
Glimpses of the modification of perovskite with graphene-analogous materials in photocatalytic applications
Saumyaprava
Acharya
,
Satyabadi
Martha
,
Prakash Chandra
Sahoo
* and
Kulamani
Parida
*
Centre for Nano Science and Nano Technology, ITER, Siksha ‘O’ Anusandhan University, Bhubaneswar-751030, Odisha, India. E-mail: kulamaniparida@soauniversity.ac.in; prakashchandrasahu@soauniversity.ac.in; Fax: +91-674-2351217; Tel: +91-674-2350181
First published on 31st July 2015
Abstract
In the 21st century, at a global level, energy and environment are the most important issues. The green approach to solve these issues is photocatalysis. In this regard, perovskite-based materials owing to their inexpensiveness and non-toxicity have attracted significant interest. The photocatalytic activity of perovskite and layered perovskite needs some modification to provide better activity than that of the neat one. The modification can be done by replacing cations, formation of heterojunctions or formation of composites. This critical review presents and discusses the current development of perovskite, layered perovskite, and their composites especially with π-conjugated carbon materials towards photocatalytic applications. After introduction of the synthesis methods of perovskite and layered perovskite as well as their properties, we focus on the description of various methods to synthesize their composites, mainly those with graphene, carbon nano-tubes and carbon nitrides. Particular importance is placed on strategies for the optimization of composite properties. Lastly, the advantages of perovskite and layered perovskite based composites in applications such as photocatalytic H2 production and pollutant degradation are described in detail.
 Saumyaprava Acharya | Ms Saumyaprava Acharya received her bachelor's degree from College of Basic Science and Humanities, OUAT, Bhubaneswar in the year 2010 and M.Sc. from Utkal University, Bhubaneswar in the year 2012. Then, she joined as a Ph.D. student (2014) under the supervision of Prof. K. M. Parida in the Centre for Nano Science and Nano Technology, ITER, SOA University. Her research area focuses on the development of layered perovskite modified π-conjugated carbon based materials for the production of hydrogen energy and environmental pollution abatement. |
 Satyabadi Martha | Dr Satyabadi Martha received his Master of Science (M.Sc.) in 2008 from Utkal University, Bhubaneswar, India. Then, he received his PhD degree from North Orissa University under Prof. Kulamani Parida. His research work focuses on the development of visible light responsive photocatalysts for hydrogen production, CO2 reduction and pollution abatement. He spent 1 year at UNIST, South Korea as a post doc. researcher with Prof. Jae Sung Lee. He has published 24 research articles in international journals and 3 national patents. Presently (from Nov. 2014), he works as an assistant professor at the Centre for Nano Science and Nano Technology, ITER, SOA University. |
 Prakash Chandra Sahoo | Dr Prakash C. Sahoo received his Master of Science (M.Sc., 2008) from Utkal University, India. He then moved to South Korea for his doctoral degree. In 2013 he completed his PhD study on resources recycling from University of Science and Technology (UST), South Korea and then started his career as a post-doc in Prof. Jay H. Lee's group at Korea Advanced Institute of Science and Technology (KAIST). He has published 14 research articles in international journals and 3 international patents. Currently (from Jan. 2015), he works as an assistant professor in Center for Nanoscience and Nanotechnology, Department of Chemistry at SOA University. |
 Kulamani Parida | Prof. K. M. Parida currently works as Professor in Chemistry and Director, Centre for Nano Science and Nano Technology, Siksha ‘O’ Anusandhan University, India. He is the author of more than 285 research articles in international journals, 18 national and international patents and also three book chapters. His research interests focus on the design and development of materials such as metal oxides, metal phosphates, metal sulfates, cationic and anionic clays, perovskites, zeolites, graphene, C3N4 and nanometal/metal oxide/complex promoted mesoporous materials, naturally occurring materials such as manganese nodules, manganese nodules leach residue, manganese oxides of natural origin and for energy and environmental applications. |
1. Introduction
Sunlight is the most abundant clean source of energy. Photocatalysts convert this energy into clean hydrogen energy by splitting water, and decompose harmful organic and inorganic pollutants. Significant progress in this regard led to the development of photocatalysts applicable to a wide region of the solar spectrum. Among the various materials that have been developed so far, perovskite based materials have attracted much attention due to their versatile applications.1Typically perovskites are binary metal oxides with general formula ABO3. The crystal structure in ABO3 has cubic structure with a space group Pmm or monoclinic or triclinic, and symmetry is distorted orthorhombic, rhombohedral or tetragonal.1 The larger A-site metal cation is often coordinated to 12 oxygen anions and the B-site cation is occupying octahedral interstices in the oxygen framework.2 Sometimes the oxygen atom is replaced by the halides resulting in the ABX3 type of perovskite. Among ABX3 perovskites, special attention has been paid to those with the ideal cubic structures or structures that are slightly distorted by cation displacements. Additionally, groups formed by compounds with hexagonal-close-packed structures which are closely related to the cubic ones showed interesting properties. The ABX3 materials can occur in various forms such as A1+B2+X3, A2+B4+X3 and A3+B+3X3.3 Due to the large stability range of the structure the oxygen content and the defect can vary with the composition of the binary metal oxide. Likewise, the layered perovskites consist of infinite 2D slabs of the ABO3 type structure which are separated by some motifs (Fig. 1). The stability of the perovskite is summarized by the Goldschmidt tolerance factor, t:
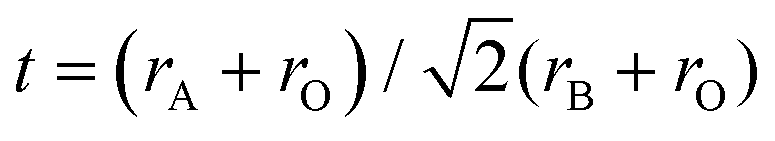
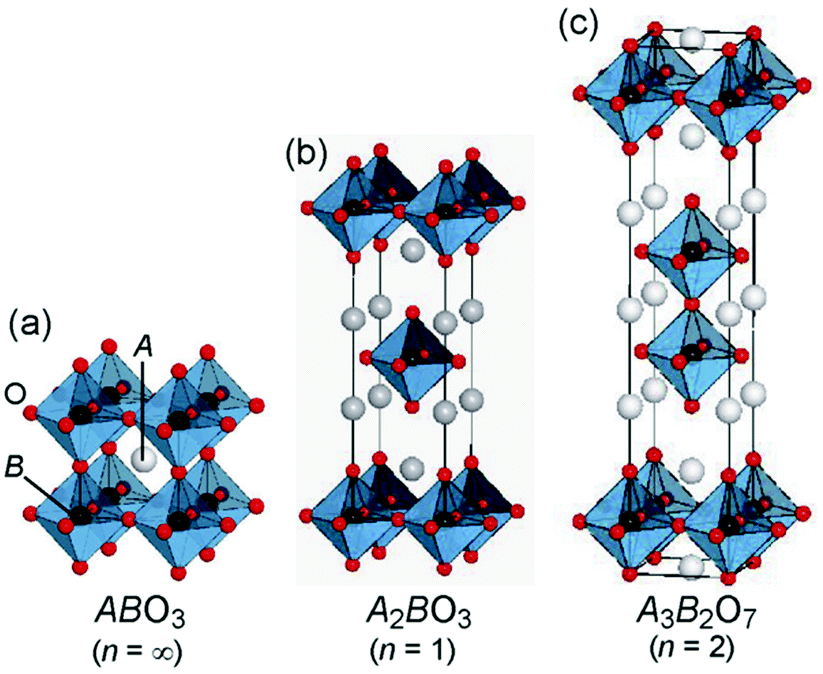 | ||
| Fig. 1 General structure of perovskite and layered perovskite (reproduced from ref. 8). | ||
For the perovskite structure the Goldschmidt tolerance factor lies between 0.75 and 1.00.5 A large combination of A and B cations forms a stable perovskite structure. A, B and oxygen are partially substituted by suitable ions based on electro-neutrality arguments.6,7 The presence of multiple oxidation states of metals is generally responsible for the catalytic activity of perovskite type oxides.
The performance of the perovskite and layered perovskite can be improved towards photocatalysis when ‘A’ and ‘B’ ions are partially substituted by suitable species. In perovskite and layered perovskite structures, the A-site ion is normally an alkaline earth or rare earth metal and the B-site ion could be a 3d, 4d or 5d transition metal element.
In the past few years, several perovskite and layered perovskite type oxides such as LaFeO3, SrTiO3, NaTaO3, K2SrTa2O7, YInO3, and KNbO3 have been found to show photocatalytic activity under visible light irradiation towards hydrogen production and environmental pollutant degradation.9–14 However, the photocatalytic activity of these oxides is still far from being satisfactory. This is due to the wide band gap and low surface area of perovskite. So, it needs some modification to enhance the photocatalytic activity. The modification can be done by several methods: doping, surface sensitization, phase/morphological control, noble metal loading, heterostructural construction etc.
Many reviews and books have been published on photocatalytic water splitting and organic contaminant degradation. In this critical review, we focus on the recent development of the composites of perovskite and layered perovskite with π-conjugated carbon materials towards photocatalytic hydrogen energy production and degradation of organic compounds. After introducing the synthesis of nano-scaled perovskite and layered perovskite composites with π-conjugated carbon materials, particular attention is focused on their surface and bulk characterization as well as applications towards photocatalytic reactions.
2. Brief sketch of the properties of graphene analogous materials
2.1. Graphene
Graphene is a thin layered honeycomb lattice consisting of a two-dimensional single-layer sheet of sp2-hybridized carbon atoms. For the first time, Stoller et al. reported that graphene is a single layer of graphite in 1986.15 It remains one of the emerging materials having superior properties, such as high electrical conductivity (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 V−1 s−1 at room temperature), high surface area (around 2600 m2 g−1), being optically transparent, etc.,15 which forces the photocatalytic researcher to make its hybrid with other semiconductor metal oxides. Graphene shows high crystal quality which helps the charge carriers to travel thousands of interatomic distances without scattering. Recently, intensive effort was made to apply graphene materials in electronic, optoelectronic, capacitor, and sensing applications. The primary role of graphene toward this composite semiconductor photocatalyst system is to delocalize the photo-generated electron through its π network, which inhibits the recombination process and hence improves the photocatalytic performance (Fig. 2). In this regard our group has reported various graphene hybrid materials such as RGO–VPO,16 reduced graphene oxide/InGaZn mixed oxide17 and CaTiO3 graphene nanocomposites,18 FeOOH-nanorod-RGO composite,19 α-Fe2O3 nanorod/RGO composite,20 and Gd(OH)3 nanorod-RGO composite.21
000 m2 V−1 s−1 at room temperature), high surface area (around 2600 m2 g−1), being optically transparent, etc.,15 which forces the photocatalytic researcher to make its hybrid with other semiconductor metal oxides. Graphene shows high crystal quality which helps the charge carriers to travel thousands of interatomic distances without scattering. Recently, intensive effort was made to apply graphene materials in electronic, optoelectronic, capacitor, and sensing applications. The primary role of graphene toward this composite semiconductor photocatalyst system is to delocalize the photo-generated electron through its π network, which inhibits the recombination process and hence improves the photocatalytic performance (Fig. 2). In this regard our group has reported various graphene hybrid materials such as RGO–VPO,16 reduced graphene oxide/InGaZn mixed oxide17 and CaTiO3 graphene nanocomposites,18 FeOOH-nanorod-RGO composite,19 α-Fe2O3 nanorod/RGO composite,20 and Gd(OH)3 nanorod-RGO composite.21
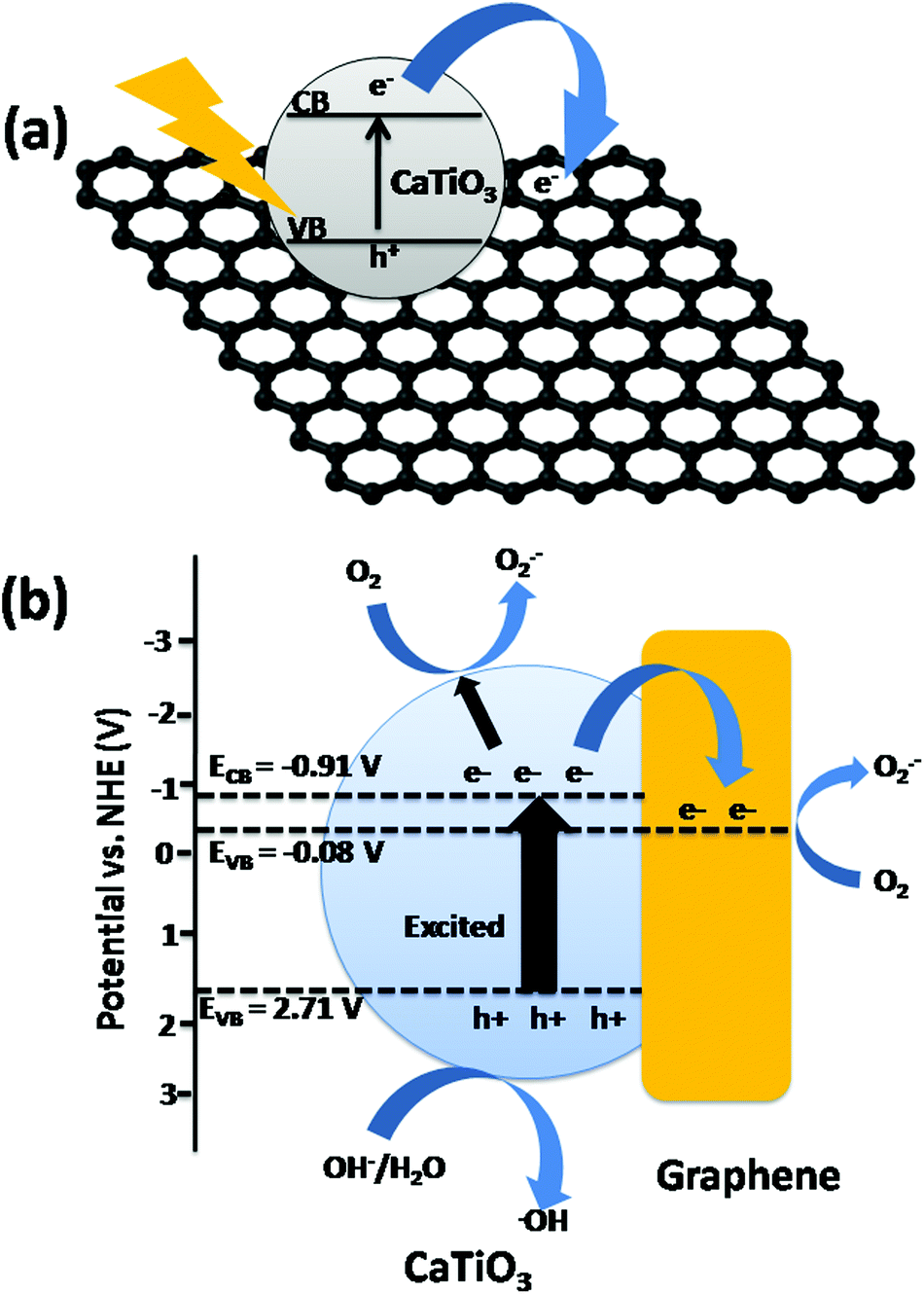 | ||
| Fig. 2 Schematic diagram of the dye degradation mechanism by the graphene–CaTiO3 composite. | ||
Xian et al.18 reported that the degradation of the methyl orange dye is highly enhanced by CaTiO3 when it makes a composite with graphene. The electron–hole recombination rate decreased in the composite, resulting in an enhancement of the photocatalytic activity. When light falls on the catalyst having energy higher than the band gap energy, electrons are excited from the valence band to the conduction band, creating electron–hole pairs. The electron from the conduction band is channelized into the graphene sheet, hence the electron–hole recombination rate decreases, resulting in an enhancement of the photocatalytic activity. The conduction band potential of CaTiO3 was calculated to be −0.91 V vs. the normal hydrogen electrode (NHE) using the following relation: ECB = X − Ee − 0.5Eg, where X represents the absolute electro-negativity of CaTiO3, Ee represents the energy of free electrons on the hydrogen scale and Eg represents the band gap energy of CaTiO3. The potential of graphene is about −0.08 V vs. NHE, which is positive to the conduction band potential of CaTiO3. This means that the transfer of photo-generated electrons from the conduction band of CaTiO3 to graphene is thermodynamically favorable. The charge transfer process reduces the electron–hole recombination process and the photo-generated holes are now available to react with OH−/H2O to generate the photo-catalytically active species, ˙OH radicals. In addition, photo-generated electrons are expected to react with dissolved O2 in the reaction medium to produce superoxide (O2˙−). The O2˙− is also a photo-catalytically active species, but it obviously plays a minor role in the present dye degradation.18
2.2. Carbon nanotubes (CNTs)
Carbon nanotubes (CNTs) are the one dimensional form of graphene. Graphene sheets are folded to form CNTs. The first such structure to be discovered was the C60 molecule by Kroto et al.22 CNTs exist as single walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs) with adjacent shell separation of ∼0.34 nm, diameters of ∼1 nm and high length/diameter ratios. CNTs have extraordinary mechanical, electrical, thermal, optical and chemical properties. They have 200× the strength and 5× the elasticity of steel, and 1000× the current capacity, 15× the thermal conductivity and 5× the electrical conductivity of copper. CNTs typically have diameters ranging from <1 nm up to 50 nm.22,23The unique electrical properties such as high current density (109 A cm−2) and π-channelized electrons make the CNTs a good matrix for the composites with metal oxides, perovskites, metal sulfides etc.23,24 Gray et al. reported the composite of TiO2–CNTs where electrons can be easily channelized through the CNTs and delay the electron–hole recombination process.25Fig. 3 shows the overall charge transfer mechanism in CdS/CNT suspension under visible light. Through the photo-electrochemical system the charge transfer from the conduction band of CdS to CNTs has been studied by Kim et al.26 The conduction band of CNTs lies at a more positive level compared to that of the CdS, hence the electrons can be transferred from CdS to CNTs.
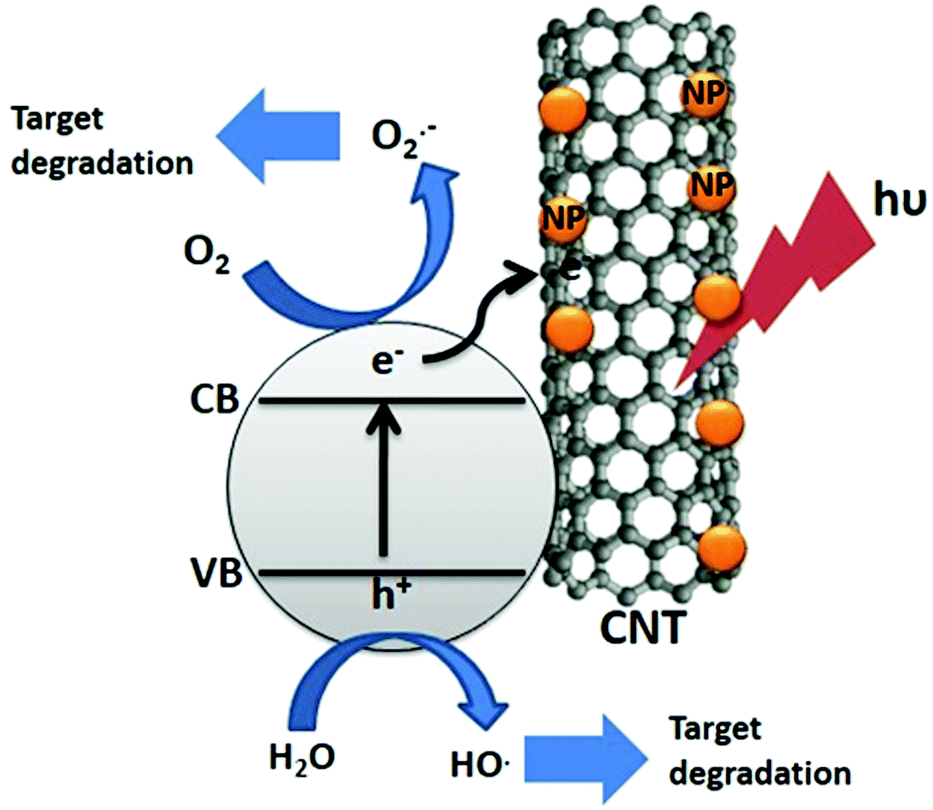 | ||
| Fig. 3 Schematic diagram of the dye degradation mechanism by the CNT–CdS composite. | ||
2.3. Graphitic carbon nitride (g-C3N4)
Graphitic carbon nitride (g-C3N4) is a metal free visible light induced semiconductor having a band gap of 2.7 eV and can absorb blue light up to 450 nm. g-C3N4 possesses stacked 2-dimensional graphite-like planar structure containing π-conjugated systems having a distance between two layers of 0.326 nm. It provides a framework for the electron to easily delocalize throughout its π-network (Fig. 4). Therefore, in a photocatalytic reaction, g-C3N4 plays an important role in the form of a composite, a heterojunction or as a dopant.27–31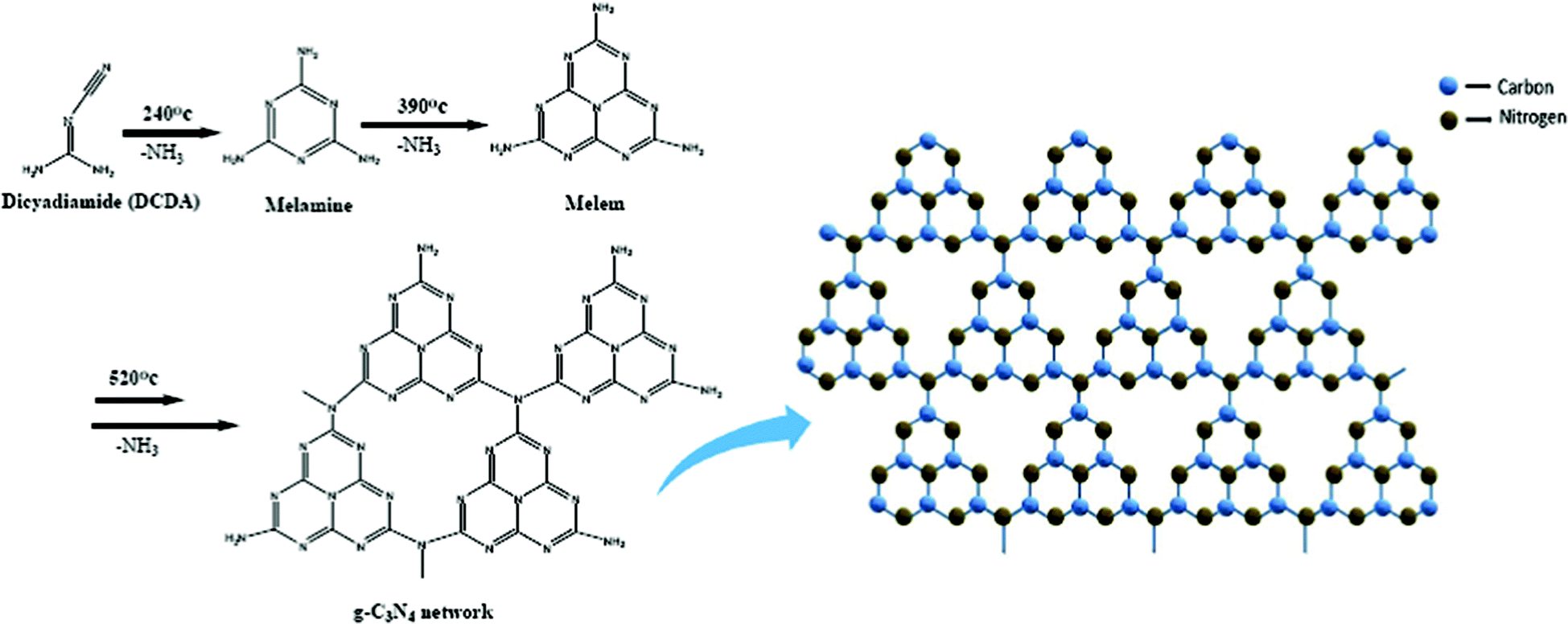 | ||
| Fig. 4 The synthesis of g-C3N4via the polymerization of melamine (Left) and Schematic representation of graphitic carbon nitride network (Right) (reproduced from ref. 60). | ||
Various morphologies of g-C3N4 can be obtained by a simple template modeling. Among different forms of g-C3N4, mesoporous carbon nitride has large surface area and is a suitable semiconductor towards the application of heterojunction construction.32 Both the triazine and tri-s-triazine rings act as the tectonic units in a single layer of g-C3N4.43–59 The modification of –NH–, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–, –NH2 groups existing on the surface of g-C3N4 is highly responsible for its enhanced optical properties.33–35 Gillan et al.36 reported that g-C3N4 is stable in water, THF, ethanol, diethyl ether, toluene etc. due to its stacking layers.37 The structure of g-C3N4 is just similar to the flake like structure of graphite, which has been proven by the XRD.38–42
N–, –NH2 groups existing on the surface of g-C3N4 is highly responsible for its enhanced optical properties.33–35 Gillan et al.36 reported that g-C3N4 is stable in water, THF, ethanol, diethyl ether, toluene etc. due to its stacking layers.37 The structure of g-C3N4 is just similar to the flake like structure of graphite, which has been proven by the XRD.38–42
According to Chen et al.61 Bi2MoO6/g-C3N4 shows enhanced photo-catalytic activity under visible light irradiation. The valence band and conduction band potentials of g-C3N4 were estimated at 1.58 eV and −1.12 eV62,63 and for Bi2MoO6 those are 2.31 eV and −0.32 eV64 respectively (Fig. 5). When light falls on Bi2MoO6/g-C3N4 composite photocatalysts, electron–hole pairs resulted due to the narrow forbidden bandwidth corresponding to visible light. The enhanced photocatalytic activity is due to the transfer of photo-generated electrons in the CB of g-C3N4 to the Bi2MoO6 and the holes in the VB of Bi2MoO6 would inject to that of g-C3N4 (Fig. 5). Owing to the less negative CB potential of Bi2MoO6 nano-plates than EO2/˙O2− = −0.046 eV/NHE, these electrons staying on the CB of Bi2MoO6 nano-plates can react with oxygen molecules absorbed on the surface of hybrids to produce radicals ˙O2− that directly oxidize organic molecules or indirectly oxidize them via transforming to OH˙ radicals by a two-electron oxidation pathway. Therefore, g-C3N4 plays an important role to suppress the electron–hole recombination and enhance the photocatalytic activity.
 | ||
| Fig. 5 Schematic diagram of the charge carrier transfer process and the possible photocatalytic mechanism of the 20 wt% Bi2MoO6/g-C3N4 hybrid composites on exposure to visible light illumination (reproduced from ref. 61). | ||
Among the three π-conjugated carbon materials (graphene, CNTs and g-C3N4), graphene is considered an excellent supporting matrix for photocatalysis due to its strong π-interactions and good electrical conductivity. The electrons can be easily channelized through the sheet and hence delay the electron–hole recombination rate. Though CNTs are a derivative of graphene, their π-interactions are less extensive than those of graphene. Finally, due to the low binding sites in g-C3N4, π-conjugation is less extensive in comparison to graphene. This is because, during the electron channelization all the lone pairs of the nitrogen atoms don't take part as some of the lone pairs are perpendicular to the plane of the g-C3N4 sheets (Table 1).
| Characteristics | Graphene | g-C3N4 | Carbon nanotubes | |
|---|---|---|---|---|
| Single walled CNTs | Multi-walled CNTs | |||
| Surface area (g m−2) | 2600 | 10 | 1315 | 100 |
| Electrical conductivity (m2 V−1 s−1) | 15000 |
|||
| Band gap (eV) | — | 2.7 | — | Low band gap |
| π-Conjugation | More extensive | Less extensive | Moderate | Moderate |
| Photocatalytic activity | No | Yes | No | No |
| Reference | 15 | 32 | 133 | 133 |
3. Composite of perovskites with graphene-analogous materials: a unique approach towards tuning the photocatalytic activity
The quick recombination of photo-generated charge carriers is a major limitation for achieving the high photocatalytic efficiency. Combining graphene, g-C3N4, CNTs etc. with semiconductor photocatalysts to develop novel nano-composite with enhanced photocatalytic performance has received increasing attention. Due to the high surface area and remarkable electrical conductivity, it can be used as an excellent supporting matrix to efficiently suppress the growth of semiconductor particles and facilitate the transportation of the generated electrons in a π-conjugated material-semiconductor composite. The combination of the semiconductor with π-conjugated materials results in band gap narrowing and hence enhances the photocatalytic activity. Introduction of π-conjugated materials to perovskite can narrow the band gap of perovskite, red shift the absorption edge to the visible region and so collect solar radiation in a wider spectral range. Perovskite/layered perovskite-π-conjugated composites were prepared through various methods such as the solvothermal process, photo reduction, the reverse co-precipitation method, the polyacrylamide route and the in situ method. Table 2 summarizes the different approaches for the preparation of graphene–perovskite nano-composites.| Materials | Methods | Reaction conditions | Reference |
|---|---|---|---|
| Graphene/YInO3 | Solvothermal process | High temp., and pressure, longer time period | 88 |
| SrTiO3–graphene | Photo reduction | Nitrogen atmosphere, irradiation from low pressure mercury lamp | 70 |
| Graphene–BiFeO3 | Reverse co-precipitation method | Amounts of KOH, high pressure and temperature | 86 |
| LaMnO3–graphene | Sol–gel method | Alkaline medium, surface active agent | 77 |
| CaTiO3–graphene | Polyacrylamide route | Acidic medium, amount of glucose | 18 |
| N2 doped Sr2Ta2O7–graphene | Solid state reaction | Stirring | 66 |
| ZnFe2O4–graphene | Conventional method | Autogenous pressure, temperature, long time reaction | 71 |
| Bi25FeO40–graphene | Hydrothermal method | pH, amount of KOH, pressure temperature, long time reaction | 68 |
| Graphene–γBi2MoO6 | Hydrothermal method | Pressure and temperature | 67 |
| Graphene–La2Ti2O7 | Precipitation method | UV-irradiation | 65 |
| g-C3N4/BiFeO3 | Conventional method | Sonication, temperature | 73 |
| SrTiO3–C3N4 | Calcination method | Temperature | 31 |
| BaTiO3–g-C3N4 | Solid state method | Calcination | 29 |
| g-C3N4–NaTaO3 | Conventional method | Sonication, stirring, temperature | 30 |
| g-C3N4–Bi2MoO6 | Conventional method followed by thermal treatment | Stirring, temperature | 61, 74 |
| g-C3N4/N-doped SrTiO3 | Solid state method | Calcination | 75 |
| Cr-doped SrTiO3–g-C3N4 | Solid state method | Temperature | 76 |
3.1. Conventional synthesis method
Perovskite/layered-perovskite–graphene composites can be prepared in a conventional way. Graphene and the material were ultrasonicated to enhance their surface interactions and the mixture was dried to obtain the composite. Xian et al. prepared CaTiO3–graphene composites through the conventional method.18 CaTiO3 nanoparticles and graphene were dispersed in absolute ethanol with a ratio of 9:1 and ultrasonically treated. The obtained mixture was dried at 60 °C in a thermostat during which ethanol was vaporized, leaving behind CaTiO3 nanoparticles well anchored on graphene sheets. Sr2Ta2O7−xNx–GO, graphene–γBi2MoO6 and graphitic C3N4–NaTaO3 composites can be obtained through a similar process.30,65–67 Halide perovskite like CH3NH3PbI3–graphene composites is also synthesized through such a conventional way. The conventional method is the easiest technique for the preparation of the composite as it involves only sonication and heating at low temperature.
3.2. Hydrothermal synthesis method
In this synthetic method, the reaction is carried out hydrothermally at higher pH using alkali as the sacrificial agent. Jiang et al. prepared the Bi25FeO40–graphene composite through this method using Bi(NO3)3·5H2O and Fe(NO3)3·9H2O as precursors.68 Graphene oxide was synthesized by the method reported by Hummers and Offeman.72 Bi25FeO40–graphene composite photocatalysts with varied graphene contents (5, 10, 20, 30 wt%) were synthesized by the alkaline hydrothermal method. 2.4 g of Bi(NO3)3·5H2O and 2.0 g of Fe(NO3)3·9H2O were added to 5 mL of 1 M HNO3. Using 6 M KOH the pH of the solution was adjusted to 10 under stirring to obtain Bi25FeO40 as a precipitate. Required amounts of GO and Bi25FeO40 were then suspended into 60 mL of distilled water and sonicated for 30 min at room temperature followed by filtration and repeated washing with distilled water. The resulting brown coloured precipitate was then added to 40 mL of 12 M KOH solution with 0.1 g KNO3. The suspension was transferred to a Teflon lined stainless autoclave and heated at 160 °C for 24 h. The product was isolated by filtration, washed with distilled water and absolute ethanol several times and dried at 60 °C for 12 h in a vacuum oven to obtain the Bi25FeO40–graphene composite. Recently, Lee et al.69 reported on LaNiO3 and graphene composites prepared through the hydrothermal method. LaNiO3 was synthesized using the starting materials La(NO3)3·6H2O and Ni(NO3)2·6H2O in de-ionized water and the pH was adjusted using alkaline medium. The resulting solution was autoclaved at 220 °C and the product was calcined at 800 °C for 3 h. Finally, the LaNiO3–CNT composite was prepared via an injection chemical vapor phase deposition method as shown in Fig. 6.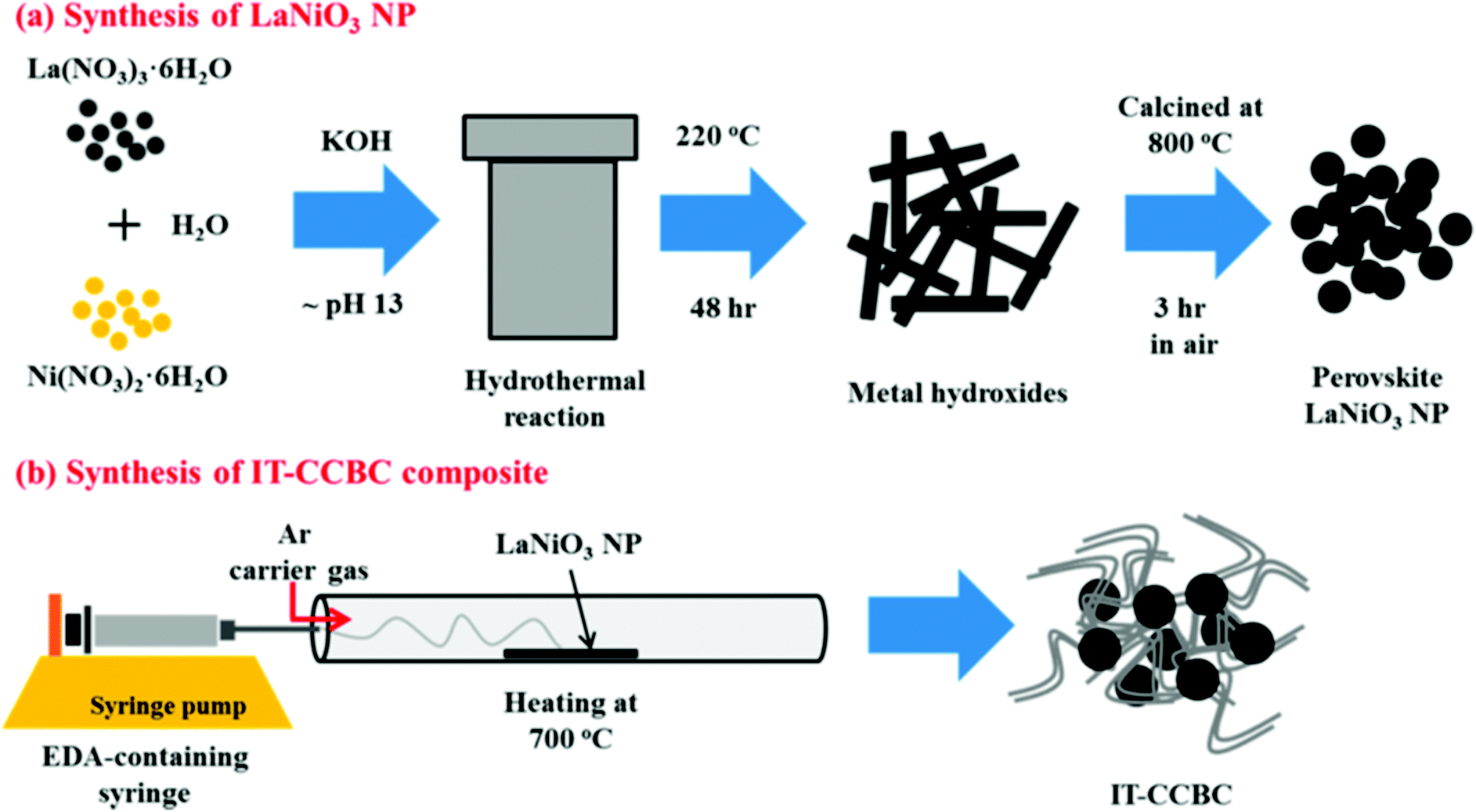 | ||
| Fig. 6 Schematic illustrations of (a) the hydrothermal synthesis of LaNiO3 NPs and (b) injection of CVD preparation of an intertwined core–corona structured bifunctional catalyst (IT-CCBC) (reprinted with permission from ref. 69. Copyright the American Chemical Society, 2015). | ||
3.3. Photocatalytic reduction method
During the photocatalytic reduction process, graphene and perovskite/layered perovskite after sonication are exposed to UV-light irradiation under mild stirring. Xian et al. synthesized SrTiO3–graphene composite through this route.70 Graphene having a weight fraction of 2.5%–10% was suspended in 50 mL distilled water and ultrasonicated for 30 minutes. 0.1 g of SrTiO3 nanoparticles and 0.0125 g ammonium oxalate were added to the resultant suspension and stirred for 10 minutes. After that, the mixture was purged with nitrogen and exposed to UV-light irradiation under mild stirring from a 15 W low pressure mercury lamp for 5 h. During this process, the color of the solution changed from brown to black indicating the reduction of graphene oxide. By centrifugation the product was isolated and dried in a thermostat drying oven at 60 °C for 4 h. Wang et al. reported the synthesis of graphene–La2Ti2O7 composites through a photocatalytic reduction method.65 In this process the ethanol solution acts as a hole scavenger and helps in the reduction of graphene oxide.3.4. Calcination method
The calcination method is a typical solid state reaction where the precursors (perovskite/layered perovskite and graphene/gC3N4) are ground and calcined at high temperature to obtain the composite. Irvine et al. obtained the SrTiO3–gC3N4 composite using SrTiO3 powder.31 The SrTiO3 powder and urea were taken in the weight ratio of 4:1, ground for 1 h using a mortar and a pestle and calcined in a sealed alumina crucible at elevated temperatures (400 °C and 600 °C) to obtain the composite.
3.5. In situ synthesis of composite materials
During the in situ process, the material and the composite were synthesized simultaneously. Wang et al. synthesized ZnFe2O4–graphene composites through this process.71 Graphene oxide was prepared from graphite according to the method reported by Hummers and Offeman.72 To synthesize the composite through this method GO and the mixture of Zn(NO3)2 and Fe(NO3)3 solutions were dispersed in the solvent (ethanol) separately. After stirring both the suspensions were mixed together and hydrothermally treated to obtain the composite.4. An in-depth analysis of composites through surface and bulk characterization
4.1. Crystal structure determination
Perovskite/layered perovskites consist of slabs of the ABO3 type structure which are separated by some motifs. The layered perovskite type materials are confirmed through different characterizations. The crystal structures of the composite materials were determined by the XRD technique. Huang et al. confirmed the structure of K2La2Ti3O10 as a layered perovskite compound.77 The K2La2Ti3O10 consists of a negatively charged lanthanum titanate perovskite layer and interlayer K+ ions. The adjacent triple perovskite sheets, La2Ti3O10, are stacked with a displacement by 1/2 along the (0 0 1) direction. The lanthanum ion occupies the 12-fold site in the center of the perovskite lattice. Likewise, the K/HSr2TaxNb3−xO10 compound consists of Ta(Nb)O6 perovskite sheets with Sr ions filling in the interstices of the octahedron of Ta(Nb)O6 and the K+ ion in the interlayer.78 In two adjacent perovskite slabs, eight oxygens of Ta(Nb)O6 octahedron coordinated to the K+ ion. The XRD data of K/HSr2TaxNb3xO10 indicate that the obtained powders are highly crystalline and free from impurities (Fig. 7).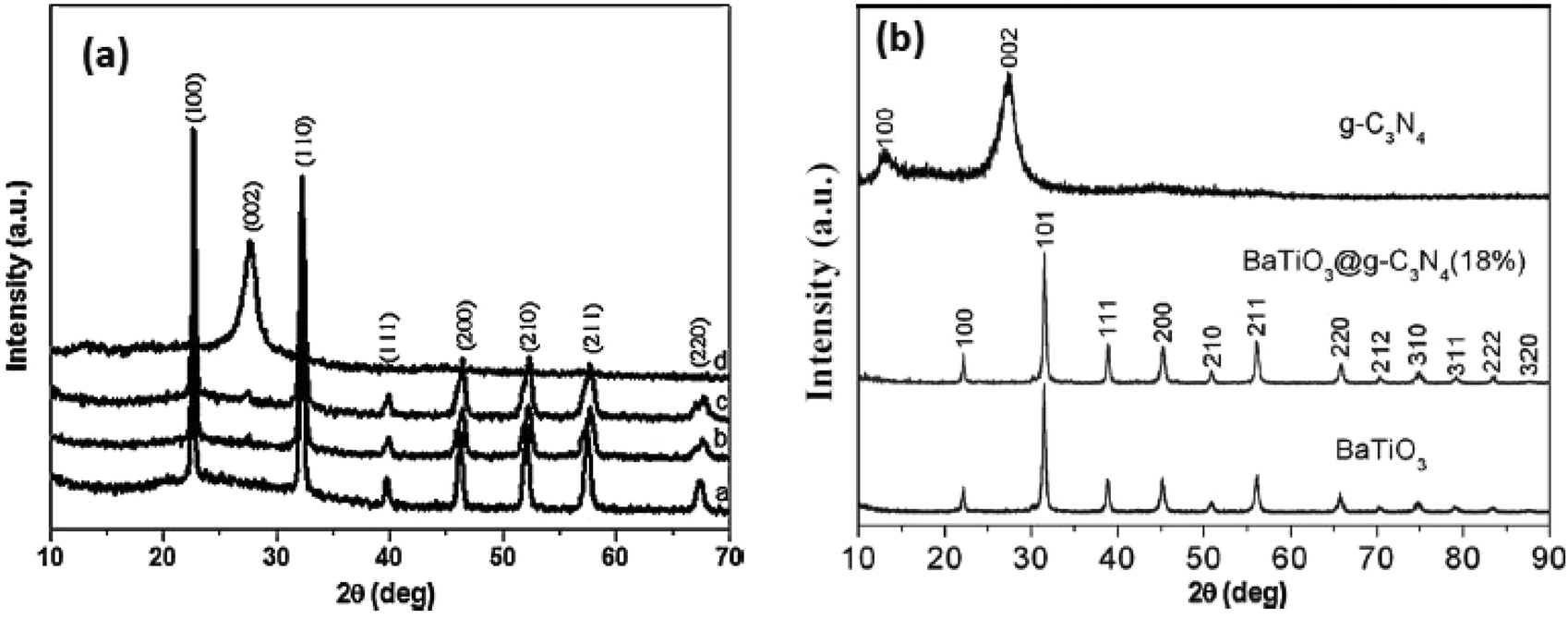 | ||
| Fig. 7 (a) XRD patterns of the prepared g-C3N4, NaTaO3 and g-C3N4/NaTaO3 photocatalysts; (b) XRD patterns of g-C3N4, BaTiO3 and BaTiO3@g-C3N4 (18%) samples (reproduced from ref. 30, license no. 3678120097745 and ref. 29, license no. 3678180169216). | ||
Volonakis et al. explained the interaction of MAPbI3 with graphene in the following way. From the X-ray diffraction pattern they found that MAPbI3 crystals exhibit strong peaks associated with the (110) and (001) reflections.79–81,130 According to Volonakis et al. the composite constructed graphene–MAPbI3 interfaces with the perovskite exposing the (110) and (001) faces. As both the surfaces of graphene and perovskite are non-polar they found that the corresponding films can be seen as an alternation of MAI and PbI2 layers as if the structure was obtained via layer-by-layer deposition using the standard precursors.82–84 In graphene–perovskite composites some structural changes occur by suppression of the octahedron in the first MAPbI3 monolayer in contact with graphene. The shift of the center of mass is compensated by a displacement of 0.1 Å of the first Pb sub-lattice towards graphene. Hence some distortion occurs at PbI6 octahedron at the interface (Fig. 8).
 | ||
| Fig. 8 Atomic model of interactions of MAPbI3 and graphene (reprinted with permission from ref. 130. Copyright the American Chemical Society, 2015). | ||
In a composite, the perovskite and layered perovskites should be well dispersed on the surface of the graphene. Usually, during composite formation some chemical and physical bonds are formed between the semiconductor and the π-conjugated carbon system. An et al. reported that in the graphene–BiFeO3 composite the hybridization of BiFeO3 with graphene results in the Fe–O–C bonds at the surface of the BiFeO3 nanoparticles.85 The strong covalent nature of C–O bonds and charge transfer from the oxygen (ligand) to the Fe ions (metal) may be further reduced in BiFeO3, which results in a decrease in the band gap of the BiFeO3–graphene composite. Occasionally, the purity of the composite depends upon the concentration of reagents used. For instance, Li et al. reported that the purity of the BiFeO3 depends on the concentration of the KOH used during the preparation process.86 This can be investigated from the XRD data of the graphene–BiFeO3 composite. Sometimes during the formation of the composite, the perovskite loses their crystalline structure. Shanker et al. reported that the crystallinity of NaTaO3 decreases in the g-C3N4–NaTaO3 composite.30 Similarly, when LaMnO3 formed a composite with graphene, from the XRD data it was found that the crystallinity decreased indicating the interaction of LaMnO3 with graphene.77
4.2. Surface morphology of the composites
The shape and size of the composite photocatalysts were determined from SEM and TEM images. From the SEM image of CaTiO3 particles (Fig. 9), it was revealed that the particles are nearly spherical in shape, with a size distribution majorly in the range of 31–42 nm. The average particle size is ∼36 nm. The TEM image of CaTiO3–graphene composites confirmed that CaTiO3 nanoparticles are well assembled on the graphene sheet.18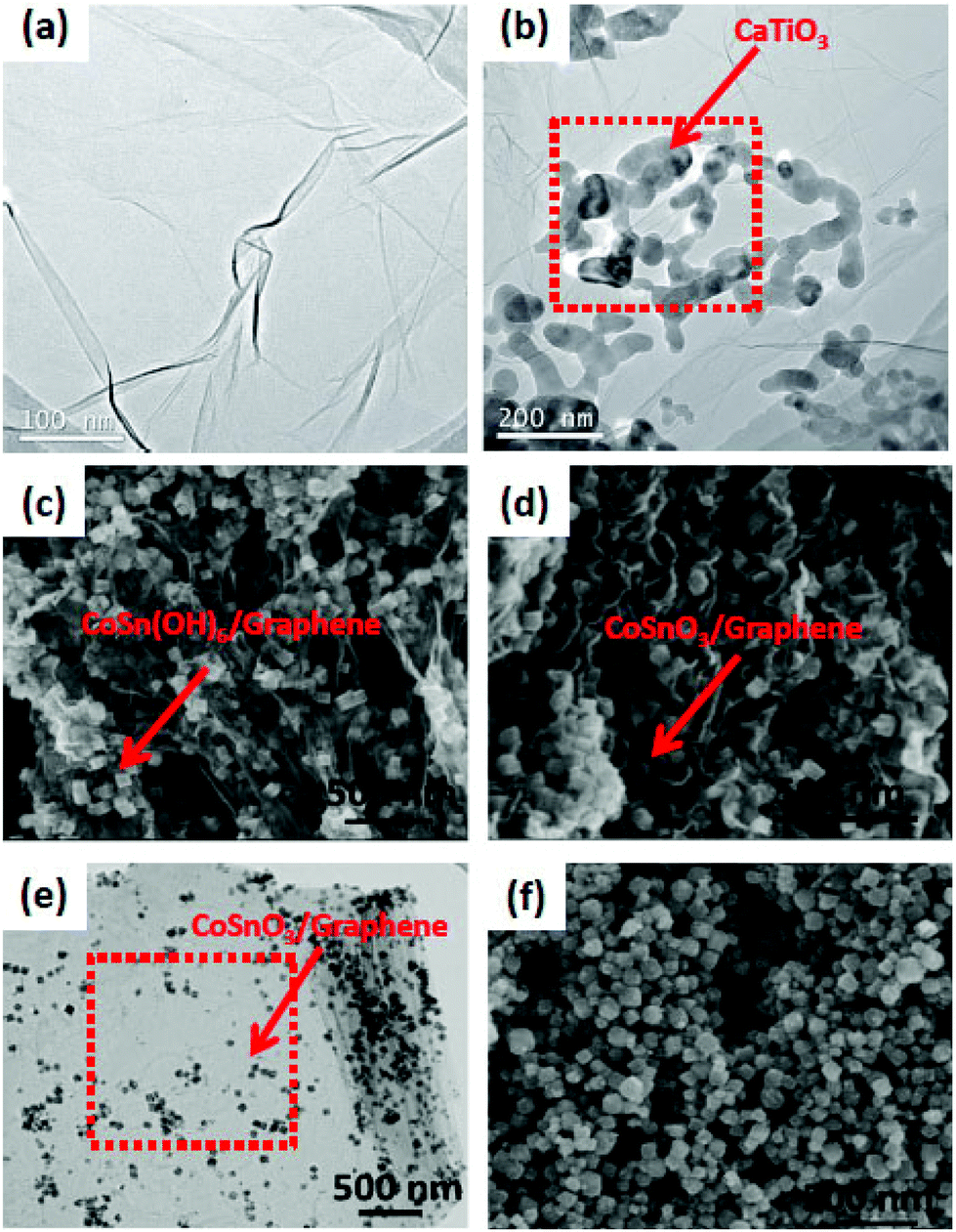 | ||
| Fig. 9 (a) and (b) TEM images of pure graphene and CaTiO3–graphene composites, respectively; SEM images of (c) CoSn(OH)6/G and (d) CoSnO3/G, (e) TEM image of CoSnO3/G, (f) SEM image of bare CoSnO3 (figure reproduced from ref. 18, © The Royal Swedish Academy of Sciences. Reproduced by permission of IOP Publishing. All rights reserved. And from ref. 106, license no. 3678200529395). | ||
From the TEM images of YInO3–graphene, it is found that the composite shows a fine rod or spherical shape having a diameter of about 100 nm.87,88 A large degree of porosity is found in the composite as the nanoparticles are linked end to end to form a net like structure. The super-heated gases released from the combustion reaction cause high porosity in the composite and also the net structure prevents aggregation of the YInO3 composite. The BET surface area of the YInO3–graphene composite is found to be more than that of pure YInO3 and the BET surface increases gradually from 8.58 to 19.29 m2 g−1 with increasing the content of graphene. YInO3 nanoparticles are well dispersed on graphene surfaces, so there are good electronic interactions between the YInO3 nanoparticle and graphene sheets, hence enhancing the charge carrier separation and photocatalytic activity of graphene–YInO3 nanocomposites.88 The TEM image of graphene oxide indicates that it has a typical two-dimensional sheet structure with crumpled feature and the TEM image of the SrTiO3 particles reveals that the particles are nearly spherical in shape with an average size of about 55 nm. The TEM image of the SrTiO3–graphene (10%) composites shows that the SrTiO3 particles are well assembled onto the graphene sheet. From TEM images it is found that the ZnFe2O4 nano-crystals with diameter 7–10 nm are homogeneously decorated on the exfoliated graphene surfaces. The formation route to anchor ZnFe2O4 nanoparticles on to the exfoliated GO sheets may be proposed as the intercalation and adsorption of zinc and iron ions into the layered GO sheets. ZnFe2O4 nano-crystals anchored on the surface of graphene sheets look like a thin film and have good dispersion behavior. In the case of pure La2Ti2O7, the nano-sheets are rectangular in shape whereas in the composite the La2Ti2O7 nanosheets are intercalated into the layer to layer spacing of the exfoliated graphene sheets. These interactions enable the electron transfer from La2Ti2O7 to exfoliated graphene sheets during the UV-induced reduction process. Li et al.47 reported that CoSnO3/G exhibits dense morphology inside a unique sandwich structure. The CoSnO3 NPs in the CoSnO3/G sandwich is homogeneously dispersed in between graphene due to the confining effect. In contrast, the bare CoSnO3 NPs are connected with each other and tend to aggregate without the immobilizing effect of graphene. Im et al.83 reported the fabrication process of the BaTiO3NP–CNT composite. Fig. 10 shows cross-sectional scanning electron microscopy (SEM) images of a 250 μm thick p-NC that is sandwiched between the top and bottom metal-coated plastic substrates. A magnified cross-sectional SEM image shows that the BaTiO3NPs and the MW-CNTs are well distributed in the PDMS matrix. The BaTiO3 NPs generate piezoelectric potential under external stress and act as an energy generating source. CNTs act as a dispersant, a stress reinforcing agent, and a conducting functional material in nanocomposite generator (NCG) devices.
 | ||
| Fig. 10 (a) Cross-sectional SEM image of a nanocomposite generator (NCG) device. (b) Magnified cross-sectional SEM photograph of the p-NC. (c) An SEM image of BaTiO3 NPs synthesized by a hydrothermal method. The inset shows a Raman spectrum obtained from BaTiO3 NPs. (d) The MW-CNTs have a diameter of 20 nm and a length of 2 μm. The inset shows a typical Raman shift of the MW-CNTs with large D bands. (e) HRTEM images of the multi-walled carbon nanotube/perovskite nanocomposite (CMC). Carbon nanotubes were grown on the surface of the perovskite particles. (f) HRTEM images of multi-walled carbon nanotubes/perovskite nanocomposite material (CMC). Carbon nanotubes are enclosing the metal oxide particles (reprinted with permission from ref. 131 and 132. Copyright the American Chemical Society, 2002 & 2012). | ||
4.3. Evaluation of optical properties of the composites
The main approach for the composite formation of perovskite and layered perovskite is to minimize the band gap energy of the compound. This can be quantitatively obtained from UV-Vis DRS by different methods. Generally, the direct or indirect band gap of semiconducting photocatalysts is determined by Tauc's plots.86 The reduced band gap energy of the perovskite and layered perovskite by the formation of the composite with π-conjugated carbon material is discussed later in the second part of the review. Huang et al. reported that pure LaMnO3 has a band gap energy of 2.82 eV which is reduced to 2.51 eV in the composite of LaMnO3–graphene (Fig. 11(a)).77 Many researchers have reported that the band gap energy decreases with the formation of the composite of metal oxide and mixed metal oxide with the π-conjugated carbon system. | ||
| Fig. 11 (a) Tauc's plots of (αhυ)1/2versus hν of LaMnO3–graphene and LaMnO3 (reproduced from ref. 125). (b) PL spectra of YInO3, Pt0.5/YInO3 and G/YInO3 nanocomposites (the excited wavelength is 365 nm) (reproduced from ref. 121). | ||
Photoluminescence is another useful technique to study the charge carrier separation rate of semiconductor materials. In the case of a graphene modified perovskite/layered perovskite material, graphene can effectively control the recombination of charge carriers. It has been studied from the PL spectra that the composites of perovskite/layered perovskite with a π-conjugated carbon system like CNTs or graphene exhibited a lower rate of recombination of the electron–hole pair than neat material. An et al.85 showed that BiFeO3 showed stronger PL emission than that of BiFeO3/graphene composites. It suggests that the recombination rate of charge carriers is significantly decreased in the case of BiFeO3/graphene composite materials. Xian et al.70 have also reported that the rate of recombination of electron–hole pairs is slower in the case of SrTiO3–graphene composites than that for neat SrTiO3. Gao et al.121 also reported the same for the YInO3–Graphene composite. So, it can be concluded that the modification of graphene could significantly check the recombination rate of charge carriers and enhanced the photocatalytic activity.
5. Application of perovskite and layered perovskite composites
5.1. Photocatalytic hydrogen energy evolution
The production of hydrogen energy has become an urgent scientific challenge that has to be addressed in order to fulfill the huge energy demand. For solving this issue, the most promising approach is photocatalytic water splitting assisted by a semiconductor. In spite of intensive research studies in this field, the number of semiconductor photocatalysts that have decent photocatalytic activity under visible-light illumination is still limited and hence it can be described as the ‘Holy Grail of Chemistry’.89 As the photocatalytic activity of the photocatalysts significantly depends on the concentration of electrons and holes, the development of visible-light responsive photocatalysts with a moderate rate of recombination of charge carriers is a great challenge in this field. For photocatalytic water splitting into H2 and O2 the band gap of the photocatalyst should have a larger than standard Gibbs free energy change (1.23 eV) and should have a conduction band with potential more negative than that for water reduction and a valence band with potential more positive than that for water oxidation.2 Water splitting using sun light has attracted intense research interest since 1972.87 Several research articles have been published on photocatalytic water splitting using the semiconducting materials.87,88,90–108 The first semiconductor used for the photocatalytic water splitting was TiO2 and due to the large band gap energy (3.1 eV) TiO2 does not efficiently split water under visible light irradiation without any modifications.87 The modification can be done by doping metals and non-metals to TiO2 to obtain the visible-light response of TiO2109–113 (Fig. 12).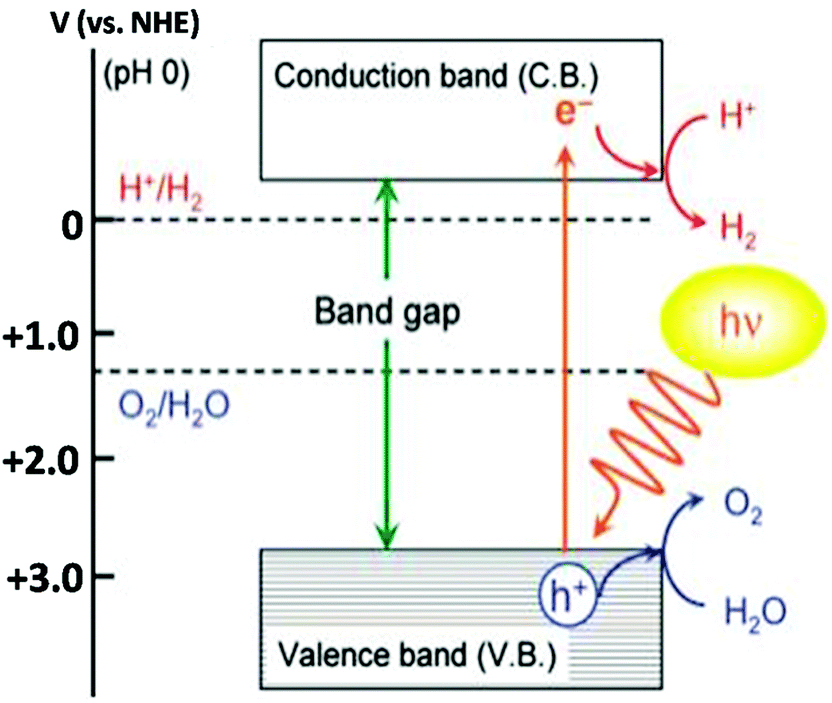 | ||
| Fig. 12 Basic principle of the photocatalytic overall water splitting process (reproduced from ref. 114). | ||
Based on the crystal structure and chemical component characteristics, different modification strategies (such as chemical component adjustment, micro/nano-structure adjustment, and local lattice structure adjustment) were employed to develop ABO3 photocatalysts with improved performance for water splitting and organic pollutant degradation. Perovskites such as MTaO3 (M = Na, K, Li, Ag), NaSbO3, LaInO3, LaFeO3etc. are used for photocatalytic reactions.8,108–113 Some perovskites showed high photocatalytic activity for hydrogen and oxygen evolution under UV-light irradiation. Due to the wide band gap under visible light irradiation these materials have less utility. By doping foreign material or replacing cations, one can tailor the electronic structure and enhance the visible light response range. Thermodynamically, the water splitting reaction is an uphill reaction with a highly positive change in Gibbs free energy.
| H2O → 1/2O2 + H2 (ΔG = +237.2 kJ mol−1, 1.23 eV) |
In water splitting, the change in Gibbs free energy (+237.2 kJ mol−1) should be overcome by the photon energy. Because the electrochemical decomposition of water is a two electron stepwise process, electrons and holes are generated on the photocatalytic surface by absorbing solar energy. When a photo-semiconductor absorbs light, photons with energies greater than 1.23 eV are needed for water splitting. Due to this absorption photoelectrons excited to the conduction band (CB) and holes remain in the valence band (VB). After that, the second step in photochemical water splitting consists of charge separation and the migration of photo-generated electron–hole pairs from the bulk of the semiconductor towards the reaction sites on the photocatalyst surface. The final step of the photocatalytic process involves the surface chemical reactions (Fig. 13).115
 | ||
| Fig. 13 Main processes in photocatalytic water splitting (reproduced from ref. 116). | ||
Oxidation H2O + 2h+ → 2H+ + 1/2O2
Reduction 2H+ + 2e → H2
Overall reaction H2O → 1/2O2 + H2
Konta et al. found that Ru, Rh and Ir doped SrTiO3 possessed intense absorption bands in the visible light region due to excitation from the discontinuous levels formed by the dopants to the conduction band of the SrTiO3 host.117 Sometimes to maintain the charge balance, the strategy of co-doping was generally adopted. (Cr, Sb), (Cr, Ta), (Cr, Ta) are co-doped to SrTiO3 and applied to hydrogen energy evolution under visible light irradiation. (Cr, Sb) co-doped SrTiO3 has a band gap of 2.4 eV and shows intense absorption bands in the visible light region.118 Kato et al. developed AgMO3 (M = Ta and Nb) photocatalysts by replacing Na+ in NaMO3 (M = Ta and Nb) with Ag+. AgTaO3 and AgNbO3 having a band gap of 3.4 and 2.8 eV respectively only had a UV-light response.119 Replacing Na+ ions by Ag+ ions enhances the light response range and hence the ability to evolve H2 or O2 from water.
Due to a wide band gap, layered perovskite type materials have weak photocatalytic activity. The band gap energy of the layered perovskite was estimated to be 4.1 eV. Machida et al. reported that the co-catalyst NiO works as a H2 evolution site in MLnTa2O7 (M = Cs, Rb, Na, or H; Ln = La, Pr, Nd, or Sm).120 In photocatalysts, the photo-generated electrons have to cross the interface between the photocatalyst and the loaded NiO co-catalyst. The co-catalyst (NiOx) enhances the photocatalytic activity of MLnTa2O7 (Table 3). Activity decreased in the following order: Rb > Cs > Na > H. Furthermore, doping the noble metals enhances the photocatalytic activity. KSr2TaxNb3−xO10 has a very low photocatalytic activity; nearly no H2 evolution was observed whereas HSr2TaxNb3−xO10 has excellent photocatalytic activity. But by intercalating Pt to KSr2TaxNb3−xO10, the compound shows an enhanced photocatalytic activity. On changing the ratio of niobium to tantalum, the band gap energy of the photocatalyst with layered perovskite structure was also controlled. The photocatalytic H2 evolution rate of HSr2TaxNb3−xO10 reached up to 1168 cm3 g−1.
| Anion loading | Unloaded/μmol h−1 | Loaded/μmol h−1 | ||
|---|---|---|---|---|
| H2 | O2 | H2 | O2 | |
| Rb | 3.3 | 1.3 | 55.5 | 26.3 |
| Na | 2.5 | 1.0 | 55.3 | 22.7 |
| H | 1.5 | 0.7 | 5.3 | 2.3 |
The photocatalytic activity of the perovskite and layered perovskite can be enhanced by the formation of composites with graphene. Gao et al. showed that the formation of COO− metal bonds in YInO3(YIO) and graphene composites helps in narrowing the band gap in the composite.121 Therefore the composite of graphene and YInO3 could effectively absorb visible light and hence exhibit improved photocatalytic activity. The photocatalytic H2 evolution of the photocatalyst graphene–YInO3 nano-composites was studied under visible light irradiation using sacrificial reagents Na2SO3 and Na2S. The photocatalytic activity of the composite was found to be 32 times more than that of pure YInO3 (Fig. 14). The presence of a small amount of graphene also increases the photocatalytic activity of the composite from 66.55 μmol h−1 g−1 to 400.4 μmol h−1 g−1 with 0.5% graphene. Further, with an increase in graphene weight percentage the photocatalytic activity decreases due to the trade-off between the excellent charge transfer capability of graphene and its detrimental effect on visible light absorption. In a new approach, Mukherji et al. reported that homogeneous nitrogen doping to layered perovskite–GO composite results in better photocatalytic activity under visible light irradiation.122 Doping of nitrogen to UV active Sr2Ta2O7 minimizes the band gap thus allowing visible light absorption.
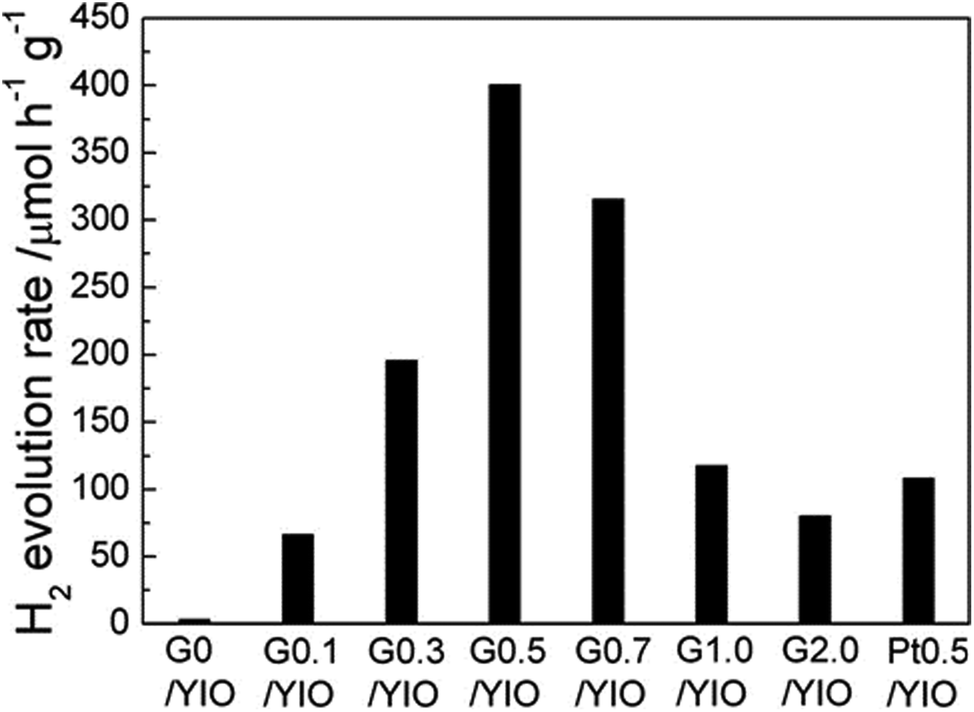 | ||
| Fig. 14 H2 evolution rate of graphene–YInO3 nanocomposites (G/YInO3) with various contents of graphene under visible light irradiation (reproduced from ref. 121). | ||
5.2. Photocatalytic degradation of pollutants
For solving the environmental issues, perovskite and layered perovskite with their composites play an important role in the degradation of organic contaminants and dyes. Photocatalytic degradation is a green technology to solve these issues. Ultimately, for the destruction of synthetic organic species resistant to conventional methods, research activities centered on advanced oxidation processes (AOPs). AOPs are based on the generation of reactive species such as hydroxyl radicals that quickly and non-selectively oxidize a broad range of organic pollutants.123,124i. Absorption of efficient photons by a photocatalytic semiconductor
Semiconductor + hν → e−CB + h+VB
ii. Oxygen ion-sorption (the first step of oxygen reduction; oxygen's oxidation state changes from 0 to 1/2)
O2 + e−CB → O˙−2
iii. OH− neutralized by holes to produce OH˙ radicals
(H2O ⇔ H+ + OH−) + h+VB → H+ + OH˙
iv. O˙−2 radicals neutralized by 2 protons
O˙−2 + H+ → HO˙2
v. Formation of hydrogen peroxide
2HO˙−2 → H2O2 + O2
vi. Reduction of oxygen by decomposition of hydrogen peroxide
H2O2 + e− → OH˙ + OH−
vii. Organic pollutants oxidized via successive attack of OH˙ radical
R + OH˙ → R˙ + H2O
viii. Organic pollutants degraded by holes
R + h+ → R+˙ → degradation products
Introduction of graphene into perovskite can narrow the band gap of perovskite, red shift the absorption edge to the visible region and so it can collect solar radiation in a wider spectral range. Huang et al.77 showed that the photocatalytic activity of LaMnO3 increases when it forms composites with graphene. The photocatalytic activity was investigated by acid red A degradation under irradiation of a 300 W xenon lamp; under lighting for 6 h, the photocatalyst LaMnO3–graphene degrades almost 99.8% of the dye.
The photocatalytic activity of LaMnO3–graphene is greatly improved due to the following three reasons:
(a) Introduction of graphene into LaMnO3 expands the light response range of LaMnO3 which causes an enhancement of the photocatalytic activity.
(b) For graphene based nanocomposites, graphene acts as an electron acceptor and transporter. So the excited electrons of LaMnO3 can transfer from the conduction band to graphene. Both the electron accepting and transporting properties of graphene in the LaMnO3–graphene composite can suppress charge recombination to achieve a higher photocatalytic rate.
(c) The synergistic effects of graphene and LaMnO3 improve the photocatalytic quantum efficiency.
In the BiFeO3–graphene composite Li et al. showed that the activity also depends upon the sacrificial agent used.86 In the case of BiFeO3–graphene the photocatalytic activity depends on the amount of KOH used. Below 2 M KOH, 4 M KOH, 6 M KOH concentrations, some impurities like Bi, Fe2O3, Bi25FeO40 and Bi25FeO40 with BiFeO3 were found. But above 8 M KOH concentration, BiFeO3 was obtained. With an increase in the KOH concentration, the photocatalytic activity subsequently reduced from ∼2.14 eV for BG4 to 1.87 eV for BG12 as shown in Table 4 (here, BG denotes a mixture of BiFeO3 prepared by the hydrothermal method and graphene; BG-sg denotes BiFeO3–graphene composites synthesized via a two-step method without the presence of OH groups).
| Concentration of KOH (M, mol L−1) | Phase composition | Band gap (eV) | Kinetic rate (102 min−1) | |
|---|---|---|---|---|
| BG4 | 4 | Bi25FeO40 | 2.14 | 0.50 |
| BG6 | 6 | Bi25FeO40, BiFeO3 | 2.04 | 0.68 |
| BG8 | 8 | BiFeO3 | 1.98 | 0.79 |
| BG10 | 10 | BiFeO3 | 1.78 | 0.92 |
| BG12 | 12 | BiFeO3 | 1.87 | 0.96 |
| BG mix | 10 | BiFeO3 | 2.24 | 0.31 |
| BG-sg | 0 | BiFeO3 | 2.18 | 0.45 |
The enhanced coupling in between BiFeO3 nanoparticles and graphene results in consistent narrowing of band gaps. The coupling is made by OH groups adsorbed on the surface of graphene. Congo red is adsorbed on the surface due to the large π-conjugation plane of graphene. As a result, the BiFeO3–graphene composite exhibited high photocatalytic activity performance under visible light irradiation. Using porous graphene the photocatalytic properties of the composite significantly improved. An et al. reported that porous graphene–BiFeO3 has a high specific surface area of about 35.07 m2 g−1 which is nearly 5 times larger than that of BiFeO3 (7.50 m2 g−1).85 As a result it makes an easy framework for electron transfer in the graphene–BiFeO3 composite. Under optimal conditions, the rate constant of tetrabromobisphenol-A degradation with the graphene–BiFeO3 composite as a catalyst was 1.19 min−1, which is 5.43 or 3.68 fold that obtained with BiFeO3 and graphene as a catalyst, respectively.
On increasing the graphene content, the SrTiO3–graphene composite shows enhanced light absorption over the whole wavelength range. The photocatalytic activity of the composite was studied using the azo dye acid orange 7 (AO7) degradation under UV-light irradiation. The highest photocatalytic activity with 88% degradation of AO7 was obtained using 7.5 wt% of graphene after 6 h of irradiation.70 As compared to bare SrTiO3 nanoparticles the SrTiO3–graphene nanocomposite shows enhanced photocatalytic activity under UV-light irradiation for AO7 degradation (schematic of the mechanism is given in Fig. 15).
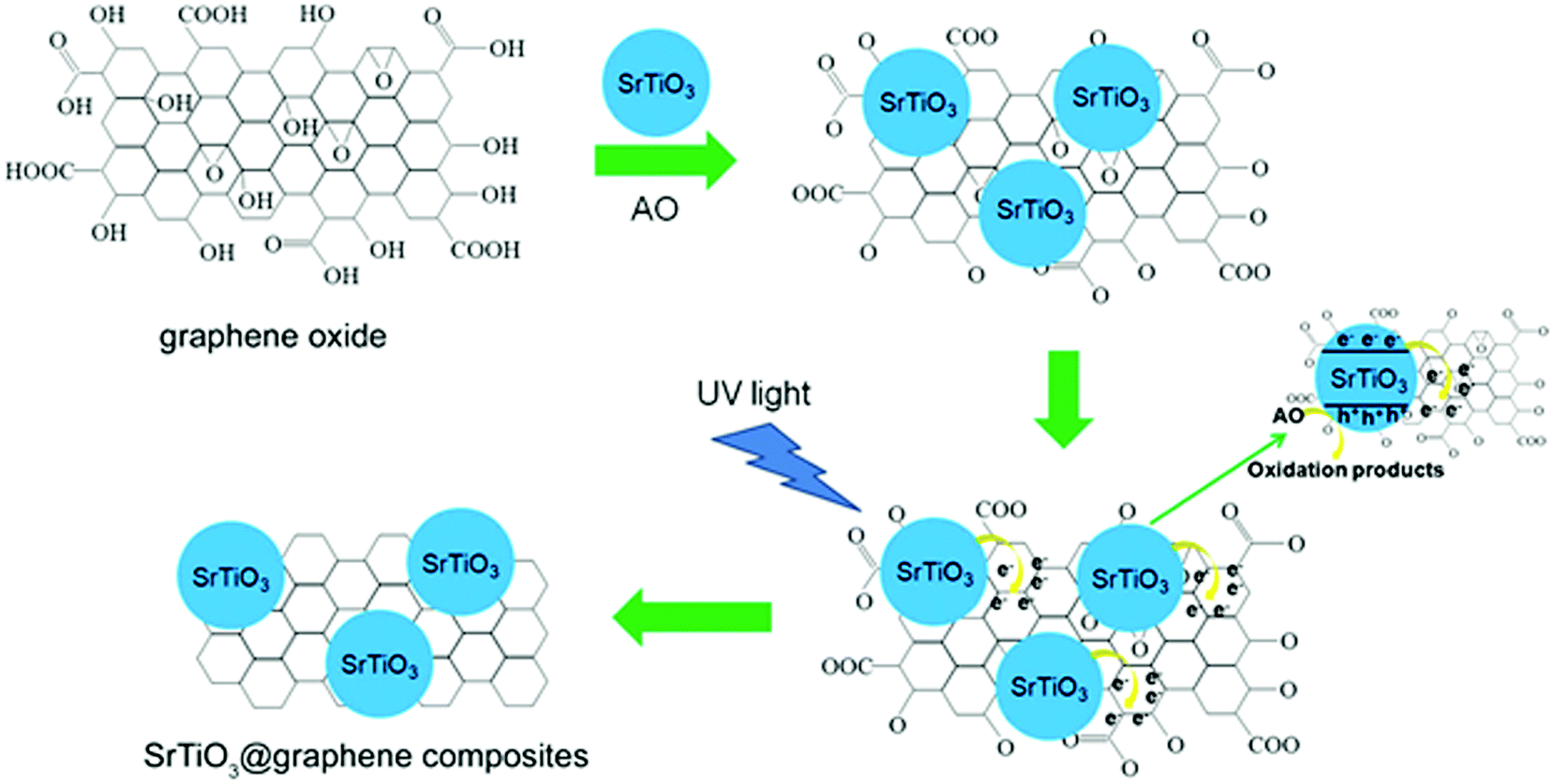 | ||
| Fig. 15 Schematic illustration of the photocatalytic reduction process of graphene oxide by UV light-irradiated SrTiO3 nanoparticles (reproduced from ref. 70). | ||
Layered perovskite has relatively high photocatalytic activity than those of bulk type metal oxides such as TiO2, ZnO etc. Introduction of a π-conjugated system such as graphene into layered perovskite enhances the photocatalytic activity very strongly. Jiang et al. studied the photocatalytic activity of Bi25FeO40–graphene for the degradation of methylene blue under visible light irradiation using a 500 W xenon lamp through UV cut-off filters to completely remove any irradiation below 420 nm and to ensure illumination by visible light only.68 From the UV-Vis diffuse reflectance spectra (DRS) the band gap of Bi25FeO40 can be estimated to be 1.7 eV according to the absorption band edge at 730 nm. Unfortunately, for pure Bi25FeO40 the absorption is much stronger than that of the Bi25FeO40–graphene system. So, it is difficult to calculate the band gap energy from DRS data. In the absence of a photocatalyst the methylene blue degradation was negligible but in the presence of a photocatalyst the degradation efficiencies were found to be 64.9%, 28.0% and 28.7% for Bi25FeO40–(20) graphene, Bi25FeO40 and BiFeO3 respectively. To investigate the synergetic effect between Bi25FeO40 and graphene sheets, photocurrent measurements were also carried out. Due to the synergetic effect caused by graphene addition, the separation efficiency of the photo-generated electrons and holes in Bi25FeO40–(20) graphene is markedly improved in comparison with Bi25FeO40. Increasing the methylene blue concentration from 0.06 mM to 0.62 mM results in an increase in the initial methylene blue degradation rate from 0.037 to 0.16 mM gcat−1 h−1; whereas the rate was almost remaining constant with further increase in the MB concentration. The photocatalytic activity also depends upon the graphene content. The amount of methylene blue degraded by 50.0, 58.2, 64.9, and 92.8%, respectively, over Bi25FeO40–(5) graphene, Bi25FeO40–(10) graphene, Bi25FeO40–(20) graphene and Bi25FeO40–(30) graphene. In comparison with BiFeO3 and Bi25FeO40, the composite Bi25FeO40–graphene shows higher photocatalytic activity for methylene blue degradation under UV light irradiation. For methylene blue degradation other composites such as ZnFe2O4–graphene71 and graphene–γBi2MoO667 also show effective results. The near transparent graphene sheets are exfoliated fully and decorated homogeneously with ZnFe2O4 nanocrystals. During the hydrothermal treatment graphene oxide was reduced to graphene in the presence of a reducing agent.125–127 The band gap energy of ZnFe2O4 was found to be 1.9 eV as reported by Jang et al.128 The absorbance of methylene blue at 464 nm was nearly unchanged after irradiation of light for 90 min. But the photo-degradation rate of methylene blue was increased by 20% for the ZnFe2O4–graphene (0.2) composite. Due to its magnetic properties, ZnFe2O4–graphene nanocrystals can be separated magnetically after use. Similarly, the graphene–Bi2MoO6 composite showed 4 times higher activity i.e. 80% methylene blue degraded under visible light irradiation compared to Bi2MoO6. The degradation rate also depends upon the crystallinity of the perovskite. In the case of BaZrO3, with an increase in the crystallinity of BaZrO3 the degradation rate of methyl orange increases. A significant number of studies have been done on the CNT–metal oxide composite for photocatalytic applications; however the photocatalytic activity of the CNT–perovskite composite is still unexplored. Lan et al. reported that the photocatalytic activity of the Zn–Al–In mixed metal oxides was enhanced in the composite of the Zn–Al–In mixed metal oxides/carbon nanotubes.129
The photocatalytic activity of the perovskite and layered perovskite also becomes enhanced when it makes a composite with gC3N4. For instance, the photocatalytic activities of the SrTiO3–g-C3N4, BaTaO3–g-C3N4, NaTaO3–g-C3N4 composites have significant effect on dye degradation. By making a composite of these materials with g-C3N4, the band gap energy is effectively minimized. From the UV-Vis DRS the band gap energy of g-C3N4 and BaTiO3 is found to be nearly 2.7 eV and 3.15 eV, respectively, while the composite shows a minor shift in the absorption edge, indicating minor changes in the band gap energy. However, the photocatalytic activity towards methyl orange (MO) degradation was 76% after 6 h irradiation of sun light in the presence of the composite and showed excellent reusability. The mechanism behind the photocatalytic reaction is that the photo-generated electrons from CB of g-C3N4 migrate to the conduction band of BaTiO3 and simultaneously the holes are transferred from the VB of BaTiO3 to that of VB g-C3N4 (Fig. 11). This mechanism effectively separated e− and h+ pairs to increase the availability of electrons.29–31 Likewise, in the case of g-C3N4–NaTaO3, the photocatalytic degradation of Rhodamine B (RhB) was significantly enhanced in comparison to pure g-C3N4 and NaTaO3. The composite has excellent photocatalytic activity under both UV-Vis and visible light irradiation and also does not lose its photocatalytic activity after 5 runs, showing excellent reusability (Fig. 16).
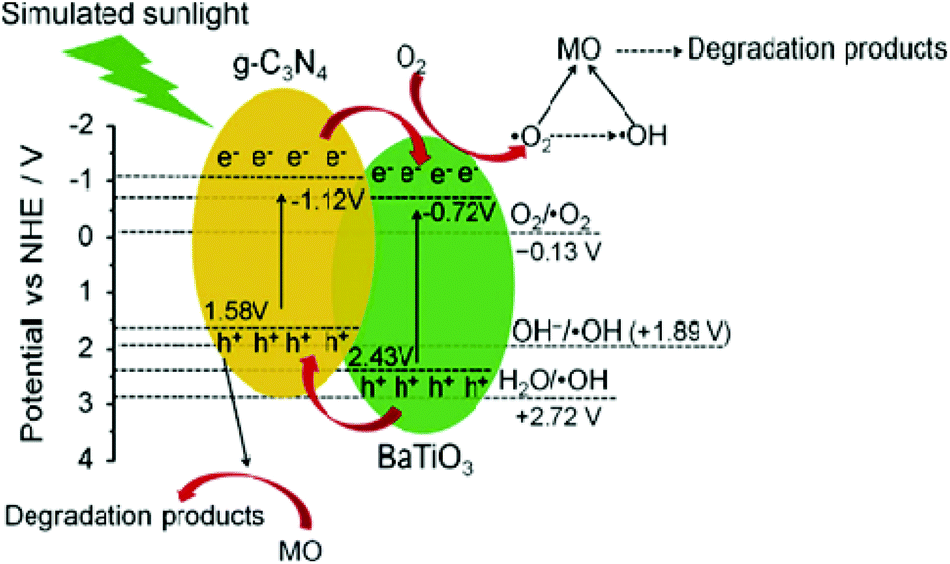 | ||
| Fig. 16 Schematic illustration of the possible photocatalytic mechanism of BaTiO3–g-C3N4 composites toward the degradation of methyl orange (reproduced from ref. 29, license no. 3678180169216). | ||
6. Conclusion and outlook
In summary, by introducing graphene, CNTs and g-C3N4, the optical and electronic properties of semiconductor photocatalysts can be modified. Modification of semiconductor photocatalysts with graphene/CNTs can improve the light absorption capacity of the material due to black body properties of these carbonaceous materials. Incorporation of graphene into the composites can significantly improve the charge separation efficiency of semiconductor photocatalysts. The charge separation efficiency significantly improved the photocatalytic activities such as organic pollutant degradation and water splitting activity. Although the present trend in this area of research could be able to develop suitable photocatalysts with significant activity in the visible area, the efficiency is still too far away from real and practical applications. For the practical utility of this technology, our ultimate target is to obtain minimum 10% efficiency at 600 nm. Therefore, the developed photocatalysts must have a narrow band gap of around 2 eV. Finally, many areas in photocatalysis are still unexplored and further research is necessary in this field. To modify a new type of semiconductor photocatalyst, detailed theoretical investigations and kinetics studies to obtain a clear idea of the material are very much necessary (Table 5).| Abbreviation | Full forms |
|---|---|
| 2D | 2-Dimensional |
| RGO | Reduced graphene oxide |
| GO | Graphene oxide |
| VPO | Vanadium phosphate |
| NHE | Normal hydrogen electrode |
| E e | Energy of free electrons |
| E g | Band gap energy |
| CNT | Carbon nanotube |
| SWCNT | Single walled carbon nanotube |
| MWCNT | Multi-walled carbon nanotube |
| g-C3N4 | Graphitic carbon nitride |
| XRD | X-ray diffraction |
| CB | Conduction band |
| VB | Valence Band |
| eV | Electron volt |
| IT-CCBC | Intertwined core–corona structured bifunctional catalyst |
| CVD | Chemical vapour deposition |
| UV-Vis | Ultra violet-visible |
| PL | Photoluminescence |
| SEM | Scanning electron microscopy |
| SAED | Selected area electron diffraction |
| TEM | Transmission electron microscopy |
| HRTEM | High-resolution transmission electron microscopy |
| CMC | Nanocomposite |
| NP | Nanoparticle |
| FE-SEM | Field emission scanning electron microscopy |
| XPS | X-ray photoelectron spectroscopy |
| NCG | Nanocomposite generator |
References
- J. Shi and L. Guo, Prog.Nat.Sci.: Mater. Int., 2012, 22, 592 CrossRef PubMed.
- S. Keav, S. K. Matam, D. Ferri and A. Weidenkaff, Catalysts, 2014, 4, 226 CrossRef PubMed.
- E. Mooser and W. B. Pearson, Acta Crystallogr., 1959, 12, 1015 CrossRef CAS.
- http://en.wikipedia.org/wiki/Goldschmidt_tolerance_factor (accessed on 18.03.2015).
- V. M. Goldschmidt and D. G. Krystallochemie, Die Naturwissenschaften, 1926, 8, 477–485 CrossRef.
- S. Gebbinghaus, H. P. Abicht, R. Dronskowski, T. Müller, A. Reller and A. Weidenkaff, Prog. Solid State Chem., 2009, 37, 173 CrossRef PubMed.
- S. Yoon, A. E. Maegli, L. Karvonen, S. K. Matam, A. Shkabko, S. Riegg, T. Grobmann, S. G. Ebbinghaus, S. Pokrant and A. Weidenkaff, J. Solid State Chem., 2013, 206, 226 CrossRef CAS PubMed.
- Y. Tsujimoto, K. Yamaura and E. T. Muromachi, Appl. Sci., 2012, 2, 206 CrossRef CAS PubMed.
- K. M. Parida, K. H. Reddy, S. Martha, D. P. Das and N. Biswal, Int. J. Hydrogen Energy, 2010, 35, 12161–12168 CrossRef CAS PubMed.
- Y. Liu, L. Xie, Y. Li, R. Yang, J. L. Qu, Y. Q. Li and X. G. Li, J. Power Sources, 2008, 183, 701 CrossRef CAS PubMed.
- C. C. Hu, C. C. Tsai and H. Teng, J. Am. Ceram. Soc., 2009, 92, 460 CrossRef CAS PubMed.
- K. Shimizu, Y. Tsuji, T. Hatamachi, K. Toda, T. Kodama, M. Sato and Y. Kitayama, Phys. Chem. Chem. Phys., 2004, 6, 1064 RSC.
- J. J. Ding, J. Bao, S. Sun, Z. L. Luo and C. Gao, J. Comb. Chem., 2009, 11, 523 CrossRef CAS PubMed.
- U. A. Joshi, J. S. Jang, P. H. Borse and J. S. Lee, Appl. Phys. Lett., 2008, 92, 242106 CrossRef PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- G. C. Behera, K. M. Parida and P. K. Satapathy, RSC Adv., 2013, 3, 4863 RSC.
- S. Martha, D. K. Padhi and K. M. Parida, ChemSusChem, 2014, 7, 585 CrossRef CAS PubMed.
- T. Xian, H. Yang and Y. S. Huo, Phys. Scr., 2014, 89, 115801 CrossRef.
- D. K. Padhi and K. M. Parida, J. Mater. Chem. A, 2014, 2, 10300 CAS.
- G. Kumar Pradhan, D. Kumar Padhi and K. M. Parida, ACS Appl. Mater. Interfaces, 2013, 5, 9101 Search PubMed.
- D. K. Padhi, G. C. Pradhan, K. M. Parida and S. K. Singh, Chem. Eng. J., 2014, 255, 78 CrossRef CAS PubMed.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162 CrossRef CAS PubMed.
- V. Gupta and T. A. Saleh, Synthesis of Carbon Nanotube-Metal Oxides Composites, Adsorption and Photo-degradation, Carbon Nanotubes - From Research to Applications, ed. S. Bianco, InTech, 2011, ISBN: 978-953-307-500-6, DOI:10.5772/18009.
- F. Dakhlaghi and M. Daghazadeh-meshgi, Effect of Cr and Co contents in the perovskite type (lacr(1-x)co(x)o3) catalysts on the characteristics of carbon nanotubes synthesized by CVD method, EU, Olomouc, Czech Republic, 2010, vol. 10, 12–14 Search PubMed.
- B. K. Vijayan, N. M. Dimitrijevic, D. F-Shapiro, J. Wu and K. A. Gray, ACS. Catal., 2012, 2, 223 CrossRef CAS.
- Y. K. Kim and H. Park, Energy Environ. Sci. 20114685–694 Search PubMed.
- H. Yu, S. Zhang and F. Peng, Mater. Res. Bull., 2014, 56, 19 CrossRef PubMed.
- Y. Xu and W. Zhang, ChemCatChem, 2013, 5, 2343 CrossRef CAS PubMed.
- T. Xian, H. Yang, L. J. Di b and J. F. Dai, J. Alloys Compd., 2015, 622, 1098 CrossRef CAS PubMed.
- S. Kumar, B. Kumar, T. Surendar and V. Shanker, Mater. Res. Bull., 2014, 49, 310 CrossRef CAS PubMed.
- X. Xu, G. Liu, C. Randorn and J. T. S. Irvine, Int. J. Hydrogen Energy, 2011, 36, 13501 CrossRef CAS PubMed.
- X. H. Li and M. Antonietti, Chem. Soc. Rev., 2013, 42, 6593–6604 RSC.
- E. Z. Lee, Y. S. Jun, W. H. Hong, A. Thomas and M. M. Jin, Angew. Chem., Int. Ed., 2010, 49, 9706–9710 CrossRef CAS PubMed.
- S. Barman and M. Sadhukhan, J. Mater. Chem., 2012, 22, 21832–21837 RSC.
- E. Z. Lee, S. U. Lee, N. S. Heo, G. D. Stucky, Y. S. Jun and W. H. Hong, Chem. Commun., 2012, 48, 3942–3944 RSC.
- E. G. Gillan, Chem. Mater., 2000, 12, 3906–3912 CrossRef CAS.
- Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- J. H. Yang, X. T. Wu, X. F. Li, Y. Liu, M. Gao, X. Y. Liu, L. N. Kong and S. Y. Yang, Appl. Phys. A, 2011, 105, 161–166 CrossRef CAS.
- H. J. Yan, Y. Chen and S. M. Xu, Int. J. Hydrogen Energy, 2012, 37, 125–133 CrossRef CAS PubMed.
- J. Fu, B. B. Chang, Y. L. Tian, F. N. Xi and X. P. Dong, J. Mater. Chem. A, 2013, 1, 3083–3090 CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
- T. Tyborski, C. Merschjann, S. Orthmann, F. Yang, M.-Ch. Lux-Steiner and T. Schedel-Niedrig, J. Phys.: Condens. Matter, 2012, 24, 162201 CrossRef CAS PubMed.
- J. Sehnert, K. Baerwinkel and J. Senker, J. Phys. Chem. B, 2007, 111, 10671–10680 CrossRef CAS PubMed.
- A. Snis and S. F. Matar, Phys. Rev. B: Condens. Matter, 1999, 15, 10855–10863 CrossRef.
- G. M. Rignanese, J. C. Charlier and X. Gonze, Phys. Rev. B: Condens. Matter, 2002, 66, 205416 CrossRef.
- B. V. Lotsch and W. Schnick, Chem. Mater., 2006, 18, 1891–1900 CrossRef CAS.
- C. Li, C. B. Cao and H. S. Zhu, Mater. Lett., 2004, 58, 1903–1906 CrossRef CAS PubMed.
- Y. G. Yoon, B. G. Pfrommer, F. Mauri and S. G. Louie, Phys. Rev. Lett., 1998, 15, 3388–3391 CrossRef.
- E. Kroke and M. Schwarz, Coord. Chem. Rev., 2004, 248, 493–532 CrossRef CAS PubMed.
- I. Alves, G. Demazeau, B. Tanguy and F. Weill, Solid State Commun., 1999, 109, 697–701 CrossRef CAS.
- V. N. Khabashesku, J. L. Zimmerman and J. L. Margrave, Chem. Mater., 2000, 12, 3264–3270 CrossRef CAS.
- J. Ortega and O. F. Sankey, Phys. Rev. B: Condens. Matter, 1995, 4, 2624–2627 CrossRef.
- J. E. Lowther, Phys. Rev. B: Condens. Matter, 1999, 18, 11683–11686 CrossRef.
- M. Mattesini, S. F. Matar and J. Etourneau, J. Mater. Chem., 2000, 10, 709–713 RSC.
- A. Y. Liu, Phys. Rev. B: Condens. Matter, 1994, 14, 10362–10365 CrossRef.
- Y. Zhang, H. Sun and C. F. Chen, Phys. Rev. B: Condens. Matter, 2006, 73, 144115 CrossRef.
- J. L. Zimmerman, R. Williams, V. N. Khabashesku and J. L. Margrave, Nano Lett., 2001, 1, 731–734 CrossRef CAS.
- Y. Miyamoto, M. L. Cohen and S. G. Louie, Solid State Commun., 1997, 102, 605–608 CrossRef CAS.
- E. Kroke, M. Schwarz, E. Horath-Bordon, P. Kroll, B. Noll and A. D. Norman, New J. Chem., 2002, 26, 508–512 RSC.
- Y. Zhang, T. Mori and J. Ye, Sci. Adv. Mater., 2012, 4, 282–291 CrossRef CAS PubMed.
- W. Chen, G.-R. Duan, T.-Yu Liu, S.-M. Chen and X.-H. Liu, Mater. Sci. Semicond. Process., 2015, 35, 45–54 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- K. Dai, L. H. Lu, C. H. Liang, Q. Liu and G. P. Zhu, Appl. Catal., B, 2014, 156–157, 331–340 CrossRef CAS PubMed.
- M. C. Long, W. M. Cai and H. Kisch, Chem. Phys. Lett., 2008, 461, 102–105 CrossRef CAS PubMed.
- C. Wu, Y. Zhang, S. Li, H. Zheng, H. Wang, J. Liu, K. Li and H. Yan, Chem. Eng. J., 2011, 178, 468 CrossRef CAS PubMed.
- A. Mukherji, B. Seger, G. Qing (Max) Lu and L. Wang, ACS Nano, 2011, 5, 3483 CrossRef CAS PubMed.
- F. Zhou, R. Shi and Y. Zhu, J. Mol. Catal. A: Chem., 2011, 340, 77 CrossRef CAS PubMed.
- A. Sun, H. Chen, C. Song, F. Jiang, X. Wang and Y. Fu, RSC Adv., 2013, 3, 4332 RSC.
- D. U. Lee, H. W. Park, M. G. Park, V. Ismayilov and Z. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 902–910 CAS.
- T. Xian, H. Yang, L. Di, J. Ma, H. Zhang and J. Dai, Nanoscale Res. Lett., 2014, 9, 327 CrossRef PubMed.
- Y. Fu and X. Wang, Ind. Eng. Chem. Res., 2011, 50, 7210 CrossRef CAS.
- W. Hummers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- X. Wanga, W. Maoa, J. Zhang, Y. Han, C. Quan, Q. Zhang, T. Yang, J. Yang, X. Li and W. Huang, J. Colloid Interface Sci., 2015, 448, 17–23 CrossRef PubMed.
- T. Yan, Q. Yan, X. Wang, H. Liu, M. Li, S. Lu, W. Xu and M. Sun, Dalton Trans., 2015, 44, 1601 RSC.
- S. Kumar, S. Tonda, A. Baruah, B. Kumar and V. Shanker, Dalton Trans., 2014, 43, 16105 RSC.
- M. Yang and X. Jin, J. Wuhan Univ. Technol., Mater. Sci. Ed. 2014291111–1116 Search PubMed.
- Y. Huang, Y. Wei, S. Cheng, L. Fan, Y. Li, J. Lin and J. Wu, Sol. Energy Mater. Sol. Cells, 2010, 94, 761 CrossRef CAS PubMed.
- Y. Huang, Y. Li, Y. Wei, M. Huang and J. Wu, Sol. Energy Mater. Sol. Cells., 2011, 95, 1019 CrossRef CAS PubMed.
- J. H. Heo, S. H. Im, J. H. Noh, T. N. Mandal, C.-S. Lim, J. A. Chang, Y. H. Lee, H.-j. Kim, A. Sarkar and M. K. Nazeeruddin, et al., Efficient Inorganic–Organic Hybrid Heterojunction Solar Cells Containing Perovskite Compound and Polymeric Hole Conductors, Nat. Photonics, 2013, 7, 486–489 CrossRef CAS PubMed.
- Y. Wang, B. G. Sumpter, J. Huang, H. Zhang, P. Liu, H. Yang and H. Zhao, Density Functional Studies of Stoichiometric Surfaces of Orthorhombic Hybrid Perovskite CH3NH3PbI3, J. Phys. Chem. C, 2015, 119, 1136–1145 CAS.
- J. Haruyama, K. Sodeyama, L. Han and Y. Tateyama, Termination Dependence of Tetragonal CH3NH3PbI3 Surfaces for Perovskite Solar cells, J. Phys. Chem. Lett., 2014, 5, 2903–2909 CrossRef CAS.
- T. Baikie, Y. Fang, J. M. Kadro, M. Schreyer, F. Wei, S. G. Mhaisalkar, M. Grätzel and T. J. White, Synthesis and Crystal Chemistry of the Hybrid Perovskite (CH3NH3)PbI3 for Solid-state Sensitised Solar Cell Applications, J. Mater. Chem. A, 2013, 1, 5628–5641 CAS.
- J.-H. Im, C.-R. Lee, J.-W. Lee, S.-W. Park and N.-G. Park, 6.5% Efficient Perovskite Quantum-dot-sensitized Solar Cell, Nanoscale, 2011, 3, 4088–4093 RSC.
- P. Docampo, F. C. Hanusch, S. D. Stranks, M. Döblinger, J. M. Feckl, M. Ehrensperger, N. K. Minar, M. B. Johnston, H. J. Snaith and T. Bein, Solution Deposition-conversion for Planar Heterojunction Mixed Halide Perovskite Solar Cells, Adv. Energy Mater., 2014, 4, 1400355 Search PubMed.
- J. An, L. Zhu, N. Wang, Z. Song, Z. Yang, D. Du and H. Tang, Chem. Eng. J., 2013, 219, 225 CrossRef CAS PubMed.
- Z. Li, Y. Shen, Y. Guan, Y. Hu, Y. Lin and C. Nan, J. Mater. Chem. A, 2014, 2, 1967 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- J. S. Lee, Catal. Surv. Asia, 2005, 9, 217 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- Report of the DOE Basic Energy Workshop on Solar Energy Utilization, Argonne Nat'l Laboratories, 2005.
- S. Martha, K. H. Reddy and K. M. Parida, J. Mater. Chem. A, 2014, 2, 3621 CAS.
- J. S. Lee, Catal. Surv. Asia, 2005, 9, 217 CrossRef CAS.
- J. M. Lehn, J. P. Sauvage and R. Ziessel, Nouv. J. Chim., 1980, 4, 623 CAS.
- S. Sato and J. M. White, Ind. Eng. Chem. Prod. Res. Dev., 1980, 19, 542 CrossRef CAS.
- K. Domen, S. Naito, M. Suma, T. Onishi and K. Tamaura, J. Chem. Soc., Chem. Commun., 1980, 543 RSC.
- J. Nozik, Annu. Rev. Phys. Chem., 1978, 29, 189 CrossRef.
- M. Grtzel, Energy Resources through Photochemistry and Catalysis, Academic Press, New York, 1983 Search PubMed.
- N. Serpone and E. Pelizzetti, Photocatalysis, Wiley, New York, 1989 Search PubMed.
- Z. Zou and H. Arakawa, J. Photochem. Photobiol., A, 2003, 158, 145 CrossRef CAS.
- M. Anpo, S. Dohshi, M. Kitano, Y. Hu, M. Takeuchi and M. Matsuoka, Annu. Rev. Mater. Res., 2005, 35, 1 CrossRef CAS.
- A. Kudo, Int. J. Hydrogen Energy, 2006, 31, 197 CrossRef CAS PubMed.
- W. Zhang, S. B. Park and E. Kim, Energy Fuels, 2006, 295 Search PubMed.
- K. Maeda, K. Teramura, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, Pure Appl. Chem., 2006, 78, 2267 CAS.
- W. Shanguan, Sci. Technol. Adv. Mater., 2007, 8, 76 CrossRef PubMed.
- Y. Cao, L. Zhang, D. Tao, D. Huo and K. Su, Electrochim. Acta, 2014, 132, 483 CrossRef CAS PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851 CAS.
- K. Maeda, K. Teramura and K. Domen, Catal. Surv. Asia, 2007, 11, 145 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992 CrossRef CAS.
- D. W. Hwang, H. G. Kim, J. S. Lee, J. Kim, W. Li and S. H. Oh, J. Phys. Chem. B, 2005, 109, 2093 CrossRef CAS PubMed.
- M. Anpo and M. Takeuchi, J. Catal., 2003, 216, 505 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454 CrossRef CAS PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851 CAS.
- P. K. Farsoiya and D. A. Prasad, Int. J. Eng. Res. Appl. (IJERA) Search PubMed , ISSN: 2248-9622, National Conference on Advances in Engineering and Technology (AET-29th March 2014).
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992 CrossRef CAS.
- T. Ishii, H. Kato and A. Kudo, J. Photochem. Photobiol., A, 2004, 163, 181 CrossRef CAS.
- H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2002, 106, 12441 CrossRef CAS.
- M. Machida, K. Miyazaki, S. Matsushima and M. Arai, J. Mater. Chem., 2003, 13, 1433 RSC.
- J. Ding, W. Yan, W. Xie, S. Sun, J. Bao and C. Gao, Nanoscale, 2014, 2299 RSC.
- A. Mukherji, B. Seger, G. Qing (Max) Lu and L. Wang, ACS Nano, 2011, 5, 3483 CrossRef CAS PubMed.
- S. Martha, A. Nashim and K. M. Parida, J. Mater. Chem. A, 2013, 1, 7816 CAS.
- K. M. Parida, S. Martha, D. P. Das and N. Biswal, J. Mater. Chem., 2010, 20, 7144 RSC.
- J. Hu, J. Ma, L. Wang and H. Huang, J. Alloys Compd., 2014, 583, 539 CrossRef CAS PubMed.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380 CrossRef CAS PubMed.
- D. H. Yoo, V. C. Tran, V. H. Pham, J. S. Chung, N. T. Khoa, E. J. Kim and S. H. Hahn, Curr. Appl. Phys., 2011, 11, 805 CrossRef PubMed.
- J. S. Jang, P. H. Borse, J. S. Lee, O. S. Jung, C. R. Cho, E. D. Jeong, M. G. Ha, M. S. Won and H. G. Kim, Korean Chem. Soc., 2009, 30, 1738 CrossRef CAS.
- M. Lan, G. Fan, W. Sun and F. Li, Appl. Surf. Sci., 2013, 282, 937 CrossRef CAS PubMed.
- G. Volonakis and F. Giustino, J. Phys. Chem. Lett., 2015, 6, 2496–2502 CrossRef CAS PubMed.
- A. Weidenkaff, S. G. Ebbinghaus and T. Lippert, Chem. Mater., 2002, 14, 1797–1805 CrossRef CAS.
- D. Xu, P. Lu, P. Dai, H. Wang and S. Ji, J. Phys. Chem. C, 2012, 116, 3405 CAS.
- A. Peigney, C. Laurent and E. Flahaut, Carbon, 2001, 39, 507 CrossRef CAS.
| This journal is © the Partner Organisations 2015 |