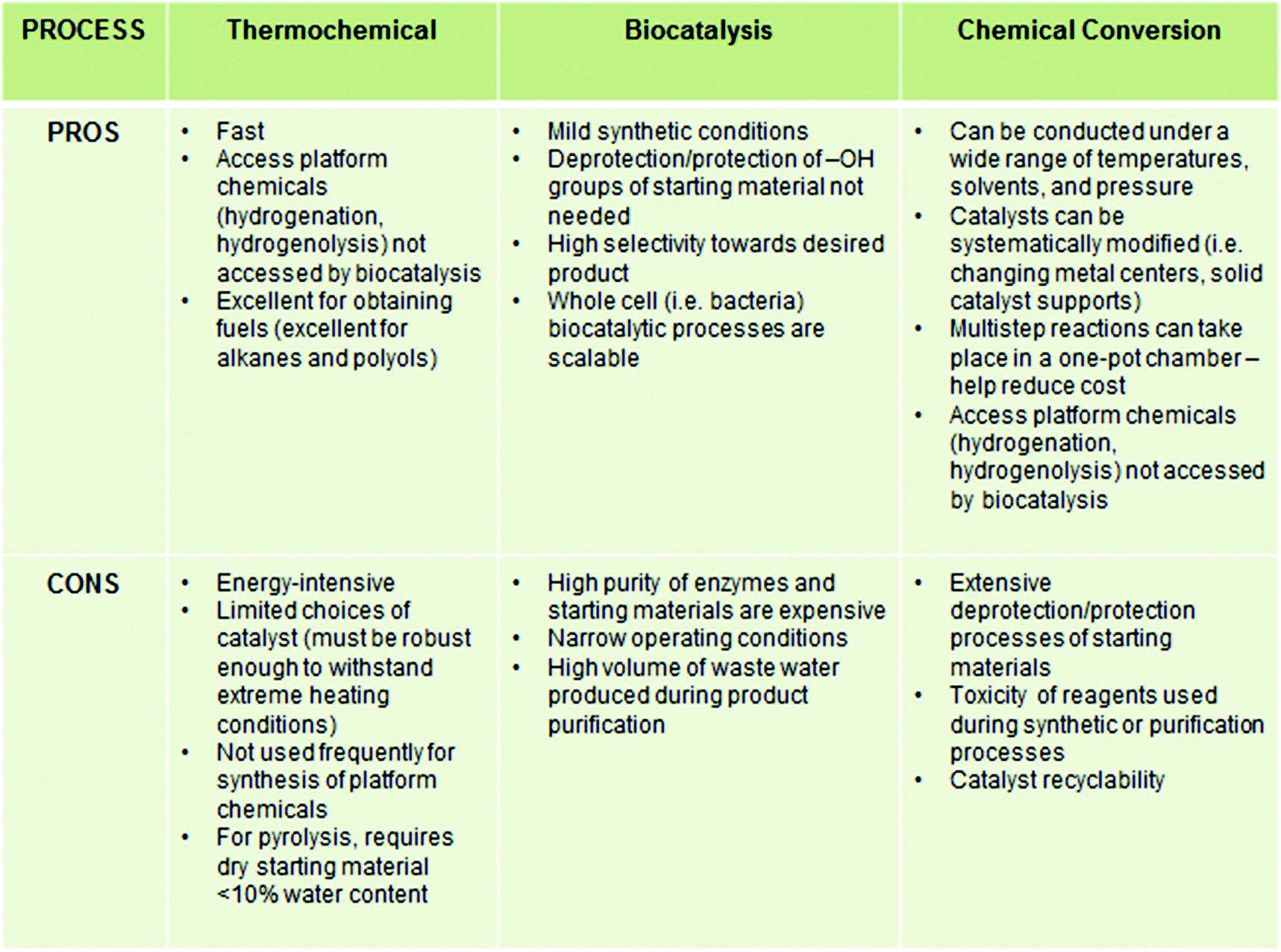 | ||
| Chart 2 General advantages and disadvantages of different biomass conversion methods. | ||
3.1. Thermochemical processes
Thermochemical methods are comprised of both non-catalytic and catalytic processes, carried out at high temperature (500–800 °C). Non-catalytic methods include gasification (syngas production), pyrolysis (to mixture of oxygenates), liquefaction, and supercritical treatment. To convert biomass to hydrocarbons and polyols, heterogeneous Ru/C and Pt/C catalysts are primarily used. Owing to the high solubility of sugars and some portions of polysaccharides in water, aqueous phase reforming of carbohydrates at high temperature (>500 °C) with heterogeneous metal catalysts, such as Pt or Ni–Sn on SiO2 or Al2O3 supports, have also been applied to the synthesis of bio-alkanes from sorbitol.22 The byproduct gases (CO2 and H2) formed are further converted to methane. Huber et al. observed that product selectivity in these processes can be controlled by tailoring the catalyst (changing the metal center, solid support) and by changing the reaction conditions (temperature, pH of the media, concentration of biomass).23 The isolation of pure compounds in these processes is often difficult. Thermochemical methods have recently been reviewed by Dumesic et al.5 and a detailed discussion is outside the scope of the present review.3.2. Biocatalysis
Biocatalysis of carbohydrate materials mainly entail enzymatic hydrolysis and fermentation.24–26 Intrinsic merits of these pathways include moderate reaction conditions, efficient conversion and low toxicity. Pretreatment of the biomass, using dilute acid and alkali, helps facilitate subsequent enzymatic hydrolysis by anaerobic microorganisms, enabling the production of useful chemicals such as lactic acid, succinic acid, isoprene and aliphatic primary (C2–C4) alcohols. However, a significant amount of waste water is produced which needs to be purified and recycled, often posing a limitation for larger scale application of this process.27 The large scale industrial application of biocatalysis is also precluded by high operational cost and lack of recyclability. In comparison, both chemical and thermochemical approaches are markedly cheaper. The recent development in this emerging field of research has been documented by recent reviewers and will not be discussed further.25,283.3. Chemical conversion
Starting in the beginning of the 21st century, the conversion of carbohydrate biomass became a major, multidisciplinary field of study. Both catalytic and non-catalytic processes have been extensively utilized in the chemical conversion of carbohydrates.In 1894, Ostwald defined a catalyst as “[a] substance that accelerates the rate of a chemical reaction, without [itself] being consumed in the reaction”. Catalysts are typically classified according to the phases of the reactants and products involved in the chemical reaction. When all reactants, products and catalysts remain in the same phase throughout the reaction, the catalyst is described as a homogeneous catalyst. Although wide applications of homogeneous catalysts are limited by the difficulty of product separation and catalyst recyclability, well-defined homogeneous catalysts such as organometallic or inorganic compounds are often employed for in-depth analysis of product selectivity, reaction kinetics, mechanistic studies and structure–activity relationships. In contrast, heterogeneous systems typically consist of a solid catalyst and reactants and products in either liquid or gaseous states. Solid adsorbents, such as metal oxides, zeolites, silica, insoluble polymers and resins, have been used as catalyst solid supports, which enable easier product separation and reuse of the catalyst. As an added bonus, solid-supported heterogeneous systems are often more stable at high reaction temperatures and pressures and provide a high concentration of active sites. For industrial applications, heterogeneous systems provide significant advantages over homogenous systems. However, the characterization of the active sites of heterogeneous catalysts and various intermediates can be challenging and costly. Typically, a wide range of surface characterization techniques is required to elucidate the catalysts’ structure, morphology, and atomic composition, all parameters which influence catalyst reactivity and longevity.
As the untreated polysaccharides are insoluble in aqueous media, even aqueous solutions of simple inorganic catalysts should be considered as heterogeneous systems, containing at least two different phases of reactants and products. To facilitate the conversion of polysaccharides, a non-catalytic acidic or alkaline pretreatment is necessary, as described earlier in section 2.5., to separate cellulose and hemicellulose fractions from lignin and to obtain cellulose hydrolysates which are more miscible with the catalyst during subsequent reactions. Mineral acids, such as sulfuric acid (H2SO4) and hydrochloric acid (HCl), have been used for non-catalytic conversions of biomass. Ionic liquids emerged as an alternative highly polar medium for biomass pretreatment. Chlorides, nitrates, acetates, and other basic anions present in ionic liquids solubilize cellulose by breaking the existing H-bonded structural network of cellulose and forming new, intermolecular hydrogen bonds with the cellulosic hydroxyl groups. The dissolved cellulose can be precipitated with the addition of an anti-solvent (acetone or water) and this precipitate can be retained for further conversion. Although there has been significant progress in using ionic liquids for biomass conversion, there are also considerable problems associated with their separation and reuse.
In addition to carbohydrate “cracking”, it is possible to chemically modify polysaccharides using common one-pot modification processes (oxidation, etherification, esterification) in which the –OH groups are functionalized while the backbone is kept intact. In this context the degree of polymerization, of the polysaccharide is an important factor. The degree of polymerization is representative of the average length of the polysaccharide, i.e. the number of monomers present in the backbone. A decrease in this number during the chemical modification process indicates degradation of the polysaccharide. The extent of functionalization of the OH– groups on present in starch or cellulose is often referred to as the degree of substitutions or the number of substitutions per monomer unit. A number of review articles describing the recent advances in this field have been published.29–32 The introduction of functional groups changes the carbohydrates’ macroscopic properties such as crystallinity, solubility, hydrophilicity and the ease of biodegradation. This approach is often considered more “sustainable” or “greener” than chemical cracking into fine chemicals, as these modification processes are typically one-step and do not necessitate extensive purification or product isolation steps. These modified biopolymers can be used for high volume applications in industries producing paper, lubricants, resins, coatings, packaging materials, surfactants and textiles.
In this review, the chemical processes have been organized according to the various types of catalysts involved, with particular emphasis on the overall activity, product selectivity and robustness or stability of the catalysts. We believe that fundamental understanding of the existing chemical processes will be extremely beneficial for the development of new synthetic strategies and technologies used to convert carbohydrate biomass into useful platform chemicals. The carbohydrate conversion pathways for the syntheses platform chemicals are outlined in Scheme 1.
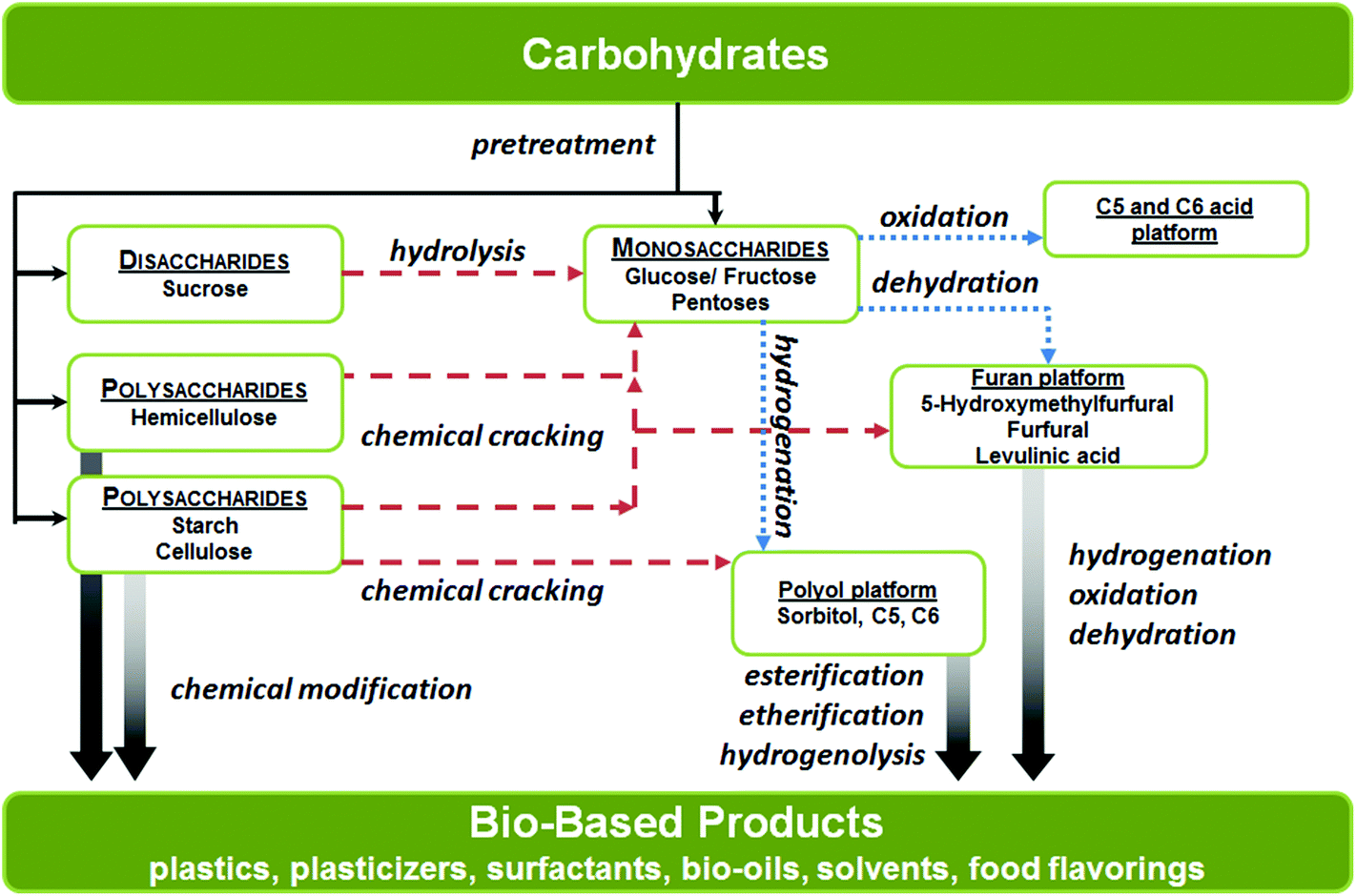 | ||
| Scheme 1 An outline of biomass conversion via chemical means. | ||
4. Chemical conversions of mono- and disaccharides
Glucose is the most widely used monosaccharide and has applications ranging from food and pharmaceutical industries to packaging and specialty chemicals.33 Currently, glucose is produced industrially by the enzymatic hydrolysis of corn. Numerous studies have been directed towards the production of glucose from less expensive sources of biomass (e.g. wood and agricultural waste). Sucrose is the most abundant disaccharide and is present in high quantities in vegetables, fruits, honey etc. Sucrose provides the major source of energy in human diets and is a common additive in commercial food products. However, the costs of synthesizing platform/commodity chemicals directly from sucrose are generally high because of the limited product selectivity of current chemical conversion pathways. As a result, sucrose still primarily serves as a raw material to supply glucose and fructose feedstocks. In this section, we summarize the most common chemical transformations of both mono- and disaccharides.4.1. Hydrolysis of sucrose
Sucrose can be hydrolyzed into a mixture of glucose and fructose by employing aqueous acid solutions acetic acid or hydrochloric acid, as shown in Scheme 2. Strongly acidic sulfonic resins34 have been industrially applied for the hydrolysis of sucrose, but this process often produces a mixture of furanic byproducts due to undesired dehydration of the monosaccharides formed, even at relatively low temperatures (25–70 °C). The use of ion exchange resins, based on polymer supports (e.g. polystyrene cross-linked with divinylbenzene) have also been applied for the conversion of sucrose to the corresponding monosaccharides.35 Buttersack et al. used porous Y zeolites as active catalysts for the hydrolysis of sucrose. Using a Si/Al ratio of 110, 90% conversion of sucrose was reported with 90% selectivity towards glucose and fructose formation over the furanic byproducts.36 Other studies performed on HY zeolite catalysts emphasize their high selectivity towards the production of glucose and fructose. The amount of byproduct formed in these processes can be as low as 100 ppm at 70 °C.37 In contrast, heteropolyacids and metallic oxides supported on silica showed poor activity and low conversion (50–60% at 80 °C, 3 h).38,39 The major limitations of this process are the commonly observed byproduct formation, the use of expensive zeolite or ion exchange resins and their stability in acidic media. A few recent processes have been patented in which up to 97% conversion and up to 98% selectivity towards monosaccharide formation were observed, but have not yet been commercialized.40 Thus, further research has been directed towards the development of inexpensive, robust, as well as selective catalysts.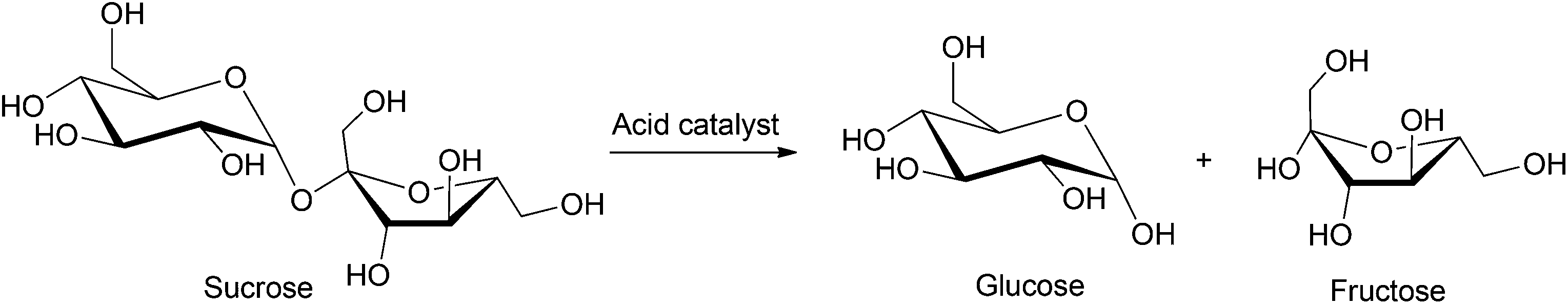 | ||
| Scheme 2 Hydrolysis of sucrose into glucose and fructose. | ||
4.2. Glucose isomerization
The isomerization of glucose into fructose is an important step in the synthesis of furan platform chemicals, such as HMF, furfural, and food additives such as high fructose corn syrup. The isomerization of the glucose unit present in lactose, a disaccharide composed of galactose and glucose, is widely used in the pharmaceutical industry to convert lactose into its ketose form, lactulose, to obtain a series of sugar based acids and alcohols. Common industrial processes involve pre-hydrolysis of cellulosic biomass into simple aldoses, followed by enzyme or base catalyzed isomerization process.41 Further dehydration of fructose provides access to the furan platform. These processes are limited by low equilibrium constants (Keq ∼ 1, at 25 °C) and in a typical isomerase-catalyzed process pure glucose is converted into an equilibrium mixture of 42 mol% fructose, 50 mol% glucose and 8 mol% of other sugars. One of the primary focuses of researchers has been to develop robust chemical catalysts for the isomerization of aldoses into the corresponding ketoses. In this section, we have summarized the current developments in both homo and heterogeneous processes. | ||
| Scheme 3 Base-catalyzed and Lewis acid-catalyzed glucose isomerization. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol ratio of glucose to Sn4+ yielded a mixture of 46 wt% glucose, 31 wt% fructose and 9 wt% mannose within 30 min at 110 °C. It was further shown that large pore size and isolated Sn(IV) sites located within the porous network facilitated the isomerization process, whereas sulfonic acid functionalized Brønsted acidic zeolites mainly yielded alkylglucopyranosides. The Sn-beta catalyst could be recycled up to 5 times and worked even in highly acidic mediums.48 This catalyst could also be used for the isomerization of lactose to lactulose, with a 93% selectivity, using 1 wt% solution of lactose and a 20:1 mol ratio of lactose:Sn at 100 °C.50
:1 mol ratio of glucose to Sn4+ yielded a mixture of 46 wt% glucose, 31 wt% fructose and 9 wt% mannose within 30 min at 110 °C. It was further shown that large pore size and isolated Sn(IV) sites located within the porous network facilitated the isomerization process, whereas sulfonic acid functionalized Brønsted acidic zeolites mainly yielded alkylglucopyranosides. The Sn-beta catalyst could be recycled up to 5 times and worked even in highly acidic mediums.48 This catalyst could also be used for the isomerization of lactose to lactulose, with a 93% selectivity, using 1 wt% solution of lactose and a 20:1 mol ratio of lactose:Sn at 100 °C.50
Riisager and coworkers compared the catalytic activities of commercially available Y, beta, ZSM-5 and mordenite zeolites for a two-step isomerization of glucose into fructose in methanol. In the first step, glucose is isomerized into fructose; the fructose formed in situ is converted into methyl fructoside to prevent further reactions at the carbonyl center. Then, in the second step, the fructoside is hydrolyzed by water to obtain fructose.51 A high yield of 55% fructose was observed using large pore H-USY zeolite in methanol, (Si/Al = 6) after 1 h reaction at 120 °C. The presence of aqueous impurities lowered the yield of fructose in these processes. Davis et al. explored the effects of solvent on the catalytic activity of large pore beta zeolites containing Ti4+ ions and found that in methanol glucose predominantly isomerizes into sorbose via intramolecular C5–C1 hydride shift.52 In contrast, in aqueous media, fructose was obtained as the major product via a C2–C1 hydride shift. Davis and coworkers also showed that the stereochemistry remains preserved at the non-participating carbon centers, yielding 73% enantiomeric excess of sorbose and 85% enantiomeric excess of D-fructose from optically pure D-glucose of 92% enantiomeric excess.
For large scale chemical conversions, heterogeneous catalysts are often preferred over homogeneous systems, but the development of Lewis acidic zeolite catalysts for the isomerization of sugars in aqueous media is still in its infancy. A few enzymatic isomerization processes have been commercialized, but are hampered by a narrow range of working conditions (pH, temperature) and the need for high substrate purity, which result in high process costs.53 For facile synthesis of platform chemicals via isomerization of aldoses, the one-step dehydration of the resulting ketose is highly desirable. A one-pot reaction, which enables both isomerization and dehydration cannot be achieved with acid-sensitive isomerases. However, more robust chemical catalysts may be able to withstand the necessary acidic conditions for a one-pot approach.
4.3. Glucose epimerization
Epimerization is the process where chirality of only one of the multiple stereogenic centers in a molecule is changed. Hence, epimers are diastereomers which differ from one another in their configurations at only one chiral center. Epimerization is widely applied for the synthesis of the rare sugars from commonly available epimers, such as L-ribose from L-arabinose, D-lyxose from D-xylose, and D-mannose from D-glucose. These monosaccharides are used as antidiabetics and immunosuppressants in the pharmaceutical industry. Large scale processes are generally catalyzed by epimerases. These microbial and enzymatic processes are equilibrium controlled; in a typical glucose epimerization process a thermodynamic mixture of 2.3:1 mol ratio of glucose to mannose is produced. The enzymatic processes are, however, expensive and require precise reaction conditions. In effect, researchers are developing processes with chemical catalysts that can operate under a wider range of conditions and allow easier recycling, in order to address the issue of cost.
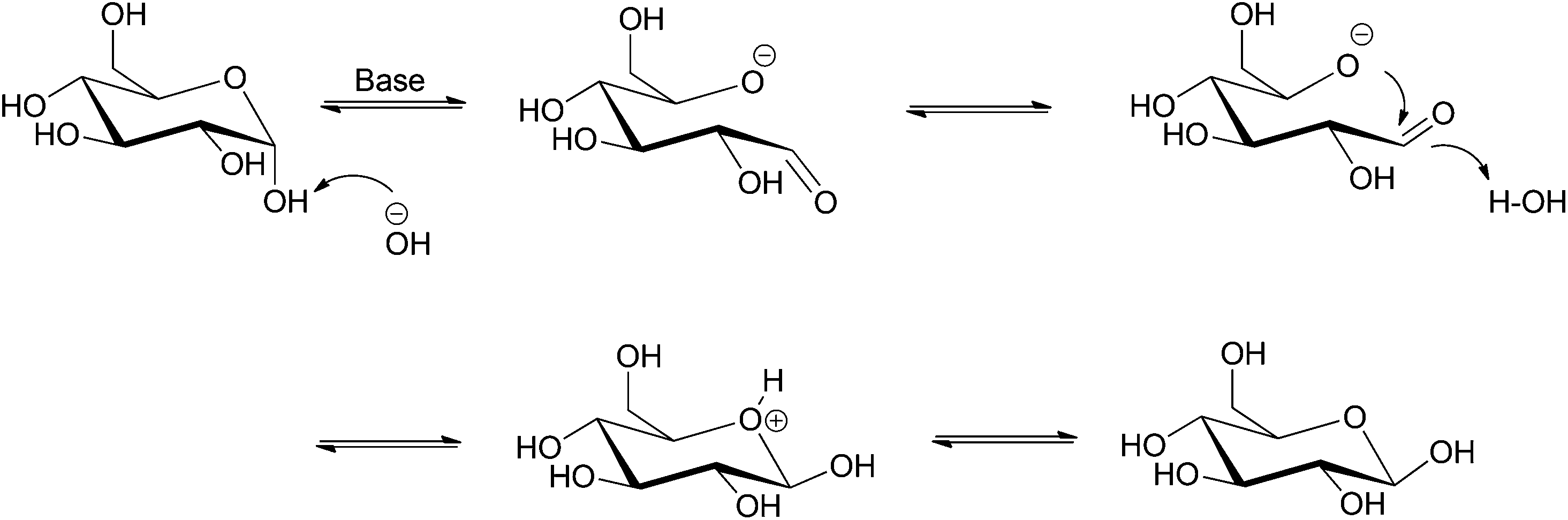 | ||
| Scheme 4 Base-catalyzed epimerization of glucose. | ||
:84, within 1 h, at 85 °C.58 Molybdenum-based polyoxometalate catalysts of the molecular formula Ag3PMo12O40, H3PMo12O40, Sn0.75PMo12O40, have recently been reported by Nikolla and coworkers for the epimerization of aldoses, with high reactivity and selectivity in aqueous media.59 Using H3PMo12O40 and a 10 wt% aqueous solution of the aldoses with a mol ratio of 1:50 for Mo: aldose, the observed conversions were 31% for glucose, 73% for mannose with ∼90% selectivity to the corresponding monomer, after 1 h of reaction at 100 °C.
Due to the overall high production cost of the biochemical processes, more research has been directed to the development of highly active chemical catalyst and the use of biocompatible metal centers instead of Sn4+ will also improve the chances of wide scale application of these catalysts in pharmaceutical industry.
4.4. Oxidation of mono- and disaccharides
Oxidation of the mono/disaccharide aldehyde functionality to a carboxylic acid produces useful chemical intermediates (see Scheme 5). The biodegradable C6 acid platform chemicals such as gluconic acid, glucaric acid and 2-keto-gluconic acids, are routinely used in the food, pharmaceutical, and paper industries. Due to the hydrophilic nature of sugars these transformations are generally carried out in aqueous media using a continuous flow of air as the oxidant. | ||
| Scheme 5 Oxidation of glucose into C6 acids. | ||
Oxidation of biomass-derived disaccharides, such as sucrose, has been well-known since early 1980s.68 In an early report, Van Bekkum et al. used NaIO4, KIO4, and NaOBr as oxidants to convert sucrose into sucrose carboxylate salts. These carboxylates could be potentially used to sequester group 2 metal cations from aqueous solutions. Selective electrocatalytic oxidation at the C-1 or C-6 centers of sucrose has also been reported69,70 and will not be discussed in the current review. The disaccharide unit remains intact during electrocatalytic oxidation.
A more recent development in this field is the use of sodium hypochlorite and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) to generate the oxidant nitrosonium ion in situ.71 In this process, both mono- and disaccharides can be oxidized with 60–90% yield at relatively low temperature (<5–15 °C) and low catalyst loading (0.5 mol% TEMPO, 2 mol% peracid). The oxidant used in this process, however, must be regenerated, using secondary oxidizing agents such as NaOCl/NaBr, NaOCl, peracetic acid, or oxone (potassium monoperoxypersulfate).72 The use of strong secondary oxidants and halogenated reagents limits the practical application of this system. A wide range of studies have been directed towards finding environment friendly method to regenerate the oxidant.73,74
| Oxidized product | Catalyst | Reaction condition | Conversion (%) | Selectivity (%) | References |
|---|---|---|---|---|---|
| Gluconic acid | Pd/C, 2 wt% | 60 °C, pH 9, 7 h | 95 | 96 | 80 |
| Gluconic acid | Pt/C, 5 wt% | 60 °C, pH 9, 7 h | 85 | 77 | 80 |
| Gluconic acid | Pd/Al2O3 | 50 °C, pH 9 | 100 | 95 | 81 |
| Gluconic acid | Pd–Bi/C, 5 wt% | 40 °C, pH 9 | 99 | 99.8 | 82 |
| Gluconic acid | Ru/C, 5 wt% | 50 °C, pH 9.5 | 28.6 | 26 | 84 |
| Gluconic acid | Ru–Pd–Bi/C, 5 wt% | 50 °C, pH 9.5 | 44 | 36 | 84 |
| Gluconic acid | Au/activated C, 0.9% | 100 °C, pH 9.5 | 99 | 95 | 88 |
| Gluconic acid | Au/Al2O3 | 50 °C, pH 9.5, 5–10 d, | 71 | 100 | 94 |
| Gluconic acid | Au/Al2O3, 0.3 wt% | 50 °C, pH 9.5, 1 equiv. H2O2 | 99 | 99 | 94 |
| Glucaric acid | Pt/C | 4–65 °C, pH < 11.0 | 55 | 57 | 104 |
| Glucaric acid | Pt–Au/TiO2 4 wt% | 119 °C, 28 bar O2 | 71 | 45 | 105 |
Several parameters, which affect the conversion of glucose to gluconic acid, have been studied. These include: catalyst deposition pathways, catalyst particle sizes, various types of catalyst supports, the rate of oxygen diffusion, pH of the solution, and glucose to catalyst ratios.19,76 Gallezot et al. showed that the use of an oxophilic Bi promoter with supported Pt and Pd catalysts prevents catalyst deactivation due to O2 binding and over oxidation of the metal center, thereby enhancing the overall catalytic activity.82 Following this approach a wide variety of heterogeneous catalysts83–87 with Bi, Te, and Tl promoters, i.e. Pd–Pt–Bi/C,83 Ru–Pd–Bi/C,84 Pd–Te/SiO2 and Pd–Te/Al2O3,85 have been employed for the selective oxidation of glucose into gluconic acid. Au catalysts supported on activated carbon, silica, metal oxides, polymers, cellulose also have shown potential towards large scale commercial application.88–97 The Au systems are often more active at low catalyst loadings but have an increased propensity towards catalyst leaching during the successive recycling steps. Further studies have been directed to improve catalyst longevity and recyclability.98–100
The heterogeneous oxidation of glucose to 2-ketogluconic acid101,102 and glucaric acid103–105 is often limited by side reactions such as C–C bond cleavage, which leads to a mixture of degradation products. The catalyst systems generally employed are Pt/C, Pt–Pb/C and Pt–Bi/C for the synthesis of 2-ketogluconic acid, while Pt/C, Pt/SiO2, and Pt–Au/SiO2 are used for the synthesis of glucaric acid. The main limitations of this process are the lower yields of the oxidized products and their separation from the byproducts generated by the degradation of glucose. In an alternative approach, Baiker and coworkers investigated the oxidation of L-sorbose into 2-ketogluconic acid. The use of tributylphosphine or hexamethylenetetramine promoters, in combination with Pt/C catalysts showed up to 95% selectivity towards 2-ketogluconic acid formation, at 30% conversion of L-sorbose.106,107
The oxidation of other pentose and hexose sugars also provides access to the aldonic acid platforms; aldonic acids are ubiquitous in the food, personal care, and pharmaceutical industries. Among the series of heterogeneous catalysts investigated for the selective production of aldonic acids, solid supported monometallic Au and bimetallic Au–Pd catalysts showed highest reactivity, with selectivity towards aldonic acids formation reaching nearly 100%.108–110 The oxidation of disaccharides, such as lactose, maltose and sucrose, by heterogeneous catalysts has also been thoroughly investigated.19,71,111 Under the reaction conditions commonly followed, the oxidation selectively occurs at the primary hydroxyl groups. The mono-, di-, and tricarboxylated products (1, 2, 3) commonly generated in this process are shown in Fig. 2. The selective monocarboxylation of sucrose over di112- or tricarboxylation73,113 has also been achieved by suitable modifications of the heterogeneous catalyst systems employed.
 | ||
| Fig. 2 Observed mono-, di- and tricarboxylation of sucrose. | ||
The selective oxidation of lactose into lactobionic acid and 2-ketolactobionic acid, as shown in Fig. 3, has garnered significant attention. Reactivity of a wide range of monometallic (Ru, Ni, Pd, Pt, Au) and bimetallic (Pd–Bi, Pt–Bi and Au–Pd) catalysts on various catalyst supports (activated carbon, Al2O3, SiO2 and CeO2) have been investigated for lactose oxidation.19,76,79 The two-step oxidation of lactose, on supported Pd and Pd–Bi/C catalysts at pH 7 can produce 2-ketolactobionic acid via lactobionic acid formation.114,115 However, lack of selectivity (≤80%) and limited conversion (50–70%) are often encountered in this reaction. The Au catalysts supported on SiO2 and CeO2 show nearly 100% selectivity towards lactobionic acid (at 65 °C, pH 9), with highest percent conversion of lactose.116,117 In contrast, the second oxidation step in which lactobionic acid is converted into 2-ketolactobionic acid is often less selective.118
 | ||
| Fig. 3 Lactobionate derivatives obtained after the oxidation of lactose. | ||
In an alternative approach various types of carbohydrate-based biomass were converted to formic acid, under mild conditions using a Keggin-type polyoxometalate (H5PV2Mo10O40) catalyst. The oxidation processes were carried out in aqueous media, using 30 bar O2, at 80 °C. For simple hexoses, almost 98% conversion was observed; isolated yields of formic acid were approximately 50%.119–122 Significant breakthroughs have been reported although both the product selectivity and reaction rate need to be improved for wide scale application of this process.
4.5. Furan oxidation
Furans can easily be oxidized to obtain a wide range of useful chemicals and chemical intermediates. The production of furans is achieved by sugar dehydration and is described in detail in section 4.6.One of the most important platform chemicals which is produced by oxidation of furans is 2,5-furandicarboxylic acid (FDCA), as shown in Scheme 6. FDCA is being targeted as a biorenewable replacement of terephthalic acid, a monomer used for the synthesis of plastic polyethylene terephthalate (PET). Experts believe that replacement of terephthalic acid with FDCA can reduce carbon emissions significantly.123 Direct synthesis of FDCA from sugars or biomass is difficult, and FDCA is most commonly obtained, in excellent yields, by oxidizing 5-hydroxymethylfurfural. However, it is important to note that HMF is also quite expensive to produce industrially. As shown in Table 2, catalytic systems used for furan oxidation often include solid-supported metals with sodium hydroxide under moderate to high oxygen pressure.
 | ||
| Scheme 6 Furan oxidation of HMF. | ||
| Starting material | Product | Catalyst | Yield (%) | Conditions/notes | References |
|---|---|---|---|---|---|
| HMF | FDCA | Au-CeO2 or Au-TiO2 | >99 | 65 °C, water, NaOH, 10 bar O2 | 124 |
| HMF | FDCA | Co(OAc)2/Zn(OAc)2/Br− with Au-TiO2 | 60 | 90 °C, 4.5 h, acetic acid, 1 bar O2 | 126 |
| HMF | HMFCA | Au-Cu on TiO2 support | 69 | 60 °C, 4 h, NaOH, 10 bar O2 | 125 |
| HMF | FDCA | Au-Cu on TiO2 support | 31 | 60 °C, 4 h, NaOH, 10 bar O2 | 125 |
| HMF | DHMTHF | Ru/solid support | 91 | 130 °C, 12 h, NaOH, 27.6 bar O2, 1-butanol–water | 127 |
| HMF | DFF | Ru/C | 84 | 110 °C, water, 20 bar O2 | 128 |
| HMF | MA | VO(acac)2 | 52 | Conv: 99%, 90 °C, 4 h, CH3CN, 10 bar O2 | 129 |
Corma et al. employed ceria-supported gold nanoparticles to convert HMF into FDCA with great success; yields of FDCA were achieved at >99%, although the catalyst recyclability was relatively poor (20% drop in yield over 4 cycles). By changing the catalyst support (iron oxide, titanium oxide, carbon) and reaction times, Corma et al. were also able to tune the gold catalysts to produce either HMFCA or FDCA.124 In a later study, Cavani and co-workers synthesized a bimetallic Au–Cu catalyst on titanium oxide, and was able to improve catalyst robustness.125 Product yield did not decrease after 6 cycles, unlike the monometallic gold catalyst. However, it should be noted that the oxidative ability of the catalyst was reduced significantly as the preferential product produced was HMFCA, not FDCA. Abu-Omar et al. also used Au-TiO2 particles, but also added a homogeneous co-catalyst, Co(OAc)2/Zn(OAc)2/Br− and trifluoroacetic acid.126 Using this combination of catalysts and acids, Abu-Omar and coworkers were able to tune the reaction to produce different amounts of DFF (2,5-diformylfuran), FFCA (furan formyldicarboxylic acid) and FDCA by changing the amounts of Zn(OAc)2, bromide, and trifluoroacetic acid added to the solution.
Researchers have also employed other precious metals, besides gold, as catalysts for furan oxidation. Dumesic and coworkers studied Ru, Pd, and Pt on carbon in conjunction with MgO and Al2O3 co-catalysts and their effects on the oxidation of HMF.127 When MgO and Al2O3 were added to the reaction, the selectivity of the reaction improved preferentially towards 2,5-dihydromethyltetrahydrofuran (DHMTHF) in both cases (the yield increased from 45%, when no co-catalyst was present, to 85–90% in the presence of co-catalyst). Cho et al. also employed Ru/γ-alumina catalysts to convert HMF to DFF, with high selectivity and yield.128
There are fewer examples of solely homogeneous catalysis used for furan oxidation. Du et al. studied a series of homogeneous metal catalysts (FeSO4, CuSO4, V(acac)2) and found that the vanadium catalyst was capable of converting HMF into maleic anhydride at 52% yield, although with a large amount of byproducts such as CO2, formic acid, and smaller amounts of oxidized furans such as DFF.129
4.6. Glycosylation of sugars
Glycosylation refers to the formation of an acetal linkage when the carbonyl group (glycosyl donor) present in a sugar unit reacts with an alcoholic –OH moiety (glycosyl acceptor). Glycosylation of fatty alcohols, which are obtained from fatty acids, is a common synthetic strategy used to produce biorenewable alkylpolyglucosides (APGs), one of the most popular surfactants in personal care products and detergents since 1980s.33,130,131 The glycosylation process can be catalyzed by both acid or base. The acid-catalyzed reaction, often used in industrial level, is also known as the Fischer glycosylation,132 which produces an equilibrium mixture of alkyl furanosides (kinetic product), alkyl pyranosides (thermodynamic product), and the desired oligomerized alkyl polyglycosides (see Scheme 7). In a typical two-step process, butanol participates as both solvent and reactant and aids the miscibility of the long chain alcohols.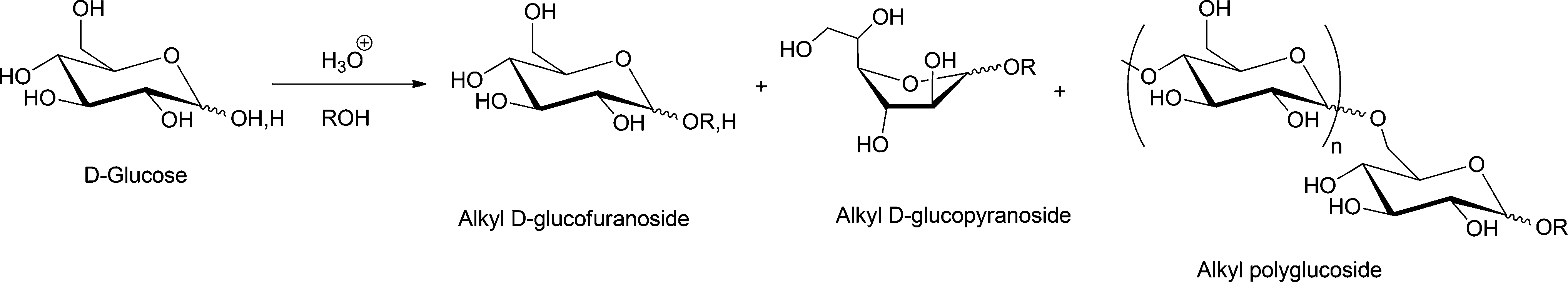 | ||
| Scheme 7 Commonly observed products in Fischer glycosylation reactions. | ||
Base-catalyzed Koegins–Knorr glycosylation processes generally involve the activation of monosaccharides at the anomeric carbon center, while rest of the hydroxyl groups present in the sugar molecule are completely or partially protected (see compound 4, Scheme 8).19,133,134 The base-catalyzed processes are highly selective towards conversion at the anomeric carbon, to yield the corresponding alkyl D-glycofuranoside 5.
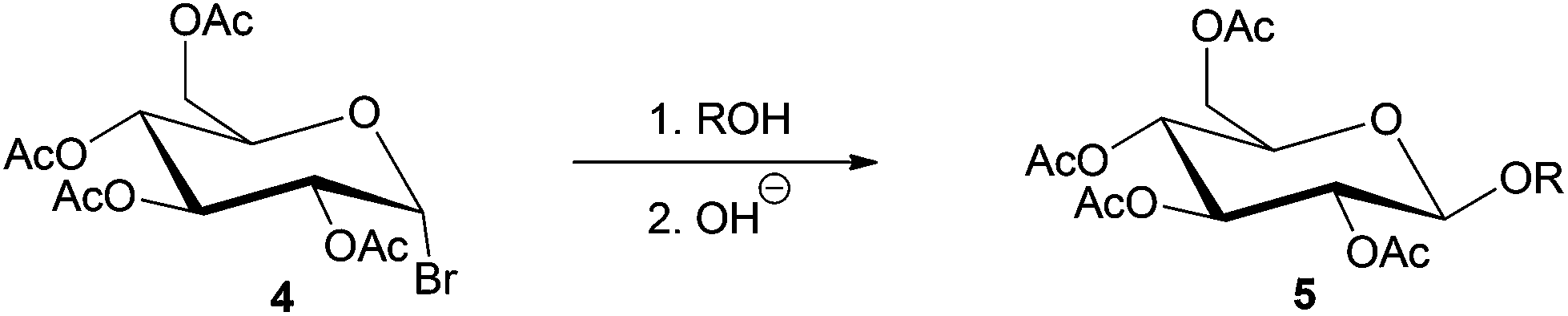 | ||
| Scheme 8 Stereoselective synthesis of glycosides by Koenigs–Knorr glycosylation (R = alkyl group).19 | ||
Boronic acid or metal catalyzed glycosylation138,139 of protected monosaccharides has been further extended to catalytic stereo- and regioselective monoglycosylation of C-6 polyols, to obtain oligosaccharides, as shown in Scheme 9.140–143 Using Ph2SnCl2 (10 mol% relative to sorbitol) catalyst, 99% conversion of sorbitol to the glucoside 7 was observed.140
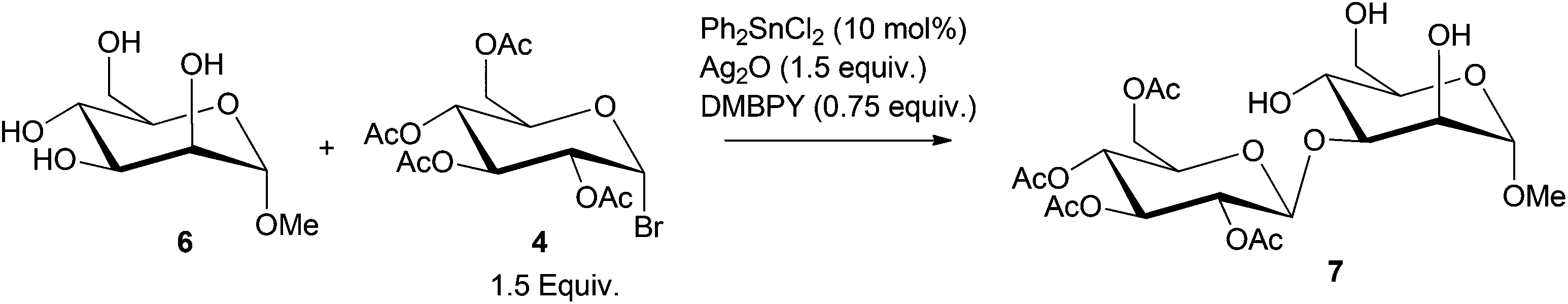 | ||
| Scheme 9 Monoglycosylation of C-6 polyols, using Sn(IV) catalysts. | ||
An “open glycosylation” process which allows regio-controlled substitution without extensive protection–deprotection steps is a highly attractive alternative route for commercial production of the glycosides. Moitessier and coworkers have prepared a H-bonding protecting group, which influences the overall rate of conversion of glucose into the glycosides.142,144
In a recent study, an organocatalyzed direct glycosylation of unprotected and unactivated monosaccharides was reported by Mahrwald and coworkers (see Scheme 10).145 These reactions were carried out in isopropanol, which is also the glycoside acceptor, while using a catalyst system comprising of 10 mol% of PPh3 and 10 mol% of carbon tetrabromide. The yield observed in these reactions depends on the monosaccharide used. The authors report 77% conversion of ribose and 75% conversion of deoxyribose, whereas the percent conversion of arabinose and xylose were very low. Additionally, because the products are difficult to purify and these homogenous catalytic systems are hard to recycle, it is challenging to scale up these processes for practical applications.
 | ||
| Scheme 10 Organocatalyzed direct glycosylation of unprotected and unactivated monosaccharides. | ||
Enzymatic glycosylation of alcohols using glycosyltransferases have been widely applied and these processes offer the advantages of ambient reaction conditions, regio or stereoselective glycosylation without extensive protection and deprotection steps.141 However, the need to use highly pure substrates, and waste water treatment add up to the cost of enzyme catalyzed glycosylation. More research is thus addressed to the limitations observed in chemical conversion processes- the use of large excess of alcohols, the need to carry out the reactions at high temperature in the presence of mineral acids, and the stability and recyclability of zeolites in aqueous media. Although significant research has been focused on the chemical synthesis of alkyl-glycosides, due to these inherent limitations, enzymatic glycosylation still remains the preferred method of synthesis for these materials.164
4.7. Reduction of sugars: catalytic hydrogenation
Reduction of carbohydrate carbonyl groups by metal catalyzed hydrogenation provides access to the polyol platform (see Scheme 11). Polyols, such as sorbitol and mannitol, are widely used as additives in various food and personal care products. Every year approximately 8 × 105 metric tons of sorbitol is produced for commercial applications.165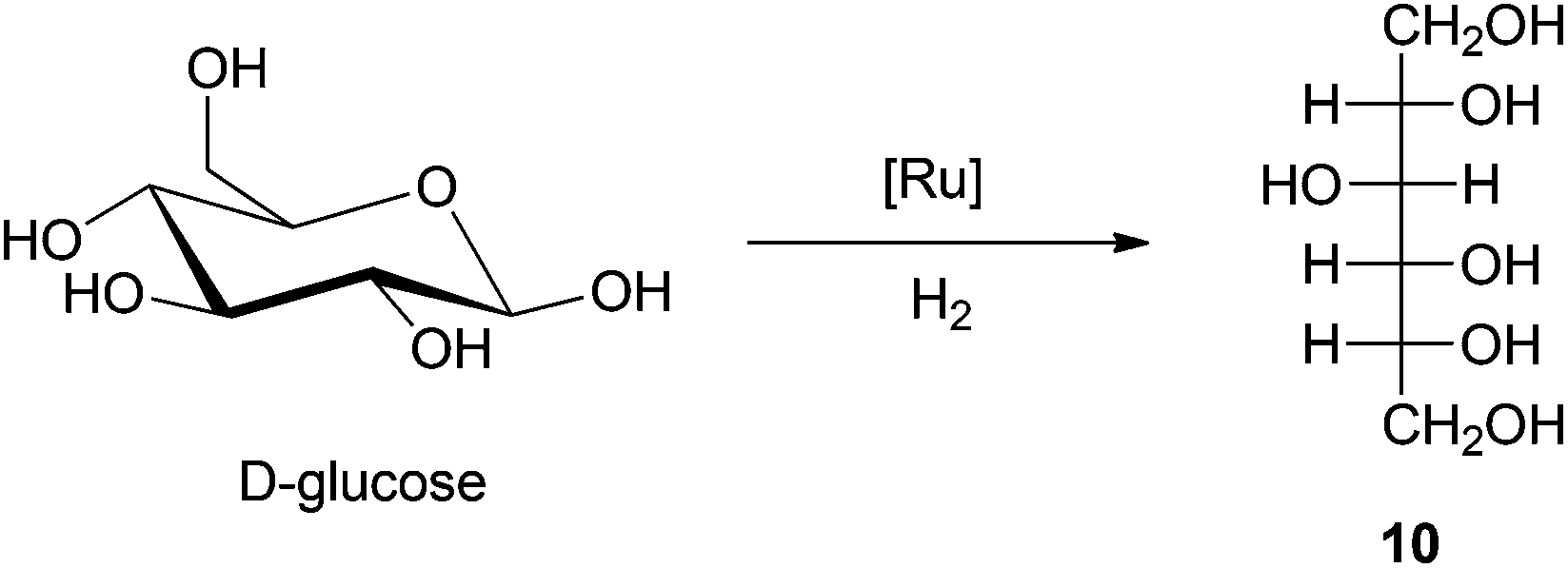 | ||
| Scheme 11 Conversion of D-glucose to sorbitol via hydrogenation. | ||
In another study, Yang also used a ruthenium catalyst, RuCl3, in the presence of hydriodic acid and hydrogen gas, to convert sugars (xylose, glucose, fructose) into tetrahydrofuran derivatives (2,5-dimethyltetrahydrofuran and 2-methyltetrahydrofuran), which can be used as biofuels.170–172 In an in-depth study of the mechanism, Yang and Grochowski found that both the acid (HI) and the catalyst (rhodium chloride) play important roles in the dehydration and hydrogenation of fructose.173 Additional processes which yield biofuels from sugars and carbohydrates have been covered in a recent publication by Dutta et al.174
| Starting material | Product | Catalyst | Yield (%) | Conditions | References |
|---|---|---|---|---|---|
| Fructose | 5-Methylfurfural | RhCl3 in hydriodic acid (HI) | 64 | 90 °C, 1 h, toluene–water biphasic system, 20.6 bar H2 | 184 |
| Xylose | Xylitol | Ru on TiO2–NiO2 support | 99.8 | 120 °C, 120 min, deoxygenated water, 55 bar O2 | 185 |
| Glucose | Sorbitol | Pt nanoparticles on activated carbon cloth | 99.5 | 100 °C, water, 80 bar H2 | 181 |
| Fructose | Mannitol | Ru/C | 40 | 100 °C, 20 h, water, 100 bar H2 | 186 |
| Glucose | Sorbitol | Ru/MWNT | 62.5 | 100 °C, 120 min, water, 40 bar H2 | 187 |
4.8. Furan reduction: catalytic hydrogenation
Furans can also be hydrogenated to yield a wide range of useful chemicals and chemical intermediates. Synthesis of furans is described in the following section. A recent review, by Nakagawa et al., summarizes the various hydrogenation techniques used for furan reduction.188 Catalytic systems for furan hydrogenation typically involve heterogeneous catalysts (Cu–Cr, Ni/O/SiO2, Pd/SiO2, etc.); conversions are high (generally >99%) although product identity and yields vary (10–80%), depending on the catalytic system and the furan starting material used.4.9. Dehydration of sugars
The dehydration of monosaccharides can produce a number of highly desirable platform chemicals; among them furfural (FUR), 5-hydroxymethylfurfuryl (HMF), levulinic acid, and lactic acid. A scheme outlining the furan platform, which includes the compounds FUR, HMF, and LA, is included (Scheme 12). These chemicals and/or their immediate derivatives have a huge potential for use in a wide array of applications. However, product selectivity and product purity are the primary barriers to using biomass as a source for these chemicals; in effect, researchers are very actively addressing these issues by using a variety of different catalytic systems to combat these issues. | ||
| Scheme 12 Furan platform from carbohydrates. | ||
| Category | Catalytic systems | Common Examples | Ref. |
|---|---|---|---|
| Heterogeneous | Sulfonated acid catalysts | HZM, SBA-15 (propylsulfonic acids), Nafion catalysts | 187,194,195,204,225–232 |
| Metal salts on solid supports | Pd, Ru, Rh, Cu on Carbon, TiO2, or in sol–gel systems | 124,127–129,196,210,230,231,233–238 | |
| Zeolites | Mordenites | 200,239–241 | |
| Aluminosilicate clays | HT (hydrotalcite) | 42,242,243 | |
| Homogeneous | Mineral acids or Brønsted Acids | HCl, H2SO4, HI, HBr, HNO3 | 203,224,244–246 |
| Water-soluble Lewis acids | Al2O3, CrCl3 | 126,129,173,184,224,247–251 | |
| Ionic Liquids | Ionic liquids only | [BMIM][Cl], [EMIM][Cl] | 252–256 |
| Ionic liquids and homogeneous catalysts | IL and IL-soluble metal salts | 229,257–260 | |
| Ionic liquids and heterogeneous catalysts | IL and sulfonated acid catalysts, metal salts on solid supports, zeolites, clays | 234,261 |
Researchers have found that by using biphasic acidic systems (another homogeneous catalytic system), they are able to improve the yield of both FUR and HMF dramatically.189 Biphasic systems commonly include an acidic aqueous layer, and an organic layer (MIBK, THF, DMSO, etc.). The furan dissolves into the organic layer, reducing the contact time of the acid-sensitive furan with the acid, leading to a reduction in byproduct formation and helps maintain overall reaction yield/selectivity. Metal salts, such as CrCl3 and AlCl3·6H2O, in biphasic systems also serve as effective catalysts.
Levulinic acid (LA) is a C5 compound that can be synthesized from glucose, fructose, or 5-hydroxymethylfurfural (5-HMF), as shown in Scheme 12. Because of its abundance and lower cost, glucose is the preferred starting material, which is a similar case for HMF. LA can also be synthesized from cellulosic biomass, which will be discussed in section 6.1. Levulinic acid is an extremely versatile platform chemical that can be further modified to produce organic solvents (γ-valerolactone (GVL)),190–197 fuels (2-methyltetrahydrofuran (2-MTHF)),198 food flavorings and fragrances (ethyl levulinate (EL)),199–203 chemical intermediates (succinic acid),204 bisphenol acids,205–207 and plasticizers (diols).208–210 Generally, the open-chain form of glucose undergoes dehydration, losing two waters to form HMF. The HMF intermediate is ultimately hydrolyzed and eliminates formic acid to form levulinic acid. Fructose yields the same products, although the reaction takes place via a cyclic route. A detailed reaction mechanism for the synthesis of levulinic acid has been provided in an excellent review written by Rackemann and Doherty.211
Levulinic acid has been synthesized in good yields (up to 83%) using mineral acid catalysts, such as sulfuric acid and hydrochloric acid, using relatively mild temperatures ranging from 95–240 °C.211 These mineral acids can then be removed from the product via vacuum distillation and steam stripping. Mineral acids, however, are highly corrosive and require special processing equipment.212 The toxicity of mineral acids has prompted some investigation into heterogeneous catalysts as possible replacements – however, both conversion and yields obtained thus far are poor.
Lactic acid is another product of glucose/fructose dehydration. Lactic acid serves as the building block of poly(lactic acid), the second largest volume bio-plastic.213,214 Currently, fermentation is the primary industrial process used to synthesize lactic acid.214 While the fermentation process can produce calcium-lactate salt (a precursor of lactic acid) in high yield and selectivity (95%), the multi-step workup is complex and expensive. In the past few years, a handful of new chemical routes have been explored although a majority of research is still focused on fermentation. Metal salts, including Al3+ and Cr3+, and lanthanide triflates have also been used to convert glucose and sucrose, a disaccharide, to lactic acid, albeit with lower yields (20–50%).215,216
Heterogeneous catalysts which have been extensively studied for HMF and FUR production include solid-supported metals, aluminosilicates, zeolites, and amberlyst acidic resins.218 These solid acid catalysts are capable of converting 50–99% of sugar dehydration, and yields up to 82% product; reaction temperatures are mild, and generally fall under 200 °C, as summarized in Table 5. Biphasic systems are commonly used to ease separation of the product from the catalytic system.184 Solvent choice varies; while there is a “green” impetus to use water, the reactivity of HMF and FUR with water usually requires the dimethylsulfoxide or poly(vinylpyrollidone) additives to suppress the byproduct formation of polyketones and insoluble humins.17
| Starting material | Product | Catalyst | Yield (%) | Conditions/reaction | Ref. |
|---|---|---|---|---|---|
| Xylose | FUR | AlCl3/[BMIM][Cl] | 82 | 160 °C, microwave | 262 |
| Fructose | HMF | CSS/[BMIM][Cl] | 83 | 80 °C, 10 min, microwave, CSS: carbonaceous sulfonated carbon | 261 |
| Glucose | HMF | AlCl3 | 68 | Conv: 91%, 170 °C, 40 min, NaCl saturated water, HCl | 263 |
| Glucose | HMF | [EMIM][Cl] | 41 | Conv: 95%, 120 °C, 3 h, boric acid | 260 |
| Fructose | HMF | [EMIM][Cl] | 78 | Conv: 95%, 120 °C, 3 h, boric acid | 260 |
| Sucrose | HMF | [EMIM][Cl] | 66 | Conv: 95%, 120 °C, 3 h, boric acid | 260 |
| Fructose | HMF | DVB–SO3H/[BMIM][Cl] | 93 | 75 °C, 20 min, DVB–SO3H: sulfonated divinylbenzene macroporous strong acid gel | 261 |
| Glucose | HMF | PTA/MIL-101/[EMIM][Cl] | 2 | Conv: 21%, 100 °C, 3 h, PTA/MIL-101: phosphotungstic acid H3PW12O40/chromium–carboxylate cubic structure (MOF) | 264 |
| Fructose | HMF | PTA/MIL-101/[EMIM][Cl] | 63 | Conv: 84%, 80 °C, 60 min | 264 |
| Fructose | HMF | Sn-β | 71 | Conv: 89%, 130 °C, 9 min, GVL–water, microwave, Sn-β: zeolite | 265 |
| Glucose | HMF | Sn-β, Amb-70 | 63 | Conv: 90%, 30 min, GVL–water, microwave | 265 |
| Fructose | HMF | GTS | 99 | Conv: 99%, 160 °C, 60 min, THF–DMSO, GTS: sulfonated carbonaceous material | 266 |
| Fructose | HMF | FSIL | 81 | 100 °C, 60 min, H2O–MIBK, FSIL: fiber supported ionic liquid, [PPFFy][HSO4−] | 267 |
Heterogeneous Sn-zeolites have been used to convert sucrose to methyl lactate in methanol with a 60% yield.219 Another heterogeneous catalyst, a polymer composed of imidazole and epichlorohydrin units, has been used to convert glucose into lactic acid at 63% yield and 99% conversion. It is not, however, possible to extract the polymer catalyst from the reaction, and subsequent recycling of the polymer eventually reduces its activity; a decrease from 60 to 50% yield of lactic acid is observed over six cycles.220
One major problem often encountered in using heterogeneous catalysts is a lack in robustness. Furan yields typically fall after the catalysts are reused, although recent studies have shown improvements in catalyst recyclability.221 Post-reaction SEM images and XRD analyses of the catalysts, typically show a change in pore size for acidic resins or nanoparticle growth for solid supported metal catalysts.200 As such, a significant portion of biomass conversion studies are dedicated to improving the robustness and to lengthen the lifetime of these catalysts.
4.10. Esterification of sugars
Fatty acid esters of mono- and disaccharides are an important class of biodegradable emulsifiers, surfactants and low calorie substitutes for edible fat.268 The sorbitan esters find application as non-toxic, non-irritating emulsifiers in the food, pharmaceutical, and cosmetic industries. Reduced monosaccharides, such as sorbitol can also undergo esterification reaction with fatty acids under and base or acid-catalyzed condensation conditions.269 An example of this process is depicted in Scheme 13, where acetyl-protected sorbitol 11 is converted to the corresponding stearate ester 12, under acid catalyzed condition. Alternatively, transesterification of fatty acid esters with sugars or reduced sugars can also produce the corresponding sugar esters.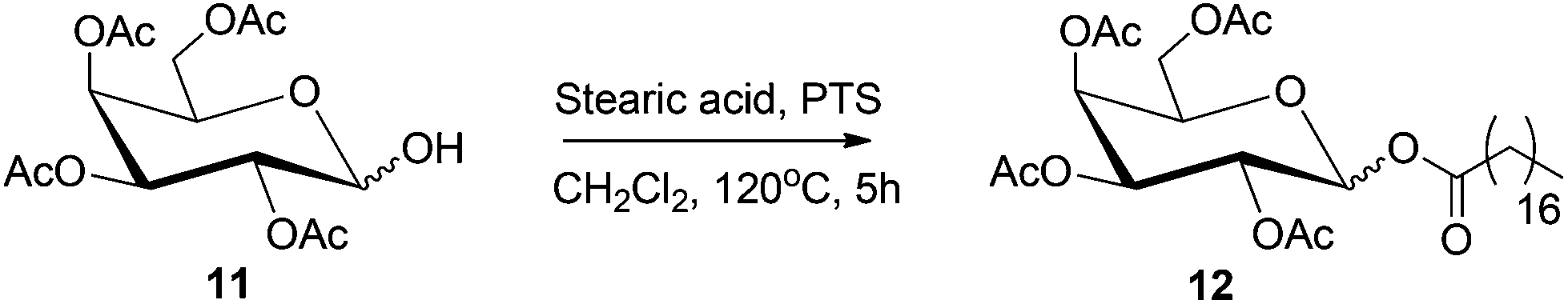 | ||
| Scheme 13 Acid catalyzed esterification of protected sorbitol. | ||
At high temperatures, sorbitol undergoes acid-catalyzed dehydration reaction to produce a mixture of anhydrides, 1,4-sorbitan, 2,5-sorbitan, and isosorbide. Consequently, acid catalyzed esterification reaction produces an equilibrium mixture of mono-, di-, and triesters of various anhydrides of sorbitol. To avoid the complex product distribution, a two-step esterification has often been adopted. In the first step sorbitol is dehydrated to sorbitan by concentrated phosphoric acid, at 180 °C, followed by the esterification of sorbitan with fatty acid at a higher temperature.270
Esterification of sucrose can also be carried out using base catalysis, although high temperatures (150–200 °C) and highly polar solvents, such as DMF or DMSO, are required to dissolve sucrose.276,277 Esterification with acyl chlorides following Mitsunobu reaction condition has also been reported by Tate et al.278,279 Selective esterifications at 6,6′, 2,2′ or 3,3′ positions have also been reported using N-acylthiazolidine thiones or metal complex reagents.280
Transesterification of fatty acid esters with sucrose has been observed by employing cyclic guanidine 4,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) supported on mesoporous silica as catalyst.282,283 By using an excess amount of fatty acid methyl ester (4 equiv.) with respect to glucose, a 92% conversion of glucose with a preferential formation of the monoesters was observed. At 110 °C in DMSO, the ratio of mono- and diesters obtained was 69:14.
In most cases, the enzymatic synthesis of sugars esters is preferred over chemical synthesis because of the higher regio-and stereoselectivity of the former approach. Additionally, the chemical routes often require the use of organic solvents and produces colored products. The resulting product must go through an additional bleaching or purification step before it is ready for further use. Thus, the development of new chemical processes involving less toxic chemical reagents is necessary for sugar ester synthesis.
4.11. Etherification of sugars
Amphiphilic ethers of mono- and disaccharides, containing long hydrophobic alkyl chains, are an emerging class of biodegradable, biorenewable, non-ionic surfactants.284,285 Amphiphilic short chain mono- and dialkyl ethers of isosorbide have also found application in waterborne paints.285 Sodium salt of dodecyl isosorbide sulfate is used as a new class of biorenewable anionic surfactants.286 The sugar-based C6-polyol platforms are commonly obtained by the reduction of hexoses (e.g. isosorbide, isomannide, isoidide) and disaccharides (e.g. sucrose), as discussed in 4.5. of this review. These polyols are further converted to their corresponding ethers by using both homogeneous and heterogeneous catalytic techniques.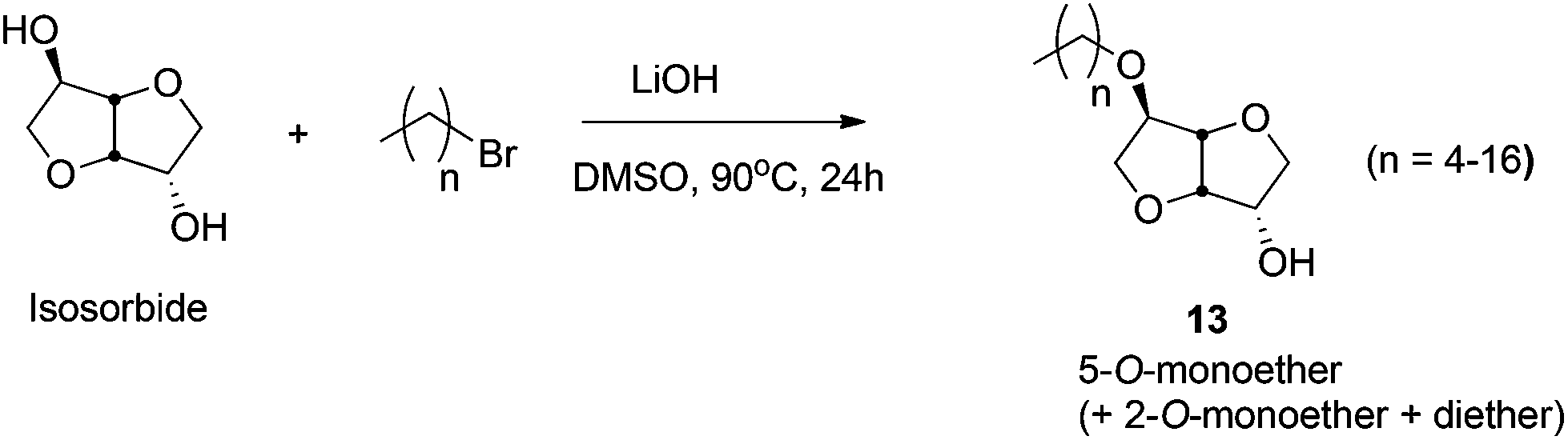 | ||
| Scheme 14 Conversion of isosorbide into amphiphilic ethers. | ||
Isosorbide has also been further functionalized to obtain O-alkylated or O-arylated β-iodoethers by Berini and coworkers.289 This approach follows iodoetherification of isosorbide derived glycals with a series of O-nucleophiles, in the presence of n-iodosuccinimide. Iodine is then removed from the resulting trans-iodo ethers and acetates to obtain the isosorbide-ethers with moderate to good yield. Additionally, aryl and fluorinated enol ethers of sorbitol and pentaerythritol have recently been synthesized.290,291 Hexose sugar and isosorbide based crown ethers, containing the monosaccharide unit in conjugation with a macrocyclic ring, have been studied by Rapi and coworkers.292 These materials have been applied as chiral ligands and auxiliary in various asymmetric transformations.
The homogeneous etherification of sucrose is generally carried out in water or highly polar organic solvents such as DMF or DMSO, at moderate temperature (100–110 °C). Ring opening of epoxides can be carried out by sucrose using tertiary amines or N-methyl imidazole as base, in the presence of a surfactant such as cetyltrimethylammonium bromide (CTAB) at 100 °C.293,294 The surfactant promotes miscibility of hydrophobic long chain epoxides and hydrophilic sucrose. Drawbacks of this approach are poor the miscibility of hydrophobic and hydrophilic reactants and the resulting low to moderate yields (20–60%).
Hill and coworkers carried out etherification of sucrose by telomerization with conjugated dienes such as butadiene, using Pd(acac)2 catalyst at 70 °C.295 The Pd(II) catalyzed telomerization process has been extended to various carbohydrate building blocks (sorbitol, sorbitan, isosorbide, isomannide) and typically produces a mixture of mono- and diethers, (14, 15, 16) as shown in Scheme 15, using isosorbide as the sugar-derivative. It was shown that the addition of 1 M NaOH to the catalyst decreases the amount of byproduct formation. In similar conditions Pd/TPPTS catalyst led to 73% conversion of sucrose, with a 2:1 ratio of mono- and dioctadienyl ethers.296 The effect of reaction conditions, such as temperature, base, concentrations of catalysts and substrates, on the extent of conversion and product selectivity have been studied in details.297–301 In a recent report Molinier et al. carried out 99% conversion of isosorbide into a mixture of monoethers while using 0.2 mol% aqueous Pd(II) catalyst with 0.8% TPPTS ligand, at 80 °C. The observed ratio of 2-O substituted monoether, 14 and the 5-O substituted monoether, 15 was 51:37, with 12% diether formation.298
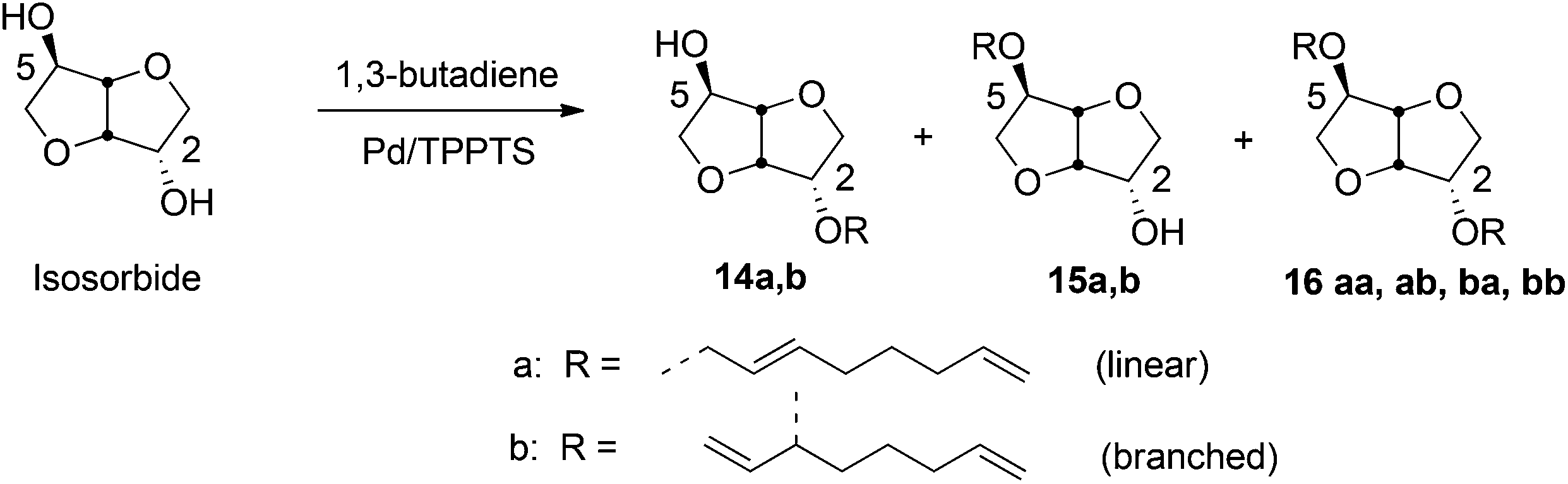 | ||
| Scheme 15 Telomerization of isosorbide with butadiene. | ||
Lipophilic bio-based ethers have found numerous applications in paints, inks, de-icers, etc. The use of expensive Pd catalysts and toxic organic solvents used in their preparation pose a severe limitation for further commercialization processes.
4.12. Hydrogenolysis of sugars and sugar alcohols
Monosaccharides and the polyols obtained after hydrogenation can undergo both C–C and further C–O bond cleavage processes to produce C2 and C3 polyols. Catalytic hydrogenolysis of hexoses produces a range of useful chemical intermediates such as ethylene glycol, glycerol, 1,2-propanediol, ethanol, and propanol. The hydrogenolysis of sugars has recently been reviewed by Palkovits et al.12 and Gallezot et al.76 These reactions are generally carried out in aqueous conditions, often at moderately high temperature (300–350 °C) and high pressure of H2 (7–20 MPa), in both continuous and batch reactors. A basic promoter (NaOH or Ca(OH)2) is often added to enhance catalytic activity by suppressing catalyst deactivation. Supported Ru, Ni, and Pd catalysts favor C–C bond cleavage, a mechanism which produces a mixture of C2 and C3 polyols.304–309 Extensive research has been directed towards optimizing the reaction conditions for hydrogenolysis. This includes further study of the metal catalysts, the use of aqueous and organic solvents, and the investigation of the effects of pH on the product selectivity of the reaction. In a recent study, a series of Al2O3 supported metal catalysts were used for the hydrogenolysis of xylitol. The rate of this reaction, at 200 °C, followed the order Ru > Co > Pt > Ni > Pd.210 The use of multimetallic Pt–ReOx/C, Ir–ReOx/SiO2 catalysts for aqueous phase reduction of sorbitol offers a wide range of product selectivity by producing a complex mixture of gaseous CO2 (40–67%) and reduced products containing C1–C6 alkanes, C2–C6 polyols, mannitol, cyclic ethers (isosorbide, sorbitan), organic carbonyl compounds, and acids.310 Further modifications on the catalyst system and the reaction conditions led to selective formation of cyclic ethers or polyols over gaseous alkanes. Recently the multimetallic Pt–ReOx/C catalyst has also been applied for the dehydroxylation of biorenewable angelica lactone to obtain C7–C10 hydrocarbons with 72% yield.311 Heterogeneous dual CuO–ZnO catalysts are more selective towards dehydroxylation over C–C bond cleavage,312 leading to the formation of C4 and higher molecular weight polyols (e.g. C4–C6 alcohols such as butanol, butanediol, hexanediol, hexanetriol, and hexane tetrols) from mono and disaccharides and reduced sugar alcohols. In contrast the Cu/SiO2 catalyst favors hydrogenolysis of xylitol with 100% conversion and 37% selectivity towards the C2–C3 diol.313The observed product selectivity thus, depends on the reaction conditions and often remains a challenge. In these processes, tailoring of the catalyst system leads to a mixture of compound, instead of a pure product. Further application of the resulting products is thus often limited to usage as bulk feedstocks. The mixtures of C4–C6 polyols have found application in the synthesis of polyurethanes and resins (see Table 6).
| Starting material | Product | Catalyst | Reaction conditions | Conversion (%) | Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|
| Xylitol | C2–C3 glycol | Ni/C | 0.06–1 g Ni/C, 10.5% catalyst loading 0.26 g Ca(OH)2, 200 °C, 40 bar H2, 40 g 10 wt% xylitol aqueous solution, 1 h | 20 | 33 | 309 |
| Xylitol | C2–C3 glycol | Cu/MgO | 0.2 g reduced catalyst, 0.3 g Ca(OH)2, 200 °C, 40 bar H2, 40 g 10 wt% xylitol aqueous solution, 2 h | 41 | 32 | 313 |
| Xylitol | C2–C3 glycol | Cu/SiO2, | 0.2 g reduced catalyst, 0.3 g Ca(OH)2, 200 °C, 40 bar H2, 40 g 10 wt% xylitol aqueous solution, 2 h | 29 | 37 | 313 |
| Sorbitol | C6 polyols | CuO–ZnO | CuO–ZnO (33:65), 180 °C, 130 bar H2 pH 7, 21 wt% sorbitol |
92 | 63 | 312 |
5. Chemical conversions of polysaccharides: starch and cellulose
5.1. Chemical cracking
Chemical conversions of polysaccharides involve an initial deconstruction of the ligninocellulosic backbone, followed by further chemical transformations of the resulting hydrolysates. In some cases, the chemical cracking of polysaccharides, starch, and cellulose can directly yield useful secondary chemicals; for example, it is possible to generate FUR, HMF, and LA – all key chemicals in the furan platform – directly from polysaccharides.314 Chemical cracking is primarily achieved using heterogeneous systems or by using ionic liquids in conjunction with either homogeneous or heterogeneous catalysts.315Mineral acids – non-catalytic conversion media – have also very effectively been used to directly convert raw biomass into levulinic acid in commercialized processes. Researchers have had significant success as yields range between 50–80%.211 By-product formation, however, makes purification of the resulting levulinic acid expensive. Also, a recent publication has reported significantly improved yields of levulinic acid by coupling the use of mineral acids with microwave heating – authors were able to convert 100% of the starting material (cellulose) with a 90% yield of levulinic acid. No additional purification was needed to obtain pure levulinic acid.316 It is also possible to synthesize alkyl levulinates, used for as fragrances, directly from biomass by simply including linear, primary alcohols in the reaction medium.317 Although safety concerns regarding the use of strong acids has prompted researchers to study alternative, heterogeneous systems for the conversion of biomass to levulinic acid, heterogeneous alternatives, thus far, have yielded poorer results.
Commercial production of furfural uses a combination of mineral acids and steam stripping;212 however, a primary drawback of this system is that in the presence of water and strong acid, the furfural can potentially continue to react to form byproducts such as humin.218 As such, researchers are actively investigating how to improve the yield of furfural – and other acid-sensitive furans (HMF) – by using alternative systems (heterogeneous and catalytic ionic liquid systems, see Table 7 for examples of IL systems). Abu-Omar and co-workers used a biphasic system to convert raw biomasses (corn stover, pinewood, poplar, cellulose) to other furfural in 29–66% yields often with HMF as a major side product (yield 6–42%).318
| Starting material | Product | Catalyst | Yield (%) | Conditions/notes | Ref. |
|---|---|---|---|---|---|
| Cellulosic biomass (corn stover) | FUR | AlCl3·6H2O | 51 | Conv: 61% (includes sugar–furan byproducts), 140 °C, 60 min, NaCl, H2O–THF, microwave | 184 |
| Inulin | HMF | Ionic-liquid based polyoxometalate salt (solid catalyst) | 76 | DMSO, 20 °C, 2 h, trace water, microwave | 368 |
| Cellulose | Furans and reduced sugars | CrCl3 | Furans: 58 | Conversion: 88%, 160 °C, 10 min, trace water, microwave | 258 |
| Sugars: 11 | |||||
| Cellulose | Ethylene glycol and polyols | Ru/MC | EG: 59 | Conv: 100%, 30 min, 60 bar H2, MC: mesoporous carbon support | 238 |
| Polyols: 41 | |||||
| Starch | Levulinic Acid | [MIMPS]H2PW12O40 | 49 | Conv: 100%, water, MIBK, [MIMPS]H2PW12O40 is a heteropolyacid ionic liquid | 369 |
| Cellulose | HMF | Zr(O)Cl3–CrCl3 in [BMIM][Cl] | 47 | Conv: ∼82%, DMA-LiCl, 120 °C, 5 min | 222 |
Cellulose can also be converted into lactic acid, using metal chloride salts (AlCl3, CrCl3),215 vanadyl cations (VO2+),319 and lanthanide triflates (Er(OTf)3).320 In a 2013 study, Wan and co-workers discovered that Pb2+ ions were particularly effective (when compared to other metal cations) at converting ball-milled cellulose into lactic acid.321 However, it is important to note that besides the toxicity of the catalyst, the cellulose loading was low (∼1 wt%), and the temperature was relatively high (290 °C).
Few examples of the direct conversion of cellulose to HMF, without the aid of IL, exist. Wang and co-workers employed a micellar, solid heteropolyacid catalyst perforated with surfactant (Cr[(DS)H2PW12O40]3) to hydrolyze 77% of cellulose with a 52.7% yield of HMF. This is an excellent result and comparable to yields obtained by IL systems.323 The presence of the surfactant, dodecyl sulfate (DS), improves both the cellulose hydrolysis and HMF yield significantly – if only the heteropolyacid (Cr[H2PW12O40]3) is used, conversion and yield drop about 15%. It appears that the presence of the DS solubilizes the cellulose, and draws it into the micelle, enabling better adsorption and consequently, better interaction of the cellulose and the HPA catalyst. By-products of the reaction included formic acid, acetic acid, and levulinic acid, enabling an efficient separation (90% HMF extracted) of the less polar and less water-soluble HMF using methyl isobutyl ketone (MiBK) as the extracting solvent. The catalyst was also easily extracted and could be recycled with only a slight drop in activity over 6 cycles.
Researchers have dedicated a significant amount of time to finding heterogeneous catalysts to synthesize levulinic acid from biomass substrates, although these alternatives have yet to match mineral acid systems in terms of yield and selectivity. Heterogeneous catalysts, with sulfonic-acid groups, have been applied to the synthesis of levulinic acid with moderate success. While the selectivity/yield of these catalysts can be quite impressive, reaching a high of yield/selectivity of 76%, it is important to keep in mind that to achieve these yields, the substrate concentration must be kept quite low (i.e. 40 mg of substrate in 2 mL of water, 0.2 wt%).324,325,336
Heterogeneous hydrogenation and hydrogenolysis of starch and cellulose to obtain polyols and other sugar derivatives have been widely studied by various research groups. These reactions are preceded by a hydrolysis process, to depolymerize the polysaccharide into the corresponding monosaccharides.326 This often is a two stage process – an initial acidic or enzymatic hydrolysis, followed by hydrogenation and hydrogenolysis of the resulting monosaccharides (as described in the conversions of monosaccharides in sections 4.5. and 4.10.). A one-pot combined approach has also been developed where the monosaccharides generated in situ, by acidic hydrolysis of polysaccharides are converted into polyols and other sugar derivatives such as hexitan and isohexide. The one-pot approach is typically carried out by using “bifunctional” metal catalysts, supported on Brønsted acidic zeolites (H-USY-3% Ru). These reactions generally take place at higher temperature (190–200 °C) to promote hydrolysis of polysaccharides. In these processes, starch is generally converted into C6 polyols such as sorbitol and mannitol or a mixture of both. In contrast the chemical cracking of cellulose offers access to a diverse group of sugars derivatives including the C4–C6 polyols, ethylene and propylene glycol, isosorbide, sorbitan and the HMF platform. The one pot chemical cracking of cellulose and starch has recently been reviewed by Gallezot Palkovits, Sels and their respective coworkers.12,76,327,328 Therefore, this section is dedicated to a comparative description of the existing processes. For the conversion of both starch and cellulose into C6 polyols Ru, Pt, Ni catalysts supported on Brønsted acidic zeolites (H-USY, H-ZSM-5, H mordernite) have been applied.329–332 A Ni catalyst supported on ZSM-5 zeolite converted microcrystalline cellulose into sorbitol, with 91% selectivity at 49% conversion.330 A nickel catalyst supported on carbon nanofiber (Ni-CNF) recently reported by Sels and coworkers showed a total of 76% yield of C6-polyols, including with 69% sorbitol and 7% mannitol, at 93% conversion of cellulose.333 Further studies confirmed that the surface density of metal catalyst versus the Brønsted acid sites, prepared by HNO3 mediated oxidation of CNF, is crucial to control the product selectivity. The relatively high density of Ni atom on the surface of CNF and low density of Brønsted acid sites are optimal for the observed high selectivity towards polyol platform. Additionally, solid acid,334–339 Lewis acids (Al2O3, ZnO)231,340–344 heteropolyacids and zeolites (H4SiW12O40, H2WO4)345–348 have also been employed as catalyst for the conversion of starch and cellulose. The product selectivity between the polyols or the sorbitan and isosorbide platforms, is dependent on the nature of metal catalyst, the acidic support and the reaction conditions, (i.e. temperature and the ratio of catalyst to substrate). The Ru, Pt, and Ni catalysts supported on zeolite, alumina or activated carbon, produce mainly C6 polyols, whereas, the Ni-doped tungsten catalysts and the heteropolyacid supports favor the formation of C2–C3 diols as the major product. Upon the addition of a solid acid catalyst with Ru/C catalyst, further dehydration of the C6 polyols is observed. In this system in situ generated glucose and sorbitol showed partial degradation to produce insoluble condensation byproducts, at temperature >200 °C. Increasing the concentration of Ru/C, cellulose and the solid acid suppressed the amount of byproduct formation and produced a mixture of hexitans and isohexides with 52% selectivity towards isosorbide.340,347,349
Sulfonated resins,350 heteropoly acids351,352 or activated carbon catalysts functionalized with sulfonic acid groups have successfully been applied for one pot hydrolysis of cellulose into glucose and its subsequent conversion into alkyl glucosides, by using C1–C4 alcohols as the glucoside acceptor.353 The catalyst system showed partial degradation after consecutive reuse.
Direct conversion of cellulose and starch (2nd generation bio-feedstock) can provide access to a diverse range of chemical platforms: C6 polyols, the corresponding dehydration products (sorbitan, other hexitan isomers, isohexides), C2–C3 polyols. The product selectivity thus plays a crucial role for useful implementation of cellulose as feedstock. Further mechanistic analysis of the combined cellulose hydrolysis-hydrogenation/hydrogenolysis processes is required to understand the difference in product selectivity and to develop catalysts suitable for commercial applications of these processes. Further mechanistic analysis of the combined cellulose hydrolysis–hydrogenation/hydrogenolysis processes is required to understand the difference in product selectivity and to develop catalysts suitable for commercial applications of these processes.
Many researchers are currently using ionic liquids to directly convert cellulose into HMF. Generally, it is necessary to marry the use of ionic liquids with a metal complex and microwave irradiation in order to achieve respectable conversion, selectivity, and yields of HMF from cellulosic biomass.
The primary drawback of ionic liquids is their high cost, part of which is driven by the difficulty in purifying the ionic layer for reuse. The process is long and tedious.366,367 The aqueous IL layer must be extracted, dried under heat and vacuum for several days, and then filtered to remove solids, such as unreacted biomass or humin, before being recycled.256,262,361 To improve recyclability, researchers are currently chemically modifying ionic liquids in order to facilities IL separation and reuse. In a recent study, fluorinated ionic liquid 1-(2′,2′,3′,3′,3′-pentafluoropropyl)-imidazolium chloride has shown improved recyclability as a potential reaction medium for cellulose dissolution and chemical hydrolysis.367
5.2. Chemical modification
Chemical modification pathways of starch and cellulose are comprised of three common mechanistic processes: oxidation, esterification, and etherification of the abundant OH-groups while the carbohydrate backbone is kept intact. The early reports on starch oxidation employed inexpensive transition metals salts (Cu2+, Fe2+) and common oxidizing agents (perchlorates or iodates).370 Although a few processes have been commercialized, the main drawback of this approach is the accumulation of high volumes of hazardous waste and the oxidative degradation of carbohydrates producing carbon dioxide as the byproduct. Radicals such as 2,2,6,6-tetramethylpiperidinine-1-oxyl (TEMPO) has also been used as catalyst, in combination with oxidant such as NaOCl/NaBr, NaOCl, peracids and oxone to regenerate the radical catalyst. In these processes, a selective oxidation of both starch71,371,372 and cellulose373–375 can be obtained at the C6 position (shown in Scheme 16) with 60–85% conversion. No degradation of starch or cellulose was reported.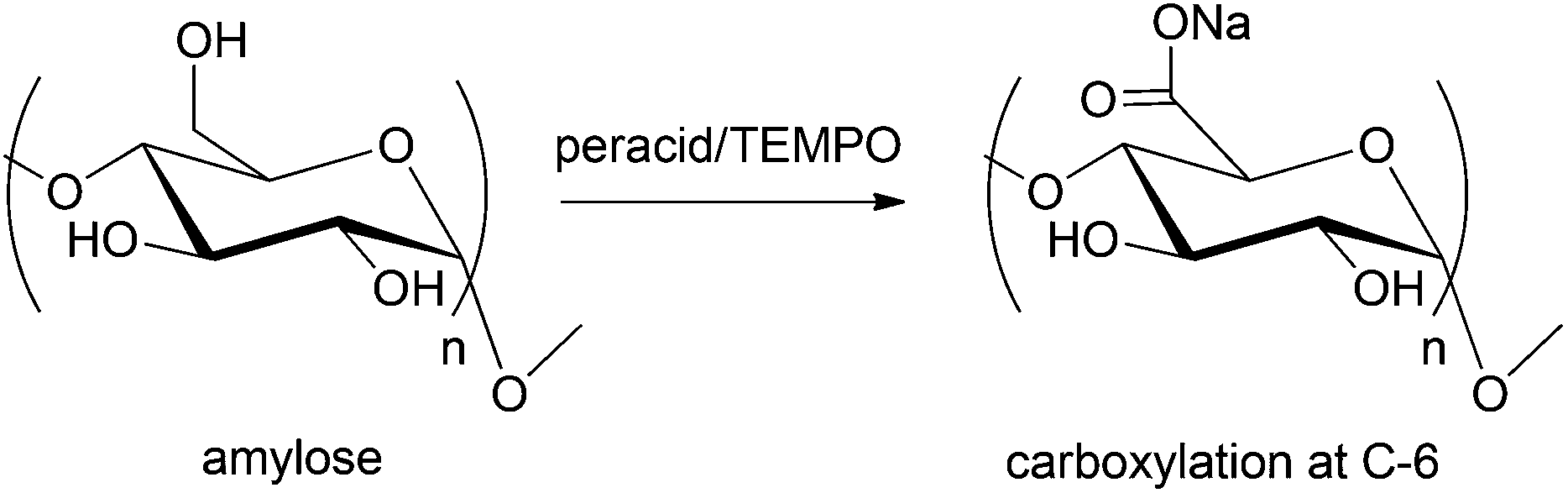 | ||
| Scheme 16 TEMPO catalyzed oxidation of starch to introduce carboxylic acid functional groups. | ||
In search of more environmentally friendly and low-cost oxidation methods of starch and cellulose, H2O2 and O2 have also been employed as oxidizing agents, often in combination with transition metal complexes (Cu, Fr, W).376–379 In similar reaction conditions, typically incorporation of 1.4 carboxyl and 6.6 carbonyl functions per 100 units of glucose, in starch is observed. A CH3ReO3/H2O2/Br− system380 was also used for oxidation of starch in acetic acid–water media, however, limited catalyst recyclability is an inherent limitation of this catalyst system. In a recent invention, native starch was converted to dialdehyde starch while using hypervalent bisacetoxyiodobenzene as the oxidant.381
The commonly followed methods to introduce hydrophobicity in starch and cellulose are esterification with fatty acid chlorides,30,382 transesterification with glycerides,383 and etherification by telomerization with butadiene, or silylation,384,385 as discussed in detail in sections 4.8. and 4.9. of this review.
The common disadvantages of the chemical modification processes is the poor mixing of hydrophilic biopolymers with organic catalysts or hydrophobic long chain fatty acids and the selectivity of the oxidation process, to avoid the degradation of the carbohydrate backbone. These reactions need to be carried out in highly polar organic solvents or in aqueous media, in the presence of a cationic surfactant, to aid the miscibility of the biopolymers with the catalyst and reagents. The oxidized, more hydrophilic polysaccharides show, enhanced solubility, adsorbent properties and extensively used for high volume applications in paper, packaging and textile industries. The hydrophobic ether and ester derivatives find application in water repellent coatings, packaging and high volume hydrophobic polymer blends.
6. Chemical conversions of hemicellulose
6.1. Chemical cracking
Hemicelluloses typically contain a large amount of xylose, a pentose sugar which is commonly used as a starting material for furans and their subsequent products (see section 4.7.). Hemicelluloses also serve as a cheap and abundant starting material for the furan platform386 – most of the methods used for the conversion of cellulose/polysaccharides to furans have already been discussed in the previous section (5.1.).6.2. Chemical modification
The most commonly employed chemical modification techniques for hemicellulose are oxidation, etherification, and esterification. Hemicellulose is susceptible to hydrolysis in strong acidic or basic media, hence the common functionalization reactions are carried out under relatively mild conditions to avoid fragmentation of the hemicellulose backbone. The TEMPO/NaOCl/NaBr system has been widely used to oxidize the carbonyl groups present on hemicellulose to carboxylic acid functionalities.391 Oxidation, followed by esterification or cationization235,392,393 decreases the crystallinity of native hemicellulose and the resulting material becomes more soluble in water. Esterification of hemicellulose has also been carried out using ionic liquids394–397 or highly polar organic solvents.398–400 Commonly employed etherification processes of hemicellulose include alkylation, sulfoalkylation, and benzylation.131,401–404 The etherification of hemicellulose increases its solubility in water, film forming capability, and anti-fouling properties. Chemically modified hemicellulose has found extensive use as additives in the paper industry as sizing and strengthening agents.7. Chemical conversion overview
As we have detailed in this review, chemical conversion processes are very versatile because of the wide selection of reaction conditions and catalysts that can be utilized to access a multitude of different platform chemicals. Because these platform chemicals are also used in downstream applications (polymers and pharmaceuticals) they must have relatively high purity. As a result, it behooves the researcher to control the process in such a way that the reaction is selective towards a single product and limits excessive by-product formation in order to prevent the need for extensive and costly purification/separation steps.Hence, reaction conditions are very important when considering the product selectivity of the reaction. A majority of studies are still primarily focused on: (1) product distribution, (2) catalyst selectivity/optimization/recyclability; (3) reduction of reaction hazards, (4) reduction of number of steps. For aqueous phase conversions of biomass, researchers commonly focus on varying (1) the temperature, (2) reaction time, (3) substrate concentration and (4) solvent. Each of these parameters is discussed in the following paragraphs. Because of the great variety of catalysts, techniques, and processing conditions involved in biomass conversions, it is extremely difficult to compare even the same substrates undergoing the same conversion process (e.g. hydrogenation, dehydration, oxidation) from one publication to the next.
Temperature optimization always plays an important role in product selectivity; generally, temperatures that are too high cause an undesired formation of byproducts, while temperatures that are too low lengthen the reaction time/reduce catalyst activity considerably. Prüße et al. found in an oxidation of glucose by Au/TiO2, that temperatures above 60 °C – with and without the presence of the gold catalyst – resulted in the degradation of glucose into carboxylic acids and aldehydes.94,95
Sels and co-workers studied the effects of substrate concentration when converting glucose to ethylene glycol. This group observed that in using higher concentrations of the starting material, glucose, the by-product formation of C3 polyols was significantly reduced. In contrast, researchers studying systems for conversion of sugar/cellulose to furans and related products, have observed the opposite trend.334 Excessive amounts of starting material, be it sugars or polysaccharides, can result in lower catalyst activity and formation of polyketones and humins via self-condensation of the furans.223
It is also extremely important to optimize the reaction time. The contact times of the starting material and its surrounding environment (solvent, catalyst) alters the product distribution profile. For example, it is consistently observed that lengthened reactions which produce HMF result in the formation of unwanted polyketone humins. Current studies often include time dependent profiles of product distribution.361
Solvent choice is also extremely important; each type of solvent used has a unique set of advantages (polarity, hydrophilicity) and disadvantages (cost, recyclability, hazard). Ionic liquids are extremely useful for polysaccharide deconstruction, and allow researchers to perform multistep reactions in one pot. Solvent choice is also extremely important in biphasic reactions and can influence the overall yield of product. The presence of an immiscible layer can keep the product and catalytic layer apart, preventing further reaction of the product.184 DMSO is also commonly used to suppress byproduct formation of polyketones in furanic reactions.17
We hope for the reader to take away a few messages from this review. There are many process-dependent variables that influence the outcome of any biomass reaction. These variables influence each reaction differently, and they should be investigated carefully. However, all of these studies appear to have these two primary goals in common: Firstly, the aim to improve the conversion and selectivity of the reaction, precluding the need for additional cost-adding purification steps; and secondly, the focus on improving the recyclability of a catalytic system. Both initiatives are cost-saving and less energy-intensive approaches that will enable biomass platform chemicals to become a more viable and attractive industrial option to pursue.
8. Conclusion
Carbohydrate biomass is primarily harvested for food and animal feed. Only a fraction of biomass is used for energy conversion, and an even smaller percentage is used to generate material goods such as clothing and chemicals. Although less than 10% of chemicals available on the market today are bio-based, both the market and the demand for bio-based chemicals have continued to grow rapidly. The United States has set a goal to have 25% of its’ chemical commodities based on biomass materials; this in turn will provide new economic opportunities, particularly for rural workers. Despite the growing demands of the chemical market, industrialists also must be aware that with an increasing global population, which is expected to reach 9 billion by 2050, that the demand for biomass-based food and animal feed will also increase. It is therefore, crucial, that use of arable land and biomass is carefully regulated because improper harvesting or over-exploitation of these resources can have profound negative impact on both society and the environment. Hence the horizon 2020 proposal aims at “the promotion of low carbon, resource efficient sustainable and competitive” biobased economy. Dusselier et al. discussed an alternative way of evaluating the practical viability of the existing platform chemicals using the atom economy and conservation of functionality as main criteria.405A range of bio-based chemicals have been targeted for large scale production or have already been commercialized; these chemicals are generated from 1st generation feedstocks such as sugar, starch, natural rubber, and plant oil. Although many of these processes rely on fermentation or thermochemical routes, a significant amount of research has also been directed towards chemical catalytic synthesis. To obtain technologically simple, low cost chemical conversion and purification processes, fermented feedstocks such as succinic acid, ethanol and isobutanol are often used as starting materials instead of more complex cellulosic biomass.406 A few notable examples of chemically converted bio-based platform chemicals, which have already been commercialized or are in commission, are: butanediol by hydrogenation of succinic acid (commercialized by BASF, technology developed by Genomatica); bioethylene via dehydration of bioethanol (commercialized by Braskem); gluconic, glucaric and adipic acids by oxidation of glucose (pilot scale production by Rennovia, Rivertop renewables); isobutylene by the dehydration of isobutanol (in commission 2014, by Global bioenergies); and isosorbide based polycarbonates by Mitsubishi chemicals. In addition, platform chemicals such as lactic acid, acrylic acid, isosorbide, ethylene glycol and furandicarboxylic (FDCA) have also been targeted as an emerging class of bio-based platform chemicals. The role of chemical catalysis for commercial production of these targeted platforms is directed by competitive technology development and economic feasibility.
As we have discussed in our review, in comparison to chemical catalysis, thermochemical methods are very energy intensive and tend to be less selective; enzymatic methods are less toxic, but are in many cases prohibitively expensive. While many chemical processes are scalable, they may not be economically feasible. In order to make these processes cost-competitive, the number of conversion and purification steps must be reduced while product selectivity must be improved significantly. Biomass proponents have also suggested that expanding biofuel manufacturing plants to include facilities for bio-based chemical commodity production will enable manufacturers to utilize the heat expended by the production of biofuels in the processing of bio-based chemicals. Ultimately, this will drive the cost of both chemicals and fuels downward. A sizable portion of ongoing research concerns the implementation of inedible biomass, namely, second generation (lignocellulosic materials: wood waste, agricultural waste, highly cellulosic rotation crops such as miscanthus, willow) and third generation (algae) bio-based feedstocks. The feasible technology development for the commercialization of these feedstocks is expected to lower any possible impact on food security.
As the body of research for carbohydrate conversion continues to expand, researchers are dedicating more of their efforts to finding safer, simpler and more effective chemical conversion technologies. These studies are bringing us closer to the goal of sustainable, bio-based technologies, which are expected to reduce our dependence on fossil fuels and also foster a better environment and society for generations to come.
Abbreviations
| FUR | Furfural |
| HMF | 5-Hydroxymethylfurfural |
| THF | Tetrahydrofuran |
| MTHF | 2-Methyltetrahydrofuran |
| FDCA | 2,5-Furandicarboxylic acid |
| SA | Succinic acid |
| FA | Formic acid |
| LA | Levulinic acid |
| MCC | Microcrystalline cellulose |
| GVL | γ-Valerolactone |
| DFF | 2,5-Diformylfuran |
| MA | Maleic anhydride |
| HMFCA | 5-Hydroxymethyl-2-carboxylfuraldehyde |
| DHMF | 2,5-Dihydroxymethylfuran |
| DHMTHF | 2,5-Dihydroxymethyltetrahydrofuran |
| [BMIM][Cl] | 1,3-Butylmethylimidazolium chloride |
| [EMIM][Cl] | 1,3-Ethylmethylimidazolium chloride |
| PTA | Phosphotungstic acid |
| FSIL | Fiber-supported ionic liquids |
| TPPTS | 3,3′,3′′-Phosphanetriyltris trisodium salt |
| TEMPO | 2,2,6,6-Tetramethylpiperidine-1-oxyl |
| MWNT | Multiwall nanotubes |
| PTS | p-Toluenesulfonate |
| APG | Alkylpolygycoside |
| MCM | Mesostructured silica |
| MIBK | Methyl isobutyl ketone |
| CSS | Carbonaceous sulfonated carbon |
| DVB | Divinylbenzene |
| GTS | Sulfonated carbonaceous material |
| DS | Dodecyl sulfate |
Acknowledgements
We thank the Department of Energy, Office of Basic Energy Science and Penn State Institutes of Energy and the Environment (PSIEE) for supporting this work. We also thank Dr Shaibal Roy for useful discussions.References
- BP Statistical Review of World Energy, http://www.bp/com/en/global/corporate/about-bp/energy-economics/statistical-review-of-world-energy-2013.html, (accessed June 2014).
-
S. Dumitriu, Polysaccharides: Structural Diversity and Functional Versatility, CRC Press, 2nd edn, 2012 Search PubMed
.
-
H. Langeveld, J. Sanders and M. Meeusen, The Biobased Economy: Biofuels, Materials and Chemicals in the Post-oil Era, Earthscan, 2012 Search PubMed
- O. R. Inderwildi and D. A. King, Energy Environ. Sci., 2009, 2, 343–346 CAS
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed
- P. Gallezot, Green Chem., 2007, 9, 295–302 RSC
-
T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, National Renewable Energy Lab., Golden, CO (US), 2004 Search PubMed
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520–540 RSC
- B. Kamm, Angew. Chem., Int. Ed., 2007, 46, 5056–5058 CrossRef CAS PubMed
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed
-
D. B. Turley, in Introduction to Chemicals from Biomass, ed. J. H. Clark and F. E. I. Deswarte, John Wiley & Sons, Ltd, 2008, pp. 21–46 Search PubMed
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed
-
Z. Fang, Pretreatment Techniques for Biofuels and Biorefineries, Springer, 2013 Search PubMed
- H. Zhao, J. H. Kwak, Y. Wang, J. A. Franz, J. M. White and J. E. Holladay, Energy Fuels, 2006, 20, 807–811 CrossRef CAS
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed
- H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740–1763 RSC
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed
- P. Gallezot, Top. Catal., 2010, 53, 1209–1213 CrossRef CAS
- J. C. Serrano-Ruiz, R. Luque and A. Sepúlveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC
- G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549–1551 CrossRef CAS PubMed
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed
- M. G. Adsul, M. S. Singhvi, S. A. Gaikaiwari and D. V. Gokhale, Bioresour. Technol., 2011, 102, 4304–4312 CrossRef CAS PubMed
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC
-
D. J. M. Hayes, in The Role of Catalysis for the Sustainable Production of Bio-fuels and Bio-chemicals, Newnes, 2013, pp. 27–60 Search PubMed
- S. Wenda, S. Illner, A. Mell and U. Kragl, Green Chem., 2011, 13, 3007–3047 RSC
- A. J. J. Straathof, Chem. Rev., 2013, 114, 1871–1908 CrossRef PubMed
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem.
Soc. Rev., 2009, 38, 2046–2064 RSC
- A. G. Cunha and A. Gandini, Cellulose, 2010, 17, 875–889 CrossRef CAS
- M. N. Belgacem, M. C. Salon-Brochier, M. Krouit and J. Bras, J. Adhes. Sci. Technol., 2011, 25, 661–684 CrossRef CAS PubMed
- N. M. L. Hansen and D. Plackett, Biomacromolecules, 2008, 9, 1493–1505 CrossRef CAS PubMed
-
J. H. Clark and F. Deswarte, in Introduction to Chemicals from Biomass, John Wiley & Sons, 2011, pp. 77–101 Search PubMed
- Javier Pérez-Maqueda, Irene Arenas-Ligioiz, Ó. López and J. G. Fernández-Bolaños, Chem. Eng. Sci., 2014, 109, 244–250 CrossRef PubMed
- H. A. Abd El-Rehim and D. A. Diaa, Carbohydr. Polym., 2012, 87, 1905–1912 CrossRef CAS PubMed
- C. Buttersack and D. Laketic, J. Mol. Catal., 1994, 94, L283–L290 CrossRef
- C. Moreau, R. Durand, F. Aliès, M. Cotillon, T. Frutz and M.-A. Théoleyre, Ind. Crops Prod., 2000, 11, 237–242 CrossRef CAS
- M. Marzo, A. Gervasini and P. Carniti, Carbohydr. Res., 2012, 347, 23–31 CrossRef CAS PubMed
- H. Iloukhani, S. Azizian and N. Samadani, React. Kinet. Catal. Lett., 2001, 72, 239–244 CrossRef CAS
-
H. Zhang, J. Yang and C. Zhang, CN/1048233, 1991
- S. G. Minasian, M. M. Whittaker and J. W. Whittaker, Biochemistry, 2004, 43, 13683–13693 CrossRef CAS PubMed
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef PubMed
- B. Y. Yang and R. Montgomery, Carbohydr. Res., 1996, 280, 27–45 CrossRef CAS
- J. F. Mendicino, J. Am. Chem. Soc., 1960, 82, 4975–4979 CrossRef CAS
- V. Choudhary, A. B. Pinar, R. F. Lobo, D. G. Vlachos and S. I. Sandler, ChemSusChem, 2013, 6, 2369–2376 CrossRef CAS PubMed
- R. Ooms, M. Dusselier, J. A. Geboers, B. O. de Beeck, R. Verhaeven, E. Gobechiya, J. A. Martens, A. Redl and B. F. Sels, Green Chem., 2014, 16, 695–707 RSC
- Y. Román-Leshkov and M. E. Davis, ACS Catal., 2011, 1, 1566–1580 CrossRef
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed
- C. Moreau, R. Durand, A. Roux and D. Tichit, Appl. Catal., A, 2000, 193, 257–264 CrossRef CAS
- R. Gounder and M. E. Davis, J. Catal., 2013, 308, 176–188 CrossRef CAS PubMed
- S. Saravanamurugan, M. Paniagua, J. A. Melero and A. Riisager, J. Am. Chem. Soc., 2013, 135, 5246–5249 CrossRef CAS PubMed
- R. Gounder and M. E. Davis, ACS Catal., 2013, 3, 1469–1476 CrossRef CAS
- P. S. Panesar and S. Kumari, Biotechnol. Adv., 2011, 29, 940–948 CrossRef CAS PubMed
-
A. E. Stütz, Glycoscience: Epimerisation, Isomerisation and Rearrangement Reactions of Carbohydrates, Springer Science & Business Media, 2001 Search PubMed
- H. Brunner and D. Opitz, J. Mol. Catal. A: Chem., 1997, 118, 273–282 CrossRef CAS
- S. J. Angyal, Carbohydr. Res., 1997, 300, 279–281 CrossRef CAS
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705–2713 CrossRef CAS
- W. R. Gunther, Y. Wang, Y. Ji, V. K. Michaelis, S. T. Hunt, R. G. Griffin and Y. Román-Leshkov, Nat. Commun., 2012, 3, 1109 CrossRef PubMed
- F. Ju, D. VanderVelde and E. Nikolla, ACS Catal., 2014, 4, 1358–1364 CrossRef CAS
- E. O. Odebunmi and A. S. Ogunlaja, Curr. Res. Chem., 2011, 3, 16–28 CrossRef CAS
- D. Keilin and E. F. Hartree, Biochem. J., 1952, 50, 331–341 CAS
- J. C. González, M. F. Mangiameli, A. C. Asis, S. Bellú and L. F. Sala, Polyhedron, 2013, 49, 84–92 CrossRef PubMed
- A. K. Singh, S. Srivastava, J. Srivastava and R. Singh, Carbohydr. Res., 2007, 342, 1078–1090 CrossRef CAS PubMed
- Z. Khan, P. S. S. Babu and Kabir-ud-Din, Carbohydr. Res., 2004, 339, 133–140 CrossRef CAS PubMed
- E. O. Odebunmi, S. A. Iwarere and S. O. Owalude, Int. J. Chem., 2006, 16, 167–176 CAS
- A. K. Singh, S. Srivastava, J. Srivastava, R. Srivastava and P. Singh, J. Mol. Catal. A: Chem., 2007, 278, 72–81 CrossRef CAS PubMed
- A. K. Singh, R. Singh, J. Srivastava, S. Rahmani and S. Srivastava, J. Organomet. Chem., 2007, 692, 4270–4280 CrossRef CAS PubMed
- M. S. Nieuwenhuizen, A. P. G. Kieboom and H. Van Bekkum, J. Am. Oil Chem. Soc., 1983, 80, 44 Search PubMed
- V. Ganesh, S. Farzana and S. Berchmans, J. Power Sources, 2011, 196, 9890–9899 CrossRef CAS PubMed
- M. Schämann and H. J. Schäfer, Electrochim. Acta, 2005, 50, 4956–4972 CrossRef PubMed
- A. E. J. de Nooy, A. C. Besemer and H. van Bekkum, Carbohydr. Res., 1995, 269, 89–98 CrossRef CAS
- P. L. Bragd, A. C. Besemer and H. van Bekkum, Carbohydr. Polym., 2002, 49, 397–406 CrossRef CAS
- S. Lemoine, C. Thomazeau, D. Joannard, S. Trombotto, G. Descotes, A. Bouchu and Y. Queneau, Carbohydr. Res., 2000, 326, 176–184 CrossRef CAS
- S. Brochette-Lemoine, D. Joannard, G. Descotes, A. Bouchu and Y. Queneau, J. Mol. Catal. A: Chem., 1999, 150, 31–36 CrossRef CAS
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127–141 CrossRef CAS
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2013, 114, 1827–1870 CrossRef PubMed
- A. Abbadi and H. van Bekkum, Appl. Catal., A, 1996, 148, 113–122 CrossRef CAS
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed
- B. T. Kusema and D. Y. Murzin, Catal. Sci. Technol., 2013, 3, 297–307 CAS
- I. V. Delidovich, O. P. Taran, L. G. Matvienko, A. N. Simonov, I. L. Simakova, A. N. Bobrovskaya and V. N. Parmon, Catal. Lett., 2010, 140, 14–21 CrossRef CAS
- X. Liang, C. Liu and P. Kuai, Green Chem., 2008, 10, 1318–1322 RSC
- M. Besson, F. Lahmer, P. Gallezot, P. Fuertes and G. Fleche, J. Catal., 1995, 152, 116–121 CrossRef CAS
- H. Bönnemann, W. Brijoux, R. Brinkmann, A. Schulze Tilling, T. Schilling, B. Tesche, K. Seevogel, R. Franke, J. Hormes, G. Köhl, J. Pollmann, J. Rothe and W. Vogel, Inorg. Chim. Acta, 1998, 270, 95–110 CrossRef
- S. Hermans and M. Devillers, Appl. Catal., A, 2002, 235, 253–264 CrossRef CAS
- I. Witońska, M. Frajtak and S. Karski, Appl. Catal., A, 2011, 401, 73–82 CrossRef PubMed
- R. Tschentscher, T. A. Nijhuis, J. van der Schaaf and J. C. Schouten, Ind. Eng. Chem. Res., 2011, 51, 1620–1634 CrossRef
- S. L. Pirard, C. Diverchy, S. Hermans, M. Devillers, J.-P. Pirard and N. Job, Catal. Commun., 2011, 12, 441–445 CrossRef CAS PubMed
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242–247 CrossRef CAS
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1–7 CrossRef CAS PubMed
- C. Ma, W. Xue, J. Li, W. Xing and Z. Hao, Green Chem., 2013, 15, 1035–1041 RSC
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef CAS PubMed
- T. Ishida, H. Watanabe, T. Bebeko, T. Akita and M. Haruta, Appl. Catal., A, 2010, 377, 42–46 CrossRef CAS PubMed
- M. Zhang, X. Zhu, X. Liang and Z. Wang, Catal. Commun., 2012, 25, 92–95 CrossRef CAS PubMed
- U. Prüße, M. Herrmann, C. Baatz and N. Decker, Appl. Catal., A, 2011, 406, 89–93 CrossRef PubMed
- U. Prüße, P. Jarzombek and K.-D. Vorlop, Top. Catal., 2012, 55, 453–459 CrossRef
- B. Gaweł, K. Lambrechts and G. Øye, Mater. Sci. Eng., B, 2012, 177, 575–580 CrossRef PubMed
- I. V. Delidovich, B. L. Moroz, O. P. Taran, N. V. Gromov, P. A. Pyrjaev, I. P. Prosvirin, V. I. Bukhtiyarov and V. N. Parmon, Chem. Eng. J., 2013, 223, 921–931 CrossRef CAS PubMed
- C. Baatz, N. Thielecke and U. Prüße, Appl. Catal., B, 2007, 70, 653–660 CrossRef CAS PubMed
- Y. Önal, S. Schimpf and P. Claus, J. Catal., 2004, 223, 122–133 CrossRef PubMed
- Y. Wang, S. V. de Vyver, K. K. Sharma and Y. Román-Leshkov, Green Chem., 2014, 16, 719–726 RSC
- A. Abbadi and H. van Bekkum, Appl. Catal., A, 1995, 124, 409–417 CrossRef CAS
- P. C. C. Smits, B. F. M. Kuster, K. van der Wiele and S. van der Baan, Appl. Catal., 1987, 33, 83–96 CrossRef CAS
- M. Besson, G. Flèche, P. Fuertes, P. Gallezot and F. Lahmer, Recl. Trav. Chim. Pays-Bas, 1996, 115, 217–221 CrossRef CAS
- P. J. M. Dijkgraaf, M. J. M. Rijk, J. Meuldijk and K. van der Wiele, J. Catal., 1988, 112, 329–336 CrossRef CAS
-
V. J. Murphy, J. Schoemaker, G. Zhu, R. Archer, G. F. Salem and E. L. Dias, US/2011/0306790, 2011
- C. Brönnimann, T. Mallat and A. Baiker, J. Chem. Soc., Chem. Commun., 1995, 1377–1378 RSC
- T. Mallat, C. Brönnimann and A. Baiker, J. Mol. Catal. A: Chem., 1997, 117, 425–438 CrossRef CAS
- B. T. Kusema, J.-P. Mikkola and D. Y. Murzin, Catal. Sci. Technol., 2012, 2, 423–431 CAS
- E. Smolentseva, B. T. Kusema, S. Beloshapkin, M. Estrada, E. Vargas, D. Y. Murzin, F. Castillon, S. Fuentes and A. Simakov, Appl. Catal., A, 2011, 392, 69–79 CrossRef CAS PubMed
- B. T. Kusema, B. C. Campo, O. A. Simakova, A.-R. Leino, K. Kordás, P. Mäki-Arvela, T. Salmi and D. Y. Murzin, ChemCatChem, 2011, 3, 1789–1798 CrossRef CAS
- R. Saliger, N. Decker and U. Prüße, Appl. Catal., B, 2011, 102, 584–589 CrossRef CAS PubMed
- L. A. Edye, G. V. Meehan and G. N. Richards, J. Carbohydr. Chem., 1994, 13, 273–283 CrossRef CAS
-
W. Fritsche-Lang, L. Leopold and M. Schlingmann, DE/1985/3535720, 1987
- H. E. J. Hendriks, B. F. M. Kuster and G. B. Marin, Carbohydr. Res., 1990, 204, 121–129 CrossRef CAS
- A. Abbadi, K. F. Gotlieb, J. B. M. Meiberg and H. van Bekkum, Appl. Catal., A, 1997, 156, 105–115 CrossRef CAS
- L.-F. Gutierrez, S. Hamoudi and K. Belkacemi, Appl. Catal., A, 2011, 402, 94–103 CrossRef CAS PubMed
- P. Mäki-Arvela, A. V. Tokarev, E. V. Murzina, B. Campo, T. Heikkilä, J.-M. Brozinski, D. Wolf and D. Y. Murzin, Phys. Chem. Chem. Phys., 2011, 13, 9268–9280 RSC
- A. V. Tokarev, E. V. Murzina, J.-P. Mikkola, J. Kuusisto, L. M. Kustov and D. Y. Murzin, Chem. Eng. J., 2007, 134, 153–161 CrossRef CAS PubMed
- R. W. Jakob Albert, Energy Environ. Sci., 2012, 5, 7956–7962 Search PubMed
- W. Wang, M. Niu, Y. Hou, W. Wu, Z. Liu, Q. Liu, S. Ren and K. N. Marsh, Green Chem., 2014, 16, 2614–2618 RSC
- R. Wölfel, N. Taccardi, A. Bösmann and P. Wasserscheid, Green Chem., 2011, 13, 2759–2763 RSC
- J. Li, D.-J. Ding, L. Deng, Q.-X. Guo and Y. Fu, ChemSusChem, 2012, 5, 1313–1318 CrossRef CAS PubMed
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS PubMed
- T. Pasini, M. Piccinini, M. Blosi, R. Bonelli, S. Albonetti, N. Dimitratos, J. A. Lopez-Sanchez, M. Sankar, Q. He, C. J. Kiely, G. J. Hutchings and F. Cavani, Green Chem., 2011, 13, 2091–2099 RSC
- B. Saha, S. Dutta and M. M. Abu-Omar, Catal. Sci. Technol., 2012, 2, 79–81 CAS
- R. Alamillo, M. Tucker, M. Chia, Y. Pagan-Torres and J. Dumesic, Green Chem., 2012, 14, 1413–1419 RSC
- C. A. Antonyraj, J. Jeong, B. Kim, S. Shin, S. Kim, K.-Y. Lee and J. K. Cho, J. Ind. Eng. Chem., 2013, 19, 1056–1059 CrossRef CAS PubMed
- Z. Du, J. Ma, F. Wang, J. Liu and J. Xu, Green Chem., 2011, 13, 554–557 RSC
- P. Foley, A. Kermanshahi pour, E. S. Beach and J. B. Zimmerman, Chem. Soc. Rev., 2012, 41, 1499 RSC
-
M. Kjellin and I. Johansson, Surfactants from Renewable Resources, John Wiley & Sons, 2010 Search PubMed
- E. Fischer, Ber. Dtsch. Chem. Ges., 1893, 26, 2400–2412 CrossRef
- R. R. Schmidt and M. Reichrath, Angew. Chem., Int. Ed. Engl., 1979, 18, 466–467 CrossRef
- P. Rosevear, T. VanAken, J. Baxter and S. Ferguson-Miller, Biochemistry, 1980, 19, 4108–4115 CrossRef CAS
- F. Bouxin, S. Marinkovic, J. L. Bras and B. Estrine, Carbohydr. Res., 2010, 345, 2469–2473 CrossRef CAS PubMed
- S. Marinkovic and B. Estrine, Green Chem., 2010, 12, 1929–1932 RSC
- C. Ludot, B. Estrine, J. L. Bras, N. Hoffmann, S. Marinkovic and J. Muzart, Green Chem., 2013, 15, 3027–3030 RSC
- C. L. Allen and S. J. Miller, Org. Lett., 2013, 15, 6178–6181 CrossRef CAS PubMed
- M. Pfaffe and R. Mahrwald, Org. Lett., 2012, 14, 792–795 CrossRef CAS PubMed
- W. Muramatsu and H. Yoshimatsu, Adv. Synth. Catal., 2013, 355, 2518–2524 CrossRef CAS
- D. S. van Es, S. Marinkovic, X. Oduber and B. Estrine, J. Surfactants Deterg., 2013, 16, 147–154 CrossRef PubMed
- N. Moitessier, P. Englebienne and Y. Chapleur, Tetrahedron, 2005, 61, 6839–6853 CrossRef CAS PubMed
- T.-Y. Huang, M. M. L. Zulueta and S.-C. Hung, Org. Biomol. Chem., 2014, 12, 376 CAS
- J. Lawandi, S. Rocheleau and N. Moitessier, Tetrahedron, 2011, 67, 8411–8420 CrossRef CAS PubMed
- S. Schmalisch and R. Mahrwald, Org. Lett., 2013, 15, 5854–5857 CrossRef CAS PubMed
- D. F. Mowery, J. Am. Chem. Soc., 1955, 77, 1667–1669 CrossRef CAS
- B. Liu and Z. Zhang, RSC Adv., 2013, 3, 12313 RSC
- A. J. J. Straathof, J. Romein, F. van Rantwijk, A. P. G. Kieboom and H. van Bekkum, Starch/Staerke, 1987, 39, 362–368 CrossRef CAS
- A. J. J. Straathof, H. van Bekkum and A. P. G. Kieboom, Starch/Staerke, 1988, 40, 229–234 CrossRef CAS
-
A. P. Rauter, N. M. Xavier, S. D. Lucas and M. Santos, in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. Horton, Academic Press, 2010, vol. 63, pp. 29–99 Search PubMed
- B. K. Gorityala, J. Ma, K. K. Pasunooti, S. Cai and X.-W. Liu, Green Chem., 2011, 13, 573–577 RSC
- M. A. Camblor, A. Corma, S. Iborra, S. Miquel, J. Primo and S. Valencia, J. Catal., 1997, 172, 76–84 CrossRef CAS
- N. M. Xavier, S. D. Lucas and A. P. Rauter, J. Mol. Catal. A: Chem., 2009, 305, 84–89 CrossRef CAS PubMed
- A. P. Rauter, T. Almeida, N. M. Xavier, F. Siopa, A. I. Vicente, S. D. Lucas, J. P. Marques, F. Ramôa-Ribeiro, M. J. Ferreira and M. Guisnet, J. Mol. Catal. A: Chem., 2007, 275, 206–213 CrossRef CAS PubMed
-
P. Gallezot and A. Kiennemann, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed
- A. Agarwal, S. Rani and Y. D. Vankar, J. Org. Chem., 2004, 69, 6137–6140 CrossRef CAS PubMed
- V. K. Rajput and B. Mukhopadhyay, J. Org. Chem., 2008, 73, 6924–6927 CrossRef CAS PubMed
- J. Augé and G. Sizun, Green Chem., 2009, 11, 1179–1183 RSC
- K. Toshima, K. Uehara, H. Nagai and S. Matsumura, Green Chem., 2002, 4, 27–29 RSC
- K. Nakajima and M. Hara, ACS Catal., 2012, 2, 1296–1304 CrossRef CAS
- K. Ikeda, Y. Ueno, S. Kitani, R. Nishino and M. Sato, Synlett, 2008, 1027–1030 CrossRef CAS PubMed
- A. Corma, S. Iborra, S. Miquel and J. Primo, J. Catal., 1998, 180, 218–224 CrossRef CAS
- A. T. J. W. de Goede, F. van Rantwijk and H. van Bekkum, Starch/Staerke, 1995, 47, 233–237 CrossRef CAS
- P. Bojarová, R. R. Rosencrantz, L. Elling and V. Křen, Chem. Soc. Rev., 2013, 42, 4774–4797 RSC
- International Starch: Sorbitol and other polyols, http://www.starch.dk/isi/glucose/sorbitol.asp, (accessed June 2014).
- P. J. Deuss, K. Barta and J. G. de Vries, Catal. Sci. Technol., 2014, 4, 1174–1196 CAS
- H. Heeres, R. Handana, D. Chunai, C. B. Rasrendra, B. Girisuta and H. J. Heeres, Green Chem., 2009, 11, 1247–1255 RSC
- S. Kolarić and V. Šunjić, J. Mol. Catal. A: Chem., 1996, 110, 189–193 CrossRef
- S. Rajagopal, S. Vancheesan, J. Rajaram and J. C. Kuriacose, J. Mol. Catal., 1992, 75, 199–208 CrossRef CAS
-
A. Sen and W. Yang, US/2011/0282079, 2013
- W. Yang and A. Sen, ChemSusChem, 2010, 3, 597–603 CrossRef CAS PubMed
- W. Yang and A. Sen, ChemSusChem, 2011, 4, 349–352 CrossRef CAS PubMed
- M. R. Grochowski, W. Yang and A. Sen, Chem. – Eur. J., 2012, 18, 12363–12371 CrossRef CAS PubMed
- S. Dutta, S. De, B. Saha and M. I. Alam, Catal. Sci. Technol., 2012, 2, 2025–2036 CAS
- B. Chen, U. Dingerdissen, J. G. E. Krauter, H. G. J. L. Rotgerink, K. Möbus, D. J. Ostgard, P. Panster, T. H. Riermeier, S. Seebald, T. Tacke and H. Trauthwein, Appl. Catal., A, 2005, 280, 17–46 CrossRef CAS PubMed
- B. W. Hoffer, E. Crezee, F. Devred, P. R. M. Mooijman, W. G. Sloof, P. J. Kooyman, A. D. van Langeveld, F. Kapteijn and J. A. Moulijn, Appl. Catal., A, 2003, 253, 437–452 CrossRef CAS
- J. Lee, D. H. K. Jackson, T. Li, R. E. Winans, J. A. Dumesic, T. F. Kuech and G. W. Huber, Energy Environ. Sci., 2014, 7, 1657 CAS
- J. Pan, J. Li, C. Wang and Z. Yang, React. Kinet. Catal. Lett., 2007, 90, 233–242 CrossRef CAS PubMed
- J. Liu, P. Bai and X. S. Zhao, Phys. Chem. Chem. Phys., 2011, 13, 3758–3763 RSC
- J. Liu, L. L. Zhang, J. Zhang, T. Liu and X. S. Zhao, Nanoscale, 2013, 5, 11044–11050 RSC
- A. Perrard, P. Gallezot, J.-P. Joly, R. Durand, C. Baljou, B. Coq and P. Trens, Appl. Catal., A, 2007, 331, 100–104 CrossRef CAS PubMed
- J. Kuusisto, J.-P. Mikkola, M. Sparv, J. Wärnå, H. Karhu and T. Salmi, Chem. Eng. J., 2008, 139, 69–77 CrossRef CAS PubMed
- V. Y. Doluda, J. Wärnå, A. Aho, A. V. Bykov, A. I. Sidorov, E. M. Sulman, L. M. Bronstein, T. Salmi and D. Y. Murzin, Ind. Eng. Chem. Res., 2013, 52, 14066–14080 CrossRef CAS
- Y. Yang, C. Hu and M. M. Abu-Omar, Green Chem., 2012, 14, 509–513 RSC
- M. Yadav, D. K. Mishra and J.-S. Hwang, Appl. Catal., A, 2012, 425–426, 110–116 CrossRef CAS PubMed
- A. W. Heinen, J. A. Peters and H. van Bekkum, Carbohydr. Res., 2000, 328, 449–457 CrossRef CAS
- T. Pan, J. Deng, Q. Xu, Y. Xu, Q.-X. Guo and Y. Fu, Green Chem., 2013, 15, 2967–2974 RSC
- Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3, 2655–2668 CrossRef CAS
- L. Hu, G. Zhao, W. Hao, X. Tang, Y. Sun, L. Lin and S. Liu, RSC Adv., 2012, 2, 11184–11206 RSC
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC
- S. M. Sen, E. I. Guerbuez, S. G. Wettstein, D. M. Alonso, J. A. Dumesic and C. T. Maravelias, Green Chem., 2012, 14, 3289–3294 RSC
- L. Xin, Z. Zhang, J. Qi, D. J. Chadderdon, Y. Qiu, K. M. Warsko and W. Li, ChemSusChem, 2013, 6, 674–686 CrossRef CAS PubMed
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC
- A. M. R. Galletti, C. Antonetti, V. De Luise and M. Martinelli, Green Chem., 2012, 14, 688–694 RSC
- C. Ortiz-Cervantes and J. J. Garcia, Inorg. Chim. Acta, 2013, 397, 124–128 CrossRef CAS PubMed
- D. J. Braden, C. A. Henao, J. Heltzel, C. C. Maravelias and J. A. Dumesic, Green Chem., 2011, 13, 1755–1765 RSC
- H. Ren, Y. Zhou and L. Liu, Bioresour. Technol., 2013, 129, 616–619 CrossRef CAS PubMed
- D. Scholz, C. Aellig and I. Hermans, ChemSusChem, 2014, 7, 268–275 CrossRef CAS PubMed
- X. Hu, Y. Song, L. Wu, M. Gholizadeh and C.-Z. Li, ACS Sustainable Chem. Eng., 2013, 1, 1593–1599 CrossRef CAS
- L. Peng, L. Lin, H. Li and Q. Yang, Appl. Energy, 2011, 88, 4590–4596 CrossRef CAS PubMed
- L.-C. Peng, L. Lin, J.-H. Zhang, J.-B. Shi and S.-J. Liu, Appl. Catal., A, 2011, 397, 259–265 CrossRef CAS PubMed
- Z. Zhang, W. Liu, H. Xie and Z. K. Zhao, Molecules, 2011, 16, 8463–8474 CrossRef CAS PubMed
- Z.-S. Liu, X.-L. Wu, K. Kida and Y.-Q. Tang, Bioresour. Technol., 2012, 119, 224–233 CrossRef CAS PubMed
- H. Choudhary, S. Nishimura and K. Ebitani, Appl. Catal., A, 2013, 458, 55–62 CrossRef CAS PubMed
- H.-F. Liu, F.-X. Zeng, L. Deng, B. Liao, H. Pang and Q.-X. Guo, Green Chem., 2013, 15, 81–84 RSC
- S. Van de Vyver, J. Geboers, S. Helsen, F. Yu, J. Thomas, M. Smet, W. Dehaen and B. F. Sels, Chem. Commun., 2012, 48, 3497–3499 RSC
- S. Van de Vyver, S. Helsen, J. Geboers, F. Yu, J. Thomas, M. Smet, W. Dehaen, Y. Román-Leshkov, I. Hermans and B. F. Sels, ACS Catal., 2012, 2, 2700–2704 CrossRef CAS
- L. Corbel-Demailly, B.-K. Ly, D.-P. Minh, B. Tapin, C. Especel, F. Epron, A. Cabiac, E. Guillon, M. Besson and C. Pinel, ChemSusChem, 2013, 6, 2388–2395 CrossRef CAS PubMed
- Z. Wu, S. Ge, C. Ren, M. Zhang, A. Yip and C. Xu, Green Chem., 2012, 14, 3336–3343 RSC
- J. Lee, Y. Xu and G. W. Huber, Appl. Catal., B, 2013, 140–141, 98–107 CrossRef CAS PubMed
- D. W. Rackemann and W. Doherty, Biofuels Bioprod. Biorefin., 2011, 34, 198–214 CrossRef
- C. M. Cai, T. Zhang, R. Kumar and C. E. Wyman, J. Chem. Technol. Biotechnol., 2014, 89, 2–10 CrossRef CAS
- M. Dusselier, P. V. Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 CAS
- M. Dusselier and B. F. Sels, Top. Curr. Chem., 2014, 353, 85–126 CrossRef
- C. B. Rasrendra, I. G. B. N. Makertihartha, S. Adisasmito and H. J. Heeres, Top. Catal., 2010, 53, 1241–1247 CrossRef CAS
- X. Wang, Y. Song, C. Huang, F. Liang and B. Chen, Green Chem., 2014, 16, 4234–4240 RSC
- A. Takagaki, S. Nishimura and K. Ebitani, Catal. Surv. Asia, 2012, 16, 164–182 CrossRef CAS
- R. Karinen, K. Vilonen and M. Niemelä, ChemSusChem, 2011, 4, 1002–1016 CrossRef CAS PubMed
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed
- X. C. Wang, Y. L. Song, C. P. Huang, J. B. Liang and B. H. Chen, Green Chem., 2014, 16, 4234–4240 RSC
- E. Lam, E. Majid, A. C. W. Leung, J. H. Chong, K. A. Mahmoud and J. H. T. Luong, ChemSusChem, 2011, 4, 535–541 CrossRef CAS PubMed
- S. Dutta, S. De, M. I. Alam, M. M. Abu-Omar and B. Saha, J. Catal., 2012, 288, 8–15 CrossRef CAS PubMed
- S. Lima, M. M. Antunes, M. Pillinger and A. A. Valente, ChemCatChem, 2011, 3, 1686–1706 CrossRef CAS
- V. Choudhary, S. H. Mushrif, C. Ho, A. Anderko, V. Nikolakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vlachos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS PubMed
- H. Choudhary, S. Nishimura and K. Ebitani, Chem. Lett., 2012, 41, 409–411 CrossRef CAS
- I. Agirrezabal-Telleria, J. Requies, M. B. Gueemez and P. L. Arias, Appl. Catal., B, 2012, 115–116, 169–178 CrossRef CAS PubMed
- J. Zhang, L. Lin and S. Liu, Energy Fuels, 2012, 26, 4560–4567 CrossRef CAS
- L. Cheng, X. Guo, C. Song, G. Yu, Y. Cui, N. Xue, L. Peng, X. Guo and W. Ding, RSC Adv., 2013, 3, 23228–23235 RSC
- J. Shi, H. Gao, Y. Xia, W. Li, H. Wang and C. Zheng, RSC Adv., 2013, 3, 7782–7790 RSC
- X. Hu, Y. Song, L. Wu, M. Gholizadeh and C.-Z. Li, ACS Sustainable Chem. Eng., 2013, 1, 1593–1599 CrossRef CAS
- Z. Wu, S. Ge, C. Ren, M. Zhang, A. Yip and C. Xu, Green Chem., 2012, 14, 3336–3343 RSC
- K. Yan, T. Lafleur, G. Wu, J. Liao, C. Ceng and X. Xie, Appl. Catal., A, 2013, 468, 52–58 CrossRef CAS PubMed
- S. K. Green, J. Lee, H. J. Kim, G. A. Tompsett, W. B. Kim and G. W. Huber, Green Chem., 2013, 15, 1869–1879 RSC
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC
- J. L. Ren, X. W. Peng, F. Peng and R. C. Sun, Adv. Mater. Res., 2011, 239–242, 463–467 CrossRef CAS
- J. Nie, J. Xie and H. Liu, J. Catal., 2013, 301, 83–91 CrossRef CAS PubMed
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef CAS PubMed
- Z. Tai, J. Zhang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2012, 48, 7052–7054 RSC
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS
- S. Leng, X. Wang, Q. Cai, F. Ma, Y. Liu and J. Wang, Bioresour. Technol., 2013, 149, 341–345 CrossRef CAS PubMed
- K. S. Arias, S. I. Al-Resayes, M. J. Climent, A. Corma and S. Iborra, ChemSusChem, 2013, 6, 123–131 CrossRef CAS PubMed
- S. Dabral, S. Nishimura and K. Ebitani, ChemSusChem, 2014, 7, 260–267 CrossRef CAS PubMed
- K. Yan, J. Liao, X. Wu and X. Xie, RSC Adv., 2013, 3, 3853–3856 RSC
- A. M. R. Galletti, C. Antonetti, V. De Luise and M. Martinelli, Green Chem., 2012, 14, 688–694 RSC
- T. Runge and C. Zhang, Ind. Eng. Chem. Res., 2012, 51, 3265–3270 CrossRef CAS
- J. van Buijtenen, J.-P. Lange, L. Espinosa Alonso, W. Spiering, R. F. Polmans and R. J. Haan, ChemSusChem, 2013, 6, 2132–2136 CrossRef CAS PubMed
- T. H. Kim, Y. J. Jeon, K. K. Oh and T. H. Kim, Korean J. Chem. Eng., 2013, 30, 1339–1346 CrossRef CAS PubMed
- L. Yan, D. D. Laskar, S.-J. Lee and B. Yang, RSC Adv., 2013, 3, 24090–24098 RSC
- T. Deng, X. Cui, Y. Qi, Y. Wang, X. Hou and Y. Zhu, Green Chem., 2012, 48, 5494–5496 CAS
- J. Deng, Y. Wang, T. Pan, Q. Xu, Q.-X. Guo and Y. Fu, ChemSusChem, 2013, 6, 1163–1167 CrossRef CAS PubMed
- C. Delhomme, L.-A. Schaper, M. Zhang-Presse, G. Raudaschl-Sieber, D. Weuster-Botz and F. E. Kuehn, J. Organomet. Chem., 2013, 724, 297–299 CrossRef CAS PubMed
- O. M. Gazit and A. Katz, Green Chem., 2012, 5, 1542–1548 CAS
- K. R. Enslow and A. T. Bell, RSC Adv., 2012, 2, 10028–10036 RSC
- M. Tan, L. Zhao and Y. Zhang, Biomass Bioenergy, 2011, 35, 1367–1370 CrossRef CAS PubMed
- J. Long, B. Guo, X. Li, Y. Jiang, F. Wang, S. C. Tsang, L. Wang and K. M. K. Yu, Green Chem., 2011, 13, 2334–2338 RSC
- J. Long, X. Li, B. Guo, F. Wang, Y. Yu and L. Wang, Green Chem., 2012, 14, 1935–1941 RSC
- B. Kim, J. Jeong, D. Lee, S. Kim, H.-J. Yoon, Y.-S. Lee and J. K. Cho, Green Chem., 2011, 13, 1503–1506 RSC
- F. Tao, H. Song and L. Chou, J. Mol. Catal., 2012, 357, 11–18 CrossRef CAS PubMed
- F. Tao, H. Song, J. Yang and L. Chou, Carbohydr. Polym., 2011, 85, 363–368 CrossRef CAS PubMed
- T. Ståhlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Chem. – Eur. J., 2011, 17, 1456–1464 CrossRef PubMed
- X. Qi, H. Guo, L. Li and R. L. Smith, ChemSusChem, 2012, 5, 2215–2220 CrossRef CAS PubMed
- L. Zhang, H. Yu, P. Wang, H. Dong and X. Peng, Bioresour. Technol., 2013, 130, 110–116 CrossRef CAS PubMed
- Y. J. Pagan-Torres, T. Wang, J. M. R. Gallo, B. H. Shanks and J. A. Dumesic, ACS Catal., 2012, 2, 930–934 CrossRef CAS
- Y. Zhang, V. Degirmenci, C. Li and E. J. M. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS PubMed
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC
- J. Wang, J. Ren, X. Liu, G. Lu and Y. Wang, AIChE J., 2013, 59, 2558–2566 CrossRef CAS
- X.-L. Shi, M. Zhang, Y. Li and W. Zhang, Green Chem., 2013, 15, 3438–3445 RSC
-
F. W. Lichtenthaler, Carbohydrates as Organic Raw Materials, Ullmann's Encyclopedia of Industrial Chemistry, 2010, pp. 583–616 Search PubMed
- J. Giacometti, N. Wolf, Z. Gomzi and C. Milin, React. Kinet. Catal. Lett., 1996, 59, 235–240 CrossRef CAS
- J. Smidrkal, R. Cervenkova and V. Filip, Eur. J. Lipid Sci. Technol., 2004, 106, 851–855 CrossRef CAS
- M. Meiland, T. Heinze, W. Guenther and T. Liebert, Tetrahedron Lett., 2009, 50, 469–472 CrossRef CAS PubMed
- M. Ogata, T. Hattori, R. Takeuchi and T. Usui, Carbohydr. Res., 2010, 345, 230–234 CrossRef CAS PubMed
- Z. Zhu, S. Li, R. Liu, J. Yuan, H. Wang, Y. Zhang and Y. Liu, Chin. J. Chem., 2010, 28, 2245–2248 CrossRef CAS
- A. Assalit, T. Billard, S. Chambert, B. R. Langlois, Y. Queneau and D. Coe, Tetrahedron: Asymmetry, 2009, 20, 593–601 CrossRef CAS PubMed
- H.-W. Shih, K.-T. Chen and W.-C. Cheng, Tetrahedron Lett., 2012, 53, 243–246 CrossRef CAS PubMed
-
K. J. Parker, R. A. Khan and K. S. Mufti, US/1976/3996206, 1976
- M. A. Cruces, F. J. Plou, M. Ferrer, M. Bernabé and A. Ballesteros, J. Am. Oil Chem. Soc., 2001, 78, 541–546 CrossRef CAS PubMed
- V. Molinier, J. Fitremann, A. Bouchu and Y. Queneau, Tetrahedron: Asymmetry, 2004, 15, 1753–1762 CrossRef CAS PubMed
- K. Baczko, C. Nugier-Chauvin, J. Banoub, P. Thibault and D. Plusquellec, Carbohydr. Res., 1995, 269, 79–88 CrossRef CAS
-
H. G. Bazin and R. J. Linhardt, US/2001/6184196, 2001
-
K. D. O. Vigier and F. Jérôme, in Carbohydrates in Sustainable Development II, ed. A. P. Rauter, P. Vogel and Y. Queneau, Springer, Berlin Heidelberg, 2010, pp. 63–92 Search PubMed
- A.-L. Le Coënt, M. Tayakout-Fayolle, F. Couenne, S. Briançon, J. Lieto, J. Fitremann-Gagnaire, Y. Queneau and A. Bouchu, Chem. Eng. Sci., 2003, 58, 367–376 CrossRef
- F. Jérôme, G. Kharchafi, I. Adam and J. Barrault, Green Chem., 2004, 6, 72–74 RSC
- A. Lavergne, Y. Zhu, V. Molinier and J.-M. Aubry, Colloids Surf., A, 2012, 404, 56–62 CrossRef CAS PubMed
- M. Durand, V. Molinier, T. Féron and J.-M. Aubry, Prog. Org. Coat., 2010, 69, 344–351 CrossRef CAS PubMed
- A. Lavergne, Y. Zhu, A. Pizzino, V. Molinier and J.-M. Aubry, J. Colloid Interface Sci., 2011, 360, 645–653 CrossRef CAS PubMed
- Y. Zhu, V. Molinier, M. Durand, A. Lavergne and J.-M. Aubry, Langmuir, 2009, 25, 13419–13425 CrossRef CAS PubMed
- Y. Zhu, M. Durand, V. Molinier and J.-M. Aubry, Green Chem., 2008, 10, 532–540 RSC
- C. Berini, A. Lavergne, V. Molinier, F. Capet, E. Deniau and J.-M. Aubry, Eur. J. Org. Chem., 2013, 1937–1949 CrossRef CAS
- S. Habib, F. Larnaud, E. Pfund, T. Lequeux, B. Fenet, P. G. Goekjian and D. Gueyrard, Eur. J. Org. Chem., 2013, 1872–1875 CrossRef CAS
- M. Tober and J. Thiem, Eur. J. Org. Chem., 2013, 566–577 CrossRef CAS
- Z. Rapi, P. Bakó, G. Keglevich, Á. Szöllősy, L. Drahos and L. Hegedűs, Carbohydr. Res., 2013, 365, 61–68 CrossRef CAS PubMed
- J. Gagnaire, G. Toraman, G. Descotes, A. Bouchu and Y. Queneau, Tetrahedron Lett., 1999, 40, 2757–2760 CrossRef CAS
- M. Danel, J. Gagnaire and Y. Queneau, J. Mol. Catal. A: Chem., 2002, 184, 131–138 CrossRef CAS
- K. Hill, B. Gruber and K. J. Weese, Tetrahedron Lett., 1994, 35, 4541–4542 CrossRef CAS
- I. Pennequin, J. Meyer, I. Suisse and A. Mortreux, J. Mol. Catal. A: Chem., 1997, 120, 139–142 CrossRef CAS
- V. Desvergnes-Breuil, C. Pinel and P. Gallezot, Green Chem., 2001, 3, 175–177 RSC
- J. Lai, S. Bigot, M. Sauthier, V. Molinier, I. Suisse, Y. Castanet, J.-M. Aubry and A. Mortreux, ChemSusChem, 2011, 4, 1104–1111 CrossRef CAS PubMed
- B. Estrine, S. Bouquillon, F. Hénin and J. Muzart, Appl. Organomet. Chem., 2007, 21, 945–946 CrossRef CAS
- P. J. C. Hausoul, P. C. A. Bruijnincx, R. J. M. KleinGebbink and B. M. Weckhuysen, ChemSusChem, 2009, 2, 855–858 CrossRef CAS PubMed
- P. J. C. Hausoul, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. M. Klein Gebbink, Pure Appl. Chem., 2012, 84 Search PubMed
- Y. Chun, S. Yan, X. Li, N. Ding, W. Zhang, P. Wang, M. Li and Y. Li, Tetrahedron Lett., 2011, 52, 6196–6198 CrossRef CAS PubMed
- R. Pierre, I. Adam, J. Fitremann, F. Jérôme, A. Bouchu, G. Courtois, J. Barrault and Y. Queneau, C. R. Chim., 2004, 7, 151–160 CrossRef CAS PubMed
- M. Banu, S. Sivasanker, T. M. Sankaranarayanan and P. Venuvanalingam, Catal. Commun., 2011, 12, 673–677 CrossRef CAS PubMed
- L. Ye, X. Duan, H. Lin and Y. Yuan, Catal. Today, 2012, 183, 65–71 CrossRef CAS PubMed
- K. L. Deutsch, D. G. Lahr and B. H. Shanks, Green Chem., 2012, 14, 1635–1642 RSC
- R. V. Chaudhari, A. Torres, X. Jin and B. Subramaniam, Ind. Eng. Chem. Res., 2013, 52, 15226–15243 CrossRef CAS
- J. ten Dam and U. Hanefeld, ChemSusChem, 2011, 4, 1017–1034 CrossRef CAS PubMed
- J. Sun and H. Liu, Catal. Today, 2014, 234, 75–82 CrossRef CAS PubMed
- Y. T. Kim, J. A. Dumesic and G. W. Huber, J. Catal., 2013, 304, 72–85 CrossRef CAS PubMed
- M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854–1857 CrossRef CAS PubMed
- B. Blanc, A. Bourrel, P. Gallezot, T. Haas and P. Taylor, Green Chem., 2000, 2, 89–91 RSC
- Z. Huang, J. Chen, Y. Jia, H. Liu, C. Xia and H. Liu, Appl. Catal., B, 2014, 147, 377–386 CrossRef CAS PubMed
- L. Hu, G. Zhao, W. Hao, X. Tang, Y. Sun, L. Lin and S. Liu, RSC Adv., 2012, 2, 11184–11206 RSC
- M. Yabushita, H. Kobayashi and A. Fukuoka, Appl. Catal., B, 2014, 145, 1–9 CrossRef CAS PubMed
- S. Tabasso, E. Montoneri, D. Carnaroglio, M. Caporaso and G. Cravotto, Green Chem., 2013, 16, 73–76 RSC
- R. Mao, Q. Zhao, G. Dima and D. Petraccone, Catal. Lett., 2011, 141, 271–276 CrossRef
- Y. Yang, C.-W. Hu and M. M. Abu-Omar, ChemSusChem, 2012, 5, 405–410 CrossRef CAS PubMed
- Z. Tang, W. Deng, Y. Wang, E. Zhu, X. Wan, Q. Zhang and Y. Wang, ChemSusChem, 2014, 7, 1557–1567 CrossRef CAS PubMed
- F.-F. Wang, C.-L. Liu and W.-S. Dong, Green Chem., 2013, 15, 2091–2095 RSC
- Y. Wang, W. Deng, B. Wang, Q. Zhang, X. Wan, Z. Tang, Y. Wang, C. Zhu, Z. Cao, G. Wang and H. Wan, Nat. Commun., 2013, 4 Search PubMed
- J. Zhang, M. Sun, X. Liu and Y. Han, Catal. Today, 2014, 233, 77–82 CrossRef CAS PubMed
- S. Zhao, M. Cheng, J. Li, J. Tian and X. Wang, Chem. Commun., 2011, 47, 2176–2178 RSC
- S. Van de Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 CAS
- R. Weingarten, W. C. Conner and G. W. Huber, Energy Environ. Sci., 2012, 5, 7559–7574 CAS
- S. Shen, B. Cai, C. Wang, H. Li, G. Dai and H. Qin, Appl. Catal., A, 2014, 473, 70–74 CrossRef CAS PubMed
- S. Vande Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS
- J. A. Geboers, S. V. de Vyver, R. Ooms, B. O. de Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 Search PubMed
- W. Deng, M. Liu, X. Tan, Q. Zhang and Y. Wang, J. Catal., 2010, 271, 22–32 CrossRef CAS PubMed
- G. Liang, H. Cheng, W. Li, L. He, Y. Yu and F. Zhao, Green Chem., 2012, 14, 2146–2149 RSC
- J. Pang, A. Wang, M. Zheng, Y. Zhang, Y. Huang, X. Chen and T. Zhang, Green Chem., 2012, 14, 614–617 RSC
- X. Wang, L. Meng, F. Wu, Y. Jiang, L. Wang and X. Mu, Green Chem., 2012, 14, 758–765 RSC
- S. Vande Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 5, 1549–1558 CrossRef CAS PubMed
- M. Mariani, F. Zaccheria, R. Psaro and N. Ravasio, Catal. Commun., 2014, 44, 19–23 CrossRef CAS PubMed
- Y. Morita, S. Furusato, A. Takagaki, S. Hayashi, R. Kikuchi and S. T. Oyama, ChemSusChem, 2014, 7, 748–752 CrossRef CAS PubMed
- Y. Zuo, Y. Zhang and Y. Fu, ChemCatChem, 2014, 6, 753–757 CrossRef CAS
- Z. Xiao, S. Jin, M. Pang and C. Liang, Green Chem., 2013, 15, 891–895 RSC
- R. Ooms, M. Dusselier, J. A. Geboers, B. O. de Beeck, R. Verhaeven, E. Gobechiya, J. A. Martens, A. Redl and B. F. Sels, Green Chem., 2014, 16, 695–707 RSC
- J. Xi, Y. Zhang, D. Ding, Q. Xia, J. Wang, X. Liu, G. Lu and Y. Wang, Appl. Catal., A, 2014, 469, 108–115 CrossRef CAS PubMed
- J. Li, H. S. M. P. Soares, J. A. Moulijn and M. Makkee, Catal. Sci. Technol., 2013, 3, 1565–1572 CAS
- F. Chambon, F. Rataboul, C. Pinel, A. Cabiac, E. Guillon and N. Essayem, ChemSusChem, 2013, 6, 500–507 CrossRef CAS PubMed
- T. Deng and H. Liu, Green Chem., 2012, 15, 116–124 RSC
- Y. Wu, F. Gu, G. Xu, Z. Zhong and F. Su, Bioresour. Technol., 2013, 137, 311–317 CrossRef CAS PubMed
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC
- Y.-B. Huang and Y. Fu, Green Chem., 2013, 15, 1095–1111 RSC
- M. Cheng, T. Shi, S. Wang, H. Guan, C. Fan and X. Wang, Catal. Commun., 2011, 12, 1483–1487 CrossRef CAS PubMed
- B. OpdeBeeck, J. Geboers, S. Vande Vyver, J. Van Lishout, J. Snelders, W. J. J. Huijgen, C. M. Courtin, P. A. Jacobs and B. F. Sels, ChemSusChem, 2013, 6, 199–208 CrossRef CAS PubMed
- S. B. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS
- P. Sun, X. Long, H. He, C. Xia and F. Li, ChemSusChem, 2013, 6, 2190–2197 CrossRef CAS PubMed
- N. Villandier and A. Corma, ChemSusChem, 2011, 4, 508–513 CrossRef CAS PubMed
- W. Deng, M. Liu, Q. Zhang and Y. Wang, Catal. Today, 2011, 164, 461–466 CrossRef CAS PubMed
- J. Chen, X. Fang, X. Duan, L. Ye, H. Lin and Y. Yuan, Green Chem., 2013, 16, 294–302 RSC
- S. Dora, T. Bhaskar, R. Singh, D. V. Naik and D. K. Adhikari, Bioresour. Technol., 2012, 120, 318–321 CrossRef CAS PubMed
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047–8050 CrossRef CAS PubMed
- S. Mateyawa, D. F. Xie, R. W. Truss, P. J. Halley, T. M. Nicholson, J. L. Shamshina, R. D. Rogers, M. W. Boehm and T. McNally, Carbohydr. Polym., 2013, 94, 520–530 CrossRef CAS PubMed
- J. B. Binder and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4516–4521 CrossRef CAS PubMed
- Y. Su, H. M. Brown, X. Huang, X. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117–122 CrossRef CAS PubMed
- Y. Su, H. M. Brown, G. Li, X. Zhou, J. E. Amonette, J. L. Fulton, D. M. Camaioni and Z. C. Zhang, Appl. Catal., A, 2011, 391, 436–442 CrossRef CAS PubMed
- P. Wang, H. Yu, S. Zhan and S. Wang, Bioresour. Technol., 2011, 102, 4179–4183 CrossRef CAS PubMed
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed
- S. Dutta, S. De, M. I. Alam, M. M. Abu-Omar and B. Saha, J. Catal., 2012, 288, 8–15 CrossRef CAS PubMed
- A. A. Dwiatmoko, J. W. Choi, D. J. Suh, Y.-W. Suh and H. H. Kung, Appl. Catal., A, 2010, 387, 209–214 CrossRef CAS PubMed
- H. Cai, C. Li, A. Wang, G. Xu and T. Zhang, Appl. Catal., B, 2012, 123–124, 333–338 CrossRef CAS PubMed
- H. Guo, X. Qi, L. Li and R. L. Smith Jr., Bioresour. Technol., 2012, 116, 355–359 CrossRef CAS PubMed
- I. A. Ignatyev, C. Van Doorslaer, P. G. N. Mertens, K. Binnemans and D. E. De Vos, ChemSusChem, 2010, 3, 91–96 CrossRef CAS PubMed
- S. M. Sen, J. B. Binder, R. T. Raines and C. T. Maravelias, Biofuels Bioprod. Biorefin., 2012, 6, 444–452 CrossRef CAS
- B. R. Caes, J. B. Binder, J. J. Blank and R. T. Raines, Green Chem., 2011, 13, 2719–2722 RSC
- J. Chen, G. Zhao and L. Chen, RSC Adv., 2014, 4, 4194–4202 RSC
- Z. Sun, M. Cheng, H. Li, T. Shi, M. Yuan, X. Wang and Z. Jiang, RSC Adv., 2012, 2, 9058–9065 RSC
- S. R. Collinson and W. Thielemans, Coord. Chem. Rev., 2010, 254, 1854–1870 CrossRef CAS PubMed
- A. e. j. de Nooy, A. c. Besemer and H. van Bekkum, Recl. Trav. Chim. Pays-Bas, 1994, 113, 165–166 CrossRef CAS
- P. L. Bragd, H. van Bekkum and A. C. Besemer, Top. Catal., 2004, 27, 49–66 CrossRef CAS
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2008, 10, 162–165 CrossRef PubMed
- S. Fujisawa, Y. Okita, H. Fukuzumi, T. Saito and A. Isogai, Carbohydr. Polym., 2011, 84, 579–583 CrossRef CAS PubMed
- M. Hirota, K. Furihata, T. Saito, T. Kawada and A. Isogai, Angew. Chem., Int. Ed., 2010, 49, 7670–7672 CrossRef CAS PubMed
- P. Parovuori, A. Hamunen, P. Forssell, K. Autio and K. Poutanen, Starch/Staerke, 1995, 47, 19–23 CrossRef CAS
- A. B. Sorokin, S. L. Kachkarova-Sorokina, C. Donzé, C. Pinel and P. Gallezot, Top. Catal., 2004, 27, 67–76 CrossRef CAS
- P. Tolvanen, A. Sorokin, P. Mäki-Arvela, S. Leveneur, D. Y. Murzin and T. Salmi, Ind. Eng. Chem. Res., 2010, 50, 749–757 CrossRef
- P. Tolvanen, P. Mäki-Arvela, A. B. Sorokin, T. Salmi and D. Y. Murzin, Chem. Eng. J., 2009, 154, 52–59 CrossRef CAS PubMed
-
R. Saladino and A. Farina, PCT/EP2009/059180, 2013
-
K. A. J. Huvaere, J.-B. F. G. Joos, A. Van and P. E. M. J. Willems, WO/2013/030264, 2013
- H. Namazi and A. Dadkhah, Carbohydr. Polym., 2010, 79, 731–737 CrossRef CAS PubMed
- T. A. Dankovich and Y.-L. Hsieh, Cellulose, 2007, 14, 469–480 CrossRef CAS
- J. Mesnager, C. Quettier, A. Lambin, F. Rataboul and C. Pinel, ChemSusChem, 2009, 2, 1125–1129 CrossRef CAS PubMed
- J. Mesnager, C. Quettier, A. Lambin, F. Rataboul, A. Perrard and C. Pinel, Green Chem., 2010, 12, 475–482 RSC
-
F. Peng, J. L. Ren, F. Xu and R.-C. Sun, in Sustainable
Production of Fuels, Chemicals, and Fibers from Forest Biomass, 2011, pp. 219–259 Search PubMed
- P. J. C. Hausoul, A. N. Parvulescu, M. Lutz, A. L. Spek, P. C. A. Bruijnincx, R. J. M. KleinGebbink and B. M. Weckhuysen, ChemCatChem, 2011, 3, 845–852 CrossRef CAS
- C. Aouf, D. Harakat, J. Muzart, B. Estrine, S. Marinkovic, C. Ernenwein and J. Le Bras, ChemSusChem, 2010, 3, 1200–1203 CrossRef CAS PubMed
- S. K. Guha, H. Kobayashi, K. Hara, H. Kikuchi, T. Aritsuka and A. Fukuoka, Catal. Commun., 2011, 12, 980–983 CrossRef CAS PubMed
- C. Li, M. Zheng, A. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383–6390 CAS
- M. R. Sierakowski, M. Milas, J. Desbrières and M. Rinaudo, Carbohydr. Polym., 2000, 42, 51–57 CrossRef CAS
- V. Kisonen, C. Xu, P. Eklund, H. Lindqvist, A. Sundberg, A. Pranovich, J. Sinkkonen, F. Vilaplana and S. Willför, Carbohydr. Polym., 2014, 99, 755–764 CrossRef CAS PubMed
- V. Bigand, C. Pinel, D. Da Silva Perez, F. Rataboul, P. Huber and M. Petit-Conil, Carbohydr. Polym., 2011, 85, 138–148 CrossRef CAS PubMed
- X. Peng, J. Ren, L. Zhong and R. Sun, Carbohydr. Polym., 2011, 86, 1768–1774 CrossRef CAS PubMed
- X. Peng, J. Ren and R. Sun, Biomacromolecules, 2010, 11, 3519–3524 CrossRef CAS PubMed
- A. Ayoub, R. A. Venditti, J. J. Pawlak, H. Sadeghifar and A. Salam, Ind. Crops Prod., 2013, 44, 306–314 CrossRef CAS PubMed
- L.-M. Zhang, T.-Q. Yuan, F. Xu and R.-C. Sun, Ind. Crops Prod., 2013, 45, 52–57 CrossRef CAS PubMed
- F.-Z. Belmokaddem, C. Pinel, P. Huber, M. Petit-Conil and D. Da Silva Perez, Carbohydr. Res., 2011, 346, 2896–2904 CrossRef CAS PubMed
- A. Salam, R. A. Venditti, J. J. Pawlak and K. El-Tahlawy, Carbohydr. Polym., 2011, 84, 1221–1229 CrossRef CAS PubMed
- V. Kisonen, P. Eklund, M. Auer, R. Sjöholm, A. Pranovich, J. Hemming, A. Sundberg, V. Aseyev and S. Willför, Carbohydr. Res., 2012, 352, 151–158 CrossRef CAS PubMed
- Z. Liu, Y. Ni, P. Fatehi and A. Saeed, Biomass Bioenergy, 2011, 35, 1789–1796 CrossRef CAS PubMed
- X. Peng, J. Ren and R. Sun, Carbohydr. Polym., 2011, 83, 1922–1928 CrossRef CAS PubMed
- H. M. Li, X. H. Chen, J. H. Zhou, Y. Han and G. W. Sun, Adv. Mater. Res., 2013, 821–822, 1060–1064 CAS
- N. Pahimanolis, A. Sorvari, N. D. Luong and J. Seppälä, Carbohydr. Polym., 2014, 102, 637–644 CrossRef CAS PubMed
- M. Dusselier, M. Mascal and B. Sels, Top. Curr. Chem., 2014, 353, 1–40 CrossRef
- Nexant , (2014, March) The Latest Thinking on Biorenewables. Presented at 9th Annual World Bio Markets.
| This journal is © The Royal Society of Chemistry 2015 |