
Designing structurally tunable and functionally versatile synthetic supercontainers†
Feng-Rong
Dai
ab,
Yupu
Qiao
b and
Zhenqiang
Wang
*b
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences Fuzhou, Fujian 350002, China
bDepartment of Chemistry, University of South Dakota, 414 East Clark Street. and Churchill-Haines Laboratories, Room 115, Vermillion, South Dakota 57069-2390, USA. E-mail: Zhenqiang.Wang@usd.edu; Fax: (+1) 605-677-6397
First published on 24th November 2015
Abstract
We describe the design of a new family of molecular containers, namely, type IV metal–organic supercontainers (MOSCs), which are constructed from the assembly of a container precursor p-tert-butylsulfonylcalix[4]arene, Co(II) or Ni(II) ion, and angular flexible dicarboxylate linkers. The combination of structural robustness and tunability of these cylinder-shaped MOSCs makes them highly attractive supramolecular hosts. The type IV MOSCs exhibit a diverse range of supramolecular functions, including their selective binding with cationic guests and their tunable activity to modulate both stoichiometric and catalytic reactions, which are not readily accessible in the molecular precursors.
 Zhenqiang Wang | Zhenqiang ‘Rick’ Wang has been an Assistant Professor in the Department of Chemistry at the University of South Dakota since August 2010. He received his B.S. degree from Peking University in 2000 and his Ph.D. degree from the University of South Florida in 2006. From February 2007 to July 2010, he was a postdoctoral researcher in the laboratory of Prof. Seth Cohen at the University of California, San Diego. He received a National Science Foundation CAREER Award in 2014, and the USD President's Award for Research Excellence (New-Mid Career Faculty) in 2015. His research program at USD focuses on investigating the synthetic and functional application aspects of a new class of supramolecular hosts known as metal–organic supercontainers (MOSCs). In his spare time, he enjoys watching comedy shows and playing soccer pickup games. |
Introduction
Molecular containers that possess well-defined nanoscale space capable of accommodating one or more guest molecules have attracted tremendous interest in recent years1–7 owing to their aesthetic appeal and their applications in a wide array of areas, such as stabilization of highly reactive species,8,9 transportation of small molecules,10 gas storage and separation,11–14 sensing,15,16 and supramolecular catalysis.17–20 Within various types of molecular containers, metal-directed coordination containers are of particular interest, thanks in large part to the reversible yet relatively robust nature of metal–ligand interactions.21,22 Recently, we reported a new class of coordination containers, termed metal–organic supercontainers (MOSCs),23–26 which were assembled from divalent metal ions, sulfonylcalix[4]arene-based container precursors,27,28 and rigid carboxylate ligands. Three prototypes of MOSCs, namely, face-directed octahedra (type I),23 edge-directed octahedra (type II),24,26 and barrel-shaped boxes (type III),25 were obtained by using trigonal, linear, and angular-planar carboxylate linkers, respectively. Related supercontainer structures based on thiacalix[4]arenes were also recently reported by the groups of Liao29,30 and Hong.31 Compared to previously known coordination containers, MOSCs are distinguished by their unique multi-pore architecture, featuring both endo and exo cavities. The presence of multiple remote binding domains in a single synthetic host is expected to provide unprecedented opportunities for designing complex functions, such as binding cooperativity and binding regulation that are widely employed in biology.32We noted that the three prototypes of MOSCs share a common design element, i.e., their formation is strictly dictated by the geometry of the carboxylate linkers and their structures are relatively rigid.33 Specifically, the assembly of type I, II, and III MOSCs is largely predetermined by the carboxylate linker possessing a C3 symmetry, a linear (i.e., 180°) geometry, and an angular-planar geometry subtending an angle of 120°, respectively. While such strict geometrical requirements permit precise construction of targeted supramolecular entities from molecular precursors, the resulting MOSCs are compositionally less flexible and limited in the scope of physical and chemical functionalities that can be accessed.
We contemplated that a design strategy that necessitates less demanding geometrical constraints would accommodate a wider range of chemical moieties and lead to structurally tunable and functionally versatile MOSCs. Herein, we describe the assembly of a new family of coordination containers, namely, type IV MOSCs, which are built from two tetranuclear units34 – each containing four divalent metal ions, one sulfonylcalix[4]arene, and one μ4-oxygen species – bridged by four structurally flexible dicarboxylate linkers (Scheme 1). We show that the high directionality of the tetranuclear units allows the construction of a family of cylinder-shaped MOSCs from a series of angular flexible dibenzoate ligands sustaining a methylene (–CH2–; L1), methyleneamino (–CH2–NH–; L2), or bismethyleneamino (–CH2–NH–CH2–; L3) spacer, providing the desired, but otherwise elusive, structural flexibility and functional versatility. We demonstrate that these MOSCs preferentially bind with positively charged guests, which can be utilized to separate cationic dyes from anionic dyes. Most importantly, the type IV MOSCs exhibit supramolecular reactivity not accessible in the molecular precursors, including their capacity to stoichiometrically modulate the isomerization of Rhodamine B35–37 and catalytically mediate Knoevenagel condensation.38,39 We further show that the capacity of the MOSCs to regulate chemical transformations can be judiciously tuned through manipulation of the carboxylate spacer, thus providing opportunities for designing novel catalytic reactivity.
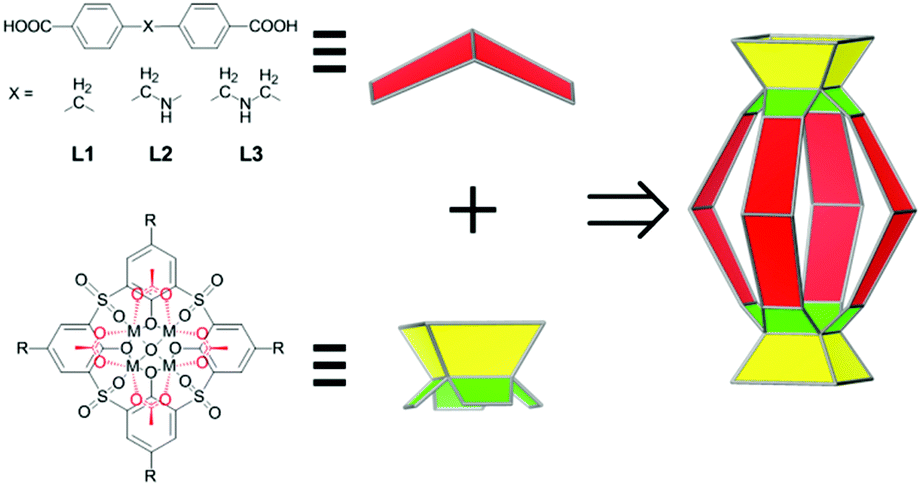 | ||
| Scheme 1 Assembly of type IV metal–organic supercontainers. | ||
Results and discussion
Synthesis and characterisation of type IV MOSCs
Three new MOSCs, namely, 1-Co, 2-Co, and 3-Ni, were obtained as single-crystalline products from the reactions of Co(II) or Ni(II) and p-tert-butylsulfonylcalix[4]arene (TBSC) with three non-rigid dicarboxylate linkers, i.e., L1, L2, and L3, respectively. Structural analysis revealed that these type IV MOSCs share a cylindrical container topology which consists of two tetranuclear units bridged by four dicarboxylate linkers that adopt a V-shaped geometry (Fig. 1). The tetranuclear units (Scheme 1), each possessing a μ4-oxygen presumed to be from a neutral water molecule, closely resemble those in the previously reported MOSCs.23–26 It is worth noting that although the linear length of the three dicarboxylate linkers increases in the order of L1 < L2 < L3, the longitudinal diameter of the cylindrical endo cavity of the resulting MOSCs shows a different trend in the solid state, following the order of 2-Co < 1-Co < 3-Ni, with the O⋯O distance between the opposing μ4-H2O molecules reaching 7.28, 8.94 and 11.04 Å (excluding the sum of the atomic van der Waals radii), respectively. This is attributed to the structural flexibility of the dicarboxylate linkers, among which ligand L2 assumes the most pronounced bending conformation. As a result, the lateral diameter of the endo cavity of 2-Co becomes the widest among the three MOSCs (Fig. 1). Owing to the non-symmetric nature of the –CH2NH– spacer in L2, 2-Co may in principle exist as several different isomers with its four N atoms arranged in either up or down configuration. This unfortunately cannot be determined by crystallographic analysis due to the high symmetry of the crystal structure. These interesting characteristics convey several important connotations that are of particular significance for designing functional versatility in MOSCs: (1) compared to other prototypes of MOSCs, the type IV congeners are indeed tolerant of a wider range of ligand conformations; (2) an array of chemical moieties are in principle compatible with and can thus be incorporated into type IV MOSCs; (3) the conformational freedom of the dicarboxylate linkers can potentially be utilized to regulate the molecular recognition capability of the MOSCs.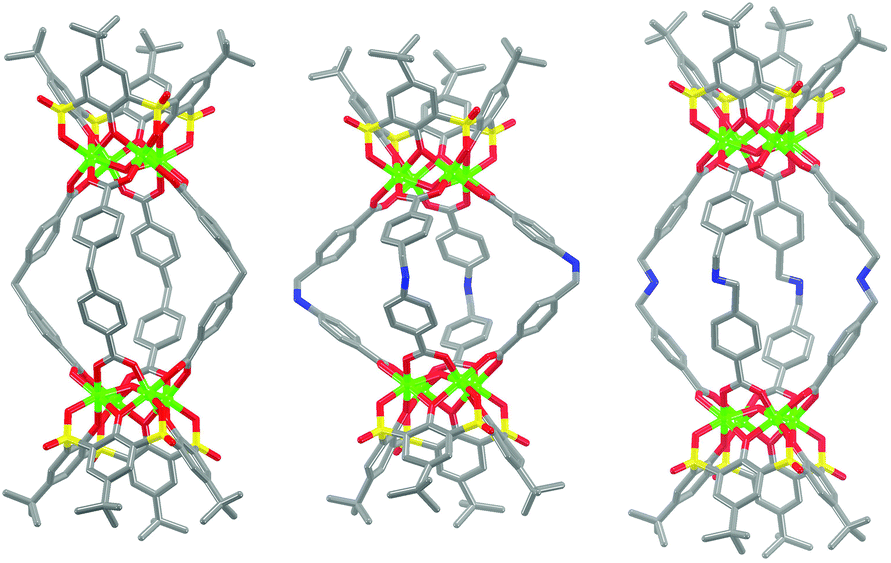 | ||
| Fig. 1 Structural representation of type IV MOSCs: (left) 1-Co (X = –CH2–), (middle) 2-Co (X = –CH2–NH–), and (right) 3-Ni (X = –CH2–NH–CH2–). Color scheme: Co/Ni, green; S, yellow; O, red; C, gray; N, blue. Hydrogen atoms are deleted for clarity. | ||
The trademark structural feature of MOSCs, i.e., the presence of both endo and exo cavities, is well retained in type IV MOSCs. Indeed, the X-ray crystal structure of 1-Co clearly illustrates its capacity to utilize both binding domains for encapsulating guest species. In the as-synthesized 1-Co crystal, a pair of N,N-dimethylformamide (DMF) molecules are trapped inside the endo cavity, while another pair of DMF molecules nestle inside the ‘super’ exo cavity formed through hydrophobic interactions among the tert-butyl groups of two TBSC units from adjacent MOSCs, resulting in an unusual one-dimensional (1D) tubular structure (Fig. 2 and S4†). Four additional DMF molecules exist outside the MOSC and 1-Co can thus be formulated as {[Co4(TBSC)(μ4-H2O)]2(L1)4(DMF)2,endo(DMF)2,exo}·4DMF. A similar tubular architecture is also observed in the crystal structure of 3-Ni (Fig. S5†), although in this case, the guest species cannot be precisely located by X-ray crystallography due to the relatively poor diffraction quality of the crystals. Interestingly, the solid-state packing of 2-Co does not follow this same pattern, possibly due to its more bending conformation, which would have rendered the tubular packing less efficient; instead, it adopts the classic body-centered cubic packing (Fig. S6†).
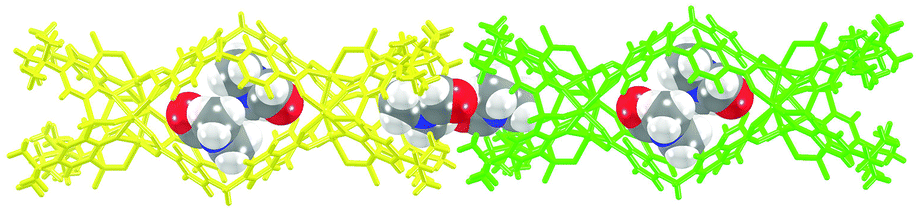 | ||
| Fig. 2 One-dimensional tubular assembly of 1-Co with the encapsulation of DMF guest molecules in both endo and exo cavities. 1-Co molecules (yellow and green) are represented in stick mode, while DMF molecules are represented in spacefilling mode. | ||
To affirm the utility of type IV MOSCs as supramolecular hosts, their compositional purity and structural robustness were examined in a number of ways. Thermogravimetric analysis (TGA) revealed that under a N2 atmosphere, 1-Co, 2-Co, and 3-Ni do not decompose until above 400 °C (Fig. S7†). Elemental analysis results suggested reasonable purity of the compounds (Table S2†). Gas adsorption analysis indicated that the MOSCs show interesting solid-state porosity and exhibit intriguing O2/N2 and CO2/N2 selectivity (Fig. S8–S13†), similar to what was previously reported for type II MOSCs, which was attributed to a size-dependent packing collapse mechanism.24 Mass spectrometry analysis suggested that the MOSCs, upon dissolution in chloroform (with moderate to good solubility in the range of 10−6 M–10−4 M), remain structurally intact (Fig. S14–S16 and Table S3†). Together, these results confirmed the chemical purity and stability of these new MOSCs and the feasibility of probing their tunable functional properties in the solution phase.
Selective binding with cationic guests
We thus selected three organic dyes (Scheme 2), namely, methylene blue (MB), eosin Y (EY), and methyl orange (MO), as representative guests to probe the molecular recognition capability of type IV MOSCs. We first examined the host–guest chemistry at the liquid–liquid interface, as it has proven to be a useful platform for providing valuable guest-binding information.24 Two immiscible solvents, i.e., water and chloroform, were chosen to afford the liquid–liquid interface due to the distinct solubility profiles of the hosts and the guests in these two solvents – while the dyes dissolve predominantly in an aqueous solution, the MOSCs are only soluble in chloroform. In the absence of MOSCs, chloroform extracted only a tiny fraction of MB and an even more negligible amount of EY or MO from the aqueous phase, as revealed by the ultraviolet-visible (UV-vis) spectra taken of the chloroform solutions after the extraction (Fig. S17†). However, when as little as 0.1 equivalent of a type IV MOSC, such as 1-Co, was added to the chloroform solution, the water-soluble MB molecules were immediately coaxed into the organic phase upon shaking and mixing of the two layers (Fig. S18†). This stark contrast is attributed to, and a vivid indication of, the robust binding of 1-Co with MB, which drives the partitioning equilibrium of the dye almost completely toward the chloroform phase, despite its otherwise insignificant chloroform solubility. | ||
| Scheme 2 Structures of three organic dyes used in this study. | ||
The favorable binding of 1-Co with MB was further validated by UV-vis titration experiments,40 which revealed a notable red-shift of the MOSC's absorption maxima (initially at ∼348 nm) upon gradual increase of the MB equivalents in chloroform (Fig. S19†); fitting the titration data to the Benesi–Hildebrand (B–H) equation41 gave rise to an apparent binding constant of (2.18 ± 0.20) × 104 (Fig. S20†), comparable to those found for type II MOSCs.24 The MB/1-Co ratio at equilibrium in chloroform was estimated to be ∼3 by comparing the MB concentrations (determined by UV-vis analysis) in the aqueous phase before and after the extraction (Table S4†). This binding equivalent suggests that the MOSC is likely interacting with two molecules of MB through its two exo cavities (i.e., one MB per cavity) and another through its endo cavity. Interestingly, 1-Co extracted neither EY nor MO in any substantial capacity under otherwise identical conditions, underlining an important bias against these two dyes (Fig. S21†). This can in principle be due to a size effect (i.e., EY, if not MO, is bulkier than MB), or a charge effect (i.e., EY and MO are anionic, whereas MB is cationic); we attribute the observed binding selectivity to the cationic nature of MB, since in control experiments, another cationic dye, Rhodamine B (vide infra), which is comparable in size to EY, can be efficiently extracted by 1-Co (Fig. S22†). Similar binding selectivity favoring cationic guests was also apparent for 2-Co, and to a lesser extent, 3-Ni42 (Fig. S23 and 24†). These results thus suggest that the electrostatic interactions between positively charged guests and electronegative oxygen atoms abundantly present in type IV MOSCs may play an important role in modulating their guest-binding affinity. A perhaps even more plausible factor contributing to the observed binding selectivity is the now widely-recognized ‘cation–π’ interaction,43 which would involve the aromatic moieties of the MOSCs interacting with the positive charges of the dyes.
Separation of dye mixtures
Encouraged by the above intriguing findings, we decided to determine whether the binding selectivity of type IV MOSCs can be utilized to separate cation–anion mixtures. Thus, two aqueous solutions containing a mixture of MB–EY (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and MB–MO (1:1), respectively, were prepared. While the color of the individual dye solutions appeared as blue (MB), orange (EY), or yellow (MO), the two mixture solutions emerged in purple (MB–EY) or green (MB–MO) color (Fig. S25†). It was noteworthy that the chloroform solubility of the dyes in the form of cation–anion mixtures appeared to be enhanced even in the absence of any MOSC (Fig. S26†), presumably due to the formation of neutral ion-pairs that are inherently more soluble in low-polarity solvents such as chloroform.
:1) and MB–MO (1:1), respectively, were prepared. While the color of the individual dye solutions appeared as blue (MB), orange (EY), or yellow (MO), the two mixture solutions emerged in purple (MB–EY) or green (MB–MO) color (Fig. S25†). It was noteworthy that the chloroform solubility of the dyes in the form of cation–anion mixtures appeared to be enhanced even in the absence of any MOSC (Fig. S26†), presumably due to the formation of neutral ion-pairs that are inherently more soluble in low-polarity solvents such as chloroform.
The separation experiments were carried out at the liquid–liquid interface with the dye mixture initially dissolved in the aqueous phase and the MOSC in the chloroform phase. Taking the MB–EY mixture as an example, in the presence of 1-Co (<0.6 equiv.) and upon shaking and allowing the two layers to fully separate, the color of the aqueous phase changed from purple to orange, whereas the chloroform phase concurrently turned into blue (Fig. 3a and S27†). These color changes clearly indicate the extraction of MB into the chloroform phase by 1-Co and the retention of EY in the aqueous phase, giving rise to effective separation of the two dyes.
 | ||
| Fig. 3 (a) Liquid–liquid extraction of methylene blue (MB)–eosin Y (EY) mixture in the absence (left) and presence (right) of 1-Co. Top layer: aqueous solution; bottom layer: CHCl3 solution. The MB–EY mixture was initially dissolved in the aqueous solution and 1-Co was dissolved in CHCl3. (b) Red curve: UV-vis spectrum of initial MB–EY mixture in aqueous solution; black curve: UV-vis spectrum of the aqueous phase after extraction by 1-Co; blue curve: UV-vis spectrum of the CHCl3 phase after extraction by 1-Co. | ||
The separation was further validated by UV-vis measurements. Prior to the extraction, the aqueous solution of the dye mixture showed two major absorption bands centered at 517 nm and 665 nm, attributed to EY and MB, respectively (Fig. 3b, red curve). Upon extraction, however, the absorption maximum at 665 nm disappeared from the aqueous phase (Fig. 3b, black curve), whereas an intense absorption band centered at 661 nm appeared in the chloroform phase (Fig. 3b, blue curve); meanwhile, the absorption maximum at 517 nm characteristic of EY in the aqueous solution remained largely unaltered (Fig. 3b, black curve). This is in sharp contrast to the liquid–liquid extraction of the same MB–EY mixture in the absence of MOSC, where both MB and EY were partially extracted to the chloroform phase, presumably as an ion pair, as indicated by the appearance of two weak but well-defined absorption bands centered at 519 nm (EY) and 654 nm (MB), respectively (Fig. S26†). These UV-vis studies thus unambiguously confirmed that MB was fully extracted into the chloroform phase by 1-Co and EY largely remained in the aqueous phase. Additionally, liquid–liquid separation was also successfully demonstrated for the MB–MO mixture using 1-Co as the extractant (Fig. S27†); efficient separation of the mixtures was achieved in a similar manner when 2-Co was used in place of 1-Co (Fig. S28†). These results together point to the promising potential of the type IV MOSCs for separation applications.
Stoichiometric modulation of Rhodamine B isomerization
In addition to exhibiting binding selectivity toward cationic guests, type IV MOSCs can also serve as ‘molecular flasks’44 for modulating chemical transformations. This was illustrated by their capacity to promote both stoichiometric and catalytic reactions, such as Rhodamine B isomerization and Knoevenagel condensation, respectively. During the course of examining Rhodamine B as a control guest as described above, it became apparent to us that the MOSCs were capable of mediating the otherwise well-documented intramolecular isomerization of the dye, which involves interconversion between the ring-opened, pink-colored cationic form (RB) and the ring-closed, colorless, neutral lactone form (known as Rhodamine B base, RBB) (Scheme 3).35–37 The isomerization is known to be influenced by several experimental parameters, including the polarity and pH of the solvent that the dye dissolves in. Specifically, higher basicity or lower polarity favors the RBB form, whereas lower basicity or higher polarity promotes the RB form. In an aqueous solution (i.e., a polar environment), the dye shows an intense pink color and strong absorption in the visible range (∼552 nm), characteristic of the cationic RB form. At the liquid–liquid (H2O–CHCl3) interface and in the absence of any MOSC, the RB form was found to remain in the aqueous phase, but only for a short amount of time (Fig. S29†). If the two layers were allowed to stay for a longer period, the intense pink color of the aqueous phase eventually faded and a very light pink color became visible in the chloroform phase (Fig. S30†). We ascribe this to the effect of the weakly polar chloroform inducing a gradual conversion of the cationic RB form to the neutral, colorless RBB form. The latter is substantially more stable and soluble in chloroform than in aqueous solution, thus driving the partition equilibrium of the dye toward the organic phase. The light pink color of the chloroform phase is due to the small percentage of the RB form in equilibrium with the RBB form (Fig. S30†). | ||
| Scheme 3 Intramolecular isomerization of Rhodamine B dye. | ||
When 1-Co was added to the chloroform phase, the color of the aqueous phase quickly faded but a rather intense pink color vividly appeared in chloroform (Fig. S31†), suggesting that the RB form was not only extracted into the chloroform phase (as described above), but it also remained predominantly as the colored, ring-opened, cationic form, despite the low polarity of chloroform which would have otherwise favored the colorless RBB form. This observation was further corroborated by UV-vis measurements taken of the chloroform phase after the extraction, which revealed a significantly increased intensity of the absorption maximum at ∼547 nm, characteristic of the RB form, compared to that of the chloroform phase in the absence of any MOSC (Fig. S32†). A similar effect was also observed when 2-Co or 3-Ni42 was present in the chloroform phase (Fig. S32†). These findings suggest that type IV MOSCs can indeed stabilize the RB form in solvents of low polarity (e.g., chloroform), most likely via encapsulating the dye molecule within their nanocavities.
To further interrogate the role that the MOSCs play in modulating the Rhodamine B isomerization, we carried out UV-vis titration experiments in homogeneous solutions, where an increasing amount of 1-Co was gradually added to a chloroform solution of the ring-closed RBB form. Upon the addition of 1-Co, the UV-vis absorption due to the ring-opened RB form quickly grew in intensity, approaching saturation upon reaching a [MOSC]/[RBB] molar ratio of one (Fig. 4 and S33†). Titrating the RBB solution with 2-Co similarly led to the formation of the RB form, albeit with a lower efficiency (Fig. 4 and S34†). Notably, when the container precursor TBSC was added as the titrant, no obvious isomerization beyond the background reaction (i.e., when chloroform was titrated instead) was observed (Fig. 4 and S35†). These findings strongly indicate that the modulation of Rhodamine B isomerization by type IV MOSCs mostly likely occurs inside the endo cavity. Furthermore, the MOSC-mediated conversion of RBB to RB in chloroform was found to be partially reversed by adding MB to the solution, as indicated by a decrease in the UV-vis absorbance attributed to the RB form (Fig. S37†). This can be explained on the basis that some of the RB cations were displaced by the competing, cationic MB molecules from the endo cavity, released into the bulk solvent, and subsequently converted back to the RBB form. In contrast, addition of MB to a TBSC-saturated Rhodamine B solution caused little change to the equilibrium (Fig. S37†). Extensive efforts have been devoted to growing single crystals of the host–guest complexes; unfortunately, weak diffraction quality of the crystals obtained has hampered an in-depth crystallographic analysis. We are currently investigating the exact mechanism of this interesting supramolecular reactivity in a number of ways, the findings of which will be communicated in due course, but several preliminary assessments can be made here: (1) we postulate that the active sites directly responsible for mediating the Rhodamine B isomerization involve the μ4-H2O located at the lower rim of the TBSC units and inside the endo cavity of the MOSC. Coordination to four divalent metal ions makes the H2O molecule a stronger Brønsted acid than usual and thus capable of releasing a proton to drive the formation of the RB form; (2) the chemical micro-environment of the binding cavity further furnishes the supramolecular reactivity. In this vein, the lower reactivity of 2-Co may be due to its more distorted endo cavity and/or its methyleneamino moieties competing for the proton; (3) the MOSC-mediated isomerization is stoichiometric and is likely a result of the MOSC's stronger binding affinity (vide supra)24 with the product (i.e., the cationic RB form) than with the substrate (i.e., the neutral RBB form), which translates to no catalytic turnover.
 | ||
| Fig. 4 Fraction of the ring-opened RB form converted from the ring-closed RBB form as a function of the titrant equivalent when a chloroform solution of the RBB form was gradually titrated with 1-Co, 2-Co, TBSC, or chloroform. The results are based on the average of three independent trials for each titrant. | ||
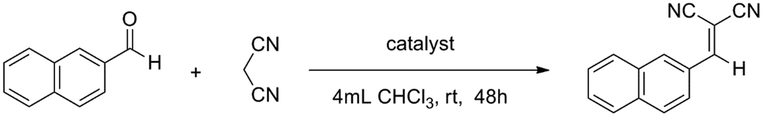
Catalytic mediation of Knoevenagel condensation
To gauge the MOSCs’ capacity to facilitate truly catalytic reactions, we examined the classic Knoevenagel condensation, a well-known C–C bond forming transformation involving a nucleophilic addition of an active hydrogen-containing compound to a carbonyl substrate, often assisted by a Lewis-base catalyst.38,39 The ease with which the methylene group in 1-Co can be modified into the methyleneamino moiety in 2-Co or the bismethyleneamino moiety in 3-Ni, without affecting the overall supercontainer architecture, makes type IV MOSCs particularly attractive as Lewis-base supramolecular catalysts.To test their catalytic activity, a chloroform solution of 2-naphthaldehyde and malononitrile was treated with 1 mol% of 1-Co, 2-Co, and 3-Ni, 4 mol% of L3 methyl diester (Me2L3), 2 mol% of TBSC, and no catalyst, respectively, under ambient conditions for 48 h. The results revealed that TBSC and 1-Co showed negligible reactivity for the reaction (Table 1), which was not unexpected, since they were not considered to contain any basic functionality. Surprisingly, despite the known Lewis basicity of its methyleneamino moieties, 2-Co manifested only a very modest activity, achieving an unassuming 14% yield. In contrast, 3-Ni was found to be a highly effective catalyst, promoting the Knoevenagel reaction nearly quantitatively (92% yield), presumably because of its stronger Lewis basicity in its bismethyleneamino groups, an aliphatic secondary amine. However, it is remarkable to note that an equivalent amount of L3 methyl diester, which contains the same aliphatic secondary amine group, gave rise to a mere 19% yield, strongly indicating the importance of the ‘cavity effect’. These findings thus highlight the exciting possibility of tuning the functional versatility of type IV MOSCs by virtue of their structural flexibility and robustness, and most importantly, the myriad of opportunities for designing novel supramolecular reactivity,45,46 an aspect we are currently actively pursuing in our laboratory.
|
|
||||||
|---|---|---|---|---|---|---|
| Catalyst: | None | TBSC | Me 2 L3 | 1-Co | 2-Co | 3-Ni |
| a Reaction conditions: 2-naphthaldehyde (0.40 mmol), malononitrile (0.40 mmol), and catalyst (1 mol% of MOSC, 4 mol% of Me2L3, or 2 mol% of TBSC) in CHCl3 (4.0 mL) at room temperature for 48 h. b Isolated yields based on two independent trials. | ||||||
| Yieldb: | 2% | 2% | 19% | 5% | 14% | 92% |
Conclusions
In summary, we have demonstrated the design and synthesis of a new family of synthetic hosts, namely, type IV MOSCs, which are shown to be structurally tunable and functionally diverse. The less stringent geometrical requirements of their design and the high chemical stability of their structure render these new supercontainers uniquely suited as ‘nano-capsules’ for modulating a wide range of molecular recognition events. The cavity-mediated binding selectivity and supramolecular reactivity of these MOSCs, which can be rationally tuned from bottom-up, provide hope for designing truly biomimetic functions, particularly in the context of molecular separation and supramolecular catalysis. We are currently addressing the exciting opportunities along these lines.Acknowledgements
This work was supported by an NSF CAREER Award (CHE 1352279) and a South Dakota Board of Regents Competitive Research Grant. The authors also acknowledge an NSF MRI Award (CHE 1229035) for the purchase of a Bruker 400 MHz NMR spectrometer.Notes and references
- D. J. Cram and J. M. Cram, Container Molecules and Their Guests, The Royal Society of Chemistry, Cambridge, England, 1997 Search PubMed.
- N. Branda, R. Wyler and J. Rebek, Science, 1994, 263, 1267–1268 CAS.
- M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469–471 CrossRef CAS.
- L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS.
- D. L. Caulder, R. E. Powers, T. N. Parac and K. N. Raymond, Angew. Chem., Int. Ed., 1998, 37, 1840–1843 CrossRef CAS.
- B. Olenyuk, J. A. Whiteford, A. Fechtenkötter and P. J. Stang, Nature, 1999, 398, 796–799 CrossRef CAS PubMed.
- P. Ballester, M. Fujita and J. Rebek Jr., Chem. Soc. Rev., 2015, 44, 392–393 RSC.
- D. J. Cram, M. E. Tanner and R. Thomas, Angew. Chem., Int. Ed. Engl., 1991, 30, 1024–1027 CrossRef.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed.
- B. Therrien, Top. Curr. Chem., 2012, 319, 35–55 CrossRef CAS PubMed.
- J. An, R. P. Fiorella, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8401–8403 CrossRef CAS PubMed.
- J. R. Li and H. C. Zhou, Nat. Chem., 2010, 2, 893–898 CrossRef CAS PubMed.
- T. Mitra, X. Wu, R. Clowes, J. T. A. Jones, K. E. Jelfs, D. J. Adams, A. Trewin, J. Bacsa, A. Steiner and A. I. Cooper, Chem. – Eur. J., 2011, 17, 10235–10240 CrossRef CAS PubMed.
- A. Avellaneda, P. Valente, A. Burgun, J. D. Evans, A. W. Markwell-Heys, D. Rankine, D. J. Nielsen, M. R. Hill, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2013, 52, 3746–3749 CrossRef CAS PubMed.
- R. Pinalli and E. Dalcanale, Acc. Chem. Res., 2013, 46, 399–411 CrossRef CAS PubMed.
- Y. Liu, X. A. Wu, C. He, Z. Y. Li and C. Y. Duan, Dalton Trans., 2010, 39, 7727–7732 RSC.
- B. C. Pemberton, R. Raghunathan, S. Volla and J. Sivaguru, Chem. – Eur. J., 2012, 18, 12178–12190 CrossRef CAS PubMed.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS PubMed.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- S. H. Leenders, R. Gramage-Doria, B. de Bruin and J. N. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- P. J. Stang, J. Am. Chem. Soc., 2012, 134, 11829–11830 CrossRef CAS PubMed.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- F.-R. Dai and Z. Wang, J. Am. Chem. Soc., 2012, 134, 8002–8005 CrossRef CAS PubMed.
- F.-R. Dai, U. Sambasivam, A. J. Hammerstrom and Z. Wang, J. Am. Chem. Soc., 2014, 136, 7480–7491 CrossRef CAS PubMed.
- F.-R. Dai, D. C. Becht and Z. Wang, Chem. Commun., 2014, 50, 5385–5387 RSC.
- N. L. Netzer, F.-R. Dai, Z. Wang and C. Jiang, Angew. Chem., Int. Ed., 2014, 53, 10965–10969 CrossRef CAS PubMed.
- H. Kumagai, M. Hasegawa, S. Miyanari, Y. Sugawa, Y. Sato, T. Hori, S. Ueda, H. Kamiyama and S. Miyano, Tetrahedron Lett., 1997, 38, 3971–3972 CrossRef CAS.
- N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291–5316 CrossRef CAS PubMed.
- M. Liu, W. P. Liao, C. H. Hu, S. C. Du and H. J. Zhang, Angew. Chem., Int. Ed., 2012, 51, 1585–1588 CrossRef CAS PubMed.
- Y. Bi, S. Du and W. Liao, Coord. Chem. Rev., 2014, 276, 61–72 CrossRef CAS.
- K. Xiong, F. Jiang, Y. Gai, D. Yuan, L. Chen, M. Wu, K. Su and M. Hong, Chem. Sci., 2012, 3, 2321–2325 RSC.
- B. Alberts, A. Johnson, J. Lewis, D. Morgan, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, 6th edn, 2015 Search PubMed.
- Q. F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS PubMed.
- T. Kajiwara, T. Kobashi, R. Shinagawa, T. Ito, S. Takaishi, M. Yamashita and N. Iki, Eur. J. Inorg. Chem., 2006, 1765–1770 CrossRef CAS.
- R. W. Ramette and E. B. Sandell, J. Am. Chem. Soc., 1956, 78, 4872–4878 CrossRef CAS.
- U. K. A. Klein and F. W. Hafner, Chem. Phys. Lett., 1976, 43, 141–145 CrossRef CAS.
- I. Rosenthal, P. Peretz and K. A. Muszkat, J. Phys. Chem., 1979, 83, 350–353 CrossRef CAS.
- E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1898, 31, 2596–2619 CrossRef CAS.
- B. List, Angew. Chem., Int. Ed., 2010, 49, 1730–1734 CrossRef CAS PubMed.
- C. A. Schalley, Analytical Methods in Supramolecular Chemistry, Wiley, 2012 Search PubMed.
- H. A. Benesi and J. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- The less pronounced effect observed for 3-Ni is attributed to its lower solubility in chloroform than 1-Co and 2-Co.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- M. Zhao, S. Ou and C. D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedures, characterisation data, and spectroscopic titrations. CCDC 1422947–1422949. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00212e |
| This journal is © the Partner Organisations 2016 |