
Self-assembly of bi-functional peptides on large-pore mesoporous silica nanoparticles for miRNA binding and delivery†
Jingxiong
Lu
a,
Hsin-Hui
Shen
*b,
Zhangxiong
Wu
c,
Bo
Wang
d,
Dongyuan
Zhao
ae and
Lizhong
He
*a
aDepartment of Chemical Engineering, Monash University, Wellington Road, Melbourne 3800, Australia. E-mail: lizhong.he@monash.edu
bDepartment of Microbiology, Monash University, Wellington Road, Melbourne 3800, Australia. E-mail: hsin-hui.shen@monash.edu
cCollege of Chemistry Chemical Engineering and Materials Science, Soochow University, Suzhou, Jiangsu 215123, P. R. China
dDepartment of Anatomy and Developmental Biology, Monash University, Wellington Road, Melbourne 3800, Australia
eDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, and Laboratory of Advanced Materials, Fudan University, Shanghai 200433, P. R. China
First published on 11th September 2015
Abstract
Bi-functional peptides were designed to have binding abilities for both silica nanoparticles and miRNAs. Non-covalent self-assembly of peptides on large pore mesoporous silica nanoparticles provides a delivery system that shows a high binding capacity for nucleic acids, strong transfection efficiency of miRNA and attractive down-regulation of protein expression.
The discovery of RNA interference (RNAi),1 a natural mechanism of gene regulation through post-transcriptional gene silencing by siRNA or miRNA, has opened numerous novel opportunities to treat a variety of protein related diseases, including cancer,2 infectious3 and autoimmune4 diseases. Recently, a first phase one trial RNAi therapy has been carried out, showing high safety and efficacy of this approach.5 Importantly, a sufficient dose of miRNA/siRNA molecules has to be delivered into cytoplasm of the target cells in order to trigger an effective gene silencing response. However, siRNAs/miRNAs cannot freely penetrate into cells due to their negative charges, and it remains a challenge to deliver a sufficient dose efficiently and conveniently.6 The recent report on the mechanism of siRNA delivery revealed that only 1–2% of the internalized siRNA actually escaped from the endocytic system7 while 70% of siRNA was exocytosed8 when delivered by one of the best delivery vehicles, lipid nanoparticles (LNPs). These studies highlighted the importance of the development of an efficient delivery system, which could enhance the endosomal escape of siRNA/miRNA after endocytosis. Significant efforts have been made to achieve such a goal.9–11
Mesoporous silica nanoparticles (MSNs)12 are attractive delivery vehicles for siRNA/miRNA due to their attractive biocompatibility, a tunable particle size and a large surface area comparing to other carriers, such as lipid nanoparticles,13 gold nanoparticles,14 and chitosan.15 Due to these advantages, there have been significant efforts to develop drug delivery systems using MSNs.16–21 In order to efficiently load miRNAs/siRNAs, surface functionalization of MSNs is required to make the silica surface positively charged. One of the most widely used functionalization methods for MSNs is amino-functionalization by amine-rich reagents, such as polyethylenimine (PEI),22 (3-aminopropyl)trimethoxysilane,22 or poly-lysine.23 However, such functionalization often requires multiple steps of chemical reactions which result in undesired surface chemistry that can be cytotoxic to cells.12
In this study, we propose a simple one-step method for the functionalization of large-pore MSNs (LP-MSNs) by non-covalent self-assembly of the designed bi-functional peptides (Scheme 1). The nontoxic bi-functional peptides can self-assemble on the silica surface while having the binding ability to siRNA/miRNA, avoiding chemical modification of silica particles. The nature of non-covalent binding also facilitates desorption of peptides and siRNA/miRNA from the MSNs for their release. The bi-functional binding peptides are designed to have two parts, one part (RGRRRRLSCRLL24 or KSLSRHDHIHHH25) having the silica binding ability and the other part (positively charged poly-lysine KKKKKKKK) having the nucleic acid binding ability. Perry et al. have suggested that the silica binding ability of the peptides is a collective contribution of ion pairing, hydrogen bonding, and other polar interactions.26 By design, these peptides have strong positive charges at neutral pH (pI > 10, see values in Table S1, ESI†), enabling efficient binding of negatively charged siRNA/miRNA under delivering conditions. As shown in Fig. S1 (ESI†), the self-assembly of bi-functional peptides on LP-MSNs was initiated by simply mixing LP-MSNs with the designed bi-functional peptides. The formed LP-MSN–peptide complexes can then bind miRNAs for their delivery into cells. The nature of non-covalent interactions between silica and the silica-binding moiety of the peptide offers the opportunity to control binding strength between silica and peptide, and existing technologies such as phage display and molecular dynamics simulation can be used to tune the peptide–silica interaction for optimized delivery and release.24,26
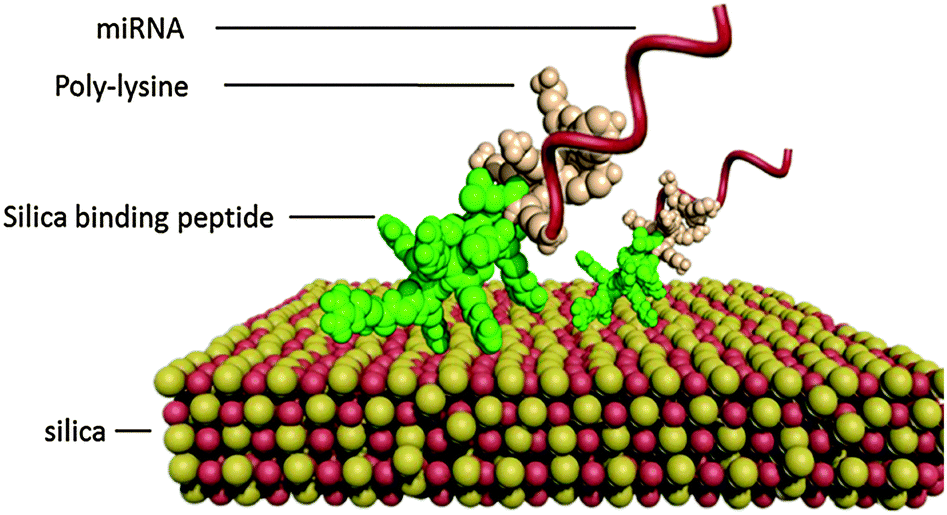 | ||
| Scheme 1 Schematic illustration of bi-functional peptides self-assemble on silica through the silica binding moiety and miRNA is bound by the poly-lysine moiety. | ||
The large-pore mesoporous silica nanoparticles (LP-MSNs) were synthesized to have the desired large pores and nanoparticle sizes.27 As shown in Fig. 1A, the LP-MSNs have a diameter of 200 ± 50 nm, and the large pores are visible in the TEM image (inset picture). BET results indicate that LP-MSN has a surface area of 219.5 ± 1.7 m2 g−1 and a pore volume of 0.72 cm3 g−1. The pore size distribution determined by the nitrogen adsorption curve (Fig. 1B) shows a pore size of ∼18.8 nm. This pore size is significantly larger than that of bi-functional peptide and miRNA. MiRNAs are short double-stranded RNAs of ∼22 bp, which give a length of 6.4 nm while bi-functional peptide is estimated to have a length scale of ca. 3–5 nm (see details of estimation in the ESI†). This large pore size is expected to enhance the binding of peptides and nucleic acids by providing not only large surface areas but also their easy access into the internal surfaces of LP-MSNs.
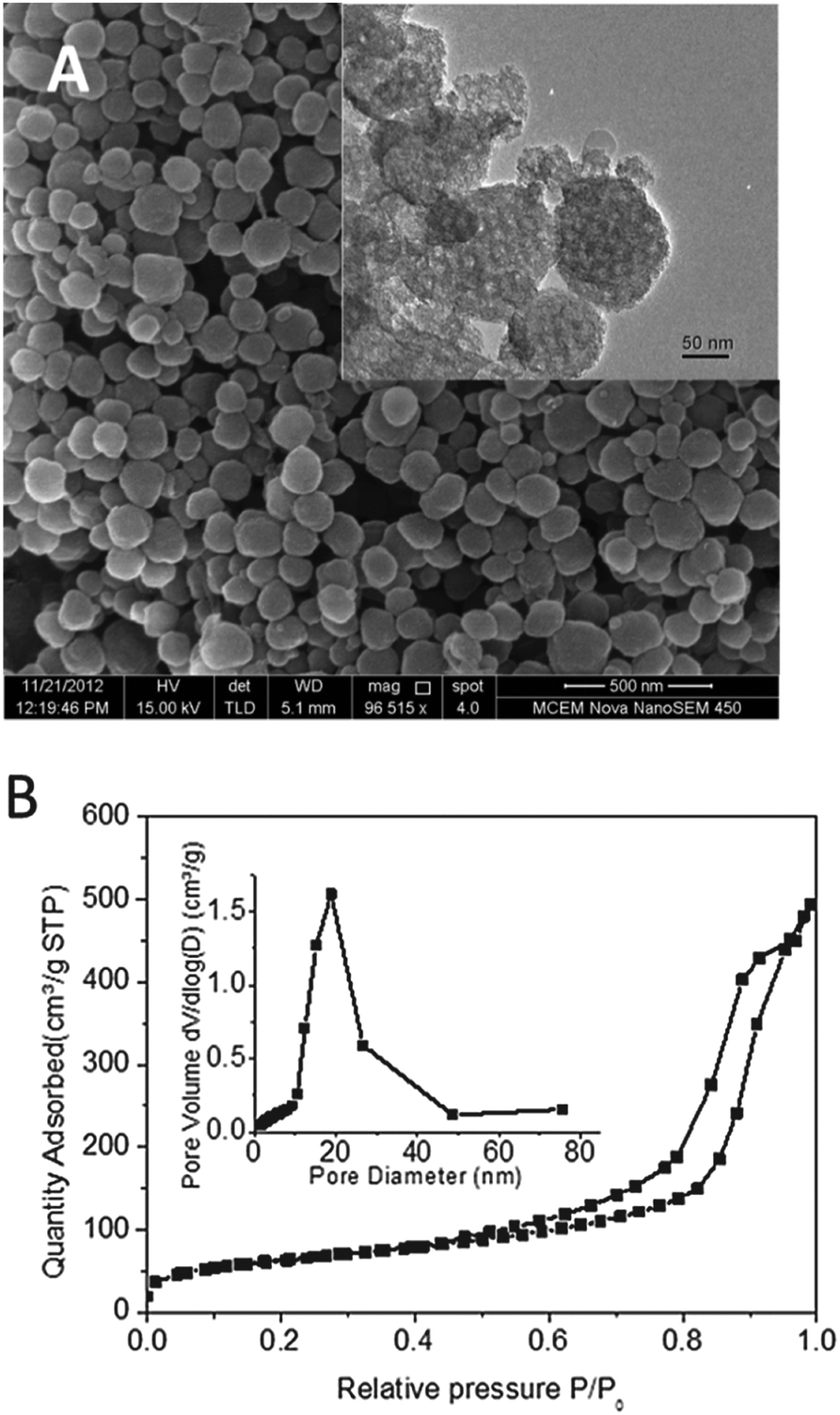 | ||
| Fig. 1 (A) SEM image and TEM image (inset) of the large-pore mesoporous silica nanoparticles (LP-MSNs). (B) N2 sorption isotherms and the inset BJH pore size distribution determined by nitrogen adsorption isotherm. | ||
Nucleic acid binding abilities of the LP-MSN–peptide complexes were examined by adsorption of CpG DNA 182628 (TCCATGACGTTCCTGACGTT) as a mimic of mature miRNA. As expected, both peptide concentration and DNA concentration influenced DNA binding. In Fig. 2A, the adsorption capacity of LP-MSNs was compared with non-porous solid silica nanoparticles (a diameter of 200 nm) and commercially available MSNs (Sigma, Australia, a particle size of around 500 nm and a pore diameter of 4 nm). The adsorptions of CpG DNA onto these three particles were measured at varied concentrations of peptide RGRRRRLSCRLLK8 while the initial concentration of CpG DNA was fixed at 0.1 mg mL−1. At low peptide concentrations, the increase of the peptide concentration enhances CpG DNA adsorption, confirming that the nucleic acid binding is facilitated by peptide (Fig. 2A). The DNA adsorption then reaches a plateau at increasing peptide concentration. The LP-MSN has a significant higher adsorption capacity for CpG DNA than the commercial MSN and the non-porous solid silica nanoparticles (2.5 and 8 times higher, respectively). In Fig. 2B, the adsorption isotherms of CpG DNA on the LP-MSN–peptide complexes formed by the self-assembly of three different peptides were compared. It shows that all three LP-MSN–peptide complexes have attractive CpG DNA binding capacities. While LP-MSN–RGRRRRLSCRLLK8 and LP-MSN–RGRRRRLSCRLL have similar binding capacities for DNA, the binding capacity of LP-MSN–KSLSRHDHIHHHK8 is slightly higher. In contrast, the LP-MSN alone without peptides cannot adsorb any CpG DNA, further confirming the importance of peptides in facilitating nucleic acid binding.
 | ||
| Fig. 2 CpG DNA adsorption onto silica nanoparticle–peptide complexes. (A) Effect of concentration of peptide RGRRRRLSCRLLK8 on adsorption of CpG DNA onto three types of silica nanoparticles. (B) Adsorption isotherms of CpG DNA onto peptide functionalized LP-MSNs. | ||
Importantly, the adsorption of CpG DNA onto LP-MSN–peptide also protects it from digestion by nuclease. The free and adsorbed CpG DNA was compared for its stability at concentrations of different DNase I. TBE-PAGE data (Fig. S2, ESI†) show that the majority of CpG DNA (1 μg) adsorbed into LP-MSN–RGRRRRLSCRLLK8 (50 μg) was still intact after digestion by 0.2 U DNase I for 1 hour at 37 °C. Under the same conditions, the free CpG DNA was fully degraded. The protection of nucleic acids from nuclease by LP-MSN–peptide complexes is beneficial for miRNA delivery because miRNAs are liable to nuclease in serum.
To evaluate the cellular uptake of miRNA facilitated by the three LP-MSN–peptide complexes, the delivery of a fluorescent dye Cy3-labelled miRNA into NRK cells was carried out. Among the three LP-MSN–peptide complexes, the LP-MSN–RGRRRRLSCRLLK8 complex (Fig. 3A) shows the strongest ability to efficiently deliver miRNA into NRK cells after 2 hours of incubation. In contrast, the other two LP-MSN–peptide complexes, LP-MSN–RGRRRRLSCRLL (Fig. 3B) and LP-MSN–KSLSRHDHIHHHK8 (Fig. 3C) didn't show obvious capability to efficiently deliver miRNA. The positive control, a lipid-based transfection reagent, Lipofectamine, also shows a significant delivery of miRNA (Fig. 3D), but it has the appearance of aggregation, indicating that the majority of miRNAs may still be in endosome after endocytosis.29 In contrast to Lipofectamine, miRNAs delivered by the LP-MSN–RGRRRRLSCRLLK8 complex (Fig. 3A) exhibit a scattered pattern inside of the NRK cell, suggesting that they may have escaped from endosome after 2 hours of uptake. Recent reports have suggested that the escape of siRNA from endosome should be enhanced to improve delivery yield because siRNA trapped inside endosome would not target cytoplasm.7,8,30 Optimized association–disassociation between carriers and nucleic acids can serve as an efficient way to enhance the endosomal escape of siRNA/miRNA.
 | ||
| Fig. 3 Cy3-labelled miRNA delivered by different vehicles into NRK cells. Red: Cy3-labelled miRNA; Blue: DAPI stained nucleus; Green: FITC-Phalloidin labelled F-actin. (A) LP-MSN–RGRRRRLSCRLLK8 complex; (B) LP-MSN–RGRRRRLSCRLL complex; (C) LP-MSN–KSLSRHDHIHHHK8 complex; (D) commercial Lipofectamine; (E) unmodified LP-MSN; (F) miRNA only. Scale bar, 10 μm. | ||
The designed sequences of the binding peptides are crucial to secure the successful delivery of miRNA into cells. First, the nucleic acid binding part (poly-lysine K8) has an important role in miRNA delivery. The LP-MSN–RGRRRRLSCRLL peptide that lacks poly-lysine (Fig. 3B) doesn't show successful delivery of miRNA while its counterpart LP-MSN–RGRRRRLSCRLLK8 does (Fig. 3A). Second, the sequence of the silica binding moiety affects miRNA delivery as evidenced by the different results between LP-MSN–RGRRRRLSCRLLK8 (Fig. 3A) and LP-MSN–KSLSRHDHIHHHK8 (Fig. 3C). Surprisingly, LP-MSN–KSLSRHDHIHHHK8, for unknown reason, doesn't efficiently deliver miRNA, although it has an attractive binding capacity for CpG DNA (Fig. 2). It is suspected that miRNAs might have been desorbed from the LP-MSN–peptide complex for both LP-MSN–KSLSRHDHIHHHK8 and MSN–RGRRRRLSCRLL systems before the complexes have been uptaken by cells. Nevertheless, the efficient delivery of miRNA by the LP-MSN–RGRRRRLSCRLLK8 system prompts us to further assess subsequent biological functions triggered by the miRNA delivery.
We first examined whether the delivery of miRNA can lead to gene silencing using miR29-b as an example of functional miRNA. The miR-29 family has been previously reported to regulate the expression of collagens including collagen 1, collagen 3, and collagen 4. Significant decrease of the expression of all collagens were observed following the delivery of miR29-b by both LP-MSN–RGRRRRLSCRLLK8 and commercial Lipofectamine. LP-MSN–RGRRRRLSCRLLK8 complexes show better gene silence for collagen 3 while the commercial Lipofectamine is better for silencing three other collagens. Protein-dependent difference between different delivery systems has been reported before.31 It is unclear why there is such a difference and further research is required to address this question.
The toxicity of LP-MSN–RGRRRRLSCRLLK8 was tested by MTT (substrate (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide)) assay (Fig. 4B). The NRK cells were incubated with LP-MSN–RGRRRRLSCRLLK8 at a series of concentrations for 24 hours, the cell viability decreases with the increase of LP-MSN–RGRRRRLSCRLLK8 concentration. However, there was no toxicity observed for LP-MSN–RGRRRRLSCRLLK8 at the working concentration (25 μg mL−1). In contrast, the commercial Lipofectamine reduced 10% of cell viability at the concentration recommended by manufacturer.
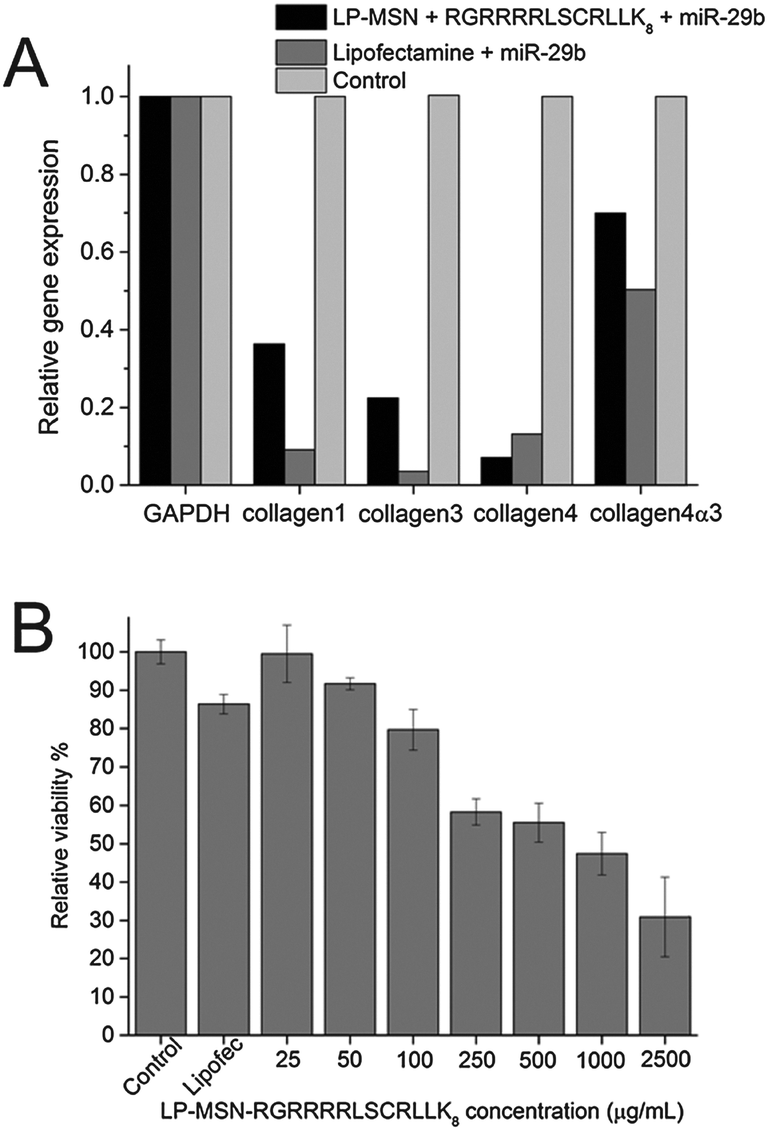 | ||
| Fig. 4 (A) Down-regulation of protein expression by miR-29b delivered by LP-MSN–RGRRRRLSCRLLK8 and Lipofectamine. (B) Cytotoxicity of LP-MSN–RGRRRRLSCRLLK8 and Lipofectamine (0.5% v/v). | ||
This study has approved the concept using bi-functional peptides for miRNA delivery. The weak affinity between the peptides and the silica surface may cause premature release of miRNA. Optimization of the peptide sequence is needed in a future study to achieve a higher affinity for silica.
Conclusions
Bi-functional peptides have been designed to non-covalently link LP-MSNs and miRNA, showing a high adsorption capacity for CpG DNA. LP-MSN–RGRRRRLSCRLLK8 can efficiently deliver miRNAs into cells and subsequently release them into cytoplasm, demonstrating a significant gene silencing function. Furthermore, the LP-MSN–peptide complexes at working concentrations showed no cytotoxicity to NRK cells. The combination of designed bi-functional peptides and LP-MSNs reported in this work offers a simple and non-toxic vehicle for efficient delivery of miRNA.Notes and references
- A. Fire, S. Q. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS PubMed.
- U. Fuchs and A. Borkhardt, in Advances in Cancer Research, ed. G. M. Hampton and K. Sikora, 2007, vol. 96, pp. 75–102 Search PubMed.
- M. Lopez-Fraga, N. Wright and A. Jimenez, Infect. Disord.: Drug Targets, 2008, 8, 262–273 CrossRef CAS.
- X. H. Wang, R. Aliyari, W. X. Li, H. W. Li, K. Kim, R. Carthew, P. Atkinson and S. W. Ding, Science, 2006, 312, 452–454 CrossRef CAS PubMed.
- J. Tabernero, G. I. Shapiro, P. M. LoRusso, A. Cervantes, G. K. Schwartz, G. J. Weiss, L. Paz-Ares, D. C. Cho, J. R. Infante, M. Alsina, M. M. Gounder, R. Falzone, J. Harrop, A. C. S. White, I. Toudjarska, D. Bumcrot, R. E. Meyers, G. Hinkle, N. Svrzikapa, R. M. Hutabarat, V. A. Clausen, J. Cehelsky, S. V. Nochur, C. Gamba-Vitalo, A. K. Vaishnaw, D. W. Y. Sah, J. A. Gollob and H. A. Burris Iii, Cancer Discovery, 2013, 3, 406–417 CrossRef CAS PubMed.
- Y. Wang, Z. Li, Y. Han, L. H. Liang and A. Ji, Curr. Drug Metab., 2010, 11, 182–196 CrossRef CAS.
- J. Gilleron, W. Querbes, A. Zeigerer, A. Borodovsky, G. Marsico, U. Schubert, K. Manygoats, S. Seifert, C. Andree, M. Stoeter, H. Epstein-Barash, L. Zhang, V. Koteliansky, K. Fitzgerald, E. Fava, M. Bickle, Y. Kalaidzidis, A. Akinc, M. Maier and M. Zerial, Nat. Biotechnol., 2013, 31, 638 CrossRef CAS PubMed.
- G. Sahay, W. Querbes, C. Alabi, A. Eltoukhy, S. Sarkar, C. Zurenko, E. Karagiannis, K. Love, D. Chen, R. Zoncu, Y. Buganim, A. Schroeder, R. Langer and D. G. Anderson, Nat. Biotechnol., 2013, 31, 653 CrossRef CAS PubMed.
- K. A. Whitehead, J. R. Dorkin, A. J. Vegas, P. H. Chang, O. Veiseh, J. Matthews, O. S. Fenton, Y. Zhang, K. T. Olejnik, V. Yesilyurt, D. Chen, S. Barros, B. Klebanov, T. Novobrantseva, R. Langer and D. G. Anderson, Nat. Commun., 2014, 5, 4277 CAS.
- D. Ma, Nanoscale, 2014, 6, 6415–6424 RSC.
- H. Lee, A. K. R. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS PubMed.
- S. B. Hartono, W. Y. Gu, F. Kleitz, J. Liu, L. Z. He, A. P. J. Middelberg, C. Z. Yu, G. Q. Lu and S. Z. Qiao, ACS Nano, 2012, 6, 2104–2117 CrossRef CAS PubMed.
- K. T. Love, K. P. Mahon, C. G. Levins, K. A. Whitehead, W. Querbes, J. R. Dorkin, J. Qin, W. Cantley, L. L. Qin, T. Racie, M. Frank-Kamenetsky, K. N. Yip, R. Alvarez, D. W. Y. Sah, A. de Fougerolles, K. Fitzgerald, V. Koteliansky, A. Akinc, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1864–1869 CrossRef CAS PubMed.
- S. H. Lee, K. H. Bae, S. H. Kim, K. R. Lee and T. G. Park, Int. J. Pharm., 2008, 364, 94–101 CrossRef CAS PubMed.
- K. A. Howard, U. L. Rahbek, X. Liu, C. K. Damgaard, S. Z. Glud, M. O. Andersen, M. B. Hovgaard, A. Schmitz, J. R. Nyengaard, F. Besenbacher and J. Kjems, Mol. Ther., 2006, 14, 476–484 CrossRef CAS PubMed.
- A. Baeza, M. Colilla and M. Vallet-Regí, Expert Opin. Drug Delivery, 2015, 12, 319–337 CrossRef CAS PubMed.
- X. Du and S. Z. Qiao, Small, 2014, 392–431 Search PubMed.
- X. Du, B. Shi, Y. Tang, S. Dai and S. Z. Qiao, Biomaterials, 2014, 35, 5580–5590 CrossRef CAS PubMed.
- X. Du, L. Xiong, S. Dai and S. Z. Qiao, Adv. Healthcare Mater., 2015, 4, 771–781 CrossRef CAS PubMed.
- Y. Kapilov-Buchman, E. Lellouche, S. Michaeli and J.-P. Lellouche, Bioconjugate Chem., 2015, 26, 880–889 CrossRef CAS PubMed.
- C. Tao, Y. Zhu, Y. Xu, M. Zhu, H. Morita and N. Hanagata, Dalton Trans., 2014, 43, 5142–5150 RSC.
- I. Y. Park, I. Y. Kim, M. K. Yoo, Y. J. Choi, M. H. Cho and C. S. Cho, Int. J. Pharm., 2008, 359, 280–287 CrossRef CAS PubMed.
- J. Liu, B. Wang, S. Budi Hartono, T. Liu, P. Kantharidis, A. P. J. Middelberg, G. Q. Lu, L. He and S. Z. Qiao, Biomaterials, 2012, 33, 970–978 CrossRef CAS PubMed.
- R. R. Naik, L. L. Brott, S. J. Clarson and M. O. Stone, J. Nanosci. Nanotechnol., 2002, 2, 95–100 CrossRef CAS PubMed.
- L. L. S. Canabady-Rochelle, D. J. Belton, O. Deschaume, H. A. Currie, D. L. Kaplan and C. C. Perry, Biomacromolecules, 2012, 13, 683–690 CrossRef CAS PubMed.
- S. V. Patwardhan, F. S. Emami, R. J. Berry, S. E. Jones, R. R. Naik, O. Deschaume, H. Heinz and C. C. Perry, J. Am. Chem. Soc., 2012, 134, 6244–6256 CrossRef CAS PubMed.
- F. Gao, P. Botella, A. Corma, J. Blesa and L. Dong, J. Phys. Chem. B, 2009, 113, 1796–1804 CrossRef CAS PubMed.
- P. S. Walker, T. Scharton-Kersten, A. M. Krieg, L. Love-Homan, E. D. Rowton, M. C. Udey and J. C. Vogel, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6970–6975 CrossRef CAS.
- I. S. Zuhorn, J. B. F. N. Engberts and D. Hoekstra, Eur. Biophys. J. Biophys. Lett., 2007, 36, 349–362 CrossRef CAS PubMed.
- Y. Wang and L. Huang, Nat. Biotechnol., 2013, 31, 610–611 Search PubMed.
- Q. Liu, R. T. Li, H. Q. Qian, J. Wei, L. Xie, J. Shen, M. Yang, X. P. Qian, L. X. Yu, X. Q. Jiang and B. R. Liu, Biomaterials, 2013, 34, 7191–7203 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Material synthesis and experimental details. See DOI: 10.1039/c5tb01133g |
| This journal is © The Royal Society of Chemistry 2015 |