
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoresponsive gelators
Emily R.
Draper
and
Dave J.
Adams
*
Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: d.j.adams@liverpool.ac.uk
First published on 19th May 2016
Abstract
Low molecular weight gels can be responsive to a range of external stimuli. The use of light as an external stimulus to modify gels is of particular interest for a number of reasons. Light is a non-invasive trigger. For example, using light it is possible to spatially target a specific area of the gel leading to patterned gel surfaces. Here, we review the different approaches that have been used to form low molecular weight gels that respond to light.
Introduction
Gels are a class of soft materials that are used in many areas, from foods to electronics. Gels consist mainly of a liquid, immobilised by a cross-linked network. The liquid can be present in very high volumes, in some cases making up 99.9% of the total mass; however, the gels still exhibit many properties of a solid. The solid network immobilising the solvent can be formed from a chemically cross-linked polymer, where the polymer chains are cross-linked irreversibly via covalent bonds (Fig. 1).1–3 Alternatively, the network can be formed from entangled high molecular weight polymers or biopolymers, such as pectins or collagen.4,5 A final class of gel is formed by the self-assembly of low molecular weight gelators (LMWG, Fig. 1).6–11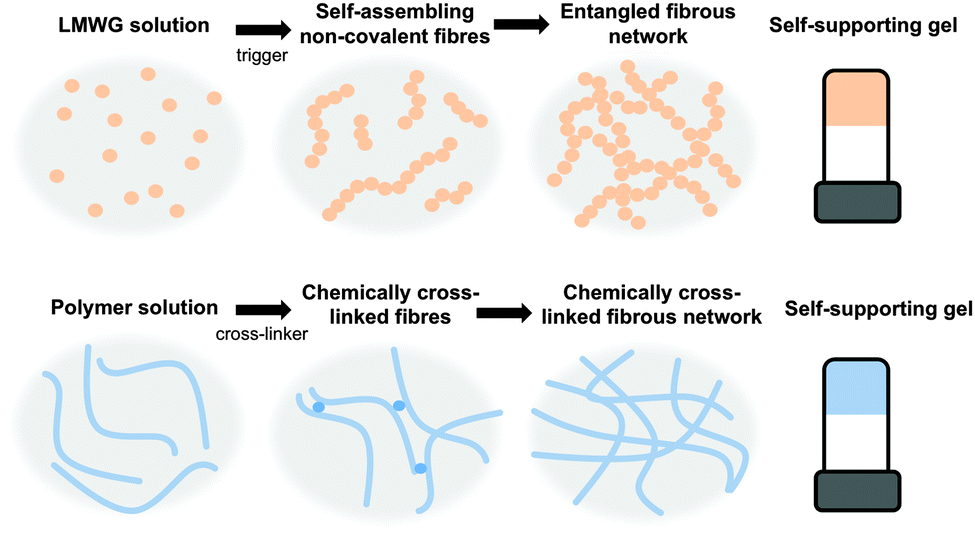 | ||
| Fig. 1 Gelation using (top) low molecular weight gelators, where the cross-links are generally formed by entanglement and (bottom) polymers, where the cross-links can be permanent chemical bonds or entanglements. | ||
These LMWG self-assemble through non-covalent interactions, including hydrogen bonding, π–π stacking, and ion pairing to form one dimensional structures such as fibres. The cross-linked network leading to the formation of gel is a result of the entanglement of these structures.
There are many different structurally diverse families of LMWG.6,10–12 These include amino acids, peptides, sugars, quaternary ammonium salts, metallic soaps, derivatised cholesterols, and hydrocarbons. To form gels, typically the LMWG are initially dissolved or suspended in the solvent (Fig. 1, top).13 These molecules are then induced to self-assemble when a suitable trigger is applied, which results in the LMWG becoming significantly less soluble. As the molecules become less soluble, they assemble into long fibres in order to minimize their interactions with the surrounding solvent (Fig. 1, top). These fibres then interact further with each other by cross-linking and entanglement resulting in the gel network (Fig. 1, top). The trigger used will depend on the system, but could be temperature (using a simple heat–cool cycle), a change in pH, a change in solvent quality (for example, by adding an anti-solvent) or addition of a specific salt.
The self-assembly of the LMWG in fibres is still poorly understood.7 A range of techniques have been used to characterise the assembly at different length-scales.14 For example, local assembly can be probed using infra-red spectroscopy or circular dichroism,14 whilst techniques such electron microscopy or small angle scattering15 gives insight into the nature of the self-assembled structures (for example if cylinders or hollow tubes are formed). Rheology can be used to gain some information about entanglement of the network and the bulk mechanical properties of the resulting gels.16,17
A key issue is the design of the LMWG. A wide range of different structures can form gels but it is difficult to predict in advance whether a molecule will be an effective gelator and, if so, exactly which solvents or solvent mixtures will be gelled.7 It is clear that directional non-covalent interactions are a pre-requisite, but these are not sufficient to always result in a gelator. A balance of solubility is clearly needed. Beyond this, a number of strategies have been employed, including synthetically iterating around the structure of a known gelator,18 using insights from crystallography,19 and computational approaches.20,21 Serendipitous discovery is also common.22
A potential advantage of gels formed from LMWG compared to chemically cross-linked polymeric hydrogelators is that the gelation is usually reversible.11 This is due to the fibrous network being held together only by non-covalent interactions. This reversibility is important when thinking about applications of the gels such as the controlled release of guest molecules or as use in sensors in logic gates.23 The weak covalent forces holding fibres together can also mean that some gels are able to quickly recover back to their original strength upon breaking.24 This so-called thixotropic behaviour means that the gels are often suitable for injection or other processing allowing them to be processed for the use in devices.
Photoresponsive gels
Low molecular weight gels can be responsive to a range of external stimuli.25–28 These gels can respond to temperature, mechanical stress, light, chemical stimulus, enzymes, pH etc. Responses to the stimuli include a change in colour, gel-to-sol transitions, gel-to-gel transitions, isomerisation, dimer formation, changes in morphology, and changes in electronic properties. This makes such gels ideal for the use in applications such as sensors, logic gates, and organic electronics.29 Post-modification of gels also allows further tuning of gel properties.30The use of light as an external stimulus to modify gels is of particular interest due to the ability to spatially target a specific area of the gel leading to patterned gel surfaces.31–34 This is useful for such applications as microfluidics, cell culture and differentiation and electronic materials. Light is also often a non-invasive trigger, so the addition of another reagent for example is not needed.
There are multiple potential responses to the application of light for photoresponsive gelators. The most common are a change in structure such that a gelator becomes a non-gelator. This results in the gel becoming a solution (Fig. 2b). Alternatively, a change in the gel network may occur, for example by a dimerization or polymerisation, often resulting the network becoming stronger (Fig. 2c). Before discussing these, it is worth noting that the aggregation of the LMWG into fibrous structures can affect the optoelectronic properties of the system.35 Aggregation of LMWG that contain aromatic groups often leads to the formation of J- or H-aggregates when fibrous structures are formed.35 This can strongly affect the absorption spectra of the system. As a result of the aggregation, a quenching of fluorescence is often also observed. However, aggregation induced emission has also been reported for a number of LMWG, where the fluorescence is enhanced on formation of the self-assembled fibres.36,37
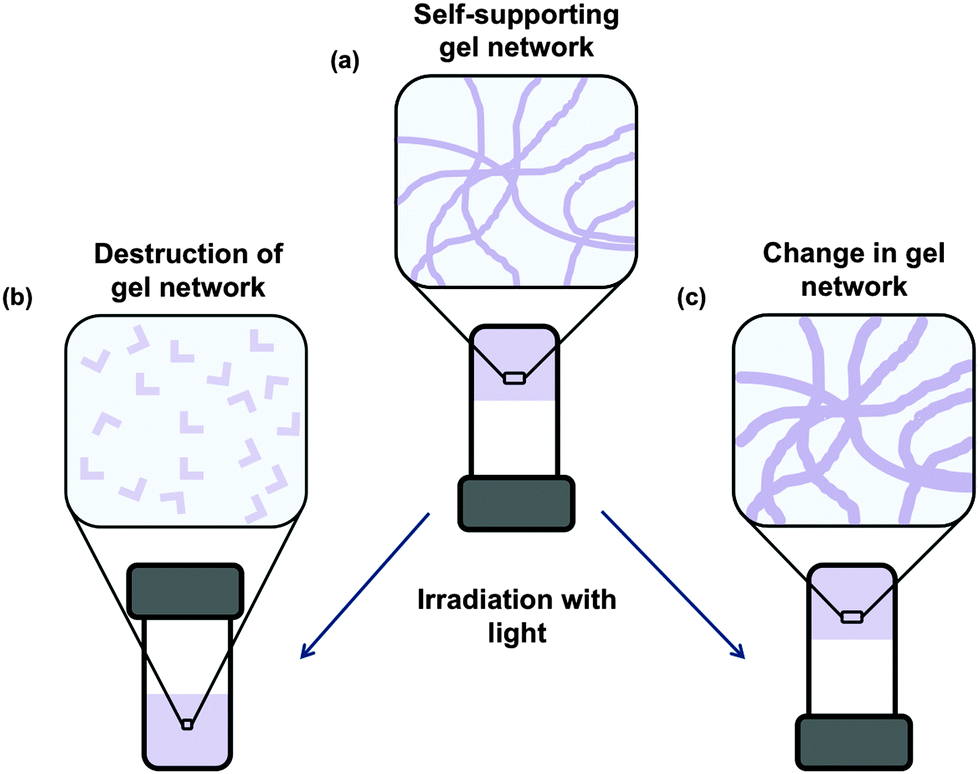 | ||
| Fig. 2 (a) Initially, a self-supporting 3D matrix of fibres is formed by the assembly of the photoresponsive LMWG. On application of light, the response of the LMWG either leads to (b) most commonly, the network falling apart, or (c) a change resulting in a strengthening of the network, for example through a dimerisation of the LMWG. | ||
Gelators that are inherently photoresponsive will contain a chromophore, which is the photoresponsive group, attached to other functional groups that drive or aid with gelation. The chromophores are usually conjugated systems that absorbs light. Hence, the conjugated systems can also aid the self-assembly by π–π stacking and (in water) hydrophobic interactions that contribute to the formation of one dimensional fibres.6,8 The chromophores collect light of a specific wavelength and convert it, leading to a photoreaction. For photoresponsive gelators, suitable and useful photoreactions include isomerisation, dimerisation, bond formation, bond cleavage and exciton formation depending on the chromophore present. The diversity in reactions allows for different applications. Here, we discuss the different approaches that have been used to form low molecular weight gels that respond to light.
Photoisomerisable gelators
Photoisomerisation is the changing of a cis isomer to the trans isomer or vice versa using the absorption of light (Fig. 3).38,39 The trans-isomers of the molecule are most often able to act as LMWG, whereas the cis-isomers tend to be non-gelators. Usually, the cis-isomer does not form gels due to change in polarisation and a less effective ability of the molecule to pack into one dimensional structures. Assuming that a gel has been formed using one isomer, the photoisomerisation commonly leads to a gel-to-sol transition, and hence the gel becoming a liquid, or a sol-to-gel transition, where a gel is formed on exposure to light. This type of response is useful for removing part of the gel to create a patterned surface. This could be especially useful when using a multi-component system so that the integrity of the gel remains but one gelator is removed from a specific area leading to different properties on specific part of the gel.40 Gel-to-sol transitions or sol-to-gel transitions can also be used in sensors such as logic gates.41,42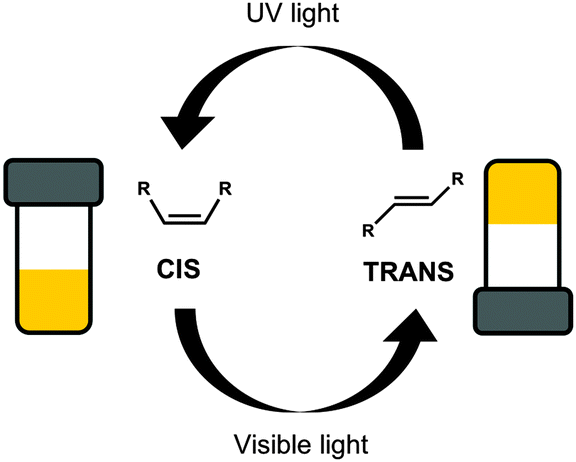 | ||
| Fig. 3 Switching between two isomers of the same molecule, the gel forming trans-isomer and the non-gelling cis-isomer, using light. | ||
Azobenzenes
Perhaps the most common photoresponsive LMWG are based on an azobenzene core.12,38,43–72 Compounds containing azobenzene moieties generally have two absorption peaks, a high-intensity peak in the UV and a lower intensity peak in the visible region. trans-Azobenzene isomerises to the cis-form when it is irradiated with UV light. The trans-isomer is more stable than the cis-isomer, meaning that the cis-to-trans isomerisation can also be achieved using heat. Hence, the use of azobenzene has the general drawback that the thermal re-isomerisation can be very quick and hence it can be difficult to maintain the cis isomer for periods of time. However, the addition of substituents to the azobenzene most often increases the lifetime of the cis-state. The photostationary state represents the equilibrium state where the composition of both isomers does not change further, no matter how long irradiation occurs (i.e. where the rates of the photo-induced cis-to-trans isomerisation is the same as the rate of photo- and heat-induced trans-to-cis isomerisation). The photostationary state for azobenzenes usually corresponds to around 80% of the cis-isomer and 20% of the trans-isomer. When the cis-isomer is irradiated with visible light (λ > 420 nm), the trans-isomer is almost entirely recovered.Azobenzene-based gelators have been extensively studied. For example, Díaz Díaz and co-workers developed a library of short peptide functionalised azobenzene LMWGs.47 These LMWG gelled a range of solvents. A trans-to-cis isomerisation took place when the gels were irradiated with UV light. This isomerisation could be followed by UV-Vis absorption spectroscopy, where the peak at 328 nm decreased in intensity, whilst a peak at 439 nm increased in intensity (Fig. 4).
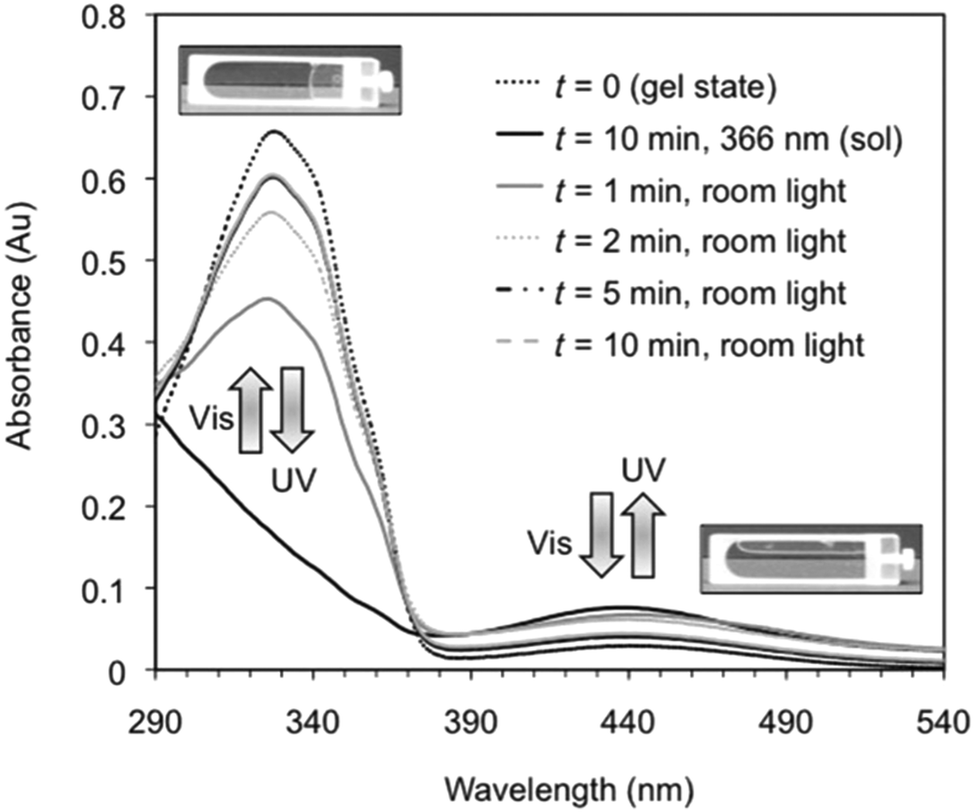 | ||
| Fig. 4 Reversible changes in the UV/Vis spectra for an organogel in toluene showing the reversible trans-to-cis isomerisation of the azobenzene moiety. On irradiation with UV light, the trans isomer changes to the cis isomer, leading to a gel-to-sol transition. Reproduced from ref. 47 with permission from the Royal Society of Chemistry. | ||
This data is typical for such azobenzene-based LMWG. On exposure to natural light, the reverse cis-to-trans isomerisation took place. This process could be repeated many times. Interestingly the relatively quick rate of isomerisation, the gel-to-sol and sol-to-gel transitions were much slower (on the order of hours). This is presumably because the length-scales involved are very different; isomerisation occurs on the molecular level, but the gels are a result of structure formation on the nano and micro-scale. This timescale is slower than some other examples, and is presumably affected by the steric environment around the azobenzenes; for example, dendritic gelators have been shown to respond much more quickly.54
Moriyama et al. mixed a discotic liquid crystal (Fig. 5a, structure 1) with a hydrogen-bonded photoresponsive gelator based on two azobenzenes (Fig. 5a, structure 2) to give a liquid crystalline gel.73 The mixed gel system was heated to 160 °C and then cooled to a different temperature. Two things happened as a result of this change in temperature: gelation of molecule 2 occurred and the liquid crystal phase of 1 also formed. When the system was cooled under UV light, cis-isomer of 2 was formed along with liquid crystals. The trans-isomer of 2 was then formed when the system was heated allowing gelation to occur after liquid crystal formation. This could be used to give two different processes, where gelation either happens first followed by liquid crystal formation, or where the liquid crystal is formed first and gelation happens afterwards. The use of the photoresponsive gelator allows two different templated materials to be made from the same molecules. The authors also managed to photopattern this system using a mask and irradiating the sample. They found where the sample was irradiated, there were aligned liquid domains whereas were the sample was covered by the mask, there was no alignment (Fig. 5b). The alignment was hence fixed by the assembled structure of 2. This alignment of liquid crystals could be of use in organic electronic materials as aligned liquid crystals have been shown to have enhanced hole mobility.74
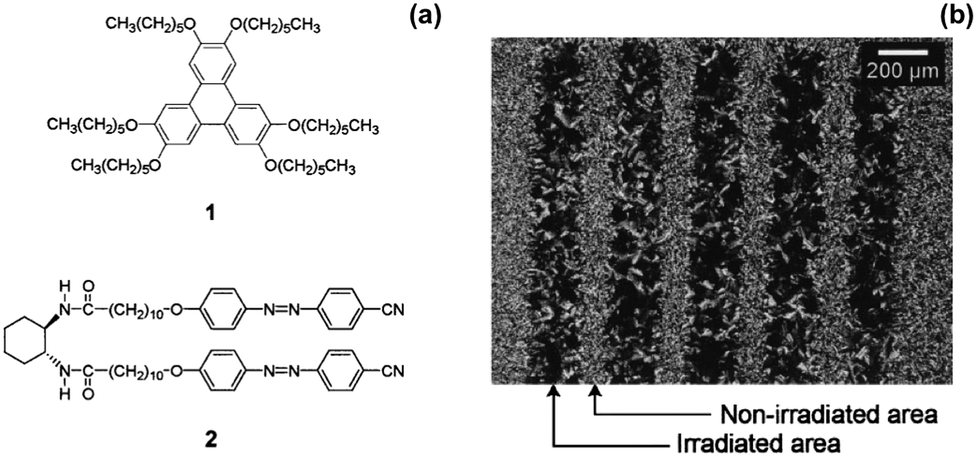 | ||
| Fig. 5 (a) Molecular structures of 1 and 2 (b) optical polarized light microscope image of a patterned sample of 1, 2 at 65 °C prepared by cooling under UV light using a photomask. Adapted from ref. 73 with permission from the Royal Society of Chemistry. | ||
As noted above, a potential problem when using azobenzene gelators is the reversibility of the trans-to-cis isomerisation. The photostationary state is not at 100% of the cis-isomer, and so normally there is a mixture of the two isomers in the gel or in the solution, which can be an issue.35 There are many examples where the reversibility of the system has been demonstrated by irradiating the cis-form with visible light. In some cases, this reversibility is not present, meaning that some groups also have to heat and sonicate solutions to regenerate the trans-form, showing that not all examples are truly photoreversible.44,66 There are also a examples of supramolecular systems where irradiation of the trans-form leads to slow or no photoisomerism.67 This is explained in terms of the packing of the azobenzene moieties in the self-assembled structures being such that the isomerisation is sterically restricted. It is also clear that when reversibility is demonstrated, almost no examples prove that the mechanical properties have been recovered. Indeed, rheological data is very rare in this field, with most demonstrations of gel-to-sol or sol-to-gel transitions being demonstrated purely by inverting vials and observing whether the samples flow or not.
Stilbenes
Stilbenes are similar in structure to azobenzenes as seen above; they differ by having a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C instead of a NN bond (Fig. 6). They act similarly to the azobenzene, with the trans-to-cis isomerisation occurring upon irradiation with UV light. Again, these have been extensively studied in the literature.40,75–78 It has been shown that the isomerisation can occur in the gel state, whilst not occurring in the solid state, which is explained by fact that the stilbene moieties are solvated effectively in the gel state.78 The cis-stilbene is less thermally stable than the trans-stilbene and so in the dark the cis-isomer can re-convert to the trans-isomer. However, the wavelength needed for the trans-to-cis isomeriation is shorter than that needed for azobenzene, and the cis-stilbene is more stable than cis-azobenzene.39
C instead of a NN bond (Fig. 6). They act similarly to the azobenzene, with the trans-to-cis isomerisation occurring upon irradiation with UV light. Again, these have been extensively studied in the literature.40,75–78 It has been shown that the isomerisation can occur in the gel state, whilst not occurring in the solid state, which is explained by fact that the stilbene moieties are solvated effectively in the gel state.78 The cis-stilbene is less thermally stable than the trans-stilbene and so in the dark the cis-isomer can re-convert to the trans-isomer. However, the wavelength needed for the trans-to-cis isomeriation is shorter than that needed for azobenzene, and the cis-stilbene is more stable than cis-azobenzene.39
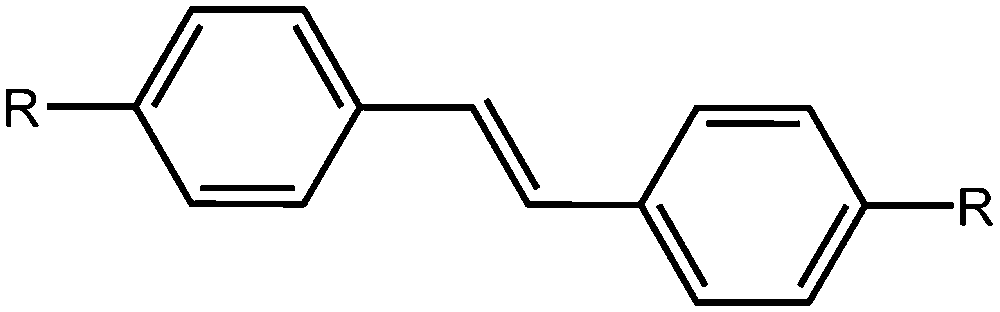 | ||
| Fig. 6 Molecular structure of trans-stilbene. | ||
Heenan and co-workers prepared organogels based on a stilbene with a gemini photo-surfactant (Fig. 7a).79 They refluxed the gelator in toluene and 0.4% N,N′-dimethylaminedodecylamine. The solution was then cooled to room temperature to form the organogel. This method of gelation gave reproducible gels, even when changing the amount of gelator in solution and the amount of N,N′-dimethylaminedodecylamine. These gels were opaque when initially formed (Fig. 7b). Upon irradiation with a mercury lamp, the gels changed to a clear solution (Fig. 7c). 1H NMR spectroscopy showed that complete conversion to the non-gelling cis-isomer occurred. They then used a mask and selectively de-gelled parts of the gel (Fig. 7d). This selective removal could be used to create patterned surfaces.
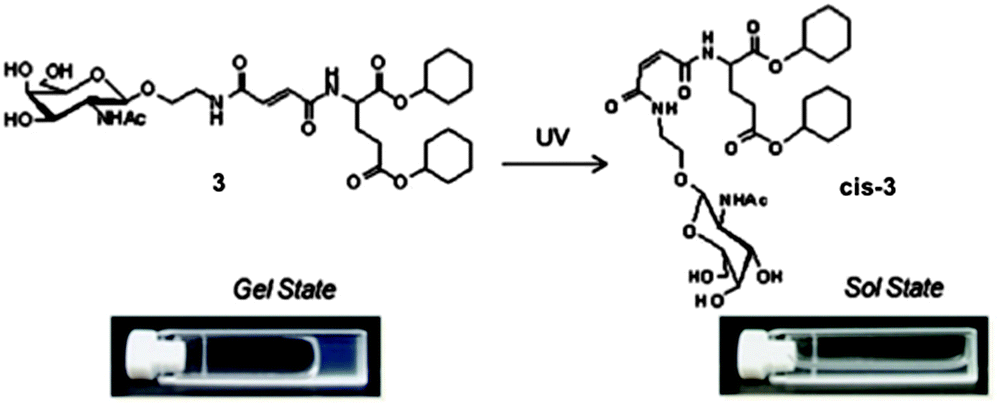 | ||
| Fig. 7 (a) Molecular structure of stilbene gemini surfactant gelator. Photographs of (a) the organogel (b) after irradiation (c) selective removal of part of the gel. Adapted from ref. 79 with the permission of the Royal Society of Chemistry. | ||
Miljanić et al. examined the gelation ability of an oxamide-based gelator bearing a stilbene functional group, varying the substituents on the stilbene.76 They prepared a series of symmetric and asymmetric gelators, with some bearing two oxamide groups and some bearing one oxamide and one long alkyl chain. They tested all the molecules in a variety of solvents. The molecule containing the long alkyl chains were more likely to gel, although not all the molecules with the alkyl chains formed gels. As noted above, this is an excellent demonstration as to how difficult it can be to design a new gelator. Again, there are few design rules for a successful gelator. For the successful LMWG, Miljanić et al. then investigated whether the corresponding cis-molecules still formed gels. Most of the cis-isomers did not gel, but one of the cis-molecules that was functionalized with an oxalamide and an alkyl chain gelled in toluene and tetralin. This is very unusual, as normally the cis-isomers do not form gel. Using this library, light-induced gelation could be achieved. Here, a solution of a non-gelling cis-isomer was irradiated to form the corresponding trans-isomer, resulting in the formation of a gel.
We have recently described the use of a stilbene LMWG as one component in a self-sorted two LMWG system.40 Here, the sequential assembly of the two components was carried out by a slow drop in the pH of the system. When the stilbene was used alone, a gel-to-sol transition was achieved by irradiation as expected. In the two component system, only the stilbene was affected by irradiation; the other component was unchanged. Hence, irradiation still lead to the isomerisation of the stilbene, but the gel maintained its integrity. We showed that this approach could be used to demonstrate using rheology that the second network was unaffected by the presence of the stilbene network that was initially present. This approach could also be used for patterning a gel structure, and allow spatial localization of specific functional groups in the network.
Alkenes
There are not many examples, but a trans-to-cis isomerisation of alkene-containing LMWG can be used in a similar fashion to the above to induce gel-to-sol transitions.80 Hamachi's group has shown a number of examples, all based around a similar structure (Fig. 8).81–83 Interestingly, they have shown that droplets can be gelled, and then the light-triggered degelation used to allow reactions and transport to occur in the droplets.81 | ||
| Fig. 8 Molecular structure of alkene-based photoresponsive LMWG. On exposure to UV light, a trans-to-cis isomerisation occurs, leading to a gel-to-sol transition. Reproduced from ref. 39 with permission of the Royal Society of Chemistry. | ||
Ring opening and closing
Another useful photoreaction is where bonds are made or broken. For photoresponsive gelators, this is most likely a ring opening or ring closing reaction.84,85 This ring opening or ring closing can have several results; these include a change in colour,86 a change in electronic properties,87 and or a gel-to-sol transition.88 The response to light makes such LMWG ideal for applications in as sensors, optical switches and displays. Molecules that can undergo ring opening reactions under UV light are often referred to as the ‘open’ molecule and ‘closed’ molecule depending on whether the ring is open or not.Dithienylethenes
Dithienylethenes undergo a photoreversible ring opening and closing and can be used to form gels.89–96 The open form of the molecule can be converted to the closed form with the use of UV light. This is reversible, with the open molecule being formed on irradiation with visible light (Fig. 9). When the molecule is closed, this restricts rotation around the cyclopentene and the thienyl groups are conjugated, giving very different electronic properties and flexibility to the open form.97 This change in conjugation and electronic properties gives interesting responses from the molecules, often leading to a change in colour. Furthermore, this reversible photoresponse can be carried out many times without loss of efficiency. Photochromic response to light and efficiency of this reaction makes these molecules useful for devices such as photo switches.89,91 | ||
| Fig. 9 Molecular structure of dithienylethene in (a) the open form and (b) the closed form. | ||
Feringa and co-workers functionalised a ring open dithienylethene with various dipeptides on the 3-position of the cyclopentene.98 By changing the amino acids in the dipeptide, the polarity of the molecules could be tuned. The gelators were dissolved at different concentrations in a phosphate buffer, or in water with the pH adjusted. The solutions were heated and sonicated to give a clear solution and, when cooled, formed weak gels which were frequency dependent. The lysine–glycine functionalised ring-open dithienylethene was then irradiated with 312 nm to form the ring-closed dithienylethene. This resulted in a change in colour of the gel from yellow to red, without a change in the rheological properties of the gel. This ring closing was reversible when using visible light >500 nm and the gel returned to the original yellow colour. This change in colour arises due to the molecule being more conjugated and so has a change in the highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO) gap. They were also able to selectively change the colour by using a photo mask. These molecules are promising for the use as a photo switch, owing to it maintaining its gel structure.
A naphthalimide and cholesterol-appended dithienylethene was synthesised by functionalising both the thienyl groups by Wang et al.99 This gave extra fluorescent properties to the molecule due to the naphthalimde group and the cholesterol group aiding with gelation. Again, when gelled, there was a photochromic response with the gel changing from a yellow to a red colour when irradiated with UV light (Fig. 10a and b). Unlike the example above, the authors showed that this molecule also responded to heat, fluoride ions and protons in both the open and closed form of the molecule. Both the open and closed form of the gel went through a gel–sol transition when heated. They showed that there was also a photochromic response when the molecule was in solution, again showing a yellow to red transition when irradiated with UV light (Fig. 10c and d). In the presence of fluoride ions, the open yellow solution changes to an orange colour due to the fluoride ion deprotonating the amide group giving a negative charge on the nitrogen and promoting a internal charge transfer and the change in colour, this was reversible upon the addition of protons (Fig. 10c and e). This orange solution also responds to the UV light and then orange colour deepens to a dark orange colour (Fig. 10e and f). This multi-responsive system would be ideal for the use in sensors and logic gates.
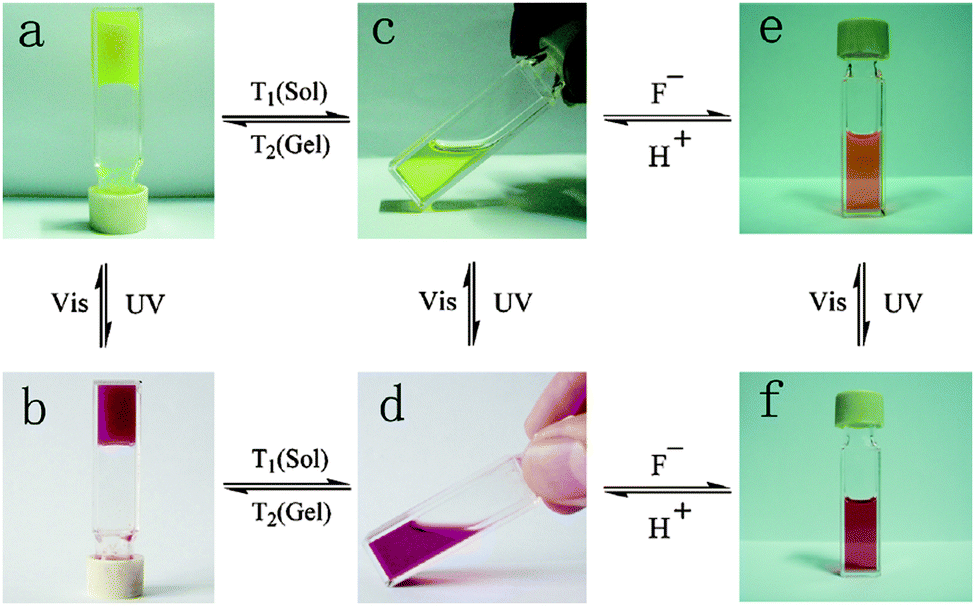 | ||
| Fig. 10 Colour change and state change upon different stimuli for a naphthalimide and cholesterol appended dithienylethene gelator. Reproduced from ref. 99 with permission of the Royal Society of Chemistry. | ||
Dithienylethene gels are very interesting materials due to them being able to maintain their rheological properties but still responding to visible and UV light. They show little fatigue after multiple cycles of ring opening/closing. The ability to be responsive to multiple stimuli also makes it possible for these materials to be used for many different applications.
Spiropyrans
Another photoresponsive gelator molecule that undergoes cleavage is spiropyrans. These molecules have a reversible photochemical cleavage of their C–O bond in the spiro unit.100 These are very closely related to spironaphthoxazines, which also have the same photochemical cleavage of the C–O bond.101 Under visible light or heat, there is a cyclisation and the C–O bond is formed to give the closed, most stable form (Fig. 11b). This can then be ring opened to the open form using UV light (Fig. 11a). The open form is normally more soluble. Hence, for LMWG based on spiropyrans, the consequence of the ring closure is a sol–gel transition as the closed form is planar, less flexible and has unfavourable π-stacking. Hence, spiropyran-based LMWG differ from many of the photoisomerisable gelators described above since there is a sol-to-gel transition under UV light as opposed to a gel-to-sol transition. There are many examples of spiropyran incorporated into polymers systems, but relatively few for low molecular weight gelators.88,102 | ||
| Fig. 11 Molecular structure of a spiropyran molecule (a) in the open form and (b) the closed form. | ||
Zhang and co-workers functionalised a spiropyran with dipeptide containing to alanine amino acids.88 The dipeptide promotes self-assembly with hydrogen-bonding and van der Waals interactions. The open form of the molecule was dissolved at neutral pH and formed a dark red gel when the pH was lowered to 3 using HCl (Fig. 12a). The closed form was unable to form a gel this way and remained as a yellow liquid. The gels formed from the open form needed to be kept away from visible light, as they became unstable. This sensitivity to visible light is not ideal for the use in applications. The gel could be irradiated with wavelengths of light >400 nm to form a yellow gelatinous precipitate (Fig. 12b), and the pH had to be altered again to form the solution, meaning this is not truly photoreversible. The yellow solution could then be irradiated with UV light to get a red gelatinous precipitate; again, the pH had to adjusted to form a gel. They also added vancomycin to the gel, which resulted in a gel-to-sol transition. This change in state is due to the ligand specific vancomycin binding to the dipeptide and increasing the solubility. Hence, this spiropyran molecule shows a multiple responses to pH, light and vancomycin.
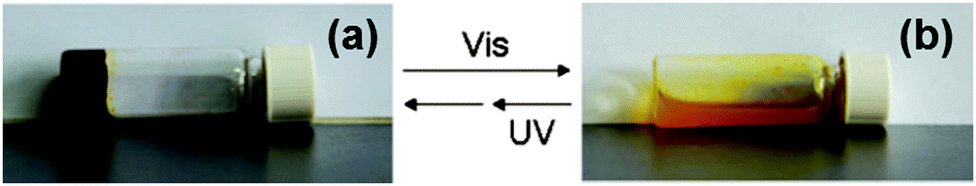 | ||
| Fig. 12 Photographs of (a) open spiropyran gel and (b) closed spiropyran solution open irradiated with different light sources. Adapted from ref. 36 with permission of the Royal Society of Chemistry. | ||
2H-Chromene
2H-Chromenes go through a photocleavage of a C–O bond upon the irradiation with UV light and ring opening the molecule (Fig. 13). This ring-opening is reversible upon heating due to the ring-closed molecule being more thermally stable than ring-opened molecule.84,86 This ring-closure occurs through an electrocyclisation. These two forms of the molecule are again referred to as the open and the closed form. The closed form is usually colourless and the ring-closed form is coloured. This change in colour makes these molecules of interest due the resistance to fatigue over multiple cycles. Despite this photochromic behaviour, there are only a few examples of these molecules being used as gelators. | ||
| Fig. 13 Molecular structure change of 2H-chromene from the (a) closed to the (b) open form. | ||
A series of 2H-chromene N-acylamino acid conjugates were reported.84,86 Both reports describe how the ring open structures were dissolved in an organic solvent and heated whilst stirring until the solution was clear; they then cooled the solutions to give colourless gels. The different molecules showed different gelation abilities and solubility in different solvents. Katritzky et al. have described how molecules with a longer chain length of the acylamino group were more likely to be LMWG.86 They also found that if the molecule contained a dipeptide rather than a single amino acid group then it was able to gel in some solvents at low temperature and at room temperature gave a gelatinous precipitate. The molecules that did gel were irradiated with 366 nm light. This caused a colour change of the gel to yellow, due to the formation of the ring open 2H-chromene. When irradiation was stopped, the gel quickly converted back to the colourless state due to the instability of the closed form. The gel was disrupted by this irradiation; this was probably due to the closed form of the molecule not being a gelator and so breaking the gel structure and therefore did not fully go into solution due to insufficient penetration depth of the UV light into the bulk sample. The authors did note that they were able to reform the gel upon heating and cooling again. This group of molecules also showed sensitivity to the presence of sodium ions as the protonated amino acid of the open form of the molecule did not gel, but the sodium salts did. This shows that the there is a delicate balance of molecular interactions needed for gelation to occur.
Photodimerisation and photopolymerisation
Photodimerisation is when two of the same molecules that are close enough or in the orientation with respect to each other form covalent bonds between each other when irradiated with light. This normally happens in conjugated molecules. In solution, this can be very difficult due to the molecules being able to freely move around and so are less likely to be in the right position for this photodimerisation to occur. However, in the gelled state, the molecules are very close to each other due to π–π stacking of the molecules, making it more likely that the molecules are close enough to each other for dimerisation to occur. Dimerisation of the gelator molecule often leads to the molecules being less soluble due to the molecule doubling in size and so becoming more hydrophobic. This decrease in solubility can lead to destruction of the gel or a decrease in the rheological properties due to the gel network being disruption of the gel network.Coumarins
Coumarins are class of molecules based on a dye molecule that is often found in plants. Dimerisation of the coumarin molecule occurs upon irradiation with light >300 nm (Fig. 14).103 | ||
| Fig. 14 The reversible photodimerisation of a coumarin from (a) the monomer to (b) the dimer. | ||
These coumarin dimerisation is reversible when light with a wavelength of <260 nm is used. The [2+2] photodimerisation of coumarins is known to occur both in solution and in the solid state,104 often to give a mixture of syn–syn, syn–anti, anti–syn and anti–anti cyclobutanes depending on how the molecules are orientated with respect to one another. A small number of coumarin-based LMWG have been reported.105–111
Parquette and co-workers designed a coumarin dipeptide hydrogelator containing two coumarin groups connected by a dipeptide.106 The two coumarin groups allowed each gelator to react with two other gelator molecules to increase the amount of cross-linking upon irradiation. Gels were prepared in water, saline or buffer and left for 24 hours before any rheology tests were done on the system. The buffer increased the amount of bundling of the fibres due to salt-induced electrostatic screening due to lysine side chains on the gelator. Photoisomerisation was carried out with 365 nm light. The hydrogel not exposed to UV light was stable for up to 3 weeks. However, upon irradiation with 365 nm light, a dark yellow precipitate was formed. After 7 days under the UV light, this precipitate became insoluble in water due to the gelators becoming cross-linked. The precipitate could be then collected by centrifugation. This cross-linked precipitate was soluble in trifluoroethanol and could be analysed. Analysis revealed the presence of uniform nanofibres (Fig. 15) around 15 nm in diameter. Rheology and microscopy revealed that two different dimerisations were occurring within the system, there were intra-fibre dimerisation within the fibres and inter-fibre dimerisation of coumarin molecules. This cross-linking stiffened the fibres and so could be used for post-modification of the hydrogel surfaces for applications such as surface patterning.
 | ||
| Fig. 15 Diagram showing the possible cross-linking within a fibre due to coumarin dimerisation of the dipeptide functionalised hydrogelator. Reproduced from ref. 106 with permission from the Centre de la Recherche Scientific (CNRS) and the Royal Society of Chemistry. | ||
We have described a rare example of irradiation leading to an increase in the rheological properties of an irradiated gel;107 in most cases, irradiation of a gel leads to a weakening of the gel or, more commonly, a gel-to-sol transition. Photodimerisation of a coumarin-dipeptide for relatively short periods of time lead to a significant increase in the storage and loss moduli of the gels. Extended irradiation however resulted in the gels becoming weaker, although they were still stronger than the original gels. This was attributed to the extended irradiation leading to sufficient stiffening of the self-assembled fibres such that the network was disrupted.
Organogels based on coumarin molecules were prepared by Yu et al.112 They investigated the gelation properties of the coumarin molecules with different length alkyl chains in a variety of solvents. A heat–cool method was used to test whether gelation could occur. They then looked at how the different solvents, and different length alkyl chain affected the morphology of the fibres formed. They observed different morphologies such as helical ribbons and spheres depending on which solvents were used, showing that the solvent used is critical to gel properties. A gel formed in cyclohexane was chosen to be studied under irradiation with >300 nm and >280 nm light. Upon irradiation with 300 nm light, the gel structure was maintained and the dimerisation was monitored by UV-Vis absorption spectroscopy, as was the reverse reaction with 280 nm. Scanning electron microscopy (SEM) revealed that morphology of the fibres changed from helical fibres to lumps upon irradiation with >300 nm and then changed to a sponge like morphology when irradiated with <280 nm. Throughout irradiation, the gel maintained its structure whilst the morphologies drastically changed.
Anthracenes
Anthracene is a molecule made of three fused benzene rings. Anthracenes undergo a [4+4] cycloaddition reaction when irradiated with >300 nm light and the reverse reaction when irradiated with light of <300 nm or with heat (Fig. 16a).113 This reaction is affected by the presence of oxygen.114 Like coumarin, there are different isomers of dianthracene that can be formed due to the position of the anthracenes with respect to each other when the cylcoaddition occurs. These are the anti head to tail, syn head to tail, anti head to head and syn head to head stereodimers (Fig. 16b). The photodimerisation of anthracene gels often leads to a gel–sol transition due to the dimer disrupting the gel network.115–120 In the gel state, photodimerisation can lead predominantly to a single photodimer, reflecting the packing of the molecules in the self-assembled fibres.115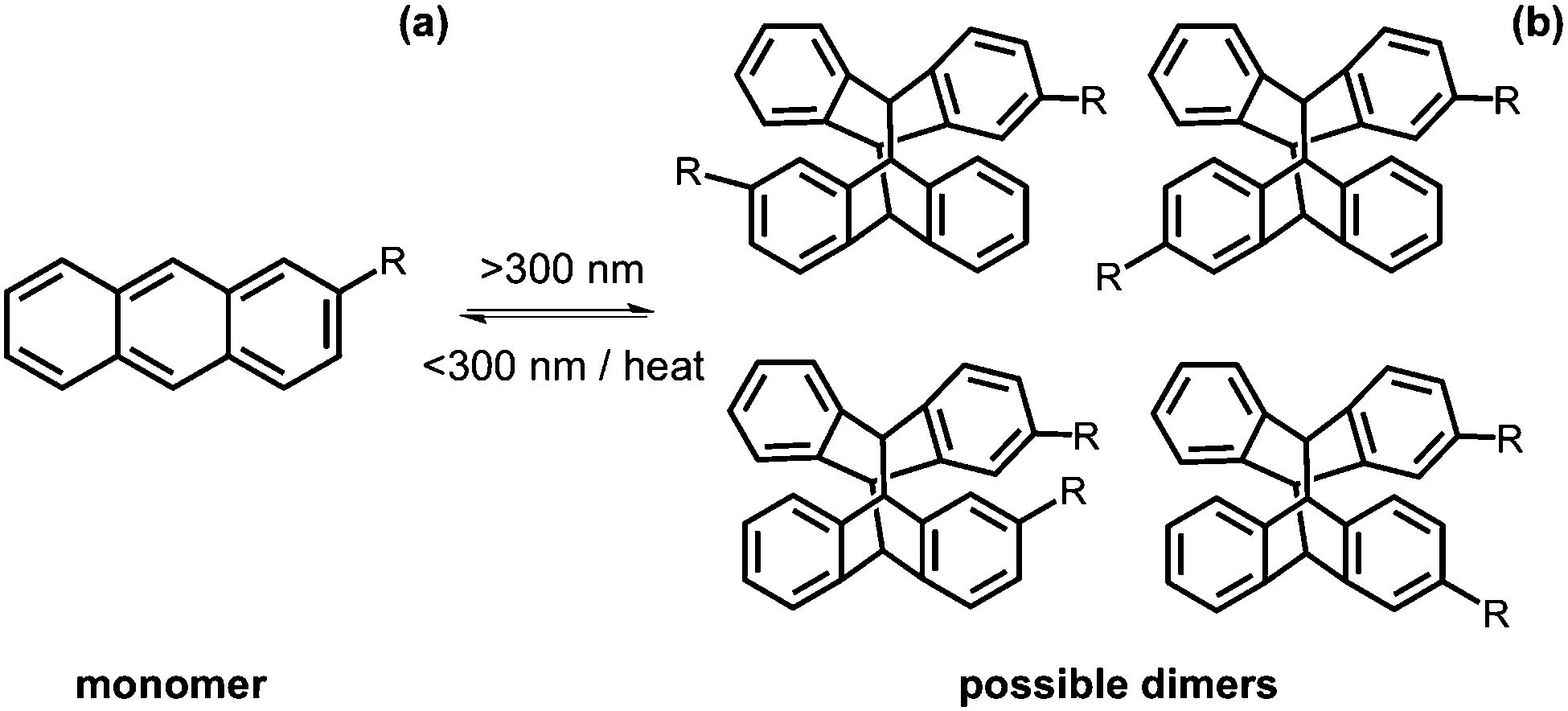 | ||
| Fig. 16 Dimerisation using >300 nm of anthracene from the (a) monomer to (b) the dimer. | ||
An anthracene modified with urea was investigated as a gelator in organic solvents.119 Several solvents were tested but the molecule was only able to gel using dichloroethane. A heat–cool method was used to form an opaque gel. This method was reversible with several heat–cool cycles (Fig. 17). Upon gelation, there was a ten times increase in fluorescence as compared to the solution and so this could have potential use as a thermal sensor. When irradiated with >300 nm light, the gel remained stable and, by NMR spectroscopy, it was seen that little dimer was formed. This could be due to the molecules not being the right orientation to each other. Photodimerisation of the urea-modified anthracene molecule in solution was monitored by 1H NMR spectroscopy and UV-Vis absorption spectroscopy. From 1H NMR spectroscopy, the change from a monomer to dimer resulted in a head to tail dimerisation. The dimer was able to form a transparent gel in cyclohexane, n-hexane, and n-heptane and shows greater gelation ability than the monomer. In addition, these dimers we stable to heat and <300 nm light and only a small portion of the gel changed to solution.
 | ||
| Fig. 17 Gelation of urea functionalised anthracene molecule in dichloroethane using a heat–cool method. Adapted from ref. 119 with permission of the Royal Society of Chemistry. | ||
Dawn et al. also investigated anthracene as organogelators.121 They used 2-anthracenecarboxylic acid to control the stereochemical formation of exclusively the head to head dimer. Gelation was achieved by the noncovalent attachment with a gelator component containing the gallic acid backbone coupled with D-alanine. This was chosen as it should enhance gelation through H-bonding and π–π stacking; it would also allow the anthracene to be close enough to allow dimerisation to occur. The incorporation of the chiral D-alanine could also induce enantioselectivity in the anthracene photoproducts via perturbation of the pre-orientation in the ground state. Cyclohexane was chosen as it had a low critical gelation concentration and was thermally reversible. The lower concentration of gelator allowed more efficient photodimerisation. When irradiated with 366 nm light, there was a gel–sol transition and from high-pressure liquid chromatography, it was shown only the head to head isomer was present. They also investigated this process at a higher temperature were they found the head to head in excess. In the solid state no isomerisation occurred and in tetrahydrofuran the head to tail isomer was formed. This system shows stereochemical control in a supramolecular assembly created by organogel systems.
Alkenes
[2+2] cycloadditions across alkenes also have potential for use in photoresponsive supramolecular gels. However, the only example appears to be that of Wang et al., who have utilised a cycloaddition reaction in a organogel to convert the fibres into hollow nanostructures.122Photopolymerisation
There are a number of examples where photopolymerisation has been carried out in LMWG containing diacetylene- or butadiene-moieties.123–134 Here, photoirradiation leads to a zipping up of the gelators (Fig. 18). This can lead to a strengthening of the gel. For example, George and Weiss have shown that organogels can be so polymerised. Before polymerisation, the gels are thermoreversible, as is typical for many such gels. However, after polymerisation of the diacetylenes, the gels are no longer thermoreversible. The gels also become highly coloured, as would be expected from the extended conjugation that results from the polymerisation. | ||
| Fig. 18 UV-polymerisation of a diacetylene-containing LMWG leads to a polymeric network being formed. | ||
In a related fashion, by addition of a ruthenium-based catalyst, a tyrosine containing LMWG was photocrosslinked on irradiation with white light (Fig. 19).135 On irradiation, the gel increased dramatically in strength as covalent cross-links are introduced into the network. As for the alkyne and butadiene cases, this photopolymerisation means that the gels are no longer reversible in nature. For this approach to be successful, the authors found that the tyrosine moieties had to be sufficiently accessible. Hence, polymerisation of their initial design where the tyrosines were close to the Fmoc group driving the hydrophobic collapse lead to deformation of the gels, which was proposed to be due to changes in the geometry around the Fmoc groups on irradiation.
 | ||
| Fig. 19 Irradiation of a tyrosine-containing LMWG in the presence of a ruthenium catalyst leads to cross-linking and a dramatic increase in gel strength. Reproduced from ref. 135 with permission of the Royal Society of Chemistry. | ||
Photocleavage
A final useful photoreaction that can be used is photocleavage. Here, the application of light results in a bond being permanently broken. This approach can be used to trigger self-assembly and gel formation. For example, Haines et al. incorporated a carboxy-2-nitrobenzyl group into a peptide sequence that is generally an effective LMWG.136 This group have established that the 20 amino acid sequence forms gels by folding into a hairpin, which undergoes lateral assembly to form fibres. The incorporation of the carboxy-2-nitrobenzyl group disrupted this assembly such that free-flowing solutions were instead formed. On irradiation and photocleavage, a gel then formed.A similar approach has been used by Muraoka et al. to form gels using a suitably functionalised peptide amphiphile (Fig. 20).137 Compound 4 (Fig. 20) forms spherical structures in water. However, on irradiation with 350 nm light, the 2-nitrobenzyl group is cleaved from the amphiphile. This results in a change in the geometry of the molecule such that 5 prefers to form nanofibres. These entangle and form a gel. Hence, the irradiation results in a sol-to-gel transition.
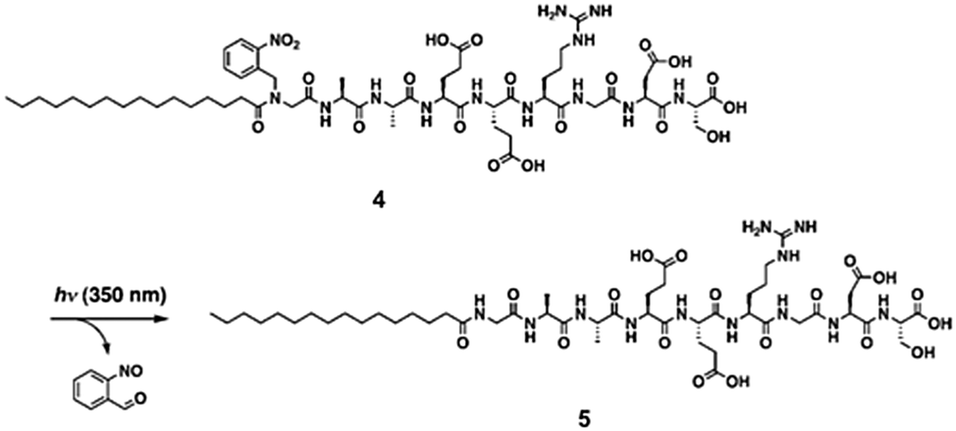 | ||
| Fig. 20 Irradiation of a peptide amphiphile results in cleavage of the 2-nitrobenzyl group and hence gelation. Reproduced from ref. 137. Copyright (2009) with permission from John Wiley and Sons. | ||
Conclusions and outlook
Photoresponsive gels in general have a variety of applications. The use of light to get a response from a system is attractive due to the speed of response, the different wavelengths giving different responses and the ability to spatially resolve this response in the material. In biomedical applications, they are a particularly interesting option to advance drug delivery systems and tissue engineering. Gels have been made that can be used for drug release upon irradiation by using gelators that undergo a gel-to-sol transition. For example, when using an isomerisible group like azobenzene, Kros and co-worker showed controllable release of proteins from the gel.138 Another way photoresponsive gels can be used is to 3D photopattern gels. Spatially resolved irradiation allows only the mechanical properties of selective areas of the gels to be affected.40 Different strength gels can be used to support the cells growth and be used for cell differentiation, which is essential for regenerative medicine.139Multi-responsive gelators have use in sensors such as logic gates.83,140 Logic gates are devices that use one or more inputs (in this case stimuli) to produce a single response. These can be used for intelligent soft materials to perform functions in response to various stimuli, and so could be used in sensors, drug delivery and organic electronics. Gels that show photochromism can be used in applications such as optical switches due to their change in colour when a light stimulus is used.141 Optical switches are made form a photoemitter (e.g. LED) and a photodetector (e.g. photodiode) mounted in a component so that the photoemitter illuminates the photodetector, but an opaque object can be inserted in a slot between them so as to break the beam. A change in the beam would give an electrical response. Gelators such as 2H-chromene and dithienylethenes can be used to form gels that exhibit different colours. The reversible photoresponsive behaviour means that they could be used in used for these applications.
Overall, photoresponsive gels show great potential for the use in a wide range of applications. The use of LMWGs allows a variety of responses due to the many different molecules that can be used to make them. We have covered the different approaches that have been used to prepare photoresponsive low molecular weight gels. Azobenzenes are the most widely used examples, although there are many other potential approaches. Most examples result in gels that become solutions on irradiation or, less commonly, solutions that form gels on irradiation. It is surprising here how few examples show rheological data for the gels. There are many examples that claim that the gel-to-sol transition (for example) can be cycled many times, but no demonstration that the gels have the same properties each time. There are also only a few examples where the rheological properties of the gels are improved on irradiation.
Acknowledgements
We thank the EPSRC for funding via a Fellowship to DA (EP/L021978/1).Notes and references
- J. Jagur-Grodzinski, Polym. Adv. Technol., 2010, 21, 27–47 CAS.
- P. P. Kalshetti, V. B. Rajendra, D. N. Dixit and P. P. Parekh, Int. J. Pharm. Pharm. Sci., 2012, 4, 1–7 CAS.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- S. J. Buwalda, K. W. M. Boere, P. J. Dijkstra, J. Feijen, T. Vermonden and W. E. Hennink, J. Controlled Release, 2014, 190, 254–273 CrossRef CAS PubMed.
- L. S. Liu, J. Kost, F. Yan and R. C. Spiro, Polymers, 2012, 4, 997 CrossRef.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- M. de Loos, J. van Esch, R. M. Kellogg and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 113, 633–636 CrossRef.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- V. J. Nebot and D. K. Smith, Functional Molecular Gels, The Royal Society of Chemistry, 2014, pp. 30–66 Search PubMed.
- J.-B. Guilbaud and A. Saiani, Chem. Soc. Rev., 2011, 40, 1200–1210 RSC.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- C. Chassenieux and L. Bouteiller, Supramol. Chem., John Wiley & Sons, Ltd, 2012 Search PubMed.
- M. L. Muro-Small, J. Chen and A. J. McNeil, Langmuir, 2011, 27, 13248–13253 CrossRef CAS PubMed.
- T. K. Adalder and P. Dastidar, Cryst. Growth Des., 2014, 14, 2254–2262 CAS.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- J. K. Gupta, D. J. Adams and N. G. Berry, Chem. Sci., 2016 10.1039/C6SC00722H.
- D. M. Zurcher and A. J. McNeil, J. Org. Chem., 2015, 80, 2473–2478 CrossRef CAS PubMed.
- A. Prasanna de Silva, Chem. – Asian J., 2011, 6, 750–766 CrossRef CAS PubMed.
- Z. Xie, A. Zhang, L. Ye, X. Wang and Z.-g. Feng, J. Mater. Chem., 2009, 19, 6100–6102 RSC.
- M. Xiong, C. Wang, G. Zhang and D. Zhang, Functional Molecular Gels, The Royal Society of Chemistry, 2014, pp. 67–94 Search PubMed.
- X. Yang, G. Zhang and D. Zhang, J. Mater. Chem., 2012, 22, 38–50 RSC.
- T. Ishi-i and S. Shinkai, in Supermolecular Dye Chemistry, ed. F. Würthner, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 119–160 Search PubMed.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- E. R. Draper and D. J. Adams, Chemoresponsive Materials: Stimulation by Chemical and Biological Signals, The Royal Society of Chemistry, 2015, pp. 332–363 Search PubMed.
- J. M. J. Paulusse and R. P. Sijbesma, Angew. Chem., Int. Ed., 2006, 45, 2334–2337 CrossRef CAS PubMed.
- S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520–1529 RSC.
- S. Yagai, T. Karatsu and A. Kitamura, Chem. – Eur. J., 2005, 11, 4054–4063 CrossRef CAS PubMed.
- A. Guerzo and J.-L. Pozzo, in Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer Netherlands, Dordrecht, 2006, pp. 817–855 Search PubMed.
- S. Khetan and J. A. Burdick, Soft Matter, 2011, 7, 830–838 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS PubMed.
- E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848–852 CrossRef CAS PubMed.
- P. A. d. Silva, Chem. – Asian J., 2011, 6, 750 CrossRef PubMed.
- A. Richter, G. Paschew, S. Klatt, J. Lienig, K.-F. Arndt and H.-J. Adler, Sensors, 2008, 8, 561 CrossRef CAS.
- M. J. Clemente, R. M. Tejedor, P. Romero, J. Fitremann and L. Oriol, New J. Chem., 2015, 39, 4009–4019 RSC.
- K. Tiefenbacher, H. Dube, D. Ajami and J. Rebek, Chem. Commun., 2011, 47, 7341–7343 RSC.
- K. Murata, M. Aoki, T. Nishi, A. Ikeda and S. Shinkai, Chem. Commun., 1991, 1715–1718 RSC.
- Y. Matsuzawa and N. Tamaoki, J. Phys. Chem. B, 2010, 114, 1586–1590 CrossRef CAS PubMed.
- P. Fatás, J. Bachl, S. Oehm, A. I. Jiménez, C. Cativiela and D. Díaz Díaz, Chem. – Eur. J., 2013, 19, 8861–8874 CrossRef PubMed.
- Y. Huang, Z. Qiu, Y. Xu, J. Shi, H. Lin and Y. Zhang, Org. Biomol. Chem., 2011, 9, 2149–2155 CAS.
- Z. L. Pianowski, J. Karcher and K. Schneider, Chem. Commun., 2016, 52, 3143–3146 RSC.
- T. M. Doran, D. M. Ryan and B. L. Nilsson, Polym. Chem., 2014, 5, 241–248 RSC.
- X. Li, Y. Gao, Y. Kuang and B. Xu, Chem. Commun., 2010, 46, 5364–5366 RSC.
- S. Yagai, T. Iwashima, K. Kishikawa, S. Nakahara, T. Karatsu and A. Kitamura, Chem. – Eur. J., 2006, 12, 3984–3994 CrossRef CAS PubMed.
- N. Koumura, M. Kudo and N. Tamaoki, Langmuir, 2004, 20, 9897–9900 CrossRef CAS PubMed.
- J. H. Kim, M. Seo, Y. J. Kim and S. Y. Kim, Langmuir, 2009, 25, 1761–1766 CrossRef CAS PubMed.
- S. Lee, S. Oh, J. Lee, Y. Malpani, Y.-S. Jung, B. Kang, J. Y. Lee, K. Ozasa, T. Isoshima, S. Y. Lee, M. Hara, D. Hashizume and J.-M. Kim, Langmuir, 2013, 29, 5869–5877 CrossRef CAS PubMed.
- Y. Ogawa, C. Yoshiyama and T. Kitaoka, Langmuir, 2012, 28, 4404–4412 CrossRef CAS PubMed.
- K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664–6676 CrossRef CAS.
- Y. Ji, G.-C. Kuang, X.-R. Jia, E.-Q. Chen, B.-B. Wang, W.-S. Li, Y. Wei and J. Lei, Chem. Commun., 2007, 4233–4235 RSC.
- W. Li, I.-s. Park, S.-K. Kang and M. Lee, Chem. Commun., 2012, 48, 8796–8798 RSC.
- S. Kume, K. Kuroiwa and N. Kimizuka, Chem. Commun., 2006, 2442–2444 RSC.
- M. Moriyama, N. Mizoshita and T. Kato, Bull. Chem. Soc. Jpn., 2006, 79, 962–964 CrossRef CAS.
- C. Wang, Q. Chen, F. Sun, D. Zhang, G. Zhang, Y. Huang, R. Zhao and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3092–3096 CrossRef CAS PubMed.
- Y. Lin, Y. Qiao, P. Tang, Z. Li and J. Huang, Soft Matter, 2011, 7, 2762–2769 RSC.
- Y. Zhou, M. Xu, J. Wu, T. Yi, J. Han, S. Xiao, F. Li and C. Huang, J. Phys. Org. Chem., 2008, 21, 338–343 CrossRef CAS.
- S. Yagai, T. Nakajima, K. Kishikawa, S. Kohmoto, T. Karatsu and A. Kitamura, J. Am. Chem. Soc., 2005, 127, 11134–11139 CrossRef CAS PubMed.
- J. K. Sahoo, S. K. M. Nalluri, N. Javid, H. Webb and R. V. Ulijn, Chem. Commun., 2014, 50, 5462–5464 RSC.
- S. Yagai, T. Karatsu and A. Kitamura, Langmuir, 2005, 21, 11048–11052 CrossRef CAS PubMed.
- M. Moriyama, N. Mizoshita, T. Yokota, K. Kishimoto and T. Kato, Adv. Mater., 2003, 15, 1335–1338 CrossRef CAS.
- F. Xie, L. Qin and M. Liu, Chem. Commun., 2016, 52, 930–933 RSC.
- N. Luisier, R. Scopelliti and K. Severin, Soft Matter, 2016, 12, 588–593 RSC.
- X. Tan, Z. Li, M. Xia and X. Cheng, RSC Adv., 2016, 6, 20021–20026 RSC.
- S. Samai, C. Sapsanis, S. P. Patil, A. Ezzeddine, B. A. Moosa, H. Omran, A.-H. Emwas, K. N. Salama and N. M. Khashab, Soft Matter, 2016, 12, 2842–2845 RSC.
- M. Moriyama, N. Mizoshita and T. Kato, Polym. J., 2004, 36, 661–664 CrossRef CAS.
- N. Mizoshita, H. Monobe, M. Inoue, M. Ukon, T. Watanabe, Y. Shimizu, K. Hanabusa and T. Kato, Chem. Commun., 2002, 428–429 RSC.
- S. Miljanić, L. Frkanec, Z. Meić and M. Žinić, Langmuir, 2005, 21, 2754–2760 CrossRef PubMed.
- S. Miljanić, L. Frkanec, Z. Meić and M. Žinić, Eur. J. Org. Chem., 2006, 1323–1334 CrossRef.
- C. Geiger, M. Stanescu, L. Chen and D. G. Whitten, Langmuir, 1999, 15, 2241–2245 CrossRef CAS.
- R. Wang, C. Geiger, L. Chen, B. Swanson and D. G. Whitten, J. Am. Chem. Soc., 2000, 122, 2399–2400 CrossRef CAS.
- J. Eastoe, M. Sanchez-Dominguez, P. Wyatt and R. K. Heenan, Chem. Commun., 2004, 2608–2609 RSC.
- L. Frkanec, M. Jokić, J. Makarević, K. Wolsperger and M. Žinić, J. Am. Chem. Soc., 2002, 124, 9716–9717 CrossRef CAS PubMed.
- S. Matsumoto, S. Yamaguchi, A. Wada, T. Matsui, M. Ikeda and I. Hamachi, Chem. Commun., 2008, 1545–1547 RSC.
- S. Matsumoto, S. Yamaguchi, S. Ueno, H. Komatsu, M. Ikeda, K. Ishizuka, Y. Iko, K. V. Tabata, H. Aoki, S. Ito, H. Noji and I. Hamachi, Chem. – Eur. J., 2008, 14, 3977–3986 CrossRef CAS PubMed.
- H. Komatsu, S. Matsumoto, S.-i. Tamaru, K. Kaneko, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5580–5585 CrossRef CAS PubMed.
- S. A. Ahmed, X. Sallenave, F. Fages, G. Mieden-Gundert, W. M. Müller, U. Müller, F. Vögtle and J.-L. Pozzo, Langmuir, 2002, 18, 7096–7101 CrossRef CAS.
- Y. Li, K. M.-C. Wong, A. Y.-Y. Tam, L. Wu and V. W.-W. Yam, Chem. – Eur. J., 2010, 16, 8690–8698 CrossRef CAS PubMed.
- A. R. Katritzky, R. Sakhuja, L. Khelashvili and K. Shanab, J. Org. Chem., 2009, 74, 3062–3065 CrossRef CAS PubMed.
- S. Xiao, T. Yi, F. Li and C. Huang, Tetrahedron Lett., 2005, 46, 9009–9012 CrossRef CAS.
- Z. Qiu, H. Yu, J. Li, Y. Wang and Y. Zhang, Chem. Commun., 2009, 3342–3344 RSC.
- M. Irie, K. Sakemura, M. Okinaka and K. Uchida, J. Org. Chem., 1995, 60, 8305–8309 CrossRef CAS.
- S. Xiao, Y. Zou, M. Yu, T. Yi, Y. Zhou, F. Li and C. Huang, Chem. Commun., 2007, 4758–4760 RSC.
- S. Yagai, K. Ishiwatari, X. Lin, T. Karatsu, A. Kitamura and S. Uemura, Chem. – Eur. J., 2013, 19, 6971–6975 CrossRef CAS PubMed.
- J. J. D. de Jong, P. R. Hania, A. Pugžlys, L. N. Lucas, M. de Loos, R. M. Kellogg, B. L. Feringa, K. Duppen and J. H. van Esch, Angew. Chem., Int. Ed., 2005, 44, 2373–2376 CrossRef CAS PubMed.
- J. J. D. de Jong, L. N. Lucas, R. M. Kellogg, J. H. van Esch and B. L. Feringa, Science, 2004, 304, 278–281 CrossRef CAS PubMed.
- J. J. D. de Jong, T. D. Tiemersma-Wegman, J. H. van Esch and B. L. Feringa, J. Am. Chem. Soc., 2005, 127, 13804–13805 CrossRef CAS PubMed.
- M. Akazawa, K. Uchida, J. J. D. de Jong, J. Areephong, M. Stuart, G. Caroli, W. R. Browne and B. L. Feringa, Org. Biomol. Chem., 2008, 6, 1544–1547 CAS.
- T. Li, X. Li, J. Wang, H. Ågren, X. Ma and H. Tian, Adv. Opt. Mater., 2016 DOI:10.1002/adom.201500694.
- O. Tosic, K. Altenhoner and J. Mattay, Photochem. Photobiol. Sci., 2010, 9, 128–130 CAS.
- J. T. van Herpt, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Chem. – Eur. J., 2014, 20, 3077–3083 CrossRef CAS PubMed.
- S. Wang, W. Shen, Y. Feng and H. Tian, Chem. Commun., 2006, 1497–1499 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1754 CrossRef CAS PubMed.
- Q. Chen, Y. Feng, D. Zhang, G. Zhang, Q. Fan, S. Sun and D. Zhu, Adv. Funct. Mater., 2010, 20, 36–42 CrossRef CAS.
- M. D'Auria and R. Racioppi, J. Photochem. Photobiol., A, 2004, 163, 557–559 CrossRef.
- K. Tanaka, Molecules, 2012, 17, 1408 CrossRef CAS PubMed.
- M. Ikeda, T. Tanida, T. Yoshii and I. Hamachi, Adv. Mater., 2011, 23, 2819–2822 CrossRef CAS PubMed.
- S. H. Kim, Y. Sun, J. A. Kaplan, M. W. Grinstaff and J. R. Parquette, New J. Chem., 2015, 39, 3225–3228 RSC.
- E. R. Draper, T. O. McDonald and D. J. Adams, Chem. Commun., 2015, 51, 12827–12830 RSC.
- Y. Huang, Y. Zhang, Y. Yuan and W. Cao, Tetrahedron, 2015, 71, 2124–2133 CrossRef CAS.
- K. Ghosh and S. Panja, RSC Adv., 2015, 5, 12094–12099 RSC.
- G.-J. Chen, P.-C. Xue, R. Lu, D.-P. Song, C.-Y. Bao, T.-H. Xu and Y.-Y. Zhao, Chem. Res. Chin. Univ., 2009, 25, 178–182 CAS.
- W. Ji, G. Liu, M. Xu, X. Dou and C. Feng, Chem. Commun., 2014, 50, 15545–15548 RSC.
- H. Yu, H. Mizufune, K. Uenaka, T. Moritoki and H. Koshima, Tetrahedron, 2005, 61, 8932–8938 CrossRef CAS.
- H. D. Becker, Chem. Rev., 1993, 93, 145–172 CrossRef CAS.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC.
- Y. Sako and Y. Takaguchi, Org. Biomol. Chem., 2008, 6, 3843–3847 CAS.
- M. Ayabe, T. Kishida, N. Fujita, K. Sada and S. Shinkai, Org. Biomol. Chem., 2003, 1, 2744–2747 CAS.
- Y. C. Lin, B. Kachar and R. G. Weiss, J. Am. Chem. Soc., 1989, 111, 5542–5551 CrossRef CAS.
- I. Furman and R. G. Weiss, Langmuir, 1993, 9, 2084–2088 CrossRef CAS.
- C. Wang, D. Zhang, J. Xiang and D. Zhu, Langmuir, 2007, 23, 9195–9200 CrossRef CAS PubMed.
- P. Terech, I. Furman and R. G. Weiss, J. Phys. Chem., 1995, 99, 9558–9566 CrossRef CAS.
- A. Dawn, N. Fujita, S. Haraguchi, K. Sada and S. Shinkai, Chem. Commun., 2009, 2100–2102 RSC.
- X. Wang, P. Duan and M. Liu, Chem. – Eur. J., 2013, 19, 16072–16079 CrossRef CAS PubMed.
- K. Inoue, Y. Ono, Y. Kanekiyo, K. Hanabusa and S. Shinkai, Chem. Lett., 1999, 429–430 CrossRef CAS.
- M. A. Markowitz, A. Singh and E. L. Chang, Biochem. Biophys. Res. Commun., 1994, 203, 296–305 CrossRef CAS PubMed.
- N. S. S. Kumar, S. Varghese, G. Narayan and S. Das, Angew. Chem., Int. Ed., 2006, 38, 6317–6321 CrossRef PubMed.
- M. George and R. G. Weiss, Chem. Mater., 2003, 15, 2879–2888 CrossRef CAS.
- M. Masuda, T. Hanada, Y. Okada, K. Yase and T. Shimizu, Macromolecules, 2000, 33, 9233–9238 CrossRef CAS.
- M. Masuda, T. Hanada, K. Yase and T. Shimizu, Macromolecules, 1998, 31, 9403–9405 CrossRef CAS.
- N. Tamaoki, S. Shimada, Y. Okada, A. Belaissaoui, G. Kruk, K. Yase and H. Matsuda, Langmuir, 2000, 16, 7545–7547 CrossRef CAS.
- S. Bhattacharya and S. N. G. Acharya, Chem. Mater., 1999, 11, 3121–3132 CrossRef CAS.
- S. R. Diegelmann, N. Hartman, N. Markovic and J. D. Tovar, J. Am. Chem. Soc., 2012, 134, 2028–2031 CrossRef CAS PubMed.
- S. R. Diegelmann and J. D. Tovar, Macromol. Rapid Commun., 2013, 34, 1343–1350 CrossRef CAS PubMed.
- L. Hsu, G. L. Cvetanovich and S. I. Stupp, J. Am. Chem. Soc., 2008, 130, 3892–3899 CrossRef CAS PubMed.
- D. W. P. M. Löwik, I. O. Shklyarevskiy, L. Ruizendaal, P. C. M. Christianen, J. C. Maan and J. C. M. van Hest, Adv. Mater., 2007, 19, 1191–1195 CrossRef.
- Y. Ding, Y. Li, M. Qin, Y. Cao and W. Wang, Langmuir, 2013, 29, 13299–13306 CrossRef CAS PubMed.
- L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025–17029 CrossRef CAS PubMed.
- T. Muraoka, C.-Y. Koh, H. Cui and S. I. Stupp, Angew. Chem., Int. Ed., 2009, 48, 5946–5949 CrossRef CAS PubMed.
- K. Peng, I. Tomatsu and A. Kros, Chem. Commun., 2010, 46, 4094–4096 RSC.
- T. D. Sargeant, C. Aparicio, J. E. Goldberger, H. Cui and S. I. Stupp, Acta Biomater., 2012, 8, 2456–2465 CrossRef CAS PubMed.
- K. Fan, J. Yang, X. Wang and J. Song, Soft Matter, 2014, 10, 8370–8375 RSC.
- M. Heilemann, E. Margeat, R. Kasper, M. Sauer and P. Tinnefeld, J. Am. Chem. Soc., 2005, 127, 3801–3806 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |