
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic strategies to nanostructured photocatalysts for CO2 reduction to solar fuels and chemicals
Donna
Chen
a,
Xingguang
Zhang
b and
Adam F.
Lee
 *b
*b
aSchool of Chemistry, Cardiff University, Cardiff, UK
bEuropean Bioenergy Research Institute, Aston University, Birmingham, UK. E-mail: A.F.Lee@aston.ac.uk; Tel: +44 (0)121 2044036
First published on 22nd May 2015
Abstract
Artificial photosynthesis represents one of the great scientific challenges of the 21st century, offering the possibility of clean energy through water photolysis and renewable chemicals through CO2 utilisation as a sustainable feedstock. Catalysis will undoubtedly play a key role in delivering technologies able to meet these goals, mediating solar energy via excited generate charge carriers to selectively activate molecular bonds under ambient conditions. This review describes recent synthetic approaches adopted to engineer nanostructured photocatalytic materials for efficient light harnessing, charge separation and the photoreduction of CO2 to higher hydrocarbons such as methane, methanol and even olefins.
 Donna Chen | Donna Hui Lin Chen relocated to Sydney, Australia, in 2003 for her BSc degree, majoring in Nanotechnology, at The University of New South Wales. She subsequently moved to the University of Sydney where she commenced a PhD at the School of Chemistry focusing on the synthesis, characterisation, testing and study of potential visible light active catalysts for H2 generation. In 2012, she took up a Research Assistant position at Cardiff University, where she initiated a CO2 photoreduction project. She is interested in technologies that minimise the environmental impact of industrial processes. |
 Xingguang Zhang | Xingguang Zhang received his Bachelor of Engineering in Chemical Engineering and Technology from Guizhou University (Guiyang, China) in 2007, Master of Engineering in Industrial Catalysis under the supervison of Professor Jun Wang in Nanjing Tech University (Nanjing, China) in 2010, and PhD under the supervision of Dr Xuebin Ke and Professor Huaiyong Zhu at Queensland University of Technology (Brisbane, Australia) in 2014. He is currently a research associate working with Professor Adam Lee and Professor Karen Wilson to develop supported metal catalysts and solid acid catalysts for clean chemicals synthesis within Aston University and UK Catalysis Hub. |
 Adam F. Lee | Adam Lee is Professor of Sustainable Chemistry and an EPSRC Leadership Fellow in the European Bioenergy Research Institute, Aston University. He holds a BA and PhD from the University of Cambridge, and following Lecturer/Senior Lecturer roles at the Universities of Hull and York respectively, has held Chair appointments at Cardiff, Warwick and Monash universities. His research addresses the rational design of nanoengineered materials for clean catalytic technologies, notably sustainable chemical synthesis and energy production, and the development of in situ methods providing molecular insight into surface reactions, for which he was awarded the 2012 Beilby Medal and Prize by the Royal Society of Chemistry. |
1. Introduction
The use of sunlight to split water as a source of electrons that can be stored in chemical bonds for the production of primary energy carriers and molecular building blocks underpins the natural world. Humanity remains highly dependent upon such elegant photosynthetic processes, developed through natural evolutionary selection, for the sustainable growth of biomass, which literally feeds our planet. However, recent technological breakthroughs offer a glimpse of a mid-term future in which anthropogenic systems will enable artificial photosynthesis with efficiencies surpassing that of plants, to deliver solar fuels and chemicals.1,2 Arguments for the development and global deployment of artificial photosynthesis technology to reduce anthropogenic CO2 emissions from their historic high,3,4 increase fuel security,5 and support a sustainable global economy and ecosystem have been recently expounded,6,7 and new mechanisms for enhancing the policy and governance profile in the context of energy sustainability sought.Solar fuels include H2 (from the reduction of H+ derived from water photolysis) or carbon-based fuels (derived from the photoreduction of CO2) such as methane, methanol or CO, and associated higher hydrocarbons via subsequent well-established industrial thermal chemistry. Artificial photosynthesis may be considered to span the genetic engineering of plant or algal crops to tailor their lignocellulosic or lipid constituents for biofuels production, synthetic biology to combine incorporate cyanobacterial proteins into chloroplasts and hence improve the rate of CO2 fixation,8 and the use of nanotechnology via inorganic or molecular systems to produce photo-electrochemical devices.9–12 In contrast to a number of competing renewable energy technologies, such as wind, tidal and photovoltaics, wherein electricity is the end product, and hence a dependency exists upon concomitant advances in storage and smart grid capabilities, the trapping of solar energy within chemical bonds provides immediate access to (potentially high) energy density carriers for transportation fuels. Water photo-oxidation for H2 production has been reviewed extensively elsewhere;13–15 hence this review focuses on photocatalytic CO2 reduction.
The potential of CO2 as a chemical feedstock via chemical and biochemical,16 electrocatalytic17,18 and photocatalytic transformations has been widely recognised, with methane19 and methanol20–23 obvious targets to replace fossil fuels in stationary power generation and transportation. Another key area in which CO2 sequestration may have a profound impact is as a carbon source for renewable polymers and plastics and solvent.24–26 In the context of polymers, the thermochemical incorporation of CO2 into polycarbonates/carbamates is well-established; however, its direct photoreduction to light (C1–C3) olefins represents a greater challenge. Olefins and their polymers are the single largest chemical commodity in the world, with annual global ethene and propene production capacity in 2012 around 156 and 80 Mt respectively.25,27 These olefins are currently obtained from non-renewable fossil fuels:28 commercial ethene and propene manufacture involves steam or catalytic cracking of naphtha, gasoil and condensates to hydrocarbon mixtures followed by distillation. Cracking crude oil to produce ethene or propene is thermodynamically unfavourable (ΔG ≈ +100 kJ mol−1), requiring high temperatures (>600 °C) to overcome the huge activation barriers to C–C cleavage (280 kJ mol−1 for kerosene conversion29). Hence steam cracking is the most energy-consuming process in chemistry, accounting for 8% of the sector's primary energy use and annual CO2 emissions of 180–200 Mt.28
1.1. Brief history and fundamental principles of photocatalytic materials for CO2 reduction
Renewable solar energy, harnessed via innovative catalysts and reactors, has the potential to photoreduce linear CO2 molecules by breaking the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds to form C–H bonds and then to yield products, such as methane, methanol or light olefins (e.g. CO2 + 2H2O ↔ CH3OH + 3/2O2; CO2 + 2H2O ↔ CH4 + 2O2; and 2CO2 + 2H2O ↔ C2H4 + 3O2), thereby creating new chemical supply chains free of current dependencies on oil, coal and natural gas and offering an economical and potentially environmentally-benign CO2 utilisation process (Fig. 1).30
O bonds to form C–H bonds and then to yield products, such as methane, methanol or light olefins (e.g. CO2 + 2H2O ↔ CH3OH + 3/2O2; CO2 + 2H2O ↔ CH4 + 2O2; and 2CO2 + 2H2O ↔ C2H4 + 3O2), thereby creating new chemical supply chains free of current dependencies on oil, coal and natural gas and offering an economical and potentially environmentally-benign CO2 utilisation process (Fig. 1).30
 | ||
| Fig. 1 Schematic of a carbon zero route to solar fuels and renewable olefins through the photocatalytic reduction of CO2. | ||
Great challenges lie in the robust chemical state of carbon atoms in CO2, which requires not only the participation of incident protons but also effective excited electrons, and thus additional reductive agents can offer better opportunities to facilitate the activation of CO2. In this regard, H2O is preferred as a reducing agent because it simultaneously takes advantage of water oxidation and CO2 fixation compared with other sacrificing reductive agents such as amines, H2, and S2−. Hence, the harvesting of input energy by means of photocatalytic materials from incident light plays a pivotal role in achieving these processes. The first study on the photocatalytic CO2 reduction was reported by Halmann in 1978, employing a Hg lamp and single crystal, p-type GaP cathode within an electrochemical cell through which CO2 was bubbled to yield formic acid, formaldehyde and methanol.31 A year later, Inoue and co-workers reported the photoelectrocatalytic reduction of CO2 to formic acid, formaldehyde, methanol and methane, over a variery of semiconductors including TiO2, ZnO, CdS, GaP, SiC, and WO3.32
These pioneering studies were followed by the development of visible-light driven photocatalysts and production of more functionalised organic moieties, e.g. CO2 photoreduction via colloidal CdS aqueous solutions to glyoxylic and acetic acids.33
Moreover, large-scale photocatalytic CO2 reduction to solar fuels or chemicals requires intensified processing and carefully engineered robust (and hence likely heterogeneous) catalysts with high accessibility, able to activate small molecules at ambient conditions.34 This challenge can only be met through novel nanotechnologies, which permit the construction of tailored materials whose structure (and function) can be precisely tuned.35–37 A practical photocatalytic chemical reactor requires appropriately selected coupled redox reactions, and the ability to efficiently harness light capable of driving the requisite electron-transfer chemistry.38 The former favours water as a green chemical reductant (mimicking natural photosynthesis), while the latter requires active catalysts either able to absorb solar energy directly or coupled to sensitisers which help gather and focus energy onto the active sites.
The optical properties of potential solid photocatalysts are dictated by the rate-limiting oxidation and reduction steps that must occur at the surface of their nanostructures:39 2H2O → O2 + 4H+ +4e−; CO2 + e− → CO2˙− (Eoredox = +1.64 V and −1.90 V, respectively, on the basis of NHE (normal hydrogen electrode) at pH = 7). Such values impose a minimum for the photoexcitation energy of electrons needed to reduce CO2, whereas few pure materials are photoresponsive to these wavelengths and catalytically active towards CO2 and water conversion, even though visible light can create sufficiently excited electrons. These insurmountable limitations necessitate the development of new photocatalytic materials particularly with specially-nanostructured antennas to harvest both UV and visible light efficiently and to initiate the chemical reactions effectively.
1.2. Strengths and challenges of nanostructured photocatalysts
Nanoscale materials often exhibit unique physicochemical which diverge widely from their bulk counterparts. Such nanomaterials, commonly synthesised in the form of nanoparticles (NPs), nanofibres (NFs) and nanotubes (NTs), in addition to more exotic topologies such as nanobelts and nanoflowers, afford high surface areas, tunable architectures and distinct electronic surface states. In particular, photocatalytic nanostructures can offer high densities of photoactive surface sites and thin-walled structures/ultrathin films able to facilitate rapid interfacial charge carrier transport to adsorbates.35,40 For many years, CO2 reduction has centred around titania-based photocatalysts, which are only able to utilise a fraction of the solar spectrum (∼2%, mainly as UVA),41–45 and require exploitation of quantum effects or doping to reduce their intrinsic band gaps,46 sometimes in combination with inorganic or organic supramolecular (biomimetic) sensitisers.47 Significant advances in artificial photosynthesis have been achieved through such photocatalytic nanoarchitectures,37,48e.g. the discovery of ‘self-repairing’ CoxPO4 catalysts producing O2 akin to the natural Mn catalyst in photosystem II,49 corrosion resistant TiO2 nanocomposites able to split water50 or reduce CO2 with H2O to yield CH4 and olefins,40,51 and the use of metal oxynitrides52 and polymeric nitrides53 for efficient water splitting under visible light. Solar CH4 production from CO2 has seen dramatic improvements in recent years, with TiO2 NTs delivering 17.5 nanomoles h−1 cm−2 mW−1 of hydrocarbons from 1 bar CO2 under sunlight, with a quantum efficiency of 0.74%,40 defined throughout this review as:where Mi is the mols of product i, ni is the number of electrons attending the production of product i, and Pm is mols of incident photons.54,55 Cu-doped titania coatings deposited on a monolithic substrate have even been reported able to deliver 117 nanomoles h−1 cm−2 m W−1 of methane.56 In addition to maximising the quantum efficiency, productivity and selectivity towards hydrocarbons, the stability of semiconductor photocatalysts under reaction conditions (notably resistance to photocorrosion57,58) and routes to their incorporation into reactor designs which facilitate efficient light absorption and mass transport,59–61 are also key considerations.

Significant developments in molecular nanocatalysts for photochemical CO2 reduction have been achieved and reviewed;38,62,63 nevertheless, improvements in the mechanical strength, physicochemical stability and photocatalytic efficiency of such materials remain necessary to meet the engineering requirements for large-scale applications. Molecular band structures control incident light absorption, electron–hole pair generation and charge carrier migration. Similarly, energy band engineering of semiconductor photocatalysts to impede the recombination of photo-excited electron–hole pairs and enhance light absorption is of paramount importance.64–66 A further challenge is the optimisation of CO2 and H2O adsorption over photocatalyst surfaces, which is poorly understood to date,67,68 and requires a thorough knowledge of the atomic structure of the photocatalyst interface. Advanced inorganic synthetic methods afford precise manipulation of exposed crystal facets, morphologies, structural periodicity, and hierarchical pore networks,69–71 in addition to the co-assembly of sensitisers or co-catalysts and promoters. Meeting these challenges will also necessitate continued advances in our fundamental understanding of photocatalytic reaction and deactivation mechanisms. This review discusses progress in the design of nanostructured solid materials CO2 to solar fuels and chemicals, with a particular focus on new synthetic routes to inorganic photocatalysts possessing well-defined physichochemical properties and the elucidation of associated structure-function relations. Materials chemistry approaches to porous and layered semiconductor architectures, and hybrid photoactive nanomaterials (e.g. plasmonic nanostructures) are also highlighted, and the most active and selective systems benchmarked.
1.3. Mechanistic complications in CO2 photoreduction
Before considering the vast array of nanomaterials employed in for CO2 photoreduction, we consider briefly some of the particular analytical challenges in evaluating photoactivity and product formation. Carbon-containing precursors and/or solvents are employed in the synthesis of most heterogeneous photocatalysts, and few studies quantify the carbon content of the resulting as-prepared materials. Such carbonaceous residues can either decompose directly upon subsequent irradiation,68 or react with photoexcited molecular species generated from CO2 or water,72 to liberate carbon-containing liquid/gas phase products. Discriminating whether such products originate solely from desired CO2 conversion and/or carbon residues is not possible by conventional chromatographic methods, and rarely accorded the attention deserved.Yui and co-workers reported that organic adsorbates such as acetic acid present over commercial, untreated P25 TiO2 were responsible for CH4 evolution as a major product during CO2 photoreduction.68 A photo-Kolbe decaboxylation mechanism was proposed that accounted for the observed methane solely in terms of acetic acid reduction: CH3COOH + H+ → CH3˙ + CO2 + H+; CH3˙ + CH3COOH → CH2COOH˙ + CH4. Pre-calcination and washing of the TiO2 suppressed methane production, with CO the dominant products; convincing evidence for the Kolbe mechanism hypothesised. Isotopic-labelling using 13CO2 over an organic-free 2 wt% Pd/TiO2 catalyst revealed the co-production of 13CH4 (unequivocally through direct photoreduction) and some 12CH4 likely originating from the CO32− species on the parent titania. In a complementary investigation, Yang et al. explored the mechanism of 13CO2 photoreduction in H2O over Cu(I)/TiO2 by means of in situ DRIFT spectroscopy.7212CO was identified from its 2115 cm−1 Cu(I)–CO signature as the primary product of reaction between photocatalytically activated adsorbed water and carbonaceous residues on the as-prepared photocatalyst surface. The residual surface carbon reflects the use of a titanium(IV) butoxide precursor and polyethylene glycol structure-directing agent, with (an unquantified amount of) carbon persisting even after the resulting sol–gel was calcined at 500 °C for 5 h. Prolonged UV irradiation of the fresh catalyst under moist air to remove carbon contaminants significantly lowered the yield of reactively-formed CO, although some 13CO was observed, evidencing true CO2 photoreduction. These studies highlight the importance of eliminating organic (and carbonate) surface residues from photocatalysts prior to CO2 photoreduction tests, via e.g. high temperature calcination, solvent washing and/or pre-exposure to UV light in the absence of CO2. Yang and co-workers also recommended that an additional control experiment be undertaken to ensure no carbon-containing products are formed in the absence of CO2 but presence of H2O. To date very few studies have acted upon this sound recommendation.
The overwhelming majority of CO2 photoreduction employs water as the electron (net hydrogen) source, and should therefore be accompanied by oxygen co-generation. Selective reduction to CO should proceed according to: CO2 + 3H2O → CO + 3H2 + 2O2 (combining 2H2O → 2H2 + O2 and CO2 + H2O → CO + H2 + O2), for which the molar ratio of (H2 + CO)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 should be 2:1. However, very few studies report such oxygen evolution, let alone endeavour to quantify it and thereby demonstrate that water is the sole electron donor. In the case of ZrO2 in aqueous NaHCO3,73 KTaO3 (ref. 74) and Ag doped ALa4Ti4O15 (A = Ca, Sr, and Ba) perovskites,75 the co-evolution of CO, H2 and O2 is reported with the predicted 2:1 stoichiometry; albeit isotopic labelling experiments with 13CO2 also revealed the formation of trace 12CO over for KTaO3 due to either 12CO2 from reactor purging or organic residues within the photoreactor. Isotope labelling, a full complement of control experiments, and careful analysis of evolved oxygen are thus critical to accurately quantify CO2 photoreduction and understand underlying reaction pathways.
:O2 should be 2:1. However, very few studies report such oxygen evolution, let alone endeavour to quantify it and thereby demonstrate that water is the sole electron donor. In the case of ZrO2 in aqueous NaHCO3,73 KTaO3 (ref. 74) and Ag doped ALa4Ti4O15 (A = Ca, Sr, and Ba) perovskites,75 the co-evolution of CO, H2 and O2 is reported with the predicted 2:1 stoichiometry; albeit isotopic labelling experiments with 13CO2 also revealed the formation of trace 12CO over for KTaO3 due to either 12CO2 from reactor purging or organic residues within the photoreactor. Isotope labelling, a full complement of control experiments, and careful analysis of evolved oxygen are thus critical to accurately quantify CO2 photoreduction and understand underlying reaction pathways.
2. Developments in nanostructured materials
2.1. Crystallinity and the role of engineered surface defects
The catalytic behaviour of photocatalysts is intrinsically linked to their surface electronic/optical properties and adsorption behaviour, all of which can (in principle) be systematically tuned through crystal structure engineering; the nature, dimensions, uniformity and termination of crystalline phases strongly influence their resultant photocatalytic efficiency.76 For instance, monoclinic sheet-like BiVO4 significantly outperformed tetragonal rod-like BiVO4 counterparts in the rate of CO2 photoreduction to C2H5OH (accompanied by O2 evolution), under both UV and visible light illumination in the presence of water.77 These monoclinic and tetragonal phases were obtained by introducing CTAB (cetyltrimethyl ammonium bromide) or PEG (polyethylene glycol), respectively, during BiVO4 synthesis. The superior photoactivity of the monoclinic BiVO4 was attributed to the more asymmetric local environment of the Bi3+ ions relative to those within the tetragonal phase. Accordingly, the Bi3+ ions exhibit stronger lone pair character in the monoclinic phase and are hence drive to form Bi–O bonds with CO2 bound as the carbonate, and consequential transfer of photogenerated electrons from the V 3d bands into the chemisorbed CO32−. It is worth noting that these two phases exhibited different UV-Vis absorbances, with the band gaps of monoclinic and tetragonal BiVO4 around 2.24 eV and 2.56 eV respectively. More recently, Li et al. demonstrated that cubic NaNbO3 was more effective than orthorhombic NaNbO3 for CO2 photoreduction.78 The cubic NaNbO3 (c-NaNbO3), which is normally only stable at T > 813 K, was successfully prepared through a furfural alcohol derived polymerisation-oxidation (FAPO) method in the presence of P123 as a surfactant stabiliser. The more common orthorhombic NaNbO3 (o-NaNbO3) phase was prepared through a polymerised complex (PC) method. While c-NaNbO3 comprised predominantly cuboid morphologies, a mixture of irregular and cuboid particles was obtained for o-NaNbO3. Both crystal phases exhibited a comparable crystallite size (c-NaNbO3 = 18.5 nm versus o-NaNbO3 = 23.1 nm) and surface area (c-NaNbO3 = 28.6 m2 g−1versus o-NaNbO3 = 26.4 m2 g−1), however, the band gap of c-NaNbO3 (3.29 eV) was smaller than that of o-NaNbO3 (3.45 eV). Under UV irradiation (300 W Xe arc lamp) in the presence of a Pt co-catalyst and water vapour, c-NaNbO3 generated almost double the amount of CH4 (0.486 μmol h−1) than that obtained from o-NaNbO3 (0.245 μmol h−1) in a gaseous, closed circulation system pressurised with 80 kPa CO2. Density functional theory (DFT) calculations ascribed this variation in photocatalytic performance to differences in conduction band (CB) energies, reflecting different octahedral ligand fields. Accordingly, electron excitation and transfer was more efficient over the high symmetry c-NaNbO3 phase than o-NaNbO3. In addition to CO (0.082 μmol h−1) and H2 (0.71 μmol h−1), trace amounts of C2H4, C2H6 and C3H8 were detected during CO2 photocatalysis over the c-NaNbO3.Numerous preparative routes have been developed to nanomaterials with controllable crystal facets such as pH regulation of the synthetic media, or post-synthesis treatments. Truong et al. selectively prepared anatase (pH 2, only 3% rutile), rutile (pH 6, only 6% of anatase) and a bi-crystalline anatase(73%)-brookite(27%) composite at pH 10.79 Under UV (500 W Xe lamp) or visible light irradiation (NaNO2 solution as a UV cut-off filter), and in the presence of a NaHCO3 solution, these materials were able to photoreduce CO2 to CH3OH, whereby the photoactivity followed the order anatase-brookite composite > rutile > anatase > commercial P25. Liu et al. also successfully prepared three different nanostructures of TiO2 polymorphs including anatase (TiA) NPs, rutile (TiR) nanoellipses and brookite (TiB) nanorods for CO2 photoreduction,80 by subjecting the as-synthesised titania to heat treatment under a He environment at 220 °C for 1.5 h to induce surface defects, such as oxygen vacancies (VO) and Ti3+ ions on these TiO2 polymorphs. Illumination by a 150 W solar simulator in the presence of CO2/water vapour revealed the untreated TiO2 polymorphs as active for CO2 photoreduction to CO and a small amount of CH4 under continuous-flow (2.0 mL min−1). Product yields decreased in the order TiA > TiB > TiR. As depicted in Fig. 2, He-treated materials TiA(He) and TiB(He) significantly outperformed their untreated counterparts, with TiB(He) exhibiting the highest photoactivity, followed by TiA(He); such annealing treatments had less impact upon the photoactivity of TiR wherein the activation barrier to surface oxygen vacancy creation was presumably higher.
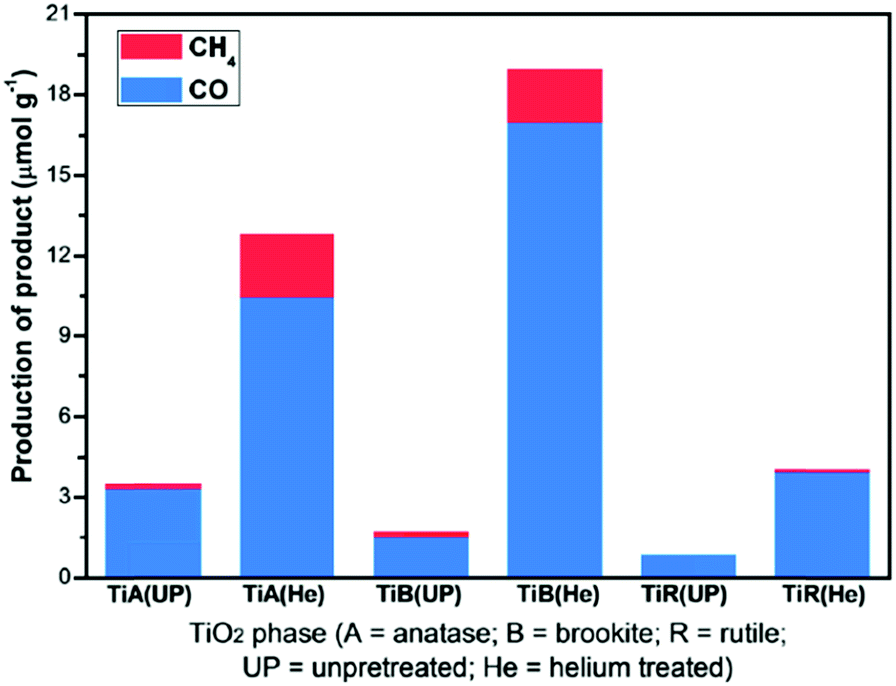 | ||
| Fig. 2 CH4 and CO productivity from untreated and He-treated TiO2 polymorphs during 6 h illumination under 150 W solar simulator in the presence of CO2/H2O vapour, in a continuous-flow mode photoreactor operating at a flow rate of 2 mL min−1. Reprinted with permission from ref. 80. Copyright 2012 American Chemical Society. | ||
In situ DRIFTS and DRUVS suggested that the relative VO:Ti3+ concentration in TiA(He) was comparable to that of TiB(He), while the surface defect concentration on TiR(He) was negligible. The higher photoactivity of TiB(He) and TiA(He) was thus attributed to surface defects, with the energy of vacancy formation for TiB (He) of 5.52 eV lower than that for anatase (5.58 eV) and rutile (5.82 eV). CO2 photoreduction over titania is clearly a strong function of the crystal structure and surface defect density; the latter postulated the active catalytic sites.
An increasing volume of research has established the influence of crystal facets upon photocatalysis; physical and chemical properties of single crystals are highly sensitive to crystallographic termination.76,81 Calculations have established that the average surface energies of low-index anatase facets decrease in the order 0.90 J m−2 (001) > 0.53 J m−2 (100) > 0.44 J m−2 for (101).82,83 The highest energy (001) surface is expected to be the most reactive, and indeed experimental studies confirm that dissociative molecular adsorption of water and methanol occur readily on the (001) surface.84,85 Consequently, efforts to control the morphology of TiO2 have focused on the synthesis of nanostructures preferentially exposing (001) facets or coexposing (001) and (101) facets86 to enhance water splitting and dye degradation.87–90 However, other studies suggest that photoreduction and photooxidation activities follow the order (001) < (101) < (010),91 with the superior performance of the (010) facet attributed to a synergy between the surface atomic structure (dictating adsorption/reactivity) and electronic band structure (dictating the CB potential).
TiO2 nanorods preferentially exposing (010) facets were therefore screened in CO2 photoreduction.91 Such nanorods were synthesised by hydrothermal treatment of a H0.68Ti1.83O3 precursor in the presence of Cs2CO3 as a pH mediator, and subsequent promotion by 1 wt% Pt. The resulting Pt-doped TiO2 nanorods outperformed P25 in CO2 photoreduction by water vapour to CH4 under 300 W Xe lamp UV irradiation in a Teflon-lined steel chamber under 0.06 MPa CO2. Enhanced activity was attributed to the unique properties of (010) facets which promote CO2 adsorption,92 specifically their more negative conduction band potential. The same group prepared hollow anatase TiO2 single crystals and mesocrystals, wherein both nanostructures were dominated by (101) facets.93 These materials were successfully synthesised by hydrothermal treatment of a Ti(SO4)2, Na3PO4 and HF solution. Fig. 3A–D shows the resulting 400 nm diameter octahedral single crystals obtained using 500 mM HF; lowering the HF concentration to 400 mM yielded hollow crystals with less prominent octahedral shapes of ∼160 nm (Fig. 3B and E). Fig. 3H reveals that these hollow crystals were single crystalline and principally (101) oriented, with a small proportion of higher-index (103) facets. Even lower HF concentrations favoured sphere-like hollow cages 150 nm diameter (Fig. 3C and F), each formed from ∼35 nm octahedral crystals, which spontaneously self-organised during crystal growth to adopt a common crystallographic orientation, resulting in ‘single crystal-like’ mesocrystals with well-defined selected area electron diffraction (SAED) patterns. Under UV irradiation (300 W Xe lamp) and in the presence of a RuO2 co-catalyst and water vapour, the hollow single crystal and mesocrystal materials titanias outperformed a non-porous single crystal by a factor of 4–5 times in photoreducing CO2 to CH4, attributed to their (i) higher surface areas (35 m2 g−1 and 42 m2 g−1 respectively) relative to the solid crystal (17 m2 g−1) and (ii) shorter diffusion length of charge carriers and concomitant suppressed bulk recombination. Fig. 3J illustrates the different electronic band structures of the single crystal, hollow single crystal and hollow mesocrystal titanias, and more negative CB potential of the hollow mesocrystal.
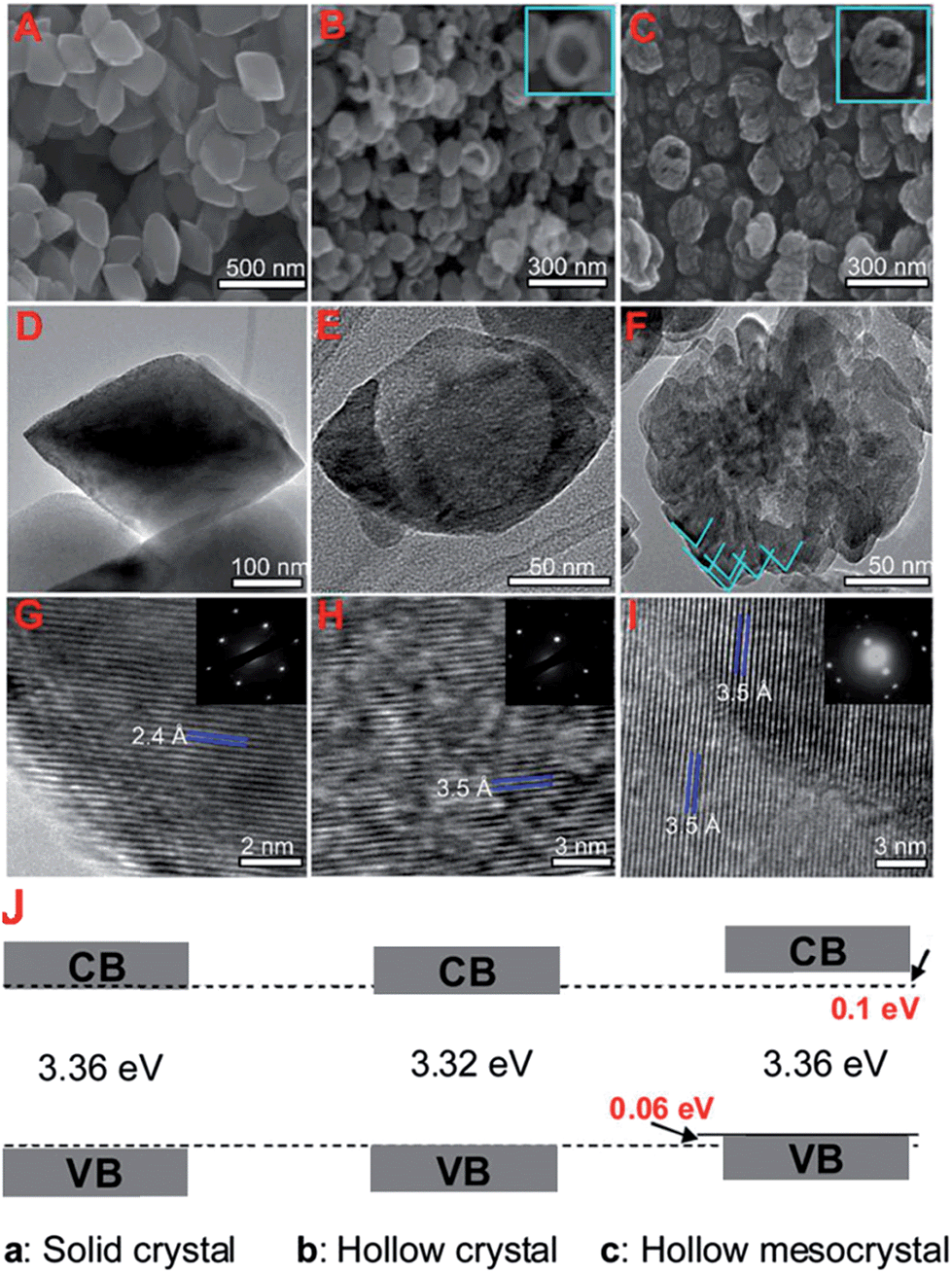 | ||
| Fig. 3 SEM images (A)–(C), TEM images (D)–(F), HRTEM images (G)–(I) and (J) electronic band structures based on UV-Vis and valence band XPS spectra of single crystal solid anatase TiO2, single crystal hollow anatase TiO2, and hollow anatase TiO2 mesocystals. Insets in (B) and (C) highlight hollow cores in the single and mesocrystal particles, insets in (G)–(I) show SAED patterns for the solid single crystal, hollow single crystal and mesocrystal anatase TiO2. Reprinted with permission from ref. 93. Copyright 2012 American Chemical Society. | ||
Adsorbate-induced restructuring of highly reactive facets remains an intrinsic problem due to their high surface energies. In this regard, reagents such HF and isopropanol have been employed as structure-directing agents to favour exposure of (001) facets over single-crystal anatase nanosheets.60 Use of such capping agents is itself problematic since they must be removed by thermochemical treatment prior to photocatalytic application (e.g. solvent extraction or calcination) which in turn may induce surface restructuring/phase changes. Fabrication of stable and uniform nanostructures terminating in specific atomic rearrangements requires continued development.
2.2. Nanostructured photocatalysts
 | ||
| Fig. 4 TEM images of (a) Zn2GeO4 nanoribbons94 and (b) In2Ge2O7 sub-nanowires (inset: corresponding SAED pattern)96 prepared by solvothermal treatment using a binary ethylenediamine/water solvent system, and Zn2GeO4 sub-nanowires prepared through solution phase route (c) at 40 °C and (d) 100 °C. Reproduced from ref. 97 with permission from The Royal Society of Chemistry. | ||
A similar synthesis was employed to prepare highly crystalline In2Ge2O7(En) hybrid sub-nanowires, as shown in Fig. 4b, with diameters around 2–3 nm.96 Although the nanowires outperformed a SSR prepared sample in CO2 photoreduction by water vapour under UV illumination (300 W Xe arc lamp) within a gas-tight Pyrex glass reactor, CO was the only photoreduction product. A low temperature solution phase route, which does not require any surfactant, was reported for the preparation of single crystalline, hexagonal Zn2GeO4 nanorods for CO2 photoreduction.97 A mixed solution of Na2GeO3 and Zn(CH3COO)2 was heated at 40–100 °C to yield Zn2GeO4 nanorods with a regular, hexagonal prism geometry. As shown in Fig. 4c and d, nanorod dimensions were strongly dependent on synthesis temperature: nanorods around 400 nm long and 50 nm wide were obtained at 40 °C, while nanorods 250 nm long and 150 nm wide formed at 100 °C. Surprisingly, the prepared nanorods contained some blind holes due to the different crystal growth rate along the c-axis, as well as nanosteps in the surface of nanorods prepared at 100 °C. The absorption band edge of nanorods was blue-shifted (4.68 eV and 4.65 eV for 40 °C and 100 °C syntheses respectively) relative to that of SSR-prepared material (4.5 eV) due to quantum size effects, restricting their use to hard UV applications. Under illumination, Zn2GeO4 nanorods outperformed those prepared by SSR towards photocatalytic H2 generation and CO2 reduction, with nanorods grown at 100 °C proving the most active for H2 evolution owing to their greater crystallinity and the presence of surface nanosteps promoting charge separation. Conversely, the 40 °C synthesised nanorods exhibited better CO2 photoreduction activity than their higher temperature analogues due to their higher surface area and CO2 adsorption capacity.
Highly ordered one-dimensional TiO2 NTs arrays have also been developed for photocatalytic applications.98–101 These nanotube arrays have increasingly gained popularity as they: (i) exhibit a relatively high interfacial surface area (because of their internal and external surfaces); (ii) promote efficient transport of electrons owing to the thin walls of NTs; (iii) efficiently retard electron–hole recombination (which can occur up to ten times slower than equivalent nanoparticle films); and (iv) demonstrate improved photon absorption efficiency attributed to strong light scattering within the nanotube arrays.98–103 The development of TiO2 NTs arrays, including their synthesis and application, has been thoroughly reviewed by Rani et al.103 spanning first generation (0.5 μm in length) to current 4th generation (∼1000 μm) technologies. Electrochemical anodisation is the preferred method for nanotube arrays synthesis since it enables the nanotube dimensions to be precisely tuned through varying synthetic parameters such as electrolyte composition and anodising time. Varghese et al. prepared N-doped TiO2 NTs arrays with pore size and wall thickness of 95 ± 13 nm and 20 ± 5 nm, respectively by anodising Ti foil in an NH4F/water/ethylene glycol electrolyte at 55 V.40 The resulting TiO2 NTs arrays were then calcined and sputter coated with large, irregular patches of Pt, Cu or both metals, as co-catalysts, albeit little information was provided on the morphology and dispersion of these promoters, with the final material containing between 0.4–0.75 at% N. Irradiation under natural sunlight and water vapour within a batch reactor, afforded CO2 photoreduction to mainly H2, CH4 and other alkanes (i.e. C2H6, C3H8, C4H10, C5H12 and C6H14), with olefins and branched paraffins also formed as minor products. Higher NTs calcination temperatures improved the photocatalytic activity, attributed to their higher crystallinity. Platinum promoted arrays generated more H2 (almost 200 ppm cm−2 h−1) than hydrocarbons, giving a total productivity of 273 ppm cm−2 h−1, whereas the copper promoted array exhibited superior hydrocarbon production (∼104 ppm cm−2 h−1). Furthermore, the Cu–TiO2 NTs arrays evolved five times more CO than Pt–TiO2 arrays. Doubly promoted NTs arrays (Fig. 5) afforded a total hydrocarbon productivity of 111 ppm cm−2 h−1 without any detectable CO. It was concluded that Pt was more active for water reduction, while Cu was more efficient at reducing CO2, their combination resulting in a strong synergy and maximal CO2 conversion to hydrocarbons, with one of the highest visible light quantum efficiencies of 0.74%.
 | ||
| Fig. 5 Product formation of N-doped TiO2 NTs arrays, calcined and sputtered coated with Pt and Cu. CO2 photoreduction was conducted in a batch reactor under natural sunlight illumination in the presence of water vapour at 44 °C. Reprinted with permission from ref. 40. Copyright 2009 American Chemical Society. | ||
The synthesis of two-dimensional WO3 nanosheets using a solid–liquid, phase arc discharge method for CO2 photoreduction was recently reported by Chen et al.104 Crystal growth was proposed to occur via the formation of a large number of 4–5 nm WO3 seeds. These nanocrystals fused together into two-dimensional nanosheets during aging, while maintained a thickness <5 nm, with preferential growth in the [100] and [010] directions. The resulting band gap of these nanosheets (2.79 eV) was larger than that of commercial WO3 powder (2.63 eV) due to quantum size effects, with their CB potential estimated to be more negative than the reduction potential for CO2/CH4 conversion. Conversely, the CB position of commercial WO3 appeared more positive than the CO2/CH4 redox potential, which should hence disfavour CO2 reduction. Indeed, visible light illumination (300 W Xe arc lamp equipped with a 420 nm cut-off filter) of the as-prepared nanosheets showed that they outperformed the commercial WO3 powder in photoreducing CO2 to CH4 in a gas-tight system. Superior nanosheet photoactivity was therefore attributed to both their ultrathin geometry, expected to promote efficient charge carrier diffusion, and their more negative conduction potential. CO2 adsorption can be enhanced through the use of alkali or alkaline earth basic oxides. Strontium promoted niobates prepared by conventional SSR have shown promise in CO2 photoreduction by water vapour, but possess extremely low surface areas and are hence unsuitable for achieving high photoactivity. An alternative hydrothermal approach affords two-dimensional SrNb2O6 nanoplates around 12 nm thick and 5–300 nm across,105 conferring a 20-fold increase in CO2 adsorption capacity and quantum efficiency of 0.065% over 10 h, an order of magnitude higher than P25. In contrast to many studies, high selectivity to CO2 reduction to CO and CH4versus competing H2O reduction to H2 was observed, alongside significant oxygen evolution. Transient photocurrent response measurements suggest that separation of photogenerated electron–hole pairs was rate-determining, with the nanoplate morphology facilitating charge-carrier diffusion to the surface and subsequent reaction with adsorbed CO2 and water.
Crystalline, porous structured Ga2O3 has been successfully synthesised by Park et al. employing a tetradecyl trimethyl ammonium bromide template.107 Field emission scanning electron microscopy (FE-SEM) and transmision electron microscopy (TEM) images (Fig. 6), revealed the co-existence of macropores and mesopores througout the resulting monodispersed material. Subsequent photocatalytic screening showed that CO2 conversion over this hierarchically porous Ga2O3 was much higher than that of reference Ga2O3 nanoparticles. Further investigations into CO2 adsorption established that the porous Ga2O3 possessed a vastly superior CO2 adsorption capacity, 300% higher than that of the bulk reference, accounting for its improved photoreduction of CO2 to CH4. Surfactant templating was also utilised to prepare mesoporous, micrometric ZnO spheres with the Pluronic P123 templating agent through hydrothermal treatment.112 Individual ZnO spheres were composed of stacks of thin ZnO flakes, which created interparticle mesoporous voids. This mesoporous architecture gave higher CO2 conversion than the equivalent non-porous ZnO, and following Cu impregnation the mesoporous ZnO favored the selective production of CH4 and CH3OH. The utility of porous structures for CO2 photoreduction to CH4 has also been reported for titania,79 albeit the surface area of the mesoporous TiO2 decreased significantly upon subsequent nitridation to generate visible light active, N-doped TiO2.
 | ||
| Fig. 6 FE-SEM images of (a) porous Ga2O3, (b) vertical cross-section of porous Ga2O3 fractured by ion beam and (c) TEM image of the porous Ga2O3. Reproduced from ref. 107 with permission from The Royal Society of Chemistry. | ||
In recent years, a new synthetic protocol has been developed to create multicomponent, mesoporous photocatalysts for CO2 reduction. Here an inorganic precursor is used to construct more thermally robust mesostructures.113 The synthetic route of Yan et al. to prepare mesoporous ZnGa2O4 is illustrated in Fig. 7A. A powdered NaGaO2 starting material was synthesised through a solid-state route, which formed colloidal particles upon dispersion in water. Weak surface repulsion between the resulting nanoparticles enabled their subsequent flocculation to form a mesoporous framework, followed by Na+ and Zn2+ ion-exchange to yield a mesoporous ZnGa2O4 (visualised in Fig. 7B). The final material remained crystalline after the ion-exchange, obviating the need for hydrothermal treatment or high temperature calcination. The final ZnGa2O4 exhibited a mean pore diameter of 3.5 nm and surface area around 110 m2 g−1. Catalytic screening demonstrated that the mesoporous ZnGa2O4 was superior to ZnGa2O4 prepared through solid-state synthesis, with photogenerated CH4 prevalent when a RuO2 co-catalyst was employed. In common with many such studies there was no justification fo the choice of co-catalyst, nor its mode of operation, which was simply presumed to improve separation of photogenerated electron–hole pairs. This synthetic procedure was further adapted to prepare a ZnAl2O4-modified, mesoporous ZnGaNO visible light photocatalyst111 from a NaGa1−xAlxO2 precursor to form an amorphous mesoporous colloid template by flocculation. Following ion-exchange with Zn2+, the resultant Zn(Ga1−xAlx)2O4 was hydrothermally treated to improve its crystallinity prior to nitridation; Al was claimed to enhance the thermal stability of the mesostructure. CO2 photoreduction was negligible over the parent ZnGaNO (with or without ZnAl2O4 modification) without Pt doping to promote electron–hole separation. The superior performance of the ZnAl2O4 modified material was attributed to improved mass transport, increased basicity and hence CO2 chemisorption, and narrowing of the band gap. Micro/mesoporous Zn2GeO4 and mesoporous zinc germanium oxynitride synthesised via the same method also evidenced superior CO2 photoconversion compared to their solid state synthesised counterparts.106,114
 | ||
| Fig. 7 (A) Schematic diagram showing the synthesis steps of preparing mesoporous ZnGa2O4 through mesoporous colloidal template and ion-exchange reaction and (B) TEM image of the resulting mesoporous ZnGa2O4 (inset: HR-TEM image of the crystal fringes). Reproduced with permission from ref. 113. Copyright 2010 Wiley. | ||
Regular mesopore voids can also be created through the self-assembly of nanoparticles wherein the mesopores are formed at the junction between nanoparticles. This approach was successfully employed by Wang et al. to prepare sub-micron TiO2–SiO2 mesoporous composites.108 These materials were obtained by atomising TiO2 and SiO2 nanocolloidal solutions of 20–45 nm particles into micron-sized droplets at elevated temperature. Solvent evaporation drove colloidal self-assembly to yield the mesoporous TiO2–SiO2 composite imaged by FE-SEM image in Fig. 8a. Cu doped variants were also produced by introducing Cu(NO3)2 into the colloidal precursor solution. Higher synthesis temperatures promoted TiO2 nanocrystal segregation at the composite surface (Fig. 8b). The porous SiO2 matrix ensured that TiO2 dispersed across the composite surfaces as ∼20 nm crystals, rather than larger agglomerates. TiO2–SiO2 composites exhibited superior photoactivity to pure TiO2 for CO2 photoreduction to CO. Activity increased with processing temperature up to 1000 °C due to an increasing density of TiO2 nanocrystals decorating the mesoporous framework, with a composite comprising 2 mol% TiO2 and 0.01 mol% Cu giving the highest rate of CO2 photoreduction. Mesoporous In(OH)3 has also been prepared through a template-free hydrothermal treatment110 resulting in the self-assembly of crystalline In(OH)3 nanocubes. The introduction of mesoporosity enhanced CO2 photoreduction to CH4 by 20-fold relative to conventional In(OH)3 crystallites attributed to the higher surface area and CO2 adsorption affinity of the mesoporous variant.
 | ||
| Fig. 8 (a) FE-SEM and (b) TEM and HR-TEM images of mesoporous TiO2–SiO2 composites prepared at 800 °C. Reproduced from ref. 108 with permission from The Royal Society of Chemistry. | ||
Graphitic carbon nitride (C3N4), a metal-free polymeric semiconductor, has gained increasing popularity in photocatalysis for visible light application.113,115–118 Switching the precursor from melamine to melamine chloride followed by rapid heating afforded porous C3N4:119Fig. 9 compares the platelet-like structure of melamine-derived C3N4 (g-C3N4) with the porous platelets obtained from the melamine chloride analogue, (p-g-C3N4) whose surface area was significantly higher (69 m2 g−1versus 1.7 m2 g−1 for g-C3N4). Despite these textural differences the non-porous g-C3N4 outperformed the porous p-g-C3N4 during CO2 the photoreduction to CO under visible light. This surprising result was attributed to the slightly broadened band gap and poorer crystallinity of p-g-C3N4, which may arise from a higher density of defects acting as electron–hole recombination centres. Porous C3N4 was also prepared by Mao et al. (denoted u-g-C3N4) from a urea precursor, which displayed superior CO2 photoreduction activity relative to melamine-derived C3N4 (m-g-C3N4).120 Akin to the findings of Dong and co-workers, porous u-g-C3N4 was less crystalline than the melamine analogue, with an order of magnitude higher surface area and slightly wider band gap. However, in this instance u-g-C3N4 exhibited superior photoactivity, presumably due to enhanced reactant adsorption and more efficient charge carrier separation. Under visible light, and in the presence of 1 M NaOH, u-g-C3N4 photoreduced CO2 to a mixture of CH3OH and C2H5OH, while m-g-C3N4 favoured C2H5OH with only trace methanol. Selective photocatalytic CO2 reduction is hence achievable through tuning of material crystallinity and microstructure.
 | ||
| Fig. 9 TEM images of (a) non-porous g-C3N4, and (b) porous p-g-C3N4. Reproduced from ref. 119 with permission from The Royal Society of Chemistry. | ||
Table 1 shows that most mesoporous semiconductors possess modest surface area, however there is scope for enhancing these through their immobilisation onto higher area supports such as mesoporous silicas.121,122 Such high area templates, albeit inert, facilitate dispersion of photoactive phases, thereby increasing the surface active site density and hence photoconversion efficiency. Yang et al. reported that TiO2 supported on SBA-15 exhibited superior photoactivity to P25 during CO2 photoreduction to CH3OH.122 The crystallite size of the dispersed TiO2 increased with loading but remained smaller than that of commercial P25, however it is unclear how much titania was incorporated with the SBA-15 pore network versus external surface, and hence performance may have been sub-optimal. Further studies revealed that higher CH3OH yields were achievable by Cu doping these TiO2/SBA-15 catalysts. Li et al. prepared TiO2/mesoporous silica composites through a one-pot, sol–gel method.113 The resulting material was more active than pure TiO2 in photororeducing CO2 to CO and CH4, with Cu addition favouring CH4 production.
| Photocatalyst | Surface area/m2 g−1 | Pore diameter/nm | Product |
|---|---|---|---|
| Ga2O3 (ref. 107) | 43 | 3.8, 5–30 | CH4 |
| 1.5 wt% Cu/ZnO112 | 22 | 31.2 | CH3OH, CH4, CO, H2 |
| 0.12 wt% Pt/TiO2 (ref. 123) | 147 | 7.8 | CH4 |
| 0.84 at% N–TiO2 (ref. 123) | 70 | 12.6 | CH4 (Pt) |
| ZnGa2O4 (ref. 113) | 110 | 3.5 | CH4 |
| ZnAl2O4/ZnGaNO111 | 53 | 15.2 | CH4 |
| Zn2GeO4 (ref. 114) | 91 | 1.2–2, 2–12 | CH4 |
| ZnGeON106 | 36 | — | CH4 |
| TiO2–SiO2 (ref. 108) | 72 | 28.3 | CO |
| In(OH)3 (ref. 110) | 65 | 9.5 | CH4 |
| u-g-C3N4 (ref. 120) | 40 | — | CH3OH, C2H5OH |
| 45 wt%TiO2–SiO2 (ref. 122) | 436 | 2–12 | CH3OH |
| 0.5%Cu/TiO2–SiO2 (ref. 113) | 386 | — | CO, CH4 |
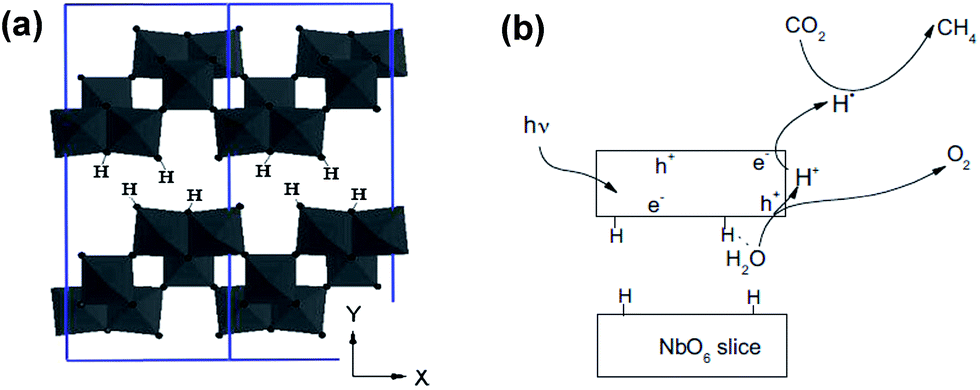 | ||
| Fig. 10 Schematic depicting (a) the layered structure of HNb3O8, and (b) proposed mechanism of CO2 photoreduction over HNb3O8. Reprinted from ref. 128, Copyright 2012, with permission from Elsevier. | ||
Expanded HNb3O8 interlayers were explored by Li et al. as a means to further boost photocatalysis through pillaring silica as a guest compound within the interlayer space.129 A HNb3O8 precursor was first prepared by conventional SSR and subsequent proton exchange. HNb3O interlayers were then expanded via n-dodecylamine, enabling access to tetraethyl orthosilicate. Calcination of the resulting material created a SiO2-pillared HNb3O8 with XRD indicating that silica pillaring increased the interlayer spacing from 0.37 nm to 2.10 nm, facilitating their visualisation by TEM (Fig. 11). Pillaring also dramatically increased the niobic acid surface area from ∼7 to 197 m2 g−1, while simultaneously loweing the band gap from 3.5 eV to 3.3 eV. Under UV irradiation (350 W Xe lamp) under water vapour, and doping with 0.4 wt% Pt promoter, this SiO2-pillared HNb3O8 proved superior to both the Nb2O5 precursor and non-pillared HNb3O8 layered structure for photoreducing CO2 to CH4 in a sealed quartz tubular reactor. Active sites of lamellar solid acids are considered to reside at the internal surfaces of interlayers.130 Accordingly, Li and co-workers proposed that this enhanced photoactivity arose from increased active site accessibility within the HNb3O8 interlayers by CO2 and H2O. However, in common with many other studies, the simultaneous variation of physical and optical properties makes it hard to elucidate the principal origin of this enhancement. SiO2-pillared HNb3O8 also outperformed the aforementioned HNb3O8 nanobelts. Additional investigations found that CH4 production was lower over SiO2-pillared KNb3O8 (prepared by cation exchange with K2CO3) than SiO2-pillared HNb3O8, suggesting that water adsorption by the solid acid variant also played an important role in driving CH4 formation.
 | ||
| Fig. 11 (a) TEM and (b) SEM images of SiO2-pillared HNb3O8. Reprinted with permission from ref. 129. Copyright 2009 American Chemical Society. | ||
Solid bases are well-established as CO2 capture agents.67,131–133 Accordingly, layered double hydroxides (LDHs) composed of [MII1−xMIIIx(OH)2]x+ cationic sheets with intercalated anions have been exploited for CO2 photoreduction.134,135 These materials are attractive because of their strong and tunable basicity, and hence good CO2 adsorption capacity. The semiconducting nature, and hence photoabsorption properties, of LDHs can be tuned to some extent through careful selection of the intralayer MII and MIII metal cations. Recent work from Sastre et al. showed that CO2 photoreduction to CH4 was enhanced by Zn/Ti or Zn/Ce LDHs,135 presumably through increased CO2 adsorption at the LDH surfaces facilitating subsequent photochemistry. Ahmed et al. prepared photoactive Zn2+/Ga3+ LDH catalysts.134 Under UV irradiation (500 W Xe arc lamp), the resulting [Zn3Ga(OH)8]2+(CO3)2−·mH2O solid base was active for reducing CO2 into CO and CH3OH as primary and secondary products respectively, employing a closed, circulating system pressurised with a mixture of 2.3 kPa CO2 and 21.7 kPa H2, i.e. highly reducing conditions wherein commercial application would require an independent photocatalytic or electrochemical hydrolysis process to supply hydrogen, or close proximity to a fossil fuel derived syn gas feed. Copper incorporation into the LDH to form a [Zn1.5Cu1.5Ga(OH)8]2+(CO3)2−·mH2O material improved selectivity towards CH3OH threefold, rendering it the major product. Control experiments in the absence of CO2 suggested that at least some of the observed reactivity was associated with the conversion of CO32− ions originating within the parent LDH, although this could not account for the total product yield. Replacing Ga3+ with Al3+ to create a [Zn3Al(OH)8]2+(CO3)2−·mH2O LDH enhanced overall photoconversion efficiency, but favoured CO formation (even after Cu2+ incorporation). These layered catalysts were reported active for 20 h continuous CO2 photoreduction, although methanol productivity declined over time. In an effort to boost selectivity towards methanol, the interlayer CO32− anions were exchanged for [Cu(OH)4]2− anions, thereby creating a [Zn1.5Cu1.5Ga(OH)8]2+(CO3)2−·mH2O material136 with a methanol yield almost three times that achievable over the carbonate counterparts. Copper was thus hypothesised as the active photocatalytic site, with the localisation of Cu sites within the interlayers facilitating their accessibility to CO2, while the replacement of CO32− with [Cu(OH)4]2− counterions significantly decreased the band gap of the resulting LDH while expanding the interlayer spacing, both factors likely contributing towards the enhanced photoactivity of the copper anion doped variant.
Highly ordered, sheaf-like, hyperbranched Zn2GeO4 superstructures were prepared in the presence of ethylenediamine through a solvothermal method by Liu et al.155 The FE-SEM image in Fig. 12a highlights 3–4 μm sheaf-like features which assembled from closely packed, well-aligned nanowires with fantails oriented toward the center and projecting radially outwards. This unique morphology was proposed to arise through a crystal-splitting growth mechanism, with the degree of splitting a function of reaction time, precursor concentration and solvent. In order to improve the visible light performance of the normally large band gap Zn2GeO4, these as-prepared fusiform bundles were subject to nitridation at 700 °C, resulting in a yellowish zinc germanium oxynitride (Zn1.7GeN1.8O) photocatalyst. While the bundle morphology of the parent Zn1.7GeN1.8O precursor was retained after this high temperature treatment, an added benefit was a concomitant significant increase in surface roughness (Fig. 12b), which doubled the Zn1.7GeN1.8O surface area relative to the Zn2GeO4 parent. Under visible light illumination (300 W Xe arc lamp equipped with a 420 nm cut-off filter), and only upon promotion with Pt or RuO2 co-catalysts, this hierarchically structured Zn1.7GeN1.8O material showed good ambient pressure CO2 photoreduction to CH4, albeit with an apparent quantum efficiency still only approaching 0.024%. It is noteworthy that the CH4 yield reached saturated around 6 h, possibly due to reversible adsorption of reactive intermediates.
 | ||
| Fig. 12 FE-SEM images of (a and b) sheaf-like Zn2GeO4 superstructure solvothermally treated for 12 h, and (c) Zn1.7GeN1.8O prepared through high temperature nitridation of Zn2GeO4 fusiform bundles. Reproduced from ref. 155 with permission from The Royal Society of Chemistry. | ||
Hexagonal nanoplate-textured micro-octahedral Zn2SnO4 were synthesised through a hydrothermal treatment in the presence of L-tryptophan.156 The resulting material comprised ordered and uniform micro-octahedra with edges of approximately 3 μm (Fig. 13a). At higher magnification, it is apparent that each micro-octahedron is densely packed with ∼50 nm thin, hexagonal nanoplates Fig. 13b. These nanoplates are vertically arranged within octahedra such that they lie parallel to neighbouring edges. The octahedral geometry was proposed a consequence of L-tryptophan impeding reaction along the [111] direction, favouring faster nucleation and growth in the [100] direction. The resulting hierarchical nanoplate/micro-octahedron morphology blue-shifted the Zn2SnO4 band gap from the bulk value of 3.67 eV to 3.87 eV, yet delivered exclusively CH4 production at 20 ppm g−1 from CO2 under UV irradiation within a gas-tight Pyrex glass cell, albeit for only 1 h in the absence of promoters. This far exceeded the photoactivity of bulk Zn2SnO4 (1.5 ppm g−1), smooth Zn2SnO4 micro-octahedra (4.7 ppm g−1) or Zn2SnO4 atactic particles (2.4 ppm g−1) prepared without L-tryptophan. This rate enhancement was ascribed to such superstructures acting as light-scattering centres promoting photon harvesting, in combination with the nanoplate morphology promoting electron transport along individual plates, and the open nanoplate-/micro-octahedron architecture which facilitated rapid molecular diffusion.
 | ||
| Fig. 13 FE-SEM images of the nanoplate/micro-octahedron Zn2SnO4 at (a) lower magnification and (b) at higher magnification. The inset of (b) shows an isolated hexagonal nanoplate. Reprinted with permission from ref. 156. Copyright 2012 American Chemical Society. | ||
Hierarchical, hollow Bi2WO6 microspheres, which were constructed from crossed nanosheets prepared through an anion exchange method by Cheng et al., successfully photoreduced CO2 to CH3OH under visible light irradiation.157 As depicted in Fig. 14, the synthesis involved anion exchange of BiOBr solid microspheres, prepared from Bi(NO3)3 and a [C12Mim]Br ionic liquid, with Na2WO6 under hydrothermal conditions at 160 °C. BiOBr and Bi2WO6 comprise crystalline layered structures consisting of alternating (Bi2O2)2+ slabs separated by intercalating anion layers. Since the solubility of Bi2WO6 was lower than that of BiOBr, thin layers of Bi2WO6 formed at the BiOBr− surfaces during the exchange process. Owing to the slower diffusion of WO42−versus Br−, a fast, continuous outflow of Br− through the Bi2WO6 shell resulted in the formation of hollow cores over a prolonged period. These 2–5 μm diameter, hollow microspheres were themselves mesoporous, with a surface area far higher than that of Bi2WO6 prepared by a solid state route (Bi2WO6 SSR). Hollow Bi2WO6 microspheres generated around 25 times more CH3OH from CO2 photoreduction by water than Bi2WO6 SSR at under visible light (300 W Xe arc lamp equipped with a 420 nm cut-off filter). This performance was notably achieved without the aid of noble metal co-catalysts. As in the previous examples, this enhanced photocatalysis was attributed to the higher surface area and CO2 adsorption affinity (eight times that of Bi2WO6 SSR) of the hierarchical structure.
 | ||
| Fig. 14 Schematic of hollow Bi2WO6 microspheres formation through anion exchange of solid BiOBr microspheres. Reproduced from ref. 157 with permission from The Royal Society of Chemistry. | ||
An exciting biomimetic approach was recently adopted by Zhou et al. to prepare hierarchical, three-dimensional perovskite titanates, ATiO3 (A = Sr, Ca, Pb), for CO2 photoreduction.158 In an effort to move one-step closer to artificial photosynthesis, fresh Cherry Blossom green leaves were employed as a structure-directing agent to prepare inorganic photocatalysts via a modified sol–gel protocol; natural leaves possess high porosity, connectivity and surface areas, tailored to facilitate efficient gas diffusion, water transport and the harnessing of solar energy. The success of this synthesis was evidenced by the FE-SEM images in Fig. 15a and b, wherein the resulting SrTiO3 exhibited a macroporous venation network characteristic of the parent leaves. Higher magnification TEM imaging revealed the network walls consisted of a mesoporous array of crystallites (Fig. 15c and d). Such biomimetic SrTiO3 and CaTiO3 architectures outperformed corresponding reference titanates in CO2 photoreduction to CO (major product) and CH4 (secondary product) during UV illumination (300 W Xe arc lamp) under water vapour in a gas closed circulation system and 80 kPa of CO2. Incorporation of Au, Ag and Cu into these hierarchical titanates enhanced their photoactivity, and also led to the evolution of trace C2H4 and C2H6, although the latter were proposed to arise via photocatalytic non-oxidative coupling of CH4 rather than direct CO2 photoreduction.159
 | ||
| Fig. 15 FE-SEM images showing (a) the cross section of the porous venation architecture of SrTiO3 (scale bar, 1 μm) (b) cross section of the porous network axially (scale bar, 10 μm), (c) magnified image of the wall of the network (scale bar, 1 μm) and (d) TEM image showing the mesoporous structure of SrTiO3 (scale bar, 2 nm). Reprinted by permission from Macmillan Publishers Ltd ref. 158, copyright 2013. | ||
The excellent performance of these leaf-architectured titanates presumably reflects their highly interconnected macroporous-mesoporous structure, which promotes gas diffusion in addition to increasing surface active sites. This sophisticated architecture may also enhance light absorption in a manner similar to that of natural leaves, by which incident light is scattered and transmitted into deeper layers of the network, thereby increasing the quantum efficiency and hence photocatalytic activity.
 | ||
| Fig. 16 Electron transfer pathway in a SC/SC hybrid system. Adapted from ref. 160, Copyright 1995, with permission from Elsevier. | ||
2.2.5.1. Inorganic hybrid photocatalysts. A number of such SC1/SC2 hybrid systems have been developed for CO2 photoreduction. Wang et al. prepared CdSe/Pt/TiO2 heterostructures using commercially available CdSe quantum dots (QDs) of two different sizes (2.5 nm and 6 nm) and commercial P25 titania.162 The resulting heterostructures, containing 0.5 at% Pt and 1 at% Cd, were active for the photoreduction of CO2 to CH4 and CH3OH (respective major and minor products) under visible light irradiation, with trace H2 and CO also reported; neither Pt/TiO2 or CdSe components were individually active under identical reaction conditions. Consequently, the observed visible light photoactivity of the semiconductor heterostructures must arise from a synergy between CdSe and TiO2. It was proposed that photoexcited electrons promoted into the CB of CdSe under visible light irradiation, were subsequently injected into the CB of TiO2, increasing their liftime and probability of transfer to CO2. However, these composites deactivated after 4–6 h irradiation, most likely due to oxidation of the QDs by the accumulated photoinduced holes in the VB of CdSe.
In other work, coupling of AgBr with P25 was also undertaken to form nanocomposites active for reducing CO2 under visible light,163 wherein the indirect band gap of AgBr, which spans wavelengths between 450–700 nm, was held responsible for the visible light activity. This hybrid was synthesised utilising a cetyltrimethylammonium bromide (CTAB) surfactant stabiliser to create well-dispersed AgBr NPs (of around 5 nm) decorating the surface of P25. The nanocomposites exhibited excellent visible light photoactivity (150 W Xe lamp with a 420 nm cut-off filter) and photostability in the presence of aqueous KHCO3, relative to pure AgBr, with CH4 as the major product together with CH3OH, CH3CH2OH and CO under high CO2 pressure (7.5 MPa). A composite containing 23.2 wt% of AgBr exhibited optimum performance, with the photocatalytic mechanism again proposed to involve visible light excitation of electrons into the AgBr conduction band and subsequent transfer into the TiO2 CB with attendant charge separation. In addition to increasing charge carrier lifetime, these hybrid structures also impeded the formation of metallic Ag through photolysis, as was observed for pure AgBr.
Qin et al. followed a similar route employing CTAB to produce 14 nm CuO/TiO2 composite nanostructures,164 although here UV irradiation in the presence of methanol (rather than water) resulted in CO2 photoreduction to methyl formate within a slurry reactor. A 1 wt% CuO/TiO2 composite calcined at 450 °C exhibited the highest photoactivity, outperforming both pure TiO2 and composites synthesised in the absence of CTAB, suggesting a role for both charge separation and particle size effects. Liu et al. also explored the synergy between copper and titania heterojunctions, employing a sol–gel route to prepare Cu2O/TiO2 composites which demonstrated superior photoactivity over pure TiO2 in photoreducing CO2 to CH4 in a stirred batch, annular quartz reactor under 1 bar of CO2 and low power (32 W) UVA illumination.165 XPS and pulse chemisorption measurements identified the presence of a two-dimensional Cu2O species decorating the surface of titania at extremely low doping levels (0.03 wt% Cu). Higher copper loadings resulted in a structural transformation to three-dimensional Cu2O crystallites, which reduced the efficiency of photoinduced electron transfer from TiO2 to Cu2O, and hence lowered their activity. TiO2 has also been coupled with iron to form FeTiO3/TiO2 hybrids able to photoreduce CO2 to CH3OH under high power UV (500 W Xe lamp) and visible light irradiation under basic conditions.166 Materials synthesised with 20 mol% Fe gave the highest rate of CO2 reduction. Under UV illumination, photoinduced electrons were proposed to transfer from the (higher energy) CB of TiO2 to that of FeTiO3, with photoinduced holes returned from the VB of FeTiO3 to that of TiO2, charge separation again inferred as improving the overall photoactivity. In contrast, under visible light irradiation, electrons were photoexcited into the FeTiO3 CB, resulting in a partially vacant FeTiO3 VB, which was charge compensated for by the consequential movement of electrons from the almost isoenergetic TiO2 VB, leaving residual holes in the TiO2 VB, and net electron–hole charge separation across the two semiconductors. Efforts have been made to couple TiO2 with CdS and Bi2S3 in order to photoreduce CO2 to CH3OH.165 Li et al. precipitated CdS or Bi2S3 onto TiO2 NTs to yield visible light active CdS/TiO2 and Bi2S3/TiO2 heterostructures, thereby delivering improved photoactivity relative to P25 or bare TiO2 NTs (although activities were not normalised to take into account the two-fold higher surface areas of the nanorod morphologies). Photoinduced electrons from the CB of CdS or Bi2S3 could be injected into that of TiO2 to achieve efficient charge separation. Nevertheless, these complex heterojunction nanorods failed to generate more CH3OH than their respective pure CdS and Bi2S3 bulk analogues.
CuO–Cu2O thin film nanorod arrays prepared by Chadimkhani et al. were recently shown to photoreduce CO2 into CH3OH under simulated solar irradiation (AM 1.5) without a co-catalyst via photoelectrocatalysis.167 Coupling of such small band gap semiconductors is advantageous in exploiting a larger fraction of the visible light region of the solar spectrum than achievable by either alone. CuO nanorods were first thermochemically grown over a Cu substrate, following which Cu2O crystallites were electrodeposited on the nanorod surfaces to generate a CuO core/Cu2O shell hybrid thin film which outperformed a compact Cu2O electrodeposited film. This enhancement may reflect more efficient transfer of photoinduced electrons from the CB of the Cu2O shell to that of the CuO core and consequent increased probability of photoinduced electrons reacting with CO2. Such a model necessitates that the tips of CuO nanorods are exposed to a CO2 saturated aqueous environment. The CuO–Cu2O thin film was reported to generate CH3OH at a potential of only −0.2 V vs. SHE, lower than the standard redox potential for CO2/CH3OH (−0.38 V vs. SHE), a feature characteristic of p-type semiconductors (such as CuO and Cu2O).
Copper oxide has also been employed by In et al. to form hollow CuO–TiO2−xNx hybrid nanocubes that are effective for photoreducing CO2 to CH4.168 As shown in Fig. 17a, Cu3N nanocubes, synthesised through the thermal decomposition of Cu(NO3)2 in octadecylamine at 240 °C, were employed as the templating agent for subsequent TiO2 deposition on their external surfaces through slow hydrolysis of titanium n-butoxide. The initial Cu3N nanocubes were uniform in shape and around 27 nm across (Fig. 17b), and underwent significant roughening following TiO2 coating (TiO2@Cu3N) as apparent in Fig. 17c. Subsequent heat treatment of the TiO2@Cu3N powder at 450 °C under air transformed these phase-separated structures into hollow CuO–TiO2−xNx hybrid nanocubes (Fig. 17d–f). The final material was composed of hollow, truncated cuboidal CuO hollow cubes of ∼57 nm diameter, decorated by small TiO2−xNx particles. FE-SEM imaging confirmed the cubic morphology and hollow core of this hybrid material (Fig. 17g–f). The authors proposed that TiO2 crystallised onto the surfaces of Cu3N during thermal processing, but did not fully encaspulate the underlying nanocubes, permitting O2 to diffuse into, and oxidise, the Cu3N surface. Assuming that the inward rate of atomic oxygen diffusion is slower than the outward diffusion of N2, high temperature treatment would drive the formation of hollow cores through the Kirkendall effect.169 As nitrogen atoms diffuse outward, they are able to react with TiO2 and hence form an oxynitride TiO2−xNx phase. Under simulated solar illumination and water vapour, this particular hybrid composite photoreduced CO2 to CH4, and C2H6, C3H8 and C4H10 as minor products. The hollow CuO–TiO2−xNx hybrid nanocubes outperformed P25, CuO (annealed Cu3N), CuO@TiO2, Cu3N and TiO2@Cu3N in the photocatalytic generation of CH3OH, likely due to their improved visible light absorption characteristics and the synergy between CuO and TiO2−xNx.
 | ||
| Fig. 17 (a) Schematic for the synthesis of hollow CuO–TiO2−xNx hybrid nanocubes, and associated TEM images of (b) Cu3N nanocubes, (c) TiO2@Cu3N nanocubes, (d)–(f) hollow CuO–TiO2−xNx hybrid nanocubes at different magnifications, and (g)–(h) FESEM images of the hollow hybrid nanocubes. Reproduced with permission from ref. 168. Copyright 2012 Wiley. | ||
2.2.5.2. Organic hybrid photocatalysts. A hybrid catalyst composed of a Nafion (perfluorinated polymer with sulfonate groups, Nf) overlayer on TiO2 with Pd co-catalyst was prepared by Kim et al. for CO2 photoreduction170 as illustrated in Fig. 18a. Pd was first impregnated onto P25, and subsequently coated with Nafion. The rationale for integrating Nafion with a photoactive semiconductor was that the superacidic polymer, which exhibits excellent proton conducting behaviour, would facilitate proton-coupled multielectron transfer (PCET) pathways during product (hydrocarbon) formation, thereby improving net CO2 reduction. It was also believed that Nafion might inhibit the reoxidation of photoreduction products, and as a fluorinated polymer, be stable towards photocorrosion and redox reactions and thus a good host support. After catalyst pre-cleaning and UV irradiation the hybrid Nf/Pd–TiO2 facilitated CO2 photoreduction to CH4, C2H6 and H2 (and trace C3H8) in a sodium carbonate aqueous solution. As shown in Fig. 18b, CO2 reduction was highly sensitive to reaction pH, with photoactivity increasing at lower pH, the higher H+ concentration aiding product generation. In the presence of Nafion, considerably more CH4 was generated at pH 3 and 11 but the effect was less prominent at pH 1. However, Nafion significantly enhanced ethane production at pH 3 and 1, with propane only observed over the Nf/Pd–TiO2 hybrid photocatalyst. The observation that Nafion favours longer chain hydrocarbons suggest that it may stabilise CO2 reduction intermediates in a manner that promotes sequential multi-electron transfer. Additional experiments with Nf/Pd–TiO2 under natural sunlight only resulted in CH4, C2H6 and C3H8 formation.
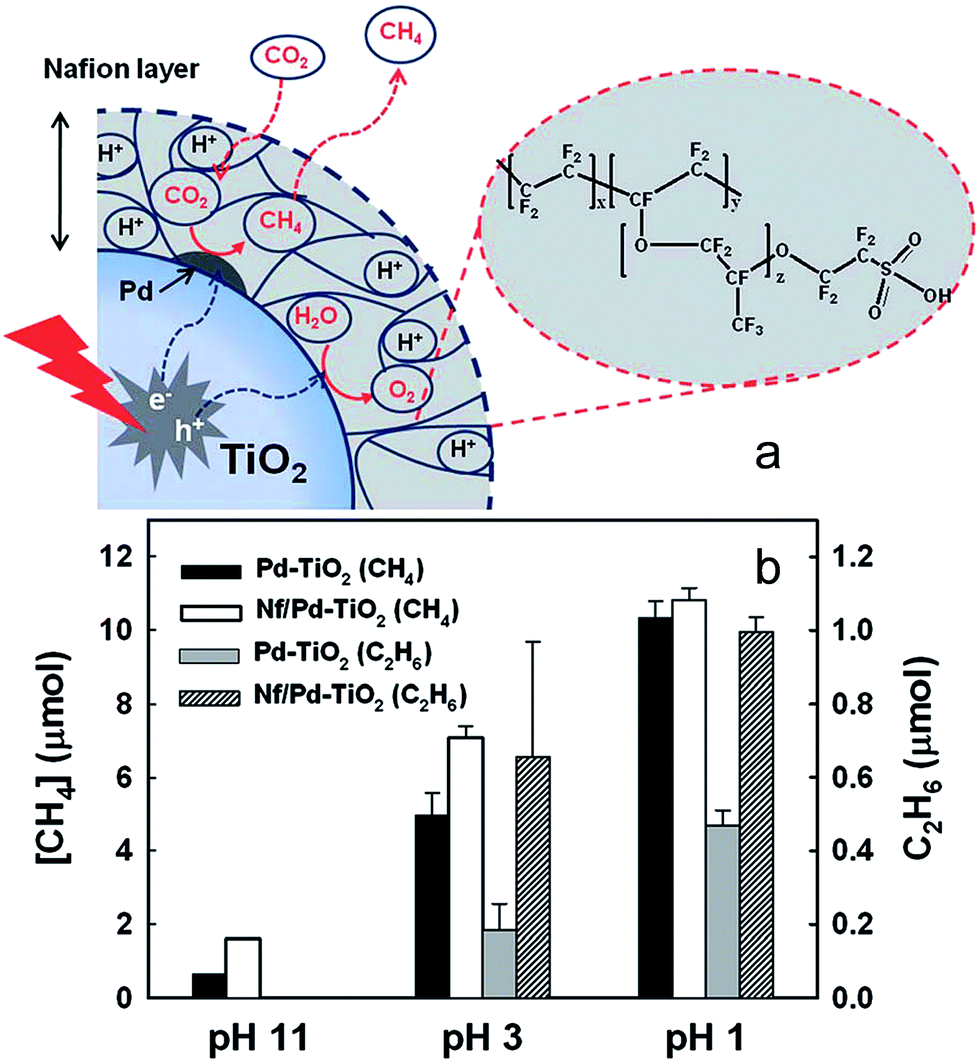 | ||
| Fig. 18 (a) Schematic of CO2 photoreduction on Nf/Pd–TiO2 nanoparticles and (b) CH4 (at 5 h) and C2H6 (at 1 h) production at different pH ranges on Pd–TiO2 and Nf/Pd–TiO2 NPs under UV illumination (λ > 300 nm) in the presence of 0.2 M Na2CO3 aqueous solution. Catalyst concentration was fixed at 1.5 g L−1 (with 1.0 wt% Pd and 0.83 wt% Nafion). Reproduced from ref. 170 with permission from The Royal Society of Chemistry. | ||
Sensitising large band gap semiconductors with transition metal complexes is another approach to designing visible light active materials.171–173 Yuan et al. recently demonstrated the utility of a Cu[(bpy)2]+ dye-sensitised TiO2 hybrid system for CO2 photoreduction.174 To obtain an air stable Cu complex, the 6- and 6′-position of the 2,2′-bipyridine (bpy) ligand were modified with a methyl group to protect the surroundings of the labile Cu(I) centre. This ligand was also designed to terminate in cyanoacetic acid moieties, thus enabling efficient anchoring of the metal complex onto the semiconductor surface. The Cu(I) complex modified P25 hybrid successfully photoreduced CO2 with water vapour into CH4 in a gas–solid reaction system under visible light illumination. It is likely that the Cu(I) complex was responsible for harnessing photons from visible light, with the resulting photoexcited electrons rapidly injected into the conduction band of the visible-inactive TiO2, thereby prolonging the lifetime of photoinduced electrons to drive CO2 reduction.
A visible light hybrid photocatalyst system, which utilised a naturally occurring enzyme CO dehydrogenase (CODH 1) to reduce CO2, was developed by Woolerton et al.175,176 This enzyme was homodimeric, such that each sub-unit contained a buried [Ni4Fe–4S] active site, wired to the protein surface through a series of [4Fe–4S] clusters that were also responsible for relaying electrons in and out of CODH 1. Fig. 19 illustrates the complete photosystem comprising CODH 1 modified, and Ru dye (RuP) sensitised, P25 NPs. During visible light irradiation, photoexcited electrons generated through metal-to-ligand charge-transfer on the RuP dye could be injected into the CB of TiO2 and transferred through the [4Fe–4S] clusters to the CODH 1 active sites, with the resulting photo-oxidised RuP regenerated via 2-(N-morpholino) ethanesulfonic acid (MES) as a sacrificial electron donor. It was proposed that the reduction process was initiated through CO2 binding as a bridging ligand between the Ni and dangling Fe atom of the enzyme. Under visible light illumination, TiO2 thus functioned as a medium for relaying electrons between the dye and enzyme. It is noteworthy that CODH 1 usually oxidises CO to CO2, but that the enzyme can be driven to catalyse the reverse reaction by applying a small overpotential.177 Further investigations revealed P25 as the preferred semiconductor with respect to ZnO, SrTiO3, rutile and anatase TiO2.175 Photoactivity was further enhanced through the addition of another sacrificial electron donor, ethylenediaminetetraacetic acid (EDTA). To improve CODH 1 attachment to titania, P25 was modified with various molecules, including polymyxin B sulphate and glutamic acid, albeit to no effect. On the other hand, functionalisation with o-phosphorylethanol led to a significant decrease in activity. Although this particular system was capable of photoreducing CO2 to CO, the photoconversion efficiency was lower than expected, and it was sensitive to O2 passivation.175,176 Photoactivity decreased after 4 h on-stream, potentially due to the difficulty in regenerating oxidised RuP, or the ineffectiveness of CODH 1 in harnessing electrons for activating CO2.
 | ||
| Fig. 19 Schematic illustrating the CODH 1 modified and Ru dye-sensitised TiO2 NPs photoactive system. Reprinted with permission from ref. 176. Copyright 2010 American Chemical Society. | ||
2.2.5.3. Carbonaceous media hybrid photocatalysts. Carbon nanomaterials, such as activated carbons, carbon monoliths, carbon NTs, graphene and graphites178,179 are promising candidates for catalyst supports, offering low cost, robust mechanical strength, high surface area, excellent porosity, chemical inertness and high electron conductivity. Moreover, carbons are amenable to the efficient recovery of precious metal promoters via combustion, and their surfaces may be readily functionalised by physical or chemical treatments, thereby modifying the resulting performance of hybrid catalysts. Significant work has been undertaken to integrate photoactive materials with carbonaceous media such as carbon NPs and graphene to deliver improve photocatalysts.
Covalently-functionalised carbon quantum dots (QDs) absorb and emit strongly in the visible light region.180–183 Because of these unique electronic properties, surface-passivated carbon QDs are a promising visible light harvesting medium. Surface-functionalised carbon NPs prepared by Cao et al. were active in photoreducing CO2 under visible light illumination.184 Sub-10 nm carbon NPs were functionalised with oligomeric poly(ethylene glycol) diamine (PEG1500N), rendering the resulting nanoparticles photoactive and water soluble. PEG-functionalised carbon QDs readily generated surface confined electrons and holes under irradiation that recombined radiatively (Fig. 20). This recombination process can be quenched by either electron donors or acceptors. Efficient charge separation and supressed radiative recombination was achieved through coated these surface-functionalised carbon NPs with Au or Pt through solution-phase photolysis; subsequent exposure to a light source would channel photogenerated electrons into the resulting metal nanoparticles, thereby prolonging the lifetime of charge carriers required for subsequent CO2 reduction. Under visible light irradiation (425–720 nm), and in the presence of water, metal decorated and surface-functionalised carbon NPs proved active for photoreducing CO2 to formic acid, although product analysis relied upon solution phase 1H and 13C NMR spectroscopy, and hence was unable to quantify the presence of potential gas phase reduction products such as light alkanes and alkenes.
 | ||
| Fig. 20 PEG-functionalised carbon nanoparticles undergoing radiative recombination (left) and noble metal-coated & PEG-functionalised carbon nanoparticles photoreducing CO2 (right). Reprinted with permission from ref. 184. Copyright 2011 American Chemical Society. | ||
As a conductive electron reservoir, carbon is also effective in channelling photoinduced electrons away from a photoactive host, thereby increasing the probability that photoexcited electrons reduce CO2 rather than recombining with holes. Ong et al. reported a hybrid catalyst, composed of CNTs grown on a Ni impregnated TiO2 composite, for CO2 photoreduction to CH4 under visible light.185 The authors first deposited 10 wt% Ni onto commercial anatase via precipitation in the presence of glycerol. Carbon NTs were subsequently grown over the calcined Ni/TiO2 through chemical vapour deposition. The band gap of the resulting hybrid material was 2.22 eV, significantly smaller than that of anatase TiO2 (3.32 eV). Hence CNT incorporation served to narrow the band gap and improve visible light absorption in the hybrid catalyst. Visible light irradiation under water vapour revealed the CNT/Ni/TiO2 hybrids were superior to either Ni/TiO2 or pure anatase TiO2, although progressive deactivation occurred after 4.5 h irradiation at which point CO2 photoreduction activity was maximal. Photoactivity of the CNT/Ni/TiO2 hybrid catalyst originated from improved visible light absorption and the excellent electron storage and transfer properties of the CNTs which helped to suppress electron–hole recombination.
Graphene, a two-dimensional planar structure of sp2 hybridised carbon, has received much attention in regard to photocatalytic applications for H2 evolution,58,163,186–194 photocurrent generation,190,194–198 and dye degradation,90,144,145,163,189,199–201 due to its outstanding electronic properties.144,197 Graphene also offers extremely high specific surface areas202 in excess of 2600 m2 g−1, a good CO2 and H2 adsorption capacity,203,204 and has been reported to sensitise large band gap semiconductors for visible light photoabsorption.205–207 However, less work has been conducted into the integration of graphene with photoactive materials for CO2 photoreduction. Nevertheless, it is known that graphene oxide (GO), an unreduced form of graphene, despite being a poor electrical conductor,195 is capable of photoreducing CO2 to CH3OH.208 Liang et al. demonstrated that the photocatalytic activity of commercial P25 is greatly enhanced for CH4 formation under both UV and visible light irradiation when used in conjunction with a low defect density, solvent-exfoliated graphene (SEG), compared to that achievable over defective, solvent-reduced graphene oxide (SGRO).206 The photoactivities of these SEG–P25 and SRGO–P25 composite films, which were synthesised by blade-coating a physical mixture of different amounts of graphene and P25 ink and subsequent annealing, are shown in Fig. 21a. Note that under UV illumination the presence of SGRO exerted little impact upon the photoactivity of TiO2, whereas graphene possessing a low defect density exhibited better electrical mobility and concomitant superior photocatalytic efficiency under either UV or visible light.
 | ||
| Fig. 21 Methane formation rate of (a) SEG–P25 and SRGO–P25 films under UV and visible light illumination, and (b) SEG–TiNS and SWCNT–TiNS films under UV (100 W Hg vapour lamp, 365 nm) and visible light irradiation (natural daylight bulb, ∼400 to 850 nm). Reprinted with permission from ref. 206 and 209. Copyright 2011 and 2012 American Chemical Society. | ||
Despite the popularity of graphene-derived materials, a direct comparison between graphene and its carbon nanotube (CNT) allotrope has not been well-explored. Further work by Liang et al. extended their studies on CO2 photoreduction catalysed by blade-coated films to examine the integration of titania nanosheets (TiNS) with SEG or single-walled carbon nanotubes (SWCNT) to form either a two-dimensional graphene–titania nanosheet composite, or a one/two-dimensional carbon nanotube–titania nanosheet composite, respectively.209Fig. 21b shows that the SEG–TiNS composites outperformed SWCNT–TiNS in photoreducing CO2 to CH4 under UV irradiation. Conversely, composites of SWCNT–TiNS demonstrated better photoactivity under visible light illumination. It was proposed that the two-dimensional interface between SEG and TiNS facilitated intimate electronic and physical coupling relative to the one-/two-dimensional interface within the SWCNT–TiNS composite. Consequently, interfacial electron transfer from TiNS to SEG was greatly improved, inhibiting the recombination of photogenerated electron–hole pairs. In contrast, the better visible light photoactivity from SWCNT–TiNS was proposed to originate from the increased optical absorption of the SWCNT component at longer wavelengths relative to that of SEG. Accordingly, it was proposed that under visible light irradiation, photoinduced electrons generated at the SWCNT surface were transferred to the TiNS substrate for subsequent CO2 photoreduction.
A series of graphene–Ta2O5 composites, with different loadings of the carbon, were prepared by Lv et al. through a hydrothermal route.207 Impregnation with a Ni/NiO co-catalyst activated these composites for CO2 photoreduction to CH3OH and H2 under UV-Vis illumination under aqueous conditions. Composites with 1 wt% graphene exhibited the highest photoactivity; in CO2-aerated water the composite produced 3.4 times and 2.3 times the amount of CH3OH and H2 respectively than achieved over pure Ta2O5. This enhanced photocatalysis was attributed to the benefits of the graphene support in promoting effective interfacial charge transfer, as illustrated in Fig. 22. Photoinduced electrons generated at the Ta2O5 surface could be effectively transported to the surfaces of either graphene or the Ni/NiO dopant for subsequent transfer and CO2 reduction.
 | ||
| Fig. 22 Schematic of potential pathways for the transport of photoinduced electrons from a Ta2O5 semiconductor nanoparticle to active CO2 photoreduction sites mediated by graphene. Reproduced from ref. 207 with permission from The Royal Society of Chemistry. | ||
Unlike the preceding materials, wherein as-synthesised graphene nanosheets were integrated with semiconductor particles through a simple impregnation/adsorption process, Tu et al. followed an alternative approach to prepare hollow spheres of alternating titania and graphene nanosheets for CO2 photoreduction.210Fig. 23 shows how the surface of a poly(methyl methacrylate) (PMMA) template was first modified with a cationic polyelectrolyte, e.g. polyethylenimine (PEI) which electrostatically attracted negatively charged Ti0.91O2 nanosheets onto their surface. Subsequent surface modification of these titania-coated PMMA beads with PEI facilitated coating by negatively charged graphene oxide (GO) nanosheets. This process was repeated multiple times to thereby prepare a PMMA core composite with five alternating layers of (PEI/Ti0.91O2/PEI/GO)5. Subsequent microwave treatment under an Ar environment decomposed the PMMA template, removed the PEI moiety, and reduced the GO to graphene (G). TEM (Fig. 23b and c) revealed that these Ti0.91O2 and graphene nanosheets alternated in a parallel, ordered lamellar arrangement, with a repeat distance estimated to be around 1.15 nm. Catalytic screening under UV-Vis illumination showed that the (G/Ti0.91O2)5 hollow spheres were superior for CO2 photoconversion versus (Ti0.91O2)5 hollow spheres (synthesised without GO) and P25 titania in a glass reactor filled with CO2 at ambient pressure, albeit the principal photolysis product was CO not CH4. A number of explanations for the excellent behaviour of these (G/Ti0.91O2)5 hollow composites were advanced. The ultrathin nature of the Ti0.91O2 nanosheets likely drove rapid transfer of charge carriers to active sites, increasing the probability of the photogenerated electrons participating in subsequent photoreduction chemistry; the compact stacking of Ti0.91O2 and graphene nanosheets would allow efficient interfacial electron transfer from the Ti0.91O2 to graphene layers, this spatial separation increasing the lifetime of photoexcited electrons. Hollow composite cores may also trap and multiply scatter incident photons, thereby improving light absorption and quantum efficiency.
 | ||
| Fig. 23 (A) Stepwise preparation of alternating TiO2 and graphene nanosheet hollow sphere assemblies, (B) TEM and (C) and HR-TEM images of (G–Ti0O2)5 hollow spheres. Scale bar of the inset in (c): 100 nm. Inset in (C) is the fast Fourier Transform pattern of the ordered lamellar structures exhibited by the (G–Ti0.91O2)5 hollow spheres. Reproduced with permission from ref. 210. Copyright 2012 Wiley. | ||
Reduced graphene oxide (RGO) has also been employed within unique heterostructured optoelectronic materials, in which “tree”-like three-dimensional hierarchical titania nanorods,211 or one dimensional ZnO nanorods212 (as wide-band-gap UV absorbing semiconductors) are contacted with CdS visible light sensitising nanoparticles via a suspended, RGO film. The resulting architectures ensure precise spatial localisation of each component and tunable charge-transport properties, facilitating a 100–150% increase in the associated photocurrent density relative to the metal oxide/chalcogenide system alone, and 25% increase in methylene blue decomposition (ZnO/RGO/CdS), and 240% increase in the applied bias to photoelectrochemical hydrogen generation efficiency for hydrogen production via water-splitting (TiO2/RGO/CdS).
2.3. Co-catalysts and plasmonic photocatalysts
Co-catalysts, typically Platinum Group and noble metals, are frequently deposited on semiconductor surfaces in order to (i) maximise the lifetime of photogenerated charge carriers, (ii) to activate reactants, and (iii) to improve the absorption of incident light.213–216 Noble metals such as Au and Ag have lower Fermi levels than their partnered semiconductors, and hence can act as electron sinks that accept photoexcited electrons,217,218 thereby retarding electron–hole recombination and increasing the availability of electrons for CO2 photoreduction. It is generally held that, when co-catalysts are employed, photoconversion efficiency varies with the co-catalyst particle size, in turn usually governed by the method of their incorporation,75,157,219 albeit the nature of this relationship (proportional or inversely proportional), magnitude and origin remains poorly studied and understood.Recent work by Iizuka et al. has shown that the photoactivity of plate-like ALa4Ti4O15 (A = Ca, Sr, and Ba) layered perovskites impregnated with a Ag co-catalyst varies with the method of co-catalyst loading.75 Silver modification of ALa4Ti4O15 with a AgNO3 precursor was achieved via: (i) wet impregnation followed by H2 reduction; (ii) in situ photodeposition; or (iii) liquid phase reduction using NaPH2O2. Under UV illumination and water, all these Ag-loaded ALa4Ti4O15 materials were active in photoreducing CO2 into CO predominantly, and a small amount of HCOOH, accompanied by H2 and O2 evolution, under a constant CO2 flow rate of 15 mL min−1. Ag loaded by liquid phase reduction demonstrated the highest photoactivity, followed by that through impregnation/H2 reduction, ahead of those photodeposited. When photodeposition was employed, Ag NPs of around 30–40 nm populated edges of the as-prepared BaLa4Ti4O15, while liquid phase reduction resulted in a homogeneous dispersion of <10 nm nanoparticles over the layered perovskite. Materials prepared via impregnation/H2 reduction produced large aggregates of ∼50 nm Ag particles, although some 10–20 nm Ag particles were observed at the edges of impregnated/reduced samples after photolysis. The basal plane and edges of plate-like BaLa4Ti4O15 were suggested as the respective active sites for photooxidation and photoreduction. Accordingly, Ag deposited at the basal plane may undergo photodissolution and redeposition at the semiconductor edges during photocatalysis, with the particle size of such redeposited Ag dependent upon the concentration of dissolved silver ions. Consequently, in situ photolysis modified the Ag particle size to increase in the order: liquid phase reduction < impregnation/H2 reduction < photodeposition. It was concluded that the photoactivity of Ag/BaLa4Ti4O15 varied inversely with Ag particle size, a function of the co-catalyst incorporation route.
TiO2 NTs arrays promoted by ultrafine Pt NPs, uniformly deposited via a rapid, microwave-assisted solvothermal approach, were investigated by Feng and co-workers.220 In this method, the crystalline nanotube arrays were first loaded in a Teflon reaction chamber containing a 0.15 M H2PtCl6/CH3OH solution. After an overnight immersion process, the reaction chamber was rapidly heated to 120 °C and held for 20 min before cooling. The resulting Pt/TiO2 NTs arrays were isolated, washed and dried prior to catalytic testing. Dark field (S)TEM shown in Fig. 24a highlights the uniform distribution of Pt NPs (with an average diameter of 3.4 nm) across the TiO2 NTs arrays. While such wet impregnation is simple to conduct and yields small Pt NPs, they exhibited a broad size distribution spanning 1.8–4.9 nm. Nevertheless, under irradiation by a solar simulator and water vapour, these Pt/TiO2 NTs arrays were superior to unpromoted TiO2 NTs arrays in photoreducing CO2 to CH4 and trace C2H6 at 45 °C. The excellent photoactivity of Pt/TiO2 NTs arrays was attributed to the high Pt nanoparticle dispersion (and hence density of surface active sites), coupled with efficient electron diffusion properties through the nanotube arrays.
 | ||
| Fig. 24 (a) Dark field (S)TEM image of ultrafine Pt NPs deposited TiO2 nanotube arrays,220 and (b) TEM image of size resolved ultrafine Pt NPs on TiO2 film. Inset: HRTEM image showing the lattice fringes corresponding to anatase TiO2 (112) and Pt (111) NPs. Reproduced from ref. 157 with permission from The Royal Society of Chemistry. | ||
In contrast, Wang et al. utilised a unique gas phase technique, tilted-target sputtering (TTS), to deposit size-controlled ultrafine Pt NPs onto a single crystalline titania thin film comprised of 1-D TiO2 columns.51 TTS enables precise control over both the flux of sputtered metal atoms and their radial distribution. Locating the TiO2 film in the outer region of the deposition flux zone, where low energy atoms participate in nanoparticle formation, permitted the selective growth of ultrafine Pt NPs on the semiconductor surface as shown by TEM in Fig. 24b; Pt NPs with an average particle size of 1.04 ± 0.08 nm were uniformly coated on anatase TiO2. It was claimed that such Pt NPs were preferentially deposited at surface defects on the titania substrate. While this gas-phase method necessitates high vacuum instrumentation, and hence is not readily scalable, it does offer Pt NPs possessing a narrow size distribution and tunable diameter (through varying the deposition time). The smallest and largest nanoparticles reported via this approach exhibited mean particle diameters of 0.63 ± 0.06 nm and 1.87 ± 0.31 nm, respectively (albeit a narrow range spanned). Subsequent exposure of Pt/TiO2 catalyst films to water vapour and UV irradiation evidenced improved photoactivity compared with P25 and an analogous bare TiO2 film for CO2 photoreduction, forming predominantly CH4 with trace CO under a slow CO2 stream in a continuous flow reactor. Optimal photoactivity was achieved for a 1.04 ± 0.8 nm Pt/TiO2 catalyst film, which delivered a remarkable combined quantum efficiency to CO and CH4 of 2.41%. The observed photoactivity enhancement reflected a combination of the high surface area and single crystalline nature of the TiO2 thin film, and excellent charge carrier separation characteristics conferred by the ultrafine Pt NPs. However, photocatalysis deteriorated after 5 h of irradiation, attributed to saturation of reactive sites by strongly-bound intermediate products, and consequent site-blocking.
Noble metals are often employed as co-catalysts to effect charge separation, but have been recently established to play an additional role as plasmonic photocatalysts in their own right, owing to the ability of gold and silver to absorb visible light through their localised surface plasmon resonance (LSPR).96,145,221–225 This latter hot topic requires an understanding of the basic principles underlying the plasmonic phenomenon. The LSPR is a collective oscillation of free electrons within metal structures, established when the natural oscillation frequency of surface electrons (induced by charge density redistribution), resonates with the frequency of incident photons. The free electrons of plasmonic Au, Ag, and Cu nanostructures can be excited by the electromagnetic field of incident visible light; as the light wavefront impinges, the electron density is polarised across these metal nanostructures, such that free conduction electrons coherently oscillate in phase with the light, creating a dynamic dipole oscillator at the metal surface (Fig. 25).225–227 Such intraband LSPR absorption (e.g. by Au 6sp electrons) induced by visible light is fundamentally different from the interband absorption caused by ultraviolet light (Au electrons excited from the 5d to 6sp bands).228
 | ||
| Fig. 25 Electromagnetic field of visible light interacting with plasmonic metal NPs: a dipole oscillator forms and resonates with the electromagnetic field of the incident visible light. Reproduced with permission from ref. 229. | ||
The strength and frequency of plasmon resonances depends on the size, shape, composition (for alloys) and local dielectric environment of the associated nanoparticles (see Fig. 26). These variations in LSPR peak positions and strengths strongly influence the photocatalytic capacities of such plasmonic nanomaterials.215,230
 | ||
| Fig. 26 The LSPR bands of plasmonic metal nanostructures with different shapes, sizes or compositions (for alloys): (a) nanoparticles of Au, Ag, and Cu; (b) Ag nanostructures with various shapes; (c) nanocubes of Ag with different cubic sizes; and (d) alloy of Au–Ag with different Au:Ag ratios. Reprinted by permission from Macmillan Publishers Ltd. Ref. 227, copyright 2011. Reproduced with permission from ref. 230. Copyright 2011 Wiley. | ||
On the basis of the literature, at least three mechanisms have been proposed to account for the enhanced visible light photoactivity of noble metal/semiconductor plasmonic photocatalyts. First, it is hypothesised that visible light photoexcited electrons at the surface of the plasmonic metal can migrate across the metal–semiconductor interface into the conduction band of the associated semiconductor, or transferred directly to adsorbates, providing a source of localised, energetic reductants.231 Second, the polarised electric field induced by the LSPR of metal NPs could enhance reactant activation over neighbouring supports by the interplay between the resonating electron cloud of the metal NPs and nearby adsorbates (polarisable or possessing polar functional groups). Finally, photoexcited electrons may not actually transfer from plasmonic metal NPs to adjacent semiconductors/reactants nor interact with them indirectly via field effects, but may rather liberate heat during relaxation of photoelectrons to their groundstate, thereby promoting thermal catalysis, although it has proven extremely difficult to qualitatively or quantitatively determine any such heating effect.232 All these models account for an increase in the photoexcited electrons available for chemical reduction.227
Plasmon-enhanced CO2 photoconversion has been recently reported,233,234 exemplified by AgX:Ag (X = Cl or Br) plasmonic NPs synthesised through a glycerol-mediated precipitation route in the presence of polyvinylpyrolidone.235 Despite their similar synthetic protocols, AgCl:Ag and AgBr:Ag exhibited cube-tetrapod-like and hexagonal nanoplates respectively. XPS and XRD confirmed the presence of metallic Ag on the as-prepared AgX:Ag NPs, with UV-Vis revealing significantly different band gaps of 3.95 (AgCl) and 3.45 eV (AgBr). When dispersed in NaHCO3 aqueous medium under visible light, the AgX:Ag plasmonic photocatalysts were active for photoreducing CO2 into CH3OH, attributed to the metallic Ag LSPR influencing the contacted AgX:Ag NPs. Recycling experiments indicated that these plasmonic catalysts were stable under the screening conditions, although in situ Ag+ reduction to additional silver metal is a plausible deactivation pathway following prolonged irradiation.
Hou et al. deposited islands of plasmonic Au NPs onto a sol–gel prepared TiO2 thin film via electron beam evaporation.148 Under visible light illumination and water vapour the resulting Au-promoted TiO2 thin film exhibited a 24-fold activity enhancement relative to a bare TiO2 thin film for CO2 photoreduction to CH4 in a stainless steel reactor at 75 °C. The low, but finite, photoactivity of the parent TiO2 film was attributed to defect-induced, sub-band gap photoexcitation. Complementary calculations suggested that direct electron transfer from Au NPs to TiO2 was not feasible under plasmonic excitation, and hence the superior photoactivity was ascribed to an increased sub-band gap charge carrier formation rate, indirectly induced by the LSPR of gold. Methane was the only reductant formed over the bare TiO2 thin film under UV irradiation, however a range of products were formed over the Au/TiO2 thin film wherein the production rate followed CH4 > C2H6 > HCHO > CH3OH. Surprisingly, the pure Au NPs were also reported to generate these aforementioned organic products, although the overall productivity was lower than that of the Au/TiO2 composite. The photocatalytic activity of Au NPs was proposed due to interband electronic transitions (and not the LSPR) wherein electrons were photoexcited to a potential more negative than the TiO2 conduction band and redox potential required to form desired reduction products. The synergy between Au NPs and the TiO2 film under UV irradiation was attributed to improved charge carrier separation.
Small Cu and Ag NPs which exhibit plasmonic effects have also been successfully loaded onto TiO2 nanorod films by an electrochemical method.219,236 Tan et al. loaded Cu NPs onto a TiO2 film via a 0.01 mM CuSO4 and 0.1 M K2SO4 mixed electrolyte at a pulse potential of −0.8 V for between 0–300 s.236 Under UV illumination and water, the resulting composite film (prepared at the optimum 100 s deposition time) demonstrated superior activity to a bare TiO2 or P25 film in photoreducing CO2 into CH4. The copper co-catalyst benefits were again ascribed to efficient charge separation due to plasmonic effects. However, it was reported that surface oxidation of Cu NPs to copper oxide occurred during photocatalyic screening. An electrochemical deposition method known as the double-potentiostatic method was also used to deposit Ag NPs onto a TiO2 nanorod film.219 This method favoured the formation of uniform and dense metallic nanoparticles due to quick nucleation and slow nanoparticle growth at high potential and low precursor concentration. Accordingly, Kong et al. loaded silver onto a TiO2 film employing a 0.1 M KNO3/0.2 mM sodium citrate/0.05 mM AgNO3 electrolyte at nucleation potentials from −1.4 V to −0.8 V for 100 s, followed by a growth potential of −0.2 V for 2400 s. TiO2/Ag films prepared at the optimum nucleation potential of −1.0 V exhibited improved photoactivity relative to a bare TiO2 film in photoreducing CO2 into CH4 under UV illumination and water vapour. Photocatalytic performance was again postulated to arise from efficient charge separation due to silver plasmonic effects. Moreover, alloy or bimetallic plasmonic nanostructures, such as Au–Pt (as shown in Fig. 27a)237 and Ag–Pt (as shown in Fig. 27b),233 have also been discovered to enhance CO2 the photoreduction by titania. In both cases, the introduction of plasmonic Au or Ag particles increased photocatalytic efficiency in terms of product selectivity and/or yield, ascribed to the LSPR effect of Au (Ag) NPs significantly increasing light absorption and charge separation of the photoexcited titania. Co-deposition of Au/Pt on TiO2 NFs considerably enhanced photocatalytic activities for both H2 generation and CO2 reduction compared to singly decorated (Pt/TiO2 or Au/TiO2) fibres. Bijith and co-workers' study demonstrates that Ag, Pt and bimetallic Ag–Pt NPs enable CH4 and CO product selectivity to be tuned:233 product yields were enhanced more than seven-fold compared with native TiO2, with a high CH4 selectivity. This was attributed to the enhanced availability of photoexcited electrons via improved electron–hole pair generation, and surface trapping of photoexcited electrons within the titania lattice, enabling the necessary accumulation of charge to drive the eight-electron CH4 forming process from adsorbed CO2.
 | ||
| Fig. 27 (a) Illustration of the photocatalytic process to produce H2 and to reduce CO2 over Au/Pt/TiO2 NFs. Reprinted with permission from ref. 237. Copyright 2010 American Chemical Society. (b) Illustration of different photoexcited charge pathways following electron–hole generation assisted by plasmonic nanoparticles over core–shell Ag@SiO2: transfer to the [a] semiconductor surface, [b] Pt NP surface, [c] Ag NP surface, and [d] bimetallic Ag–Pt surface. Reproduced with permission from ref. 233. Copyright 2013 IOP Publishing Ltd. | ||
2.4. Miscellaneous photocatalysts
Cationic and/or anionic doping is commonly employed to decrease semiconductor band gaps.238–242 Richardson et al. co-doped TiO2 with Cu and Ga via a sol–gel route to form composites that were active in CO2 photoreduction at longer wavelengths than possible utilising P25.139 The absorption band edge wavelength of doped photocatalysts increased in the order P25 < Ga/TiO2 < Cu/TiO2 < Cu, Ga/doped TiO2, with a corresponding decrease in the band of the doped-materials relative to P25. It should be noted that the absorption band edges of doped materials still resides in the UV region despite their modified optical properties. Under 200 W near-UV (350–450 nm), 350 W mid-UV (280–350 nm) or 500 W deep-UV (240–260 nm) irradiation, and immersion in a NaOH and KHCO3 aqueous medium, Cu and Ga co-doped TiO2 photocatalysts were superior to P25 and the monometal-doped titanias in photoreducing CO2 to HCOOH and other carbonic products (determined by TOC) in a continuous flow reactor at room temperature. Highest photoactivity was observed for the Cu0.78–Ga0.22/TiO2 catalyst, with Cu the proposed CO2 adsorption and reduction site for HCOOH and other organic products which include trace CH3OH and HCHO.Anion doping by Tsai et al. improved the visible light absorption properties of InTaO4. Doping was performed by nitridation of SSR prepared InTaO4 under flowing NH3 at 800 °C,243 resulting in the absorption band edge shifting to longer wavelengths in comparison to pure InTaO4. Results were attributed to hybridisation between the N 2p and O 2p orbitals, which shifts the valence band upwards, thereby narrowing the InTaO4 band gap. Under visible light illumination (390 to 770 nm) and in the presence of water, the prepared N-doped InTaO4 outperformed pure InTaO4 in photoreducing CO2 into CH3OH in a continuous flow reactor. XANES analysis indicated that N doping narrowed the band gap, and induced oxygen vacancies, the latter possibly contributing to photocatalytic enhancement. The photoactivity of the N-doped InTaO4 was further improved by the addition of a Ni@NiO core–shell co-catalyst, incorporated through a sequence of wet impregnation, chemical reduction and final calcination. Promotion by the Ni@NiO core–shell co-catalyst was ascribed to enhanced charge separation and light harvesting.
Solid solutions formed between wide and narrow band gap semiconductors have been reported to possess valence and conduction bands that provide smaller gaps than their parent materials.244–247 In general, the component semiconductors must possess the same crystal structure and similar lattice parameters. Liu et al. recently reported the application of CuxAgyInzZnkSm solid solutions as visible light photocatalysts for CO2 photoreduction.248 The band gap of this composite was a strong function of composition, and spanned 1.33–1.44 eV. Visible light irradiation (1000 W Xenon lamp at λ > 400 nm) in a NaHCO3 aqueous medium of these solid solution photocatalysts facilitated CO2 photoreduction to CH3OH in a closed glass gas circulation system. Systematic relations could not be derived between the catalyst composition and photoactivity, although photoactivity was composition dependent. A range of co-catalysts were investigated, including RuO2 and Rh1.32Cr0.66O3, although these did not promote CO2 conversion. Methanol productivity was enhanced under a H2 environment, wherein a higher density of protons was formed.
3. Conclusions and future directions
A wide variety of approaches have been developed for the synthesis of photocatalytically active materials for CO2 reduction to solar fuels and chemicals. To date, there is no standard protocol for evaluating photocatalytic performance, or single parameter that enables quantitative benchmarking of CO2 conversion efficiencies to specific carbon-containing solar fuels or chemicals. In a first step towards addressing these deficiencies, Table 2 compares a range of the nanomaterials discussed in this review, with a view to informing on which show the most promise with respect to productivity per unit catalyst mass, and thus provide a strong rationale for future development. Of course, many additional factors such as earth abundance, cost, toxicity, and scalability will also influence which materials are ultimately best suited to commercialisation. However, a step-change in photoactivity, and comprehensive mechanistic insight into the catalytic cycle and nature of the surface sites participating in CO2 adsorption and reduction, is required before such considerations should influence ongoing fundamental research efforts.| Material | Light source | T/°C | P CO2/bar | Product yield/μmol h−1 gcat−1 | Selectivity (product)/mol% | Quantum efficiency/% | Reference |
|---|---|---|---|---|---|---|---|
| ZnGa2O4 | 300 W Xe | — | 1 | 1 wt% RuO2 7245, mesoporous 762 | 100 (CH4) | — | 113 |
| Pt/TiO2 | 400 W Xe (20 mW cm−2 250–388 nm) | 22–32 | 1 | 1361 (CH4), 180 (CO) | 88.5 (CH4), 11.5 (CO) | 2.33 (CH4)0.077 (CO) | 51 |
| Zn2SnO4 | 300 W Xe | — | 1 | Bulk 17.0 (CH4), atactic 34.5, micro-octahedra 54.6, nanoplate/micro-octahedra 288.9, RuO2/Pt 1246.3 | 100 (CH4) | — | 155 |
| W18O49 | 300 W Xe(30 mW cm−2) | 70 | 1 | 11.31 (CH4, visible) 858 (CH4, UV-visible) | 95 (CH4), 5 (ethanol, acetone…) | 0.03 (CH4, visible) | 65 |
| Au/Pt–TiO2 | 500 W Xe | — | — | 114 (CH4), 20 (CO) | 85 (CH4),15 (CO) | — | 234 |
| (RuO2 + Pt)–Zn2GeO4 | 300 W Xe | RT | 1 | 25 | 100 (CH4) | — | 94 |
| Nafion layer-enhanced Pd–TiO2 | 300 W Xe (λ > 300 nm) | — | — | 21.9 (CH4), 1.03 (C2H6) | 95.5 (CH4), 4.5 (C2H6) | — | 170 |
| Pt–Cu/TiO2 | 200 W Xe (320–780 nm) | 50 | 2 | 9.8 (CH4), 5.9 (CO), 51 (H2) | 14.7 (CH4), 8.8 (CO) | — | 249 |
| 0.5 wt% Cu/TiO2–SiO2 | Xe (2.4 mW cm−2 250–400 nm) | — | — | 10 (CH4), 60 (CO) | 14.3 (CH4), 85.7 (CO) | 0.56 (CH4), 0.85 (CO) | 121 |
| 0.5 wt% Pt ZnAl2O4–ZnGaNO | 300 W Xe (λ ≥ 420 nm) | — | 1 | 9.2 | 100 (CH4) | — | 111 |
| 0.5 wt% Pt/NaNbO3 | 300 W Xe (λ > 300 nm) | 0.8 | 0% cubic 2.19 (CH4), 15 (CO), 45 (H2), 69% cubic 5.86 (CH4), 16 (CO), 42 (H2), 98% cubic 4.91 (CH4), 12 (CO), 25 (H2) | 12.7 (CH4), 87.3 (CO), 26.8 (CH4), 73.2 (CO), 29 (CH4) 71 (CO) | — | 250 | |
| AgBr/TiO2 | 150 W Xe (λ > 420 nm) | RT | 75 | 5.71 (CH4), 15.57 (CH3OH), 2.66 (C2H5OH), 6.43 (CO) | 18.8 (CH4) 51.3 (CH3OH) 8.8 (C2H5OH) | — | 163 |
| Amine-functionalised TiO2 | Xe (λ > 302) | — | — | 3.08 (CH4), 11.92 (CO) | 20.5 (CH4), 79.5 (CO) | — | 67 |
| Pt–, Au–, or Ag–TiO2 | 350 W Xe (35 mW cm−2) | 60 | — | 0.2% Pt 2.85, 0.2% Au 1.51, 0.1% Ag 0.65, TiO2 0.238 | 100 (CH4) | 0.68,0.36, 0.15, 0.06 | 123 |
| Ti0.91O2 | 300 W Xe | — | 1 | Graphene 1.14 (CH4), 8.91 (CO), Ti0.91O2 1.41 (CH4) | 11.4 (CH4), 88.6 (CO), 100 (CH4) | — | 210 |
| Cu(I) dye-sensitised TiO2 | 300 W Xenon (λ > 420 nm) | — | 1 | 0.725 | 100 (CH4) | — | 174 |
| Ga2O3 | 300 W Xe (500 mW cm−2) | — | — | 0.3935 | 100 (CH4) | 0.007 | 107 |
| RuO2–Zn2GeO4 | 300 W Xe | — | 1 | 0.39 (CH4), 1.01 (CO) | 25.5 (CH4), 74.5 (CO) | — | 97 |
| Carbon nanotubes@ Ni/TiO2 | 75 W visible daylight lamp (λ > 400 nm) | 50 | 1 | 0.145 | 100 (CH4) | — | 185 |
| Pd–TiO2 | 500 W Hg (λ > 310 nm) | 5 | 0.867 | 1% Pd 0.037 (CH4), 0.06 (C2H6), 0.04 (CO), TiO2 0.027 (CH4), 0.353 (CO) | 78.9 (CH4), 12.7 (C2H6), 8.4 (CO), 7.1 (CH4), 92.9 (CO) | — | 68 |
| N-doped ZnO | 8 W fluorescent tube 7 mW cm−2 | 50 | 2 | 0.01 (CH4), 0.03 (CH3OH), 0.04 (CO), 0.09 (H2) | 4.8 (CH4), 19.0 (CH3OH), 23.8 (CO) | — | 112 |
| CdS/TiO2, Bi2S3/TiO2 | 500 W Xe (λ ≥ 400 nm) | — | 1 | CdS 40.1, TNTs–CdS 32.2,Bi2S3 64.5, TNTs–Bi2S3 45.3, TNTs 20.5, P25 18.2 | 100 (CH3OH) | — | 165 |
| Graphene-modified NiOx–Ta2O5 | 400 W Metal halogen lamp | RT | 1 | 52 (CH3OH), 41 (H2) | 55.9 (CH3OH) | — | 207 |
| AgCl:Ag, AgBr:Ag |
300 W Xe, (λ ≥ 400 nm) | 0 | 1 | AgCl 37.7, AgBr 21.7 | 100 (CH3OH) | 1.3 0.97 | 235 |
| Bi2WO6 | 300 W Xe (λ ≥ 420 nm) | 0 | — | Hollow microspheres 16.3, bulk 0.64 | 100 (CH3OH) | — | 157 |
| g-C3N4 | 300 W Xe (λ ≥ 420 nm, 267 mW cm−2) | 20 | 1.013 | u-g-C3N4 6.28 (CH3OH), 4.51 (C2H5OH), 21.33 (O2), m-g-C3N4 3.64 (C2H5OH), 10.29 (O2), trace (CH3OH) | 19.6 (CH3OH), 14 (C2H5OH), 26.1 (C2H5OH) | 0.04 (CH3OH + C2H5OH), 0.018 (C2H5OH) | 120 |
| TiO2 | 500 W Xe (λ > 300 nm, 2.5 mW cm−2) | — | 1 | Anatase-brookite 0.59, flower-like rutile 0.32, rutile 0.30, anatase 0.19,P25 0.18 | 100 (CH3OH) | 0.0059, 0.0032, 0.0030, 0.0019, 0.0018 | 79 |
| FeTiO3/TiO2 | 500 W Xe (λ > 300 nm) | — | — | TiO2 0.175 (UV-visible), 10% FeTiO3/TiO2 0.338 (UV-visible), 20% FeTiO3/TiO2 0.462 (UV-visible), 50% FeTiO3/TiO2 0.298 (UV-visible), TiO2 0.141(visible), 10% FeTiO3/TiO2 0.319 (visible), 20% FeTiO3/TiO2 0.432 (visible), 50% FeTiO3/TiO2 0.352 (visible) | 100 (CH3OH) | — | 166 |
| ZIF-8/Zn2GeO4 | 500 W Xe | — | — | Zn2GeO4 0.14 (CH3OH), Zn2GeO4/ZIF-8 0.23 (CH3OH), 1 wt% Pt/Zn2GeO4/ZIF-8 0.27 (CH3OH) | 100 (CH3OH) | — | 95 |
| Graphene oxide (GO) | 300 W halogen lamp | 25 | — | GO-3 0.172, GO-1 0.110, GO-2 0.089 | 100 (CH3OH) | — | 208 |
| Zn–Cu–M(III) (M = Al, Ga) layered double hydroxides | 500 W Xe (UV-visible) | 32–40 | 0.023 | [Zn1.5Cu1.5Ga(OH)8]2+ 0.17 (CH3OH), 0.079 (CO),[Zn3Ga(OH)8]2+ 0.051 (CH3OH), 0.080 (CO), [Zn3Al(OH)8]2+ 0.039 (CH3OH), 0.62 (CO),[Zn1.5Cu1.5Al(OH)8]2+ 0.13 (CH3OH), 0.37 (CO) | 68.3 (CH3OH), 31.7 (CO), 38.9 (CH3OH), 61.1 (CO), 5.9 (CH3OH), 94.1 (CO), 26 (CH3OH), 74 (CO) | 0.00015 (CH3OH), 0.000019 (CO), 0.00028 (CH3OH), 0.000083 (CO), — | 134 |
| Ag/ALa4Ti4O15 (A = Ca, Ba and Sr) | 400 W Hg | RT | 1 | Ca 4.3 (HCOOH), 3.3 (CO), 18.7 (H2), 7.0 (O2), Ba 10 (HCOOH), 14.3 (CO), 33.3 (H2), 23.3 (O2), Sr 16.7 (HCOOH), 6.0 (CO), 9.0 (H2), 6.0 (O2) | HCOOH: 12.9, CO: 9.9, HCOOH: 82.3, CO: 17.7, HCOOH: 44.3, CO: 15.9 | — | 75 |
| BiVO4 | 300 W Xe (λ ≥ 400 nm, or without UV filter) | 0 | 1 | Monoclinic BiVO4 110 (visible), tetragonal BiVO4 12.5 (visible), monoclinic BiVO4 2150 (UV-visible), tetragonal BiVO4 250 (UV-visible) | 100 (C2H5OH) | — | 77 |
| Dye-sensitised + dehydrogenase TiO2 | Halogen lamp (λ > 420 nm 45 mW cm−2) | 20 | — | 287.2 | 100 (CO) | — | 176 |
| KTaO3 by SSR or solvothermal | 300 W Xe | — | — | SSR–KTaO3 0.436 (H2), 0.041 (CO), 0.173 (O2), hexane–KTaO3 0.088 (CO), 40.13 (H2), 0.4 (O2), ethanol–KTaO3 0.283 (CO), 84.7 (H2), 0.2 (O2) | 6.3 (CO), 0.2 (CO), 0.6 (CO) | — | 74 |
The optoelectronic properties of crystalline materials, and hence their photoactivity, are a strong function of the crystal structure, with different semiconductor polymorphs exhibiting disparate photocatalytic behaviour reflecting their surface atom arrangement and attendant electronic band structure. Although surface defects may serve as electron–hole recombination centres detrimental to the achievement of high quantum efficiencies, such sites can also promote visible light absorption and hence photocatalysis. Band gap engineering of earth abundant, large band gap semiconductor to facilitate visible light photocatalysis remains an important barrier to visible light absorption properties, although transition metal and p-block dopants provide a simple route to band-gap narrowing. Integrated dyes or small band gap semiconductors may also be employed as visible light sensitisors of conventional semiconductor photocatalysts, with the resulting hybrid, composite materials facilitating both UV and visible light capture and efficient charge carrier separation via preferential transfer of photoexcited electrons into one material and of holes into the other. In the latter regard, one- and two-dimensional semiconductor nanostructures, exhibiting high surface to volume ratios, can promote efficient transport of charge carriers to adsorbates during photocatalysis.
Integration of photoactive semiconductors with electrically conductive materials such as graphene offers new routes to maximising charge carrier lifetimes and consequent electron transfer to CO2. High surface area and porous materials increase the density of surface active sites available to chemically bind and activate CO2, its reactive intermediates, and water. Such nanostrutures can be created through the synthesis of semiconductors with intrinsic porosity on the micro-, meso- or macroscale, or via immobilisation (embedding) of photocatalysts onto (within) an inert, high area architecture. Three-dimensional hierarchical materials epitomise catalyst engineering, affording rapid molecular transport to/from active sites, enhanced light harvesting, and superior charge carrier transport and recombination characteristics. It seems extremely unlikely that any single semiconductor composition or structure can deliver the requisite optical, material and catalytic properties necessary to harness sunlight under ambient conditions for the rapid and selective photoreduction of CO2 into the key chemical targets of methane, methanol, formaldehyde or ethene, hence the majority of promising systems employ co-catalysts whose contributions depend strongly on their method of incorporation.
Noble metal co-catalysts in particular offer exciting possibilities for visible light absorption through plasmonic effects, and indeed have even proven efficient photocatalysts in their own right for the selective oxidation of CO, ethene and ammonia251 and reduction of nitroaromatics.252 Photoreactor design will play an increasingly important role in ensuring optimum light (and catalyst) utilisation for CO2 reduction, and enable quantitative benchmarking of different photocatalysts in terms of their absolute productivity, selectivity and quantum efficiency. Innovation is required to facilitate CO2 separation/recycle, on-stream gas/liquid product separation, and optical materials to avoid losses in transmission to the internal reactor volume.
Ultimately, the final selection and commercialisation of the most promising (i.e. highly active, selective and stable) photocatalysts will be dictated by process economics, since even the highest value oxygenate or olefin products directly derivable from CO2 will continue to face stiff competition with low cost fossil fuel sources for the foreseeable future. Indicative 2015 prices per metric ton are: methanol $420; formaldehyde $420; formic acid $500, and ethylene/propylene $800–1100. Overall process costs will be a strong function of the choice of chemical precursors, type and recyclability of solvents, (re)use of textural templates, mechanothermal processing, photocatalyst forming, and reactor operating conditions (temperature, pressure, separation and recycle). Many of the most promising photocatalysts described in this review utilise noble metal co-catalysts,40,51,94,123,161,213,220,234,249 and with common H2PtCl6 and HAuCl4 prices in excess of $10000 per kg, high turnover numbers per metal atom (>3000) would be essential to even recoup the precursor costs. An obvious target for synthetic improvement is thus maximising noble metal efficiency through e.g. lowering the size/dimensionality of associated nanostructures, or atomically dispersing noble metal atoms exclusively within the terminating layer of catalysts.253–256 Soft and hard templating methods are widely adopted in heterogeneous catalysis to introduce porosity, and hence enhance the surface density and accessibility of active sites.137 A number of photocatalysts for CO2 reduction employ such templates to similar effect, typically surfactants (e.g. TTAB,107 CTAB77,163,164 and P123 (ref. 78 and 112)) or polymers.210 While their costs are comparatively low ($150–350 per kg), in the majority of cases such surfactants are removed through combustion and hence are not recoverable, and even solvent-extraction protocols rarely recover sufficiently pure and chemically unaltered templates amenable to re-use, though ultrasonic template removal shows promise as an energy and atom efficient solution.257 The majority of synthetic strategies for nanostructure photocatalysts employ solvothermal processing, and hence comparatively low (<200 °C) temperatures (in the absence of a template calcination step) offering little scope for cost savings via energy input reduction. As with all syntheses, Green Chemistry principles provide the best general guidelines to reduce the number of reaction steps and minimise associated waste generation and the use of auxiliaries,258 in concert with lower production costs.
Acknowledgements
We thank the EPSRC (EP/G007594/4, EP/K014714/1 and EP/K014706/1) for financial support and award of a Leadership Fellowship (AFL).Reference
- J. Barber and P. D. Tran, J. R. Soc., Interface, 2013, 10, 20120984 CrossRef PubMed
.
-
Solar Fuels And Artificial Photosynthesis: Science And Innovation To Change Our Future Energy Options, Royal Society of Chemistry, 2012, http://www.rsc.org/campaigning-outreach/global-challenges/energy/ (accessed June 1st, 2015) Search PubMed
- D. Luthi, M. Le Floch, B. Bereiter, T. Blunier, J.-M. Barnola, U. Siegenthaler, D. Raynaud, J. Jouzel, H. Fischer, K. Kawamura and T. F. Stocker, Nature, 2008, 453, 379–382 CrossRef PubMed
- C. John, N. Dana, A. G. Sarah, R. Mark, W. Bärbel, P. Rob, W. Robert, J. Peter and S. Andrew, Environ. Res. Lett., 2013, 8, 024024 CrossRef
- M. Z. Jacobson, Energy Environ. Sci., 2009, 2, 148–173 CAS
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. deGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. G. Nocera, D. M. Tiede, H. Dau, W. Hillier, L. Wang and R. Amal, Energy Environ. Sci., 2013, 6, 1074–1076 Search PubMed
-
United Nations, The Future We Want: Outcome, 2012, https://sustainabledevelopment.un.org/futurewewant.html (accessed June 1st, 2015) Search PubMed
- M. T. Lin, A. Occhialini, P. J. Andralojc, J. Devonshire, K. M. Hines, M. A. J. Parry and M. R. Hanson, Plant J., 2014, 79, 1–12 CrossRef CAS PubMed
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS PubMed
- G. F. Moore and G. W. Brudvig, Annu. Rev. Condens. Matter Phys., 2011, 2, 303–327 CrossRef CAS
- R. Lomoth, A. Magnuson, M. Sjödin, P. Huang, S. Styring and L. Hammarström, Photosynth. Res., 2006, 87, 25–40 CrossRef CAS PubMed
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS PubMed
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC
- C. Costentin, M. Robert and J.-M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC
- N. Z. Muradov and T. N. Veziroglu, Int. J. Hydrogen Energy, 2005, 30, 225–237 CrossRef CAS PubMed
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed
- I. Ganesh, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS PubMed
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC
- C. S. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS PubMed
- M. Taherimehr and P. P. Pescarmona, J. Appl. Polym. Sci., 2014, 131, 41141 CrossRef PubMed
- A. I. Cooper, J. Mater. Chem., 2000, 10, 207–234 RSC
- http://www.petrochemistry.eu/about-petrochemistry/facts-and-figures.html (accessed June 2nd, 2015).
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS PubMed
- Y. Xing, W. Xie, W. Fang, Y. Guo and R. Lin, Energy Fuels, 2009, 23, 4021–4024 CrossRef CAS
- G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425–3437 RSC
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS PubMed
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS PubMed
- B. R. Eggins, J. T. S. Irvine, E. P. Murphy and J. Grimshaw, J. Chem. Soc., Chem. Commun., 1988, 1123–1124 RSC
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed
- V. L. Kuznetsov and P. P. Edwards, ChemSusChem, 2010, 3, 44–58 CrossRef CAS PubMed
- A. Kubacka, M. Fernandez-Garcia and G. Colon, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745–758 CAS
- O. K. Varghese, M. Paulose, T. J. LaTempa and C. A. Grimes, Nano Lett., 2009, 9, 731–737 CrossRef CAS PubMed
- M. Tahir and N. S. Amin, Energy Convers. Manage., 2013, 76, 194–214 CrossRef CAS PubMed
- G. Liu, N. Hoivik, K. Wang and H. Jakobsen, Sol. Energy Mater. Sol. Cells, 2012, 105, 53–68 CrossRef CAS PubMed
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217–9233 CAS
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC
- A. D. Handoko, K. Li and J. Tang, Curr. Opin. Chem. Eng., 2013, 2, 200–206 CrossRef PubMed
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef CAS PubMed
- W.-N. Wang, W.-J. An, B. Ramalingam, S. Mukherjee, D. M. Niedzwiedzki, S. Gangopadhyay and P. Biswas, J. Am. Chem. Soc., 2012, 134, 11276–11281 CrossRef CAS PubMed
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- S. Kuwabata, K. Nishida, R. Tsuda, H. Inoue and H. Yoneyama, J. Electrochem. Soc., 1994, 141, 1498–1503 CrossRef CAS PubMed
- E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342–6344 CrossRef CAS PubMed
- O. Ola and M. Mercedes Maroto-Valer, Catal. Sci. Technol., 2014, 4, 1631–1637 CAS
- L. Zhang, H. Cheng, R. Zong and Y. Zhu, J. Phys. Chem. C, 2009, 113, 2368–2374 Search PubMed
- P. D. Tran, S. K. Batabyal, S. S. Pramana, J. Barber, L. H. Wong and S. C. J. Loo, Nanoscale, 2012, 4, 3875–3878 RSC
- R. J. Braham and A. T. Harris, Ind. Eng. Chem. Res., 2009, 48, 8890–8905 CrossRef CAS
- Z. Xing, X. Zong, J. Pan and L. Wang, Chem. Eng. Sci., 2013, 104, 125–146 CrossRef CAS PubMed
- I. Rossetti, A. Villa, C. Pirola, L. Prati and G. Ramis, RSC Adv., 2014, 4, 28883–28885 RSC
- M. D. Doherty, D. C. Grills, J. T. Muckerman, D. E. Polyansky and E. Fujita, Coord. Chem. Rev., 2010, 254, 2472–2482 CrossRef CAS PubMed
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed
- H. Huang, S. Wang, Y. Zhang and P. K. Chu, RSC Adv., 2014, 4, 41219–41227 RSC
- G. Xi, S. Ouyang, P. Li, J. Ye, Q. Ma, N. Su, H. Bai and C. Wang, Angew. Chem., 2012, 124, 2445–2449 CrossRef PubMed
- A. M. Smith and S. Nie, Acc. Chem. Res., 2009, 43, 190–200 CrossRef PubMed
- Y. Liao, S.-W. Cao, Y. Yuan, Q. Gu, Z. Zhang and C. Xue, Chem.–Eur. J., 2014, 20, 10220–10222 CrossRef CAS PubMed
- T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594–2600 CAS
- G. Liu, J. C. Yu, G. Q. Lu and H.-M. Cheng, Chem. Commun., 2011, 47, 6763–6783 RSC
- W. Fan, Q. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2013, 15, 2632–2649 RSC
- Y. Izumi, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef CAS PubMed
- C.-C. Yang, Y.-H. Yu, B. van der Linden, J. C. S. Wu and G. Mul, J. Am. Chem. Soc., 2010, 132, 8398–8406 CrossRef CAS PubMed
- K. Sayama and H. Arakawa, J. Phys. Chem., 1993, 97, 531–533 CrossRef CAS
- K. Li, A. D. Handoko, M. Khraisheh and J. Tang, Nanoscale, 2014, 6, 9767–9773 RSC
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS PubMed
- A. Selloni, Nat. Mater., 2008, 7, 613–615 CrossRef CAS PubMed
- Y. Liu, B. Huang, Y. Dai, X. Zhang, X. Qin, M. Jiang and M.-H. Whangbo, Catal. Commun., 2009, 11, 210–213 CrossRef CAS PubMed
- P. Li, S. Ouyang, Y. Zhang, T. Kako and J. Ye, J. Mater. Chem. A, 2013, 1, 1185–1191 CAS
- Q. D. Truong, T. H. Le, J. Y. Liu, C. C. Chung and Y. C. Ling, Appl. Catal., A, 2012, 437, 28–35 CrossRef PubMed
- L. Liu, H. Zhao, J. M. Andino and Y. Li, ACS Catal., 2012, 2, 1817–1828 CrossRef CAS
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155409 CrossRef
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 119901 CrossRef
- X.-Q. Gong and A. Selloni, J. Phys. Chem. B, 2005, 109, 19560–19562 CrossRef CAS PubMed
- A. Vittadini, A. Selloni, F. P. Rotzinger and M. Grätzel, Phys. Rev. Lett., 1998, 81, 2954–2957 CrossRef CAS
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed
- G. Liu, H. G. Yang, X. Wang, L. Cheng, J. Pan, G. Q. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2009, 131, 12868–12869 CrossRef CAS PubMed
- J. Yu, L. Qi and M. Jaroniec, J. Phys. Chem. C, 2010, 114, 13118–13125 CAS
- B. Wu, C. Guo, N. Zheng, Z. Xie and G. D. Stucky, J. Am. Chem. Soc., 2008, 130, 17563–17567 CrossRef CAS PubMed
- W.-S. Wang, D.-H. Wang, W.-G. Qu, L.-Q. Lu and A.-W. Xu, J. Phys. Chem. C, 2012, 116, 19893–19901 CAS
- D. Barpuzary, Z. Khan, N. Vinothkumar, M. De and M. Qureshi, J. Phys. Chem. C, 2011, 116, 150–156 Search PubMed
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Fuels, 2008, 22, 2611–2618 CrossRef CAS
- W. Jiao, L. Wang, G. Liu, G. Q. Lu and H.-M. Cheng, ACS Catal., 2012, 2, 1854–1859 CrossRef CAS
- Q. Liu, Y. Zhou, J. Kou, X. Chen, Z. Tian, J. Gao, S. Yan and Z. Zou, J. Am. Chem. Soc., 2010, 132, 14385–14387 CrossRef CAS PubMed
- Q. Liu, Z.-X. Low, L. Li, A. Razmjou, K. Wang, J. Yao and H. Wang, J. Mater. Chem. A, 2013, 1, 11563–11569 CAS
- Q. Liu, Y. Zhou, Y. Ma and Z. Zou, RSC Adv., 2012, 2, 3247–3250 RSC
- S. Yan, L. Wan, Z. Li and Z. Zou, Chem. Commun., 2011, 47, 5632–5634 RSC
- Z. Liu, X. Zhang, S. Nishimoto, M. Jin, D. A. Tryk, T. Murakami and A. Fujishima, J. Phys. Chem. C, 2007, 112, 253–259 Search PubMed
- M. Ye, J. Gong, Y. Lai, C. Lin and Z. Lin, J. Am. Chem. Soc., 2012, 134, 15720–15723 CrossRef CAS PubMed
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2005, 5, 191–195 CrossRef CAS PubMed
- S. Liang, J. He, Z. Sun, Q. Liu, Y. Jiang, H. Cheng, B. He, Z. Xie and S. Wei, J. Phys. Chem. C, 2012, 116, 9049–9053 CAS
- K. Zhu, N. R. Neale, A. Miedaner and A. J. Frank, Nano Lett., 2006, 7, 69–74 CrossRef PubMed
- S. Rani, S. C. Roy, M. Paulose, O. K. Varghese, G. K. Mor, S. Kim, S. Yoriya, T. J. LaTempa and C. A. Grimes, Phys. Chem. Chem. Phys., 2010, 12, 2780–2800 RSC
- X. Chen, Y. Zhou, Q. Liu, Z. Li, J. Liu and Z. Zou, ACS Appl. Mater. Interfaces, 2012, 4, 3372–3377 CAS
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, Chem. Commun., 2015, 51, 3430–3433 RSC
- N. Zhang, S. Ouyang, T. Kako and J. Ye, Chem. Commun., 2012, 48, 1269–1271 RSC
- H.-A. Park, J. H. Choi, K. M. Choi, D. K. Lee and J. K. Kang, J. Mater. Chem., 2012, 22, 5304–5307 RSC
- W.-N. Wang, J. Park and P. Biswas, Catal. Sci. Technol., 2011, 1, 593–600 CAS
- N. Bao, L. Shen, T. Takata and K. Domen, Chem. Mater., 2007, 20, 110–117 CrossRef
- J. Guo, S. Ouyang, T. Kako and J. Ye, Appl. Surf. Sci., 2013, 280, 418–423 CrossRef CAS PubMed
- S. Yan, H. Yu, N. Wang, Z. Li and Z. Zou, Chem. Commun., 2012, 48, 1048–1050 RSC
- J. Núñez, V. A. de la Peña O'Shea, P. Jana, J. M. Coronado and D. P. Serrano, Catal. Today, 2013, 209, 21–27 CrossRef PubMed
- S. C. Yan, S. X. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye, Y. Zhou and Z. G. Zou, Angew. Chem., Int. Ed., 2010, 49, 6400–6404 CrossRef CAS PubMed
- N. Zhang, S. Ouyang, P. Li, Y. Zhang, G. Xi, T. Kako and J. Ye, Chem. Commun., 2011, 47, 2041–2043 RSC
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed
- C. Pan, J. Xu, Y. Wang, D. Li and Y. Zhu, Adv. Funct. Mater., 2012, 22, 1518–1524 CrossRef CAS PubMed
- L. Ge, F. Zuo, J. Liu, Q. Ma, C. Wang, D. Sun, L. Bartels and P. Feng, J. Phys. Chem. C, 2012, 116, 13708–13714 CAS
- G. Dong and L. Zhang, J. Mater. Chem., 2012, 22, 1160–1166 RSC
- J. Mao, T. Peng, X. Zhang, K. Li, L. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253–1260 CAS
- Y. Li, W.-N. Wang, Z. Zhan, M.-H. Woo, C.-Y. Wu and P. Biswas, Appl. Catal., B, 2010, 100, 386–392 CrossRef CAS PubMed
- H.-C. Yang, H.-Y. Lin, Y.-S. Chien, J. Wu and H.-H. Wu, Catal. Lett., 2009, 131, 381–387 CrossRef CAS
- X. Li, Z. Zhuang, W. Li and H. Pan, Appl. Catal., A, 2012, 429–430, 31–38 CrossRef CAS PubMed
- T. W. Kim, S. J. Hwang, S. H. Jhung, J. S. Chang, H. Park, W. Choi and J. H. Choy, Adv. Mater., 2008, 20, 539–542 CrossRef CAS PubMed
- T. W. Kim, S. G. Hur, S. J. Hwang, H. Park, W. Choi and J. H. Choy, Adv. Funct. Mater., 2007, 17, 307–314 CrossRef CAS PubMed
- J. Wu, Y. Cheng, J. Lin, Y. Huang, M. Huang and S. Hao, J. Phys. Chem. C, 2007, 111, 3624–3628 CAS
- X. Li, N. Kikugawa and J. Ye, Adv. Mater., 2008, 20, 3816–3819 CrossRef CAS PubMed
- X. Li, H. Pan, W. Li and Z. Zhuang, Appl. Catal., A, 2012, 413–414, 103–108 CAS
- X. Li, W. Li, Z. Zhuang, Y. Zhong, Q. Li and L. Wang, J. Phys. Chem. C, 2012, 116, 16047–16053 CAS
- J. Yoshimura, Y. Ebina, J. Kondo, K. Domen and A. Tanaka, J. Phys. Chem., 1993, 97, 1970–1973 CrossRef CAS
- J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75–87 CrossRef CAS PubMed
- J. C. Lavalley, Catal. Today, 1996, 27, 377–401 CrossRef CAS
- M. G. Plaza, C. Pevida, A. Arenillas, F. Rubiera and J. J. Pis, Fuel, 2007, 86, 2204–2212 CrossRef CAS PubMed
- N. Ahmed, Y. Shibata, T. Taniguchi and Y. Izumi, J. Catal., 2011, 279, 123–135 CrossRef CAS PubMed
- F. Sastre, A. Corma and H. Garcia, J. Am. Chem. Soc., 2012, 134, 14137–14141 CrossRef CAS PubMed
- N. Ahmed, M. Morikawa and Y. Izumi, Catal. Today, 2012, 185, 263–269 CrossRef CAS PubMed
- C. M. A. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876–3893 RSC
- J. Yu, J. Fan and L. Zhao, Electrochim. Acta, 2010, 55, 597–602 CrossRef CAS PubMed
- P. L. Richardson, M. L. N. Perdigoto, W. Wang and R. J. G. Lopes, Appl. Catal., B, 2013, 132–133, 408–415 CrossRef CAS PubMed
- H. Bai, Z. Liu and D. D. Sun, Int. J. Hydrogen Energy, 2012, 37, 13998–14008 CrossRef CAS PubMed
- F. Dong, Y. Sun, M. Fu, W.-K. Ho, S. C. Lee and Z. Wu, Langmuir, 2011, 28, 766–773 CrossRef PubMed
- Y. J. Hwang, C. H. Wu, C. Hahn, H. E. Jeong and P. Yang, Nano Lett., 2012, 12, 1678–1682 CrossRef CAS PubMed
- S. H. Ko, D. Lee, H. W. Kang, K. H. Nam, J. Y. Yeo, S. J. Hong, C. P. Grigoropoulos and H. J. Sung, Nano Lett., 2011, 11, 666–671 CrossRef CAS PubMed
- M.-S. Gui and W.-D. Zhang, Nanotechnology, 2011, 22, 265601 CrossRef PubMed
- J. Hou, R. Cao, Z. Wang, S. Jiao and H. Zhu, J. Hazard. Mater., 2012, 217–218, 177–186 CrossRef CAS PubMed
- J. Duan, W. Shi, L. Xu, G. Mou, Q. Xin and J. Guan, Chem. Commun., 2012, 48, 7301–7303 RSC
- J. Hou, Y. Qu, D. Krsmanovic, C. Ducati, D. Eder and R. V. Kumar, J. Mater. Chem., 2010, 20, 2418–2423 RSC
- J. Hou, Z. Wang, S. Jiao and H. Zhu, J. Hazard. Mater., 2011, 192, 1772–1779 CrossRef CAS PubMed
- Z. Wang, J. Hou, C. Yang, S. Jiao, K. Huang and H. Zhu, Phys. Chem. Chem. Phys., 2013, 15, 3249–3255 RSC
- L. Zhou, W. Wang, H. Xu, S. Sun and M. Shang, Chem.–Eur. J., 2009, 15, 1776–1782 CrossRef CAS PubMed
- Z. Khan, M. Khannam, N. Vinothkumar, M. De and M. Qureshi, J. Mater. Chem., 2012, 22, 12090–12095 RSC
- G. Xi, B. Yue, J. Cao and J. Ye, Chem.–Eur. J., 2011, 17, 5145–5154 CrossRef CAS PubMed
- D. Chen and J. Ye, Adv. Funct. Mater., 2008, 18, 1922–1928 CrossRef CAS PubMed
- J. Yu, W. Liu and H. Yu, Cryst. Growth Des., 2008, 8, 930–934 CAS
- Q. Liu, Y. Zhou, Z. Tian, X. Chen, J. Gao and Z. Zou, J. Mater. Chem., 2012, 22, 2033–2038 RSC
- Z. Li, Y. Zhou, J. Zhang, W. Tu, Q. Liu, T. Yu and Z. Zou, Cryst. Growth Des., 2012, 12, 1476–1481 CAS
- H. Cheng, B. Huang, Y. Liu, Z. Wang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2012, 48, 9729–9731 RSC
- H. Zhou, J. Guo, P. Li, T. Fan, Z. Di and J. Ye, Sci. Rep., 2013, 3, 1667 Search PubMed
- L. Yuliati, T. Hattori, H. Itoh and H. Yoshida, J. Catal., 2008, 257, 396–402 CrossRef CAS PubMed
- N. Serpone, P. Maruthamuthu, P. Pichat, E. Pelizzetti and H. Hidaka, J. Photochem. Photobiol., A, 1995, 85, 247–255 CrossRef CAS
- H. Park, W. Choi and M. R. Hoffmann, J. Mater. Chem., 2008, 18, 2379–2385 RSC
- C. Wang, R. L. Thompson, J. Baltrus and C. Matranga, J. Phys. Chem. Lett., 2009, 1, 48–53 CrossRef
- M. Abou Asi, C. He, M. Su, D. Xia, L. Lin, H. Deng, Y. Xiong, R. Qiu and X.-z. Li, Catal. Today, 2011, 175, 256–263 CrossRef CAS PubMed
- S. Qin, F. Xin, Y. Liu, X. Yin and W. Ma, J. Colloid Interface Sci., 2011, 356, 257–261 CrossRef CAS PubMed
- X. Li, H. Liu, D. Luo, J. Li, Y. Huang, H. Li, Y. Fang, Y. Xu and L. Zhu, Chem. Eng. J., 2012, 180, 151–158 CrossRef CAS PubMed
- Q. D. Truong, J.-Y. Liu, C.-C. Chung and Y.-C. Ling, Catal. Commun., 2012, 19, 85–89 CrossRef CAS PubMed
- G. Ghadimkhani, N. R. de Tacconi, W. Chanmanee, C. Janaky and K. Rajeshwar, Chem. Commun., 2013, 49, 1297–1299 RSC
- S.-I. In, D. D. Vaughn and R. E. Schaak, Angew. Chem., Int. Ed., 2012, 51, 3915–3918 CrossRef CAS PubMed
- W. Wang, M. Dahl and Y. Yin, Chem. Mater., 2012, 25, 1179–1189 CrossRef
- W. Kim, T. Seok and W. Choi, Energy Environ. Sci., 2012, 5, 6066–6070 CAS
- W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS PubMed
- R. Abe, K. Shinmei, K. Hara and B. Ohtani, Chem. Commun., 2009, 3577–3579 RSC
- W. Kim, T. Tachikawa, T. Majima and W. Choi, J. Phys. Chem. C, 2009, 113, 10603–10609 CAS
- Y.-J. Yuan, Z.-T. Yu, J.-Y. Zhang and Z.-G. Zou, Dalton Trans., 2012, 41, 9594–9597 RSC
- T. W. Woolerton, S. Sheard, E. Pierce, S. W. Ragsdale and F. A. Armstrong, Energy Environ. Sci., 2011, 4, 2393–2399 CAS
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS PubMed
- A. Parkin, J. Seravalli, K. A. Vincent, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2007, 129, 10328–10329 CrossRef CAS PubMed
- P. Trogadas, T. F. Fuller and P. Strasser, Carbon, 2014, 75, 5–42 CrossRef CAS PubMed
- E. Yli-Rantala, A. Pasanen, P. Kauranen, V. Ruiz, M. Borghei, E. Kauppinen, A. Oyarce, G. Lindbergh, C. Lagergren, M. Darab, S. Sunde, M. Thomassen, S. Ma-Andersen and E. Skou, Fuel Cells, 2011, 11, 715–725 CrossRef CAS PubMed
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, M. Karakassides and E. P. Giannelis, Small, 2008, 4, 455–458 CrossRef CAS PubMed
- H. Peng and J. Travas-Sejdic, Chem. Mater., 2009, 21, 5563–5565 CrossRef CAS
- H. Jiang, F. Chen, M. G. Lagally and F. S. Denes, Langmuir, 2009, 26, 1991–1995 CrossRef PubMed
- L. Cao, S. Sahu, P. Anilkumar, C. E. Bunker, J. Xu, K. A. S. Fernando, P. Wang, E. A. Guliants, K. N. Tackett and Y.-P. Sun, J. Am. Chem. Soc., 2011, 133, 4754–4757 CrossRef CAS PubMed
- W.-J. Ong, M. M. Gui, S.-P. Chai and A. R. Mohamed, RSC Adv., 2013, 3, 4505–4509 RSC
- L. Jia, D.-H. Wang, Y.-X. Huang, A.-W. Xu and H.-Q. Yu, J. Phys. Chem. C, 2011, 115, 11466–11473 CAS
- A. K. Agegnehu, C.-J. Pan, J. Rick, J.-F. Lee, W.-N. Su and B.-J. Hwang, J. Mater. Chem., 2012, 22, 13849–13854 RSC
- P. Gao, J. Liu, S. Lee, T. Zhang and D. D. Sun, J. Mater. Chem., 2012, 22, 2292–2298 RSC
- Z. Khan, T. R. Chetia, A. K. Vardhaman, D. Barpuzary, C. V. Sastri and M. Qureshi, RSC Adv., 2012, 2, 12122–12128 RSC
- J. Hou, Z. Wang, W. Kan, S. Jiao, H. Zhu and R. V. Kumar, J. Mater. Chem., 2012, 22, 7291–7299 RSC
- F. Pei, Y. Liu, S. Xu, J. Lü, C. Wang and S. Cao, Int. J. Hydrogen Energy, 2013, 38, 2670–2677 CrossRef CAS PubMed
- T. Peng, K. Li, P. Zeng, Q. Zhang and X. Zhang, J. Phys. Chem. C, 2012, 116, 22720–22726 CAS
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed
- H.-i. Kim, G.-h. Moon, D. Monllor-Satoca, Y. Park and W. Choi, J. Phys. Chem. C, 2011, 116, 1535–1543 Search PubMed
- N. Yang, J. Zhai, D. Wang, Y. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS PubMed
- I. V. Lightcap and P. V. Kamat, J. Am. Chem. Soc., 2012, 134, 7109–7116 CrossRef CAS PubMed
- P. Wang, Y. Zhai, D. Wang and S. Dong, Nanoscale, 2011, 3, 1640–1645 RSC
- Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2011, 5, 7426–7435 CrossRef CAS PubMed
- X. Pan, Y. Zhao, S. Liu, C. L. Korzeniewski, S. Wang and Z. Fan, ACS Appl. Mater. Interfaces, 2012, 4, 3944–3950 CAS
- J. Du, X. Lai, N. Yang, J. Zhai, D. Kisailus, F. Su, D. Wang and L. Jiang, ACS Nano, 2010, 5, 590–596 CrossRef PubMed
- J. Zhang, Z. Zhu, Y. Tang and X. Feng, J. Mater. Chem., 2013, 1, 3752–3756 RSC
- A. Peigney, C. Laurent, E. Flahaut, R. R. Bacsa and A. Rousset, Carbon, 2001, 39, 507–514 CrossRef CAS
- A. Ghosh, K. S. Subrahmanyam, K. S. Krishna, S. Datta, A. Govindaraj, S. K. Pati and C. N. R. Rao, J. Phys. Chem. C, 2008, 112, 15704–15707 CAS
- B. Huang, Z. Li, Z. Liu, G. Zhou, S. Hao, J. Wu, B.-L. Gu and W. Duan, J. Phys. Chem. C, 2008, 112, 13442–13446 CAS
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2012, 6, 9777–9789 CrossRef CAS PubMed
- Y. T. Liang, B. K. Vijayan, K. A. Gray and M. C. Hersam, Nano Lett., 2011, 11, 2865–2870 CrossRef CAS PubMed
- X.-J. Lv, W.-F. Fu, C.-Y. Hu, Y. Chen and W.-B. Zhou, RSC Adv., 2013, 3, 1753–1757 RSC
- H.-C. Hsu, I. Shown, H.-Y. Wei, Y.-C. Chang, H.-Y. Du, Y.-G. Lin, C.-A. Tseng, C.-H. Wang, L.-C. Chen, Y.-C. Lin and K.-H. Chen, Nanoscale, 2013, 5, 262–268 RSC
- Y. T. Liang, B. K. Vijayan, O. Lyandres, K. A. Gray and M. C. Hersam, J. Phys. Chem. Lett., 2012, 3, 1760–1765 CrossRef CAS
- W. Tu, Y. Zhou, Q. Liu, Z. Tian, J. Gao, X. Chen, H. Zhang, J. Liu and Z. Zou, Adv. Funct. Mater., 2012, 22, 1215–1221 CrossRef CAS PubMed
- P. Pathak, S. Gupta, K. Grosulak, H. Imahori and V. Subramanian, J. Phys. Chem. C, 2015, 119, 7543–7553 CAS
- B. Mukherjee, S. Gupta, A. Peterson, H. Imahori, A. Manivannan and V. Subramanian, Chem.–Eur. J., 2014, 20, 10456–10465 CrossRef CAS PubMed
- A. Iwase, H. Kato and A. Kudo, Catal. Lett., 2006, 108, 7–10 CrossRef CAS PubMed
- M. Liu, W. You, Z. Lei, G. Zhou, J. Yang, G. Wu, G. Ma, G. Luan, T. Takata, M. Hara, K. Domen and C. Li, Chem. Commun., 2004, 2192–2193 RSC
- Y. Mizukoshi, K. Sato, T. J. Konno and N. Masahashi, Appl. Catal., B, 2010, 94, 248–253 CrossRef CAS PubMed
- M. Hara, J. Nunoshige, T. Takata, J. N. Kondo and K. Domen, Chem. Commun., 2003, 3000–3001 RSC
- V. Subramanian, E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2001, 105, 11439–11446 CrossRef CAS
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed
- D. Kong, J. Z. Y. Tan, F. Yang, J. Zeng and X. Zhang, Appl. Surf. Sci., 2013, 277, 105–110 CrossRef CAS PubMed
- X. Feng, J. D. Sloppy, T. J. LaTempa, M. Paulose, S. Komarneni, N. Bao and C. A. Grimes, J. Mater. Chem., 2011, 21, 13429–13433 RSC
- Z. W. Seh, S. Liu, M. Low, S.-Y. Zhang, Z. Liu, A. Mlayah and M.-Y. Han, Adv. Mater., 2012, 24, 2310–2314 CrossRef CAS PubMed
- Q. Zhang, D. Q. Lima, I. Lee, F. Zaera, M. Chi and Y. Yin, Angew. Chem., 2011, 123, 7226–7230 CrossRef PubMed
- T. Takahashi, A. Kudo, S. Kuwabata, A. Ishikawa, H. Ishihara, Y. Tsuboi and T. Torimoto, J. Phys. Chem. C, 2012, 117, 2511–2520 Search PubMed
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676–1680 CrossRef CAS PubMed
- X. Zhou, G. Liu, J. Yu and W. Fan, J. Mater. Chem., 2012, 22, 21337–21354 RSC
- W. A. Murray and W. L. Barnes, Adv. Mater., 2007, 19, 3771–3782 CrossRef CAS PubMed
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed
- H. Zhu, X. Chen, Z. Zheng, X. Ke, E. Jaatinen, J. Zhao, C. Guo, T. Xie and D. Wang, Chem. Commun., 2009, 7524–7526 RSC
-
X. Zhang, PhD thesis, Queensland University of Technology, 2014, http://eprints.qut.edu.au/67714/ (accessed June 2nd, 2015)
- M. S. Shore, J. Wang, A. C. Johnston-Peck, A. L. Oldenburg and J. B. Tracy, Small, 2011, 7, 230–234 CrossRef CAS PubMed
- P. Wang, B. Huang, Y. Dai and M.-H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC
- M. Xiao, R. Jiang, F. Wang, C. Fang, J. Wang and J. C. Yu, J. Mater. Chem. A, 2013, 1, 5790–5805 CAS
- D. M. Bijith, J. Babu and K. G. Vinay, Nanotechnology, 2013, 24, 405402 CrossRef PubMed
- Z. Zhang, Z. Wang, S.-W. Cao and C. Xue, J. Phys. Chem. C, 2013, 117, 25939–25947 CAS
- C. An, J. Wang, W. Jiang, M. Zhang, X. Ming, S. Wang and Q. Zhang, Nanoscale, 2012, 4, 5646–5650 RSC
- J. Z. Y. Tan, Y. Fernandez, D. Liu, M. Maroto-Valer, J. Bian and X. Zhang, Chem. Phys. Lett., 2012, 531, 149–154 CrossRef CAS PubMed
- J.-J. Chen, J. C. S. Wu, P. C. Wu and D. P. Tsai, J. Phys. Chem. C, 2010, 115, 210–216 Search PubMed
- Q. Xiang, J. Yu and M. Jaroniec, Phys. Chem. Chem. Phys., 2011, 13, 4853–4861 RSC
- K. Iwashina and A. Kudo, J. Am. Chem. Soc., 2011, 133, 13272–13275 CrossRef CAS PubMed
- F. Dong, S. Guo, H. Wang, X. Li and Z. Wu, J. Phys. Chem. C, 2011, 115, 13285–13292 CAS
- S. Bingham and W. A. Daoud, J. Mater. Chem., 2011, 21, 2041–2050 RSC
- J. Zhang, Y. Wu, M. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715–726 CAS
- C.-W. Tsai, H. M. Chen, R.-S. Liu, K. Asakura and T.-S. Chan, J. Phys. Chem. C, 2011, 115, 10180–10186 CAS
- I. Tsuji, H. Kato and A. Kudo, Angew. Chem., Int. Ed., 2005, 44, 3565–3568 CrossRef CAS PubMed
- I. Tsuji, H. Kato, H. Kobayashi and A. Kudo, J. Am. Chem. Soc., 2004, 126, 13406–13413 CrossRef CAS PubMed
- K. Maeda and K. Domen, Chem. Mater., 2009, 22, 612–623 CrossRef
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed
- J.-Y. Liu, B. Garg and Y.-C. Ling, Green Chem., 2011, 13, 2029–2031 RSC
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 5776–5779 CrossRef CAS PubMed
- P. Li, H. Xu, L. Liu, T. Kako, N. Umezawa, H. Abe and J. Ye, J. Mater. Chem. A, 2014, 2, 5606–5609 CAS
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CAS
- H. Zhu, X. Ke, X. Yang, S. Sarina and H. Liu, Angew. Chem., Int. Ed., 2010, 49, 9657–9661 CrossRef CAS PubMed
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS PubMed
- J. Xing, J. F. Chen, Y. H. Li, W. T. Yuan, Y. Zhou, L. R. Zheng, H. F. Wang, P. Hu, Y. Wang, H. J. Zhao, Y. Wang and H. G. Yang, Chem.–Eur. J., 2014, 20, 2085 CrossRef CAS PubMed
- A. Bruix, Y. Lykhach, I. Matolínová, A. Neitzel, T. Skála, N. Tsud, M. Vorokhta, V. Stetsovych, K. Ševčíková, J. Mysliveček, R. Fiala, M. Václavů, K. C. Prince, S. Bruyère, V. Potin, F. Illas, V. Matolín, J. Libuda and K. M. Neyman, Angew. Chem., Int. Ed., 2014, 53, 10525–10530 CrossRef CAS PubMed
- G. Pei, X. Liu, A. Wang, A. F. Lee, M. A. Isaacs, L. Li, X. Pan, X. Yang, X. Wang, Z. Tai, K. Wilson and T. Zhang, ACS Catal., 2015, 5, 3717–3725 CrossRef CAS
- C. Pirez, K. Wilson and A. F. Lee, Green Chem., 2014, 16, 197–202 RSC
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC
| This journal is © The Royal Society of Chemistry 2015 |